
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the functional hotspots inside protein hydrophobic pockets by in situ photochemical trifluoromethylation and mass spectrometry†
Can
Lai
ae,
Zhiyao
Tang
b,
Zheyi
Liu
a,
Pan
Luo
acd,
Wenxiang
Zhang
a,
Tingting
Zhang
ae,
Wenhao
Zhang
ae,
Zhe
Dong
*b,
Xinyuan
Liu
 b,
Xueming
Yang
bcd and
Fangjun
Wang
*ace
b,
Xueming
Yang
bcd and
Fangjun
Wang
*ace
aCAS Key Laboratory of Separation Sciences for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: wangfj@dicp.ac.cn
bDepartment of Chemistry, College of Science, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: dongz@sustech.edu.cn
cState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
dInstitute of Advanced Science Facilities, Shenzhen 518107, China
eUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 11th January 2024
Abstract
Due to the complex high-order structures and interactions of proteins within an aqueous solution, a majority of chemical functionalizations happen on the hydrophilic sites of protein external surfaces which are naturally exposed to the solution. However, the hydrophobic pockets inside proteins are crucial for ligand binding and function as catalytic centers and transporting tunnels. Herein, we describe a reagent pre-organization and in situ photochemical trifluoromethylation strategy to profile the functional sites inside the hydrophobic pockets of native proteins. Unbiased mass spectrometry profiling was applied for the characterization of trifluoromethylated sites with high sensitivity. Native proteins including myoglobin, trypsin, haloalkane dehalogenase, and human serum albumin have been engaged in this mild photochemical process and substantial hydrophobic site-specific and structure-selective trifluoromethylation substitutes are obtained without significant interference to their bioactivity and structures. Sodium triflinate is the only reagent required to functionalize the unprotected proteins with wide pH-range tolerance and high biocompatibility. This “in-pocket” activation model provides a general strategy to modify the potential binding pockets and gain essential structural insights into the functional hotspots inside protein hydrophobic pockets.
Introduction
Precise modification of native proteins with site- and structure-selective chemical probes provides a powerful toolbox to explore the structure–function relationships of proteins.1,2 Protein labeling combined with mass spectrometry (MS) based structural proteomics is a promising strategy to profile the protein conformational dynamics in solution with minimal sample required.3,4 To maintain the bioactivity and high-order structures of native proteins, it is critical that the labeling reaction should be carried out in a biocompatible buffer at room temperature. At present, uncatalyzed hydrogen/deuterium exchange, fast photochemical oxidation with reactive species, and certain fast cross-linking approaches qualify.5,6 To introduce more complex chemical transformations on more diverse protein residues, a variety of stoichiometric organometallic reactions and photo-redox radical reactions have been developed.7 Although they indeed greatly explore the new chemical space for protein functionalization, concerns that heavy metals or photocatalysts may poison the original protein and interfere with its functions remain unsolved. More importantly, due to the large sizes of those catalysts/reagents, diffusion of the catalyst and reagent together into the protein internal hydrophobic pocket is extremely difficult under biocompatible conditions. This results in highly selective functionalization of hydrophilic sites on protein surfaces since they are more exposed to solutions. For palladium mediated reactions8,9 or nucleophilic substitution reactions,10 they usually prefer to react with free thiol (Cys) and amino (Lys) groups, which usually occur on the protein hydrophilic surfaces (Fig. 1A).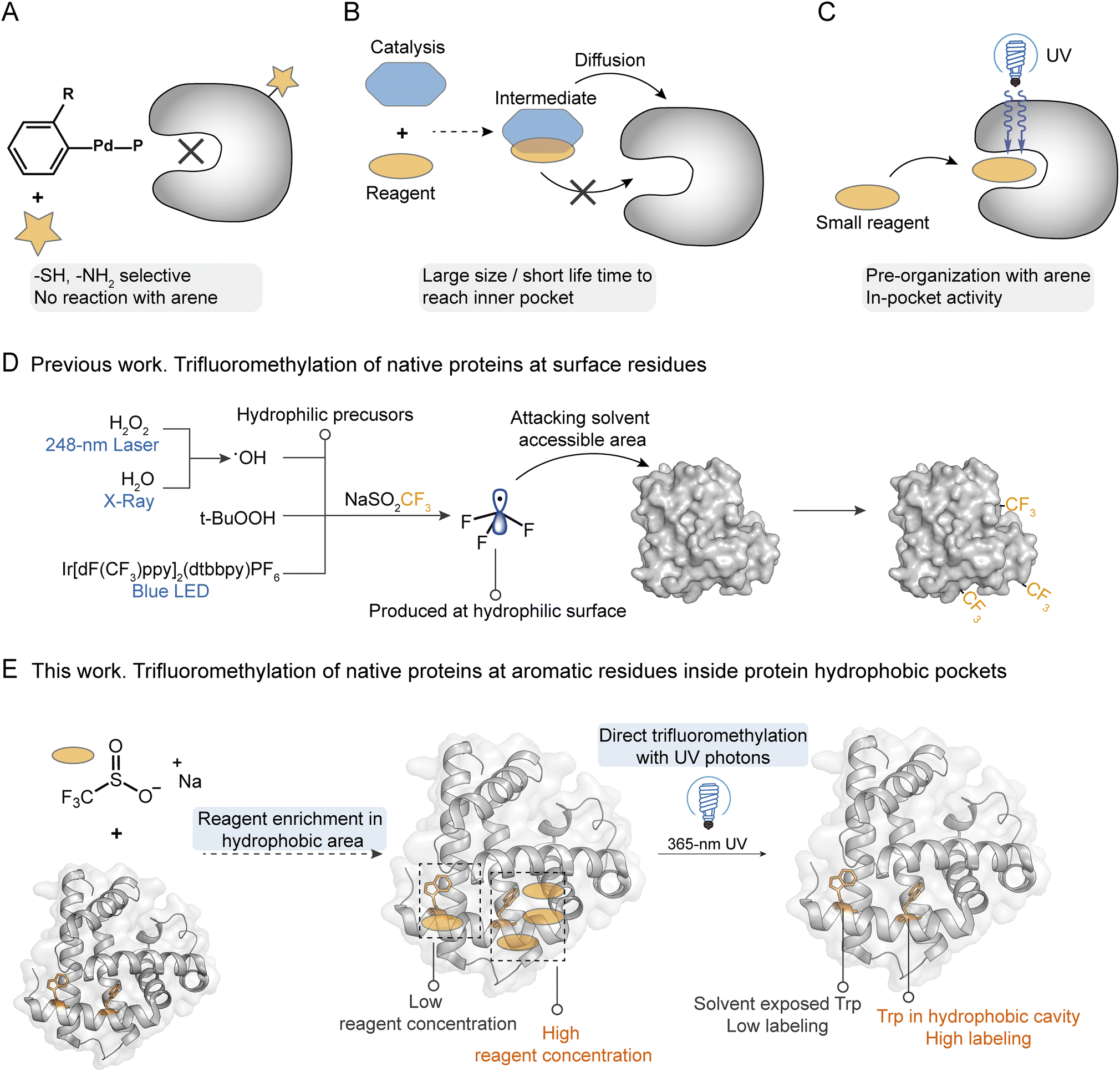 | ||
| Fig. 1 The scope of labeling techniques for the functionalization and structure-elucidation of native proteins. (A) Schematic diagram of palladium reagent engaged protein bioconjugation. (B) Schematic diagram of the structure preference for catalysis associated protein labeling. (C) Proposed strategy for the “in-pocket” photoactivation and arene modification with reagent pre-organization in the protein hydrophobic core. Schematic diagram of native protein trifluoromethylation by using Langlois' reagent from other works (D) and this work (E). | ||
Hydrophobic interaction is essential during protein folding into an ordered structure with a hydrophilic surface and hydrophobic core.11,12 Further, the protein recognition of small molecules such as drugs,13,14 ligand binding with receptors,15 and enzyme biocatalysis reactions16,17 often occur in protein hydrophobic pockets with essential functional hotspots. Recently, studies of structural biology have reported the key binding sites in the hydrophobic pockets of membrane proteins, such as vitamin K epoxide reductases (VKOR) with vitamin K antagonists,18 ATP-binding cassette (ABD) transporters with inhibitors,19 and G-protein-coupled receptors (GPCRs) with various ligands.20–22 These proteins exhibit crucial functions in the regulation of many important physiological processes, demonstrating the essential roles of probing the hotspots inside hydrophobic pockets for understanding protein–ligand interactions and designing targeted inhibitors. Many methods have been developed for hydrophobic site profiling based on X-ray crystallography, nuclear magnetic resonance spectroscopy (NMR), or computational prediction.23–25 These methods usually need the assistance of organic probes such as ethanol and acetonitrile, and the intense sample consumption and low accuracy limit their applications.25 A recent work reported the modification of hydrophobic Trp at the recognition pocket for the manipulation of interaction between histone and its reader, but a genetic strategy was needed for the introduction of Trp derivatives.13,26 Thus, it is crucial to develop a convenient technique to directly profile the functional hotspots within protein hydrophobic pockets with high sensitivity and selectivity.27
The trifluoromethyl group is the most popular fluorine-bearing substituent for biomolecule fluorination due to the great clinical success of trifluoromethylated drug candidates in the last two decades.28 Recently, 5-(trifluoromethyl)-dibenzothiophenium trifluoromethanesulfonate (Umemoto's reagent) was applied for 18F incorporation of unprotected peptides at the nucleophilic thiol side chain of Cys for positron emission tomography (PET) imaging.29 Sodium triflinate (NaSO2CF3, Langlois' reagent) and Zn(SO2CF3)2 (ZnTFMS, Baran's reagent) were further applied for the trifluoromethylation of the aromatic residues (mainly Trp and Tyr) of proteins and peptides under redox initiation by peroxides (e.g. t-BuOOH) or photocatalysts (e.g. Ir[dF(CF3)ppy]2(dtbbpy)(PF6)).30–32 A cyclic hypervalent iodine-fluoroalkyl reagent (Togni's reagent) using ascorbic acid as a reductant was also used for fluoroalkylation of aromatic residues in proteins to probe their structure alterations.33 Among these trifluoromethylation reagents, NaTFMS exhibits the highest potential for labeling native proteins owing to its water solubility and high biocompatibility,32,34 which has been applied for the fast foot-printing trifluoromethylation of protein surfaces through the photolysis of hydrogen peroxide (H2O2) by a 248 nm laser34 or water by X-rays35 to generate a hydroxyl radical (˙OH).34,35
We hypothesize that sterically small sodium triflinate reagents with high lipophilicity might be able to diffuse into the hydrophobic pockets of native proteins. Based on Dougherty's pioneering work,36 sodium cations have cation–π interactions with Phe, Tyr, and Trp, which will bring the triflinate anion and these electron-rich aromatic residues into close proximity. Upon direct photonic activation, a highly electrophilic trifluoromethyl radical37 can be generated under pH-neutral and additive-free conditions via desulfonylation. Then, a facile radical addition into an electron-rich arene will spontaneously happen on the hydrophobic site (Fig. 1C). In contrast, previously reported trifluoromethylation methods need an additional oxidant/reductant/photocatalyst to interact with the trifluoromethyl precursor for the generation of trifluoromethyl radicals. It is highly unlikely that both the oxidant/reductant/photocatalyst and trifluoromethyl precursor can enter the hydrophobic pocket at the same time (Fig. 1B). On the other hand, the pyramidal trifluoromethyl radical is one of the highest energy alkyl radicals (BDE = 107 kcal mol−1), and thus itself should have a relatively short life-time and would readily react with the weak C–H bond.38 It is also unlikely that the trifluoromethyl radical is generated in solution and then diffuses into the hydrophobic site. Taking all these facts into consideration, it is not surprising that even trifluoromethyl is such a hydrophobic functional group that the previous methods can only introduce the CF3 group at the solvent accessible residues on the external hydrophilic surfaces of proteins.32,34,35
In the current work, we describe a reagent pre-organization photochemical trifluoromethylation strategy to profile the functional sites inside the hydrophobic pockets of native proteins, which was realized with NaTFMS in biocompatible aqueous solutions just under the irradiation of 365 nm UV photons (Fig. 1E). Compared to existing methods that require hydrophilic precursors to produce trifluoromethyl radicals near protein surfaces, resulting in high trifluoromethylation efficiency at protein hydrophilic surfaces (Fig. 1D), our additive- and photocatalyst-free trifluoromethylation strategy can modify the unprotected aromatic residues inside protein hydrophobic pockets with high structure selectivity. We demonstrated that the functional hotspots of the internal hydrophobic sites in native proteins such as haloalkane dehalogenases (HLDs) and human serum albumin (HSA) could be successfully probed via this “in-pocket” activation and in situ photochemical strategy.
Results and discussion
Structure-selective trifluoromethylation of native proteins
At first, the trifluoromethylation (TFM) reactivity of protein residues was investigated by applying a series of hexapeptides containing an identical peptide backbone and different types of side chains for the photochemical TFM under identical conditions. Compared to non-irradiation controls, TFM products were mainly observed for peptides containing Trp, Tyr, Phe or His, and Trp exhibited the highest reactivity (Fig. S2†). The residue trifluoromethylation was confirmed by the mass shift of 67.987 Da (+[CF3]–[H]) in MS detection32 and the LC retention time delay resulted from the hydrophobic trifluoromethyl group.34,39 Both mono- and di-trifluoromethylation substitutes of Trp and Tyr were observed with mono-TFM as the major products (Fig. S2A and B†). Only the mono-TFM form was observed for Phe and His (Fig. S2C and D†).Then, we applied this photochemical TFM strategy to native proteins with higher-order structures (Fig. 2A). A variety of proteins with different sizes including holo-myoglobin (holo-Mb), trypsin, haloalkane dehalogenases (HLDs), and human serum albumin (HSA) were trifluoromethylated and multiple TFM products were detected (Fig. S3†). Considering that long-time UV irradiation may lead to degradation, the protein concentration was determined after TFM modification. Less than 15% loss of holo-Mb was observed after the TFM process (Fig. S4B†), suggesting the biocompatibility was within the tolerance under the reaction conditions. No great distortion was found in the proteins' structural integrity based on their charge state distribution in native MS (nMS) characterization with non-denaturing electrospray ionization (Fig. S3A and S4A†).40,41 Further, CD spectroscopy was applied for the analysis of the secondary structure of TFM holo-Mb,35 and no significant change was detected compared to holo-Mb (Fig. S5†). Furthermore, to test the impact on the bioactivity of proteins, we chosen trypsin that contains no Trp residue in its catalytic center for the functional analysis. Compared to the original trypsin, TFM-trypsin could also digest BSA into peptides with comparable cleavage efficiency (Fig. S6A and B†), and the digested BSA peptides showed no distinct type and number differences (Fig. S6C†), indicating the photochemical TFM conditions had an insignificant impact on the trypsin functional structure.
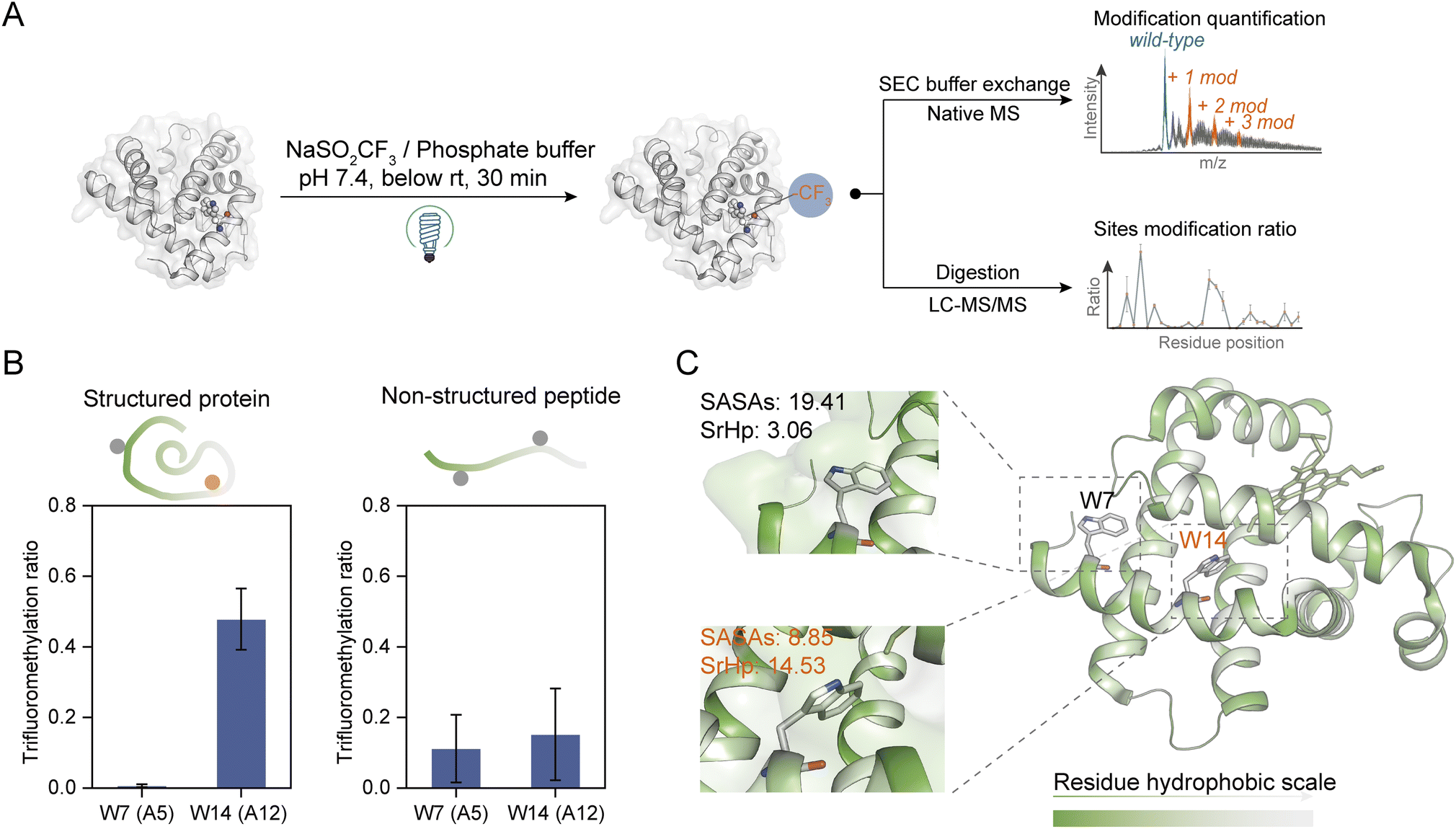 | ||
| Fig. 2 Site- and structure-selective photochemical trifluoromethylation of holo-Mb. (A) General procedure for the trifluoromethylation of native proteins. (B) TFM ratios of Trp residues in structure-maintained holo-Mb protein and non-structured tryptic Mb peptides under identical conditions (n = 3). (C) Crystal structure of holo-Mb with color-coded residue hydrophobicity and Trp residues shown in sticks (PDB: 1npf). The substrate and reagent were mixed to a final concentration of 10 μM and 50 mM, respectively, in 0.1 M phosphate buffer (PB, pH 7.4). | ||
To further investigate the structure selectivity for the trifluoromethylation of native proteins, the TFM proteins were digested and the residue TFM ratios were determined (Fig. 2A). Hydrophilicity and hydrophobicity are the factors surveyed to associate the residues' TFM propensity with their microenvironments. There are two Trp residues Trp-A5 (W7) and Trp-A12 (W14) on the same helix structure of holo-Mb with distinct solvent accessibility surface areas (SASAs) (Fig. S7 and Table S1†). Interestingly, W14 with lower SASAs exhibited a significantly higher TFM ratio than W7 (Fig. 2B, left), suggesting that the actual concentration or “local concentration” of NaTFMS around W14 should be higher than at W7 since only NaTFMS reagent and UV irradiation were engaged in this modification process. To verify this presumption, we applied non-structured tryptic Mb digests to the modification process under identical conditions as holo-Mb. Both residues showed comparatively low TFM ratios (Fig. 2B, right), indicating the TFM difference in native holo-Mb W7 and W14 had indeed resulted from structure discrepancy and NaTFMS concentration around W14 was higher than in solution. Thus, we constructed sequence hydrophobicity in the holo-Mb crystal structure.42 It showed that residues in proximity to W14 are more hydrophobic than those close to W7 (Fig. 2C). Besides, we calculated the “surrounding hydrophobicity” (SrHp) by summing the hydrophobic indices43 of residues within an 8 Å radius around W7 and W14 (Table S1†).44 It turned out W14 (13.68) has a remarkably greater SrHp than W7 (3.06), indicating W14 indeed locates in a more “hydrophobic cavity”. Considering NaTFMS contains the hydrophobic trifluoromethyl group, it's convincing that Langlois' reagent would pre-organize in the hydrophobic cavity around W14 to increase its “local concentration”,27 resulting in the higher TFM ratio than for W7. Meanwhile, the oxidation of Trp was inevitable during the photochemical TFM process (Fig. S2A†), and a higher oxidation ratio was observed with lower reagent concentration (Fig. S8A†). Thus, the Trp with a larger SASA should be more prone to oxidation than the Trp in the more hydrophobic cavity. As expected, W7 in intact holo-Mb was more oxidized than W14, but both shared similar oxidation ratios in the non-structured Mb tryptic peptides (Fig. S9A and B†). Therefore, the oxidation information of Trp can be a cross-referenced index for profiling residue microenvironments as well.
The mechanism of photochemical trifluoromethylation
To explore the mechanism behind this photochemical trifluoromethylation process, we utilized octreotide for comprehensive study. Octreotide, a cyclic therapeutic peptide of a somatostatin derivative45 containing one Trp and two Phe residues (Fig. 3B), was photochemically modified (Fig. 3A). Consistent with Trp hexapeptide, mono- and di-TFM forms were observed with the relative ratio of mono-TFM higher than that of the di-TFM form (Fig. 3C and S10A†). The modification sites were mainly Trp determined by MS/MS analysis (Fig. S10†). Furthermore, many oxidation by-products were detected with low concertation of NaTFMS (5 mM, Fig. S8A†), but the total yield of TFM-octreotide was gradually improved with the increase of reagent concentration (Fig. S8A†). Therefore, high reagent concentration can efficiently suppress the photochemical oxidation and increase the TFM modification efficiency. In addition, this method showed its compatibility in variant bio-buffer with wide pH range (Table S2†), and an aqueous solution without other salt additives seemed beneficial to higher conversion to TFM-octreotide (Fig. S8B†).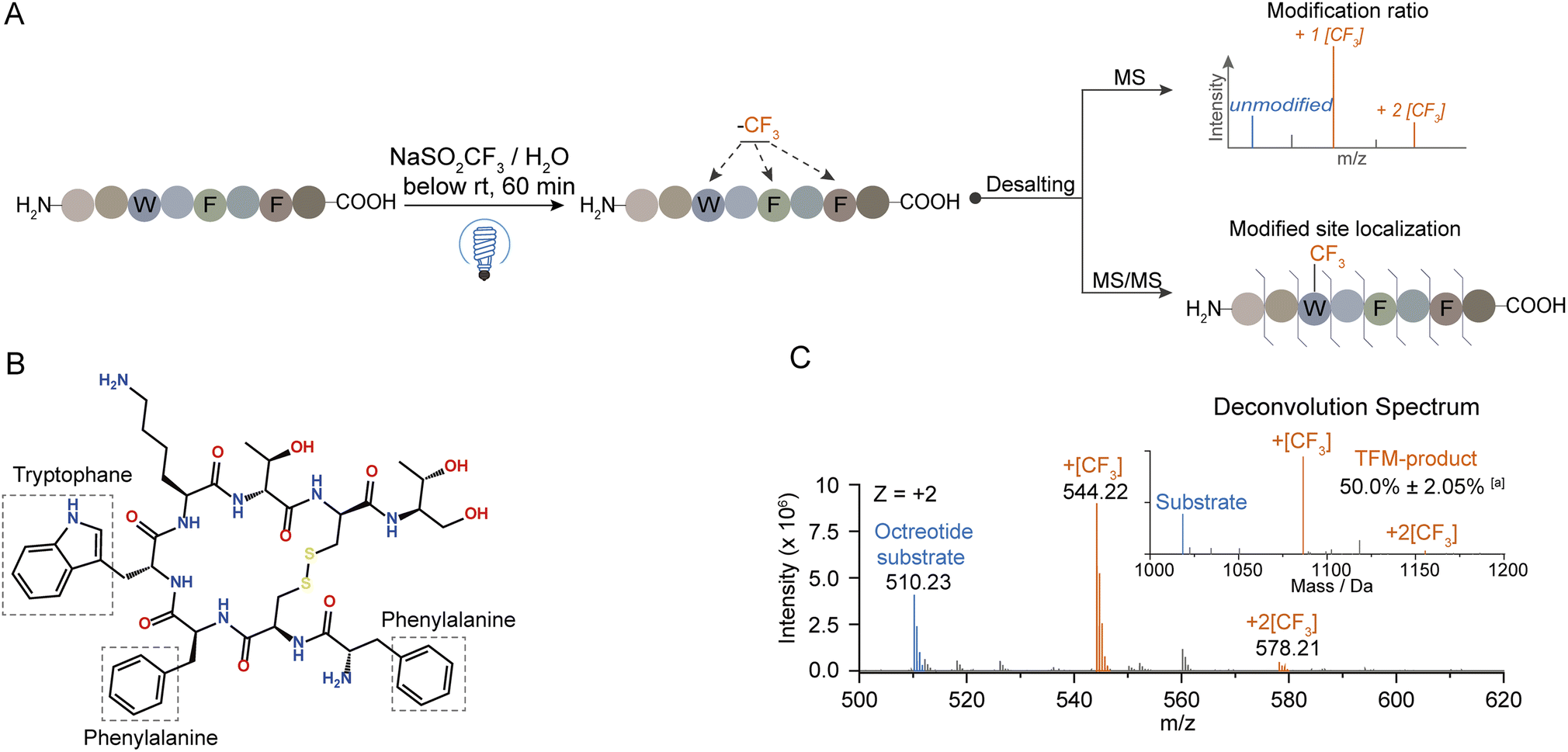 | ||
| Fig. 3 Photochemical trifluoromethylation of cyclic octreotide. (A) General procedure for the photochemical trifluoro-methylation of peptides. (B) Chemical structure of octreotide. (C) MS spectrum of trifluoromethylated octreotide; the inset in the MS spectrum is the deconvolution spectrum. aRelative intensity was calculated as the percentage of TFM product peak intensity to the total intensity of deconvoluted peaks (n = 3). The substrate and reagent were mixed in water to a final concentration of 100 μM and 150 mM, respectively. | ||
Then, we introduced N2 gas into the reaction solution to diminish the dissolved O2 (Fig. S1B†). Although the photo-oxidation by-products were efficiently suppressed after the decrease of dissolved O2, the transformation of the octreotide substrate was also sharply decreased (Fig. S11A†). This proved that oxygen gas dissolved in the water was likely to be the terminal oxidant for the overall trifluoromethylation reaction. Next, the reaction solution with H2O18 was applied to determine the oxygen source of photo-oxidation.46 Interestingly, the mono-oxidation product was still dominated with 16O (from dissolved O2), while both 16O and 18O were incorporated into the multiple-oxidation forms of products (Fig. S11B†). Therefore, the water dissolved O2 was the medium which was excited to an active state to trigger the reaction.47,48
UV-vis absorption analysis was performed to identify the photon excitation species. The results demonstrated that the peptides containing Trp, Tyr, Phe, or His all exhibited no apparent absorption to 365 nm UV photons (Fig. S12A†). In contrast, NaTFMS exhibited much higher photon adsorption from the visible to UV wavelength (Fig. S12B†). Therefore, the SO2CF3− anion was the main excitation target of 365 nm UV photons to trigger the following radical-based modifications. Interestingly, changing the reagent cation from Na+ to Zn2+ showed inferior modification efficiency even with higher concentration (Fig. 4A), indicating both the metal cation and anion were engaged in the activation of TFM modification. Then, we monitored the photon absorption of NaTFMS at 365 nm with and without the peptide substrate. No apparent changes appeared when NaTFMS was irradiated alone, but the absorption changed with the existence of octreotide, suggesting the substrate was also associated (Fig. S12C†). We speculated the NaTFMS not only served as the [CF3] source, but also associated with the substrate for the initiation of photochemical modifications and cations were involved.
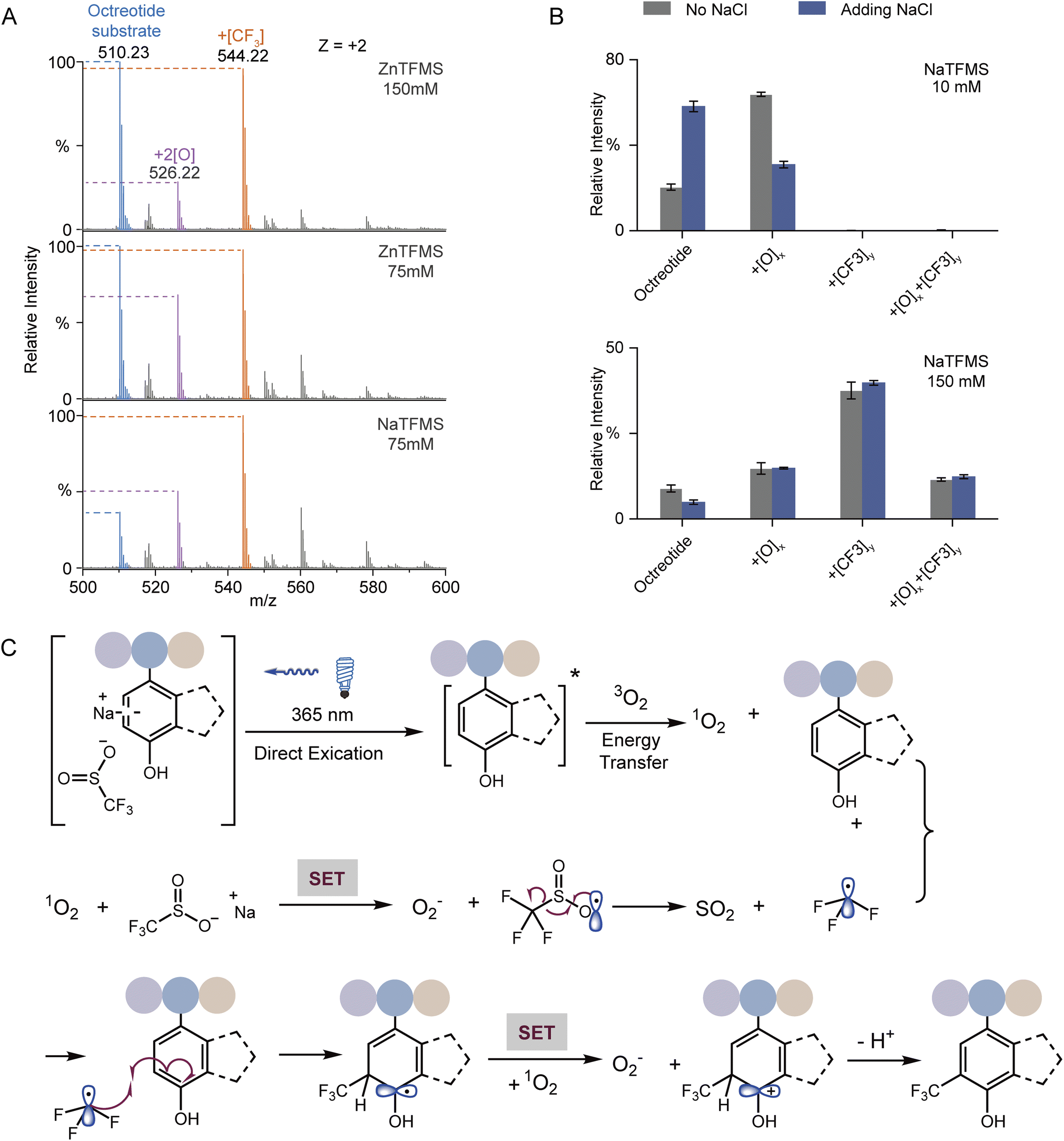 | ||
| Fig. 4 (A) Product MS spectra of octreotide reacting with NaTFMS and ZnTFMS. (B) Influence of extra Na+ on the relative MS intensity of products at low or high reagent concentration (n = 3; 150 mM NaCl was added). (C) Proposed mechanism of the protein arene photochemical trifluoromethylation process. The EDA complex of protein and sodium sulfinate can be selectively activated by 365 nm light, which will generate singlet oxygen via energy transfer. Spontaneous single electron transfer will generate a trifluoromethyl radical which will then quickly add onto the electron-rich arene. | ||
To test the effect of cations, extra NaCl was added for the modification of octreotide. The yields of products dropped sharply when Na+ was added at low reagent concentration but when a high concentration of NaTFMS was applied, the influence was negligible (Fig. 4B), indicating excessive cations would interfere with the photochemical modification at low reagent concentration in particular. We hypothesized that cation–π interaction between Na+ and Trp of octreotide occurred during the modification process.49 Cation–π interaction has been widely observed either in protein folding to maintain the structure stability50 or binding with various ligands in the functional pockets.13,26,51 Studies have shown the cation–π interaction of charged groups with aromatic residues near the hydrophobic pockets of proteins, such as glycine with Phe in glycine receptors,52 the ammonium group of acetylcholine with Trp in nicotinic acetylcholine receptors,53 and the trimethyl ammonium group of the drug iperoxo with Tyr in muscarinic acetylcholine receptors.54 Consequently, interference with the cation–π interaction between the NaTFMS reagent and Trp would disturb the modification level. Thus, we applied Baran's reagent for the modification of holo-Mb as well as adding extra NaCl. Only the oxidation form was observed for W7 and W14 when ZnTFMS was used (Fig. S13†) because of lower activation activity from Zn2+ cations (Fig. 4A). Meanwhile, adding extra NaCl led to the decreased modification ratio of W7 but no change at W14 (Fig. S13B†), further demonstrating that the NaTFMS concentration around W7 was lower than that around W14 and the reagent would pre-organize in the proteins' hydrophobic cavity (Fig. 2B).
Combining the above observations with previous reports,32,34,35,46–48,55 a possible reaction mechanism of the photochemical TFM modification was proposed (Fig. 4C). At first, the substrate in association with NaTFMS through cation–π interaction can be directly activated to the excited state (substrate*) by 365 nm photon irradiation. Then, triplet oxygen (3O2) is converted to singlet oxygen (1O2) through energy transfer between substrate* and 3O2. The active 1O2 can oxidize the –SO2CF3 anion via single electron transfer (SET) to form the TFMS radical (˙SO2CF3) and byproduct superoxide O2−, which might be the cause of some undesired oxidation products. Subsequently, the ˙SO2CF3 radical will undergo a well-documented β-scission to generate trifluoromethyl radicals (˙CF3) which would preferentially add into the electron-rich arene. The resulting dearomatized aryl radical would be further oxidized by oxygen and then re-aromatized to form TFM products. Consequently, for the TFM of native proteins, the reagent would pre-organize in the protein hydrophobic pocket, and then, arene residues in the pocket could give high TFM modification ratios through a cation–π mediated chemical process upon UV irradiation (Fig. 1E).
Functional hotspot profiling of native proteins
Inspired by the structure selectivity, we gained insights into the TFM ratio changes after the removal of ligand heme from holo-Mb. Compared to TFM holo-Mb, the charge distribution of TFM apo-Mb stays unchanged (Fig. 5A), indicating the removal of heme won't lead to the loss of the compact structure.33,34 The Mb hydrophobic pocket for heme binding locates between helix E and helix F,56 thus, the TFM ratios of W7 (Trp-A5), F43 (Phe-CD2), F46 (Phe-CD5), H64 (His-E7), and H93 (His-F8) around this pocket were significantly increased after heme removal (Fig. 5B and C). The increased TFM ratios at F43, H64, and H93 can be attributed to the reduction of steric hindrance from heme, leading to the increase of the local concentration of NaTFMS in the hydrophobic pocket (Fig. 5D). The differences of W7 and F46 indicate that the removal of heme would induce their conformation to change to a more hydrophobic area. Interestingly, H64 and H93 are also involved in gaseous ligand binding such as O2, CO, and NO to the holo-Mb vacant iron as reported by previous X-ray crystallography and computational simulation studies.56,57 Compared to the reported method that modification differences mainly rely on the SASA variations,33,34 these observations add new insights into the conformational changes involving the hydrophobic microenvironment around reactive residues, suggesting our method can be applied as complementary for probing functional residues in hydrophobic pockets.27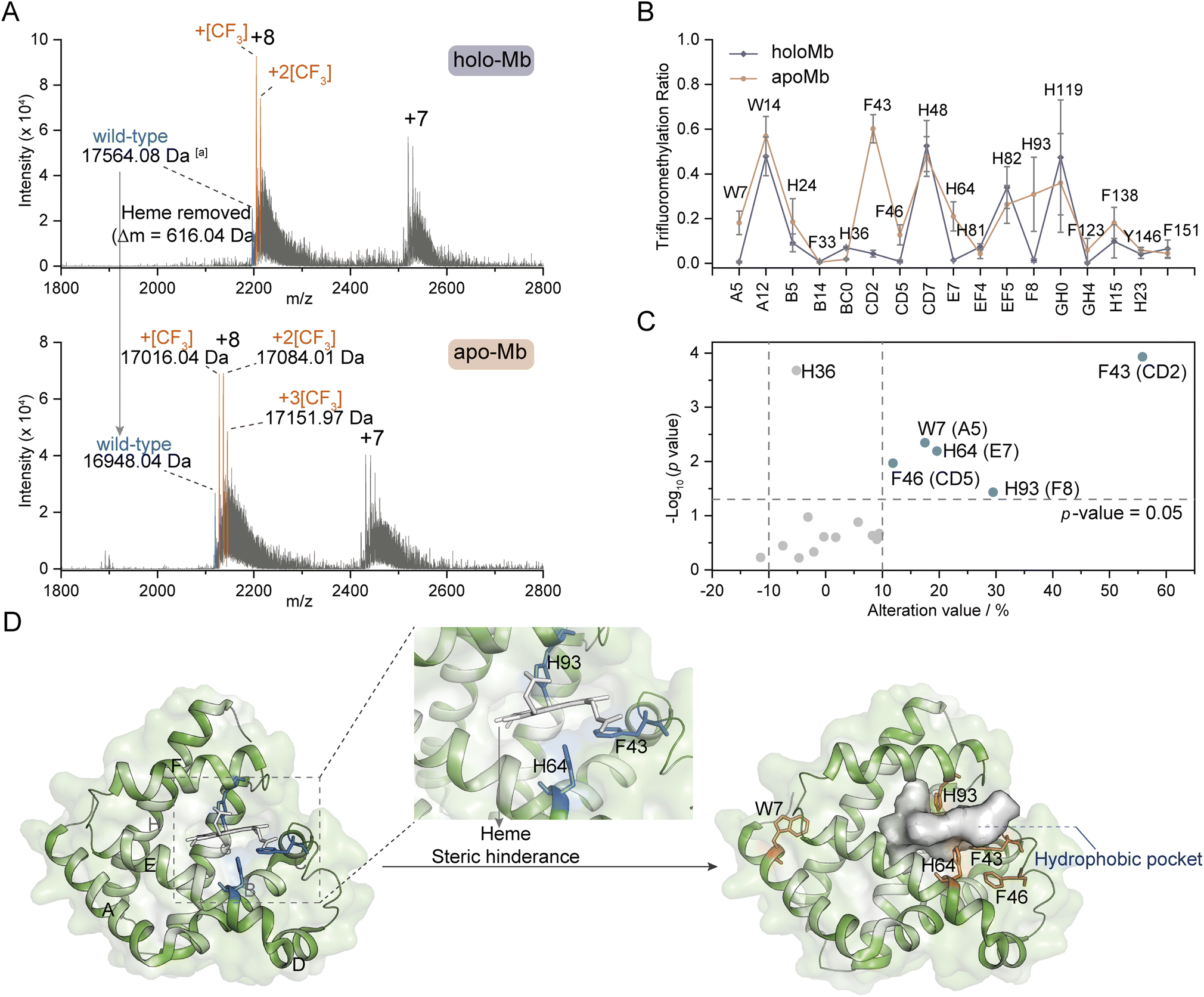 | ||
| Fig. 5 Ligand-binding hotspot profiling in Mb. (A) Native MS spectra of trifluoromethylated holo- and apo-Mb. (B) TFM ratio of modified residues in holo- and apo-Mb (n = 3) with the topological helical position given along the x-axis. (C) Volcano plot of TFM ratio variations after removing the heme ligand from holo-Mb. (D) Crystal structures of holo-Mb with highlighted W7, F43, F46, H64, and H93 residues in sticks (PDB: 1npf). aProtein molecular weight was calculated by the m/z of the monotopic peak and corresponding charge state. The substrate and reagent were mixed to a final concentration of 10 μM and 50 mM, respectively, in 0.1 M phosphate buffer (PB, pH 7.4). | ||
Aromatic residues have been identified as prominent sites at the protein interfaces, with π–π, anion–π, and cation–π interactions playing crucial roles in the ligand stabilization.58 Furthermore, the aromatic rings frequently serve as anchor points within the hydrophobic cavities for ligand binding. Consequently, it is imperative to profile the key aromatic sites located within protein hydrophobic pockets. We further investigated the modification ratios of HLDs and HSA residues to confirm if this photochemical TFM strategy could be used to probe the functional hotspots in hydrophobic cavities (Fig. 6A and B). HLDs are hydrolytic enzymes that convert a variety of haloalkane compounds to alcohols with co-factor water.59 The catalysis of hydrophobic haloalkane molecules occurs within the hydrophobic pocket of HLDs, with five key residues playing pivotal roles in this process, including N38, D108, W109, E132, and H272. Notably, the aromatic residue W109, in conjunction with N38, functions as an essential stabilization site for the halogen anion during the conversion of haloalkane to alcohol60 (Fig. S14†). Apparently, among the identified TFM Trp residues, W109 had the highest TFM ratio (Fig. 6A). Inspection into the microenvironments of these Trp residues showed W109 is spatially close to the hydrophobic pocket61 (Fig. 6C, left) and has the highest SrHp (Table S1†). Besides, both TFM (Fig. 6A) and oxidation ratios (Fig. S9C†) of W207 were much lower despite it being adjacent to W109 (Fig. 6C, middle), resulting from its lower SrHp (Table S1†) and the steric hindrance of being deeply buried with lower SASAs (Fig. 6C, top right). Meanwhile, W140 with the highest SASAs and lowest SrHp (Table S1†) exhibited low TFM reactivity as a consequence of its surface being exposed to solvent (Fig. 6C, bottom right). Even with the highest SASAs, W140 exhibited a low oxidation ratio (Fig. S9C†) possibly due to the cation–π interaction with Q152 interfering with the Trp reactivity (Fig. 6C, bottom right and S13B†). Based on these findings, we are convinced that the aromatic residues in the more hydrophobic pocket have higher TFM ratios than those deeply buried inside protein or exposed at the protein hydrophilic surface.
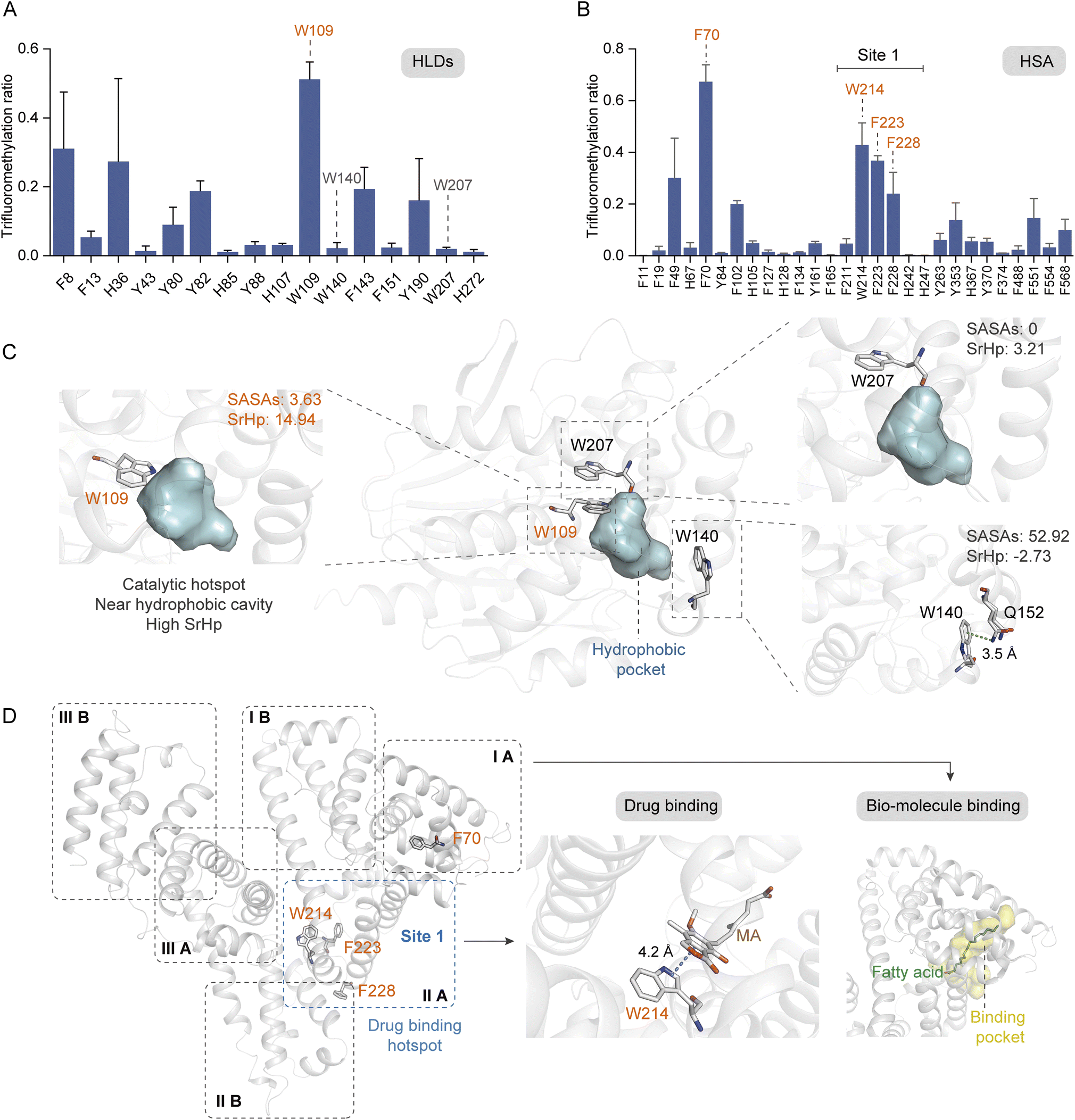 | ||
| Fig. 6 Probing the hydrophobic hotspots in native proteins. Site trifluoromethylation ratios of HLDs (A) and HSA (B) (n = 3). (C) Crystal structure of HLDs with microenvironments of TFM Trp residues shown in detail (PDB: 1mj5). (D) Crystal structure of HSA (PDB: 1bj5) and its binding sites for drug (PDB: 7wkz) and fatty acid (PDB: 1e7i). The concentrations of proteins and reagent applied in trifluoromethylation reactions were HLDs 15 μM, HSA 10 μM, and NaTFMS 50 mM, respectively, in 0.1 M phosphate buffer (PB, pH 7.4). | ||
HSA is the most abundant protein in human plasma and serves as a crucial carrier for the transport of numerous small-molecular compounds.62 Many hydrophobic regions are present within the HSA structure (Fig. S15†), functioning as binding pockets for both endogenous hormones and lipids, as well as exogenous drugs such as warfarin.63,64 It is expected that the arene residues locating adjacent to these binding pockets would exhibit greater reactivity of trifluoromethylation. As a result, four residues with high TFM ratios were profiled (Fig. 6B), of which W214, F223, and F228 were located in the subdomain IIA and F70 was located in subdomain IA (Fig. 6D, left). Subdomains IA and IIA form a narrow hydrophobic cavity upon the binding of fatty acid methylene tails, serving as the major binding sites for fatty acids with different chain lengths64 (Fig. 6D, right). Subdomain IIA contains the largest drug binding pocket (site 1) for the combination of variant drugs, in which W214, the only Trp residue in HSA, plays a key role in ligand combination.63 A recent study has reported the stacking interaction of W214 with an immunosuppressive drug, mycophenolic acid (MA)65 (Fig. 6D, middle). As such, residue W214 was traditionally applied for the profile of interaction between HSA and small molecules based on the tryptophane fluorescence response. The high efficiency of TFM at these crucial binding sites clearly demonstrates the ability of our photochemical TFM strategy in probing functional hotspots inside protein hydrophobic pockets and can be applied for profiling essential interactions between proteins and ligands.
Conclusion
In summary, hydrophobic pockets are crucial functional domains of proteins for the recognition and binding of ligands, substrates, and pharmaceutical molecules. We have demonstrated a convenient reagent pre-organization and in situ photochemical strategy for the direct structure-selective trifluoromethylation of residues inside proteins' hydrophobic pockets using a 365 nm UV lamp without adding catalysts and additives. The mechanism behind the reaction reveals that direct photochemical trifluoromethylation is triggered through association of the substrate with the NaTFMS reagent through cation–π interaction under UV irradiation to activate oxygen from triplet O2 to singlet O2 to produce trifluoromethyl radicals. The site TFM ratio differences show high structure selectivity, revealing that the NaTFMS reagent would pre-organize in protein hydrophobic pockets and the aromatic residues in the pockets would undergo in situ trifluoromethylation upon UV photon irradiation. Consequently, residues in more hydrophobic regions with less steric hindrance would obtain higher trifluoromethylation ratios. The structure selectivity of this photochemical trifluoromethylation method exhibits great potential for the protein structure–function characterization, as well as the discovery of functional hotspots in essential protein hydrophobic pockets for ligand interactions. Nevertheless, this method is a slow reaction indeed compared to the previous fast foot-printing techniques,34,66 thus thorough investigation and validation of the perturbance on protein activity and structure is needed.67 Though experiments have been conducted on model TFM proteins for the exploration of function and structure changes, more types of proteins including membrane proteins and protein complexes should be investigated in the future to expand its applications in profiling the hydrophobic hotspots of more complex bio-systems.Methods
Photochemical trifluoromethylation of unprotected peptides
100 μL peptide aqueous solution (200 μM) was mixed with 100 μL NaTFMS (300 mM) in water to a final concentration of peptide (100 μM) and NaTFMS (150 mM) respectively. The mixed solution was transferred to a quartz bottle and irradiated under four 365 nm UV lamps (approximately 3 cm between the bottle and the lamp) for 60 min in a water bath to keep the reaction temperature below room temperature (Fig. S1A†). After modification, the products were desalted with homemade C18 SPE tips, lyophilized, and stored at −80 °C for further analysis.Photochemical trifluoromethylation of native proteins
100 μL protein solution (20 μM) was mixed with 100 μL NaTFMS (100 mM) in 10× PB buffer (0.1 M phosphate, pH 7.4) (trifluoromethylation of trypsin was carried out in 1 mM HCl) to a final concentration of protein (10 μM) and NaTFMS (50 mM) respectively. The mixed solution was transferred to a quartz bottle and irradiated under four 365 nm UV lamps (approximately 3 cm between the bottle and the lamp) for 30 min in a water bath to keep the reaction temperature below room temperature. After reaction, part of the sample solution was buffer exchanged from 10× PB to 50 mM CH3COONH4 solution using a Micro Bio-Spin column (trypsin was desalted using a 3K ultrafiltration tube with 1 mM HCl) and centrifugation at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 5 min before further native MS characterization. The other part was desalted with homemade SPE tips packed with PS-DVB beads and lyophilized for further digestion.
000g for 5 min before further native MS characterization. The other part was desalted with homemade SPE tips packed with PS-DVB beads and lyophilized for further digestion.
Protein expression and digestion
A haloalkane dehalogenase (linB, PDB: 1MJ5) plasmid constructed with a His-tag at the C-terminus was obtained from the Public Protein/Plasmid Library (Jiangsu, China); protein expression and purification were based on a reported method.68 The trifluoromethylated protein samples were digested and analyzed by LC-MS/MS to determine the modification sites. Briefly, the lyophilized protein samples were re-dissolved into 100 mM NH4HCO3 (pH = 8.0) buffer containing 8 M urea, reduced and alkylated with TCEP and IAA. Then, the protein samples were diluted 4 times with 100 mM NH4HCO3 (pH 8.0) buffer and trypsin was added in a substrate/enzyme ratio of 50:1 (w/w), followed by incubation at 37 °C overnight. For proteins without disulfide bonds, lyophilized protein samples were directly re-dissolved into NH4HCO3 buffer followed by trypsin digestion. For haloalkane dehalogenase, the lyophilized powder was redissolved in 100 mM NH4HCO3 (pH 8.0) buffer containing 2 mM CaCl2 and chymotrypsin was added in a substrate/enzyme ratio of 25:1 (w/w), followed by incubation at 37 °C for 4 hours. After digestion, the samples were purified with C18 SPE tips, lyophilized, and stored at −80 °C for further LC-MS/MS analysis.
LC-MS/MS analysis
The lyophilized peptide samples were re-dissolved into 0.1% FA/H2O to 0.01 μg μL−1 for trifluoromethylated protein digests. An Orbitrap Fusion Lumos Tribrid MS (Thermo Fisher, USA) coupled to a Vanquish Flex HPLC system (Thermo Fisher, USA) was applied for LC-MS/MS analysis of the peptide samples. In brief, 10 μL sample was loaded onto a trap column (3 cm × 150 μm i.d.) packed with C18 AQ beads (3 μm, 120 Å) at a flow rate of 5 μL min−1 of 100% mobile phase A for 10 min, then the sample was analyzed using a separation column (30 cm × 150 μm i.d.) packed with C18 AQ beads (2.4 μm, 120 Å) at 500 nL min−1 with a binary RP separation gradient from 5% to 30% mobile phase B for 40 min. The MS acquisition was set to positive mode in a data-dependent analysis (DDA) manner. A resolution of 60000 was set for MS1 scans; the MS2 scans were operated at “top-speed” mode (3 seconds) with a resolution of 15000. The intensity threshold was 3 × 104; the precursor ions with charge state of 2 to 7 were isolated and subjected to the HCD cell with normalized energy of 30%, and the dynamic exclusion time was enabled with an exclusion duration of 60 s.
MS dataset analysis
The MS datasets were deconvoluted by using the Xtract algorithm of FreeStyle with the default settings for native proteins and charge range from 1 to 2 for octreotide.The LC-MS/MS datasets of trifluoromethylated proteins were searched against a lab-built protein database with Thermo Proteome Discoverer (version 2.4.1.15). For trifluoromethylated proteins, the variable modifications were set as mono-oxidation (+[O]) (+15.995 Da) to Met and Trp, di-oxidation (+2[O]) (+31.990 Da) to Trp, mono-trifluoromethylation (+[CF3]) (+67.987 Da) to Trp, Tyr, Phe and His, di-trifluoromethylation (+2[CF3]) (+135.975 Da) to Trp and Tyr, mono-trifluoromethylation-mono-oxidation (+[CF3] + [O]) (+83.982 Da) and mono-trifluoromethylation-di-oxidation (+[CF3] + 2[O]) (+99.977 Da) to Trp, and the default variable modification of acetylation to the N-terminus (+42.011 Da) was also added. The static modification was carbamidomethyl (+57.021 Da) to Cys. The enzyme was selected as chymotrypsin (FLMWY, full) for haloalkane dehalogenase samples and trypsin (KR, full) for other protein digests. The number of maximum missed cleavage site was 2 for both trypsin and chymotrypsin. The validation was based on the q-value of concatenated target/decoy selection with a strict FDR of 0.01. Other parameters were set as default values.
The calculation of the relative intensity of trifluoromethylation products by using MS and protein site modification ratios by using MS/MS was based on the following equations:
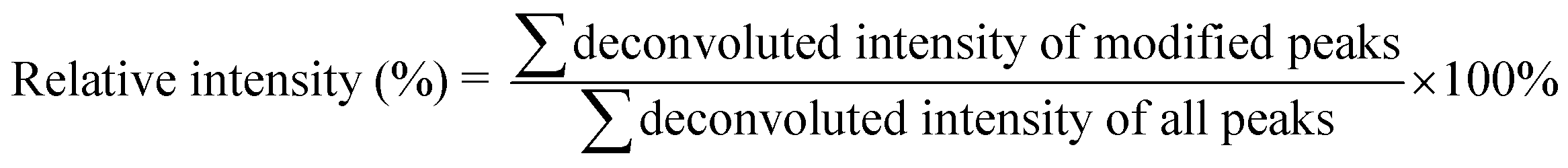 | (1) |
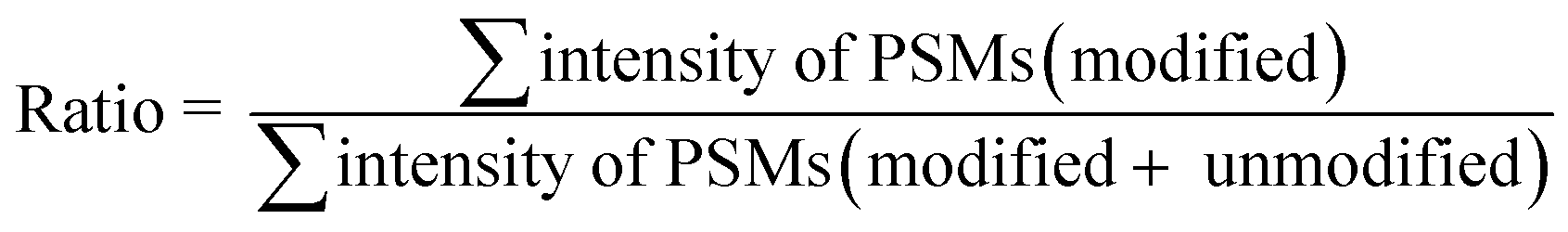 | (2) |
Only residues with ≥10 PSMs (peptide spectrum matches) in database matching were counted for the calculation of the site modification ratio with eqn (2).
Data availability
All data supporting the conclusion of this study are available in the main text and/or the supplementary materials. The LC-MS/MS datasets and database search results have been deposited to the ProteomeXchange Consortium via the PRIDE69 partner repository with the dataset identifier PXD045153.Author contributions
Conceptualization: F. W. and C. L.; methodology: C. L., Z. T., and Z. L.; formal analysis: C. L., P. L., W. Z., T. Z., and Z. D.; investigation: C. L., W. Z., Z. D., and F. W.; resources: X. Y., X. L., Z. D., W. Z., and Z. L.; supervision: F. W., X. Y., X. L., and Z. D.; project administration, F. W.; funding acquisition, F. W., X. Y., X. L., and Z. D.; writing-original draft: F. W. and C. L.; writing-review & editing: F. W., Z. D., and C. L.Conflicts of interest
The authors declare they have no competing interests.Acknowledgements
We acknowledge the financial support from the National Key R&D Program of China (2019YFE0119300, 2022YFC3400502, and 2022YFC3401100), National Natural Science Foundation of China (92253304 and 32088101), Guangdong Innovative & Entrepreneurial Research Team Program (No. 2021ZT09C278), and the grant from DICP (DICPI202242). The authors acknowledge the technological support from the biological mass spectrometry station of Dalian Coherent Light Source.References
- T. Tamura and I. Hamachi, Chemistry for Covalent Modification of Endogenous/Native Proteins: From Test Tubes to Complex Biological Systems, J. Am. Chem. Soc., 2019, 141, 2782–2799 Search PubMed
.
- B. Josephson, C. Fehl, P. G. Isenegger, S. Nadal, T. H. Wright, A. W. J. Poh, B. J. Bower, A. M. Giltrap, L. Chen, C. Batchelor-McAuley, G. Roper, O. Arisa, J. B. I. Sap, A. Kawamura, A. J. Baldwin, S. Mohammed, R. G. Compton, V. Gouverneur and B. G. Davis, Light-driven post-translational installation of reactive protein side chains, Nature, 2020, 585, 530–537 CrossRef CAS PubMed
- Y. Bai, Z. Liu, Y. Li, H. Zhao, C. Lai, S. Zhao, K. Chen, C. Luo, X. Yang and F. Wang, Structural Mass Spectrometry Probes the Inhibitor-Induced Allosteric Activation of CDK12/CDK13-Cyclin K Dissociation, J. Am. Chem. Soc., 2023, 145, 11477–11481 CrossRef CAS PubMed
- X. R. Liu, M. M. Zhang and M. L. Gross, Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications, Chem. Rev., 2020, 120, 4355–4454 CrossRef CAS PubMed
- E. V. Petrotchenko and C. H. Borchers, Protein Chemistry Combined with Mass Spectrometry for Protein Structure Determination, Chem. Rev., 2022, 122, 7488–7499 CrossRef CAS PubMed
- Y. Zhou, Z. Liu, M. Yao, J. Chen, Y. Xiao, G. Han, J.-R. Shen and F. Wang, Elucidating the Molecular Mechanism of Dynamic Photodamage of Photosystem II Membrane Protein Complex by Integrated Proteomics Strategy, CCS Chem., 2022, 4, 182–193 CrossRef CAS
- X. Chen, B. Josephson and B. G. Davis, Carbon-Centered Radicals in Protein Manipulation, ACS Cent. Sci., 2023, 9, 614–638 CrossRef CAS PubMed
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Organometallic palladium reagents for cysteine bioconjugation, Nature, 2015, 526, 687–691 CrossRef CAS PubMed
- H. H. Dhanjee, I. Buslov, I. W. Windsor, R. T. Raines, B. L. Pentelute and S. L. Buchwald, Palladium–Protein Oxidative Addition Complexes by Amine-Selective Acylation, J. Am. Chem. Soc., 2020, 142, 21237–21242 CrossRef CAS PubMed
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Π-Clamp-mediated cysteine conjugation, Nat. Chem., 2016, 8, 120–128 CrossRef CAS PubMed
- M.-C. Wu and T. Yow Tsong, Local Hydrophobicity in Protein Secondary Structure Formation, J. Phys. Soc. Jpn., 2013, 82, 114801 CrossRef
- K. Asija and C. M. Teschke, A Hydrophobic Network: Intersubunit and Intercapsomer Interactions Stabilizing the Bacteriophage P22 Capsid, J. Virol., 2019, 93, e00727 CrossRef CAS PubMed
- H. Zhao, C. Liu, W. Ding, L. Tang, Y. Fang, Y. Chen, L. Hu, Y. Yuan, D. Fang and S. Lin, Manipulating Cation-π Interactions with Genetically Encoded Tryptophan Derivatives, J. Am. Chem. Soc., 2022, 144, 6742–6748 CrossRef CAS PubMed
- Q. Zhang, X. Wu, J. Zhou, L. Zhang, X. Xu, L. Zhang, Q. You and L. Wang, Design, synthesis and bioevaluation of inhibitors targeting HSP90-CDC37 protein-protein interaction based on a hydrophobic core, Eur. J. Med. Chem., 2021, 210, 112959 CrossRef CAS PubMed
- A. Balliu and L. Baltzer, Exploring Non-obvious Hydrophobic Binding Pockets on Protein Surfaces: Increasing Affinities in Peptide–Protein Interactions, ChemBioChem, 2017, 18, 1396–1407 CrossRef CAS PubMed
- I. Roterman, K. Stapor and L. Konieczny, New insights on the catalytic center of proteins from peptidylprolyl isomerase group based on the FOD-M model, J. Cell. Biochem., 2023, 124, 818–835 Search PubMed
- L. Bai and H. Li, Structural insights into the membrane chaperones for multi-pass membrane protein biogenesis, Curr. Opin. Struct. Biol., 2023, 79, 102563 Search PubMed
- S. Liu, S. Li, G. Shen, N. Sukumar, A. M. Krezel and W. Li, Structural basis of antagonizing the vitamin K catalytic cycle for anticoagulation, Science, 2021, 371, eabc5667 Search PubMed
- F. A. Thélot, W. Zhang, K. Song, C. Xu, J. Huang and M. Liao, Distinct allosteric mechanisms of first-generation MsbA inhibitors, Science, 2021, 374, 580–585 CrossRef PubMed
- C. Mao, P. Xiao, X.-N. Tao, J. Qin, Q.-T. He, C. Zhang, S.-C. Guo, Y.-Q. Du, L.-N. Chen, D.-D. Shen, Z.-S. Yang, H.-Q. Zhang, S.-M. Huang, Y.-H. He, J. Cheng, Y.-N. Zhong, P. Shang, J. Chen, D.-L. Zhang, Q.-L. Wang, M.-X. Liu, G.-Y. Li, Y. Guo, H. E. Xu, C. Wang, C. Zhang, S. Feng, X. Yu, Y. Zhang and J.-P. Sun, Unsaturated bond recognition leads to biased signal in a fatty acid receptor, Science, 2023, 380, eadd6220 CrossRef CAS PubMed
- Y. Zhuang, Y. Wang, B. He, X. He, X. E. Zhou, S. Guo, Q. Rao, J. Yang, J. Liu, Q. Zhou, X. Wang, M. Liu, W. Liu, X. Jiang, D. Yang, H. Jiang, J. Shen, K. Melcher, H. Chen, Y. Jiang, X. Cheng, M.-W. Wang, X. Xie and H. E. Xu, Molecular recognition of morphine and fentanyl by the human μ-opioid receptor, Cell, 2022, 185, 4361–4375 Search PubMed
- L.-H. Zhao, Q. He, Q. Yuan, Y. Gu, X. He, H. Shan, J. Li, K. Wang, Y. Li, W. Hu, K. Wu, J. Shen and H. E. Xu, Conserved class B GPCR activation by a biased intracellular agonist, Nature, 2023, 621, 635–641 CrossRef CAS PubMed
- G. F. Mayol, L. A. Defelipe, J. P. Arcon, A. G. Turjanski and M. A. Marti, Solvent Sites Improve Docking Performance of Protein–Protein Complexes and Protein–Protein Interface-Targeted Drugs, J. Chem. Inf. Model., 2022, 62, 3577–3588 CrossRef CAS PubMed
- E. Liepinsh and G. Otting, Organic solvents identify specific ligand binding sites on protein surfaces, Nat. Biotechnol., 1997, 15, 264–268 CrossRef CAS PubMed
- J. P. Arcon, L. A. Defelipe, C. P. Modenutti, E. D. López, D. Alvarez-Garcia, X. Barril, A. G. Turjanski and M. A. Martí, Molecular Dynamics in Mixed Solvents Reveals Protein–Ligand Interactions, Improves Docking, and Allows Accurate Binding Free Energy Predictions, J. Chem. Inf. Model., 2017, 57, 846–863 Search PubMed
- H. Zhao, L. Tang, Y. Fang, C. Liu, W. Ding, S. Zang, Y. Chen, W. Xu, Y. Yuan, D. Fang and S. Lin, Manipulating Cation−π Interactions of Reader Proteins in Living Cells with Genetic Code Expansion, J. Am. Chem. Soc., 2023, 145, 16406–16416 CrossRef CAS PubMed
- S. E. Biehn, P. Limpikirati, R. W. Vachet and S. Lindert, Utilization of Hydrophobic Microenvironment Sensitivity in Diethylpyrocarbonate
Labeling for Protein Structure Prediction, Anal. Chem., 2021, 93, 8188–8195 CrossRef CAS PubMed
- M. Cheng, C. Guo and M. L. Gross, The Application of Fluorine-Containing Reagents in Structural Proteomics, Angew. Chem., Int. Ed., 2020, 59, 5880–5889 CrossRef CAS PubMed
- S. Verhoog, C. W. Kee, Y. Wang, T. Khotavivattana, T. C. Wilson, V. Kersemans, S. Smart, M. Tredwell, B. G. Davis and V. Gouverneur, (18)F-Trifluoromethylation of Unmodified Peptides with 5-(18)F-(Trifluoromethyl)dibenzothiophenium Trifluoromethanesulfonate, J. Am. Chem. Soc., 2018, 140, 1572–1575 CrossRef CAS PubMed
- N. Ichiishi, J. P. Caldwell, M. Lin, W. Zhong, X. Zhu, E. Streckfuss, H. Y. Kim, C. A. Parish and S. W. Krska, Protecting group free radical C-H trifluoromethylation of peptides, Chem. Sci., 2018, 9, 4168–4175 RSC
- Y. W. Bo Ding, Y. L. C. Song, L. Yin, J. Yuan, Y. Ren, A. Lei and C.-W. Chiang, Selective Photoredox Trifluoromethylation of TryptophanContaining Peptides, Eur. J. Org Chem., 2019, 7596–7605, DOI:10.1002/ejoc.201901572
- M. Imiolek, G. Karunanithy, W. L. Ng, A. J. Baldwin, V. Gouverneur and B. G. Davis, Selective Radical Trifluoromethylation of Native Residues in Proteins, J. Am. Chem. Soc., 2018, 140, 1568–1571 Search PubMed
- L. Fojtík, J. Fiala, P. Pompach, J. Chmelík, V. Matoušek, P. Beier, Z. Kukačka and P. Novák, Fast Fluoroalkylation of Proteins Uncovers the Structure and Dynamics of Biological Macromolecules, J. Am. Chem. Soc., 2021, 143, 20670–20679 CrossRef PubMed
- M. Cheng, B. Zhang, W. Cui and M. L. Gross, Laser-Initiated Radical Trifluoromethylation of Peptides and Proteins: Application to Mass-Spectrometry-Based Protein Footprinting, Angew Chem. Int. Ed. Engl., 2017, 56, 14007–14010 CrossRef CAS PubMed
- M. Cheng, A. Asuru, J. Kiselar, G. Mathai, M. R. Chance and M. L. Gross, Fast Protein Footprinting by X-ray Mediated Radical Trifluoromethylation, J. Am. Soc. Mass Spectrom., 2020, 31, 1019–1024 CrossRef CAS PubMed
- D. A. Dougherty, Cation-π Interactions in Chemistry and Biology: A New View of Benzene, Phe, Tyr, and Trp, Science, 1996, 271, 163–168 CrossRef CAS PubMed
- B. Kaboudin, M. Ghashghaee, A. Bigdeli, A. Farkhondeh, M. Eskandari and H. Esfandiari, Recent Advances on the Application of Langlois' Reagent in Organic Transformations, ChemistrySelect, 2021, 6, 12998–13014 CrossRef CAS
- J. Gong and P. L. Fuchs, Alkynylation of C−H Bonds via Reaction with Acetylenic Triflones1, J. Am. Chem. Soc., 1996, 118, 4486–4487 CrossRef CAS
- N. S.-R. Charlène Gadais, E. Chelain, J. Pytkowicz and T. Brigaud, Tailored Approaches towards the Synthesis of L-S-(Trifluoromethyl)cysteine- and L-TrifluoromethionineContaining Peptides, Eur. J. Org Chem., 2017, 246–251, DOI:10.1002/ejoc.201601318
- S. Y. Ro, L. F. Schachner, C. W. Koo, R. Purohit, J. P. Remis, G. E. Kenney, B. W. Liauw, P. M. Thomas, S. M. Patrie, N. L. Kelleher and A. C. Rosenzweig, Native top-down mass spectrometry provides insights into the copper centers of membrane-bound methane monooxygenase, Nat. Commun., 2019, 10, 2675 CrossRef PubMed
- J. Zhang, G. Reza Malmirchegini, R. T. Clubb and J. A. Loo, Native top-down mass spectrometry for the structural characterization of human hemoglobin, Eur. J. Mass Spectrom., 2015, 21, 221–231 CrossRef CAS PubMed
- J. Kyte and R. F. Doolittle, A simple method for displaying the hydropathic character of a protein, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS PubMed
- D. Eisenberg, E. Schwarz, M. Komaromy and R. Wall, Analysis of membrane and surface protein sequences with the hydrophobic moment plot, J. Mol. Biol., 1984, 179, 125–142 Search PubMed
- G. D. Rose, A. R. Geselowitz, G. J. Lesser, R. H. Lee and M. H. Zehfus, Hydrophobicity of amino acid residues in globular proteins, Science, 1985, 229, 834–838 CrossRef CAS PubMed
- S. Capone, I. Kieltsch, O. Flogel, G. Lelais, A. Togni and D. Seebach, Electrophilic S-Trifluoromethylation of Cysteine Side Chains in alpha- and beta-Peptides: Isolation of Trifluoronethylated Sandostatin (R) (Octreotide) Derivatives, Helv. Chim. Acta, 2008, 91, 2035–2056 CrossRef CAS
- X. R. Liu, M. M. Zhang, B. Zhang, D. L. Rempel and M. L. Gross, Hydroxyl-Radical Reaction Pathways for the Fast Photochemical Oxidation of Proteins Platform As Revealed by (18)O Isotopic Labeling, Anal. Chem., 2019, 91, 9238–9245 CrossRef CAS PubMed
- B. Sun, P. Huang, Z. Yan, X. Shi, X. Tang, J. Yang and C. Jin, Self-Catalyzed Phototandem Perfluoroalkylation/Cyclization of Unactivated Alkenes: Synthesis of Perfluoroalkyl-Substituted Quinazolinones, Org. Lett., 2021, 23, 1026–1031 CrossRef CAS PubMed
- X. Zhu, Y. Liu, C. Liu, H. Yang and H. Fu, Light and oxygen-enabled sodium trifluoromethanesulfinate-mediated selective oxidation of C–H bonds, Green Chem., 2020, 22, 4357–4363 RSC
- S. Yamada, Cation-pi Interactions in Organic Synthesis, Chem. Rev., 2018, 118, 11353–11432 CrossRef CAS PubMed
- J. P. Gallivan and D. A. Dougherty, Cation-π interactions in structural biology, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS PubMed
- D. T. Infield, A. Rasouli, G. D. Galles, C. Chipot, E. Tajkhorshid and C. A. Ahern, Cation-π Interactions and their Functional Roles in Membrane Proteins, J. Mol. Biol., 2021, 433, 167035 CrossRef CAS PubMed
- A. P. Stephan, P. H. Ariele, L. P. Kerry, W. L. Joseph, A. L. Henry, A. D. Dennis and C. R. L. Sarah, A Cation-π Interaction at a Phenylalanine Residue in the Glycine Receptor Binding Site Is Conserved for Different Agonists, Mol. Pharmacol., 2011, 79, 742 Search PubMed
- W. Zhong, J. P. Gallivan, Y. Zhang, L. Li, H. A. Lester and D. A. Dougherty, From ab initio quantum mechanics to molecular neurobiology: a cation–π binding site in the nicotinic receptor, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12088–12093 CrossRef CAS PubMed
- A. C. Kruse, A. M. Ring, A. Manglik, J. Hu, K. Hu, K. Eitel, H. Hübner, E. Pardon, C. Valant, P. M. Sexton, A. Christopoulos, C. C. Felder, P. Gmeiner, J. Steyaert, W. I. Weis, K. C. Garcia, J. Wess and B. K. Kobilka, Activation and allosteric modulation of a muscarinic acetylcholine receptor, Nature, 2013, 504, 101–106 Search PubMed
- L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C. J. Li, Simple and Clean Photoinduced Aromatic Trifluoromethylation Reaction, J. Am. Chem. Soc., 2016, 138, 5809–5812 CrossRef CAS PubMed
- A. Ostermann, R. Waschipky, F. G. Parak and G. U. Nienhaus, Ligand binding and conformational motions in myoglobin, Nature, 2000, 404, 205–208 CrossRef CAS PubMed
- J. S. Olson, Kinetic mechanisms for O2 binding to myoglobins and hemoglobins, Mol. Aspects Med., 2022, 84, 101024 CrossRef CAS PubMed
- E. Lanzarotti, L. A. Defelipe, M. A. Marti and A. G. Turjanski, Aromatic clusters in protein–protein and protein–drug complexes, J. Cheminf., 2020, 12, 30 CAS
- Y. Nagata, Y. Ohtsubo and M. Tsuda, Properties and biotechnological applications of natural and engineered haloalkane dehalogenases, Appl. Microbiol. Biotechnol., 2015, 99, 9865–9881 CrossRef CAS PubMed
- T. Koudelakova, S. Bidmanova, P. Dvorak, A. Pavelka, R. Chaloupkova, Z. Prokop and J. Damborsky, Haloalkane dehalogenases: biotechnological applications, Biotechnol. J., 2013, 8, 32–45 Search PubMed
- R. H. B. Smith, A. Dar, A. Schlessinger, PyVOL: a PyMOL plugin for visualization, comparison, and volume calculation of drug-binding sites, bioRxiv, 2019, preprint, DOI:10.1101/816702.
- L. Leboffe, A. di Masi, F. Polticelli, V. Trezza and P. Ascenzi, Structural Basis of Drug Recognition by Human Serum Albumin, Curr. Med. Chem., 2020, 27, 4907–4931 CrossRef CAS PubMed
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, Structural Basis of the Drug-binding Specificity of Human Serum Albumin, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed
- A. A. Bhattacharya, T. Grüne and S. Curry, Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin, J. Mol. Biol., 2000, 303, 721–732 CrossRef CAS PubMed
- K. Yamasaki, H. Teshima, R. Yukizawa, K. Kuyama, K. Tsukigawa, K. Nishi, M. Otagiri and A. Kawai, Structural Basis of the Change in the Interaction Between Mycophenolic Acid and Subdomain IIA of Human Serum Albumin During Renal Failure, J. Med. Chem., 2023, 66, 951–961 CrossRef CAS PubMed
- A. B. Moyle, M. Cheng, N. D. Wagner and M. L. Gross, Benzoyl Transfer for Footprinting Alcohol-Containing Residues in Higher Order Structural Applications of Mass-Spectrometry-Based Proteomics, Anal. Chem., 2022, 94, 1520–1524 CrossRef CAS PubMed
- A. B. Moyle, N. D. Wagner, W. J. Wagner, M. Cheng and M. L. Gross, Workflow for Validating Specific Amino Acid Footprinting Reagents for Protein Higher Order Structure Elucidation, Anal. Chem., 2023, 95, 10119–10126 CrossRef CAS PubMed
- J. Chen, A. Wang, B. Liu, Y. Zhou, P. Luo, Z. Zhang, G. Li, Q. Liu and F. Wang, Quantitative Lysine Reactivity Profiling Reveals Conformational Inhibition Dynamics and Potency of Aurora A Kinase Inhibitors, Anal. Chem., 2019, 91, 13222–13229 Search PubMed
- Y. Perez-Riverol, J. Bai, C. Bandla, D. García-Seisdedos, S. Hewapathirana, S. Kamatchinathan, D. J. Kundu, A. Prakash, A. Frericks-Zipper, M. Eisenacher, M. Walzer, S. Wang, A. Brazma and J. A. Vizcaíno, The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences, Nucleic Acids Res., 2022, 50, D543–d552 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05106d |
| This journal is © The Royal Society of Chemistry 2024 |