
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromoting the formation of metal–carboxylate coordination to modulate the dimensionality of ultrastable lead halide hybrids†
Yilin
Jiang
,
Jinlin
Yin
,
Ruonan
Xi
and
Honghan
Fei
 *
*
Shanghai Key Laboratory of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, 1239 Siping Rd., Shanghai 200092, China. E-mail: fei@tongji.edu.cn
First published on 19th January 2024
Abstract
Crystal engineering of metal halide hybrids is critical to investigate their structure–property relationship and advance their photophysical applications, but there have been limited efforts to employ coordination chemistry to precisely control the dimensionality of metal halide sublattices. Herein, we present a coordination–assembly synthetic strategy developed for the rational modulation of lead halide dimensionality, realizing the transition from 2D to 3D architectures. This manipulation is achieved by utilizing three organocarboxylates featuring the identical cyclohexane backbone unit. Specifically, the 1,4-cyclohexanedicarboxylate and 1,2,4,5-cyclohexanetetracarboxylate ligands facilitate the formation of quasi-2D layered structures, characterized by weakly corrugated and strongly corrugated lead halide layers, respectively. Importantly, the introduction of the 1,2,3,4,5,6-cyclohexanehexacarboxylate ligand results in coordination architectures featuring 3D lead chloride/bromide sublattices. The formation of the 3D coordination architectures templated by the 1,2,3,4,5,6-cyclohexanehexacarboxylate ligand affords extended wavelength coverage and superior carrier transport properties compared to their quasi-2D layered analogues. Importantly, both the 2D and 3D lead halide-based coordination polymers exhibit high aqueous stability over a wide pH range, outperforming the conventional ionic-bound lead halides. Notably, the chemically stable 3D lead bromide exhibits efficient photocatalytic ethylbenzene oxidation with the conversion rate of 498 μmol g−1 h−1, substantially higher than its 2D lead bromide counterparts. This work highlights the important role of coordination chemistry in the rational design of metal halide hybrids, which is crucial for advancing their photophysical properties and applications.
Introduction
In recent years, lead halide hybrids have garnered significant attention due to their exceptional photophysical properties, including strong light absorption, wide wavelength coverage, tunable bandgaps, high carrier mobility and long carrier lifetimes.1,2 Among them, three-dimensional (3D) lead halide hybrids have emerged as a class of semiconductor photocatalysts,3–5 including photocatalytic C(sp3)–H oxidation.6,7 The C(sp3)–H bond is one of the most common bonds in organic compounds and its selective oxidation is essential in pursuit of many high value-added chemicals.8–11 For example, FAPbBr3 perovskite was reported to activate the C(sp3)–H bond under solar light irradiation based on the fabrication of a three-component solar photocatalyst cell.12 However, the practical applications of a vast majority of 3D lead halide hybrids are hindered by their instability issues under various physical and chemical conditions, even in the presence of ambient humidity.13,14 On the other hand, quasi-2D layered lead halide hybrids exhibit higher moisture stability compared to their 3D counterparts, but suffer from larger bandgaps and lower carrier mobility owing to carrier confinement within the 2D lead halide layers.15 Consequently, there is a pressing need to address the trade-off between the instability of 3D hybrids and the localized charge carriers of 2D layered lead halides.A variety of synthetic strategies have been developed to control the dimensionality of lead halide sublattices at the molecular level.16 For example, incorporating organoammonium cations with large molecular size and high rigidity often leads to the formation of low-dimensional lead halide hybrids.17–19 In addition, a commonly employed approach involves varying synthesis conditions such as precursor ratios, temperatures and solvents, which often serendipitously result in the formation of lead halide hybrids with different dimensionalities.20–24 More recently, the inclusion of guest molecules (e.g. H2O, NH3 and MeOH) into the parent lattice of lead halides has been discovered as a successful approach to isolate the inorganic halide connectivity, leading to the formation of low-dimensional lead halide structures.25–28 Although less explored in the literature, rational synthetic approaches based on coordination chemistry to modulate the dimensionality of lead halides remain limited.29
Our group focused on the synthesis of lead halide-based coordination polymers, which are extended frameworks consisting of lead halide inorganic components and coordinating anionic ligands as the organic components.30–33 By employing this general synthetic strategy, we have significantly advanced the structural integrity and intrinsic stability of lead halide hybrids, primarily attributed to the formation of 3D coordination networks instead of ionically bound structures.34–36 This class of extended coordinative architectures display excellent aqueous stability across a wide pH range and even under boiling conditions.30,37 However, the vast majority of these structures adopt quasi-2D layered arrangements with tunable layer corrugation and thickness.30,38,39 It is challenging to achieve the rational transition from 2D to 3D in lead halide sublattices for this class of coordination polymers, but it is a compelling pursuit to discover the 3D analogues combining the outstanding photophysical properties and high aqueous stability.
Herein, we have successfully achieved precise control over the dimensionality, transitioning from 2D to 3D lead halide sublattices for the coordination polymers. This was realized by employing three organocarboxylates that share the identical cyclohexane backbone unit. The use of 1,4-cyclohexanedicarboxylate (cdc2−) and 1,2,4,5-cyclohexanetetracarboxylate (ctc4−) led to the formation of quasi-2D layered structures with weakly corrugated and strongly corrugated features, respectively. Intriguingly, the incorporation of 1,2,3,4,5,6-cyclohexanehexacarboxylate (chc6−) gives rise to two chemically stable 3D lead chloride/bromide coordination architectures that exhibit remarkable aqueous pH stability. Moreover, our investigation involving band structure studies, Hall effect measurements and ultrafast transient absorption (TA) spectroscopy indicates that the 3D structures templated by chc ligands demonstrate wide wavelength coverage and enhanced carrier-transport characteristics compared to their quasi-2D analogues. In addition, since developing C(sp3)–H activation under mild conditions is crucial for direct conversion of abundant hydrocarbons,8,9,40,41 we have employed the photocatalytic benzyl C(sp3)–H oxidation as a model reaction to evaluate their photocatalytic performance. Notably, the 3D lead bromide coordination polymer loaded with trace amount of the Pt co-catalyst exhibits the highest ethylbenzene conversion rates of 498 μmol g−1 h−1 with a selectivity of 61%, substantially outperforming the 2D lead bromide counterparts.
Results and discussion
Synthesis and crystal structures
TJU-10(Cl) ([Pb2Cl2]2+(cdc2−), TJU = Tongji University) and TJU-11(Br) ([Pb2Br2]2+(cdc2−)) are synthesized according to our earlier reports,38 affording slightly corrugated [Pb2X2]2+ (X = Cl−/Br−) coordinatively pillared by the interlamellar cdc2− linkers (Fig. 1a and b). The systematic enhancement of Pb2+-carboxylate coordination within these lead halide coordination polymers was achieved by the replacement of the original dicarboxylates with the cyclohexane-based ligands containing four and six carboxylates (Fig. 1). Solvothermal synthesis of PbCl2 and ctc4− in H2O/MeCN at 120 °C affords the colorless block-shaped crystals of [Pb3Cl2]4+(ctc4−)·3H2O, which we denote as TJU-13(Cl). X-ray crystallography reveals that TJU-13(Cl) is crystallized in the monoclinic crystal system with the P21/c space group (Table S1†). TJU-13(Cl) consists of strongly wavy [Pb3Cl2]4+ slabs with the high corrugation amplitude of ∼18.4 Å along the b-axis, substantially larger than that of TJU-10(Cl) (∼3.5 Å) (Fig. S1†). This pronounced corrugation primarily originates from the increased Pb2+-carboxylate coordination linkages introduced by the incorporation of ctc4− ligands. Notably, three of the four carboxylates in each ctc4− ligand coordinate to a single [Pb3Cl2]4+ layer, inducing a profound layer corrugation effect, while the remaining carboxylate group connects with the adjacent inorganic layer (Fig. 1c). Each [Pb3Cl2]4+ layer is composed of an ordered array of 6-membered [Pb3Cl3]3+ rings linked by inter-cluster Pb–Cl bonds, defining the inorganic layer along the bc plane (Fig. S2†). In addition, the isostructural bromide analogue ([Pb3Br2]4+(ctc4−)·3H2O, TJU-13(Br)) was successfully synthesized by substitution of PbCl2 with PbBr2 as the precursor (Fig. 1d and Table S1†). TJU-13(Br) also consists of strongly wavy [Pb3Br2]4+ slabs, which are coordinatively pillared by the interlamellar ctc ligands (Fig. 1d). The ordered arrays of 6-membered [Pb3Br3]3+ rings are connected by inter-cluster Pb–Br bonds that propagate indefinitely along the bc plane, which comprise each [Pb3Br2]4+ layer (Fig. S2†).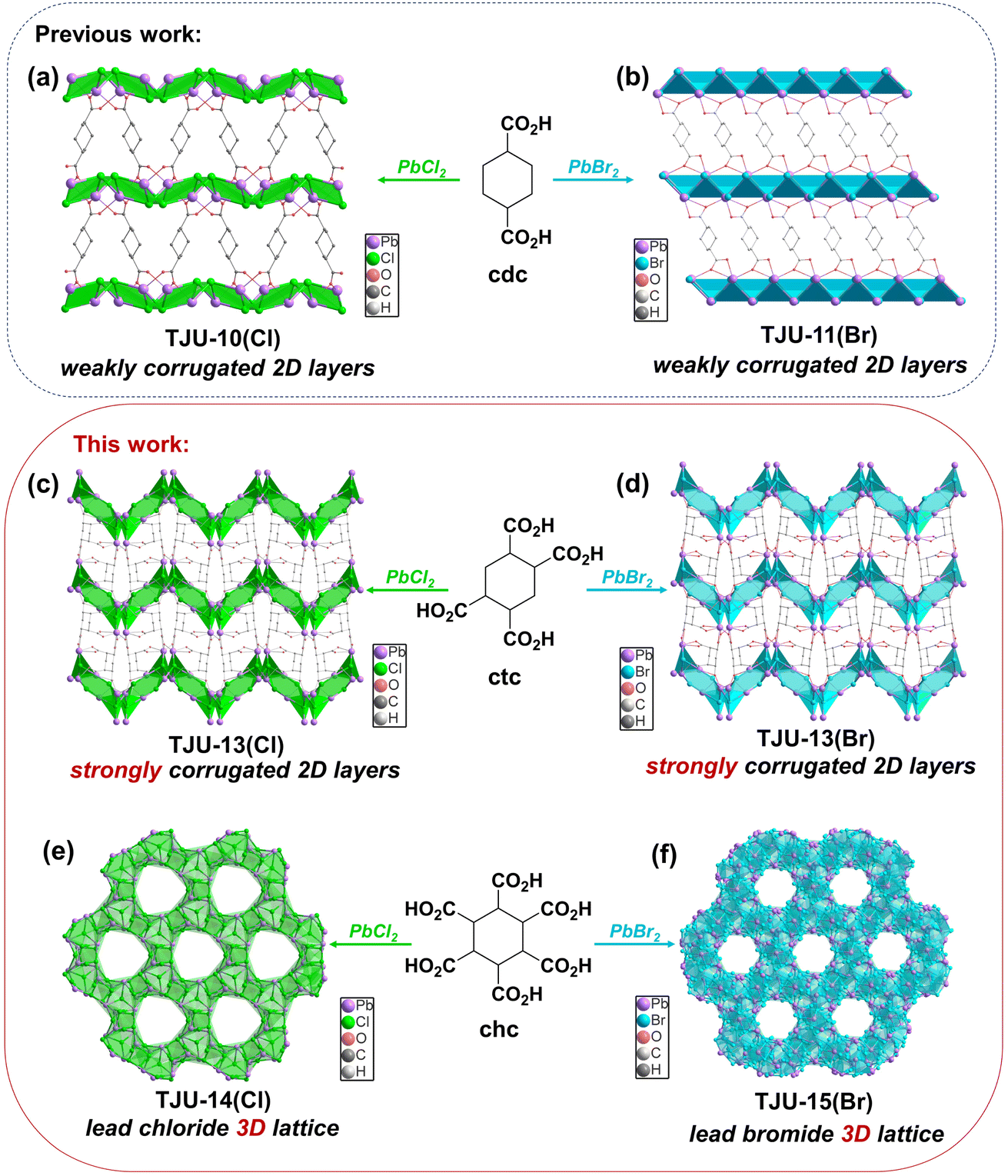 | ||
| Fig. 1 Synthetic scheme of TJU-10(Cl), TJU-11(Br), TJU-13(Cl), TJU-13(Br), TJU-14(Cl) and TJU-15(Br) and their templating organocarboxylate ligands. (a, b) Crystallographic view of weakly corrugated 2D [Pb2X2]2+ (X = Cl−/Br−) layers in TJU-10(Cl) (a) and TJU-11(Br) (b). (c, d) Crystallographic view of strongly corrugated 2D [Pb3X2]4+ (X = Cl−/Br−) layers in TJU-13(Cl) (c) and TJU-14(Br) (d). (e, f) Crystallographic view of 3D [Pb12Cl17(OH)]6+ lattice in TJU-14(Cl) (e) and 3D [Pb15Br24]6+ lattice TJU-15(Br) (f), chc ligands are omitted for clarity. The coordination geometry of Pb2+ centers is shown in the polyhedron. The green polyhedra represent lead chloride units and the blue polyhedra represent lead bromide units. | ||
Despite the pronounced corrugation observed in TJU-13, the interlayer spacing of ∼13 Å was unfavorable for efficient carrier transport. Therefore, the lead halide dimensionality modulation from 2D to 3D is further investigated by using chc6− as the templating ligand. Solvothermal reaction of PbCl2 and chc in deionized H2O afforded colorless block-shaped crystals of [Pb12Cl17(OH)]6+(chc6−), denoted as TJU-14(Cl). X-ray crystallography studies revealed that TJU-14(Cl) is crystallized in the trigonal crystal system with the space group of P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c1 (Table S2†). TJU-14(Cl) comprises edge- and vertex-sharing PbCl5 and PbCl6 units extending in three dimensions, defining a 3D inorganic [Pb12Cl17(OH)]6+ framework (Fig. 1e and S3†). The 3D inorganic hybrid features an array of honeycomb-like channels along the c-axis, with an open aperture diameter of ∼9.4 Å but accommodating the chc6− ligands. Notably, all six carboxylates in the chc6− ligands coordinatively bonded with the Pb2+ centers, forming an array of 12-membered-ring channels. This phenomenon suggests the crucial role of an increasing number of carboxylate groups in organic ligands for the 2D to 3D dimensionality modulation. Both crystallographically independent Pb2+ centers adopt eight-coordinate geometries, with one coordinating to five Cl− anions, two carboxylate oxygens and one μ3-OH, while the other coordinating with six Cl− anions and two carboxylate oxygens.
c1 (Table S2†). TJU-14(Cl) comprises edge- and vertex-sharing PbCl5 and PbCl6 units extending in three dimensions, defining a 3D inorganic [Pb12Cl17(OH)]6+ framework (Fig. 1e and S3†). The 3D inorganic hybrid features an array of honeycomb-like channels along the c-axis, with an open aperture diameter of ∼9.4 Å but accommodating the chc6− ligands. Notably, all six carboxylates in the chc6− ligands coordinatively bonded with the Pb2+ centers, forming an array of 12-membered-ring channels. This phenomenon suggests the crucial role of an increasing number of carboxylate groups in organic ligands for the 2D to 3D dimensionality modulation. Both crystallographically independent Pb2+ centers adopt eight-coordinate geometries, with one coordinating to five Cl− anions, two carboxylate oxygens and one μ3-OH, while the other coordinating with six Cl− anions and two carboxylate oxygens.
The substitution of PbCl2 with PbBr2 in our synthetic protocol resulted in the formation of a coordination polymer with a more densely packed 3D lead bromide topology ([Pb15Br24]6+(chc6−), namely TJU-15(Br)). TJU-15(Br) crystallizes in the highly symmetric cubic crystal system with the Pa-3 space group (Table S2†). TJU-15(Br) also consists of edge- and vertex-sharing PbBr5 and PbBr6 units extending in three dimensions to define the inorganic framework and the chc ligand coordinatively bridge the lead centers within the pore cages (Fig. 1f). The inorganic sublattice of TJU-15(Br) exhibits higher overall occupancy and a more densely packed structure compared to TJU-14(Cl). Each chc6− ligand comprises 12 carboxylate oxygens, all of which form coordination bonds with the Pb2+ centers. This arrangement results in the effective spatial separation of individual ligands within the inorganic skeleton. Half of the crystallographically independent Pb atoms exhibit coordination to six bridging Br atoms, forming the [PbBr6]4− octahedron as for perovskites, while the other Pb atoms adopt a seven-coordinate environment with two carboxylate oxygens and five bromides. The Pb–Br bond distances (2.920–3.310 Å) are well within the accepted range for a covalent Pb–Br bond, suggesting that TJU-15(Br) indeed occupies a 3D lead bromide sublattice.
All of the four new lead halide coordination polymers in this work, TJU-13(Cl), TJU-13(Br), TJU-14(Cl), and TJU-15(Br), can be synthesized with high yields (>75%) and high phase purity, evidenced by C/H elemental analysis and powder X-ray diffraction (PXRD) patterns. All of the experimental PXRD matched well with the theoretical patterns simulated from single-crystal data (Fig. S4†). Thermogravimetric analysis (TGA) and ex situ thermodiffraction suggest the thermal stability of TJU-13(Cl) and TJU-13(Br) up to 120 °C (Fig. S5 and S6†). The increased lead halide dimensionality and higher charge of chc6− ligands synergistically render the improved thermal stability up to 200 °C for TJU-14(Cl) and 220 °C for TJU-15(Br), respectively (Fig. 2a and b and S5†). The chemical stability studies were performed by incubating the as-synthesized crystals in aqueous solutions over a wide range of pH (4–10) at room temperature for 24 h. All of our four coordination polymers retain high crystallinity and demonstrate negligible loss in mass balance, superior to the conventional ionically bound lead halide hybrids (Fig. 2a, b and S6†).
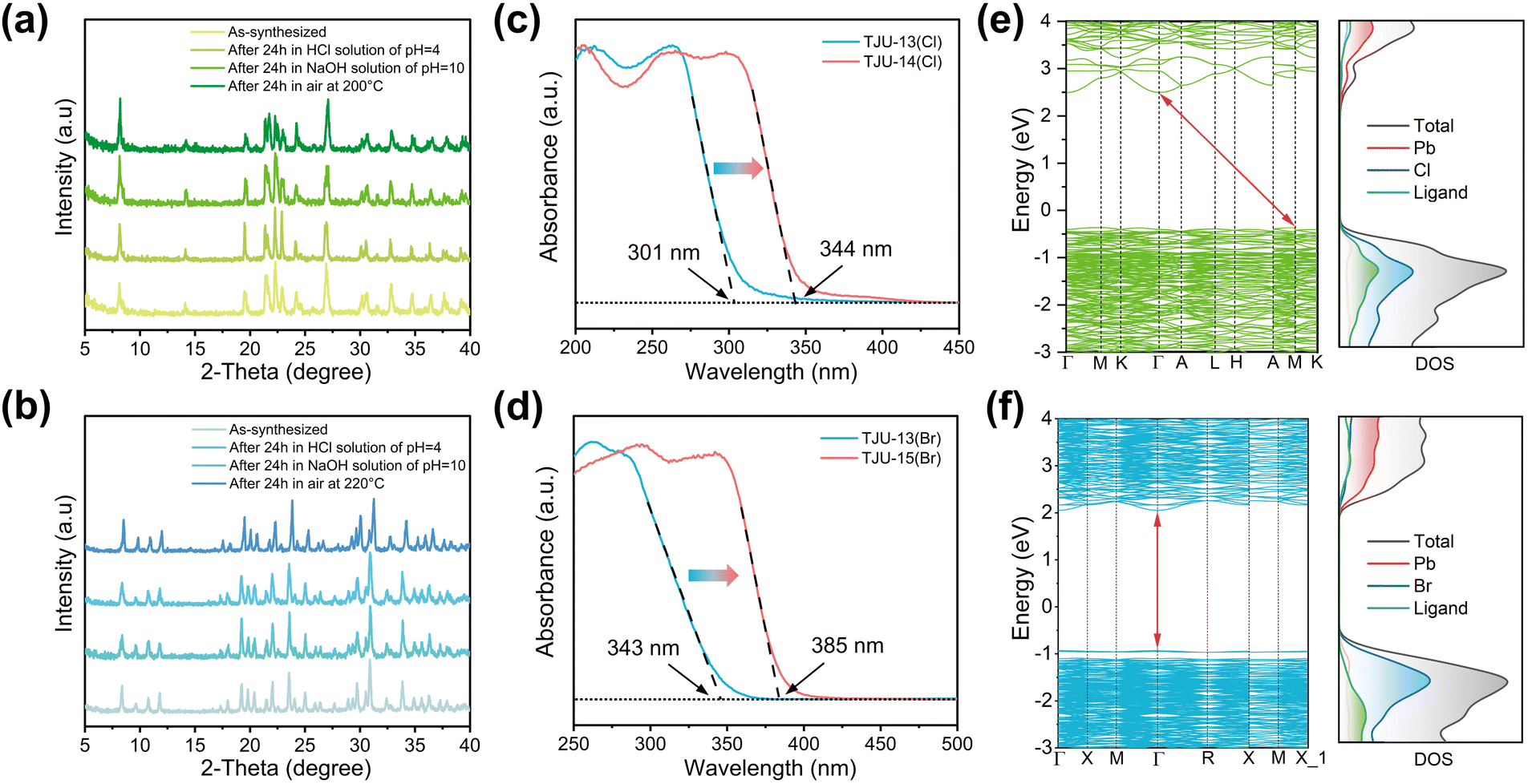 | ||
| Fig. 2 (a,b) PXRD of TJU-14(Cl) (a) and TJU-15(Br) (b) before and after thermal and chemical treatment. (c) UV-vis diffuse reflectance spectroscopy of 2D-TJU-13(Cl) and 3D-TJU-14(Cl). (d) UV-vis diffuse reflectance spectroscopy of 2D-TJU-13(Br) and 3D-TJU-15(Br). (e) Calculated band structures and DOS of TJU-14(Cl). (f) Calculated band structures and DOS of TJU-15(Br). | ||
Band structures
We continue to investigate the electronic band structures of these coordination polymers by experimental studies and theoretical calculations. Ultraviolet-visible (UV-vis) diffusion reflectance spectroscopy revealed notable red-shifts in absorption edges for both chlorides and bromides as a result of the structural transition (Fig. 2c and d). Specifically, TJU-15(Br), the 3D bromide counterpart, exhibited the absorption extending up to 385 nm, while the quasi-2D layered TJU-13(Br) displayed an absorption edge of 343 nm. According to the Tauc plot analysis, the bandgap energies (Eg) were determined to be 3.50 eV for the 3D chloride (TJU-14(Cl)) and 3.29 eV for the 3D bromide (TJU-15(Br)) (Fig. S7 and S8†).The band structures of TJU-14(Cl) and TJU-15(Br) were further studied by the density functional theory (DFT) calculations using the Perdew–Burke–Ernzerhof (PBE) functional, which demonstrated the calculated bandgaps to be 3.45 eV for TJU-14(Cl) and 2.97 eV for TJU-15(Br), respectively (Fig. 2e and f). Both values matched well with the experimental bandgaps (Fig. 2c and d). In addition, TJU-14(Cl) possessed an indirect bandgap configuration, with the conductive band minimum (CBM) located at G point and the valence band maximum (VBM) at M point, agreeing with the experimental Tauc plot (Fig. 2e and S7†). Meanwhile, both frontier orbitals of TJU-15(Br) were located at G point, suggesting the direct bandgap nature (Fig. 2f and S8†). Furthermore, the calculated projected density of states (pDOS) of both coordination polymers showed that the VBMs were mainly contributed by the Cl 3p orbitals for TJU-14(Cl) or the Br 4p orbitals for TJU-15(Br), which elucidated the broader absorption wavelength coverage for the bromide over its chloride counterpart (Fig. 2e and f). Meanwhile, the ligands partially contribute to the VBM, owing to the Pb–carboxylate coordinative behavior. The CBMs of both materials were primarily dominated by the Pb 6p orbitals, analogous to many ionic bound lead halide hybrids.
Ultraviolet photoelectron spectroscopy (UPS) suggested the ionization energies, defined as the energy differences between the VBMs and the vacuum levels, to be 6.11 eV for TJU-13(Br) and 6.64 eV for TJU-15(Br) (Fig. S9†). The energy level positions of their VBMs were determined to be 1.96 eV for TJU-13(Br) and 2.55 eV for TJU-15(Br), respectively, both of which were measured below the Fermi level (Ef). After converting the energy potentials into volts with respect to the normal hydrogen electrode (NHE), the frontier orbitals of TJU-13(Br) were found to be 1.61 V for VBM and −1.82 V for CBM (vs. NHE, pH = 7) while the VBM and CBM of TJU-15(Br) are 2.14 V and −1.15 V (vs. NHE, pH = 7) (Fig. S10†). The CBMs of both materials were more negative than the redox potentials of O2/˙O2− (−0.28 V vs. NHE, pH = 7), indicating their potentials for aerobic photooxidation of ethylbenzene.
Carrier-transport characteristics
To elucidate the effect of the 2D to 3D dimensionality control on charge separation and transport dynamics, we have performed photoluminescence (PL) spectroscopy, ultrafast transient absorption (TA) spectroscopy and AC Hall effect measurements for both 2D and 3D materials. First, steady-state PL studies demonstrated that the emission maxima are located at 530 nm (2.34 eV) for TJU-13(Br) and 630 nm (1.97 eV) for TJU-15(Br), respectively (Fig. 3a). The ∼0.37 eV red-shift in PL was consistent with the energy differences (∼0.39 eV) of absorption edges between TJU-13(Br) and TJU-15(Br) (Fig. 2d), suggesting that both 2D and 3D bromide structures exhibited an analogous radiative recombination of carriers.42,43 Both PL profiles featured broad emission with large full width at half-maximum (0.56 eV for TJU-13(Br) and 0.63 eV for TJU-15(Br)) and large Stokes shifts of 1.41–1.59 eV. The large Stokes-shifted broadband emissions are widely known to originate from the strong electron–phonon coupling by the interactions between electrons and lattice vibrations.44–46 The time-resolved PL decay of TJU-15(Br) was well fitted by a bi-exponential function, revealing a short-lived component (τ1 = 20.3 ns; 24.8%) and a long-lived component (τ2 = 148.7 ns; 75.2%), probably corresponding to free excitons and self-trapped excitons (STEs), respectively (Fig. S12†).47,48 Importantly, the PL decay of TJU-15(Br) with the 3D lead bromide sublattice rendered an average lifetime of 57.9 ns, which was substantially longer than that of the quasi-2D layered TJU-13(Br) (32.1 ns) (Fig. 3b). This implied that the radiative carrier recombination is largely suppressed by the crystal engineering of the 2D to 3D lead bromide dimensionality.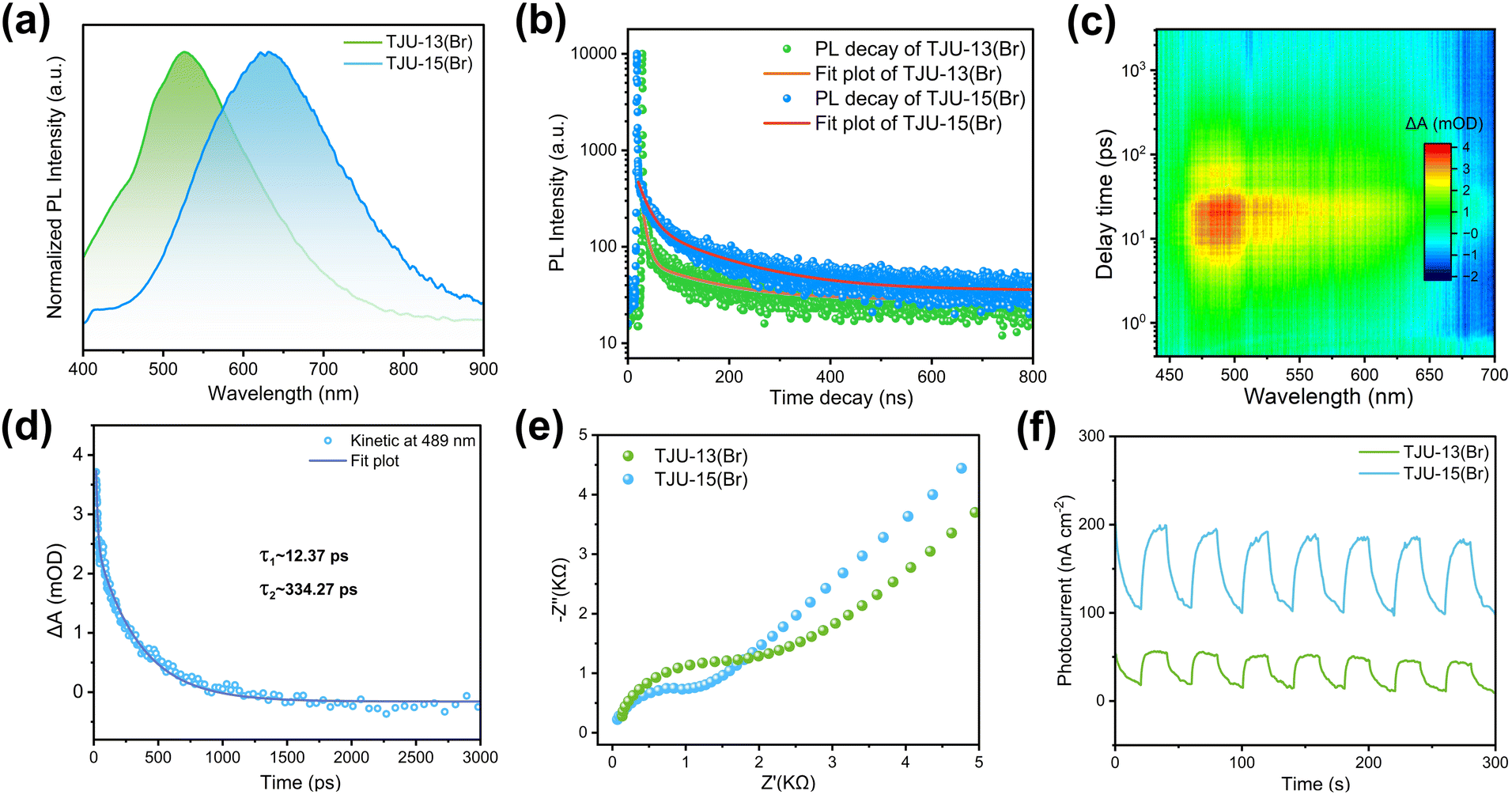 | ||
| Fig. 3 (a) Normalized PL spectra of TJU-13(Br) and TJU-15(Br). (b) Photoluminescence decay curve and fitting plot of TJU-13(Br) and TJU-15(Br), respectively. (c) Two-dimensional pseudo-color TA plot of TJU-15(Br). (d) TA kinetics of TJU-15(Br). (e) Nyquist plots of TJU-13(Br) and TJU-15(Br). (f) Transient photocurrent densities of TJU-13(Br) and TJU-15(Br) under light irradiation. | ||
AC Hall effect measurement at room temperature provided further insights into the carrier dynamics of the materials. We have measured the carrier mobility of three independent samples at different current densities from 10 nA to 1 mA, and reported the mean and error to guarantee the good statistical accuracy. The quasi-2D TJU-13(Br) exhibited a carrier mobility of ∼3.37 ± 0.13 cm2 V−1 s−1 while the 3D bromide counterpart (TJU-15(Br)) demonstrated an enhanced carrier mobility of ∼3.83 ± 0.22 cm2 V−1 s−1, despite both materials having analogous carrier concentrations (Table 1). These values again confirmed that the high-dimensional lead bromide hybrid favors efficient charge separation and transport. The carrier transport length (LD) was calculated to be in the range of 0.45–1.21 μm for TJU-15(Br) based on the equation LD = (kBT/e × μ × τ)1/2, where kB is the Boltzmann's constant, e is the electron charge, T is the absolute temperature, μ is the Hall carrier mobility, and τ is the carrier lifetime.
| Material | Band gap [eV] | Mobility [cm2 V−1 s−1] | Carrier concentration [cm−3] | TA lifetimes [ps] | PL lifetimes [ns] |
|---|---|---|---|---|---|
| TJU-13(Br) | 3.43 | 3.37 | 3.96 × 1016 | τ 1 = 108.51 | τ 1 = 9.19 |
| τ 2 = 555.77 | τ 2 = 131.35 | ||||
| TJU-15(Br) | 3.29 | 3.83 | 2.87 × 1016 | τ 1 = 12.37 | τ 1 = 20.31 |
| τ 2 = 334.27 | τ 2 = 148.73 |
Ultrafast femtosecond TA spectroscopy (the pump laser is set as 350 nm, above the band gap, the range of probe wavelength is 400–700 nm) was further employed to gain deeper insight of the carrier transport of TJU-15(Br). The pseudo-color TA spectra of TJU-15(Br) revealed a broad positive photoinduced absorption (PIA) band ranging from 460 to 650 nm, in good agreement with the formation of self-trapped states (Fig. 3c).49,50 The gradually decreased intensity of PIA signals at longer probe delay times (∼1 ns) suggested a detrapping process. The TA dynamics of TJU-15(Br) were fitted using a global bi-exponential equation based on the decay curve, affording two time constants of 12.4 ps and 334.3 ps, respectively (Fig. 3d). The ultrafast time component τ1 represents the rapid trapping of free excitons,51 while the component τ2 is attributed to the nonradiative recombination of the STEs.52,53 Both components were consistent with the STE mechanism, providing further evidence for TJU-15(Br)'s intrinsic large-Stokes shifted broadband emission (Fig. 3a and b). In addition, electrochemical impedance spectroscopy (EIS) and transient photocurrent responses on TJU-13(Br) and TJU-15(Br) were further investigated to compare the charge separation/transfer properties between 2D and 3D structures. The 3D analogue, TJU-15(Br), exhibited a smaller Nyquist plot diameter and higher photocurrent intensity over multiple on–off cycles, strongly demonstrating that the charge transport increased via the modulation of the lead halide dimensionality (Fig. 3e and f).
Photocatalytic benzyl C(sp3)–H activation
Since C(sp3)–H activation is one important and challenging reaction in organic chemistry, we have employed the benzyl C(sp3)–H photooxidation as a model reaction to evaluate the photocatalytic performances of lead halide coordination polymers. Photocatalytic ethylbenzene oxidation was performed in a closed reaction system at room temperature by using 1 atm O2 as the oxidant. Photocatalytic products were determined by gas chromatography-mass spectroscopy (GC-MS) with naphthalene as the internal standard. Among the three lead bromide photocatalysts, TJU-15(Br) exhibited the highest photocatalytic performance with the ethylbenzene conversion rate of 245 μmol g−1 h−1, superior to the 2D lead bromide analogues (i.e. TJU-11(Br) and TJU-13(Br)) (Fig. S13 and Table S3†).In order to enhance photogenerated carrier separation and transport, we have performed the photodeposition of Pt nanoparticles on three bromide photocatalysts using K2PtCl4 as the precursor. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) indicated the Pt loadings of 0.43 wt% for TJU-11(Br), 0.35 wt% for TJU-13(Br), and 0.44 wt% for TJU-15(Br), respectively (Table S4†). Importantly, the crystallinity of all three coordination polymers was well retained after the Pt nanoparticle deposition, as evidenced by PXRD (Fig. S14, S15 and S16†). For TJU-15(Br), transmission electron microscopy (TEM) revealed successful photodeposition of Pt nanoparticles with a particle size distribution of 4–12 nm (Fig. S17†). High-resolution TEM images indicated a d-spacing of ∼0.226 nm, corresponding to the (111) lattice planes of face-centered cubic (fcc) Pt (Fig. S17†). Energy-dispersive X-ray spectroscopy (EDS) elemental mapping further confirmed the presence of Pb, Br and Pt in a single crystal of TJU-15(Br) (Fig. S18†).
Among the three lead bromide photocatalysts, Pt0.44@TJU-15(Br) exhibited the highest photocatalytic performances with the ethylbenzene conversion rate of 326 μmol g−1 h−1, similarly superior to the 2D lead bromide analogues (i.e., Pt0.43@TJU-11(Br) and Pt0.35@TJU-13(Br)) (Fig. 4a and Table S3†). In order to investigate the effect of particle size on the catalytic activity, we first tested the particle sizes of the three samples. Fig. S19† shows similar distributional center positions and trends of the three catalysts. This excludes the effect of differences in particle size on catalytic activity.
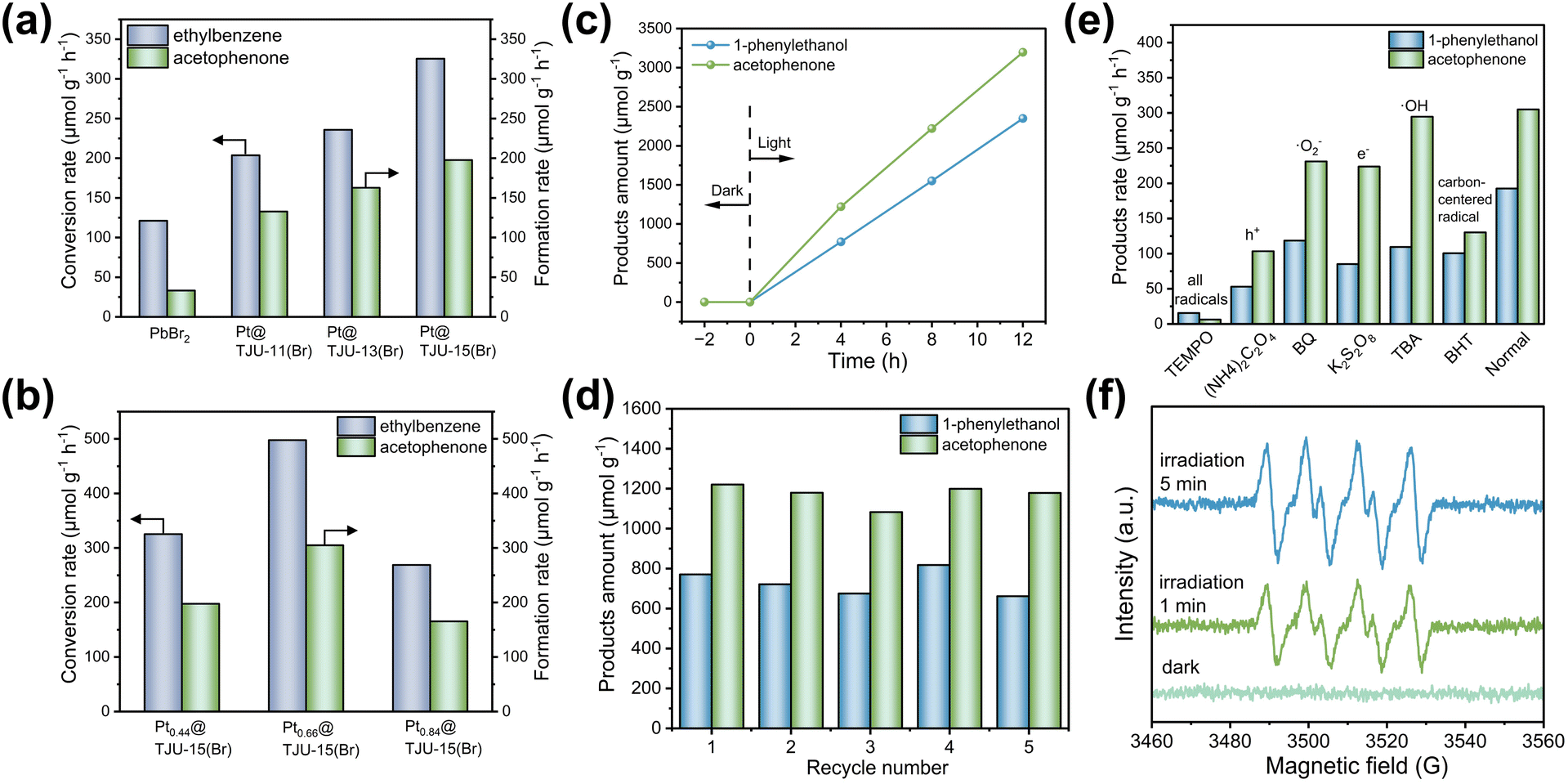 | ||
| Fig. 4 Photocatalytic ethylbenzene oxidation performances of PbBr2, Pt@TJU-11(Br), Pt@TJU-13(Br), Pt@TJU-15(Br) (a) and photocatalytic ethylbenzene oxidation performances of Pt0.44@TJU-15(Br), Pt0.66@TJU-15(Br), Pt0.84@TJU-15(Br) (b). Conditions: photocatalyst (0.02 g), 2 mL ethylbenzene and 1 mL CH3CN, 1 atm O2, 293 K, AM1.5G simulated light irradiation, reaction time: 4 h. Conversion rate of ethylbenzene and the formation rate of acetophenone are shown in the figures. (c) Time dependence of photocatalytic oxidation of ethylbenzene over Pt0.66@TJU-15(Br). (d) Recyclable catalysis studies of Pt0.66@TJU-15(Br) over five consecutive runs. (e) Ethylbenzene oxidation rates with or without various radical scavengers. (f) The DMPO spin-trapping ESR spectra of Pt0.66@TJU-15(Br) for ˙O2− under different visible-light irradiation times. | ||
Importantly, control experiments with the absence of light or photocatalyst resulted in negligible turnover. In order to further verify whether Pt deposition initiates the aerobic oxidation of produced alcohols in the reaction, we have performed the control experiments using 1-phenylethanol as the substrate under the same conditions but in the absence of light. As a result, no acetophenone products were detected, ruling out the possibility of aerobic oxidation initiated by Pt species. Therefore, the modified Pt nanoparticles play a role in promoting rapid charge transfer for photocatalysis (Fig. S20†).54,55 Using PbBr2 as the photocatalyst only yielded a low ethylbenzene conversion rate of 121 μmol g−1 h−1.
The different amounts of Pt loadings on TJU-15(Br) were examined in the ethylbenzene oxidation, and the optimized loading amount was determined to be 0.66 wt% by ICP-OES. Pt0.66@TJU-15(Br) exhibited the highest ethylbenzene conversion rates of 498 μmol g−1 h−1, with a selectivity of 61% for the corresponding ketone (i.e., acetophenone) and a selectivity of 39% for the alcohol (i.e., 1-phenylethanol) (Fig. 4b and c). Long-term stability of the Pt0.66@TJU-15(Br) catalyst was investigated by recovering the used photocatalysts by centrifugation and directly performing in the consecutive catalytic runs. No obvious loss of photocatalytic activity (<8%) was observed for at least five photocatalytic cycles (Fig. 4d). The high recyclability of Pt0.66@TJU-15(Br) was further confirmed by the retention of the crystallinity and negligible leaching of Pb2+ species (∼2%) (Fig. S21†). Moreover, our lead bromide coordination polymers do not require the addition of anhydrous Na2SO4 to remove the water generated during the reaction, distinguishing them from the moisture-sensitive perovskite-type metal halide photocatalysts reported previously.56
The photocatalytic mechanism was investigated by performing control experiments using different radical scavengers (Fig. 4e). The addition of tetra-methylpiperidine N-oxide (TEMPO) as the quenching agent for all radicals completely shut down the turnover, demonstrating that the reaction was primarily governed by a free radical-based process. Using tert-butanol (TBA) as a hydroxyl radical (˙OH) scavenger had minimal effect on the photocatalytic activity, suggesting that ˙OH radicals do not play a significant role in the photocatalysis. Moreover, using 1,4-benzoquinone (BQ) as the superoxide radical (˙O2−) scavenger or K2S2O8 as the electron (e−) scavenger led to a decrease in photocatalytic performances, suggesting that the ˙O2− and electrons are involved in the photocatalytic mechanism. Indeed, the existence of ˙O2− was again confirmed by electron paramagnetic resonance (EPR) spectroscopy under visible-light irradiation, where the characteristic peaks of the DMPO-˙O2− adduct was clearly observed in the photocatalytic reaction solution (Fig. 4f). Both the carbon-centered radical (˙R) scavenger (e.g. butylated hydroxytoluene, BHT) and the hole (h+) scavenger (e.g., (NH4)2C2O4) also render an obvious loss in photocatalytic turnovers, indicating that the carbon-centered radical and the photogenerated holes are also intermediates for the oxidation of ethylbenzene. These radical studies collectively suggest that the activation of C(sp3)–H bonds to generate ˙R radicals is a key reaction step in the mechanistic pathway, followed by the reaction between ˙R and ˙O2−/O2 to yield oxidized products (Fig. S22†). Based on the above experiments, the photocatalysis mechanism is concluded in Fig. S22.† First, photoinduced electrons and holes were generated in the CB and VB of lead bromide photocatalysts under visible light irradiation. Following this, O2 molecules captured the photoelectrons to generate active ˙O2−, whereas the holes drove the challenging oxidation of C(sp3)–H bonds in ethylbenzene to form ˙R radicals. Subsequently, the as-formed ˙R intermediates reacted with ˙O2− to form oxidized products (acetophenone as the major product). The ˙R intermediates may also react with O2 to form oxidized products (acetophenone and 1-phenylethanol are generated in equal proportions).12,57,58
Conclusions
In summary, our study has successfully achieved the precise control over the inorganic dimensionality transition from 2D to 3D in lead halide-based coordination polymers, employing three organocarboxylate ligands sharing an identical cyclohexane backbone. The resulting 3D lead halide architectures templated by hexacarboxylate ligands demonstrate superior absorption wavelength coverage and enhanced carrier-transport characteristics when compared to their quasi-2D layered analogues, which are templated by dicarboxylate or tetracarboxylate ligands. The enhancement in photophysical properties has been comprehensively studied by absorption spectroscopy, PL spectroscopy, Hall effect measurements and ultrafast transient absorption (TA) spectroscopy. Notably, the coordination polymer featuring a 3D lead bromide sublattice (TJU-15(Br)) exhibits an impressive photocatalytic ethylbenzene conversion rate of 498 μmol g−1 h−1 with a selectivity of 61% for the ketone product, clearly outperforming the performance of its 2D lead bromide counterparts. Our work presents the potential of coordination chemistry as a powerful strategy for the rational design of lead halide photocatalysts with advanced photophysical characteristics.Data availability
The data that support the finding of this study are available from the corresponding author upon reasonable request.Author contributions
Y. J. and H. F. conceived the project. H. F. supervised the project and Y. J. performed the majority of experimental studies. J. Y. assisted in the crystallographic analysis. R. X. assisted in synthesis and photophysical studies. Y. J. and H. F. wrote and revised the manuscript. All authors contributed to discussion and revisions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (21971197, 22171214), the Xiaomi Young Talents Program, the Shanghai Rising-Star Program (20QA1409500), the Natural Science Foundation of Shanghai (22ZR1463200), the Recruitment of Global Youth Experts by China and the Science & Technology Commission of Shanghai Municipality (19DZ2271500).Notes and references
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- X.-K. Liu, W. Xu, S. Bai, Y. Jin, J. Wang, R. H. Friend and F. Gao, Nat. Mater., 2021, 20, 10–21 CrossRef CAS PubMed.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef PubMed.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS PubMed.
- I. Rosa-Pardo, D. Zhu, A. Cortés-Villena, M. Prato, L. De Trizio, L. Manna, R. E. Galian and J. Pérez-Prieto, ACS Energy Lett., 2023, 8, 2789–2798 CrossRef CAS PubMed.
- Y. Fan, W. Zhou, X. Qiu, H. Li, Y. Jiang, Z. Sun, D. Han, L. Niu and Z. Tang, Nat. Sustain., 2021, 4, 509–515 CrossRef.
- G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 4078–4082 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- H. Wang, C. Cao, D. Li, Y. Ge, R. Chen, R. Song, W. Gao, X. Wang, X. Deng, H. Zhang, B. Ye, Z. Li and C. Li, J. Am. Chem. Soc., 2023, 145, 16852–16861 CrossRef CAS PubMed.
- X. Xiao, Z. Ruan, Q. Li, L. Zhang, H. Meng, Q. Zhang, H. Bao, B. Jiang, J. Zhou, C. Guo, X. Wang and H. Fu, Adv. Mater., 2022, 34, 2200612 CrossRef CAS PubMed.
- H. Huang, H. Yuan, J. Zhao, G. Solís-Fernández, C. Zhou, J. W. Seo, J. Hendrix, E. Debroye, J. A. Steele, J. Hofkens, J. Long and M. B. J. Roeffaers, ACS Energy Lett., 2019, 4, 203–208 CrossRef CAS.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- Q. Fu, X. Tang, B. Huang, T. Hu, L. Tan, L. Chen and Y. Chen, Adv. Sci., 2018, 5, 1700387 CrossRef PubMed.
- N. K. Tailor, Yukta, R. Ranjan, S. Ranjan, T. Sharma, A. Singh, A. Garg, K. S. Nalwa, R. K. Gupta and S. Satapathi, J. Mater. Chem. A, 2021, 9, 21551–21575 RSC.
- Y. Liu, S. Yuan, H. Zheng, M. Wu, S. Zhang, J. Lan, W. Li and J. Fan, Adv. Energy Mater., 2023, 13, 2300188 CrossRef CAS.
- T. B. Shonde, M. Chaaban, H. Liu, O. J. Olasupo, A. Ben-Akacha, F. G. Gonzalez, K. Julevich, X. Lin, J. S. R. V. Winfred, L. M. Stand, M. Zhuravleva and B. Ma, Adv. Mater., 2023, 35, 2301612 CrossRef CAS PubMed.
- C. Zhou, H. Lin, M. Worku, J. Neu, Y. Zhou, Y. Tian, S. Lee, P. Djurovich, T. Siegrist and B. Ma, J. Am. Chem. Soc., 2018, 140, 13181–13184 CrossRef CAS PubMed.
- I. Spanopoulos, I. Hadar, W. Ke, P. Guo, S. Sidhik, M. Kepenekian, J. Even, A. D. Mohite, R. D. Schaller and M. G. Kanatzidis, J. Am. Chem. Soc., 2020, 142, 9028–9038 CrossRef CAS PubMed.
- P. M. Forster, N. Stock and A. K. Cheetham, Angew. Chem., Int. Ed., 2005, 44, 7608–7611 CrossRef CAS PubMed.
- M. Dan and C. N. R. Rao, Angew. Chem., Int. Ed., 2006, 45, 281–285 CrossRef CAS PubMed.
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 46, 4780–4795 RSC.
- J. Q. Zhao, M. F. Han, X. J. Zhao, Y. Y. Ma, C. Q. Jing, H. M. Pan, D. Y. Li, C. Y. Yue and X. W. Lei, Adv. Opt. Mater., 2021, 9, 2100556 CrossRef CAS.
- Y. Jiang, J. Yin and H. Fei, J. Mater. Chem. C, 2022, 10, 13254–13261 RSC.
- B. B. Cui, Y. Han, B. Huang, Y. Zhao, X. Wu, L. Liu, G. Cao, Q. Du, N. Liu, W. Zou, M. Sun, L. Wang, X. Liu, J. Wang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 5190 CrossRef PubMed.
- W. Ma, J. Yin, X. Chen, C. Sun, X. Song and H. Fei, Chem. Mater., 2022, 34, 4403–4413 CrossRef CAS.
- G. Yu, F. Lin, K. Zhou, S. Fang, Y. Shi, W. Liu, H. Hu, B. Ma and H. Lin, Chem. Mater., 2021, 33, 5668–5674 CrossRef CAS.
- D. Y. Li, J. H. Song, Y. Cheng, X. M. Wu, Y. Y. Wang, C. J. Sun, C. Y. Yue and X. W. Lei, Angew. Chem., Int. Ed., 2022, 61, e202206437 CrossRef CAS PubMed.
- J. Chen, S. Zhang, X. Pan, R. Li, S. Ye, A. K. Cheetham and L. Mao, Angew. Chem., Int. Ed., 2022, 61, e202205906 CrossRef CAS PubMed.
- Z. Zhuang, C. Peng, G. Zhang, H. Yang, J. Yin and H. Fei, Angew. Chem., Int. Ed., 2017, 56, 14411–14416 CrossRef CAS PubMed.
- C. Peng, Z. Zhuang, H. Yang, G. Zhang and H. Fei, Chem. Sci., 2018, 9, 1627–1633 RSC.
- C. Peng, X. Song, J. Yin, G. Zhang and H. Fei, Angew. Chem., Int. Ed., 2019, 58, 7818–7822 CrossRef CAS PubMed.
- J. Yin, Y. Yu, X. Song, Y. Jiang and H. Fei, CCS Chem., 2022, 4, 540–547 CrossRef CAS.
- C. Sun, R. Xi and H. Fei, Acc. Chem. Res., 2023, 56, 452–461 CrossRef CAS PubMed.
- Y. Jiang and H. Fei, Adv. Opt. Mater., 2022, 10, 2102148 CrossRef CAS.
- R. Xi, Y. Jiang, Y. Li, J. Yin and H. Fei, Inorg. Chem. Front., 2023, 10, 2645–2652 RSC.
- X. Song, G. Wei, J. Sun, C. Peng, J. Yin, X. Zhang, Y. Jiang and H. Fei, Nat. Catal., 2020, 3, 1027–1033 CrossRef CAS.
- J. Yin, H. Yang and H. Fei, Chem. Mater., 2019, 31, 3909–3916 CrossRef CAS.
- H. Yang, J. Yin, X. Xu and H. Fei, J. Mater. Chem. C, 2019, 7, 7090–7095 RSC.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, Chem. Sci., 2012, 3, 2812–2822 RSC.
- R. Yuan, S. Fan, H. Zhou, Z. Ding, S. Lin, Z. Li, Z. Zhang, C. Xu, L. Wu, X. Wang and X. Fu, Angew. Chem., Int. Ed., 2013, 52, 1035–1039 CrossRef CAS PubMed.
- Z. Wu, Q. Zhang, B. Li, Z. Shi, K. Xu, Y. Chen, Z. Ning and Q. Mi, Chem. Mater., 2019, 31, 4999–5004 CrossRef CAS.
- J. Li, X. Wang, Y. Tan, D. Liang, Y. Zou, L. Cai, T. Wu, K. Wen, Y. Wang, Y. Li, T. Song, L. Wang and B. Sun, Adv. Optical Mater., 2020, 8, 2001073 CrossRef CAS.
- E. R. Dohner, A. Jaffe, L. R. Bradshaw and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 13154–13157 CrossRef CAS PubMed.
- E. R. Dohner, E. T. Hoke and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 1718–1721 CrossRef CAS PubMed.
- L. Mao, P. Guo, M. Kepenekian, I. Hadar, C. Katan, J. Even, R. D. Schaller, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 13078–13088 CrossRef CAS PubMed.
- Z. Yuan, C. Zhou, Y. Tian, Y. Shu, J. Messier, J. C. Wang, L. J. van de Burgt, K. Kountouriotis, Y. Xin, E. Holt, K. Schanze, R. Clark, T. Siegrist and B. Ma, Nat. Commun., 2017, 8, 14051 CrossRef CAS PubMed.
- T. Hu, M. D. Smith, E. R. Dohner, M.-J. Sher, X. Wu, M. T. Trinh, A. Fisher, J. Corbett, X. Y. Zhu, H. I. Karunadasa and A. M. Lindenberg, J. Phys. Chem. Lett., 2016, 7, 2258–2263 CrossRef CAS PubMed.
- M. D. Smith and H. I. Karunadasa, Acc. Chem. Res., 2018, 51, 619–627 CrossRef CAS PubMed.
- L. Zhou, J.-F. Liao, Z.-G. Huang, J.-H. Wei, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, Angew. Chem., Int. Ed., 2019, 58, 15435–15440 CrossRef CAS PubMed.
- J. Xu, S. Li, C. Qin, Z. Feng and Y. Du, J. Phys. Chem. C, 2020, 124, 11625–11630 CrossRef CAS.
- Y. Zhang, F. Wang, X. Feng, Z. Zhang, K. Liu, F. Xia, W. Liang, X. Hu, Y. Ma, H. Li, G. Xing and T. Zhai, Adv. Funct. Mater., 2022, 32, 2205757 CrossRef CAS.
- L. Zhou, J.-F. Liao, Y. Qin, X.-D. Wang, J.-H. Wei, M. Li, D.-B. Kuang and R. He, Adv. Funct. Mater., 2021, 31, 2102654 CrossRef CAS.
- J.-D. Xiao, Q. Shang, Y. Xiong, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9389–9393 CrossRef CAS PubMed.
- F. Guo, Y.-P. Wei, S.-Q. Wang, X.-Y. Zhang, F.-M. Wang and W.-Y. Sun, J. Mater. Chem. A, 2019, 7, 26490–26495 RSC.
- Y. Dai, C. Poidevin, C. Ochoa-Hernandez, A. A. Auer and H. Tuysuz, Angew. Chem., Int. Ed., 2020, 59, 5788–5796 CrossRef CAS PubMed.
- Z. Zhang, Y. Yang, Y. Wang, L. Yang, Q. Li, L. Chen and D. Xu, Angew. Chem., Int. Ed., 2020, 59, 18136–18139 CrossRef CAS PubMed.
- Z.-J. Bai, Y. Mao, B.-H. Wang, L. Chen, S. Tian, B. Hu, Y.-J. Li, C.-T. Au and S.-F. Yin, Nano Res., 2023, 16, 6104–6112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional characterization. CCDC 2294271, 2294272, 2294273 and 2294304. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04969h |
| This journal is © The Royal Society of Chemistry 2024 |