
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLewis acid catalysed polymerisation of cyclopentenone†
Deepamali
Dissanayake‡
a,
Alysia
Draper‡
a,
Zhizhou
Liu‡
ab,
Neelofur
Jaunnoo
a,
Joris J.
Haven
a,
Craig
Forsyth
 a,
Alasdair I.
McKay
a,
Tanja
Junkers
*a and
Dragoslav
Vidović
*a
a,
Alasdair I.
McKay
a,
Tanja
Junkers
*a and
Dragoslav
Vidović
*a
aSchool of Chemistry, Monash University, Clayton 3800, Australia. E-mail: drasko.vidovic@monash.edu; tanja.junkers@monash.edu
bSuzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences, Suzhou 215163, China
First published on 2nd December 2023
Abstract
A modest structural change of a β-diketiminate-supported aluminium complex leads to dramatic differences in the reactivity towards cyclopentenone. While the bulkier complex efficiently executes Diels Alder transformations the smaller analogue performs unique polymerisation of this substrate. This observation appears to be unprecedented in the chemistry of Lewis acids and cyclic dienophiles as it represents a unique way to polymerise a functionalised olefin.
Functionalised polyolefins can be prepared either by post-polymerisation modifications1 of non-functionalised polymers or by (co)polymerisation of functional olefin monomers.1–4 Even though polymer post-functionalisation is presently the most utilized commercial method for the preparation of functionalised polyolefins, this approach typically requires very harsh reaction conditions, potentially leading to unwanted and abundant side reactions.4 On the other hand, creating functionalised polyolefins by polymerisation of functionalised monomers has had only very limited success to date. While radical polymerisation of functionalised olefins is generally difficult to control, transition metal catalysed polymerisation is significantly restricted due to the “polar monomer problem”.1 For example, functional group coordination and weaker olefin binding (accentuated for vinyl-based monomers) are some of the issues that restrict effective polymerisation of functionalised olefin via transition metal-based catalysis. As a result, functionalised olefins are normally copolymerised with non-polar analogues but the polar monomer incorporation percentages are usually limited to single digits (<10%). Therefore, polymer chemists have been striving towards completely new approaches for (co)polymerisation of functional olefins that would allow for higher functional group tolerance, and that can provide materials with new properties.1–4
The use of Lewis acids (LAs) for various stoichiometric and catalysed organic transformations is an extensively studied and developed field.5 However, there is growing evidence that the presence of hidden Brønsted acids (HBAs) is predominantly responsible for the reported activities of the described Lewis acids.6–9 As a result, erroneous observations were (in)advertently reported regarding the chemistry of Lewis acids. Therefore, a vigilant design and use of LA systems in organic synthesis might yet lead to unexpected and, more importantly, unique outcomes. Herein, we wish to report one of these exclusive feats as unprecedented polymerisation of 2-cyclopeten-1-one (a “vinyl-functionalised” olefin) using β-diketiminate-supported aluminium complexes leading to polycyclopentenone (a fully functionalised olefin; Scheme 1).10
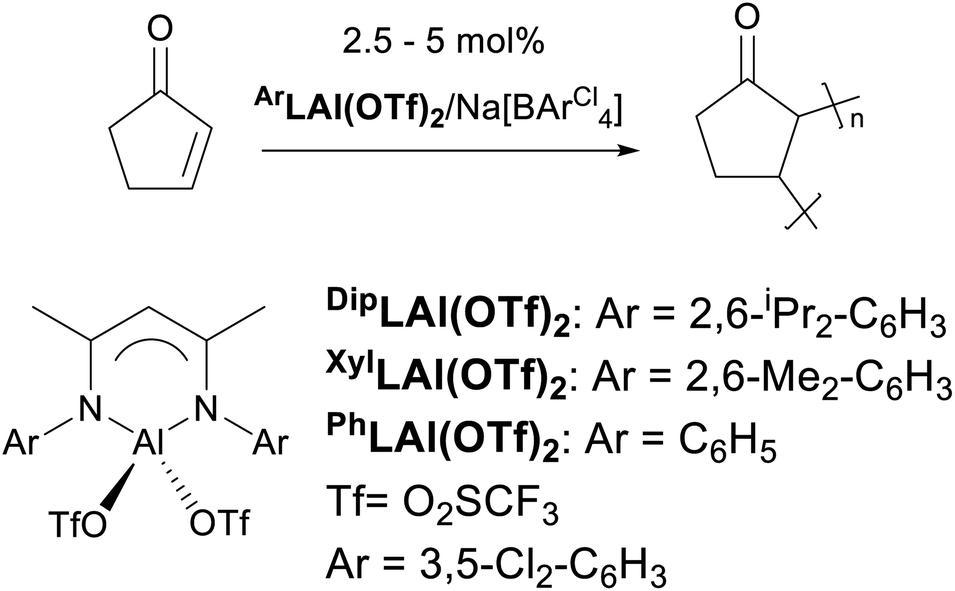 | ||
| Scheme 1 Novel type of polymerisation described herein. | ||
Our efforts in the field of LA catalysis have been focused on the development of well-characterized aluminium-based complexes and their use in several organic transformation including Diels–Alder cycloadditions and Michael additions.11–13 Our reported work on aluminium-based LA catalysts bearing a β-diketiminate ligand was focused solely on DipLAl(OTf)2 (DipL− = [(CH)(CMe)2(NDip)2]−; Dip = 2,6-diisopropylphenyl; Tf = O2SCF3; Scheme 1) complex as it formed a quite catalytically active system once mixed with Na[BArCl4] (ArCl = 3,5-dichlorophenyl).11,13 As minor modifications of β-diketiminate ligands led to dramatic changes in the reactivity of the supporting complexes,14,15 we aimed to investigate the influence of modifying the N-aryl (i.e. Dip) substituents on the overall catalytic activity of our Lewis acid system. Thus, XylLAl(OTf)2 (Xyl = 2,6-dimethylphenyl) was first prepared and its catalytic activity (when mixed with Na[BArCl4]) towards several Diels–Alder cycloadditions using 2,3-dimethylbutadiene and various enones (i.e. vinyl ketones) was investigated. The overall results are summarized in Table S1† (See the ESI†) and the most significant differences between the two catalytic systems occurring when cyclic enones i.e. cyclopentenone and cyclohexenone were examined. The Dip-containing catalytic system successfully generated the target Diels–Alder adducts for these two cyclic enones in high yields. However, the Xyl-based catalytic system struggled to convert cyclohexenone (∼24% product yield) while in the case of cyclopentenone we noticed that even though the amount of this particular enone was greatly diminished, the diene was essentially not used up at all and, hence, no trace of the target Diels–Alder adduct was detected. Upon closer examination of this reaction's 1H NMR spectrum, a set of broad signals between 1.0 and 3.5 ppm was observed (Fig. 1). Repeating the latest reaction in the absence of the diene had no influence on the observed reaction outcomes such as the consumption of the enone and the appearance of broad signals in the 1H NMR spectrum.
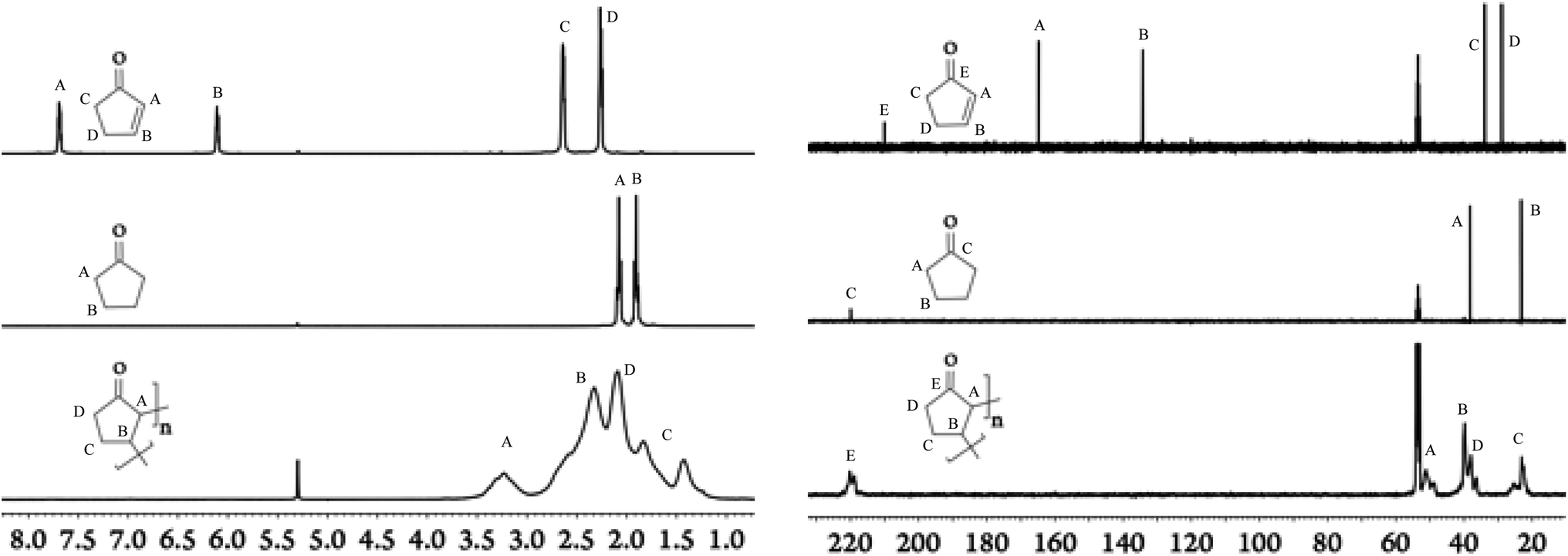 | ||
| Fig. 1 1H-NMR (left) and 13C{1H} NMR spectra (right) in parts per million (ppm) of the monomer (top), the repeating unit motif (middle), and the polymer (bottom) in deuterated dichloromethane. | ||
Due to broad characteristics of the 1H NMR signals, formation of oligomeric/polymeric nature of the new species is hinted. More evidence for this hypothesis was acquired from the 13C NMR spectrum (Fig. 1) as it also contained broad signals. In fact, when the 1H and 13C NMR spectra of this new material were compared with cyclopentenone and cyclopentanone (Fig. 1) several important conclusions could be drawn. Firstly, a broad signal at δC ∼ 220 ppm suggested that the carbonyl group was still intact which was also confirmed by IR spectroscopy revealing a strong carbonyl band at ∼ 1730 cm−1. Secondly, the position of this 13C NMR signal is virtually identical to the position of the carbonyl signal observed for cyclopentanone but about 10 ppm downfield shifted in comparison to cyclopentenone. Lastly, the absence of any signals associated with an alkene group in both 1H and 13C NMR spectra of the new material implied that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of monomeric enone was involved in the formation of this new oligomeric/polymeric material. It should be mentioned that the presence of CH2 and CH groups was also confirmed using DEPT 13C NMR spectroscopy (see Fig. S10†).
C double bond of monomeric enone was involved in the formation of this new oligomeric/polymeric material. It should be mentioned that the presence of CH2 and CH groups was also confirmed using DEPT 13C NMR spectroscopy (see Fig. S10†).
Furthermore, according to MALDI-TOF mass spectrometry the newly prepared material consisted of a structural repeat unit with the value for the molar mass of 82 g mol−1 (see Fig. S17†), which is consistent with the monomer molecular formula (C5H6O) for the repeat unit. Then, the average molar mass of the purified material, using size exclusion chromatography (SEC), was determined to be ca. 60 kDa (determined relative to polystyrene standards) with a dispersity of 1.5–2, while its decomposition temperature was established around 300 °C (via TGA – see Fig. S15†).
The abovementioned results are consistent with formation of a polymeric material having an all-carbon backbone. The disappearance of alkene signals in the product spectrum also indicated that the polymerisation occurred via the π bond of the monomer's CC double bond which is consistent with olefin polymerisation. Lastly, considering that cyclopentenone could be considered as a functionalised olefin, the resulting polymer can be described as ketone-functionalised polyolefin. Thus, this appears to be the most efficient generation of functionalised polyolefins (i.e. polymers with a significant molecular weight) via polymerisation of a “vinyl-substituted” olefin using metal-based catalysis. Further, ketone functional polymers are generally not as easily obtained in other conventional polymerisation routes, making this method very interesting for synthesis of carbonyl-rich polymer materials. Only few monomers, such as methyl vinyl ketone are known to produce polymers with similar backbone functionalities. Repeating ketone units are virtually impossible to obtain through the to--date known metal-based catalytic alkene polymerisation methods.
With the observation of a new type of polymerisation, we turned at understanding the underlying mechanism better. The nature of the polymerisation remains at this point, however, somewhat in the dark. In order to gather more information about the overall polymerisation mechanism, numerous additional experiments were performed. A step-growth mechanism could be ruled out quickly as high molecular weight is formed already during early stages of the polymerisation. It must thus follow a chain-growth mechanism, which is also in line with the observed overall dispersities. Taking several reaction aliquots at different time intervals during the polymerisation resulted in no detectable difference in the average molecular weight of the polymeric material (See Fig. S12,† and for initial kinetic studies see Fig. S11†), indicating a non-living chain growth mechanism. In line with that, the average polymer molecular mass could be influenced by the overall concentration of the reaction mixture. Lower starting monomer concentrations favour formation of shorter polymer chains (see Fig. S13†). Interestingly, increasing the catalyst loading to ≥10% resulted in the isolation of insoluble material which might suggest that only catalyst bound i.e. activated monomers can be polymerised. Increasing catalyst loading would certainly increase the number of activated monomers and may then yield branched material that forms networks rather than linear chains (see also mechanistic discussion below).
Additionally, most common Lewis acids, such as AlCl3, B(C6F5)3 and BF3OEt2, were not adequate catalysts for polymerisation as no conceivable transformation of cyclopentenone was detected under the reaction conditions used for the Xyl-based system.16 The same inactivity was observed when a soluble source of a Brønsted acid (e.g. HOTf prepared by mixing tBuCl/AgOTf6 or (C6F5)3B–OH2 (ref. 17)) was mixed with cyclopentenone. Moreover, introducing 2,6-di-tert-butylpyridine, as a quite potent proton scavenger,6 had no influence on the polymerisation reaction clearly suggesting that (hidden) Brønsted acid(s) was not responsible for the formation of the polymer material. Of course, it would be possible that the catalyst “only” acts as a radical initiator, and that the polymerisation would be a conventional radical vinyl polymerisation. This was, however, easily ruled out as heating the enone in the presence of azobisisobutyronitrile resulted in no observable polymerisation. This is not surprising considering that 1,2-disubstituted alkenes usually do not polymerise via a radical mechanism.18 In addition, using catalytic amounts of [nBu4]OTf showed no signs of the observed polymerisation which excluded a possibility of triflate anion initiating the polymerisation process.19 In fact, the presence of a significantly stronger base Na[N(SiMe3)2] in catalytic amounts also yielded no observable polymer strongly suggesting the absence of a base-initiated process. Hence, all of the abovementioned control experiments clearly favour the idea that the LA must be vital in the chain growth process, catalysing the propagation reaction itself in a non-radical pathway.
We then turned our attention to further modify our catalytic system by synthesizing the Ph-based complex i.e.PhLAl(OTf)2 (Ph = C6H5; Scheme 1). We tested the efficacy of the Dip- and Ph-based systems in the overall polymerisation and even though these two systems were capable of polymerising cyclopentenone the reaction rate, the polymer yields as well as the average polymer molecular weight were significantly reduced compared to the Xyl-based system (See Fig. S16†). These experiments signified that the presence of the ortho-Me groups at the N-aryl substituents of the catalytic system was quite beneficial for the overall polymerisation.
Moreover, we attempted to polymerise several additional substrates using the Xyl-based system in order to investigate the role of the carbonyl and the alkene groups as well as the importance of the cyclic nature of the polymerised enone (i.e. cyclopentenone). Firstly, there was no indication that either cyclopentanone or cyclopentene could be polymerised with the Xyl-based system suggesting that collective presence of these functional groups was crucial in the overall polymerisation. The same results (no polymer formation) were observed when acyclic enones, such as trans-4-phenyl-3-buten-2-one and 1,3-diphenyl-2-propenone were mixed with catalytic amounts of the Xyl-based system. On the other hand, it was possible to polymerise other cyclic enones (e.g. cyclohexenone or cycloheptenone, albeit requiring longer reaction times resulting also in reduced isolated yields). Unfortunately, high insolubility of these materials in most common organic solvents prevented their further characterization apart from FTIR spectroscopy which revealed signals around 1700 cm−1 associated with a CO functional group.5 Nevertheless, the sole formation of these polymeric materials suggested that, besides the presence of the carbonyl and alkene groups, the cyclic nature of the substrate plays a pivotal role in the polymerisation. While ring strain alone is – compared to the enthalpy gain of from the conversion of a π bond into a σ bond – not significant enough to explain the driving force of the polymerisation, it can be speculated that the ring structure plays an important role in the stability of the LA activated propagation reaction. This would also explain slower reaction rates and lower isolated yields for transformation of the other cyclic analogues due to a lesser degree of ring strain in comparison to cyclopentenone.
While all the above observations stand on their own, we tried to elucidate the polymerisation mechanism in more detail but we were unsuccessful in generating conclusive experimental evidence. What is clear from the more or less constant and instantaneously high molecular weight of the residual polymer of the polymerization is that the reaction proceeds in an uncontrolled chain growth mechanism. It hence must consist of an initiation reaction, propagation and an irreversible termination step. The overall rate of termination and the rate of initiation must be in a steady state, and overall very fast reactions that occur concomitantly throughout the whole polymerization. In Scheme 2 a plausible mechanism is portrayed that is based on Lewis acid catalysed conjugate addition whose initiation step involves the formation of an aluminium-enolate species (A). The propagation step is then manifested by intramolecular conjugate addition forming B, propagating the active chain end. The termination step is most contentious as, based on overall experimental evidence, it should have the rate as initiation as mentioned above, and be irreversible. Therefore, formation of sodium-enolate C would be less likely as it would be expected to be in an equilibrium with a propagating species, while a potential ArCl-transfer step20 involving the borate anion should produce a thermodynamically more stable system (i.e.D) that may be irreversible in its formation on the timescale of the polymerization.
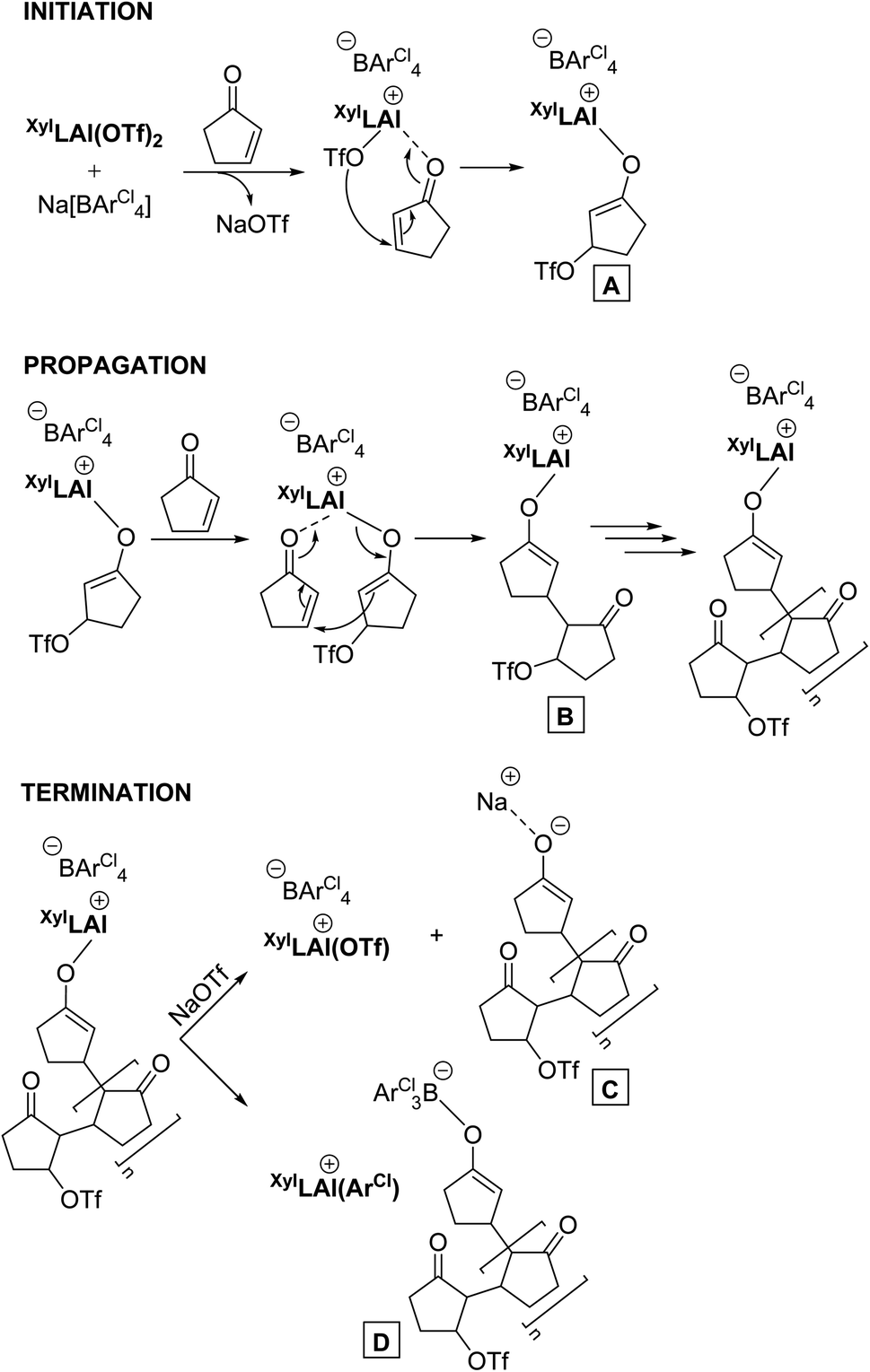 | ||
| Scheme 2 Proposed mechanism for initiation, propagation and termination in the polymerisation. | ||
Of course, this mechanism is a hypothesis only at this stage. Yet it is consistent with all experimental observations so far. Future studies will allow for better insights, and refined mechanistic understandings.
It is, however, extremely unlikely that the observed polymerisation occurs via the established mechanism for transition metal-based catalysed systems which involves alkene coordination followed by subsequent monomer insertions (Scheme 3a). The prior observations make it likely that only activated monomer can undergo chain addition, not unlike in organocatalysed ring opening polymerisation.21 This hypothesis is further supported by the fact that there was no evidence that a free olefin (e.g. 2,3-dimethylbutadiene; vide supra) is involved in polymerisation under these reaction conditions (Scheme 3b).
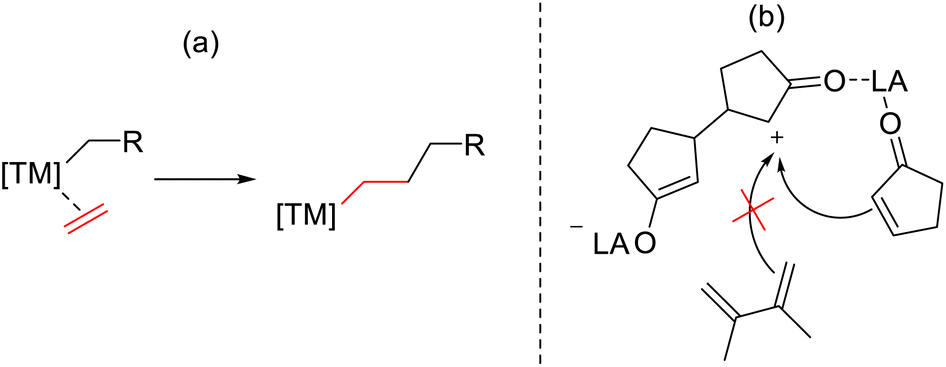 | ||
| Scheme 3 The well-known propagation step in cyclopentenone polymerisation via transition metal (TM) catalysis (a) and the absence of butadiene's involvement in the proposed propagation step involving polymerisation of cyclic enones using aluminium Lewis acids (b). | ||
An interesting feature of our proposed mechanism is that the LA remains always at the active end of growing chain, then also binding any approaching monomer. This would explain the relatively high reaction rates, and also explain the necessity of the LA activation in the propagation itself. In the absence of the ketone, the LA is not able to bind to the approaching monomer, and consequently other monomers cannot add to the chain.
In conclusion, a Lewis acid system based on aluminium has been identified as very reactive catalyst for polymerisation of cyclopentenone leading to a formation of a novel polymer. Since the monomer is considered as a functionalised olefin, the polymeric material is described as functionalised polyolefin. In fact, this is the first time that a functionalised olefin has been solely polymerised (i.e. in the absence of non-functionalised analogue(s)) using a metal-based catalyst. Preliminary mechanistic studies have suggested that monomers involved in the polymer chain growth needed to be first activated i.e. coordinated to separate Lewis acid sites. Polymerisations follow a non-living chain growth mechanism, and seem to have cationic character. We are currently working on expanding this type of polymerisation on other similar substrates primarily the acyclic analogues and on understanding the mechanism more in-depth.
Data availability
Experimental data is included in the ESI.†Author contributions
D. D., A. D. and L. Z. performed the synthetic work, J. J. H., N. J. and A. I. M. were responsible for the analysis, C. F. collected and solved the single crystal X-ray diffraction data and T. J. and D. V. supervised the project and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors thank Monash University for funding. L.Z. also acknowledges the support from the Natural Science Foundation of Jiangsu Province, China (project # BK20210105) and DV thanks the Australian Research Council (FT230100565) for funding. We also would like to acknowledge the reviewers whose insightful comments greatly assisted in the overall understanding of the described polymerisation process.Notes and references
- A. Keyes, H. E. Basbug Alhan, E. Ordonez, U. Ha, D. B. Beezer, H. Dau, Y.-S. Liu, E. Tsogtgerel, G. R. Jones and E. Harth, Angew. Chem., Int. Ed., 2019, 58, 12370 CrossRef CAS.
- C. Chen, Nat. Rev. Chem., 2018, 2, 6 CrossRef CAS.
- J. Chen, Y. Gao and T. J. Marks, Angew. Chem., Int. Ed., 2020, 59, 14726 CrossRef CAS PubMed.
- N. M. G. Franssen, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2013, 42, 5809 RSC.
- H. Yamamoto and K. Ishihara, Acid Catalysis in Modern Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- T. T. Dang, F. Boeck and L. Hintermann, J. Org. Chem., 2011, 76, 9353 CrossRef CAS PubMed.
- I. Solic, H. X. Lin and R. W. Bates, Tetrahedron Lett., 2018, 59, 4434 CrossRef CAS.
- J. D. Steen, S. Stepanovic, M. Parvizian, J. W. de Boer, R. Hage, J. Chen, M. Swart, M. Gruden and W. R. Browne, Inorg. Chem., 2019, 58, 14924 CrossRef CAS.
- J. N. Hall and P. Bollini, ACS Catal., 2020, 10, 3750 CrossRef CAS.
- There has been a report on formation of a mixture of polymeric products from dicyclopentadienone upon treatment with Et2AlCN, however, the material was not characterised: C. Alvarez, R. Pelaez and M. Medarde, Tetrahedron, 2007, 63, 2132 CrossRef CAS.
- Z. Liu, J. H. Q. Lee, R. Ganguly and D. Vidović, Chem. – Eur. J., 2015, 21, 11344 CrossRef CAS PubMed.
- Z. Liu, R. Ganguly and D. Vidović, Dalton Trans., 2017, 46, 753 RSC.
- Z. Liu and D. Vidović, J. Org. Chem., 2018, 83, 5295 CrossRef CAS PubMed.
- J. M. Smith, A. R. Sadique, T. R. Cundari, K. R. Rodgers, G. Lukat-Rodgers, R. J. Lachicotte, C. J. Flaschenriem, J. Vela and P. L. Holland, J. Am. Chem. Soc., 2006, 128, 756 CrossRef CAS PubMed.
- M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780 CrossRef CAS PubMed.
- Polymerisation of this enone was also not reported for other Lewis acids. For example, see: K. Muther, R. Frohlich, C. Muck-Lichtenfeld, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2011, 133, 12442 CrossRef CAS PubMed.
- D. C. Bradley, I. S. Harding, A. D. Keefe, M. Motevalli and D. H. Zheng, J. Chem. Soc., Dalton Trans., 1996, 3931 RSC.
- G. Odian, Principles of Polymerization, Wiley-VCH, Weinheim, Germany, 4th edn, 2004 Search PubMed.
- B. Dhakal, L. Bohe and D. Crich, J. Org. Chem., 2017, 82, 9263 CrossRef CAS PubMed.
- M. S. Ziegler, D. S. Levine, K. V. Lakshmi and T. D. Tilley, J. Am. Chem. Soc., 2016, 138, 6484 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2175768–2175770. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05186b |
| ‡ D. D., A. D. and L. Z. contributed equally to the synthetic part of this work. |
| This journal is © The Royal Society of Chemistry 2024 |