
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManganese-catalyzed base-free addition of saturated nitriles to unsaturated nitriles by template catalysis†
Subramanian
Thiyagarajan
 a,
Yael
Diskin-Posner
b,
Michael
Montag
a and
David
Milstein
*a
a,
Yael
Diskin-Posner
b,
Michael
Montag
a and
David
Milstein
*a
aDepartment of Molecular Chemistry and Materials Science, Weizmann Institute of Science, Rehovot 7610001, Israel. E-mail: david.milstein@weizmann.ac.il
bDepartment of Chemical Research Support, Weizmann Institute of Science, Rehovot 7610001, Israel
First published on 16th January 2024
Abstract
The coupling of mononitriles into dinitriles is a desirable strategy, given the prevalence of nitrile compounds and the synthetic and industrial utility of dinitriles. Herein, we present an atom-economical approach for the heteroaddition of saturated nitriles to α,β- and β,γ-unsaturated mononitriles to generate glutaronitrile derivatives using a catalyst based on earth-abundant manganese. A broad range of such saturated and unsaturated nitriles were found to undergo facile heteroaddition with excellent functional group tolerance, in a reaction that proceeds under mild and base-free conditions using low catalyst loading. Mechanistic studies showed that this unique transformation takes place through a template-type pathway involving an enamido complex intermediate, which is generated by addition of a saturated nitrile to the catalyst, and acts as a nucleophile for Michael addition to unsaturated nitriles. This work represents a new application of template catalysis for C–C bond formation.
Introduction
Nitriles are fundamental building blocks in organic synthesis, and are ubiquitous in natural products, biologically active compounds and pharmaceuticals.1 Moreover, they are invaluable intermediates in present-day chemical industry, used for the synthesis of amines, imines, amides, aldehydes, ketones, and carboxylic acid derivatives.2,3 Dinitriles are employed as primary precursors in the polymer industry, but are also used as building blocks for fine chemicals.4 For instance, the linear dinitrile glutaronitrile is an attractive intermediate for the synthesis of substituted N-heterocycles, such as piperidines, pyridines, and other amine derivatives.5 Furthermore, dinitrile compounds can be easily converted into diamines, diesters and diamides, as well as lactones, lactams, and cyclic imides.6 Nevertheless, the synthesis of dinitriles, including glutaronitrile and its substituted variants, normally requires the use of toxic cyanide or multistep processes that involve harsh conditions.Michael addition, which is a well-known type of conjugate addition reaction,7 is an established synthetic tool for generating C–C bonds.8 Nevertheless, this kind of reaction typically requires the in situ generation of carbon nucleophiles (Michael donors) through the use of strong bases, which may be incompatible with sensitive functional groups and can lead to undesired side products.9 Transition-metal-catalyzed variants of Michael-type reactions have been receiving growing attention due to their excellent selectivity and efficiency.10 Catalytic base-free Michael additions involving nitriles were pioneered in the late 1980's by Murahashi and coworkers, who reported a series of ruthenium-catalyzed reactions of activated nitrile nucleophiles featuring acidic α-methylene and α-methine groups (i.e., active methylene nitriles).11 Since then, other examples of transition-metal-mediated Michael-type reactions of activated nitriles have been reported.12 However, these processes required the use of bases or other additives, and were conducted at high temperatures. In 2013, we made our first contribution to this field, when we reported the catalytic Michael addition of benzyl cyanides to α,β-unsaturated carbonyl compounds, promoted by a pincer-type rhenium complex capable of metal–ligand cooperation (MLC), and carried out under base-free and mild conditions.13
The phenomenon of MLC, wherein both the metal center and ligand of a given complex are directly involved in bond activation, has opened new opportunities for selective activation of chemical bonds and has led to the discovery of new catalytic reactions that are atom-economical and environmentally-benign.14 Significant advances in this field have been achieved by our group, primarily using transition metal complexes of pyridine-based PNP- and PNN-type pincer ligands that exhibit aromatization/dearomatization of the pincer backbone, thereby enabling the cleavage of strong chemical bonds.14,15 Such was the reactivity of the aforementioned rhenium complex, which bears a PNP-type ligand and can activate the nitrile C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond via reversible C–C coupling with the pincer backbone and simultaneous M–N coordination to the metal center.13 The activated nitrile was found to be susceptible to electrophilic attack, either at its nitrogen or α-carbon atom, while the metal-pincer framework serves as an anchor, or template, for the nitrile substrate. This mode of activation, which we have termed “template catalysis”, has enabled the catalytic substitution of nitriles at their α-positions.16 Otten, de Vries and coworkers have also developed similar nitrile-activating systems employing pyridine-based PNP- and PNN-ruthenium pincer complexes.17
N bond via reversible C–C coupling with the pincer backbone and simultaneous M–N coordination to the metal center.13 The activated nitrile was found to be susceptible to electrophilic attack, either at its nitrogen or α-carbon atom, while the metal-pincer framework serves as an anchor, or template, for the nitrile substrate. This mode of activation, which we have termed “template catalysis”, has enabled the catalytic substitution of nitriles at their α-positions.16 Otten, de Vries and coworkers have also developed similar nitrile-activating systems employing pyridine-based PNP- and PNN-ruthenium pincer complexes.17
The application of earth-abundant transition metal catalysts in organic synthesis has been gradually expanding, in light of the high natural prevalence and low cost of these elements, as compared to noble metals – the long-established workhorses of catalysis – like the abovementioned ruthenium.18,19 Manganese, a widely-used, inexpensive base metal, caught our attention nearly a decade ago, and we have since developed a series of PNP- and PNN-type complexes of this metal, which have shown unique catalytic activity based on MLC.19 This includes template catalysis, which allowed us to accomplish a variety of new transformations involving nitriles, namely, Michael addition of unactivated nitriles to α,β-unsaturated carbonyl compounds,16 oxa- and aza-Michael additions to unsaturated nitriles,20 and hydration and α-deuteration of nitriles21 (Scheme 1A and B).
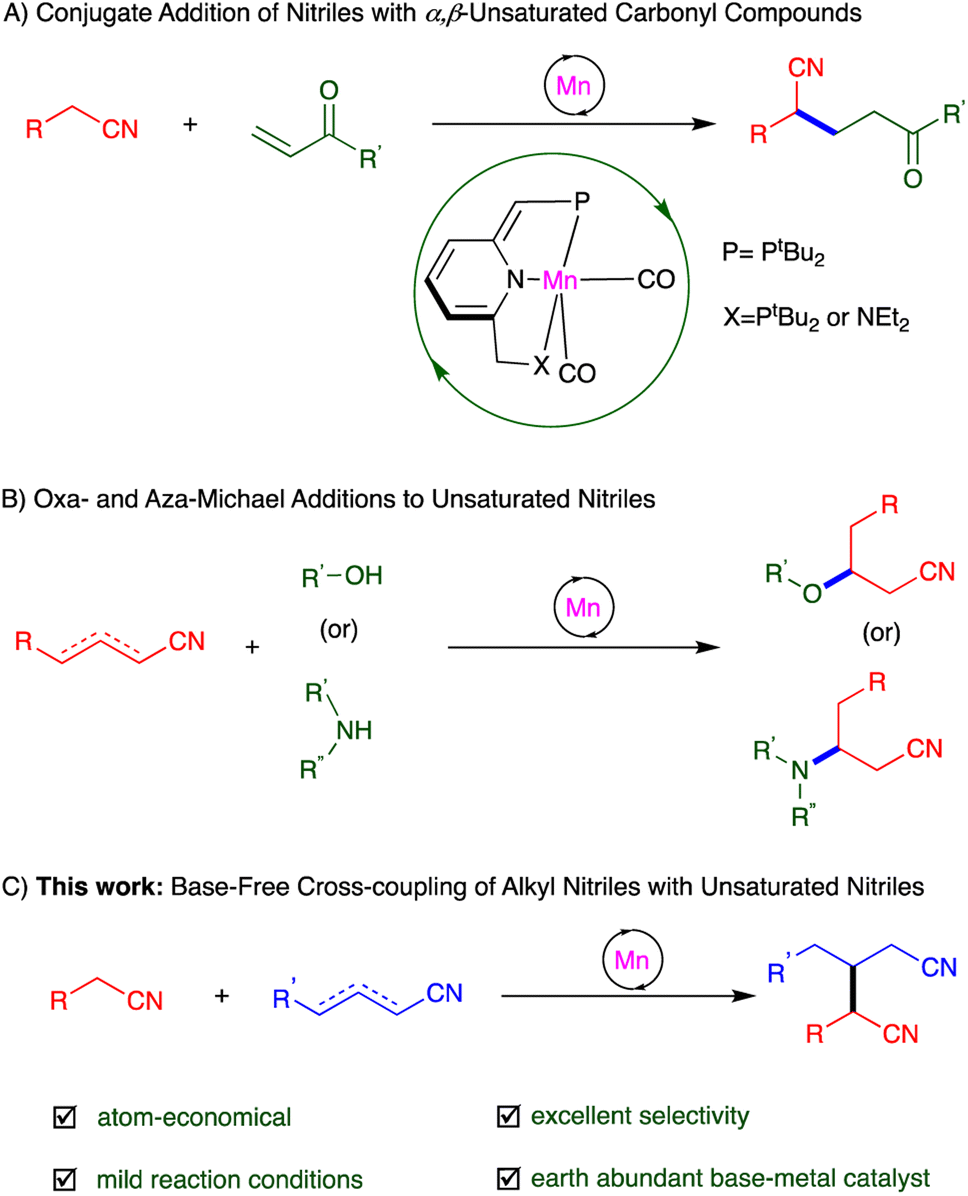 | ||
| Scheme 1 Addition reactions involving nitriles promoted by manganese pincer complexes through template catalysis. | ||
Michael additions are typically carried out using stoichiometric amounts of strong bases, which are necessary for generating Michael donors in situ, but these bases are incompatible with many functional groups, as noted above.10,12,22 Moreover, the electrophilic addition partners (Michael acceptors) used in such reactions have thus far been largely limited to α,β-unsaturated ketones, esters, amides and nitro compounds.10 By contrast, α,β- and β,γ-unsaturated nitriles have rarely been reported as Michael acceptors, because such nitriles are prone to side reactions like polymerization and self-addition.9,17b Hence, directly using unsaturated nitriles for selective C–C bond formation via Michael addition reactions is a highly challenging task. Herein, we report the synthesis of glutaronitriles through direct addition of unactivated saturated nitriles to α,β- and β,γ-unsaturated nitriles, catalyzed by a PNP-manganese pincer complex under very mild, neutral conditions (Scheme 1C). This catalytic system, which operates in the absence of base, was shown to preserve base-sensitive functional groups, and afforded a variety of dinitriles in generally good to excellent selectivity and yield. To the best of our knowledge, such base-free catalytic heteroaddition of saturated nitriles to unsaturated ones to generate dinitriles has not been previously documented.
Results and discussion
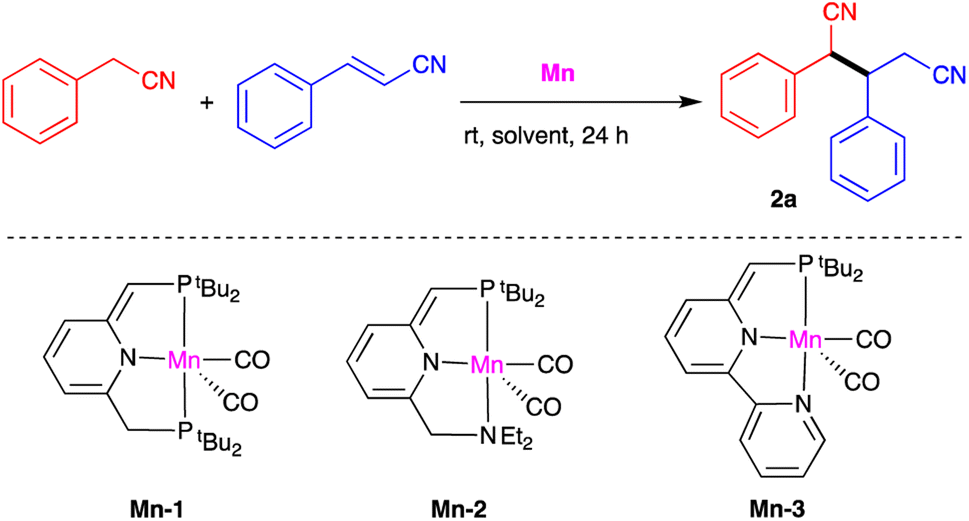
Our investigation of catalytic nitrile heteroaddition began with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of benzyl cyanide and cinnamonitrile as model substrates, and the manganese complexes Mn-1, Mn-2 and Mn-3 as potential catalysts (Table 1). Using 0.5 mol% of Mn-1 and THF as solvent, the corresponding dinitrile product 2a was obtained in quantitative yield after 24 h at room temperature (Table 1, entry 1). Gas chromatographic (GC) analysis indicated that this dinitrile comprised a 1:1 mixture of diastereomers. Reducing the catalyst loading to 0.3 mol% decreased conversion to 80% and the isolated yield to 76% under otherwise identical conditions (entry 2), and very similar results were obtained when THF was replaced with benzene (entry 3). Complexes Mn-2 and Mn-3 were both found to catalyze the addition reaction, but were less effective than Mn-1 (entries 4 and 5). It should be noted that using excess cinnamonitrile (2 equiv. vs. benzyl cyanide) did not result in double addition, and the single-addition product 2a was the only observed product, isolated in 99% yield (entry 6). Finally, a control experiment was carried out in the absence of catalyst, but no product was obtained, clearly indicating the critical role of the catalyst (entry 7).
:1 mixture of benzyl cyanide and cinnamonitrile as model substrates, and the manganese complexes Mn-1, Mn-2 and Mn-3 as potential catalysts (Table 1). Using 0.5 mol% of Mn-1 and THF as solvent, the corresponding dinitrile product 2a was obtained in quantitative yield after 24 h at room temperature (Table 1, entry 1). Gas chromatographic (GC) analysis indicated that this dinitrile comprised a 1:1 mixture of diastereomers. Reducing the catalyst loading to 0.3 mol% decreased conversion to 80% and the isolated yield to 76% under otherwise identical conditions (entry 2), and very similar results were obtained when THF was replaced with benzene (entry 3). Complexes Mn-2 and Mn-3 were both found to catalyze the addition reaction, but were less effective than Mn-1 (entries 4 and 5). It should be noted that using excess cinnamonitrile (2 equiv. vs. benzyl cyanide) did not result in double addition, and the single-addition product 2a was the only observed product, isolated in 99% yield (entry 6). Finally, a control experiment was carried out in the absence of catalyst, but no product was obtained, clearly indicating the critical role of the catalyst (entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Conversionb | Yieldc |
| a Reaction conditions: benzyl cyanide (0.3 mmol), cinnamonitrile (0.3 mmol), solvent (1 mL), catalyst (loading as indicated), stirred at room temperature for 24 h. b Conversion of benzyl cyanide was determined by GC analysis, using mesitylene as internal standard. c Yield of 2a was determined for the isolated compound after column chromatography. d 0.6 mmol (2 equiv.) of cinnamonitrile was used. | ||||
| 1 | Mn-1 (0.5 mol%) | THF | >99 | 99 |
| 2 | Mn-1 (0.3 mol%) | THF | 80 | 76 |
| 3 | Mn-1 (0.5 mol%) | Benzene | 78 | 76 |
| 4 | Mn-2 (0.5 mol%) | THF | 80 | 80 |
| 5 | Mn-3 (0.5 mol%) | THF | 72 | 70 |
| 6d | Mn-1 (1 mol%) | THF | >99 | 99 |
| 7 | — | THF | — | — |
With the optimized reaction conditions in hand, we set to explore the substrate scope of our catalytic nitrile heteroaddition system (Scheme 2; 2a is duplicated from Table 1 for comparative purposes). Reaction of benzyl cyanide with alkyl-substituted acrylonitrile derivatives, namely, trans-3-cyclopentyl-acrylonitrile and crotononitrile, afforded the corresponding products, 2b and 2c, in moderate to good yields (Scheme 2). Benzyl cyanide was also coupled with 2-pentenenitrile, but in the absence of solvent, giving dinitrile 2d in excellent yield, thereby demonstrating the efficiency of the catalytic system under solvent-free conditions. Furthermore, benzyl cyanides bearing various electron donating and withdrawing substituents on their arene rings were coupled with different unsaturated nitriles, affording the corresponding products, 2e–l, in good to excellent yields. This indicates that the present catalytic system tolerates these substituents, and this is particularly notable for the halogen and cyano functionalities. Moreover, these experiments show that catalyst Mn-1 can promote both the isomerization of allyl cyanide and its subsequent conjugate addition to saturated nitriles (2f, 2j and 2l). Importantly, base-sensitive functional groups, namely, ketone, ester, and amide, were also tolerated, and the corresponding dinitrile products 2m–o were isolated in good yields. An N-heterocyclic saturated nitrile underwent smooth heteroaddition, using allyl cyanide as its partner, to furnish dinitrile 2p in 70% yield. Similarly, an O-heterocyclic α,β-unsaturated nitrile, 2-furanacrylonitrile, was coupled with 1-naphthylacetonitrile to give the desired product 2q in 90% yield. The reaction of α-substituted benzyl cyanides with acrylonitrile proceeded efficiently to afford the desired products 2r and 2s in 99% and 68% yield, respectively. Lastly, we examined the highly challenging application of unactivated aliphatic nitriles as substrates in our catalytic system. Under the optimized catalytic conditions, acetonitrile reacted with cinnamonitrile to give the corresponding product 2t in low yield (<20%). However, employing acetonitrile as solvent, instead of THF, and increasing the catalyst loading to 5 mol%, enabled us to achieve high to quantitative product yields (2t and 2u). It should be noted that when the reaction of acetonitrile with cinnamonitrile was repeated under the same conditions, but with the manganese catalyst replaced by an equimolar amount of the strong base KOtBu, poor results were obtained (8% conversion, 5% yield of product 2t), thereby highlighting the nitrile coupling efficiency of Mn-1 relative to general base catalysis. Other unactivated nitriles, i.e., propionitrile and pentanenitrile, were coupled with vinyl nitriles, using Mn-1 under similar conditions, to afford products 2v and 2w in 47% and 49% yield, respectively (Scheme 2).
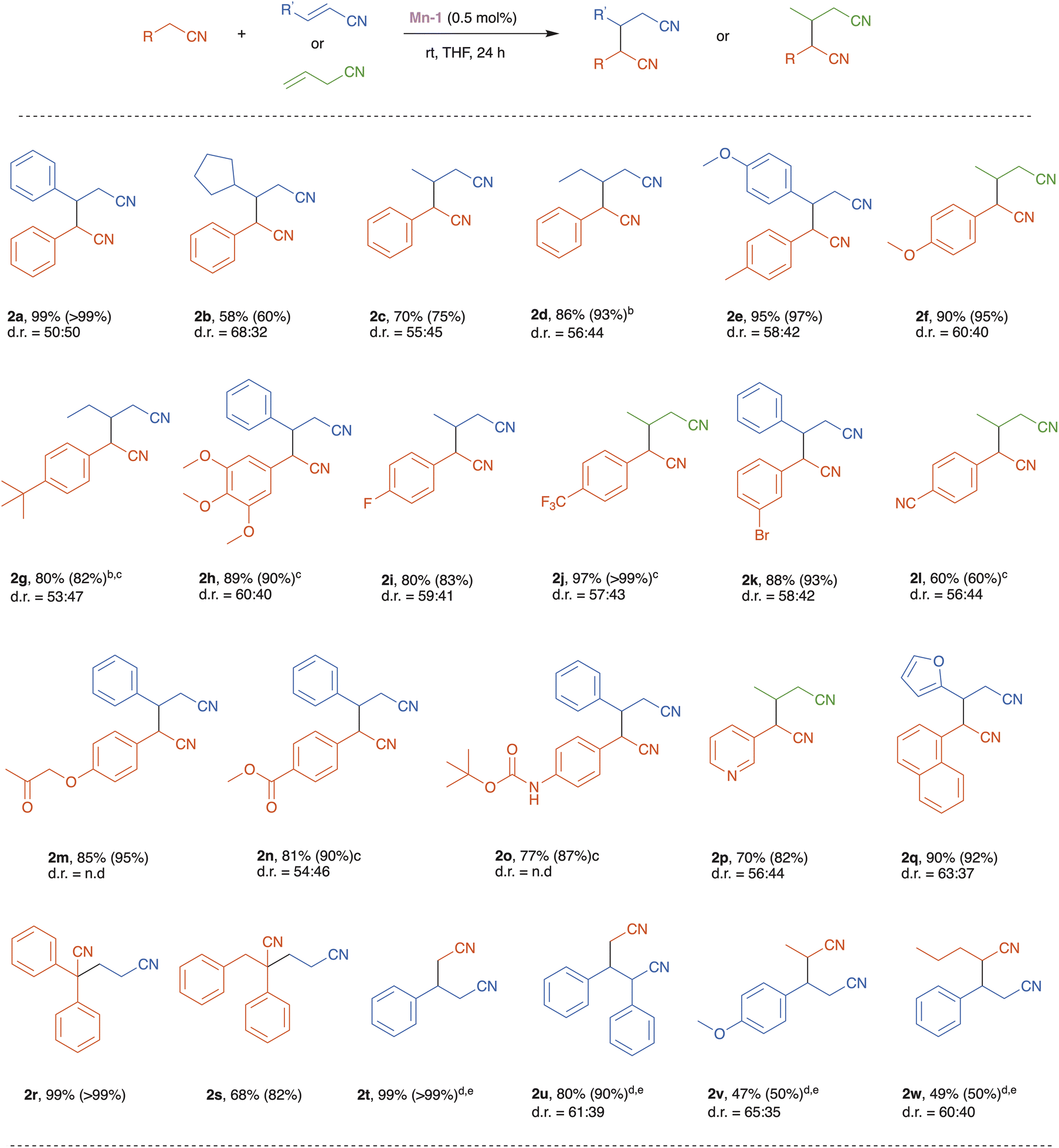 | ||
| Scheme 2 Substrate scope of the manganese-catalyzed base-free heteroaddition of nitriles. aReaction conditions: saturated nitrile (0.3 mmol), unsaturated nitrile (0.3 mmol), THF (1 mL), Mn-1 (0.5 mol%), stirred at room temperature for 24 h. Reaction yields correspond to the pure isolated products. Values in parentheses are the conversions of the saturated nitrile substrates, as determined by GC analysis using mesitylene as internal standard. bReaction performed without solvent. c2 mol% of Mn-1 was used. dMn-1 (5 mol%), saturated aliphatic nitrile (1 mL) and unsaturated nitrile (0.3 mmol) were stirred at room temperature for 48 h. eThe value in parentheses is the conversion of the vinyl nitrile, as determined by GC analysis using mesitylene as internal standard. The diastereoisomeric ratios (d.r.) noted for the various products were determined by GC or NMR spectroscopic analysis. | ||
The underlying mechanism of nitrile addition catalyzed by complex Mn-1 was probed through stoichiometric experiments (Scheme 3). A competition experiment was performed, wherein this catalyst was treated with an equimolar mixture of benzyl cyanide and cinnamonitrile, each at 5 equiv. per catalyst, in THF at room temperature (Scheme 3a). Interestingly, only benzyl cyanide reacted productively with the dearomatized complex, affording the respective rearomatized enamido complex Mn-4, whereas no cinnamonitrile complex, nor derivative thereof, was observed (see ESI†). This enamido complex has already been fully characterized, including X-ray crystallographic and density functional theory analyses, as part of our previous work on conjugate additions involving nitriles.16 In the present work, when independently-prepared Mn-4 was employed as catalyst, benzyl cyanide and cinnamonitrile were coupled to quantitatively give dinitrile 2a (Scheme 3b), thereby implying that the enamido complex is an intermediate in this addition reaction.
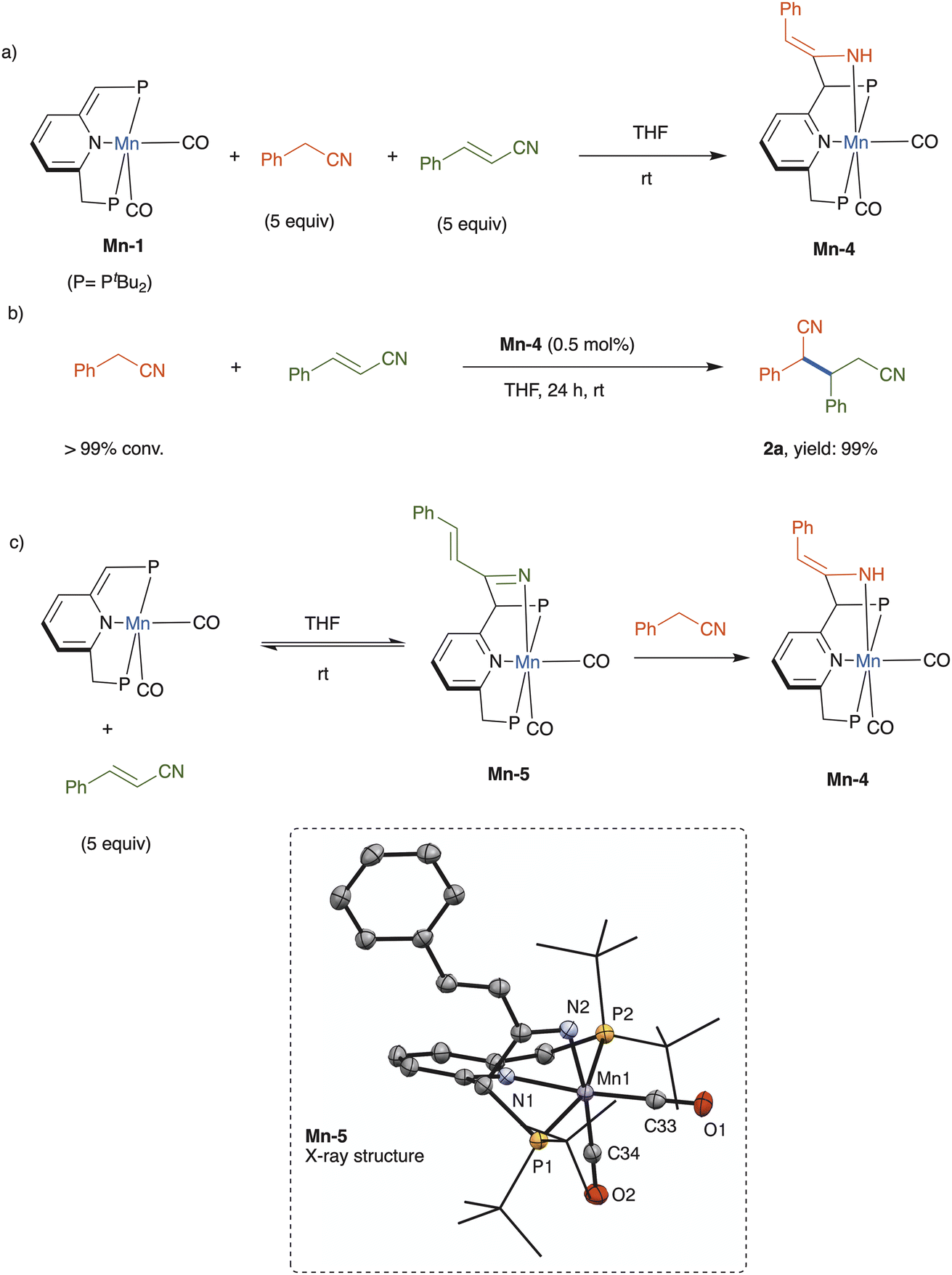 | ||
| Scheme 3 Mechanistic studies. | ||
The aforementioned competition experiment showed that Mn-1 reacts preferentially with benzyl cyanide, rather than cinnamonitrile. This is due to the higher thermodynamic stability of the generated enamine complex, which cannot be formed in the case of cinnamonitrile.23 However, in the absence of benzyl cyanide, this complex reacted with 5 equiv. of cinnamonitrile in THF to afford a new complex, the ketimido adduct Mn-5 (Scheme 3c), which was structurally identified by NMR spectroscopy and X-ray crystallography (see ESI† for full details). In THF solution, Mn-5 exists in equilibrium with Mn-1 and free cinnamonitrile, and addition of benzyl cyanide (5 equiv.) leads to quantitative formation of complex Mn-4 within minutes at room temperature (Scheme 3c).
A plausible catalytic cycle for nitrile heteroaddition by Mn-1 is proposed (Scheme 4), based on the above experimental observations and previous mechanistic studies involving this complex.16,20,21 Initially, the saturated nitrile adds across the dearomatized metal-ligand framework of Mn-1 to generate the rearomatized ketimido intermediate I, which undergoes facile tautomerization to the thermodynamically more stable enamido intermediate II. The ketimido species I′, which forms reversibly upon reaction of Mn-1 with the unsaturated nitrile, is likely an off-cycle species that is not directly involved in the catalytic mechanism. Intermediate II reacts with the unsaturated nitrile through a Michael-type addition, leading to C–C bond formation between the two species and generating the formally zwitterionic intermediate III. This, in turn, undergoes proton transfer from the N–H bond of the coordinated ketimine group to the dangling ketenimide fragment, affording intermediate IV. Finally, the dinitrile product is released from this intermediate, thereby regenerating the dearomatized complex Mn-1 and closing the catalytic cycle.
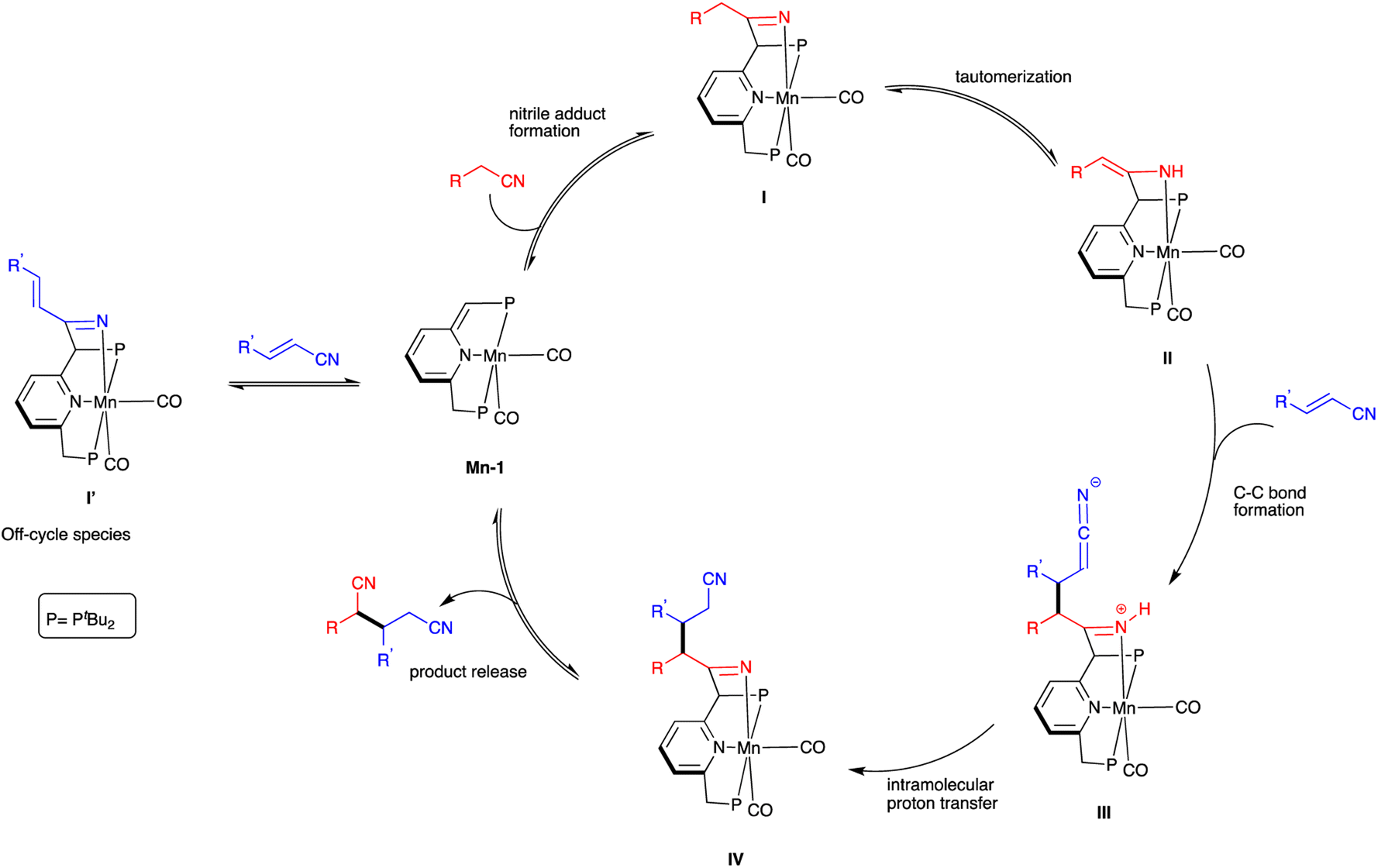 | ||
| Scheme 4 Proposed mechanism of nitrile heteroaddition catalyzed by Mn-1. | ||
Conclusions
In summary, we have presented the first example of transition-metal-catalyzed heteroaddition of benzylic and aliphatic nitriles to α,β- and β,γ-unsaturated nitriles to generate glutaronitrile derivatives. The resulting dinitrile compounds represent valuable intermediates in N-heterocycle synthesis and the polymer industry. Mechanistic investigations demonstrated the selective activation of a saturated nitrile over an unsaturated one. Moreover, the synergistic cooperation between the dearomatized PNP–Mn complex and saturated nitriles allows the generation of an enamido–manganese species, which is a key mechanistic intermediate. The new synthetic protocol outlined above provides facile means of nitrile heteroaddition at room temperature under base-free conditions, thereby allowing access to dinitrile compounds in an atom-economical, environmentally benign fashion.Data availability
The authors declare that all supporting data are available in the ESI† and from the corresponding author upon request.Author contributions
D. M. and S. T. conceived and directed the project and designed the experiments. S. T. performed all of the experiments and analyzed their results. Y. D.-P. carried out the crystallographic studies. M. M. provided insightful discussions. D. M., S. T. and M. M. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. T. is thankful to the Feinberg Graduate School of the Weizmann Institute of Science for a research fellowship.Notes and references
- (a) R. Ló pez and C. Palomo, Angew. Chem., Int. Ed., 2015, 54, 13170–13184 CrossRef PubMed; (b) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed; (c) F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597–606 RSC.
- (a) Ullmann's encyclopedia of industrial chemistry, ed B. Elvers and F. Ullmann, Wiley-VCH, Weinheim, 2011 Search PubMed; (b) H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 5th edn, 2010, ch. 10 Search PubMed.
- Reviews: (a) C. Scotti and J. W. Barlow, Nat. Prod. Commun., 2022, 17, 1–24 CrossRef; (b) A. Rakshit, H. N. Dhara, A. K. Sahoo and B. K. Patel, Chem.–Asian J., 2022, 17, e202200792 CrossRef CAS PubMed; (c) Y. Xia, zH. Jiang and W. Wu, Eur. J. Org Chem., 2021, 2021, 6658–6669 CrossRef CAS.
- (a) J. Long, R. Yu, J. Gao and X. Fang, Angew. Chem., Int. Ed., 2020, 59, 6785–6789 CrossRef CAS PubMed; (b) F. Sun, J. Gao and X. Fang, Chem. Commun., 2020, 56, 6858–6861 RSC; (c) J. Chen, P.-Z. Wang, B. Lu, D. Liang, X.-Y. Yu, W.-J. Xiao and J.-R. Chen, Org. Lett., 2019, 21, 9763–9768 CrossRef CAS PubMed; (d) Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2006, 128, 3928–3930 CrossRef CAS PubMed.
- (a) J. Long, S. Xia, T. Wang, G.-J. Cheng and X. Fang, ACS Catal., 2021, 11, 13880–13890 CrossRef CAS; (b) L. Qi, R. Li, X. Yao, Q. Zhen, P. Ye, Y. Shao and J. Chen, J. Org. Chem., 2020, 85, 1097–1108 CrossRef CAS PubMed; (c) T. Wang, Y.-N. Wang, R. Wang, B.-C. Zhang, C. Yang, Y.-L. Li and X.-S. Wang, Nat. Commun., 2019, 10, 5373 CrossRef PubMed; (d) S. Laval, W. Dayoub, L. Pehlivan, E. Métay, A. Favre-Reguillon, D. Delbrayelle, G. Mignani and M. Lemaire, Tetrahedron, 2014, 70, 975–983 CrossRef CAS; (e) R. W. Hartmann and C. Batzl, J. Med. Chem., 1986, 29, 1362–1369 CrossRef CAS PubMed.
- (a) K. Cen, M. Usman, W. Shen, M. Liu, R. Yang and J. Cai, Org. Biomol. Chem., 2022, 20, 7391–7404 RSC; (b) P.-Z. Wang, Y. Gao, J. Chen, X.-D. Huan, W.-J. Xiao and J.-R. Chen, Nat. Commun., 2021, 12, 1815 CrossRef CAS PubMed; (c) J. E. Gavagan, S. K. Fager, R. D. Fallon, P. W. Folsom, F. E. Herkes, A. Eisenberg, E. C. Hann and R. DiCosimo, J. Org. Chem., 1998, 63, 4792–4801 CrossRef CAS.
- P. Perlmutter and J. E. Baldwin, Conjugate Addition Reactions in Organic Synthesis, Elsevier Science, Amsterdam, 2013 Search PubMed.
- (a) T. Tokoroyama, Eur. J. Org Chem., 2010, 2009–2016 CrossRef CAS; (b) A. Michael, J. Prakt. Chem., 1887, 35, 349–356 CrossRef.
- F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035–2078 CrossRef CAS PubMed.
- (a) K. Zheng, X. Liu and X. Feng, Chem. Rev., 2018, 118, 7586 CrossRef CAS PubMed; (b) C. Hui, F. Pu and J. Xu, Chem.–Eur. J., 2017, 23, 4023 CrossRef CAS PubMed.
- (a) S.-I. Murahashi, T. Naota, H. Taki, M. Mizuno, H. Takaya, S. Komiya, Y. Mizuho, N. Oyasato and M. Hiraoka, J. Am. Chem. Soc., 1995, 117, 12436–12451 CrossRef CAS; (b) T. Naota, H. Taki, M. Mizuno and S.-I. Murahashi, J. Am. Chem. Soc., 1989, 111, 5954–5955 CrossRef CAS.
- (a) N. Zhang, C. Zhang, X. Hu, X. Xie and Y. Liu, Org. Lett., 2021, 23, 6004–6009 CrossRef CAS PubMed; (b) S. Nakamura, A. Tokunaga, H. Saito and M. Kondo, Chem. Commun., 2019, 55, 5391–5394 RSC; (c) K. Ebitani, K. Motokura, K. Mori, T. Mizugaki and K. Kaneda, J. Org. Chem., 2006, 71, 5440–5447 CrossRef CAS PubMed.
- M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018 CrossRef CAS PubMed.
- (a) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (c) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed; (d) D. Milstein, Philos. Trans. R. Soc., A, 2015, 373, 20140189 CrossRef PubMed; (e) C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed; (f) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- (a) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (b) S. Kar and D. Milstein, Chem. Commun., 2022, 58, 3731–3746 RSC.
- A. Nerush, M. Vogt, U. Gellrich, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6985–6997 CrossRef CAS PubMed.
- (a) B. Guo, J. G. de Vries and E. Otten, Chem. Sci., 2019, 10, 10647–10652 RSC; (b) L. E. Eijsink, S. C. P. Perdriau, J. G. de Vries and E. Otten, Dalton Trans., 2016, 45, 16033–16039 RSC; (c) S. Perdriau, D. S. Zijlstra, H. J. Heeres, J. G. de Vries and E. Otten, Angew. Chem., Int. Ed., 2015, 54, 4236–4240 CrossRef CAS PubMed.
- (a) Y. Wang, M. Wang, Y. Li and Q. Liu, Chem, 2021, 7, 1180–1223 CrossRef CAS; (b) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed; (c) A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS; (d) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494–1502 CrossRef CAS PubMed; (e) P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495–2495 CrossRef CAS PubMed; (f) I. Bauer and H. J. Knoelker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- Selected examples: (a) U. K. Das, A. Kumar, Y. Ben-David, M. A. Iron and D. Milstein, J. Am. Chem. Soc., 2019, 141, 12962–12966 CrossRef CAS PubMed; (b) P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2019, 141, 12202–12206 CrossRef CAS PubMed; (c) S. Chakraborty, P. Daw, Y. Ben David and D. Milstein, ACS Catal., 2018, 8, 10300–10305 CrossRef CAS PubMed; (d) N. A. Espinosa-Jalapa, A. Kumar, G. Leitus, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11722–11725 CrossRef CAS PubMed; (e) S. Chakraborty, U. K. Das, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11710–11713 CrossRef CAS PubMed; (f) S. Chakraborty, U. Gellrich, Y. Diskin-Posner, G. Leitus, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 4229–4233 CrossRef CAS PubMed.
- S. Tang and D. Milstein, Chem. Sci., 2019, 10, 8990–8994 RSC.
- Q. Q. Zhou, Y. Q. Zou, S. Kar, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 10239–102455 CrossRef CAS PubMed.
- M. M. Al-Arab, H. D. Tabba, I. A. Abu-Yousef and M. M. Olmstead, Tetrahedron, 1988, 44, 7293–7302 CrossRef CAS.
- Our previous work has shown that the reaction of Mn-1 with benzyl cyanide readily forms enamido complex Mn-4, wherein the enamido moiety is stabilized by conjugation to an aromatic ring (see ref. 16). We have also demonstrated that propionitrile reacts with Mn-1 to predominately give the corresponding ketamido complex (imine intermediate), rather than an enamido one (enamine intermediate), because propionitrile cannot stabilize the latter through conjugation. Thus, our previous results clearly indicate that the complex bearing the imine intermediate is less stable, and is in equilibrium with the dearomatized manganese complex Mn-1 and free nitrile.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2224607 and 2224950. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04935c |
| This journal is © The Royal Society of Chemistry 2024 |