
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMo-doping heterojunction: interfacial engineering in an efficient electrocatalyst for superior simulated seawater hydrogen evolution†
Zuo-Ming
He‡
ab,
Chun-Xiao
Zhang‡
d,
Si-Qi
Guo‡
e,
Peng
Xu
a,
Yuan
Ji
a,
Si-Wei
Luo
a,
Xiang
Qi
 a,
Yun-Dan
Liu
*a,
Ning-Yan
Cheng
*e,
Shi-Xue
Dou
f,
Yun-Xiao
Wang
*fg and
Bin-Wei
Zhang
*bc
a,
Yun-Dan
Liu
*a,
Ning-Yan
Cheng
*e,
Shi-Xue
Dou
f,
Yun-Xiao
Wang
*fg and
Bin-Wei
Zhang
*bc
aHunan Key Laboratory of Micro-Nano Energy Materials and Devices, Xiangtan University, Xiangtan 411105, PR China. E-mail: liuyd@xtu.edu.cn
bSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, PR China. E-mail: binwei@cqu.edu.cn
cCenter of Advanced Electrochemical Energy, Institute of Advanced Interdisciplinary Studies, Chongqing 400044, PR China
dSchool of Physics and Optoelectronic Engineering, Shandong University of Technology, Zibo 255000, PR China
eInformation Materials and Intelligent Sensing Laboratory of Anhui Province, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Institutes of Physical Science and Information Technology, Anhui University, Hefei 230601, PR China. E-mail: ningyancheng@ahu.edu.cn
fInstitute of Energy Materials Science (IEMS), University of Shanghai For Science and Technology, Shanghai, 200093, China
gInstitute for Superconducting and Electronic Materials, Australian Institute of Innovative Materials, University of Wollongong, North Wollongong, New South Wales 2500, Australia. E-mail: yunxiao@uow.edu.cn
First published on 6th December 2023
Abstract
Exploring economical, efficient, and stable electrocatalysts for the seawater hydrogen evolution reaction (HER) is highly desirable but is challenging. In this study, a Mo cation doped Ni0.85Se/MoSe2 heterostructural electrocatalyst, Mox-Ni0.85Se/MoSe2, was successfully prepared by simultaneously doping Mo cations into the Ni0.85Se lattice (Mox-Ni0.85Se) and growing atomic MoSe2 nanosheets epitaxially at the edge of the Mox-Ni0.85Se. Such an Mox-Ni0.85Se/MoSe2 catalyst requires only 110 mV to drive current densities of 10 mA cm−2 in alkaline simulated seawater, and shows almost no obvious degradation after 80 h at 20 mA cm−2. The experimental results, combined with the density functional theory calculations, reveal that the Mox-Ni0.85Se/MoSe2 heterostructure will generate an interfacial electric field to facilitate the electron transfer, thus reducing the water dissociation barrier. Significantly, the heteroatomic Mo-doping in the Ni0.85Se can regulate the local electronic configuration of the Mox-Ni0.85Se/MoSe2 heterostructure catalyst by altering the coordination environment and orbital hybridization, thereby weakening the bonding interaction between the Cl and Se/Mo. This synergistic effect for the Mox-Ni0.85Se/MoSe2 heterostructure will simultaneously enhance the catalytic activity and durability, without poisoning or corrosion of the chloride ions.
Introduction
Hydrogen fuel is considered as an ideal alternative to traditional fossil resources due to its superior energy density of 142 kJ g−1 and its environmentally friendly nature.1–3 Water splitting is an economical strategy to generate hydrogen. However, current mature water electrolysis technology typically relies on high-purity water.4,5 If this water electrolysis were to be commercialized, it would place a significant strain on freshwater resources.6 Seawater, which accounts for approximately 96.5% of the Earth's water,7 shows promise as a natural electrolyte. Nevertheless, direct seawater electrolysis is still in its early stages and faces significant challenges, including the poisoning effect of non-innocent ions such as chlorine anions, and the low kinetics of the hydrogen evolution reaction (HER).8–10 Furthermore, these will occur in the chlorine evolution reaction (CER) in an acid environment, and they compete with the oxygen evolution reaction (OER).11 It is widely accepted that in alkaline conditions, the CER can be efficiently blocked, and the OER usually exhibits a reasonable performance.12,13 Moreover, the possibility of seawater splitting in alkaline conditions has been widely demonstrated in recent years.13–15 Platinum (Pt)-based materials are regarded as the state-of-art HER catalysts,16,17 however, the scarcity and expensiveness of Pt limit its wide application.18–20 In addition, the presence of chlorine anions in seawater will also poison Pt-based catalysts, which decreases the electrochemical performance for seawater splitting.21 Therefore, it is crucial to explore earth-abundant, efficient, and non-poisonous electrocatalysts for HERs in seawater.A great deal of effort has been made in developing efficient non-precious HER electrocatalysts over the past several decades.22–24 Among these candidates, transition-metal chalcogenide heterostructural electrocatalysts have attracted significant attention due to their lower cost and outstanding electrochemical properties.25–27 For example, Ni-based heterogeneous catalysts have attracted much attention as they have strong interfacial effects and unique electronic structures.28 Although significant research has been made to improve the activity of HER, Ni-based selenides still suffer from the poisoning of the chlorine ions and poor stability.29 However, the use of MoSe2 as a HER catalyst has been extensively investigated because of its low Gibbs' free energy for hydrogen adsorption on the Mo-edge of MoSe2.30,31 Thus, rational integration of NiSex and MoSe2 to construct a heterostructure can optimize the electrocatalytic activity for HER.32 However, to the best of our knowledge, so far, the corresponding work on the NiSex/MoSe2 heterostructures as HER electrocatalysts in the seawater environment has seldom been studied in detail.
In this work, novel Mox-Ni0.85Se/MoSe2 heterostructures were successfully achieved using the edge epitaxy of atomic MoSe2 nanosheets on Mo-doped Ni0.85Se using a one-pot selenization process of the precursor NiMoO4 nanosheets. Such Mox-Ni0.85Se/MoSe2 heterostructures exhibited efficient electrocatalytic behavior in alkaline simulated seawater, with a low overpotential of 110 mV at 10 mA cm−2. Significantly, the Mox-Ni0.85Se/MoSe2 catalyst could be effective against the poisoning and corrosion of chloride ions, and displayed an impressive electrochemical stability over 80 h in alkaline simulated seawater. A deep insight into the outstanding electrochemical performance mechanism was obtained that showed that the built-in interfacial electric field of the Mox-Ni0.85Se/MoSe2 heterostructure would improve the charge transfer, and that the Mo cation could modify the local electronic environment of Ni0.85Se via the formation of sp3d2 hybridization in the Ni–Se octahedral geometry, and sp3d hybridization in the Mo–Se square pyramidal geometry, thus weakening the bonding interaction between the Cl–Se and Cl–Mo.
Results and discussion
Synthesis and characterization of Mox-Ni0.85Se/MoSe2
The Mox-Ni0.85Se/MoSe2 heterostructures anchored on carbon cloth (CC) were prepared by hydrothermal selenization of NiMoO4 with adding Ni sources, a reducing agent (NaBH4), and additional Mo sources, as shown in Fig. 1a. Scanning electron microscopy (SEM) images (Fig. S1, ESI†) and X-ray diffraction spectra (XRD, Fig. S2, ESI†) of NiMoO4 nanosheets indicated that they were vertically supported on a carbon fiber with a highly open structure made up of interwoven nanosheets. To optimize the morphology and performance, a series of different amounts of additional Mo sources were employed during the hydrothermal selenization process (Fig. S3, ESI†). It was clearly seen that when the weight of Na2MoO4·2H2O was below 60 mg, the obtained samples could maintain the nanosheet structure. When the additional weight of Na2MoO4·2H2O was 60 mg, the obtained sample (named as Mox-Ni0.85Se/MoSe2) shows a thin nanosheet structure (Fig. 1b, S3e and f, ESI†). When adding excess Mo sources, a large number of nanoparticles accumulated. Their HER performance was studied and the results are shown in Fig. S4 (ESI),† and the Mox-Ni0.85Se/MoSe2 showed the best HER performance among these samples.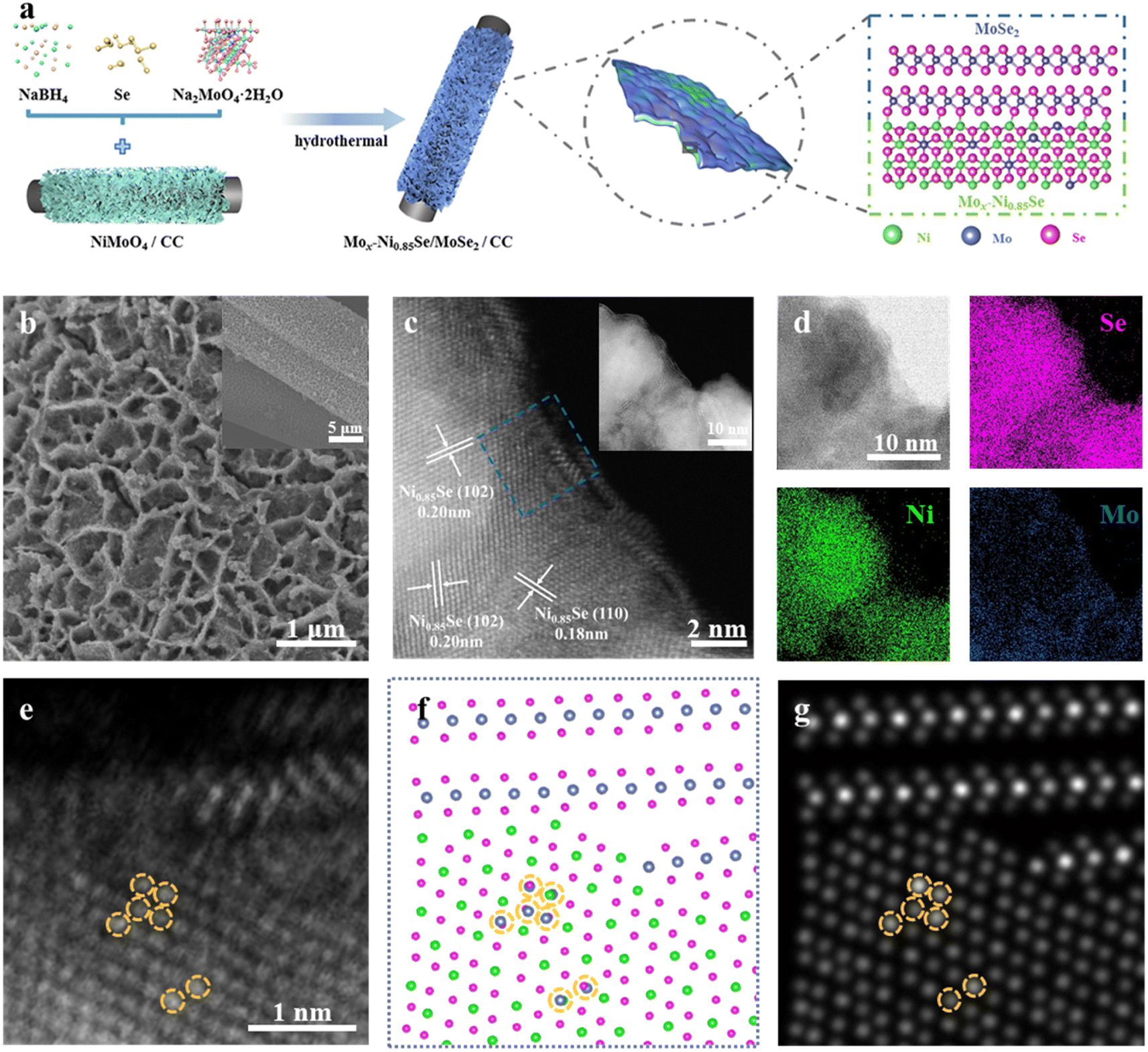 | ||
| Fig. 1 The synthetic scheme and characterization of Mox-Ni0.85Se/MoSe2. (a) A schematic illustration for the synthesis of Mox-Ni0.85Se/MoSe2. Low (inset) and high magnification (b) SEM, and (c) HAADF-STEM images of Mox-Ni0.85Se/MoSe2. (d) Bright field STEM image and corresponding EDS elemental mapping of Mox-Ni0.85Se/MoSe2. (e)-(g) From left to right are experimental HAADF-STEM image, structure model (Blue: Mo atom; Green: Ni atom; Red: Se atom), and simulated HAADF-STEM image of the selected area in (c), respectively. | ||
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of Mox-Ni0.85Se/MoSe2 (Fig. 1c) showed the epitaxially grown heterostructure of Ni0.85Se/MoSe2, i.e., the epitaxially growing atomic MoSe2 nanosheets at the edge of the Ni0.85Se nanoparticles. It was clearly seen that the epitaxial growth of a few atomic layers of MoSe2 corresponded with elemental mapping results (Fig. 1d). The high-resolution STEM (HR-STEM) image clearly showed the lattice fringes of the inter-nanoparticle with a spacing of 0.20 and 0.18 nm, which corresponded to the (102) and (110) crystal planes of the Ni0.85Se, respectively. This result also demonstrated the heterostructure of the Ni0.85Se/MoSe2. Additionally, the HAADF-STEM image of Mox-Ni0.85Se/MoSe2 in Fig. 1e also shows bright dots in the Ni0.85Se nanoparticles. As the atomic number of Mo was the highest among these three elements, the bright dots could be attributed to Mo. Thus, it was concluded that the Ni0.85Se nanoparticles had been doped by the Mo cations. The model of the Mo cation doping Mox-Ni0.85Se was also identified by simulating its atomic resolution HR-STEM image using QSTEM software (Fig. 1e–g and S5, ESI†).33 The Mo cation doping is circled in yellow in Fig. 1e–g. These results also demonstrate the Mo cation doping of the Mox-Ni0.85Se structure. We also prepared pure NiSe2 and MoSe2 as comparison and the results are shown in Fig. S6 and S7 (ESI).† Then, the XRD of these samples were employed to investigate their crystal structure and phase composition. As shown in Fig. S8 (ESI),† the two distinct peaks at about 26° and 43° were assigned to the carbon cloth. It was also noted that the MoSe2 exhibited an amorphous structure, which also suggested its atomic layer structure. The peaks of NiSe2 were in agreement with the standard card of NiSe2 (PDF#65-1843). The Mox-Ni0.85Se/MoSe2 also presented the typical Ni0.85Se (PDF#18-0888), but the peak of (101) at 33.5° was positive shifted, indicating the successful Mo cation doping in the lattice of Ni0.85Se. To characterize the composition in the heterojunction, the Raman spectra were utilized and are shown in Fig. S9 (ESI).† The peak (Ag) at 237 cm−1 was attributed to the Se–Se stretching mode of NiSe2.34 The Mox-Ni0.85Se/MoSe2 presented three peaks of 139, 235, and 284 cm−1, which were attributed to the phonon mode of MoSe2.35 In addition, there was also a faint peak at ∼510 cm−1, which was attributed to the unique stretch mode of the Ni–Se of Ni0.85Se with a hexagonal structure.34
Synchrotron X-ray absorption spectroscopy (XAS) (Fig. 2a–h) was performed to further investigate the charge states and the local structure of Mox-Ni0.85Se/MoSe2. Fig. 2a shows the Ni K-edge X-ray absorption near edge structure (XANES) spectra for Mox-Ni0.85Se/MoSe2, NiSe2, and Ni foil. It can be clearly seen that the white-line intensity of Mox-Ni0.85Se/MoSe2 was much lower than that of NiSe2, suggesting that its chemical state was lower than that of NiSe2. In addition, the Mox-Ni0.85Se/MoSe2 also shifted to a higher energy than the Ni foil, revealing that it had a higher chemical valence than Ni0. These results demonstrated that the Ni chemical state of Mox-Ni0.85Se/MoSe2 was between 0 and +2. Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra of the Ni K-edge for these samples are shown in Fig. 2d. The main peaks at 1.71 Å and 2.17 Å can be assigned to the Ni–Se coordination in NiSe2, and the Ni–Ni coordination in Ni foil, respectively.36 As for Mox-Ni0.85Se/MoSe2, the Ni–Se coordination exhibited was similar to that of NiSe2, but the shift was in a positive direction, which was caused by the different bond lengths in Mox-Ni0.85Se/MoSe2.36–38 The Mo K-edge XANES spectrum of Mox-Ni0.85Se/MoSe2 (Fig. 2b) showed a noticeable shift in the higher energy region compared to that of MoSe2, which suggested there was a lower Mo chemical valency. The Mo EXAFS spectrum (Fig. 2e) of Mox-Ni0.85Se/MoSe2 showed the main peaks at 1.16 Å and 2.11 Å which were assigned to the Mo–Se coordination and Mo–Mo coordination, respectively.39 The Mo–Mo peak of Mox-Ni0.85Se/MoSe2 was in agreement with that of MoSe2 (2.11 Å), but the Mo–Se peak of Mox-Ni0.85Se/MoSe2 is positive shift 0.06 Å than that of MoSe2 (1.22 Å), which could be attributed to the Mo cation doping in the Ni0.85Se. The Se K-edge XANES for the Mox-Ni0.85Se/MoSe2 in Fig. 2c showed that it was more similar to that of MoSe2, which suggested that they shared similar coordination environments to the Se element. The Se EXAFS spectra (Fig. 2f) indicated that the chemical environment of Se in Mox-Ni0.85Se/MoSe2 was similar to that in MoSe2. To more clearly probe the coordination structures of the Mox-Ni0.85Se/MoSe2 epitaxially grown heterostructure, wavelet transforms (WT) with a high resolution in both the K and R spaces of the Ni K-edge and the Mo K-edge EXAFS oscillations were further performed and the results are shown in Fig. 2g, h and S10 (ESI).† Both the Ni and Mo in the Mox-Ni0.85Se/MoSe2 showed a certain degree of deviation, which indicated the change of the element coordination environment, resulting in the fast electron transfer in the heterostructure.40,41 These results indicated that the heterogeneous structure of Mox-Ni0.85Se/MoSe2 may form an interfacial electric field, which facilitates the electron transfer.
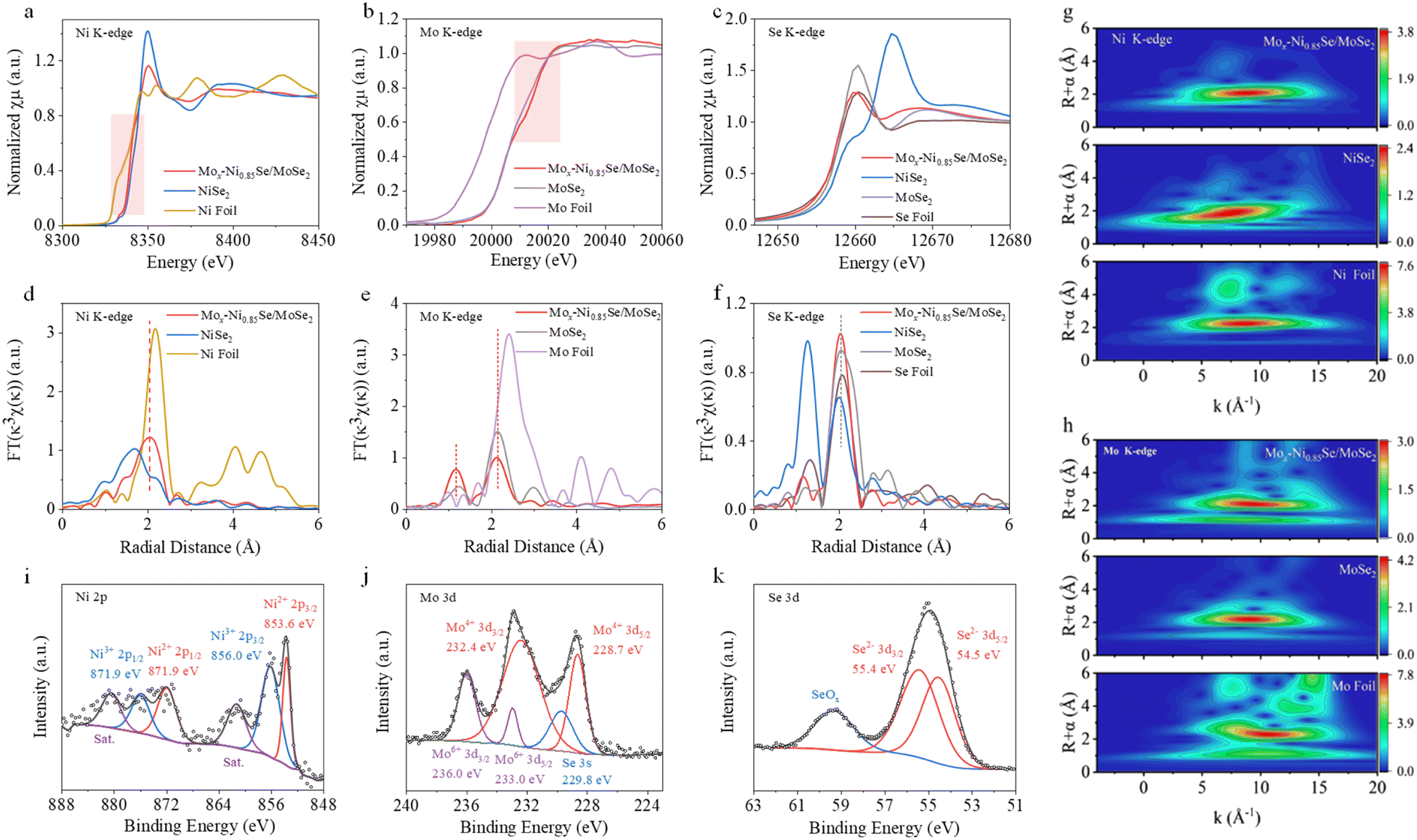 | ||
| Fig. 2 Characterizations of Mox-Ni0.85Se/MoSe2. (a)–(c) The XANES spectra of Ni, Mo, and the Se K-edge, and (d)–(f) the corresponding FT-EXAFS spectra for Mox-Ni0.85Se/MoSe2, NiSe2, MoSe2, Ni foil, Mo foil, and Se foil. The wavelet transform of (g) the Ni K-edge and (h) the Mo K-edge EXAFS for Mox-Ni0.85Se/MoSe2, NiSe2, Ni foil, MoSe2, and Mo foil. The XPS spectra of (i) Ni 2p, (j) Mo 3d, and (k) Se 3d for Mox-Ni0.85Se/MoSe2. | ||
The X-ray photoelectron spectroscopy (XPS) measurements (Fig. 2i–k, and S11, ESI†) were employed to further investigate the chemical and phase compositions. The Ni 2p3/2 XPS spectra of the Mox-Ni0.85Se/MoSe2 exhibited two peaks located at 853.6 eV (Ni2+ 2p3/2) and 856.1 eV (Ni3+ 2p3/2), which showed a slight decrease of ∼0.2 eV when compared with that of pure Ni0.85Se.42 The Mo 3d5/2 XPS spectra (Fig. 2j) can be divided into Mo4+ 3d5/2 (228.7 eV) and Mo6+ 3d5/2 (233.0 eV), and the Mo4+ 3d5/2 showed an obvious increase of ∼0.3 eV in Mox-Ni0.85Se/MoSe2 compared to that of pure MoSe2.43 This shift strongly suggested the presence of an interfacial electric field that serves to expedite the transfer of electrons from the MoSe2 atomic layer to the Ni0.85Se matrix. This observation significantly bolsters the evidence supporting the interaction between MoSe2 and Ni0.85Se within the Mox-Ni0.85Se/MoSe2 heterostructure. Moreover, the Se 3d spectrum showed two large peaks at 54.5 and 55.4 eV, which were attributed to Se2− 3d5/2 and Se2− 3d3/2. In addition, the binding energy of 59.5 eV could be attributed to SeOx resulting from the oxidation of the Se surface.1,32
The electrocatalytic HER performance of Mox-Ni0.85Se/MoSe2
The HER performance of Mox-Ni0.85Se/MoSe2 was evaluated in 1 M KOH with high-purity water and alkaline simulated seawater (1 M KOH and 0.5 M NaCl) using a three-electrode device (Fig. 3, and S12–S21, ESI†). It can be clearly seen that the Mox-Ni0.85Se/MoSe2 exhibited excellent HER performance in these two electrolytes, and only needed 110 mV to achieve 10 mA cm−2 in both 1 M KOH and alkaline simulated seawater. In contrast, NiSe2 and MoSe2 showed 147 mV@10 mA cm−2 and 211 mV@10 mA cm−2 in 1 M KOH, respectively. As for alkaline simulated seawater, NiSe2 and MoSe2 showed 160 mV@10 mA cm−2 and 232 mV@10 mA cm−2, respectively. It was noted that, to a certain extent, the NiSe2 could endure the Cl− poisoning, whereas the activity of MoSe2 decreased significantly in the presence of Cl−. The physical mixed NiSe2 and MoSe2 (named as NiSe2 + MoSe2) showed 126 mV@10 mA cm−2 (1 M KOH) and 132 mV@10 mA cm−2 (alkaline simulated seawater), suggesting that the physical mixture cannot efficiently endure the Cl− poisoning. These results demonstrated that the Mox-Ni0.85Se/MoSe2 heterostructure showed an excellent performance in alkaline simulated seawater which should be attributed to the heterostructure structure. In addition, the Mo cation in Mox-Ni0.85Se/MoSe2 will also modify its local electronic structure, and thus enhanced the electron transfer between Mox-Ni0.85Se and MoSe2 in the heterostructure.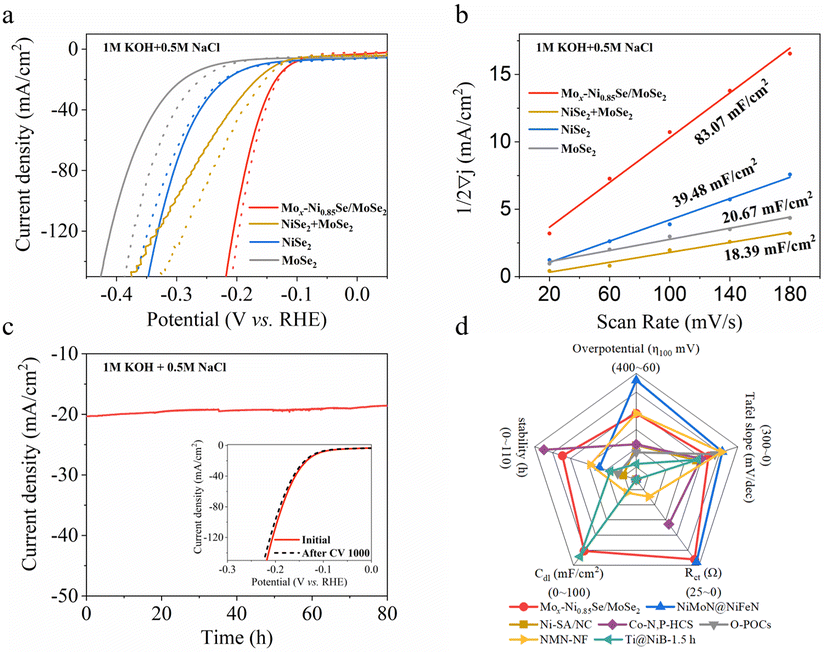 | ||
| Fig. 3 The electrochemical properties of Mox-Ni0.85Se/MoSe2 catalyst. (a) Polarization curves. (b) The capacitive currents at −0.5 V vs. RHE as a function of the scan rate for the obtained catalysts. (c) Chronopotentiometry test results of the Mox-Ni0.85Se/MoSe2 catalyst at 20 mA cm−2 for 80 h (inset: LSV curves before and after 1000 cycles) in alkaline simulated seawater (1 M KOH + 0.5 M NaCl). (d) The electrochemical performance comparison between Mox-Ni0.85Se/MoSe2 and previously reported values obtained in alkaline simulated seawater, found in the literature . | ||
The Tafel slope values of Mox-Ni0.85Se/MoSe2 in 1 M KOH and alkaline simulated seawater (Fig. S14, ESI†) are the lowest among these catalysts and were 70.12 and 86.19 mV dec−1, respectively. This indicated the favorable kinetics of the Mox-Ni0.85Se/MoSe2 during the electrocatalytic hydrogen production process, even in the presence of Cl−. Moreover, the value of the Tafel slope of Mox-Ni0.85Se/MoSe2 is between the Volmer mechanism (120 mV dec−1) and the Heyrovský mechanism (40 mV dec−1), which indicated that it most likely obeys the Volmer–Heyrovský mechanism.1,44,45 This was in agreement with previous reports where nickel chalcogenides were found to be efficient to produce had in the water dissociation process (Volmer step), resulting in Had migrating to the neighboring active site of MoSe2 to undergo the Heyrovský mechanism to produce H2O molecules.44,46,47 The electrochemical impedance spectroscopy (EIS) at an overpotential of 250 mV revealed that the Mox-Ni0.85Se/MoSe2 has the lowest charge transfer resistance of 1.749 Ω in 1 M KOH (Fig. S15, ESI†) and 1.508 Ω in alkaline simulated seawater (Fig. S16, ESI†) when compared with the other three electrocatalysts. This implied its quick charge transfer capability, which could be attributed to the built-in interfacial electric field of Ni0.85Se/MoSe2 heterostructure and the Mo cation doping, and this was in agreement with the XAS and XPS results.
To obtain insights into the electrochemical surface area (ECSA) of the materials, the electrochemical double-layer capacitance (Cdl) values were estimated and are shown in Fig. 3b, S17 and S18 (ESI).† The Mox-Ni0.85Se/MoSe2 had the highest Cdl value of 92.97 mF cm−2 in 1 M KOH, and this indicated that it had a larger electrocatalytic active surface area. However, this decreased to 83.07 mF cm−2 in alkaline simulated seawater. This result suggested that the Cl− will slightly influence the electrochemistry of Mox-Ni0.85Se/MoSe2. The previously mentioned results demonstrated that the Mox-Ni0.85Se/MoSe2 was an excellent electrocatalyst even in alkaline simulated seawater. However, in the practical application of electrocatalysts, stability is another important parameter, especially in the alkaline simulated seawater environment, due to the fact that the Cl− can cause electrocatalyst poisoning and corrosion. The cyclic voltammetry (CV) was performed for 1000 cycles and the i–t mode was performed at 20 mA cm−2 for a continuous 80 h Mox-Ni0.85Se/MoSe2 in alkaline simulated seawater (Fig. 3c). It can be clearly seen that after 80 h, the current density of Mox-Ni0.85Se/MoSe2 only showed a slight decrease, as shown in the inset of Fig. 3c, after 1000 CV cycles, and the linear sweep voltammetry (LSV) curve of Mox-Ni0.85Se/MoSe2 was almost unchanged. Moreover, after 1000 CV cycles, the Mox-Ni0.85Se/MoSe2 can maintain its structure (Fig. S21, ESI†). Both these results illustrated that the Mox-Ni0.85Se/MoSe2 has the ability for anti-Cl ion corrosion, and a great performance even in alkaline simulated seawater. The radar chart (Fig. 3d) and the comparison with results reported in the literature (Table S1, ESI†) showed that the integrated HER performance of Mox-Ni0.85Se/MoSe2 exhibited the best HER performance in alkaline simulated seawater.
Thus, the role of Mo cation doping has two distinct functions: firstly, it enhances the charge transfer capability of the built-in interfacial electric field within the Ni0.85Se/MoSe2 heterostructure; secondly, the Mo cation doping can also modulate the local electronic state of Ni, effectively enabling the resistance against chloride ions in alkaline seawater solutions and safeguarding the electrode materials from chloride ion corrosion.
Mechanism of cation tuning
The first-principles calculations were adopted to evaluate the effects of Mo absorption, and the MoSe2/NiSe heterojunction. The absorption energies of H2O on NiSe and Mo-NiSe (Fig. S22, ESI†) were −0.88 and −0.70 eV, respectively. The water dissociation barrier for NiSe was 3.02 eV (Fig. S23a, ESI†), however, the Mo cation doping of Mo-NiSe increased it to 3.08 eV. This indicated that the Mo substitution did not directly enhance the water dissociation. As for the MoSe2/NiSe heterojunction, its water dissociation barrier was 2.72 eV, which was much lower than that of NiSe (3.02 eV) as shown in Fig. S23b (ESI),† indicating the great performance of MoSe2/NiSe for hydrogen evolution. The lower water dissociation barrier of the MoSe2/NiSe heterojunction could be attributed to its built-in interfacial electric field, which facilitated the charge-transfer.The resistance to Cl− corrosion for these samples was evaluated and the results are shown in Fig. 4. The absorption energy of H2O around the Cl adatom on the surface of the NiSe + Cl increased from −0.88 eV to −0.19 eV, indicating that the Cl absorption hinders the H2O absorption. Fig. 4a–c show that the Cl atom can only be absorbed on the top of the Se atom on the surface of NiSe (referred to as NiSe + Cl), whereas the Cl atoms can be absorbed on both the Se and Mo on the surface of Mo-NiSe (referred to as Mo-NiSe + Cl1 and Mo-NiSe + Cl2, respectively). The absorption energies (Gabs) were −0.54, −0.06, and −0.26 eV for NiSe + Cl, Mo-NiSe + Cl1, and Mo-NiSe + Cl2, respectively, as shown in Fig. 4g. The increased Gabs when compared to NiSe indicated that there was a resistance to Cl-ion corrosion after the Mo doping of NiSe. To explain this phenomenon, the charge density difference (ρdiff) and the projected density of states (PDOS) were calculated (Fig. 4 and S24, ESI†).
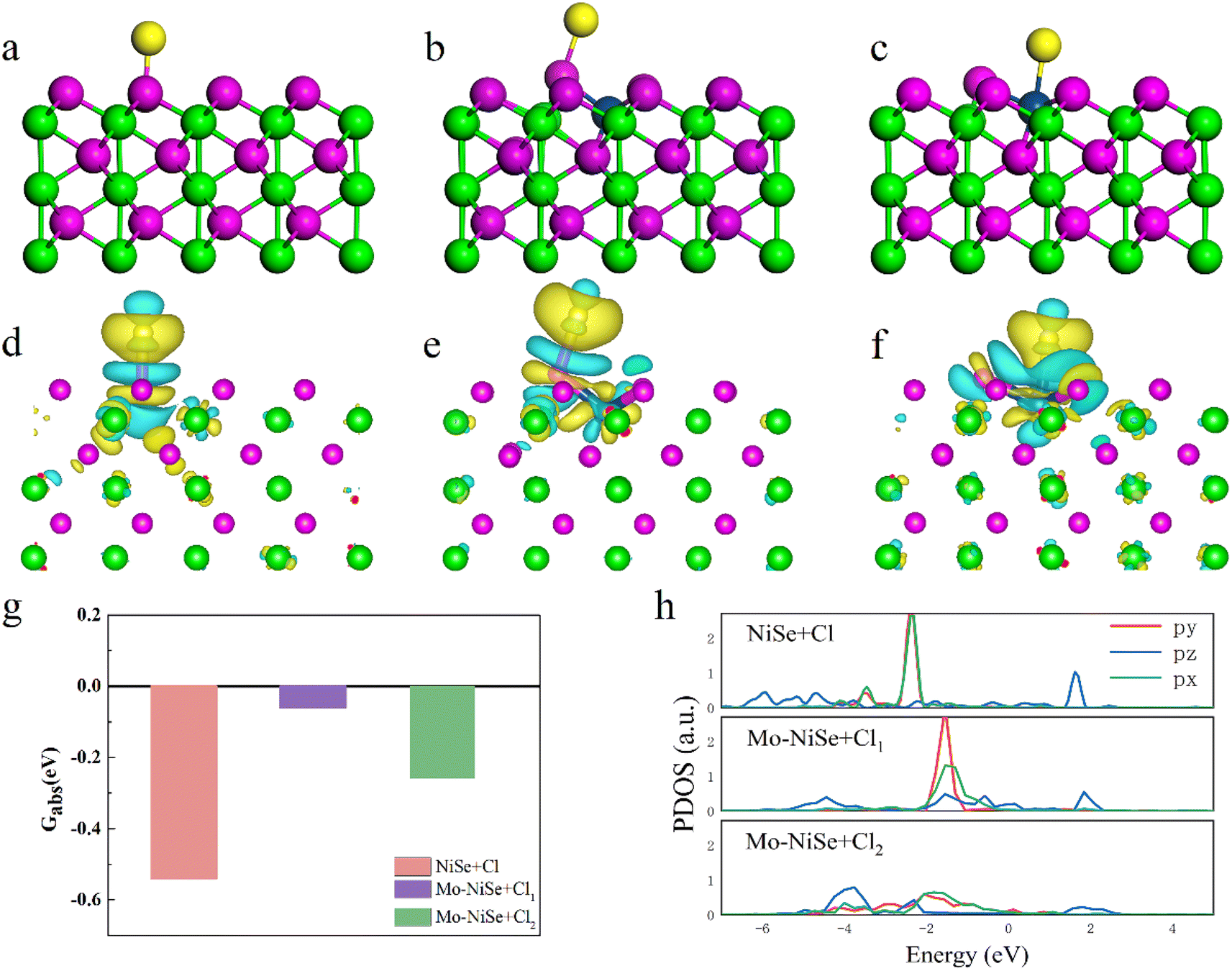 | ||
| Fig. 4 Mechanistic studies using DFT. (a)–(c) Schematic diagrams of the atomic structure of NiSe + Cl, Mo-NiSe + Cl1, and Mo-NiSe + Cl2. (d)-(f) Distribution of ρdiff in NiSe + Cl, Mo-NiSe + Cl1, and Mo-NiSe + Cl2. The isosurface level is set to 10−3. (g) and (h) The absorption energies and the PDOS of the Cl adatom in NiSe + Cl, Mo-NiSe + Cl1, and Mo-NiSe + Cl2, respectively. | ||
As shown in Fig. 4d–f, a polar covalent bond was formed between the Cl adatom and the Se atom in both NiSe + Cl and Mo-NiSe + Cl1, whereas an ionic bond was formed between the Cl adatom and the Mo atom in Mo-NiSe + Cl2. From Fig. S24 (ESI),† it can be seen that the bonding interaction arose from the resonance between the Cl_pz and Se_pz states in both NiSe + Cl and Mo-NiSe + Cl1, whereas the bonding interaction corresponded to the coupling between the Cl_pz and the Mo_dz2 in Mo-NiSe + Cl2. Compared to NiSe + Cl, a considerable shifting of the bonding state towards the Fermi level was found in Mo-NiSe + Cl1 and Mo-NiSe + Cl2 (Fig. 4h), and this indicated a weakening of the bonding interaction strength between the Cl adatom and the surface atom. The variation in the strength of the bonding interaction is related to the difference in the coordination environment between Mo and Ni, which modifies the crystal field splitting and orbital hybridization. As shown in Fig. 4a, the Ni atom bonds with six neighboring Se atoms, forming an octahedral geometry. In this octahedral geometry, the d orbitals form sp3d2 hybridization, and the Ni exhibits a valency of +2. However, the Mo atom bonds with five neighboring Se atoms (Fig. 4b), forming a square pyramidal geometry. In this structure, the d orbitals form sp3d hybridization, and the Mo exhibits a valency of +3. The variation of the valency influences the charge transfer and weakens the bonding interaction between the Cl and Se in Mo-NiSe + Cl1. In the Mo-NiSe + Cl2, the Cl–Mo direction did not align with the Mo_4d hybrid orbitals, indicating that the Cl–Mo direction was not suitable for electron pairing to form a chemical bond. Consequently, only a weak ionic bond was formed between the Cl and Mo atoms in Mo-NiSe + Cl2.
Conclusions
In summary, a convenient strategy to synthesize Mox-Ni0.85Se/MoSe2 nanosheet network heterostructures supported on carbon cloth has been successfully developed by the selenization of the NiMoO4 nanosheets precursor. The STEM and XAS results demonstrate that the atomic MoSe2 nanosheets were grown epitaxially at the edge of the Mox-Ni0.85Se, and the Mo cation doping tuned the local electronic environment of the Mox-Ni0.85Se/MoSe2 heterostructures. This exhibits an excellent performance for HER in alkaline simulated seawater with a low overpotential of 110 mV at the current density of 10 mA cm−2, and shows almost no sign of waning even after 80 h at 20 mA cm−2. The excellent HER performance in alkaline simulated seawater could be attributed to the built-in interfacial electric field of the Mox-Ni0.85Se/MoSe2 heterostructure and Mo cation doping, that improves the electron transfer between adsorbed species and Mox-Ni0.85Se/MoSe2. Moreover, the Mo cation doping also weakens the bonding interaction between Cl–Se and Cl–Mo, thereby protecting it against the poisoning and erosion of chloride ions.Data availability
The authors confirm that the data supporting the findings of this study are available within the article ESI.†Author contributions
ZH, CZ, and SG contributed equally to this manuscript. BZ and YL supervised this study. BZ, YL, and ZH conceived the idea. ZH and YJ planned and carried out the experiments, and collected and analyzed the experimental data. SG and NC performed SEM and TEM characterizations. SL performed the Raman characterizations. CZ conducted the theoretical calculations. BZ and ZH wrote the paper. All the authors have discussed the results and wrote the paper together.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 12004321, 22279011, 52201003), the Fundamental Research Funds for the Central Universities (Grant No. 2023CDJXY-046), the Scientific Research Project of Hunan Provincial Education Department (Grant No. 23B0157), and the Program for Changjiang Scholars and Innovative Research Team in University (Grant No. IRT17R91).Notes and references
- Y. Ji, W. J. Luo, Y. D. Liu, Z. M. He, N. Y. Cheng, Z. Zhang, X. Qi, J. X. Zhong and L. Ren, Bifunctional o-CoSe2/c-CoSe2/MoSe2 heterostructures for enhanced electrocatalytic and photoelectrochemical hydrogen evolution reaction, Mater. Today Chem., 2022, 23, 100724 CrossRef CAS.
- M. Zhu, Q. Yan, Y. Q. Xue, Y. D. Yan, K. Zhu, K. Ye, J. Yan, D. X. Cao, H. J. Xie and G. L. Wang, Free-standing P-doped NiSe2/MoSe2 catalyst for efficient hydrogen evolution in acidic and alkaline media, ACS Sustain. Chem. Eng., 2021, 10, 279–287 CrossRef.
- L. Wu, F. H. Zhang, S. W. Song, M. H. Ning, Q. Zhu, J. Q. Zhou, G. H. Gao, Z. Y. Chen, Q. C. Zhou and X. X. Xing, Efficient alkaline water/seawater hydrogen evolution by a nanorod-nanoparticle-structured Ni-MoN catalyst with fast water-dissociation kinetics, Adv. Mater., 2022, 34, 2201774 CrossRef CAS.
- J. X. Guo, Y. Zheng, Z. P. Hu, C. Y. Zheng, J. Mao, K. Du, M. Jaroniec, S. Z. Qiao and T. Ling, Direct seawater electrolysis by adjusting the local reaction environment of a catalyst, Nat. Energy, 2023, 8, 264–272 CAS.
- M. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Seawater electrolysis for hydrogen production: a solution looking for a problem?, Energy Environ. Sci., 2021, 14, 4831–4839 RSC.
- S. r. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, Direct electrolytic splitting of seawater: opportunities and challenges, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- B. H. Suryanto, Y. Wang, R. K. Hocking, W. Adamson and C. Zhao, Overall electrochemical splitting of water at the heterogeneous interface of nickel and iron oxide, Nat. Commun., 2019, 10, 5599 CrossRef CAS.
- J. N. Hausmann, R. Schlögl, P. W. Menezes and M. Driess, Is direct seawater splitting economically meaningful?, Energy Environ. Sci., 2021, 14, 3679–3685 RSC.
- F. Sun, J. S. Qin, Z. Y. Wang, M. Z. Yu, X. H. Wu, X. Sun and J. S. Qiu, Energy-saving hydrogen production by chlorine-free hybrid seawater splitting coupling hydrazine degradation, Nat. Commun., 2021, 12, 4182 CrossRef CAS.
- Y. Y. Guo, Y. H. Cao, J. D. Lu, X. R. Zheng and Y. D. Deng, The concept, structure, and progress of seawater metal-air batteries, Microstructures, 2023, 3, 2023038 CrossRef.
- X. Y. Lu, J. Pan, E. Lovell, T. H. Tan, Y. H. Ng and R. Amal, A sea-change: Manganese doped nickel/nickel oxide electrocatalysts for hydrogen generation from seawater, Energy Environ. Sci., 2018, 11, 1898–1910 RSC.
- Y. Kuang, M. J. Kenney, Y. Meng, W.-H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li and L. Wang, Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, Design criteria, operating conditions, and nickel–iron hydroxide catalyst materials for selective seawater electrolysis, ChemSusChem, 2016, 9, 962–972 CrossRef CAS.
- W. J. Zang, T. Sun, T. Yang, S. B. Xi, M. Waqar, Z. K. Kou, Z. Y. Lyu, Y. P. Feng, J. Wang and S. J. Pennycook, Efficient hydrogen evolution of oxidized Ni-N3 defective sites for alkaline freshwater and seawater electrolysis, Adv. Mater., 2021, 33, 2003846 CrossRef CAS.
- D. Liu, H. Q. Ai, M. P. Chen, P. F. Zhou, B. W. Li, D. Liu, X. Y. Du, K. Lo, K. Ng and S. P. Wang, Multi-phase heterostructure of CoNiP/CoxP for enhanced hydrogen evolution under alkaline and seawater conditions by promoting H2O dissociation, Small, 2021, 17, 2007557 CrossRef CAS.
- B. W. Zhang, L. Ren, Z. F. Xu, N. Y. Cheng, W. H. Lai, L. Zhang, W. Hao, S. Q. Chu, Y. X. Wang and Y. Du, Atomic Structural Evolution of Single-Layer Pt Clusters as Efficient Electrocatalysts, Small, 2021, 17, 2100732 CrossRef CAS.
- B. W. Zhang, H. L. Yang, Y. X. Wang, S. X. Dou and H. K. Liu, A comprehensive review on controlling surface composition of Pt-based bimetallic electrocatalysts, Adv. Energy Mater., 2018, 8, 1703597 CrossRef.
- B. W. Zhang, Y. X. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Fabrication of superior single-atom catalysts toward diverse electrochemical reactions, Small Methods, 2019, 3, 1800497 CrossRef.
- J. H. Zheng, J. L. Zhang, G. Li, J. M. Zhang, B. W. Zhang, Y. X. Jiang and S. G. Sun, Tuning atomic Pt site surface on PtAu alloy toward electro-oxidation of formic acid, Mater. Today Energy, 2022, 27, 101028 CrossRef CAS.
- J. H. Zheng, G. Li, J. M. Zhang, N. Y. Cheng, L. F. Ji, J. Yang, J. L. Zhang, B. W. Zhang, Y. X. Jiang and S. G. Sun, General strategy for evaluating the d-band center shift and ethanol oxidation reaction pathway towards Pt-based electrocatalysts, Sci. China: Chem., 2023, 66, 279–288 CrossRef CAS.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Electrolysis of low-grade and saline surface water, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian and W. C. Cheong, Rational design of single molybdenum atoms anchored on N-doped carbon for effective hydrogen evolution reaction, Angew. Chem., 2017, 129, 16302–16306 CrossRef.
- J. Mahmood, F. Li, S. M. Jung, M. S. Okyay, I. Ahmad, S. J. Kim, N. Park, H. Y. Jeong and J. B. Baek, An efficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS.
- Y. Jiao, Y. Zheng, K. Davey and S. Z. Qiao, Activity origin and catalyst design principles for electrocatalytic hydrogen evolution on heteroatom-doped graphene, Nat. Energy, 2016, 1, 1–9 Search PubMed.
- Y. Q. Zhao, B. Jin, Y. Zheng, H. Y. Jin, Y. Jiao and S. Z. Qiao, Charge state manipulation of cobalt selenide catalyst for overall seawater electrolysis, Adv. Energy Mater., 2018, 8, 1801926 CrossRef.
- B. S. Zhang, W. W. Xu, S. Liu, X. Chen, T. F. Ma, G. H. Wang, Z. Y. Lu and J. Sun, Enhanced interface interaction in Cu2S@ Ni core-shell nanorod arrays as hydrogen evolution reaction electrode for alkaline seawater electrolysis, J. Power Sources, 2021, 506, 230235 CrossRef CAS.
- C. Z. Wang, M. Z. Zhu, Z. Y. Cao, P. Zhu, Y. Q. Cao, X. Y. Xu, C. X. Xu and Z. Y. Yin, Heterogeneous bimetallic sulfides based seawater electrolysis towards stable industrial-level large current density, Appl. Catal., B, 2021, 291, 120071 CrossRef CAS.
- Y. Li, X. Bao, D. Chen, Z. Wang, N. Dewangan, M. Li, Z. Xu, J. Wang, S. Kawi and Q. Zhong, A Minireview on Nickel-Based Heterogeneous Electrocatalysts for Water Splitting, ChemCatChem, 2019, 11, 5913–5928 CrossRef CAS.
- R. Andaveh, A. Sabour Rouhaghdam, J. Ai, M. Maleki, K. Wang, A. Seif, G. Barati Darband and J. Li, Boosting the electrocatalytic activity of NiSe by introducing MnCo as an efficient heterostructured electrocatalyst for large-current-density alkaline seawater splitting, Appl. Catal., B, 2023, 325, 122355 CrossRef CAS.
- L. Zhang, T. Wang, L. Sun, Y. J. Sun, T. W. Hu, K. W. Xu and F. Ma, Hydrothermal synthesis of 3D hierarchical MoSe2/NiSe2 composite nanowires on carbon fiber paper and their enhanced electrocatalytic activity for the hydrogen evolution reaction, J. Mater. Chem. A, 2017, 5, 19752–19759 RSC.
- X. Q. Wang, B. J. Zheng, B. Wang, H. Q. Wang, B. C. Sun, J. R. He, W. L. Zhang and Y. F. Chen, Hierarchical MoSe2-CoSe2 nanotubes anchored on graphene nanosheets: A highly efficient and stable electrocatalyst for hydrogen evolution in alkaline medium, Electrochim. Acta, 2019, 299, 197–205 CrossRef CAS.
- J. Y. Xue, F. L. Li, B. B. Chen, H. B. Geng, W. Zhang, W. Y. Xu, H. W. Gu, P. Braunstein and J. P. Lang, Engineering multiphasic MoSe2/NiSe heterostructure interfaces for superior hydrogen production electrocatalysis, Appl. Catal., B, 2022, 312, 121434 CrossRef CAS.
- C. Koch, Determination of core structure periodicity and point defect density along dislocations, Arizona State University, 2002 Search PubMed.
- Z. D. Huang, B. Xu, Z. G. Li, J. W. Ren, H. Mei, Z. N. Liu, D. G. Xie, H. B. Zhang, F. Dai and R. Wang, Accurately Regulating the Electronic Structure of NixSey@ NC Core–Shell Nanohybrids through Controllable Selenization of a Ni-MOF for pH-Universal Hydrogen Evolution Reaction, Small, 2020, 16, 2004231 CrossRef CAS.
- S. Setayeshgar, M. Karimipour, M. Molaei, M. R. Moghadam and S. Khazraei, Synthesis of scalable 1T/2H–MoSe2 nanosheets with a new source of Se in basic media and study of their HER activity, Int. J. Hydrogen Energy, 2020, 45, 6090–6101 CrossRef CAS.
- S. L. Han, Y. N. Hao, Z. Y. Guo, D. S. Yu, H. J. Huang, F. Hu, L. L. Li, H. Y. Chen and S. J. Peng, Self-supported N-doped NiSe2 hierarchical porous nanoflake arrays for efficient oxygen electrocatalysis in flexible zinc-air batteries, Chem. Eng. J., 2020, 401, 126088 CrossRef CAS.
- Y. C. Chu, C. J. Chang, Y. P. Zhu, S. C. Lin, C. W. Tung, T. L. Chen and H. M. Chen, Anionic effects on metal pair of Se-doped nickel diphosphide for hydrogen evolution reaction, ACS Sustain. Chem. Eng., 2019, 7, 14247–14255 CrossRef CAS.
- S. J. Shen, Z. P. Lin, K. Song, Z. P. Wang, L. G. Huang, L. H. Yan, F. Q. Meng, Q. H. Zhang, L. Gu and W. W. Zhong, Reversed active sites boost the intrinsic activity of graphene-like cobalt selenide for hydrogen evolution, Angew. Chem., 2021, 133, 12468–12473 CrossRef.
- I. S. Kwon, I. H. Kwak, J. Y. Kim, T. T. Debela, Y. C. Park, J. Park and H. S. Kang, Concurrent Vacancy and Adatom Defects of Mo1–xNbxSe2 Alloy Nanosheets Enhance Electrochemical Performance of Hydrogen Evolution Reaction, ACS Nano, 2021, 15, 5467–5477 CrossRef CAS PubMed.
- Y. Cheng, H. R. Guo, X. P. Li, X. Wu, X. H. Xu, L. R. Zheng and R. Song, Rational design of ultrahigh loading metal single-atoms (Co, Ni, Mo) anchored on in-situ pre-crosslinked guar gum derived N-doped carbon aerogel for efficient overall water splitting, Chem. Eng. J., 2021, 410, 128359 CrossRef CAS.
- W. Wan, Y. Zhao, S. Wei, C. A. Triana, J. Li, A. Arcifa, C. S. Allen, R. Cao and G. R. Patzke, Mechanistic insight into the active centers of single/dual-atom Ni/Fe-based oxygen electrocatalysts, Nat. Commun., 2021, 12, 5589 CrossRef CAS.
- D. Chen, J. W. Shen, X. Li, S. A. Cao, T. Li, W. Luo and F. Xu, Ni0. 85Se hexagonal nanosheets as an advanced conversion cathode for Mg secondary batteries, J. Energy Chem., 2020, 48, 226–232 CrossRef.
- H. Tang, H. Huang, X. Wang, K. Wu, G. Tang and C. Li, Hydrothermal synthesis of 3D hierarchical flower-like MoSe2 microspheres and their adsorption performances for methyl orange, Appl. Surf. Sci., 2016, 379, 296–303 CrossRef CAS.
- H. R. Inta, S. Ghosh, A. Mondal, G. Tudu, H. V. Koppisetti and V. Mahalingam, Ni0. 85Se/MoSe2 interfacial structure: an efficient electrocatalyst for alkaline hydrogen evolution reaction, ACS Appl. Energy Mater., 2021, 4, 2828–2837 CrossRef CAS.
- R. Qin, J. G. Hou, C. X. Xu, H. X. Yang, Q. X. Zhou, Z. Z. Chen and H. Liu, Self-supporting Co0.85Se nanosheets anchored on Co plate as highly efficient electrocatalyst for hydrogen evolution reaction in both acidic and alkaline media, Nano Res., 2020, 13, 2950–2957 CrossRef CAS.
- X. Zhang, Y. Y. Zhang, Y. Zhang, W. J. Jiang, Q. H. Zhang, Y. G. Yang, L. Gu, J. S. Hu and L. J. Wan, Phase-controlled synthesis of 1T-MoSe2/NiSe heterostructure nanowire arrays via electronic injection for synergistically enhanced hydrogen evolution, Small Methods, 2019, 3, 1800317 CrossRef.
- H. Ding, Q. Y. Jiao, H. F. Lv, K. Xu, Q. Y. Xing, M. Chen, W. S. Chu, X. J. Wu and Y. Q. Guo, Promoting the water reduction reaction of transition metal dichalcogenides in a basic electrolyte by interface engineering, J. Mater. Chem. A, 2018, 6, 17488–17494 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05220f |
| ‡ These authors contributed equally: Z. M. He, C. X. Zhang, S. Q. Guo. |
| This journal is © The Royal Society of Chemistry 2024 |