
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgramming interchangeable and reversible heterooligomeric protein self-assembly using a bifunctional ligand†
Soyeun
Son
 and
Woon Ju
Song
*
and
Woon Ju
Song
*
Department of Chemistry, College of Natural Sciences, Seoul National University, Seoul 08826, Republic of Korea. E-mail: woonjusong@snu.ac.kr
First published on 23rd January 2024
Abstract
Protein design for self-assembly allows us to explore the emergence of protein–protein interfaces through various chemical interactions. Heterooligomers, unlike homooligomers, inherently offer a comprehensive range of structural and functional variations. Besides, the macromolecular repertoire and their applications would significantly expand if protein components could be easily interchangeable. This study demonstrates that a rationally designed bifunctional linker containing an enzyme inhibitor and maleimide can guide the formation of diverse protein heterooligomers in an easily applicable and exchangeable manner without extensive sequence optimizations. As proof of concept, we selected four structurally and functionally unrelated proteins, carbonic anhydrase, aldolase, acetyltransferase, and encapsulin, as building block proteins. The combinations of two proteins with the bifunctional linker yielded four two-component heterooligomers with discrete sizes, shapes, and enzyme activities. Besides, the overall size and formation kinetics of the heterooligomers alter upon adding metal chelators, acidic buffer components, and reducing agents, showing the reversibility and tunability in the protein self-assembly. Given that the functional groups of both the linker and protein components are readily interchangeable, our work broadens the scope of protein-assembled architectures and their potential applications as functional biomaterials.
Introduction
Protein–protein interactions (PPIs) are crucial in maintaining various cellular functions, including signal transduction, metabolic regulation, and immune responses.1,2 PPIs exhibit remarkable accuracy and selectivity even within highly crowded intracellular environments.3 A single protein can also engage in multiple PPIs with different partners,4via static or transient processes.5–7The creation and precise adjustment of novel protein–protein interactions (PPIs) can be employed for various potential applications, including proteome exploration,8,9 biocatalytic cascade reaction,10,11 allosteric control, and sensing.12,13 Self-assembled protein architectures can be created by applying protein symmetry, shape/sequence complementarity, and chemical bonding interactions with small molecules or metal ions.14–17 In many cases, a single protein is employed as a building block to minimize the number and complexity of protein interfaces. Then, the chemical space for artificially designed protein-assembled structures and functions remains limited. Alternatively, self-assembly of two protein components can be achieved through gene fusion,18,19 disulphide bond,20 gene fusion and covalent linkage,21 host–guest interaction,22 and computational design.23–26 However, their components are not easily interchangeable unless their gene constructs or PPIs are redesigned from scratch.
Small molecules can serve as synthetically adjustable adaptors for protein self-assembly. Previous studies have harnessed non-covalent interactions between specific proteins and small molecules,27 such as heme,28 biotin,29 and sugar.30 While these naturally occurring interactions are limited, synthetic small-molecule libraries,31–34 including inhibitors and modulators, can serve as abiological binders to recognize and connect specific proteins of interest. Besides, a maleimide could be an alternative versatile moiety applicable to various proteins having surface-exposed cysteine residues via thiol–ene bioconjugation. In this context, various heterobifunctional molecules have been synthesized to facilitate the development of proteolysis-targeting chimera for clinical applications.35,36 Nevertheless, to the best of our knowledge, such a synthetic approach has not been applied for inducing protein self-assembly.
We herein report a rationally designed bifunctional linker capable of connecting two proteins of interest in an easily exchangeable manner. One end of the linker contains an inhibitor that specifically recognizes the first protein when the other end contains a maleimide to connect to the second protein. This linker allows two different macromolecules to create asymmetric PPIs, thereby determining the overall protein size, shape, and function of the resulting heterooligomeric assemblies. The two distinct terminal groups of the linker prevent the formation of homooligomers, imparting directionality to the heteroassembly. Moreover, both protein components are readily interchangeable, as long as they exhibit binding affinity to the binder molecule at one end or possess a cysteine residue on the protein surface. Thus, the bifunctional ligand is effective and versatile in leveraging its intrinsic chemical nature to accommodate various protein building blocks for self-assembly and significantly broadening the chemical space of artificially engineered protein-assembled architectures.
Results and discussion
Selection of building-block proteins
We employed four genetically, structurally, and functionally unrelated proteins as building-blocks to demonstrate that this strategy can induce multiple heterooligomeric assemblies. The first one is a Zn-containing human carbonic anhydrase II (CA; 30 kDa) (Fig. 1a and S1†) because several inhibitors have been developed for this metalloenzyme.37–40 In particular, benzenesulfonamide (BzS) (Fig. 1b) is valuable due to its feasibility in synthetic modification and suitable levels of binding affinity (9–640 nM)38,41 for monitoring both assembly and disassembly events. For the second protein, we selected 2-keto-3-deoxygluconate aldolase from a thermophilic archaeon Sulfolobus solfataricus (KDGA)42 (Fig. 1c and S2†); it is a D2 homotetrameric protein (133 kDa) with sufficient expression yields and thermal stability for this work. | ||
| Fig. 1 Heterooligomeric protein assembly. (a) Human carbonic anhydrase II (CA) (PDB code 3CA2). (b) Bifunctional ligands, BzSn. The benzenesulfonamide (BzS) and maleimide are colored in blue and red, respectively. (c) 2-Keto-3-deoxygluconate aldolase (KDGA) (PDB code 1W37). The overall sizes of the building-block proteins are depicted with scale bars in (a) and (c). (d) A schematic representation of the heteroassembly of three components, BzS5, KDGA, and CA. | ||
Design of bifunctional linkers
To facilitate the assembly of CA and KDGA, we designed bifunctional linkers composed of three parts: BzS, ethylene glycol, and maleimide (Fig. 1b). Ethylene glycol provides solubility and an adjustable length to connect the two proteins. The bifunctional linkers were prepared through a ten-step synthesis (Scheme 1, details are listed in the ESI†), allowing for the substitution of BzS and adjustment of the ethylene glycol length with other protein components in future studies. Since the active site of CA is buried 15 Å from the surface, the linker required at least three repeating units of the ethylene glycol groups. Thus, we synthesized two bifunctional linkers with five (BzS5) or three (BzS3) ethylene glycol repeats, which provided either sufficient or close-fitting distances between the two proteins. | ||
| Scheme 1 Synthetic procedure of bifunctional linkers, BzS5 (b; n = 5) and BzS3 (a; n = 3). (i): Toluene, Ar, 90 °C; (ii): DCC, dry DMF, 0 °C to r.t.; (iii): p-TsCl, dry THF, 0 °C to r.t.; (iv): NaN3, abs. EtOH, 0 °C to reflux; (v): PPh3, THF/H2O, 0 °C to r.t.; (vi): Boc2O, DCM, 0 °C to r.t.; (vii): 1, DIAD, PPh3, dry THF, 0 °C to r.t.; (viii): TFA, DCM, r.t.; (ix): 2, DIPEA, dry DMF, 0 °C to r.t.; (x): dry anisole/dry MeCN, reflux. | ||
KDGA bioconjugation with bifunctional linkers
We examined the X-ray crystal structure of KDGA to determine the optimal position for introducing a cysteine residue. Although pre-existing ones might not entirely participate in thiol–ene bioconjugation, we prepared a double mutant (C120A/C150A or KDGA*) to eliminate any potential interference (Fig. S2e†). We anticipated that E288C mutation would be suitable for attaching BzSn (n = 5 or 3) not to generate steric clash upon the binding of two CA proteins to each side of KDGA (Fig. 1d). High-resolution electrospray ionization mass spectrometry (HR-ESI-MS) and thiol assay results indicated that all four cysteine residues in a tetramer were successfully conjugated with the bifunctional linkers, yielding KDGA*-E288C|BzS5 (Fig. S3 and Table S5†).Self-assembly of CA and KDGA enzymes
We investigated the self-assembly of CA and KDGA using dynamic light scattering (DLS) (Fig. 2). The hydrodynamic diameters of the wild-type proteins, CA and KDGA, in TAPS buffer were determined to be 5.1 ± 0.30 nm and 9.7 ± 0.94 nm, respectively (Fig. 2a). These sizes are comparable to the calculated values (4.7 nm and 8.3 nm) using the X-ray crystal structures and WinHydroPro10.43 The hydrodynamic diameter of KDGA*-E288C|BzS5 (10 ± 1.2 nm) was similar to that of the wild-type protein, indicating that attaching a flexible linker marginally altered the overall protein size.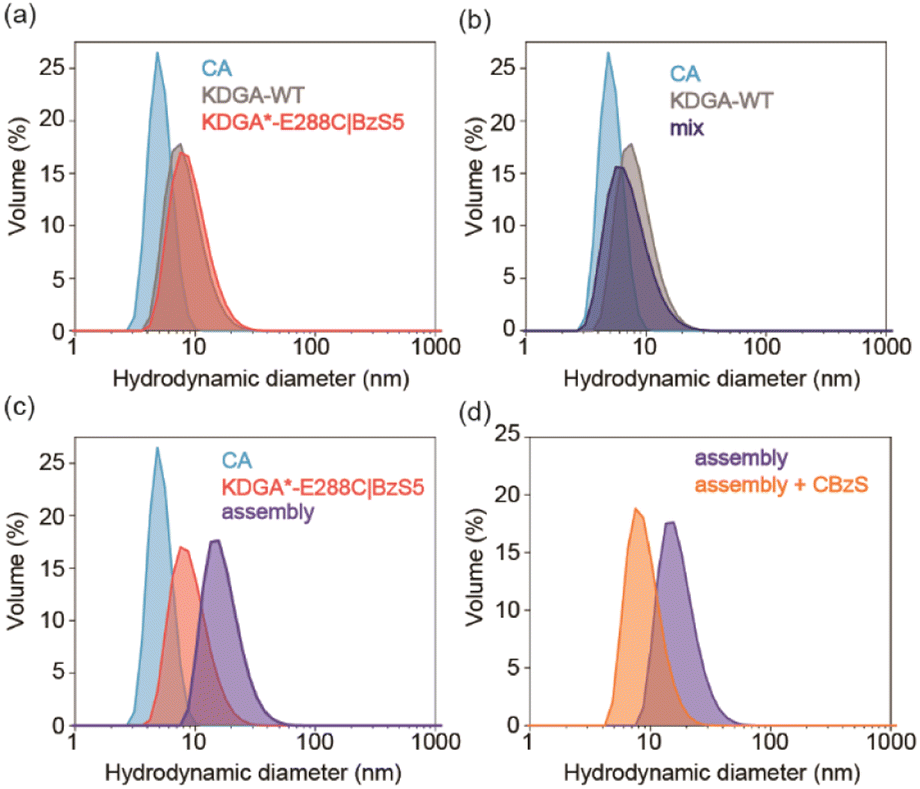 | ||
| Fig. 2 Dynamic light scattering (DLS) of heterooligomeric protein assembly. (a) CA, KDGA, and KDGA*-E288C|BzS5 alone (10 μM monomer). Before and after mixing CA and (b) KDGA or (c) KDGA*-E288C|BzS5. (d) The addition of CBzS (10 μM) to the CA/KDGA assembly in 50 mM borate, pH 8.0 buffer. The protein samples in (a)–(c) were prepared in 50 mM TAPS, pH 8.0, 100 mM Na2SO4 buffer. | ||
Mixing the wild-type proteins CA and KDGA overnight at room temperature without the linker did not induce any spectral changes (Fig. 2b). However, mixing KDGA*-E288C|BzS5 with CA (CA/KDGA) in equal monomeric ratios immediately generated an opaque solution with white precipitates. When we mixed the samples at 4 °C for 15 min to reduce nonspecific aggregation, the solution was transparent but yielded protein complexes with an increased hydrodynamic diameter of 13 ± 0.60 nm (Fig. 2c). The size was comparable to the simulated value (11 nm) using the programs AutoDock Vina,44 HADDOCK,45 and WinHydroPro10, assuming that all added CA proteins bound to a homotetramer of KDGA*-E288C (Fig. 1d and S4†). Therefore, these results indicate that CA and KDGA, two structurally and functionally unrelated proteins, form heterooligomers via specific interactions between BzS5 and the Zn cofactor in CA.
We investigated whether protein assembly could be reversed by adding CA inhibitors, such as 4-carboxybenzenesulfonamide (CBzS). Notably, we used borate buffer (pH 8.0) because CBzS and CA alone created small yet detectable aggregates in TAPS, but not in borate buffer (Fig. S5†); heteroassembly was similarly observed in borate buffer with a slight size increase (14 ± 0.11 nm), possibly due to a buffer-specific effect in determining hydrodynamic diameters46 (Fig. 2d). The addition of CBzS reverted the size of the pre-assembled heterooligomers back to 9.2 ± 0.21 nm, reporting that CBzS competes with the BzS5 linker for CA binding and reversibly releases the KDGA component. The reversibility of heterooligomer self-assembly and disassembly are akin to how small molecules transmit chemical signals to macromolecules and regulate intermolecular interactions in biological systems.
Characterization of CA/KDGA heterooligomeric enzymes
The molecular interactions between CA and its inhibitor function as the primary driving force for protein self-assembly and determine the fractions of bound CA in the heteroassembly. Thus, the hydrolytic activity of unbound CA with p-nitrophenyl acetate (pNPA)47 could be a sensitive and accurate readout to quantitatively analyse how many protein components participate in self-assembly and how strongly these PPIs occur (Fig. 3a). The specific activity and catalytic efficiency of the wild-type protein CA towards pNPA were measured to be 5.3 ± 0.34 μmol min−1 mg−1 and 2500 ± 290 M−1 s−1, respectively (Fig. S6†), consistent with previous reports.48 The addition of unmodified wild-type KDGA protein did not affect the catalytic activity of CA.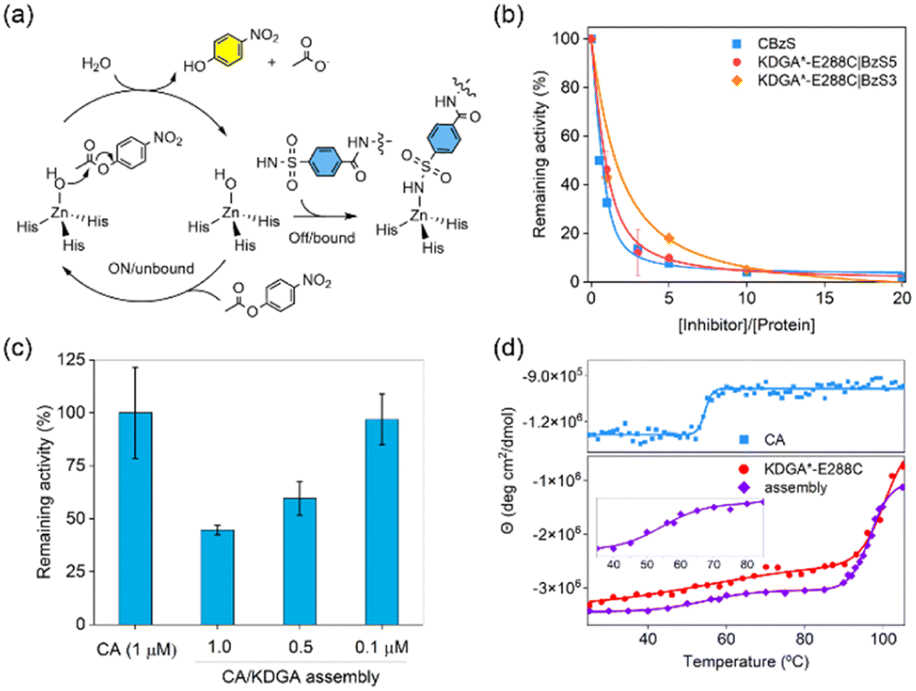 | ||
Fig. 3 Quantitative analysis of heterooligomeric protein self-assembly. (a) A reaction scheme of CA with pNPA before (catalytically on/inhibitor-unbound state) and after (off/bound) the addition of BzS-containing molecules. (b) The specific activity of CA (1.0 μM) with pNPA upon the addition of CBzS or KDGA*-E288C-conjugated with BzS5 or BzS3 linkers. (c) The specific activity of CA or 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixing of the CA/KDGA assemblies at various concentrations. (d) Temperature-dependent circular dichroism analysis of CA, KDGA*-E288C, and the 1:1 ratio of the assembled species (5 μM monomer in total). :1 mixing of the CA/KDGA assemblies at various concentrations. (d) Temperature-dependent circular dichroism analysis of CA, KDGA*-E288C, and the 1:1 ratio of the assembled species (5 μM monomer in total). | ||
When KDGA*-E288C|BzS5 (1 μM) was added at a 1:1 monomer ratio to CA, esterase activity was reduced by 54% (Fig. 3b). The incomplete shutdown of CA activity can be attributed to the modest binding affinity of the BzS and 10-fold lower protein concentration than in DLS experiments described above. A 5-fold excess of KDGA*-E288C|BzS5 to CA led to 90% inactivation of the esterase activity, which led to the estimated Ki value of BzS5 attached to the KDGA protein to be 370 ± 53 nM, using the Dynafit.49 This number is only marginally altered from the Ki value of CBzS (170 ± 37 nm), suggesting that the molecular functionalization and attachment to a macromolecule barely perturbed the binding affinity to the CA as shown previously.50–52 Based on these quantitative analyses, we estimated that 94% of the CA and KDGA*-E288C|BzS5 proteins would participate in the heteroassembly when 10 μM of each monomer was mixed for the DLS experiments shown in Fig. 2c.
Next, we replaced the CA with other isoforms to determine whether the binding affinity of BzS can be quantitatively translated to protein oligomerization. CA1 or CA12 have 60% and 36% sequence identity to CA, respectively, and exhibit significantly different binding affinities (Ki values) to a BzS analog, p-azo benzenesulfonamide: 1015 nM, 92 nM, and 87 nM in CA1, CA, and CA12, respectively.53 The mixing of KDGA*-E288C|BzS5 protein with CA12, having a comparable binding affinity to the BzS-like molecule, yielded a similar size growth to CA. In contrast, CA1, with at least a 10-fold weaker binding affinity than CA, led to a negligible alteration in the hydrodynamic diameter of the protein upon mixing with KDGA*-E288C|BzS5 (Fig S7†). These results indicate that the binding affinity of the BzS moiety serves as the primary driving force for protein self-assembly. These data also suggest that heteroassembly occurs with high selectivity for protein building blocks. When we switched to the shorter linker (BzS3), KDGA*-E288C|BzS3 exhibited a lower level of CA inhibition, resulting in an estimated Ki value of 1.2 ± 0.06 μM (Fig. 3b). These data suggest that BzS5 exhibits the optimal length to accommodate the formation of an asymmetric CA–KDGA interface for self-assembly. In addition, we measured the hydrolytic activities of the CA/KDGA assembly after serial dilutions (Fig. 3c). Presumably due to the reversibility of BzS-driven PPIs, the net catalytic activities were inversely proportional to the final protein concentrations and consistent with the simulation using the Ki value. These results indicate that the association and dissociation of the heteroassembly are reversible and kinetically labile.
Furthermore, we assessed the secondary structures and thermal stabilities of the protein components and self-assembled heterooligomers by circular dichroism (CD) spectroscopy. Each protein component (CA and KDGA*-E288C) showed α-helical structures, and the spectrum was retained after protein heterooligomerization (Fig. S8a†), suggesting that the flexible linker enforced the two proteins in proximity with no significant structural perturbation. Temperature-dependent CD spectral changes at 210 nm demonstrated that CA and KDGA showed melting temperatures (Tm) at 54.5 °C and 96.7 °C, respectively, consistent with the reported values (Fig. 3d).54,55 The mixing of CA and KDGA*-E288C|BzS5, where 68% of the BzS is expected to be bound to the CA, revealed two discrete Tm values, 54.7 °C and 100 °C. Because DLS data showed no temperature dependence below the Tm of CA (Fig. S8b†), these data suggest that each component protein retained its native thermal stability and behaved independently while being physically connected. These results contrast with other designed protein assemblies with increased thermal stability.56–58 The distinction could be attributed to the discrete assembly mechanism; BzS5 containing ethylene glycol chains provides structural flexibility and sufficient distance between the two component proteins. Thus, this unique feature could benefit the formation of self-assembled protein architectures where their individual biochemical properties need to be preserved in an additive manner.
Self-assembly of dimeric CA and KDGA enzymes
To diversify protein heterooligomers, we designed a dimer of CA (dCA) by placing a cysteine residue at the E187 position (E187C) while removing the pre-existing cysteine (C206S) (Fig. 4a).59 The double mutant was obtained as a mixture of monomers and dimers (Fig. S9b and c†), and treatment with 5 molar equiv. of cupric chloride60,61 to the CA monomer, followed by size exclusion chromatography led to the isolation of dCA (Fig. S9†). It showed the hydrodynamic diameter of 8.5 ± 0.52 nm (Fig. 4b), comparable with the calculated value from the manually docked dimer structure using the PyMOL program (7.5 nm).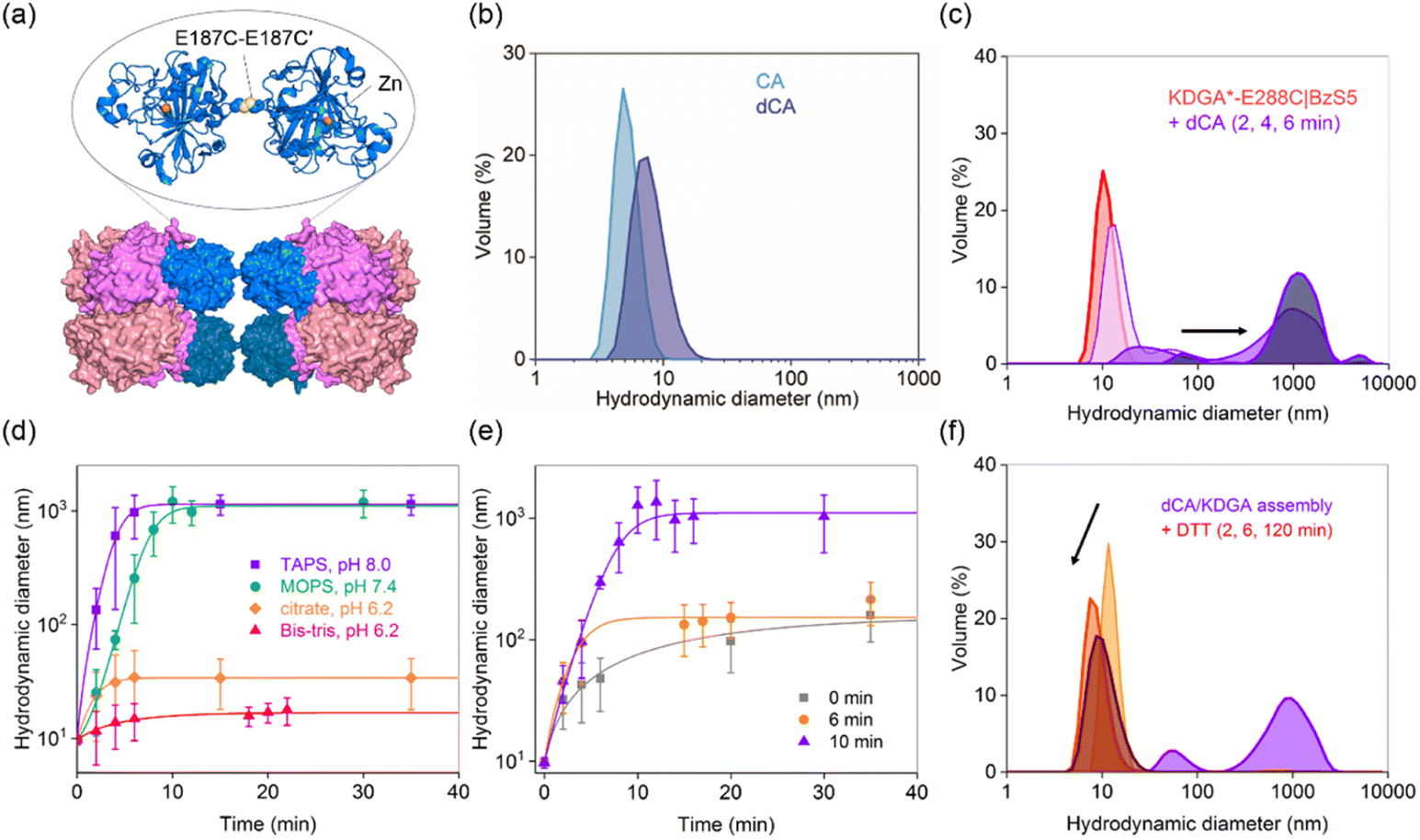 | ||
| Fig. 4 Redesign of dimeric CA (dCA) and its heterooligomer with BzS5-conjugated KDGA. (a) A model of dCA and KDGA*-E288C|BzS5 assembly. DLS data of (b) dCA (10 μM) and (c) dCA/KDGA assembly with KDGA*-E288C|BzS5 in an equimolar ratio (10 μM monomer) before and after mixing for 2–6 min. (d) Buffer-dependent dCA/KDGA assembly. (e) dCA/KDGA assembly in TAPS buffer (pH 8.0) upon the addition of citrate at various time points (final pH = 7.9). (f) Disassembly of dCA/KDGA assembly upon adding DTT (10 mM). | ||
Next, we mixed dCA and KDGA*-E288C|BzS5 (dCA/KDGA) in TAPS pH 8.0 buffer at an equivalent monomer ratio, which immediately generated large species with a hydrodynamic diameter of 1200 ± 140 nm (Fig. 4c). Prolonged incubation ultimately led to the formation of white protein precipitates. The analogous dCA/KDGA assembly was observed in pH 7.4 buffer (Fig. 4d), but not in pH 6.2 citrate and Bis–Tris buffers; the size of the assembled products was reduced to 20 ± 0.64 nm, which roughly corresponds to the size of two dCA binding to one KDGA*-E288C|BzS5 tetramer. These data suggest that the pH-dependent BzS binding affinity to CA62 is operative as the primary force for PPI formation.
We further investigated the dCA/KDGA assembled products by measuring the hydrolytic activities with pNPA (Fig. S6 and S10†); dCA alone showed comparable activity to that of the wild-type monomeric protein, and the addition of KDGA without the linker did not perturb the esterase activity of dCA. However, KDGA*-E288C|BzS5 reduced the activity of dCA, with its binding affinity calculated to be 290 ± 18 nM. The value was comparable to that of monomeric CA, supporting that PPIs between dCA and KDGA occur through a direct coordination of the BzS group to the zinc ion with similar degrees of thermodynamic driving forces to those with CA/KDGA.
Previous studies reported that a metal-chelator, reducing agent, or protease can trigger protein disassembly as external stimuli.63–65 Similarly, we explored whether such changes could regulate the heteroassembly. The addition of 13 mM citrate (26% v/v) prior to or during dCA/KDGA assembly in TAPS buffer significantly perturbed the PPI formation (Fig. 4e). Because the net pH change was minimal (pH 7.9), these results were attributed to citrate acting as a competing metal-coordinating ligand.66 Thus, citrate presumably functioned both as an acidic buffer component and a metal chelator, when we initiated the self-assembly in 50 mM citrate, pH 6.2, 100 mM Na2SO4 buffer (Fig. 4d). In contrast, no size reduction was observed, when citrate was added to the fully assembled product in TAPS buffer (Fig. 4e). It is possible that the heteroassembly might subsequently form nonspecific hierarchical structures and lower the accessibility of citrate into the active site pockets of CA.
In contrast, reducing agents, such as dithiothreitol (DTT), can reduce the size of the pre-mixed dCA and KDGA*-E288C|BzS5 in TAPS buffer (Fig. 4f). The fraction of large species grown for 4 min decreased significantly and dissociated into each component protein, suggesting that the disulfide bond in dCA or thiol–ene bioconjugation was reverted, possibly reflecting the reducing environments. This dynamic and responsive feature is also reminiscent of how chemical stimuli regulate natural macromolecular assembly and disassembly.
Heteroassembly of other building block proteins
Next, we replaced KDGA with two alternative components, a 279 kDa homohexameric aminoglycoside acetyltransferase (AT) from Bacillus anthracis67 and an 1850 kDa homo-60-meric encapsulin (Enc) from Thermotoga maritima, as the self-assembly partner for dCA (Fig. 5a). We previously used AT for homooligomeric assembly68 and metalloenzyme design,69 benefiting from its thermal stability and global symmetry. Enc also exhibited excellent thermal stability.70,71 More importantly, both proteins differ from KDGA in size (7, 12, and 21 nm in diameter), symmetry (D2, D3, and I), and shape (square, triangular, and cage), allowing us to validate the scope and versatility of the current study. The X-ray crystal structure of AT indicated that the K44 position is suitable for BzS5 attachment (Fig. 5a and S11a†) owing to its exposure and distance from the subunit interfaces. Thus, we introduced a K44C mutation, followed by the removal of pre-existing cysteine residues (C133S, C139S, and C244S mutations, AT*-K44C). Enc possesses three native cysteine residues (C123, C141, and C197), although only C123 is exposed to the surface and is available for bioconjugation (Fig. S12a and Table S5†). Therefore, we used the wild-type protein Enc without any modification. | ||
| Fig. 5 Expanded heterooligomeric protein assemblies. (a) The size comparison of 2-keto-3-deoxygluconate aldolase (KDGA, PDB code 1W37), aminoglycoside acetyltransferase (AT, PDB code 3N7Z) and encapsulin (Enc, PDB code 7MU1). K44 residue in AT and the pre-existing C123 residue in Enc are colored in yellow and used for the bio-conjugation with BzS5. DLS data upon the mixing of dCA with (b) AT*-K44C|BzS5 and (c) Enc|BzS5. TEM images and modelled structures of dCA complexed with (d) AT*-K44C|BzS5 and (e) Enc|BzS5. The scale bars in (d) and (e) indicate 20 nm. | ||
The hydrodynamic diameters of the bioconjugated AT and Enc alone were 11 ± 0.22 nm and 26 ± 0.81 nm, respectively (Fig. 5b and c). Coincubation of AT or Enc with dCA led to no perturbation in size when a bifunctional linker was absent (Fig. S13†). However, AT*-K44C|BzS5 and Enc|BzS5 (dCA/AT and dCA/Enc, respectively) led to the formation of large species with hydrodynamic diameters of 1100 ± 550 nm and 1000 ± 160 nm, respectively. These data indicate that the macromolecular interaction and formation of heterooligomers is spontaneous and rapid, and the building blocks can be readily replaced to create novel and diverse protein-based architectures using the identical bifunctional linker.
Finally, we conducted transmission electron microscopy (TEM) of dCA/AT and dCA/Enc heterooligomers. We incubated dCA and AT*-K44C|BzS5 and Enc|BzS5 overnight at 4 °C in TAPS buffer. Although the rapid formation of white precipitates and ∼100-fold sample dilution for grid deposition might have perturbed the size and structure of self-assembled species, several TEM images revealed the heterooligomers, such as three AT linked with five or six dCA proteins (Fig. 5d) and one or two Enc associated with two or four dCA proteins (Fig. 5e). The snapshots demonstrated how two structurally and functionally unrelated proteins spatially pair via specific chemical interactions. Given that the molecular weight of the linker is ∼8000 times smaller than that of the resulting heterooligomeric protein complexes, these data reflect the selective and robust Zn-coordinate covalent interaction of BzS binding to dCA assisted by the flexible and sufficiently long linker. Although the resulting protein complexes are heterogeneous in length and size, and further investigation in improving the linker design is required, our study demonstrates that a rationally designed bifunctional linker can effectively induce heterooligomers of various proteins in a readily exchangeable, repetitive, and reproducible manner.
Conclusion
We have demonstrated a versatile and efficient method for constructing a wide-range of two-component protein self-assemblies by utilizing an inhibitor-derived bifunctional linker. Each end of the linker serves as a specific molecular adaptor, facilitating connections between two protein components. The adaptability of this approach allows for an easy substitution of protein components, as long as they exhibit comparable binding affinity to the inhibitor or possess surface-exposed cysteine residues. Consequently, we successfully generated four distinct combinations of protein assemblies using a single bifunctional linker and four structurally and functionally distinct proteins, all without the need to optimize the protein–protein interfaces for each pair. This study significantly broadens the repertoire of self-assembled protein architectures with high precision and practicality. This work also highlighted the potential of merging synthetic and biochemical components to diversify artificial protein complexes, opening up a wide array of applications in the design of protein-based functional materials. With the ongoing discovery of small-molecule libraries that interact with specific proteins or enzymes with more variations in binding affinity, function, and structural rigidity, bifunctional linkers can be readily and extensively applied to more diverse protein components for various applications, such as biosensors, biocatalysts, and the construction of high-order hierarchical structures.Data availability
Biochemical experimental procedures, synthetic procedures and characterization of the bifunctional linker, ESI figures and tables, and NMR spectra of the synthesized molecules are available in the ESI.†Author contributions
S. Son: conceptualization, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing. W. J. Song: conceptualization, funding acquisition, project administration, supervision, validation, writing – original draft, writing – review & editing. S. S. and W. J. S. designed the project, S. S. performed the experiments, and S. S. and W. J. S. wrote the paper.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Creative-pioneering re-searchers program from Seoul National University (SNU), National Research Foundation (NRF) from Korea government (NRF-2022R1A2C4001207). The authors are also grateful to Prof. Byeong-Hyeok Sohn and Saero Kim for the guidance in TEM experiments and Prof. Hyun-Woo Rhee and Chang Ryul Choi for HR-ESI-MS experiments.Notes and references
- S. Jones and J. M. Thornton, Principles of protein–protein interactions, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13–20 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Self-assembly at all scales, Science, 2002, 295, 2418–2421 CrossRef CAS.
- S. L. Speer, C. J. Stewart, L. Sapir, D. Harries and G. J. Pielak, Macromolecular crowding is more than hard-core repulsions, Ann. Rev. Biophys., 2022, 51, 267–300 CrossRef CAS.
- R. Pricer, J. E. Gestwicki and A. K. Mapp, From fuzzy to function: the new frontier of protein–protein interactions, Acc. Chem. Res., 2017, 50, 584–589 CrossRef CAS PubMed.
- I. M. A. Nooren and J. M. Thornton, Diversity of protein–protein interactions, EMBO J., 2003, 22, 3486–3492 CrossRef CAS PubMed.
- J. R. Perkins, I. Diboun, B. H. Dessailly, J. G. Lees and C. Orengo, Transient protein–protein interactions: structural, functional, and network properties, Structure, 2010, 18, 1233–1243 CrossRef CAS PubMed.
- O. Keskin, N. Tuncbag and A. Gursoy, Predicting protein–protein interactions from the molecular to the proteome level, Chem. Rev., 2016, 116, 4884–4909 CrossRef CAS PubMed.
- C. M. Crews, Targeting the undruggable proteome: the small molecules of my dreams, Chem. Biol., 2010, 17, 551–555 CrossRef CAS.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Proteome-wide covalent ligand discovery in native biological systems, Nature, 2016, 534, 570–574 CrossRef CAS.
- V. Köhler, Y. M. Wilson, M. Dürrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knörr, D. Häussinger, F. Hollmann, N. J. Turner and T. R. Ward, Synthetic cascades are enabled by combining biocatalysts with artificial metalloenzymes, Nat. Chem., 2013, 5, 93–99 CrossRef.
- D. G. G. McMillan, S. J. Marritt, M. A. Firer-Sherwood, L. Shi, D. J. Richardson, S. D. Evans, S. J. Elliott, J. N. Butt and L. J. C. Jeuken, Protein–protein interaction regulates the direction of catalysis and electron transfer in a redox enzyme complex, J. Am. Chem. Soc., 2013, 135, 10550–10556 CrossRef CAS.
- T. Tamura and I. Hamachi, Recent progress in design of protein-based fluorescent biosensors and their cellular applications, ACS Chem. Biol., 2014, 9, 2708–2717 CrossRef CAS PubMed.
- A. Quijano-Rubio, H.-W. Yeh, J. Park, H. Lee, R. A. Langan, S. E. Boyken, M. J. Lajoie, L. Cao, C. M. Chow, M. C. Miranda, J. Wi, H. J. Hong, L. Stewart, B.-H. Oh and D. Baker, De novo design of modular and tunable protein biosensors, Nature, 2021, 591, 482–487 CrossRef CAS PubMed.
- J. Zhu, N. Avakyan, A. Kakkis, A. M. Hoffnagle, K. Han, Y. Li, Z. Zhang, T. S. Choi, Y. Na, C.-J. Yu and F. A. Tezcan, Protein assembly by design, Chem. Rev., 2021, 121, 13701–13796 CrossRef CAS PubMed.
- C. Lv, X. Zhang, Y. Liu, T. Zhang, H. Chen, J. Zang, B. Zheng and G. Zhao, Redesign of protein nanocages: the way from 0D, 1D, 2D to 3D assembly, Chem. Soc. Rev., 2021, 50, 3957–3989 RSC.
- L. Shao, J. Ma, J. L. Prelesnik, Y. Zhou, M. Nguyen, M. Zhao, S. A. Jenekhe, S. V. Kalinin, A. L. Ferguson, J. Pfaendtner, C. J. Mundy, J. J. De Yoreo, F. Baneyx and C.-L. Chen, Hierarchical materials from high information content macromolecular building blocks: construction, dynamic interventions, and prediction, Chem. Rev., 2022, 122, 17397–17478 CrossRef CAS PubMed.
- B. Maity, M. Taher, S. Mazumdar and T. Ueno, Artificial metalloenzymes based on protein assembly, Coord. Chem. Rev., 2022, 469, 214593 CrossRef CAS.
- A. Ledesma-Fernandez, S. Velasco-Lozano, J. Santiago-Arcos, F. López-Gallego and A. L. Cortajarena, Engineered repeat proteins as scaffolds to assemble multi-enzyme systems for efficient cell-free biosynthesis, Nat. Commun., 2023, 14, 2587 CrossRef CAS PubMed.
- J. C. Sinclair, K. M. Davies, C. Vénien-Bryan and M. E. M. Noble, Generation of protein lattices by fusing proteins with matching rotational symmetry, Nat. Nanotechol., 2011, 6, 558–562 CrossRef CAS.
- J. W. Soon, K. Oohora and T. Hayashi, A disulphide bond-mediated hetero-dimer of a hemoprotein and a fluorescent protein exhibiting efficient energy transfer, RSC Adv., 2022, 12, 28519–28524 RSC.
- W. H. Jeong, H. Lee, D. H. Song, J.-H. Eom, S. C. Kim, H.-S. Lee, H. Lee and J.-O. Lee, Connecting two proteins using a fusion alpha helix stabilized by a chemical cross linker, Nat. Commun., 2016, 7, 11031 CrossRef CAS PubMed.
- R. Wang, S. Qiao, L. Zhao, C. Hou, X. Li, Y. Liu, Q. Luo, J. Xu, H. Li and J. Liu, Dynamic protein self-assembly driven by host–guest chemistry and the folding–unfolding feature of a mutually exclusive protein, Chem. Commun., 2017, 53, 10532–10535 RSC.
- J. B. Bale, S. Gonen, Y. Liu, W. Sheffler, D. Ellis, C. Thomas, D. Cascio, T. O. Yeates, T. Gonen, N. P. King and D. Baker, Accurate design of megadalton-scale two-component icosahedral protein complexes, Science, 2016, 353, 389–394 CrossRef CAS.
- Z. Chen, S. E. Boyken, M. Jia, F. Busch, D. Flores-Solis, M. J. Bick, P. Lu, Z. L. VanAernum, A. Sahasrabuddhe, R. A. Langan, S. Bermeo, T. J. Brunette, V. K. Mulligan, L. P. Carter, F. DiMaio, N. G. Sgourakis, V. H. Wysocki and D. Baker, Programmable design of orthogonal protein heterodimers, Nature, 2019, 565, 106–111 CrossRef CAS PubMed.
- L. J. M. Lemmens, J. A. L. Roodhuizen, T. F. A. de Greef, A. J. Markvoort and L. Brunsveld, Designed asymmetric protein assembly on a symmetric scaffold, Angew. Chem., Int. Ed., 2020, 59, 12113–12121 CrossRef CAS.
- J. Laniado, K. A. Cannon, J. E. Miller, M. R. Sawaya, D. E. McNamara and T. O. Yeates, Geometric lessons and design strategies for nanoscale protein cages, ACS Nano, 2021, 15, 4277–4286 CrossRef CAS PubMed.
- J. C. T. Carlson, S. S. Jena, M. Flenniken, T. Chou, R. A. Siegel and C. R. Wagner, Chemically controlled self-assembly of protein nanorings, J. Am. Chem. Soc., 2006, 128, 7630–7638 CrossRef CAS PubMed.
- Y. Hitomi, T. Hayashi, K. Wada, T. Mizutani, Y. Hisaeda and H. Ogoshi, Interprotein electron transfer reaction regulated by an artificial interface, Angew. Chem., Int. Ed., 2001, 40, 1098–1101 CrossRef CAS PubMed.
- K. Oohora, S. Burazerovic, A. Onoda, Y. M. Wilson, T. R. Ward and T. Hayashi, Chemically programmed supramolecular assembly of hemoprotein and streptavidin with alternating alignment, Angew. Chem., Int. Ed., 2012, 51, 3818–3821 CrossRef CAS.
- F. Sakai, G. Yang, M. S. Weiss, Y. Liu, G. Chen and M. Jiang, Protein crystalline frameworks with controllable interpenetration directed by dual supramolecular interactions, Nat. Commun., 2014, 5, 4634 CrossRef CAS PubMed.
- M. Volgraf, P. Gorostiza, R. Numano, R. H. Kramer, E. Y. Isacoff and D. Trauner, Allosteric control of an ionotropic glutamate receptor with an optical switch, Nat. Chem. Biol., 2006, 2, 47–52 CrossRef CAS.
- J. A. Day and S. M. Cohen, Investigating the selectivity of metalloenzyme inhibitors, J. Med. Chem., 2013, 56, 7997–8007 CrossRef CAS PubMed.
- C. Sheng, G. Dong, Z. Miao, W. Zhang and W. Wang, State-of-the-art strategies for targeting protein–protein interactions by small-molecule inhibitors, Chem. Soc. Rev., 2015, 44, 8238–8259 RSC.
- M. Békés, D. R. Langley and C. M. Crews, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef.
- S. L. Schreiber, The rise of molecular glues, Cell, 2021, 184, 3–9 CrossRef CAS.
- S. Zheng, Y. Tan, Z. Wang, C. Li, Z. Zhang, X. Sang, H. Chen and Y. Yang, Accelerated rational PROTAC design via deep learning and molecular simulations, Nat. Mach. Intell., 2022, 4, 739–748 CrossRef.
- C. T. Supuran, Carbonic anhydrases: novel therapeutic applications for inhibitors and activators, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS.
- J. Zhao, A. Kajetanowicz and T. R. Ward, Carbonic anhydrase II as host protein for the creation of a biocompatible artificial metathesase, Org. Biomol. Chem., 2015, 13, 5652–5655 RSC.
- R. Mehta, M. H. Qureshi, M. K. Purchal, S. M. Greer, S. Gong, C. Ngo and E. L. Que, A new probe for detecting zinc-bound carbonic anhydrase in cell lysates and cells, Chem. Commun., 2018, 54, 5442–5445 RSC.
- C. B. O'Herin, Y. W. Moriuchi, T. A. Bemis, A. J. Kohlbrand, M. D. Burkart and S. M. Cohen, Development of human carbonic anhydrase II heterobifunctional degraders, J. Med. Chem., 2023, 66, 2789–2803 CrossRef PubMed.
- V. M. Krishnamurthy, G. K. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein–ligand binding, Chem. Rev., 2008, 108, 946–1051 CrossRef CAS.
- A. Theodossis, H. Walden, E. J. Westwick, H. Connaris, H. J. Lamble, D. W. Hough, M. J. Danson and G. L. Taylor, The structural basis for substrate promiscuity in 2-keto-3-deoxygluconate aldolase from the Entner-Doudoroff pathway in Sulfolobus solfataricus, J. Biol. Chem., 2004, 279, 43886–43892 CrossRef CAS.
- A. Ortega, D. Amorós and J. G. de la Torre, Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models, Biophys. J., 2011, 101, 892–898 CrossRef CAS PubMed.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Computational protein–ligand docking and virtual drug screening with the AutoDock suite, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS PubMed.
- C. Dominguez, R. Boelens and A. M. J. J. Bonvin, HADDOCK: A protein–protein docking approach based on biochemical or biophysical information, J. Am. Chem. Soc., 2003, 125, 1731–1737 CrossRef CAS.
- S. Brudar and B. Hribar-Lee, Effect of buffer on protein stability in aqueous solutions: a simple protein aggregation model, J. Phys. Chem. B, 2021, 125, 2504–2512 CrossRef CAS PubMed.
- J. A. Verpoorte, S. Mehta and J. T. Edsall, Esterase activities of human carbonic anhydrases B and C, J. Biol. Chem., 1967, 242, 4221–4229 CrossRef CAS PubMed.
- J. F. Krebs, J. A. Ippolito, D. W. Christianson and C. A. Fierke, Structural and functional importance of a conserved hydrogen bond network in human carbonic anhydrase II, J. Biol. Chem., 1993, 268, 27458–27466 CrossRef CAS.
- P. Kuzmič, Program DYNAFIT for the analysis of enzyme kinetic data: Application to HIV proteinase, Anal. Biochem., 1996, 237, 260–273 CrossRef.
- D. P. Martin, Z. S. Hann and S. M. Cohen, Metalloprotein–inhibitor binding: human carbonic anhydrase II as a model for probing metal–ligand interactions in a metalloprotein active site, Inorg. Chem., 2013, 52, 12207–12215 CrossRef CAS PubMed.
- A. Jain, S. G. Huang and G. M. Whitesides, Lack of effect of the length of oligoglycine-derived and oligo(ethylene-glycol)-derived para-substituents on the affinity of benzenesulfonamides for carbonic anhydrase II in solution, J. Am. Chem. Soc., 1994, 116, 5057–5062 CrossRef CAS.
- K. Aggarwal, T. P. Kuka, M. Banik, B. P. Medellin, C. Q. Ngo, D. Xie, Y. Fernandes, T. L. Dangerfield, E. Ye, B. Bouley, K. A. Johnson, Y. J. Zhang, J. K. Eberhart and E. L. Que, Visible light mediated bidirectional control over carbonic anhydrase activity in cells and in vivo using azobenzenesulfonamides, J. Am. Chem. Soc., 2020, 142, 14522–14531 CrossRef CAS PubMed.
- D. Pagnozzi, N. Pala, G. Biosa, R. Dallocchio, A. Dessì, P. K. Singh, D. Rogolino, A. Di Fiore, G. De Simone, C. T. Supuran and M. Sechi, Interaction studies between carbonic anhydrase and a sulfonamide inhibitor by experimental and theoretical approaches, ACS Med. Chem. Lett., 2022, 13, 271–277 CrossRef CAS.
- C. L. Buchanan, H. Connaris, M. J. Danson, C. D. Reeve and D. W. Hough, An extremely thermostable aldolase from Sulfolobus solfataricus with specificity for non-phosphorylated substrates, Biochem. J., 1999, 343, 563–570 CrossRef CAS.
- B. S. Avvaru, S. A. Busby, M. J. Chalmers, P. R. Griffin, B. Venkatakrishnan, M. Agbandje-McKenna, D. N. Silverman and R. McKenna, Apo-human carbonic anhydrase II revisited: implications of the loss of a metal in protein structure, stability, and solvent network, Biochemistry, 2009, 48, 7365–7372 CrossRef CAS PubMed.
- J. D. Brodin, J. R. Carr, P. A. Sontz and F. A. Tezcan, Exceptionally stable, redox-active supramolecular protein assemblies with emergent properties, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2897–2902 CrossRef CAS PubMed.
- L. Zhao, Y. Li, T. Wang, S. Qiao, X. Li, R. Wang, Q. Luo, C. Hou, J. Xu and J. Liu, Photocontrolled protein assembly for constructing programmed two-dimensional nanomaterials, J. Mater. Chem. B, 2018, 6, 75–83 RSC.
- W. Bai, C. J. Sargent, J.-M. Choi, R. V. Pappu and F. Zhang, Covalently-assembled single-chain protein nanostructures with ultra-high stability, Nat. Commun., 2019, 10, 3317 CrossRef PubMed.
- E. T. Mack, P. W. Snyder, R. Perez-Castillejos and G. M. Whitesides, Using covalent dimers of human carbonic anhydrase II to model bivalency in immunoglobulins, J. Am. Chem. Soc., 2011, 133, 11701–11715 CrossRef CAS PubMed.
- I. S. M. Lee, M. Suzuki, N. Hayashi, J. Hu, L. J. Van Eldik, K. Titani and M. Nishikimi, Copper-dependent formation of disulfide-linked dimer of S100B protein, Arch. Biochem. Biophys., 2000, 374, 137–141 CrossRef.
- G. Y. Kreitman, J. C. Danilewicz, D. W. Jeffery and R. J. Elias, Copper(II)-mediated hydrogen sulfide and thiol oxidation to disulfides and organic polysulfanes and their reductive cleavage in wine: mechanistic elucidation and potential applications, J. Agric. Food Chem., 2017, 65, 2564–2571 CrossRef CAS PubMed.
- P. W. Taylor, R. W. King and A. S. V. Burgen, Influence of pH on the kinetics of complex formation between aromatic sulfonamides and human carbonic anhydrase, Biochemistry, 1970, 9, 3894–3902 CrossRef CAS.
- E. Golub, R. H. Subramanian, J. Esselborn, R. G. Alberstein, J. B. Bailey, J. A. Chiong, X. Yan, T. Booth, T. S. Baker and F. A. Tezcan, Constructing protein polyhedra via orthogonal chemical interactions, Nature, 2020, 578, 172–176 CrossRef CAS.
- Y. Suzuki, G. Cardone, D. Restrepo, P. D. Zavattieri, T. S. Baker and F. A. Tezcan, Self-assembly of coherently dynamic, auxetic, two-dimensional protein crystals, Nature, 2016, 533, 369–373 CrossRef CAS.
- J. E. Miller, Y. Srinivasan, N. P. Dharmaraj, A. Liu, P. L. Nguyen, S. D. Taylor and T. O. Yeates, Designing protease-triggered protein cages, J. Am. Chem. Soc., 2022, 144, 12681–12689 CrossRef CAS.
- J. E. Coleman, Mechanism of action of carbonic anhydrase: substrate, sulfonamide, and anion binding, J. Biol. Chem., 1967, 242, 5212–5219 CrossRef CAS PubMed.
- K. D. Green, T. Biswas, C. Chang, R. Wu, W. Chen, B. K. Janes, D. Chalupska, P. Gornicki, P. C. Hanna, O. V. Tsodikov, A. Joachimiak and S. Garneau-Tsodikova, Biochemical and structural analysis of an Eis family aminoglycoside acetyltransferase from Bacillus anthracis, Biochemistry, 2015, 54, 3197–3206 CrossRef CAS PubMed.
- M. Yang and W. J. Song, Diverse protein assembly driven by metal and chelating amino acids with selectivity and tunability, Nat. Commun., 2019, 10, 5545 CrossRef CAS.
- S.-M. Jung, M. Yang and W. J. Song, Symmetry-adapted synthesis of dicopper oxidases with divergent dioxygen reactivity, Inorg. Chem., 2022, 61, 12433–12441 CrossRef CAS PubMed.
- B. J. LaFrance, C. Cassidy-Amstutz, R. J. Nichols, L. M. Oltrogge, E. Nogales and D. F. Savage, The encapsulin from Thermotoga maritima is a flavoprotein with a symmetry matched ferritin-like cargo protein, Sci. Rep., 2021, 11, 22810 CrossRef CAS.
- I. Boyton, S. C. Goodchild, D. Diaz, A. Elbourne, L. E. Collins-Praino and A. Care, Characterizing the dynamic disassembly/reassembly mechanisms of encapsulin protein nanocages, ACS Omega, 2022, 7, 823–836 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Biochemical experimental procedures, synthetic procedures and characterization of the bifunctional linker, supplementary figures and tables, and NMR spectra of the synthesized molecules. See DOI: https://doi.org/10.1039/d3sc05448a |
| This journal is © The Royal Society of Chemistry 2024 |