
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligase-mediated synthesis of CuII-responsive allosteric DNAzyme with bifacial 5-carboxyuracil nucleobases†
Yusuke
Takezawa
 *,
Hanci
Zhang
,
Keita
Mori
,
Lingyun
Hu
and
Mitsuhiko
Shionoya
*
*,
Hanci
Zhang
,
Keita
Mori
,
Lingyun
Hu
and
Mitsuhiko
Shionoya
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: takezawa@chem.s.u-tokyo.ac.jp; shionoya@chem.s.u-tokyo.ac.jp
First published on 20th January 2024
Abstract
A CuII-responsive allosteric DNAzyme has been developed by introducing bifacial 5-carboxyuracil (caU) nucleobases, which form both hydrogen-bonded caU–A and metal-mediated caU–CuII–caU base pairs. The base sequence was logically designed based on a known RNA-cleaving DNAzyme so that the caU-modified DNAzyme (caU-DNAzyme) can form a catalytically inactive structure containing three caU–A base pairs and an active form with three caU–CuII–caU pairs. The caU-DNAzyme was synthesized by joining short caU-containing fragments with a standard DNA ligase. The activity of caU-DNAzyme was suppressed without CuII, but enhanced 21-fold with the addition of CuII. Furthermore, the DNAzyme activity was turned on and off during the reaction by the addition and removal of CuII ions. Both ligase-mediated synthesis and CuII-dependent allosteric regulation were achieved by the bifacial base pairing properties of caU. This study provides a new strategy for designing stimuli-responsive DNA molecular systems.
Introduction
The highly sophisticated molecular recognition abilities of nucleic acids based on complementary hydrogen-bonded base pairing have led to a dramatic growth in the research field recognized as DNA nanotechnology.1 Numerous DNA nanodevices, sensors, and molecular machines have been created by controlling DNA hybridization and structures in response to stimuli such as DNA/RNA binding, pH changes, and light irradiation.2 Metal ions also serve as external stimuli to regulate the DNA structure and function, particularly by exploiting metal-mediated unnatural base pairing.3 Metal-mediated artificial base pairs are formed between two opposing ligand-type nucleobase analogs by complexation with a bridging metal ion. Metal-mediated base pairing generally stabilizes DNA duplexes, thus controlling DNA hybridization in a metal-dependent manner.Aiming to switch DNA functions efficiently by metal complexation, we have recently established a new concept of metal-mediated base pair switching of bifacial 5-modified pyrimidine nucleobases.4–7 Bifacial bases such as 5-hydroxyuracil (UOH)4,5 and 5-carboxyuracil (caU)6 are designed to form metal-mediated self-base pairs (e.g., UOH–GdIII–UOH) in the presence of certain metal ions and to form Watson–Crick-like base pairs with a natural nucleobase in DNA duplexes (e.g., UOH–A). Based on the switching between UOH–A and UOH–GdIII–UOH base pairs, GdIII-triggered DNA strand displacement reactions were demonstrated and GdIII-mediated control of DNA tweezer structures and DNAzyme functions was successfully achieved.5
In this study, a metal-responsive DNAzyme was newly developed by utilizing bifacial 5-carboxyuracil (caU) bases as metal binding sites (Fig. 1). DNAzymes are DNA molecules with catalytic activity that have been widely applied for the development of DNA-based biosensors and molecular machines.8 Of particular interest is the rational design of allosteric DNAzymes whose activity can be controlled in response to specific stimuli. Such stimuli-responsive DNAzymes are versatile components for building up deformable DNA nanoarchitectures as well as DNA reaction networks. Metal-responsive DNAzymes have been developed previously by incorporating metal-mediated base pairs such as CuII-mediated hydroxypyridone base pairs (H–CuII–H).9 The bifacial caU bases used in this study not only form hydrogen-bonded caU–A base pairs, but also CuII-mediated caU–CuII–caU base pairs with high metal selectivity.6 The caU–CuII–caU base pairing significantly stabilizes the DNA duplexes (ΔTm = +30.7 °C for a duplex with three caU–CuII–caU pairs), whereas the caU–A base pairs are destabilized by the addition of CuII ions (ΔTm = −7.3 °C for a duplex with three caU–A pairs by using 6 equiv. of CuII ions). Therefore, carefully designed DNAzymes containing caU bases were expected to exhibit good responsiveness to CuII ions. To prepare DNAzyme strands containing multiple caU nucleotides, enzymatic synthesis using a DNA ligase was also investigated. A standard T4 DNA ligase was utilized because it was reported to tolerate modified base pairs and backbones.9b,d,10 Thus, we expected that caU-containing short fragments could be enzymatically ligated to give long DNA strands modified with caU bases.
 | ||
| Fig. 1 Ligase-mediated synthesis of a CuII-responsive DNAzyme containing bifacial 5-carboxyuracil (caU) bases, which form hydrogen-bonded caU–A and metal-mediated caU–CuII–caU base pairs. U in the figure represents a caU base. | ||
Results and discussion
The CuII-responsive allosteric DNAzyme was logically designed by modifying the base sequence of the known RNA-cleaving NaA43 DNAzyme11 (Fig. 2a) in a manner similar to the GdIII-responsive DNAzyme with UOH bases.5 The NaA43 DNAzyme was chosen because it does not require metal cofactors that can be trapped by common chelators used to selectively remove CuII ions for reversible regulation of DNAzyme activity (vide infra). Since duplex stabilization is more pronounced when three or more consecutive caU–CuII–caU pairs are used,6 we decided to incorporate three caU–CuII–caU pairs into the parent NaA43 DNAzyme. Three pairs of caU bases were introduced into the stem region and the surrounding bases (shown in orange) were redesigned to form a different secondary structure in the absence of CuII ions. The caU-modified DNAzyme (caU-DNAzyme) was expected to undergo a structural change upon addition of CuII ions, from a catalytically inactive structure with three caU–A base pairs to an active form with three caU–CuII–caU base pairs (Fig. 2b). The plausible secondary structure was simulated using the NUPACK software12 (Fig. S1†). The caU bases were replaced with natural T bases to calculate the structure in the absence of CuII, and the potential caU–CuII–caU pairs were changed to G–C base pairs to simulate the structure in the presence of CuII. The results indicated that both the inactive and the active structures can be stably formed via the formation of caU–A and caU–CuII–caU base pairs, respectively.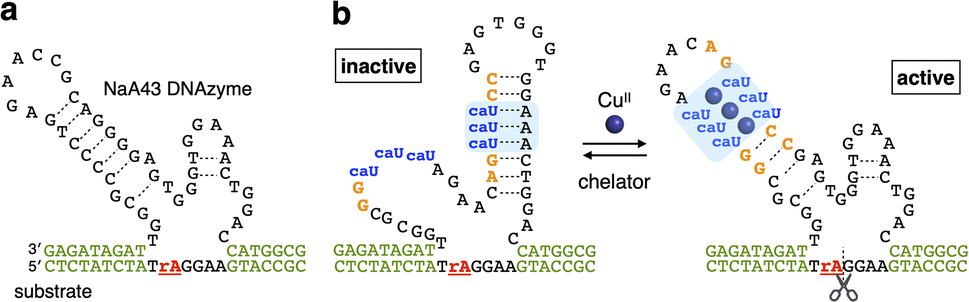 | ||
| Fig. 2 Molecular design of a CuII-responsive allosteric DNAzyme with caU nucleobases (caU-DNAzyme). (a) Sequence of the parent RNA-cleaving DNAzyme (NaA43 DNAzyme). (b) Sequence of caU-DNAzyme. Both the active structure with caU–CuII–caU base pairs and the inactive structure with caU–A base pairs are shown. “rA” in the substrate indicates an adenosine ribonucleotide at the cleavage site. | ||
A caU-modified DNAzyme strand (58-nt) was synthesized by ligating short DNA fragments (Fig. 3a). The caU-containing strands can be chemically synthesized based on the conventional phosphoramidite chemistry,6 but the chemical synthesis requires an additional protecting group on the carboxylate of the caU bases. Although the coupling yield is sufficiently high, incomplete deprotection often makes purification of long oligonucleotides containing multiple caU bases very difficult. Therefore, we expected that the ligase-mediated synthesis would be a suitable strategy to synthesize caU-modified DNAzymes. Since caU nucleobases can form caU–A base pairs with adenine bases on the complementary DNA, caU-modified oligonucleotides were expected to assemble on the splint strand and to be ligated by a standard T4 DNA ligase. A DNA tetramer 5′-caUcaUcaUG-3′ (1) containing three caU nucleotides was used to introduce three consecutive caU bases into the resulting DNA strands. A natural nucleotide G was added at the 3′-terminal so that an additional G–C base pairing would facilitate the hybridization of tetramer 1 to the splint DNA 5. The caU-containing fragment 1 was prepared using an automated DNA synthesizer according to the reported procedure.6 Prior to the ligation reaction, fragments 1, 2, and 3 were treated with T4 polynucleotide kinase (T4 PNK) to introduce a phosphate group at the 5′ end (step i). After all the DNA fragments were hybridized to splint 5 (step ii), a ligation reaction was performed using T4 DNA ligase (step iii). Reaction products were analyzed by denaturing polyacrylamide gel electrophoresis (PAGE) (Fig. 3b). After incubation at 16 °C for 18 h, fragment 4 was efficiently consumed and a low mobility band appeared. The mobility of the new band was nearly identical to that of a chemically synthesized T-DNAzyme strand (58-nt) in which the caU nucleotides of the caU-DNAzyme were replaced with natural thymidines (T). Comparing the band intensities, the reaction yield reached 70% or more, confirming that the ligase reaction was proceeding well. The formation of the desired caU-DNAzyme strand was confirmed by MALDI mass spectrometry after isolation ([M–2H]2−: calcd 9150.31, found 9150.67, Fig. S2†). It was demonstrated that T4 PNK and T4 DNA ligase successfully phosphorylated and ligated the short strand 1 (5′-caUcaUcaUG-3′) despite the presence of a modified caU base at the 5′ end. Since the desired product can be easily isolated by denaturing PAGE, the ligase-mediated synthesis was shown to be a powerful alternative method to incorporate multiple caU bases into functional DNA sequences.
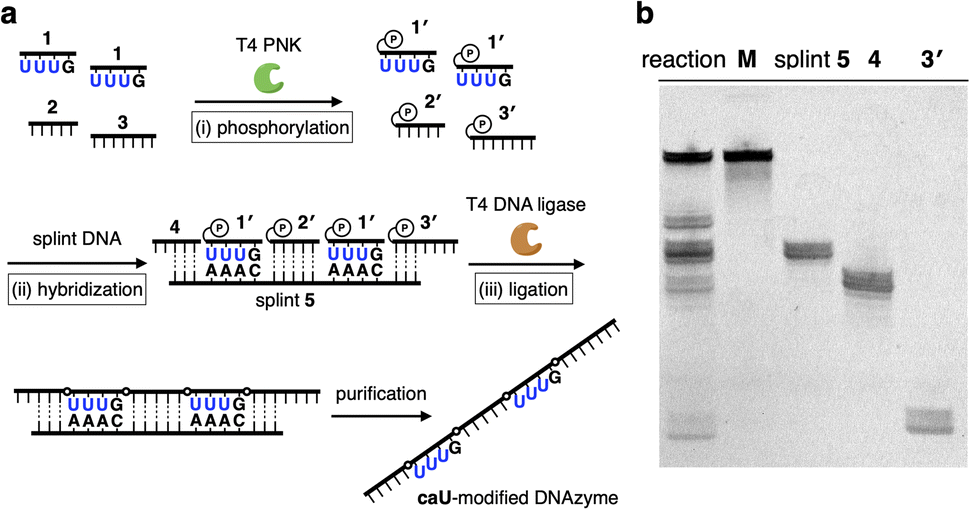 | ||
| Fig. 3 Ligase-mediated synthesis of DNA strands containing caU nucleobases. (a) Reaction scheme. (i) A short DNA strand 1 containing three caU bases (5′-caUcaUcaUG-3′) was phosphorylated by T4 polynucleotide kinase (T4 PNK). (ii) DNA fragments were hybridized with a splint DNA strand 5. (iii) The DNA fragments were ligated by T4 DNA ligase. [1′] = 30 μM, [2′] = [3′] = 12 μM, [4] = [5] = 10 μM, [ligase] = 2 U per μL in quick ligation buffer, 16 °C, 18 h. U in the figure represents a caU base. (b) Denaturing PAGE analysis of the reaction products. The bands were detected after SYBR gold staining. M: chemically synthesized T-DNAzyme, which has T bases instead of caU bases. | ||
Using the resulting caU-modified DNAzyme strand, we examined whether the catalytic activity of caU-DNAzyme can be regulated in response to the addition of CuII ions. DNAzyme-catalyzed RNA-cleaving reactions were performed using 10 equiv. of substrates labeled with a fluorescent dye (FAM). Substrate cleavage was quantitatively evaluated by denaturing PAGE analysis (Fig. S3†). Fig. 4a compares the RNA cleavage reaction catalyzed by caU-DNAzyme in the presence of varying concentrations of CuII ions. In the absence of CuII ions, the RNA-cleaving activity of the caU-DNAzyme was greatly suppressed. The DNAzyme activity was found to be enhanced by the addition of CuII ions; the highest activity was observed when 9 equiv. of CuII ions were added. Time-course analysis further confirmed the CuII-dependent activation of caU-DNAzyme (Fig. 4b and S4†). Under the same conditions, the catalytic activity of the unmodified NaA43 DNAzyme was reduced by adding CuII ions (Fig. S5†). A control T-DNAzyme containing natural T bases in place of caU showed no RNA-cleaving activity both in the absence and presence of CuII ions. These results clearly demonstrate that the addition of CuII ions enhances the catalytic activity of the caU-modified DNAzyme. In the presence of CuII ions, the caU-DNAzyme cleaved approximately 70% of the substrate (i.e., 7 equiv.) in 20 h, confirming that the modified DNAzyme maintains multiple turnover ability.
 | ||
| Fig. 4 (a) RNA-cleaving activity of caU-DNAzyme in the presence of varying concentrations of CuII ions. The fractions of the cleaved substrate after a 6 h reaction are shown (see also Fig. S4†). (b) RNA-cleaving activity of caU-DNAzyme and the parent NaA43 DNAzyme in the absence and the presence of 9 equiv. of CuII. T-DNAzyme, in which all caU bases are replaced with natural T bases, was used as a control. [DNAzyme] = 1.0 μM, [substrate] = 10 μM, [CuSO4] = 0, 9.0 μM in 10 mM HEPES (pH 7.0), 100 mM NaCl, 25 °C. (c) CuII-mediated change in the hybridization partners of the caU-containing strand (1U). (d) Native PAGE analysis of the hybridization product in the presence of varying amounts of CuII ions. [DNA] = 2 μM each in 10 mM HEPES (pH 7.0) and 100 mM NaCl. The samples were annealed prior to the analysis. FAM detection. | ||
It is most likely that the activity of caU-DNAzyme was switched based on the changes in the base-pairing partners of the caU bases. This was supported by a model experiment using a FAM-labeled strand containing three caU bases in the middle (1U) (Fig. 4c and S6†). The DNA 1U was annealed with two complementary strands 2U and 2A containing three caU or A bases, respectively, and the hybridization products were analyzed by native PAGE (Fig. 4d). Under CuII-free conditions, only the duplex 1U·2A with caU–A pairs was formed. In the presence of CuII ions, the duplex 1U·2U with caU–CuII–caU pairs was formed (up to about 60%). These results clearly show the CuII-mediated change in the hybridization partners, which is the driving force behind the allosteric regulation of the caU-DNAzyme.
The catalytically active form contains three caU–CuII–caU base pairs, but the maximum activity was observed in the presence of 9 equiv. of CuII ions (Fig. 4a). This inconsistency can be explained by the stability of the caU–A base pairs in addition to the caU–CuII–caU pairs.6 Melting experiments (Fig. S7†) showed that the model 15-bp duplex 1U′·2U, containing three caU–caU pairs, exhibited the highest melting temperature (Tm) in the presence of 3 equiv. of CuII ions, due to the quantitative formation of caU–CuII–caU pairs. On the other hand, the stability of duplex 1U′·2A′ with three caU–A pairs decreases with increasing amounts of CuII ions, possibly due to the binding of CuII ions to the caU bases.6 The difference in the Tm values of duplexes 1U′·2U and 1U′·2A′ was maximal when more than 3 equiv. of CuII were added. In fact, the hybridization experiments (Fig. 4d) showed that an excess of CuII ions are required to change the hybridization partners of the caU bases. Therefore, the requirement for an excess of CuII ions to activate the caU-DNAzyme suggests that the DNAzyme functions through both CuII-mediated destabilization of the caU–A pairs (inactive state) and CuII-mediated caU–CuII–caU base pair formation (active state) exactly as designed.
The apparent first-order rate constants (kobs) for the DNAzyme reactions were estimated from the initial rates (Table 1). The catalytic activity of caU-DNAzyme was found to increase by approximately 21-fold with the addition of 9 equiv. of CuII ions. The maximum activity of the caU-DNAzyme (kobs = 4.2 × 10−2 h−1) was lower than that of the unmodified NaA43 DNAzyme (kobs = 4.7 × 10−1 h−1). This may be due not only to the incomplete transformation into the active state, but also to the structural distortion caused by the caU–CuII–caU base pairs.6 As indicated by circular dichroism (CD) analysis in the previous study,6caU–CuII–caU base pairing can unwind the stem duplex to some extent. Introducing the caU base at a position more distant from the catalytic core would reduce the negative effect on the DNAzyme activity. It is noteworthy that the CuII-mediated activation of caU-DNAzyme (21-fold) was much more efficient than that of a CuII-responsive H-modified DNAzyme (5.9-fold), which was developed by incorporating an H–CuII–H base pair into the same NaA43 DNAzyme. The results clearly show that the bifacial caU nucleobases, which form both hydrogen-bonded caU–A and metal-mediated caU–CuII–caU base pairs, are useful for metal-responsive switching of DNA functions.
| DNAzyme | k obs/h−1 | Ratio | |
|---|---|---|---|
| CuII+ | CuII– | ||
| a A NaA43 DNAzyme modified with a pair of hydroxypyridone (H) nucleobases.9b b In the presence of 9 equiv. of CuII ions. c In the presence of 1 equiv. of CuII ions. | |||
| caU-DNAzyme | 4.3 × 10−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
2.0 × 10−3 | 21 |
| NaA43 DNAzyme | 4.7 × 10−1b |
8.5 × 10−1 | 0.56 |
| T-DNAzyme | <1.0 × 10−3b |
<1.0 × 10−3 | — |
| H-modified DNAzymea | 2.8 × 10−1c |
4.7 × 10−2 | 5.9 |
We further carried out the DNAzyme reactions in the presence of HgII ions that can mediate caU–HgII–caU base pairing6 (Fig. S8†). In contrast to CuII ions, the addition of HgII ions did not activate caU-DNAzyme at all. Note that the activity of the unmodified NaA43 DNAzyme was significantly reduced by the addition of HgII ions. This is probably due to the undesired binding of HgII ions to the natural T bases. These results clearly demonstrate the advantage of using metal ions that do not interact strongly with natural bases (e.g., CuII), especially with long oligonucleotides such as DNAzymes.
The RNA-cleaving activity of caU-DNAzyme was reversibly regulated in response to CuII ions during the reaction (Fig. 5). The reaction was initiated in the absence of CuII ions, and after 4 h, CuII ions (9 equiv.) were added. The reaction rate was immediately increased to kobs = 3.9 × 10−2 h−1, which is comparable to the rate observed when the reaction was initiated with CuII ions (Fig. 5a). In a similar manner, removal of CuII ions was shown to inactivate caU-DNAzyme. Addition of the chelating agent EDTA or CuII-binding peptide (GHK)13 in equimolar amounts with CuII ions (9 equiv.) immediately slowed the reaction (kobs = 2.5 × 10−3 h−1 and 5.3 × 10−3 h−1, respectively) (Fig. 5b). The addition of sodium ascorbate, which can reduce CuII to CuI, also decreased the activity of caU-DNAzyme to kobs = 3.6 × 10−3 h−1 (Fig. S9†). These results demonstrate that the activity of caU-DNAzyme can be rapidly switched by the addition, removal, and reduction of CuII ions under isothermal conditions. Alternate addition of CuII ions (9 equiv.) and EDTA (9 equiv.) cycled the on–off switching of caU-DNAzyme (Fig. 5c). The kobs values in each step demonstrate a clear switching in the caU-DNAzyme activity in response to CuII (Fig. 5d).
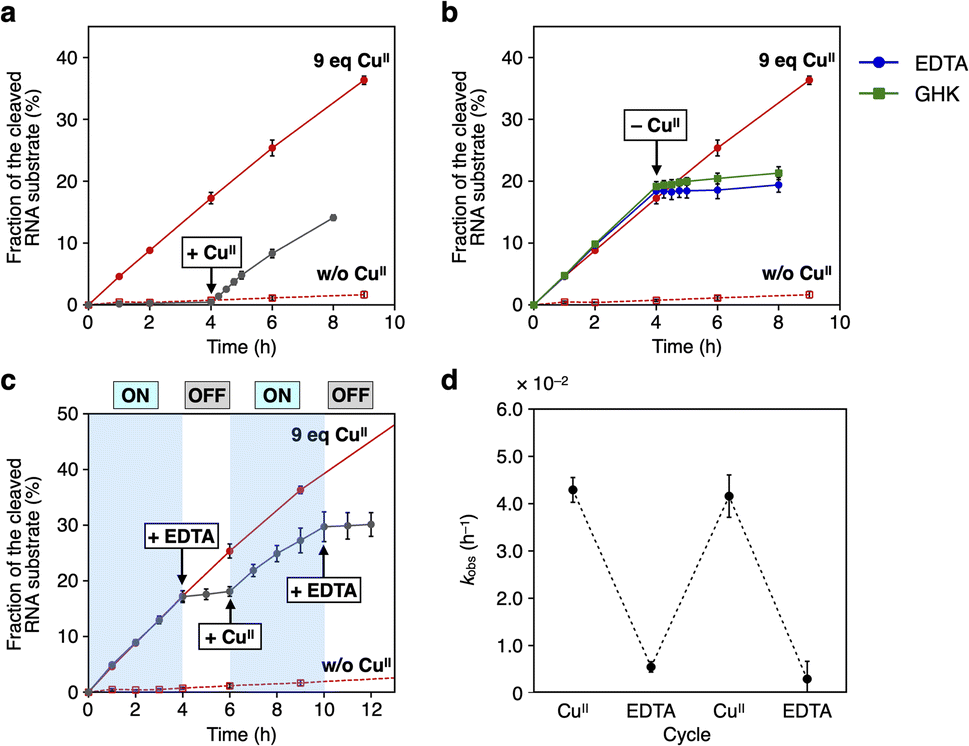 | ||
| Fig. 5 CuII-dependent regulation of the RNA-cleaving activity of caU-DNAzyme. (a) Activation of caU-DNAzyme by the addition of CuII ions (9 equiv.). (b) Deactivation of caU-DNAzyme by the removal of CuII ions with a chelating agent EDTA or a CuII-binding tripeptide (GHK) (9 equiv.). (c) Iterative switching of the DNAzyme activity. CuII (9 equiv.) and EDTA (9 equiv.) were alternately added. [DNAzyme] = 1.0 μM, [substrate] = 10 μM, 25 °C. The activities of caU-DNAzyme in the absence (red dotted lines) and presence of CuII ions (red solid lines) are also shown. (d) Apparent first-order rate constant (kobs) for each step. N = 3. Error bars indicate standard errors. | ||
Conclusions
In summary, a CuII-responsive allosteric DNAzyme was developed by introducing 5-carboxyuracil (caU) nucleobases into a known DNAzyme sequence. The caU-modified DNAzyme (caU-DNAzyme) was enzymatically synthesized by joining short caU-containing fragments with a standard T4 DNA ligase. The ligase-mediated synthesis was possible because the caU base was structurally similar to the natural T base and could form a Watson–Crick-like base pair with the A base on the splint DNA. The base sequence of caU-DNAzyme is logically designed to form both the catalytically inactive structure by caU–A base pairing and the active form by metal-mediated caU–CuII–caU base pairing. The activity of caU-DNAzyme was enhanced 21-fold by the addition of CuII ions and could be turned on and off during the reaction by the addition and removal (or reduction) of CuII ions. These results demonstrate that the caU-modified DNAzyme was allosterically regulated through metal-mediated base-pair switching between caU–A and caU–CuII–caU. The use of CuII is essential to induce base-pair switching of the caU base. The caU base can form other types of metal-mediated base pairs such as caU–HgII–T, caU–AgI–C, and caU–CuII–G.6 Therefore, caU-modified DNAs are expected to be further applied in constructing more complex DNA systems responsive to multiple metal ions.This study confirms that metal-responsive DNA systems can be logically designed based on metal-mediated base-pair switching of bifacial caU nucleobases. The strategic design of caU-modified DNAzymes is expected to be applied to other types of DNAzymes14 and other functional DNAs as well. Ligase-mediated synthesis provides a simple way to incorporate caU bases into longer DNA sequences and is advantageous for sequence screening. Thus, it is suggested that the incorporation of bifacial caU bases is a powerful strategy for creating a variety of CuII-responsive DNA molecular systems. The range of metal ions used could be expanded by developing other types of bifacial nucleobases with a different metal coordinating functionality at the 5-position of pyrimidine bases. In fact, GdIII-responsive DNA systems have been developed using cognate UOH nucleobases.5 The bifacial 5-modified pyrimidine bases are expected to be introduced into DNA via the ligase-mediated synthesis, similar to the case with caU. Therefore, metal-mediated base-pair switching of bifacial nucleobases and the ligase-mediated synthesis have the potential to be versatile tools for building DNA-based stimuli-responsive systems such as biosensors, molecular machines, and computing devices. Further applications of bifacial caU nucleobases to metal-triggered operation of DNA nanoarchitectures and DNA logic circuits are currently under investigation.
Data availability
All the data supporting this study are included in the main text and the ESI.†Author contributions
Y. T. and M. S. conceived and directed the study. H. Z. and K. M. performed the experiments and analyzed the data with the aid of Y. T. and L. H. All the authors prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP18H02081 and JP21H02055 to Y. T., JP21H05022 to M. S., MEXT KAKENHI Grant Numbers JP21H00384 (Molecular Engine), JP21H05866, and JP23H04399 (Molecular Cybernetics) to Y. T., and JP16H06509 (Coordination Asymmetry) to M. S., and Grant-in-Aid for JSPS Fellows to K. M. (JP21J11325) and to L. H. (JP21J11332), Japan. We would like to thank Ms Yoko Shimada for her cooperation in the experiment.Notes and references
- (a) N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2018, 3, 17068 CrossRef CAS; (b) M. Madsen and K. V. Gothelf, Chem. Rev., 2019, 119, 6384–6458 CrossRef CAS PubMed; (c) E. Del Grosso, E. Franco, L. J. Prins and F. Ricci, Nat. Chem., 2022, 14, 600–613 CrossRef CAS PubMed.
- (a) H. Ramezani and H. Dietz, Nat. Rev. Genet., 2020, 21, 5–26 CrossRef CAS PubMed; (b) S. Lu, J. Shen, C. Fan, Q. Li and X. Yang, Adv. Sci., 2021, 8, 2100328 CrossRef CAS PubMed; (c) F. Wang, X. Liu and I. Willner, Angew. Chem., Int. Ed., 2015, 54, 1098–1129 CrossRef CAS PubMed; (d) S. Murata, T. Toyota, S. M. Nomura, T. Nakakuki and A. Kuzuya, Adv. Funct. Mater., 2022, 32, 2201866 CrossRef CAS.
- (a) Y. Takezawa, J. Müller and M. Shionoya, Chem. Lett., 2017, 46, 622–633 CrossRef CAS; (b) Y. Takezawa and M. Shionoya, Acc. Chem. Res., 2012, 45, 2066–2076 CrossRef CAS PubMed; (c) S. Naskar, R. Guha and J. Müller, Angew. Chem., Int. Ed., 2020, 59, 1397–1406 CrossRef CAS PubMed; (d) Y. Tanaka, J. Kondo, V. Sychrovský, J. Šebera, T. Dairaku, H. Saneyoshi, H. Urata, H. Torigoe and A. Ono, Chem. Commun., 2015, 51, 17343–17360 RSC; (e) Y. Takezawa and M. Shionoya, in Modern Avenues in Metal-Nucleic Acid Chemistry (Metal Ions In Life Science), vol. 25, ed. J. Müller and B. Lippert, CRC Press, Boca Raton, 2023, pp. 257–289 Search PubMed; (f) Y. Takezawa and M. Shionoya, in Handbook of Chemical Biology of Nucleic Acids, ed. N. Sugimoto, Springer, Singapore, 2023, pp. 2645–2683 Search PubMed.
- (a) Y. Takezawa, K. Nishiyama, T. Mashima, M. Katahira and M. Shionoya, Chem.–Eur. J., 2015, 21, 14713–14716 CrossRef CAS PubMed; (b) K. Nishiyama, Y. Takezawa and M. Shionoya, Inorg. Chim. Acta, 2016, 452, 176–180 CrossRef CAS; (c) K. Nishiyama, K. Mori, Y. Takezawa and M. Shionoya, Chem. Commun., 2021, 57, 2487–2490 RSC.
- Y. Takezawa, K. Mori, W.-E. Huang, K. Nishiyama, T. Xing, T. Nakama and M. Shionoya, Nat. Commun., 2023, 14, 4759 CrossRef CAS PubMed.
- Y. Takezawa, A. Suzuki, M. Nakaya, K. Nishiyama and M. Shionoya, J. Am. Chem. Soc., 2020, 142, 21640–21644 CrossRef CAS PubMed.
- K. Mori, Y. Takezawa and M. Shionoya, Chem. Sci., 2023, 14, 1082–1088 RSC.
- (a) S. K. Silverman, Trends Biochem. Sci., 2016, 41, 595–609 CrossRef CAS; (b) M. Liu, D. Chan and Y. Li, Acc. Chem. Res., 2017, 50, 2273–2283 CrossRef CAS; (c) E. M. McConnell, I. Cozma, Q. Mou, J. D. Brennan, Y. Lu and Y. Li, Chem. Soc. Rev., 2021, 50, 8954–8994 RSC; (d) Z. Huang, X. Wang, Z. Wu and J.-H. Jiang, Chem.–Asian J., 2022, 17, e202101414 CrossRef CAS PubMed.
- (a) Y. Takezawa, T. Nakama and M. Shionoya, J. Am. Chem. Soc., 2019, 141, 19342–19350 CrossRef CAS PubMed; (b) T. Nakama, Y. Takezawa, D. Sasaki and M. Shionoya, J. Am. Chem. Soc., 2020, 142, 10153–10162 CrossRef CAS PubMed; (c) Y. Takezawa, L. Hu, T. Nakama and M. Shionoya, Angew. Chem., Int. Ed., 2020, 59, 21488–21492 CrossRef CAS PubMed; (d) T. Nakama, Y. Takezawa and M. Shionoya, Chem. Commun., 2021, 57, 1392–1395 RSC; (e) S. C. Rajasree, Y. Takezawa and M. Shionoya, Chem. Commun., 2023, 59, 1006–1009 RSC; (f) Y. Takezawa, L. Hu, T. Nakama and M. Shionoya, Chem. Commun., 2024, 60, 288–291 RSC.
- (a) R. Hili, J. Niu and D. R. Liu, J. Am. Chem. Soc., 2013, 135, 98–101 CrossRef CAS PubMed; (b) D. Kong, W. Yeung and R. Hili, J. Am. Chem. Soc., 2017, 139, 13977–13980 CrossRef CAS PubMed; (c) Y. Lei, J. Washington and R. Hili, Org. Biomol. Chem., 2019, 17, 1962–1965 RSC; (d) D. Kestemont, M. Renders, P. Leonczak, M. Abramov, G. Schepers, V. B. Pinheiro, J. Rozenski and P. Herdewijn, Chem. Commun., 2018, 54, 6408–6411 RSC; (e) J. Riedl, Y. Ding, A. M. Fleming and C. J. Burrows, Nat. Commun., 2015, 6, 8807 CrossRef CAS PubMed; (f) N. Sabat, A. Stämpfli, S. Hanlon, S. Bisagni, F. Sladojevich, K. Püntenerc and M. Hollenstein, ChemRxiv, 2023 DOI:10.26434/chemrxiv-2023-vwwt5.
- S.-F. Torabi, P. Wu, C. E. McGhee, L. Chen, K. Hwang, N. Zheng, J. Cheng and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5903–5908 CrossRef CAS PubMed.
- J. N. Zadeh, C. D. Steenberg, J. S. Bois, B. R. Wolfe, M. B. Pierce, A. R. Khan, R. M. Dirks and N. A. Pierce, J. Comput. Chem., 2011, 32, 170–173 CrossRef CAS PubMed.
- L. Pickart, J. H. Freedman, W. J. Loker, J. Peisach, C. M. Perkins, R. E. Stenkamp and B. Weinstein, Nature, 1980, 288, 715–717 CrossRef CAS PubMed.
- (a) R. R. Breaker and G. F. Joyce, Chem. Biol., 1995, 2, 655–660 CrossRef CAS PubMed; (b) J. W. Liu, A. K. Brown, X. L. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056–2061 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05042d |
| This journal is © The Royal Society of Chemistry 2024 |