
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecular adjuvants repurpose antibiotics towards Gram-negative bacterial infections and multispecies bacterial biofilms†
Rajib
Dey
a,
Sudip
Mukherjee
a,
Riya
Mukherjee
a and
Jayanta
Haldar
 *ab
*ab
aAntimicrobial Research Laboratory, New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bengaluru 560064, Karnataka, India. E-mail: jayanta@jncasr.ac.in
bSchool of Advanced Materials, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bengaluru 560064, Karnataka, India
First published on 15th November 2023
Abstract
Gram-negative bacterial infections pose a significant challenge due to two major resistance elements, including the impermeability of the outer membrane and the overexpression of efflux pumps, which contribute to antibiotic resistance. Additionally, the coexistence of multispecies superbugs in mixed species biofilms further complicates treatment, as these infections are refractory to most antibiotics. To address this issue, combining obsolete antibiotics with non-antibiotic adjuvants that target bacterial membranes has shown promise in combating antibacterial resistance. However, the clinical translation of this cocktail therapy has been hindered by the toxicity associated with these membrane active adjuvants, mainly due to a limited understanding of their structure and mechanism of action. Towards this goal, herein, we have designed a small molecular adjuvant by tuning different structural parameters, such as the balance between hydrophilic and hydrophobic groups, spatial positioning of hydrophobicity and hydrogen bonding interactions, causing moderate membrane perturbation in bacterial cells without any toxicity to mammalian cells. Moderate membrane perturbation not only enhances the internalization of antibiotics, but also increases the intracellular concentration of drugs by hampering the efflux machinery. This revitalises the efficacy of various classes of antibiotics by 32–512 fold, without inducing toxicity. The leading combination not only exhibits potent bactericidal activity against A. baumannii biofilms but also effectively disrupts mature multispecies biofilms composed of A. baumannii and methicillin-resistant Staphylococcus aureus (MRSA), which is typically resistant to most antibiotics. Importantly, the combination therapy demonstrates good biocompatibility and excellent in vivo antibacterial efficacy (>99% reduction) in a skin infection model of A. baumannii. Interestingly, A. baumannii shows reduced susceptibility to develop resistance against the leading combination, underscoring its potential for treating multi-drug resistant infections.
Introduction
The increasing incidence of infectious diseases and the associated mortality caused by drug-resistant infections have created a significant concern in public healthcare.1,2 A recent report indicates that approximately 5 million people succumb to drug-resistant infections, with a majority of these infections caused by Gram-negative superbugs, such as carbapenem-resistant A. baumannii, P. aeruginosa, and Enterobacteriaceae spp., which have been designated as critical priority pathogens by the World Health Organization (WHO).3 The emergence of these life-threatening pathogens in both community-associated and hospital-acquired infections has posed a serious challenge to the healthcare system.4 The prevalence of multispecies bacterial co-infections and secondary bacterial infections among hospitalized COVID patients has further exacerbated the situation.5,6 This menace is amplified by the slow pace of development, approval, and translation of novel antibiotics, rendering these infections resilient to almost all existing antibiotic classes and demanding innovative alternatives. Resistance mechanisms in Gram-negative bacteria are broadly classified into: (1) impermeability caused by the outer membrane consisting of lipopolysaccharides (LPSs); (2) overexpression of efflux pumps driven by membrane potential; (3) production of antibiotic degrading enzymes such as β-lactamases (including serine-β-lactamase and metallo-β-lactamase); and (4) target mutations.7 Among these, impermeability of the outer membrane and the transmembrane efflux pumps contribute to multi-drug resistance in Gram-negative pathogens, rendering most conventional antibiotics ineffective.8 Recently, membrane-targeting antibiotic adjuvants have emerged as a promising strategy to tackle antimicrobial resistance (AMR) by repurposing and revitalizing obsolete antibiotics.9–12 Many of these adjuvants draw inspiration from naturally occurring antimicrobial peptides (AMPs).13,14 These cationic amphiphilic peptides, which directly target bacterial membranes, not only exhibit direct microbicidal action but also enhance the intracellular concentration of antibiotics, acting as potentiators.15,16However, the current use of cationic lipophilic compounds as permeabilization enhancers to sensitize resistant antibiotics is hindered by their toxicity towards mammalian cells.17,18 Therefore, a detailed understanding of the optimal amphiphilicity in the structure such as the balance between hydrophobicity and hydrophilic moieties, spatial positioning of hydrophobic groups and the influence of hydrogen bonding on structural parameters, is crucial.19–21 Furthermore, the overexpression of efflux machinery is another major cause of acquired resistance to antibiotics.22,23 While several effective efflux pump disruptors have been reported, their severe toxicity has limited their applicability as antibiotic adjuvants.24,25 A detailed understanding of the structural parameters and mechanistic insights of these membrane potential-targeting compounds is scarce in the literature. In the last two decades, different small molecular mimics of antimicrobial peptides (SMAMPs) have been developed as antibiotic adjuvants to repurpose and rejuvenate obsolete antibiotics. As examples, SPR741 (developed by Spero Therapeutics) and Nylexa (developed by NovaBiotics) are in the initial stage of clinical trials to be used as potentiators with a wide range of antibiotics.26,27 Recently, another three drugs approved for non-antimicrobial indications, namely 5-fluorouracil, fluspirilene, and Bay 11-7082, resensitized a clinical strain of A. baumannii to azithromycin.28
In this study, we investigated the synergistic activity of a small molecular adjuvant to repurpose and revitalize different resistant antibiotics including rifampicin, fusidic acid, minocycline and chloramphenicol against Gram-negative pathogens (Fig. 1A). The structural features of the compound were designed to obtain a weak to moderate membrane-perturbing and membrane-depolarizing adjuvant without inducing toxicity towards mammalian cells. The adjuvant enhances the intracellular concentration of antibiotics, and the synergistic effect was validated through checkerboard assays against various multi-drug resistant Gram-negative bacteria and their clinical isolates. The bactericidal activities of the leading combinations were investigated against NDM-1 producing pathogens using time-kill kinetics. The biocompatibility of the leading combinations and the small molecule alone were assessed against mammalian cell lines and through in vivo systemic toxicity, respectively. Furthermore, the efficiency of the leading combination in disrupting preformed biofilms was studied. In addition to biofilm-mediated infections, multispecies bacterial infections pose an intense threat to healthcare practices. Multispecies biofilms are prevalent in nature and are particularly prominent in dental abscesses, cystic fibrosis, and diabetic wounds. Therefore, the antibacterial and anti-biofilm efficacy of the small molecular adjuvant-based combination therapy was evaluated against a co-culture and biofilm of MRSA and NDM-1 producing A. baumannii, which represents a multispecies bacterial infection. Importantly, the in vivo antibacterial activity of the leading combination therapy was investigated using a mouse model of skin infection caused by NDM-1 producing Gram-negative pathogens. Finally, the frequency of resistance development for the combination was assessed.
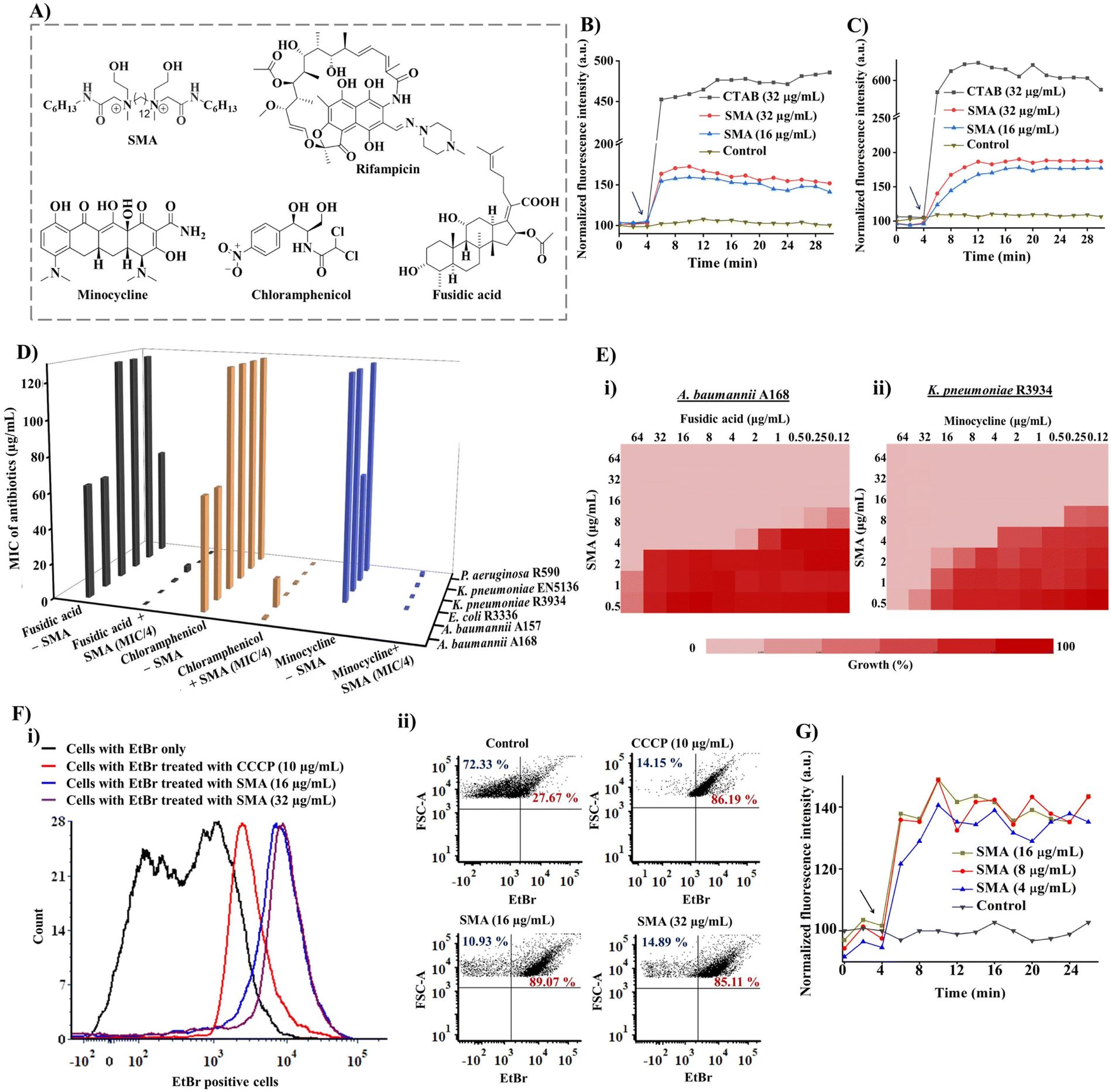 | ||
| Fig. 1 (A) Chemical structures of SMA and resistant antibiotics sensitized by the adjuvant; (B) outer membrane permeabilization (by using N-phenyl-1-naphthylamine (NPN) fluorescent dye); and (C) cytoplasmic membrane depolarization (by using a DiSC3(5) fluorescence probe) by SMA at different sub-MIC concentrations against NDM-1 producing bacteria A. baumannii A168. Surfactant CTAB (cetyltrimethylammonium bromide) was taken as a positive control. Arrow indicates the time of compound addition; (D) 3D flow chart representing the potentiation efficacy of SMA with fusidic acid, chloramphenicol, and minocycline against various drug-resistant Gram-negative superbugs; (E) chequerboard assay revealing synergistic efficacy of (i) SMA and fusidic acid against A. baumannii A168, (ii) SMA and minocycline against K. pneumoniae R3934; (F) efflux pump inhibition assay; (i) the number of parent cells used in the EtBr assay; (ii) SMA activated a dose-dependent increase in ethidium bromide (EtBr) accumulation in A. baumannii A168 bacteria. Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) was taken as a positive control. Fluorescence intensity was measured using a flow cytometer; (G) minocycline uptake assay upon treatment with different concentrations of SMA against NDM-1 producing K. pneumoniae R3934. | ||
Results and discussion
Design and synthesis of small molecular adjuvant (SMA)
The small molecular adjuvant, SMA, was carefully designed to incorporate specific structural features that enhance its interaction with the lipid bilayer of Gram-negative bacteria. The presence of the outer membrane in these bacteria creates an additional barrier that restricts the permeability of antibiotics. To overcome this challenge, hexyl pendant long chains with spacer hydrophobicity were introduced in the molecular design. The strategic distribution of hydrophobic moieties in both the spacer and pendent regions was fine-tuned in our previous study, as the spatial positioning of hydrophobicity plays a crucial role in improving the selectivity of membrane-targeting compounds.15,21 In addition, two ethanol moieties were introduced to achieve optimal amphiphilicity and to facilitate superior interaction with the outer membrane of Gram-negative bacteria through hydrogen bonding. The rational design of SMA considered these structural parameters to ensure its effectiveness as a membrane-targeting adjuvant. The synthesis of SMA was performed on a large scale (gram scale) using a straightforward three-step synthetic route, as outlined in Scheme S1.† Detailed synthetic procedures can be found in the ESI (Fig. S1–S3),† providing comprehensive information on the synthesis of SMA.Overall, the design and synthesis of SMA involved careful consideration of hydrophobicity, spatial positioning of hydrophobic groups, and the incorporation of ethanol moieties to achieve optimal amphiphilicity and facilitate effective interaction with the outer membrane of Gram-negative bacteria.
Membrane targeting mechanism of SMA against Gram-negative bacteria
The initial exploration of SMA's limited antibacterial activity against Gram-negative superbugs prompted us to investigate its potential to enhance the efficiency of resistant antibiotics against multi-drug resistant Gram-negative strains. To unravel the mechanism of SMA's interaction with bacterial membranes, we conducted membrane depolarization and bacterial outer membrane permeabilization assays on Gram-negative bacteria.The bacterial membrane potential is reliant on the proton-motive force (PMF), which comprises two components: trans-membrane electrical potential (ΔΨ) and trans-membrane pH gradient (ΔpH).29 We assessed bacterial membrane depolarization using the fluorescent dye 3,3′ dipropylthiadicarbocyanine iodide (DiSC3(5)). In energized cells, this dye accumulates at the cytoplasmic membrane and undergoes self-quenching, resulting in low fluorescence intensity. However, if the bacterial membrane potential is disrupted, the dye is released into the extracellular environment, leading to an increase in fluorescence intensity. Additionally, we evaluated the outer membrane permeabilization using N-phenyl-1-naphthylamine (NPN). Since NPN cannot penetrate the lipopolysaccharide (LPS) layer, it cannot enter the bacterial phospholipid layer under normal conditions. However, in a perturbed membrane, NPN readily translocates into the bacterial phospholipid layer, resulting in an escalation of fluorescence intensity. We employed the DiSC3(5) assay and NPN assay to investigate the extent of membrane-perturbing properties of the SMA compound against A. baumannii A168 bacteria (Fig. 1B and C). As a positive control, we used the surfactant CTAB, which exhibited strong outer membrane permeabilization, evident from a sharp increase in fluorescence intensity. In contrast, treatment with SMA resulted in a slight increase in fluorescence intensity in the NPN assay, indicating moderate outer membrane permeabilization against A. baumannii A168. Similarly, SMA induced moderate cytoplasmic membrane depolarization compared to CTAB in the DiSC3(5) assay. Overall, SMA, a small amphiphilic molecule, demonstrated a modest membrane-perturbing nature, which can aid in potentiation of antibiotics that are unable to cross the outer membrane or are expelled by efflux pumps.
These findings shed light on the membrane targeting mechanism of SMA and its potential to facilitate the action of antibiotics by perturbing the bacterial membrane, allowing the entry of otherwise restricted antibiotics and overcoming efflux pump-mediated resistance.
In vitro potentiation ability against drug-resistant Gram-negative superbugs
To assess the in vitro synergistic activity of SMA with various resistant antibiotics, we conducted chequerboard assays against a range of multi-drug resistant Gram-negative bacteria, including New Delhi metallo-β-lactamase-1 (NDM-1) producing strains. Initially, we determined the minimum inhibitory concentrations (MICs) of SMA and the antibiotics against the tested pathogens. SMA exhibited limited activity against most Gram-negative pathogens, with MIC values ranging from 64–256 μg mL−1, except for the A. baumannii A157 strain, where it displayed an MIC of 16 μg mL−1. The relatively poor activity of SMA may be attributed to its moderate membrane-perturbing nature.Next, we evaluated the potentiation ability of SMA against multi-drug resistant Gram-negative superbugs, (including NDM-1 producing strains, through chequerboard assays with four different classes of antibiotics: rifampicin, minocycline, chloramphenicol, and fusidic acid (Fig. 1D and Table 1)). Rifampicin and fusidic acid, both hydrophobic antibiotics commonly used for treating tuberculosis and Gram-positive infections, respectively, are typically impermeable to the Gram-negative bacterial outer membrane due to the presence of LPSs.30 Conversely, bacteria have developed resistance against other antibiotics such as minocycline and chloramphenicol through the efflux pump mechanism (e.g., TetA and AcrAB-TolC) present in the cytoplasmic membrane.31 SMA demonstrated the ability to potentiate these four antibiotics against various multidrug-resistant Gram-negative superbugs.
| Bacterial strain | SMA MIC (μg mL−1) | Antibiotic MIC (μg mL−1) | MIC of antibiotic (μg mL−1) in the presence of SMA | FICIa | ||
|---|---|---|---|---|---|---|
| MIC/4 | MIC/8 | MIC/16 | ||||
| a FICI – Fractional inhibitory concentration index (determined at MICSMA/16 to MICSMA/4). b ND – not determined. | ||||||
| Rifampicin | ||||||
| A. baumannii R674 | 128 | 64 | 0.25 | 0.25 | 1 | 0.08–0.25 |
| A. baumannii A168 | 256 | 16 | 0.031 | 0.031 | 0.062 | 0.05–0.25 |
| A. baumannii A157 | 16 | 1 | 0.031 | 0.062 | 0.5 | 0.16–0.31 |
| E. coli R3336 | 256 | 512 | 0.125 | 0.25 | 1 | 0.05–0.25 |
| K. pneumoniae R3934 | 128 | 512 | 0.25 | 0.25 | 1 | 0.06–0.25 |
| K. pneumoniae ATCC BAA2146 | 64 | 16 | 0.125 | 0.5 | NDb | 0.16–0.26 |
| K. pneumoniae EN5136 | 64 | 8 | 0.125 | 0.5 | 2 | 0.27–0.31 |
| P. aeruginosa R590 | 128 | 128 | 0.125 | 0.125 | 0.125 | 0.06–0.25 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Fusidic acid | ||||||
| A. baumannii R674 | 128 | 64 | ND | ND | ND | ND |
| A. baumannii A168 | 256 | 64 | 0.125 | 0.125 | 0.5 | 0.08–0.13 |
| A. baumannii A157 | 16 | 64 | 0.25 | 2 | 16 | 0.16–0.31 |
| E. coli R3336 | 256 | >512 | 1 | 8 | ND | 0.14–0.25 |
| K. pneumoniae R3934 | 128 | 512 | 4 | 16 | ND | 0.26–0.38 |
| K. pneumoniae ATCC BAA2146 | 64 | >256 | 0.125 | ND | ND | 0.25–0.31 |
| K. pneumoniae EN5136 | 64 | >256 | 0.125 | 2 | 32 | 0.14–0.25 |
| P. aeruginosa R590 | 128 | 64 | 0.5 | 4 | ND | 0.19–0.26 |
|
||||||
| Chloramphenicol | ||||||
| A. baumannii R674 | 128 | >128 | ND | ND | ND | ND |
| A. baumannii A168 | 256 | 64 | 1 | 8 | 16 | 0.25–0.31 |
| A. baumannii A157 | 16 | 64 | 16 | 32 | ND | 0.5–0.51 |
| E. coli R3336 | 256 | >128 | 0.125 | 0.25 | 1 | 0.14–0.25 |
| K. pneumoniae R3934 | 128 | >128 | 1 | 4 | ND | 0.16–0.31 |
| K. pneumoniae ATCC BAA2146 | 64 | >256 | 0.125 | 64 | ND | 0.25–0.37 |
| K. pneumoniae EN5136 | 64 | >256 | 0.125 | 32 | ND | 0.38–0.51 |
| P. aeruginosa R590 | 128 | >128 | 1 | 4 | ND | 0.25–0.35 |
|
||||||
| Minocycline | ||||||
| A. baumannii R674 | 128 | 8 | 0.125 | 0.25 | 1 | 0.19–0.31 |
| A. baumannii A168 | 256 | ND | ND | ND | ND | ND |
| A. baumannii A157 | 16 | ND | ND | ND | ND | ND |
| E. coli R3336 | 256 | 32 | 0.125 | 0.125 | 0.5 | 0.08–0.19 |
| K. pneumoniae R3934 | 128 | 64 | 0.125 | 0.5 | 4 | 0.09–0.13 |
| K. pneumoniae ATCC BAA2146 | 64 | 16 | 0.125 | 1 | 16 | 0.19–0.26 |
| K. pneumoniae EN5136 | 64 | 16 | 0.125 | 0.5 | 1 | 0.13–0.26 |
| P. aeruginosa R590 | 128 | 8 | 1 | 4 | 8 | 0.38–0.56 |
SMA significantly reduced the MIC values of rifampicin by 32-2024-fold at its sub-MIC concentration against all tested Gram-negative bacteria, indicating its potential for repurposing this anti-tuberculosis antibiotic (Table 1, Fig. S4†). Notably, SMA sensitized fusidic acid, commonly prescribed for treating Gram-positive bacterial infections, against drug-resistant Gram-negative bacteria, with a fractional inhibitory concentration index (FICI) value of <0.4. In the case of NDM-1 producing bacterium E. coli R3336, the combination therapy reduced the MIC of fusidic acid by 512-fold. Against another NDM-1 producing pathogen A. baumannii A168, the combination of SMA at an MIC/8 concentration lowered the MIC of fusidic acid to 0.125 μg mL−1 (512-fold reduction) (Fig. 1D, E(i), and S5†).
Furthermore, SMA showed promising synergy with chloramphenicol, reducing the MIC values of the antibiotic by 4-512-fold against most tested pathogens (Table 1, Fig. 1D and S6†). In the case of the NDM-1 producing bacteria K. pneumoniae R3934 strain, the combination of SMA at an MIC/8 concentration and chloramphenicol resulted in a MIC reduction to 4 μg mL−1 (Fig. 1D). However, SMA only exhibited an additive effect with chloramphenicol against the A. baumannii A157 strain, with a FICI value >0.5. Additionally, SMA sensitized minocycline by reducing the MIC values to ≤1 μg mL−1, with an FICI value of <0.5, against the tested Gram-negative pathogens (Table 1 and Fig. 1D). Specifically, SMA at an MIC/8 concentration decreased the MIC value of minocycline to 0.5 μg mL−1 (by 128-fold reduction) and 1 μg mL−1 (16-fold reduction) against NDM-1 producing bacteria K. pneumoniae R3934 and K. pneumoniae BAAATCC2146, respectively (Table 1, Fig. 1E(ii), S7†). In summary, our findings validate that SMA can potentiate rifampicin and fusidic acid and restore the efficacy of minocycline and chloramphenicol against critical drug-resistant Gram-negative bacteria, leveraging its membrane-active nature. These results reinforce our initial premise and underscore the potential of SMA as an adjunct therapy to combat drug-resistant Gram-negative superbugs.
Inhibition of efflux pumps
Bacterial efflux pumps play a significant role in the rapid emergence of drug resistance, with many of these pumps located in the cytoplasmic membrane, such as the resistance-nodulation-cell division (RND) superfamily and major facilitator superfamily (MFS), which are regulated by membrane potential.22 Given that SMA exhibited a moderate membrane depolarization effect, resulting in a change in membrane potential, we investigated its direct impact on bacterial efflux pumps.To assess the efflux inhibition properties of SMA, we performed an ethidium bromide (EtBr) accumulation assay using flow cytometry in A. baumannii A168 (NDM-1+). EtBr is a substrate of the RND efflux machinery and is expelled from healthy bacterial cells.32 Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) was taken as a positive control, which abolishes the entire proton-motive force resulting in the disruption of efflux machinery, while cells stained with EtBr only served as the negative control. CCCP at 10 μg mL−1 directly inhibited the efflux pump and increased the intracellular concentration of EtBr. Similarly, our potentiator SMA, at different concentrations (16 μg mL−1 and 32 μg mL−1), significantly increased the number of EtBr-stained cells, confirming the efflux pump inhibition by SMA (Fig. 1F(i) and (ii)). Consequently, the interference of the efflux machinery by SMA will lead to increased intracellular antibiotic concentrations and alleviation of drug resistance.
Therefore, the inhibitory effect of SMA on efflux pumps offers a promising mechanism to counteract drug resistance, as it enhances the intracellular concentration of antibiotics and combats the efflux-mediated resistance mechanism.
Accumulation of antibiotics inside bacteria
To further investigate the impact of SMA on antibiotic accumulation inside the bacterial cells, we examined the uptake of minocycline, a model antibiotic known to be resistant due to the activity of transmembrane efflux pumps. Minocycline exhibits fluorescence upon binding to the 30S subunit of bacterial ribosomes but is expelled by bacterial efflux pumps.33 In the presence of the potentiator SMA, we performed a minocycline uptake assay in NDM-1-producing bacteria, specifically K. pneumoniae R3934. Remarkably, the addition of SMA resulted in an increase in fluorescence intensity, confirming the enhanced accumulation of minocycline inside the bacterial cells (Fig. 1G). This observation underscores the ability of SMA to elevate the intracellular concentration of antibiotics, reinforcing the potency of combination therapy involving SMA. The enhanced intracellular antibiotic concentration achieved through the use of SMA holds great promise for overcoming efflux-mediated resistance mechanisms and improving the efficacy of antibiotic treatments. This strategy represents a significant advancement in mitigating the challenges posed by antimicrobial resistance.Bactericidal kinetics and visual turbidity assay
To demonstrate the bactericidal efficacy of the combination therapies, we conducted time-kill kinetics experiments using SMA in combination with three obsolete antibiotics (fusidic acid, chloramphenicol, or minocycline) against New Delhi metallo-β-lactamase-1 (NDM-1) producing Gram-negative pathogens. In addition to spread plating the treated bacterial cell suspensions onto solid agar media, we employed a visual turbidity test to further validate the effectiveness of the combination therapy (Fig. 2A and B). | ||
| Fig. 2 Bactericidal activity of combination therapies, (A) time-kill kinetics of the combinations consisting of (i) SMA and fusidic acid against A. baumannii A168, (ii) SMA and minocycline against K. pneumoniae R3934, and (iii) SMA and chloramphenicol against K. pneumoniae R3934, the asterisk indicates complete killing and the detection limit of experiment was <50 CFU mL; (B) visual turbidity of the combination of SMA and fusidic acid against A. baumannii A168; biofilm disruption ability against the preformed biofilm of A. baumannii A168; (C) percentage of biomass after treatment through crystal violet staining; (D) viability of the biofilm-embedded bacteria and dispersed cells after treatment; (E) 3D reconstruction of z-stack images of the biofilm after treatment, by imaging by confocal laser scanning microscopy through co-staining with SYTO-9 (green fluorescence; stained both live and dead cells) and PI (red fluorescence; stained dead cells); merged images represented with thickness. Scale bar: 10 μm. | ||
The combination of SMA (16 μg mL−1) and fusidic acid (4 μg mL−1) completely eliminated NDM-1 producing A. baumannii A-168 within 12 h, resulting in a 5.3log reduction in bacterial count. Notably, the growth media containing the SMA and fusidic acid cocktail showed no visual turbidity, and there were no bacterial colonies on the solid agar. In contrast, bacterial suspensions treated with SMA (16 μg mL−1) or fusidic acid (8 μg mL−1) alone displayed turbidity in the solution and abundant bacterial growth on the agar plate (Fig. 2A(i) and 2B).
Similarly, the combination of SMA (8 μg mL−1) and minocycline (4 μg mL−1) completely eradicated NDM-1 producing K. pneumoniae R3934 within 12 h, resulting in a 5.2log reduction in bacterial burden. In contrast, when SMA (8 μg mL−1) or minocycline (4 μg mL−1) was used as individual treatments, the bacterial count increased by ∼8log, similar to the untreated control. The visual turbidity experiment and spread plating of the treated bacterial suspension also confirmed the potent efficacy of the SMA-minocycline combination against NDM-1 producing K. pneumoniae strains (Fig. 2A(ii) and S8A†).
Furthermore, the combinations of SMA (32 μg mL−1) with chloramphenicol (4 μg mL−1) and SMA (16 μg mL−1) with chloramphenicol (16 μg mL−1) completely eliminated the tested NDM-1 producing bacteria K. pneumoniae R3934 within 6 h and 12 h, respectively, resulting in a 5.4log reduction in bacterial burden. In contrast, treatment with chloramphenicol alone at a concentration of 16 μg mL−1 increased bacterial viability to 9log, similar to the untreated control, within 12 h (Fig. 2A(iii) and S8B†).
In summary, the SMA molecule effectively repurposed various classes of antibiotics against critical NDM-1 producing Gram-negative pathogens, leading to complete eradication of bacterial burden in the solution phase. Based on the promising results, we selected the combination therapy of SMA and fusidic acid for further studies, as it demonstrated the potential to repurpose fusidic acid, which is typically prescribed for treating Gram-positive bacterial infections, against multi-drug resistant (MDR) Gram-negative superbugs.
Biofilm disruption ability against NDM-1 producing bacteria
Biofilm-mediated infections pose a serious threat to global health as they are resistant to conventional antibiotics. Biofilms are composed of millions of bacterial cells surrounded by a self-generated extracellular polymeric matrix (EPM).7 Unfortunately, the rigid EPS matrix renders most conventional antibiotics ineffective against biofilms. This situation is further exacerbated when dealing with biofilms formed by multidrug-resistant Gram-negative pathogens. Therefore, there is a critical need to develop potent anti-biofilm agents.To address this challenge, we investigated the biofilm disruption efficacy of the leading combination therapy of SMA and fusidic acid against NDM-1 producing A. baumannii A168 biofilms. Crystal violet staining confirmed that the combination therapy of SMA (16 μg mL−1) and fusidic acid (4 μg mL−1) effectively disrupted the preformed biofilm of NDM-1 producing A. baumannii. In contrast, treatment with fusidic acid alone did not help penetrate the preformed biofilm (Fig. 2C). Additionally, we quantified the viability of bacteria embedded within the biofilm. Interestingly, SMA and fusidic acid alone did not significantly reduce the cell count in the preformed biofilm. However, the combination of SMA (16 μg mL−1) and fusidic acid (4 μg mL−1), as well as SMA (16 μg mL−1) and fusidic acid (8 μg mL−1), resulted in 2.5log (>99% killing) and 3log (∼99.9% killing) reduction in viable bacterial count within the NDM-1 producing A. baumannii biofilm-embedded bacteria, respectively (Fig. 2D). Moreover, this combination therapy exhibited potent bactericidal efficacy against biofilm-dispersed cells, including metabolically dormant cells. The combination of SMA (16 μg mL−1) and fusidic acid (4 μg mL−1) reduced the bacterial burden by 5.6log in the dispersed cells of the A. baumannii A-168 biofilm (Fig. 2D).
Furthermore, we visualized the biofilm disruption ability and eradication of biofilm-embedded bacteria using confocal laser scanning microscopy (CLSM) with instantaneous staining using SYTO-9 (green fluorescence) and propidium iodide (PI: red fluorescence) dyes. SYTO-9 stains both live and dead bacterial cells, while PI specifically stains only dead cells. In the control biofilm, all cells were stained green, confirming the presence of live cells, and the biofilm thickness measured 8.5 μm. In the case of individual treatment with SMA or fusidic acid, all cells were green fluorescence-stained, indicating the presence of live cells, and the biofilm thickness measured 7.2 μm and 8.1 μm, respectively. However, treatment with the combination therapy of SMA and fusidic acid remarkably reduced the thickness of the preformed biofilm to 1.2 μm (Fig. 2E). In contrast, the combination-treated biofilm-embedded bacteria exhibited staining with both green and red fluorescence, confirming the killing of viable bacteria within the biofilm. Overall, SMA not only facilitated the disruption of the preformed biofilm but also repurposed fusidic acid to eliminate biofilm-embedded NDM-1 producing A. baumannii bacteria.
Antibacterial efficacy of combination therapy against multispecies bacterial co-culture
Multispecies bacterial co-infections are commonly observed under conditions such as lung infections, cystic fibrosis, and diabetic wound infections.34 When treating these critical multispecies bacterial co-infections, doctors often prescribe combination therapies using different types of antibiotics. However, conventional antibiotics are increasingly becoming ineffective in tackling these co-infections, leading to the emergence of drug resistance.35 Therefore, novel strategies are needed to address this challenge. In this study, we evaluated the efficacy of the leading combination therapy against a multispecies bacterial co-culture comprising Gram-positive and Gram-negative bacteria. SMA demonstrated potent anti-staphylococcal activity against vancomycin-susceptible and vancomycin-resistant strains (Table S1†). Furthermore, SMA repurposed fusidic acid to target MDR Gram-negative bacteria. Consequently, we investigated the effectiveness of the combination of SMA and fusidic acid against a multispecies co-culture of MRSA ATCC33591 and NDM-1 producing A. baumannii A168 bacteria. The anti-MRSA agent SMA (16 μg mL−1), both alone and in combination with fusidic acid (8 μg mL−1), completely eliminated the MRSA bacterial burden in the co-culture, resulting in a 5.2log reduction within 12h. Fusidic acid exhibited a static effect against the MRSA bacterial burden in the co-culture with A. baumannii. The well-known antibiotic vancomycin (16 μg mL−1) reduced the viability of MRSA bacteria by 0.7log within 12 h. Surprisingly, both SMA and fusidic acid alone increased the viability of A. baumannii A168 to 8.2log within 12 h in the co-culture with MRSA. Similarly, the viability of A. baumannii in the co-culture treated with colistin (8 μg mL−1) also increased to 8log within 12 h. Remarkably, the combination of SMA and fusidic acid achieved a ∼2.2log reduction (>99% killing) in the A. baumannii bacterial burden within the co-culture with MRSA (Fig. 3A(i)). The visual turbidity test also confirmed the potent efficacy of the leading combination (Fig. 3A(ii)). In conclusion, the combination therapy of SMA and fusidic acid demonstrated effectiveness in eliminating the multispecies bacterial co-culture.
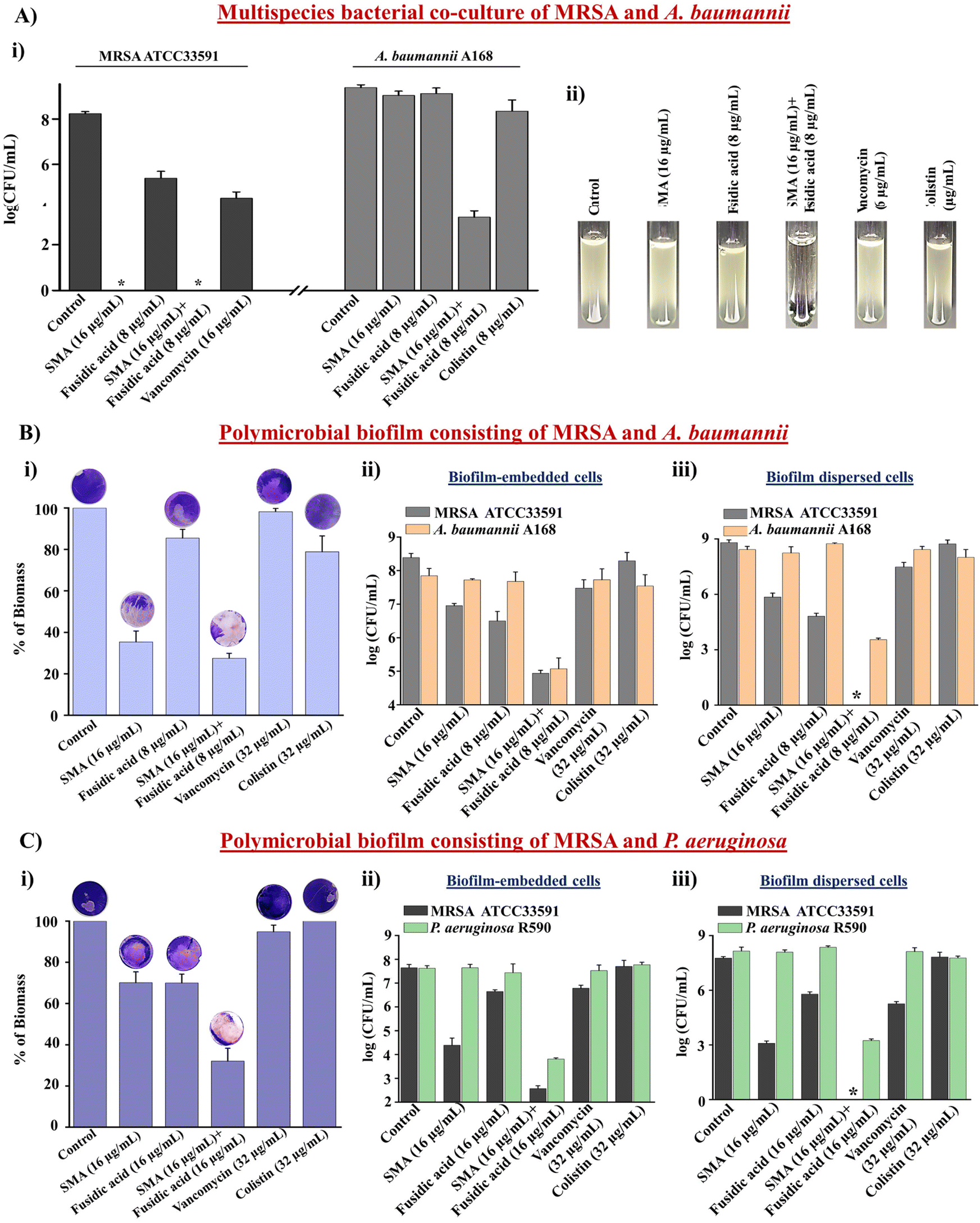 | ||
| Fig. 3 (A) Bactericidal efficacy of the combination of SMA + fusidic acid against multispecies bacterial co-culture of MRSA ATCC33591 and A. baumannii A168; (i) viable bacterial count in multispecies bacterial co-culture, and (ii) visual turbidity test of multispecies bacterial co-culture, asterisks indicate complete killing; (B) anti-biofilm efficacy of the combination of SMA + fusidic acid against the polymicrobial biofilm consisting of both MRSA ATCC33591 and A. baumannii A168, (i) percentage of biomass after treatment, (ii) no. of viable biofilm-embedded bacteria, and (iii) no. of viable bacteria in biofilm dispersed cells; (C) anti-biofilm efficacy of the combination of SMA + fusidic acid against the polymicrobial biofilm consisting of both MRSA ATCC33591 and P. aeruginosa R590, (i) percentage of biomass after treatment, (ii) no. of viable biofilm-embedded bacteria, and (iii) no. of viable bacteria in biofilm dispersed cells, asterisks indicate complete killing. The detection limit of the experiment was <50 CFU mL−1. | ||
Anti-biofilm efficacy of combination therapy against the multispecies bacterial biofilm
The significant reduction observed in the multispecies bacterial co-culture experiment prompted us to evaluate the efficacy of the lead cocktail (fusidic acid + SMA) against more complex multispecies bacterial biofilms. A preformed biofilm of MRSA ATCC33591 and NDM-1 producing A. baumannii A-168 was grown on coverslips and subsequently treated with the combination therapy. Crystal violet staining confirmed that SMA and fusidic acid individually reduced the biofilm biomass by 70% and 17% respectively. The control antibiotics, vancomycin and colistin, reduced the biofilm biomass by 7% and 22%, respectively. In contrast, the combination of SMA (16 μg mL−1) and fusidic acid (8 μg mL−1) achieved a remarkable 75% reduction in the biomass of the multispecies biofilm (Fig. 3B(i)). Moreover, the combination treatment of SMA (16 μg mL−1) and fusidic acid (8 μg mL−1) eliminated 3.2log MRSA bacterial burden within the multispecies biofilm. Individually, the anti-MRSA agents SMA and fusidic acid achieved reductions of 1.4log and 1.7log, respectively, in the MRSA bacterial burden within the biofilm. The blockbuster antibiotic, vancomycin (32 μg mL−1), only achieved a 1log reduction in the MRSA bacterial burden within the preformed biofilm. Additionally, the combination therapy effectively reduced the A. baumannii bacterial burden by 3.2log within the multispecies biofilm. In contrast, SMA and fusidic acid individually were ineffective against A. baumannii bacteria embedded in the multispecies biofilms. Similarly, colistin (32 μg mL−1) failed to eliminate the biofilm-embedded A. baumannii bacteria (Fig. 3B(ii)). Furthermore, this combination therapy demonstrated potent bactericidal efficacy against the dispersed cells within the multispecies biofilm, targeting various phases of bacterial cells. The combination of SMA (16 μg mL−1) and fusidic acid (8 μg mL−1) achieved complete elimination of the bacterial burden (8.5log reduction) of MRSA and reduced the A. baumannii A168 bacterial burden by 4.6log in the dispersed cells of the mixed-species biofilm (Fig. 3B(iii)). Similarly, the combination of SMA and fusidic acid effectively disrupted the preformed multispecies biofilm of MRSA and P. aeruginosa R590, resulting in a 70% reduction in biomass (Fig. 3C(i)). Furthermore, it successfully killed both MRSA and P. aeruginosa bacteria within the biofilm, achieving a 5.1log reduction for MRSA and a 3.8log reduction for P. aeruginosa (Fig. 3C(ii)). Additionally, the combination therapy demonstrated efficacy against the dispersed cells, resulting in a 7.7log reduction for MRSA and a 5.9log reduction for P. aeruginosa (Fig. 3C(iii)). In contrast, vancomycin (32 μg mL−1) and colistin (32 μg mL−1) treatments were ineffective against this multispecies bacterial biofilm comprising MRSA and P. aeruginosa. In conclusion the cocktail therapy of SMA and fusidic acid has the potential to effectively combat biofilm-mediated multispecies bacterial infections.
In vitro cytotoxicity of lead combinations
Furthermore, we investigated the cytotoxicity of the leading combinations against the human embryonic kidney cell line (HEK- 293T) using the LIVE/DEAD assay, which involved staining with calcein-AM (green fluorescence) and PI (red fluorescence). The combination of SMA (16 μg mL−1) and fusidic acid (32 μg mL−1) treated cells exhibited green staining only, indicating that the cells were alive, similar to the control group. In contrast, cells treated with Triton-X were solely stained with PI, confirming cell death. Therefore, the lead combination therapy demonstrated remarkable biocompatibility with mammalian cells (Fig. 4A). It is worth noting that the cytotoxicity of SMA alone has been previously reported.21 | ||
| Fig. 4 (A) Biocompatibility of the leading combinations against the mouse HEK-293T cell line by performing the LIVE/DEAD assay through staining with calcein-AM (green fluorescence; stained live cells) and PI (red fluorescence; stained dead cells); merged channel images are represented; scale bar: 50 μm; (B) in vivo systemic toxicity of SMA through different routes of administration in the mouse model. (C) Frequency of resistance growth by NDM-1 producing bacteria A. baumannii A168 against the leading combinations; (D) in vivo antibacterial efficacy of the combination consisting of SMA and fusidic acid; (i) the experimental design of the mouse skin infection model with A. baumannii A168 bacteria, (ii) number of viable bacteria in the skin tissue sample upon treatment, and (iii) mice infected skin tissue histology. Scale bar: 200 μm. In untreated and SMA and fusidic acid treatment cases, severe to moderate inflammatory cell infiltration is shown (asterisks), whereas the combination treatment tissue sample and colistin tissue sample revealed minimum inflammatory cell infiltration (asterisks). Arrows indicate the keratin layer; sebaceous glands (SG); hair follicles (HF); adipose tissue (AT). | ||
In vivo systemic toxicity of SMA
To assess the therapeutic applicability, we investigated the in vivo systemic toxicity of SMA by administering different dosages via intra-peritoneal and subcutaneous injections, following the guidelines provided by OECD 425. In the case of intra-peritoneal administration, SMA exhibited a considerable LD50 (lethal dose for 50% mortality) of 130 mg kg−1. However, when administered subcutaneously at a dosage of 175 mg kg−1, all mice survived without any adverse effects. Therefore, the LD50 of SMA for subcutaneous administration was determined to be >175 mg kg−1. These results indicate that SMA exhibits biocompatibility for both systemic and topical applications in combination therapy (Fig. 4B).Resistance frequency
The emergence of drug resistance poses a significant obstacle to the development and approval of new antimicrobials. In order to address this concern, we determined the frequency of resistance development against the lead combination therapy of SMA and fusidic acid, using NDM-1 producing A. baumannii A168 bacteria. 100 μL of bacterial suspension of various concentrations (ranging from 109 CFU to 105 CFU) were spread-plated on nutrient agar plates containing either the combination of SMA and fusidic acid, fusidic acid alone, or colistin and then incubated for 24 h. At a bacterial concentration of 3 × 106 CFU on the plate containing 512 μg mL−1 (8 × MIC) of fusidic acid alone, we observed the presence of 40 resistant mutants, resulting in a frequency of resistance of A. baumannii against fusidic acid alone at 1.3 × 10−5. On the other hand, when the combination therapy of SMA (16 μg mL−1) and fusidic acid (4 μg mL−1) was tested against bacterial suspensions of 3 × 108 CFU, we observed 51 resistant mutants, yielding a frequency of resistance against the combination therapy at 1.7 × 10−7. Similarly, the presence of 55 resistant A. baumannii mutants indicated a resistance frequency against colistin at 8 μg mL−1 (8 × MIC) of 1.8 × 10−8 (Fig. 4C). These results collectively indicate that NDM-1 producing A. baumannii bacteria are less prone to developing spontaneous resistance against the combination of SMA and fusidic acid compared to fusidic acid alone, even at very high concentrations. This may be attributed to the enhanced intracellular concentration of the antibiotics in the presence of SMA, which potentially contributes to the reduced resistance development.In vivo antibacterial efficacy of combination therapy
The in vivo antibacterial activity of the lead combination therapy comprising SMA and fusidic acid was evaluated using a murine skin infection model. Briefly, the skin of the mice was infected with ∼106 CFU mL−1 (per mouse) of NDM-1 producing A. baumannii A168. After 4 h of infection, the infected skin was treated with different antimicrobial agents for up to 4 days, with a single dosage administered each day. On day five, all animals were sacrificed and skin tissues were collected and homogenised in saline to determine the bacterial viability in the infected tissues (Fig. 4D(i)). In the untreated group (washed with saline daily), the bacterial burden was increased to ∼8.7 × 108 CFU g−1. Treatment with only SMA (40 mg kg−1) resulted in a modest reduction of 0.2log compared to the untreated case. However, the individual treatment of fusidic acid failed to decrease the bacterial burden in the infected skin, with a count of viable bacteria at ∼8.8 × 108 CFU g−1, similar to the untreated case. Interestingly, the combination therapy of SMA and fusidic acid exhibited potent in vivo antibacterial efficacy. In mice treated with the combination of SMA (40 mg kg−1) and fusidic acid (40 mg kg−1), the bacterial viability of NDM-1 producing bacteria A. baumannii was reduced by ∼2.5log compared to the untreated case. Colistin also demonstrated superior activity, reducing the bacterial viability by ∼2.1log in this murine skin infection model (Fig. 4D(ii)). Furthermore, histopathological studies were conducted on the tissue samples from all groups. In the untreated group and in mice treated with SMA or fusidic acid individually, the corresponding tissue samples exhibited severe to moderate infiltration of inflammatory cells and damaged cells. In contrast, the combination-treated tissues showed a normal appearance of the keratin layer, adipose tissue, and sebaceous glands, with minimal infiltration of inflammatory cells (Fig. 4D(iii)). Collectively, these results indicate that the combination therapy of SMA and fusidic acid demonstrates potent efficacy in combating NDM-1 producing bacterial-induced topical infections, surpassing the effects of individual treatments. These findings hold great promise for the development of an innovative antibacterial strategy to address infections caused by multi-drug resistant Gram-negative pathogens.
Conclusions
Combining antibiotics with membrane-targeting molecules represents a frontline approach in the battle against antimicrobial resistance (AMR), as targeting the bacterial membrane offers a smart alternative. In this study, we have demonstrated the remarkable potentiation efficacy of a small amphiphilic molecule called SMA against various classes of obsolete antibiotics, including rifampicin, minocycline, fusidic acid, and chloramphenicol, against a range of multi-drug resistant Gram-negative bacteria. The combination of SMA and antibiotics exhibited potent bactericidal activity against NDM-1 producing planktonic Gram-negative bacterial cells. Moreover, it effectively eradicated bacteria embedded within the preformed biofilms of NDM-1 producing A. baumannii. SMA was found to inhibit bacterial efflux pumps, leading to increased accumulation of antibiotics inside bacterial cells. Given the escalating bacterial resistance, treating multispecies bacterial infections has become a critical challenge in the healthcare system. However, the combination of SMA (an anti-MRSA agent) and fusidic acid demonstrated potent bactericidal activity against both MRSA and NDM-1 producing A. baumannii in multispecies bacterial co-cultures and their preformed biofilms. Importantly, the lead combination exhibited biocompatibility with mammalian RAW cell lines. In vivo studies using a mouse model demonstrated the high LD50 value of SMA, indicating its safety for systemic use. Furthermore, the combination therapy exhibited excellent antibacterial efficacy in a mouse skin infection model. Notably, A. baumannii exhibited slow development of resistance against the combination therapy. Overall, this combination therapy holds enormous potential as a novel strategy to combat antimicrobial resistance in Gram-negative superbugs.Ethical statement
The Institutional Bio-Safety Committee of the Jawaharlal Nehru Centre for Advanced Scientific Research approved the antibacterial studies, which also included hemolytic activities (JNC/IBSC/2020/JH-12). The in vivo experiments were conducted in accordance with the guidelines approved by the Jawaharlal Nehru Centre for Advanced Scientific Research's Institutional Animal Ethics Committee (IAEC) (201/Go/ReBi/S/2000/CPCSEA).Data availability
The authors declare that all data supporting the findings of this study are available within the article and ESI,† and raw data files are available from the corresponding authors upon request.Author contributions
The work and the experiments were designed by R. D. and J. H. Cell toxicity assay was performed by R. M. All other studies were conducted by R. D., S. M., and R. M. All the data were analyzed by R. D., R. M., and J. H. The manuscript was written by R. D., S. M., and J. H. All the authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. D. acknowledges CSIR for a Senior Research Fellowship (SRF). S. M. thanks JNCASR for funding. The authors acknowledge Rohana Veterinary Diagnostic Laboratory (Bangalore, Karnataka, India) and R. V. Metropolis Clinical Laboratory (Bangalore, Karnataka, India) for assistance in in vivo studies. R. D. thanks Geetika Dhanda for various helpful scientific discussions.References
- GBD 2019 Antimicrobial Resistance Collaborators, Lancet, 2023, 400, 2221 Search PubMed.
- C. Ghosh, P. Sarkar, R. Issa and J. Haldar, Alternatives to conventional antibiotics in the era of antimicrobial resistance, Trends Microbiol., 2019, 27, 323 CrossRef CAS PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans and Y. Carmeli, WHO Pathogens Priority List Working Group, Lancet Infect. Dis., 2018, 18, 318 CrossRef PubMed.
- R. A. Garibaldi, J. Hosp. Infect., 1999, 43, S9 CrossRef.
- C. D. Russell, C. J. Fairfield, T. M. Drake, L. Turtle, R. A. Seaton, D. G. Wootton, L. Sigfrid, E. M. Harrison, A. B. Docherty, T. I. De Silva, C. Egan, R. Pius, H. E. Hardwick, L. Merson, M. Girvan, J. Dunning, J. S. Nguyen-Van-Tam, J. S. Openshaw, J. K. Baillie, M. G. Semple and A. Ho, Lancet Microbe, 2021, 2, e354 CrossRef CAS PubMed.
- N. Shafran, I. Shafran, H. Ben-Zvi, S. Sofer, L. Sheena, I. Krause, A. Shlomai, E. Goldberg and E. H. Sklan, Sci. Rep., 2021, 11, 12703 CrossRef CAS PubMed.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, A. G. Robles, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell and S. Hackett, Lancet, 2022, 399, 629 CrossRef CAS.
- C. R. MacNair, J. M. Stokes, L. A. Carfrae, A. A. Fiebig-Comyn, B. K. Coombes, M. R. Mulvey and E. D. Brown, Nat. Commun., 2018, 9, 458 CrossRef.
- M. Vaara, Molecules, 2019, 24, 249 CrossRef PubMed.
- E. van Groesen, C. J. Slingerland, P. Innocenti, M. Mihajlovic, R. Masereeuw and N. I. Martin, ACS Infect. Dis., 2021, 7, 2746 CrossRef CAS PubMed.
- P. Brown, E. Abbott, O. Abdulle, S. Boakes, S. Coleman, N. Divall, E. Duperchy, S. Moss, D. Rivers, M. Simonovic, J. Singh, S. Stanway, A. Wilson and M. J. Dawson, ACS Infect. Dis., 2019, 5, 1645 CrossRef CAS PubMed.
- M. Song, Y. Liu, X. Huang, S. Ding, Y. Wang, J. Shen and K. Zhu, Nat. Microbiol., 2020, 5, 1040 CrossRef CAS PubMed.
- J. M. Stokes, C. R. MacNair, B. Ilyas, S. French, J.-P. Côté, C. Bouwman, M. A. Farha, A. O. Sieron, C. Whitfield, B. K. Coombes and E. D. Brown, Nat. Microbiol., 2017, 2, 17028 CrossRef PubMed.
- A. Cannatelli, S. Principato, O. L. Colavecchio, L. Pallecchi and G. M. Rossolini, Front. Microbiol., 2018, 9, 1808 CrossRef.
- M. M. Konai and J. Haldar, ACS Infect. Dis., 2020, 6, 91 CrossRef CAS PubMed.
- G. Blankson, A. K. Parhi, M. Kaul, D. S. Pilch and E. J. LaVoie, Eur. J. Med. Chem., 2019, 178, 30 CrossRef CAS PubMed.
- G. D. Wright, Trends Microbiol., 2016, 24, 862 CrossRef CAS.
- H. Douafer, V. Andrieu, O. Phanstiel and J. M. Brunel, J. Med. Chem., 2019, 62, 8665 CrossRef CAS.
- M. Chang, K. V. Mahasenan, J. A. Hermoso and S. Mobashery, Acc. Chem. Res., 2021, 54, 917 CrossRef CAS.
- R. Dey, S. Mukherjee, S. Barman and J. Haldar, Macromol. Biosci., 2021, 21, 2100182 CrossRef CAS PubMed.
- R. Dey, K. De, R. Mukherjee, S. Ghosh and J. Haldar, MedChemComm, 2019, 10, 1907 RSC.
- P. J. F. Henderson, C. Maher, L. D. H. Elbourne, B. A. Eijkelkamp, I. T. Paulsen and K. A. Hassan, Chem. Rev., 2021, 121, 5417 CrossRef CAS.
- M. A. Webber and L. J. V. Piddock, J. Antimicrob. Chemother., 2003, 51, 9 CrossRef CAS.
- D. Ma, D. N. Cook, J. E. Hearst and H. Nikaido, Trends Microbiol., 1994, 2, 489 CrossRef CAS PubMed.
- A. Cauilan and C. Ruiz, Pathogens, 2022, 11, 1409 CrossRef CAS PubMed.
- D. Corbett, A. Wise, T. Langley, K. Skinner, E. Trimby, S. Birchall, A. Dorali, S. Sandiford, J. Williams, P. Warn, M. Vaara and T. Lister, Antimicrob. Agents Chemother., 2017, 61, e00200 CrossRef CAS.
- M. Besouw, R. Masereeuw, L. van den Heuvel and E. Levtchenko, Drug Discovery Today, 2013, 18, 785 CrossRef CAS PubMed.
- G. Dhanda, Y. Acharya and J. Haldar, ACS Omega, 2023, 8, 10757 CrossRef CAS.
- K. Klobucar and E. D. Brown, Curr. Opin. Chem. Biol., 2022, 66, 102099 CrossRef CAS PubMed.
- B. J. Berry, A. J. Trewin, A. M. Amitrano, M. Kim and A. P. Wojtovich, J. Mol. Biol., 2018, 430, 3873 CrossRef CAS PubMed.
- D. G. J. Larsson and C. F. Flach, Nat. Rev. Microbiol., 2022, 20, 257 CrossRef CAS.
- S. J. Howard, M. Catchpole, J. Watson and S. C. Davies, Lancet Infect. Dis., 2013, 13, 1001 CrossRef PubMed.
- G. Dhanda, R. Mukherjee, D. Basak and J. Haldar, ACS Infect. Dis., 2022, 8, 1086 CrossRef CAS PubMed.
- J. R. Gaston, A. O. Johnson, K. L. Bair, A. N. White and C. E. Armbruster, Infect. Immun., 2021, 89, e00652 CrossRef CAS.
- B. M. Peters, M. A. Jabra-Rizk, G. A. O'May, J. W. Costerton and M. E. Shirtliff, Clin. Microbiol. Rev., 2012, 25, 193 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, supplementary figures for characterization and biological assays. See DOI: https://doi.org/10.1039/d3sc05124b |
| This journal is © The Royal Society of Chemistry 2024 |