
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMono- and bis-Pd(II) complexes of N-confused dithiahexaphyrin(1.1.1.1.1.0) with the absorption and aromaticity modulated by Pd(II) coordination, macrocycle contraction and ancillary ligands†
Meng
Sun
a,
Yongshu
Xie
 *ab,
Glib
Baryshnikov
c,
Chengjie
Li
a,
Feng
Sha
a,
Xinyan
Wu
a,
Hans
Ågren
d,
Shijun
Li
b and
Qizhao
Li
*a
*ab,
Glib
Baryshnikov
c,
Chengjie
Li
a,
Feng
Sha
a,
Xinyan
Wu
a,
Hans
Ågren
d,
Shijun
Li
b and
Qizhao
Li
*a
aKey Laboratory for Advanced Materials, Frontiers Science Center for Materiobiology and Dynamic Chemistry, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: yshxie@ecust.edu.cn; zhaolq@ecust.edu.cn
bCollege of Material, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou, 311121, China
cDepartment of Science and Technology, Laboratory of Organic Electronics, Linköping University, SE-601 74 Norrköping, Sweden
dDepartment of Physics and Astronomy, Uppsala University, SE-751 20 Uppsala, Sweden
First published on 3rd January 2024
Abstract
To further enrich the coordination chemistry of hexaphyrins and probe the underlying property–structural correlations, N-confused dithiahexaphyrin(1.1.1.1.1.0) (1) with 26 π-electron Hückel aromaticity was synthesized. Based on its unprecedented two unsymmetrical cavities, five palladium complexes 2, 3, 4-Ph, 4-Cl and 5 have been successfully synthesized under various coordinations. Thus, two mono-Pd(II) complexes 2 and 3 with the Pd(II) atom coordinated in the two different cavities were obtained by treating 1 with palladium reagents PdCl2, and (PPh3)2PdCl2 respectively. On this basis, bis-Pd(II) complexes 4-Ph and 4-Cl were synthesized by treating 2 and 3 with (PPh3)2PdCl2 and PdCl2, respectively. As a result, both 4-Ph and 4-Cl contain two Pd(II) atoms coordinated within the two cavities, with one of the Pd(II) atoms further coordinated to a triphenylphosphine ligand in addition to an anionic ancillary ligand of Ph− and Cl−, respectively. Notably, a further contracted mono-Pd(II) complex 5 was synthesized by treating 1 with Pd(PPh3)4 by eliminating one of the meso-carbon atoms together with the corresponding C6F5 moiety. These complexes present tunable 26 π aromaticity and NIR absorption up to 1060 nm. This work provides an effective approach for developing distinctive porphyrinoid Pd(II) complexes from a single porphyrinoid, without resorting to tedious syntheses of a series of porphyrinoid ligands.
Introduction
As a class of tetrapyrrole macrocyclic compounds, porphyrins and their metal complexes, namely, metalloporphyrins exist widely in nature and play important roles in materials science and life activities, owing to their rich photophysical, electronic and catalytic properties.1–6 In recent years, the tetrapyrrolic frameworks of porphyrins have been modified to afford various porphyrin analogues termed as porphyrinoids.7–21 Thus, N-confused porphyrins,10,11 core-modified porphyrins22–27 and expanded porphyrins28–30 have been developed by incorporating an α,β′-linked pyrrole, using heteroatoms like O, S and Se in place of the N atoms, and employing more than four pyrrolic units, respectively (Fig. 1a–c). As a result, these porphyrinoids exhibit much richer coordination behavior. For example, hexaphyrins may coordinate to either one or two metal ions within the two cavities, and the coordinated metal ions exhibit rich oxidation states.31–48 In addition, the N-confused pyrrole unit may trigger interesting macrocycle transformations and thus further modulate the coordination behavior.49–55 These metal complexes are promising for applications in various areas like catalytic, photoelectric and biomedical materials.56–67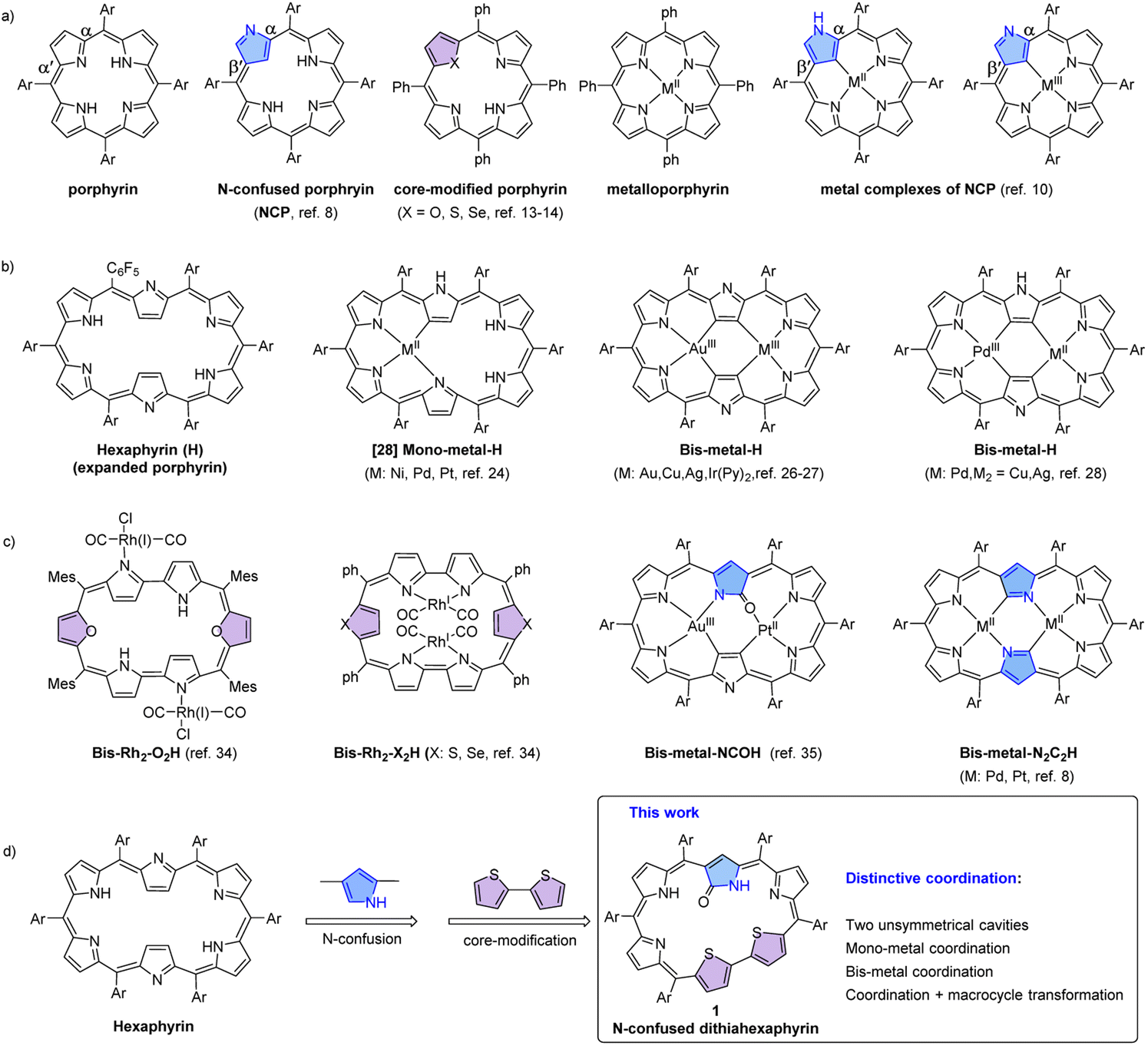 | ||
| Fig. 1 Chemical structures of free bases and metal complexes of porphyrin and its tetrapyrrolic analogues (a), hexaphyrin (b) and its analogues (c), and N-confused dithiahexaphyrin (d). | ||
To further enrich the coordination chemistry of hexaphyrins and probe the underlying property–structural correlations for porphyrinoid complexes, herein we report N-confused dithiahexaphyrin 1, which has been synthesized by simultaneously incorporating an N-confused pyrrole unit and a bithiophene unit into a hexaphyrin framework (Fig. 1d). As a result, 1 possesses two coordination cavities with different sizes and coordinating atoms employable for metal coordination. Considering that rich structural diversity has been reported for Pd(II) complexes of porphyrinoids involving Pd(II)–C and Pd(II)–N coordination bonds,50,51 and the presence of the Pd(II)–C bonds may facilitate cross-coupling reactions and macrocycle interconversions,68–701 was treated with various palladium reagents to afford a series of Pd(II) complexes. Interestingly, five mono-Pd(II) and bis-Pd(II) complexes 2, 3, 4-Ph, 4-Cl and 5 have been synthesized. Thus, successive coordination of 1 with PdCl2 and (PPh3)2PdCl2 in CH2Cl2 and MeOH afforded 2 and 4-Ph, respectively. Interestingly, when the reaction sequence was reversed, complexes 3 and 4-Cl were obtained, respectively. As a result, the Pd(II) atom in the mono-Pd(II) complexes 2 and 3 is coordinated in the two different cavities, respectively. On the other hand, different ancillary anionic ligands Ph− and Cl− are observed for bis-Pd(II) complexes 4-Ph and 4-Cl, respectively. Notably, the treatment of 1 with Pd(PPh3)4 in CH2Cl2 afforded a third mono-Pd(II) complex 5, with the macrocycle further contracted by eliminating one of the meso-carbon atoms and the corresponding C6F5 moiety. These results provide examples for synthesizing unprecedented palladium complexes with tunable structures and properties from a single porphyrinoid ligand with the advantages of avoiding tedious syntheses of a series of porphyrinoid ligands.
Results and discussion
Initially, N-confused dithiahexaphyrin(1.1.1.1.1.0) (1) was conveniently synthesized as a violet solid in the yield of 12% by condensing N-confused bilane with bithiophene diol in the presence of BF3·Et2O, followed by oxidation with DDQ (see ESI†). The high-resolution mass spectrometry (HRMS) of 1 shows a pseudo molecular ion peak at m/z = 1332.0127 ([M]+, calcd for C59H13F25N4OS2: 1332.0126) (Fig. S2†), indicative of a dithiahexaphyrin(1.1.1.1.1.0) structure with the attachment of an oxygen atom (Scheme 1). With 1 in hand, it was coordinated with Pd(II) under various conditions. When 1 was coordinated with the respective Pd(II) reagents of PdCl2 and (PPh3)2PdCl2 in a mixed solvent of CH2Cl2/MeOH, mono-Pd complexes 2 and 3 were obtained as blue and dark blue solids in the yields of 45% and 24%, respectively (Scheme 1). The HRMS of 2 and 3 reveals pseudo molecular ion peaks at m/z = 1435.9018 ([M]+, calcd for C59H11F25N4OPdS2: 1435.9023) (Fig. S3†) and m/z = 1733.9787 ([M]+, calcd for C77H27ClF25N4OPPdS2: 1733.9784) (Fig. S4†), respectively, indicative of mono-Pd(II) coordination structures of 2 and 3 and additional ancillary coordination in 3 (vide infra). On this basis, it may be anticipated that the mono-Pd(II) complexes can be further coordinated with alternative Pd(II) reagents to afford the corresponding bis-Pd(II) complexes. Thus, 2 and 3 were further coordinated with (PPh3)2PdCl2 and PdCl2, respectively. As a result, bis-Pd(II) complexes 4-Ph and 4-Cl were obtained as green solids in the yields of 31% and 37%, respectively. Although 4-Ph can also be obtained by treating 2 with Pd(PPh3)4, and 4-Cl can also be obtained by treating 3 with other palladium salts like Pd(PPh3)4, (PPh3)2PdCl2 and Pd(OAc)2, the yields are all very low. The HRMS of bis-Pd complex 4-Ph reveals a molecular ion peak at m/z = 1879.9299 ([M]+, calcd for C83H30F25N4OPPd2S2: 1879.9297) (Fig. S5†), consistent with a bis-Pd complex structure containing ancillary PPh3 and phenyl ligands. Whereas, the pseudo molecular ion peak observed for 4-Cl at m/z = 1887.8573 ([M]+, calcd for C77H25ClF25N4OPPd2S2: 1887.8588) reveals a bis-Pd structure with ancillary PPh3 and chloro ligands (Fig. S6†). The only structural difference between 4-Ph and 4-Cl lies in their ancillary ligands, but they could not be transformed into each other even after repeated trials, indicative of strong coordination of the ancillary anionic ligand, which is thus difficult to substitute. Inspired by the syntheses of mono-Pd(II) complexes 2 and 3 by employing different Pd(II) reagents, 1 was further coordinated with additional Pd(II) reagents. Interestingly, when Pd(PPh3)4 was used to coordinate with 1 in CH2Cl2, another mono-Pd(II) complex 5 were obtained as purple solid in the yield of 15%. The HRMS of 5 reveals a pseudo molecular ion peak at m/z = 1257.9174 ([M]+, calcd for C52H12F20N4OPdS2: 1257.9179) (Fig. S7†), which reveals the elimination of a C7F5 moiety, compared with 2. These data indicate that the hexaphyrin ligand has been further contracted by eliminating one of the meso-carbon atoms as well as the corresponding C6F5 moiety on the basis of the mono-Pd(II) complex 2. Based on these results, a plausible reaction mechanism has been proposed for the generation of 5 (Fig. S8†), which involves the coordination of a Pd(0) atom within the cavity together with a PPh3 ligand. Similar to the synthesis of 5 from the reaction of Pd(PPh3)4 with 1 in CH2Cl2, 5 was also detected when PPh3 was added to the solution of 1 and Pd(OAc)2 in CH2Cl2. In contrast, 5 could not detected when PPh3 was replaced by more bulky phosphine ligands like BINAP, Sphos, (tBu)3PHBF4, and DPEphos. These results indicate that the coordination of the Pd(0) atom together with the bulky ligand may be hampered by serious steric hindrance induced by the bulky ligand. Considering that complex 3 still possesses a noncoordinated cavity similar to that of free base 1, it could be anticipated that 3 may undergo a reaction similar to that of 1 to afford a contracted product similar to 5. Thus, complex 3 was treated with Pd(PPh3)4, but no contracted product could be obtained, which may be related to the difficulty in Pd(0) coordination because the cavity cannot be easily adapted to satisfy the coordination requirement of Pd(0) due to the more rigid structure of complex 3 than that of free base 1. This inference is also evidenced by the very low yield of 4-Cl obtained from the coordination of 3 with Pd(PPh3)4 (vide supra). Although complex 5 still possesses a noncoordinated cavity, no bis-Pd(II) complex could be obtained from 5, which may be related to the insufficient size of the contracted structure.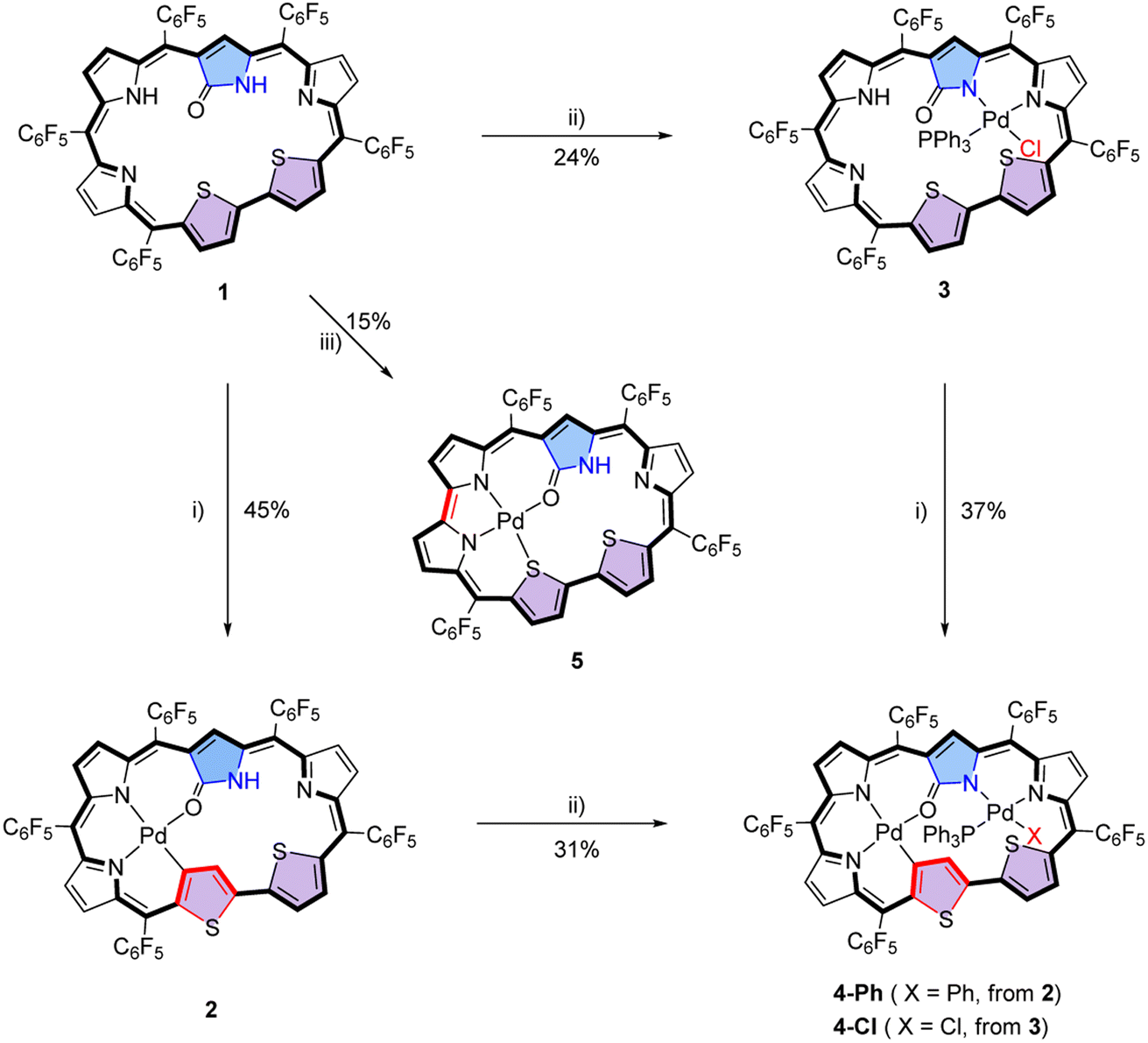 | ||
| Scheme 1 Syntheses of complexes 2, 3, 4-Ph/4-Cl and 5, conditions: (i) PdCl2 in CH2Cl2/MeOH with NaOAc; (ii) (PPh3)2PdCl2 in CH2Cl2/MeOH with NaOAc; and (iii) Pd(PPh3)4 in CH2Cl2. | ||
Consistent with the HRMS data of 1 (vide supra), its 1H NMR spectrum in CDCl3 shows two NH signals in the relatively high field region (δ = 1.28 and 1.50 ppm) and eleven CH protons in the relatively low field region (δ = 8.34–10.70 ppm) (Fig. S11†), indicative of an aromatic macrocycle.71 Consistently, single crystal X-ray diffraction analysis reveals that 1 possesses a dithiahexaphyrin(1.1.1.1.1.0) macrocycle containing three normal pyrroles, an N-confused pyrrole and a bithiophene unit, with an oxygen atom attached to the α-position of the confused pyrrole C (Fig. 2a, b and S23†), which is also consistent with the HRMS data (vide supra). The C1–O1 bond length of 1.208 Å is typical for a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond. Within the macrocycle, H1 and H2 are hydrogen-bonded to O1 and N3, with N1⋯O1 and N2⋯N3 distances of 2.766 and 2.715 Å, respectively.
O double bond. Within the macrocycle, H1 and H2 are hydrogen-bonded to O1 and N3, with N1⋯O1 and N2⋯N3 distances of 2.766 and 2.715 Å, respectively.
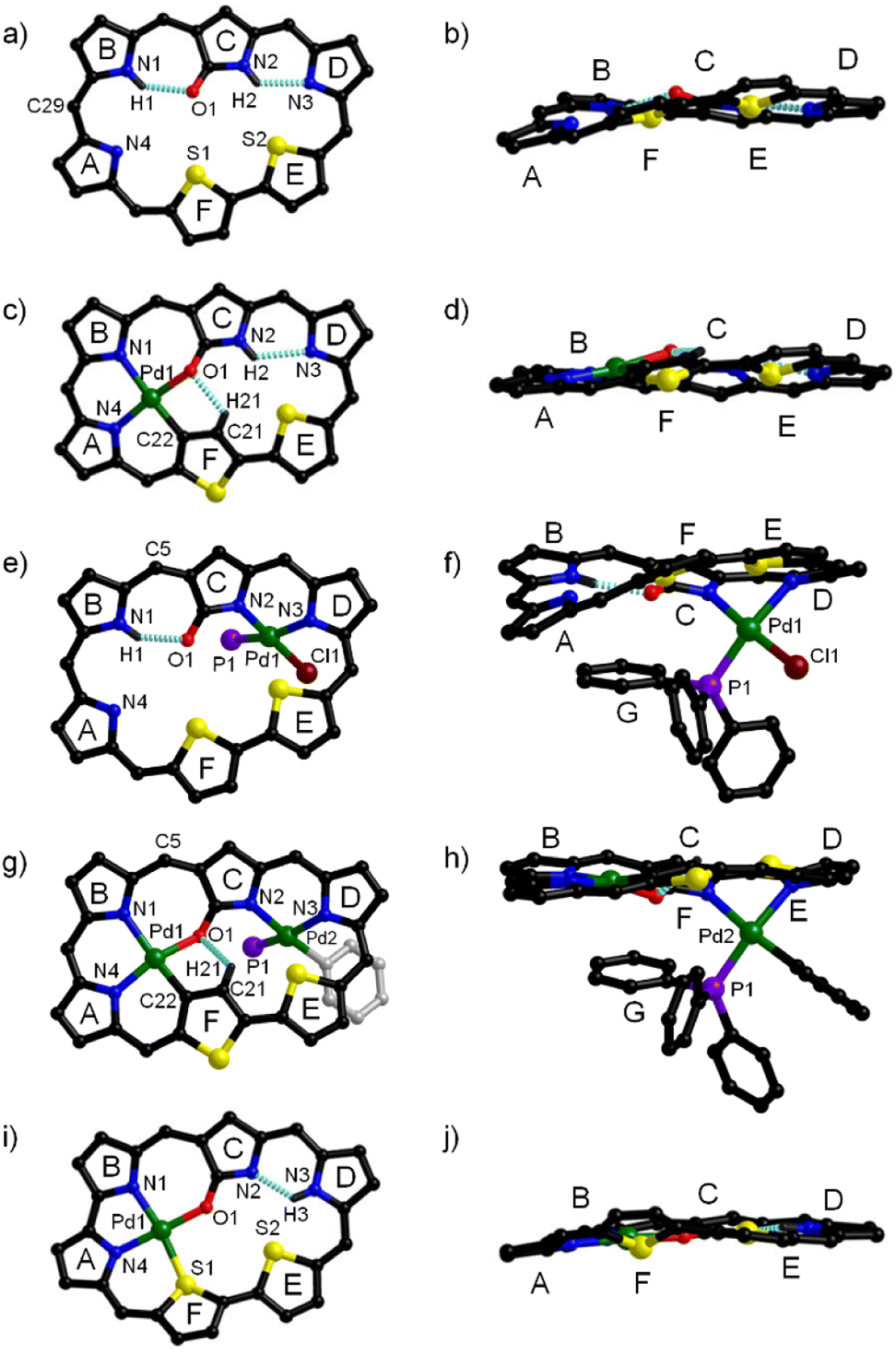 | ||
| Fig. 2 Complementary views of the molecular structures of 1 (a) and (b), 2 (c) and (d), and 3 (e) and (f), 4-Ph (g) and (h), and 5 (i) and (j). For clarity, all the C6F5 groups, solvents, and the hydrogen atoms attached to the carbon atoms are omitted. In addition, the phenyl groups in the PPh3 ligands are omitted in figures (e) and (g). The dotted lines represent intramolecular hydrogen bonds. | ||
The 1H NMR spectrum of Pd(II) complex 2 in CDCl3 (Fig. S13†) shows nine CH signals in the low field region of δ = 8.93–10.50 ppm, an NH proton in the high field region at δ = −0.93, and a CH proton at δ = −2.85. Compared with free base 1, 2 exhibits one fewer inner NH, two fewer peripheral CH protons and one more inner CH, indicating that one NH is deprotonated, and one of the heterocycles is inverted with one of its β-CH moieties deprotonated upon coordination with Pd(II). This inference is consistent with the crystal structure (vide infra). The 1H NMR spectrum of 3 exhibits eleven peripheral CH protons in the low field region (δ = 8.46–11.28 ppm) and one NH signal at δ = 0.28 ppm (Fig. S15†), indicating that one of the NH moieties in 1 has been deprotonated. Meanwhile, fifteen additional protons are observed with δ = 4.29–6.23 ppm, indicating the coordination of a PPh3 ligand in the shielding region of the dithiahexaphyrin macrocycle, which is consistent with the crystal structure (vide infra). Consistent with the HRMS and NMR data, the crystal structure of 2 unambiguously reveals the coordination of a palladium atom within the cavity composed of rings A, B, C and F. Notably, thiophene ring F is inverted with one of the C–H moieties coordinated in the deprotonated form, and the other C–H remains intact within the macrocycle (Fig. 2c, d and S24†). The coordination bond lengths lie in the range of 1.980–2.086 Å. Within the macrocycle, H2 and H21 are hydrogen-bonded to N3 and O1, with N2⋯N3 and O1⋯C21 distances of 2.839 and 2.735 Å, respectively. In contrast to 2, the crystal structure of 3 reveals the coordination of Pd(II) in another cavity composed of rings C, D, E, and F, with the Pd1 atom coordinated to the N2 and N3 from pyrroles C and D, respectively, in addition to two auxiliary ligands of Cl− and PPh3 (Fig. 2e, f and S25†). The distances of Pd1–N2, Pd1–N3, Pd1–P1 and Pd1–Cl lie in the range of 2.010–2.289 Å. Within the macrocycle, H1 is hydrogen-bonded to O1, with the N1⋯O1 distance of 2.680 Å. Notably, a PPh3 ligand is located in the shielding facial zone, which is consistent with the 1H NMR data (vide supra).
The 1H NMR spectrum of bis-Pd complex 4-Ph shows nine CH protons in the low field region (δ = 9.01–10.75 ppm) and one CH proton at δ = −3.78 ppm (Fig. S17†), indicating that further coordination of 2 with the second Pd(II) atom is accompanied with the deprotonation of an NH moiety within the second cavity. Notably, twenty protons appear in the region of δ = 2.27–6.38 ppm, assignable to an ancillary phenyl ligand in addition to a PPh3 ligand in the shielding zone, which is consistent with the HRMS data (vide supra). The 1H NMR spectrum of 4-Cl shows nine CH in the low field region (δ = 9.10–10.66 ppm) and one CH proton in the high field region (δ = −4.08 ppm) (Fig. S19†), which is similar to that of 4-Ph, indicating that the dithiahexaphyrin ligand in 4-Cl coordinates in the mode identical to that in 4-Ph. However, only fifteen protons appear in the region of δ = 4.22–6.23 ppm, which is consistent with the coordination of a chloro ligand instead of the phenyl ligand in 4-Ph, as revealed by the HRMS data (vide supra). Consistent to the HRMS and NMR data, single crystal X-ray diffraction analysis reveals that 4-Ph possesses a bis-Pd coordination structure, with two Pd(II) atoms coordinated within the two cavities. Pd1 is coordinated in a NNOC environment donated by rings A, B, C and F, and the inverted thiophene ring F is coordinated in the deprotonated form like that observed in complex 2. Meanwhile, Pd2 is coordinated to two N atoms donated by pyrrole rings C and D (Fig. 2g, h and S26†). In addition, Pd2 is also coordinated to an ancillary PPh3 ligand as well as a phenyl ligand, different from that of a chloro ligand in 3. The coordination bond lengths for Pd1 and Pd2 lie in the ranges of 1.851–2.032 Å and 1.999–2.263 Å, similar to those of 2 and 3, respectively. Within the cavity, H21 is hydrogen-bonded to O1, with the C21⋯O1 distance of 2.279 Å. Obviously, the ancillary PPh3 and Ph− ligands are located in the shielding facial zone, consistent with the 1H NMR data (vide supra). Although the single crystals of 4-Cl could not be obtained, 4-Cl seems to have a bis-Pd(II) coordination structure similar to that of 4-Ph considering the similar NMR characters (vide supra), with a chloro ligand coordinated in place of the Ph− ligand.
The 1H NMR spectrum of 5 exhibits eleven CH protons in the low field region and one NH proton in the high field region (Fig. S20 and S21†), which is consistent with the mono-Pd(II) coordination structure inferred from the HRMS data (vide supra) with one meso-carbon and the corresponding C6F5 moiety eliminated. Consistently, the crystal structure of 5 reveals that the meso-carbon atom between pyrrole rings A and B and the corresponding C6F5 moiety have been eliminated, and thus these two pyrrole units are directly linked, and thus a Pd(II) atom is coordinated in an ONNS environment contributed by pyrrole rings A, B, C and thiophene ring F (Fig. 2i, j and S27†). The coordination bond lengths lie in the range of 1.906–2.331 Å. Within the macrocycle, H3 is hydrogen-bonded to N2, with the N2⋯N3 distance of 2.550 Å, which is obviously smaller than the corresponding value in 2 (2.839 Å). Similarly, the respective distances of 3.297 and 4.249 Å for S2⋯N2 and O1⋯N3 are also obviously smaller than the corresponding values of 3.476 and 4.797 Å observed for complex 2. These data indicate that elimination of the meso-carbon atom facilitates the coordination of the sulphur atom, resulting in the shrinking of the remaining noncoordinated cavity, which is unfavourable for further coordination of an additional Pd(II) atom. Consistent to this expectation, we failed to synthesize any bis-Pd complex from 5 irrespective of the palladium reagents used.
It is obvious that the conformation and planarity of the hexaphyrin macrocycles can be effectively modulated by metal coordination. To assess the planarity of the macrocycles, the root mean squared deviation (RMSD) values for all the atoms in the macrocycle can be calculated from the crystal data as a quantitative index.72,73 As a result, the RMSD value of 1 was calculated to be 0.404 Å, similar that of 0.402 Å obtained for the corresponding all-pyrrole hexaphyrin(1.1.1.1.1.0).74 Upon coordination of Pd(II) in the cavity composed of A, B, C and F rings, the RMSD value of 2 was calculated to be 0.319 Å, indicating that the coordination results in enhanced planarity. On the other hand, the coordination of Pd(II) in the other cavity results in an obviously larger RMSD value of 0.568 Å for 3, indicating more severe distortion of the macrocycle compared with that of 1, which may be related to the good flexibility of the hexaphyrin macrocycle and the steric hindrance associated with the ancillary ligands. However, the bis-Pd(II) coordination in 4-Ph results in improved planarity with the RMSD value of 0.301 Å, which is smaller than those of 2 (0.319 Å) and 1 (0.404 Å). This observation indicates that the bis-Pd(II) coordination enhances both the rigidity and the planarity of the hexaphyrin macrocycle. Upon meso-carbon elimination and coordination of the cavity composed of A, B, C and F rings, 5 exhibits the smallest RMSD value of 0.296 Å among these compounds, indicating the best planarity of the macrocycle.
Based on the crystal structure studies, the absorption spectra of the compounds were measured in CH2Cl2 (Fig. 3), which exhibit aromatic features and near-infrared (NIR) absorption. The absorption spectrum of 1 exhibits an intense Soret-like band at 552 nm and two weak Q-like bands at 679 nm and 835 nm, with the band edge of ca. 1000 nm. The unsymmetrical mono-Pd(II) complexes 2 and 3 exhibit split Soret-like bands and three weak Q-like bands with the band edges extended to ca. 1050 nm. Similarly, mono-Pd(II) complex 5 also shows split Soret-like bands, but the band edge is blue-shifted to ca. 980 nm relative to those of 2 and 3, because of the contracted conjugation structure. On the other hand, the relatively more symmetrical bis-Pd complex 4-Ph exhibits an intense Soret-like band at 622 nm, and three weak Q-like bands at 760 nm, 870 nm and 984 nm. Compared with 1, the band-edge of 4-Ph is about 60 nm red-shifted with the absorption tail extended to ca. 1060 nm. Similarly, 4-Cl exhibits a similar absorption spectrum with an intense Soret-like band at 622 nm, and three weak Q-like bands at 768 nm, 862 nm and 974 nm. Compared with 1, the band-edge of 4-Cl is about 40 nm red-shifted with the absorption tail extended to ca. 1040 nm. Complexes 2, 3, 4-Ph and 4-Cl all show red-shifted absorption due to the narrower energy gaps relative to 1, which may derive from the electronic interaction between the metal atoms and the macrocyclic ligand. Furthermore, the absorption spectra of these compounds simulated by time-dependent density functional theory (TD-DFT) calculations roughly agree with the experimental results (Fig. S28–S33†).
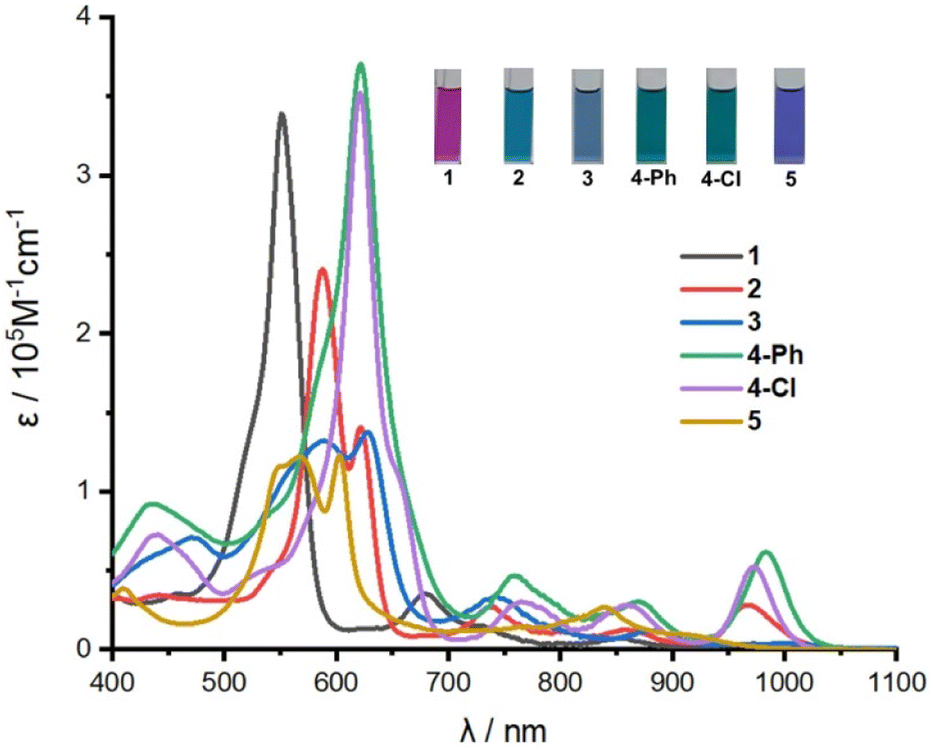 | ||
| Fig. 3 Absorption spectra of 1, 2, 3, 4-Ph, 4-Cl, 5 in CH2Cl2. The inset shows the photographs of the corresponding solutions in CH2Cl2. | ||
To further probe the aromaticity of the compounds, the anisotropy of the induced current density (ACID) plot, nucleus-independent chemical shift (NICS) and harmonic oscillator model for aromaticity (HOMA) were calculated. Thus, the ACID plots of the compounds all display clockwise ring current along the hexaphyrinoid macrocycle (Fig. S34–S39†), consistent with their 26 π aromatic characters revealed by the 1H NMR data and the crystal structures. The NICS(0) value of −10.3 obtained for 1 is less negative than that of −16.7 obtained for the corresponding all-pyrrole hexaphyrin(1.1.1.1.1.0), indicative of weakened aromaticity of 1, which can be attributed to the introduction of the weakly aromatic bithiophene unit. The NICS(0) values of palladium complexes 2, 3, 4-Ph, 4-Cl and 5 were calculated to be −13.4, −13.8, −12.8, −12.2 and −14.2 respectively, more negative than that of 1, indicating stronger aromaticity of the Pd(II) complexes (Fig. S40†). Consistently, the HOMA values of 2, 3, 4-Ph and 5 were found to be 0.62, 0.77, 0.65 and 0.84, respectively, larger than that of 0.60 calculated for 1 (Fig. S23–S27†). These results indicate that the Pd(II) coordination within the cavities of 1 are favourable for improving the planarity of the macrocycle and thus enhancing the aromatic character. Furthermore, coordination of Pd(II) within the contracted macrocycle afforded the best planarity and strongest aromaticity of 5 among all the complexes.
Notably, different from hexaphyrin(1.1.1.1.1.1) and hexaphyrin(1.1.0.1.1.0),33–36 dithiahexaphyrin(1.1.1.1.1.0) 1 exhibits a highly unsymmetrical structure due to the incorporation of N-confused pyrrole and bithiophene units, which endows 1 with diverse Pd-complexation behaviour. Different from the 28 π Möbius-aromaticity, nonaromaticity and 24 π Hückel-antiaromaticity observed for the Pd(II) complexes of hexaphyrin(1.1.1.1.1.1) and hexaphyrin(1.1.0.1.1.0), respectively,33–36 the corresponding Pd(II) complexes of 1 all show 26 π Hückel-aromaticity, which may be related to the relatively good stability of 26 π Hückel-aromaticity for the rigid skeletons incorporating the bithiophene unit. Consistently, the molar extinction coefficients of 1 and its Pd-complexes are noticeably higher than those of hexaphyrin(1.1.1.1.1.1), hexaphyrin(1.1.0.1.1.0) and their Pd-complexes.
Conclusions
In summary, we synthesized 26 π-electron aromatic N-confused dithiahexaphyrin 1 (1.1.1.1.1.0) by incorporating an N-confused pyrrole and a bithiophene into the hexaphyrin framework. Because of the presence of two unsymmetrical cavities, 1 exhibits interesting coordination behaviour, and five palladium complexes have been synthesized under various conditions. Thus, treating 1 with PdCl2 and (PPh3)2PdCl2 afforded two mono-Pd(II) complexes 2 and 3, respectively, with the Pd(II) atom coordinated in the different cavities. On this basis, bis-Pd(II) complexes 4-Ph and 4-Cl were synthesized by treating 2 and 3 with (PPh3)2PdCl2 and PdCl2, respectively. As a result, both 4-Ph and 4-Cl contain two Pd(II) atoms coordinated within the two cavities, with one of the Pd(II) atoms further coordinated to a phosphine ligand as well as an anionic ancillary ligand of Ph− and Cl−, respectively. These results indicate that the Pd(II) complexes can be modulated by the reaction sequence of the palladium reagents employed. Notably, treating 1 with Pd(PPh3)4 afforded a third mono-Pd(II) complex 5, with the Pd(II) atom coordinated within a further contracted hexaphyrin macrocycle which has been generated from 1 by eliminating one of the meso-carbon atoms and the corresponding C6F5 moiety. These complexes present 26 π aromaticity and NIR absorption up to 1060 nm, and the absorption can be effectively modulated by Pd(II) coordination, macrocycle contraction and the ancillary ligand coordination. This work may provide an effective approach for synthesizing a series of novel porphyrinoid palladium complexes with tunable structures and properties by employing a single porphyrinoid ligand, which may avoid tedious syntheses of a series of porphyrinoid ligands.Data availability
Experimental and theoretical details supporting the statements of this article are included in the ESI.†Author contributions
Conceptualization: Q. Li, Y. Xie; methodology: C. Li, F. Sha, X. Wu, S. Li; synthesis and test: M. Sun; theoretical calculations: G. Baryshnikov, H. Ågren; writing–original draft: M. Sun; writing–review & editing: Y. Xie, Q. Li.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work at ECUST was financially supported by NSFC (22131005, 21971063, 22201074, 22075077), the Fundamental Research Funds for the Central Universities, Shanghai Rising-Star Program (23QA1402100), and Natural Science Foundation of Shanghai (23ZR1415600, 22ZR1416100). G. B. thanks for the Swedish National Infrastructure for Computing (SNIC) resources at the High-Performance Computing Center North (HPC2N) and the financial support of the Swedish Research Council (2018-05973, 2020-04600). The authors thank the Research Center of Analysis and Test of East China University of Science and Technology for compound characterization.Notes and references
- D. W. Thuita and C. Brückner, Chem. Rev., 2022, 122, 7990–8052 CrossRef CAS PubMed.
- P. Jiang, T. Zhao, J. Rong, B. Yin, Y. Rao, M. Zhou, L. Xu and J. Song, Chin. Chem. Lett., 2021, 32, 2562–2566 CrossRef CAS.
- M. Hou, W. Chen, J. Zhao, D. Dai, M. Yang and C. Yi, Chin. Chem. Lett., 2022, 33, 4101–4106 CrossRef CAS.
- J. Zhang, L. Gong, X. Zhang, M. Zhu, C. Su, Q. Ma, D. Qi, Y. Bian, H. Du and J. Jiang, Chem.–Eur. J., 2020, 26, 13842–13848 CrossRef CAS PubMed.
- I. Yadav, J. K. Sharma, M. Sankar and F. D'Souza, Chem.–Eur. J., 2023, 29, e202301341 CrossRef CAS PubMed.
- S.-Y. Liu, N. Kishida, J. Kim, N. Fukui, R. Haruki, Y. Niwa, R. Kumai, D. Kim, M. Yoshizawa and H. Shinokubo, J. Am. Chem. Soc., 2023, 145, 2135–2141 CrossRef CAS PubMed.
- T. Chatterjee, V. S. Shetti, R. Sharma and M. Ravikanth, Chem. Rev., 2017, 117, 3254–3328 CrossRef CAS PubMed.
- M. Toganoh and H. Furuta, Chem. Rev., 2022, 122, 8313–8437 CrossRef CAS PubMed.
- R. Misra and T. K. Chandrashekar, Acc. Chem. Res., 2008, 41, 265–279 CrossRef CAS PubMed.
- H. Maeda, Y. Ishikawa, T. Matsuda, A. Osuka and H. Furuta, J. Am. Chem. Soc., 2003, 125, 11822–11823 CrossRef CAS PubMed.
- P. J. Chmielewski, L. Latos-Grażyński and T. Glowiak, Angew. Chem., Int. Ed. Engl., 1994, 33, 779–781 CrossRef.
- N. Tripathi, A. Sinha and M. Ravikanth, Org. Biomol. Chem., 2023, 21, 6617–6623 RSC.
- F. D'Souza, Angew. Chem., Int. Ed., 2015, 54, 4713–4714 CrossRef.
- T. D. Lash, Chem. Rev., 2017, 117, 2313–2446 CrossRef CAS PubMed.
- S. Shimizu, Chem. Rev., 2017, 117, 2730–2784 CrossRef CAS PubMed.
- Y. Matano, Chem. Rev., 2017, 117, 3138–3191 CrossRef CAS PubMed.
- T. Sarma and P. K. Panda, Chem. Rev., 2017, 117, 2785–2838 CrossRef CAS PubMed.
- T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584–2640 CrossRef CAS PubMed.
- B. Szyszko, M. J. Białek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839–2909 CrossRef CAS PubMed.
- J. Setsune, Chem. Rev., 2017, 117, 3044–3101 CrossRef CAS PubMed.
- Q. Li, M. Ishida, C. Li, G. Baryshnikov, F. Sha, B. Zhu, X. Wu, H. Ågren, H. Furuta and Y. Xie, CCS Chem., 2023, 5, 1332–1342 CrossRef CAS.
- K. Hurej, M. Pawlicki, L. Szterenberg and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2016, 55, 1427–1431 CrossRef CAS PubMed.
- A. Ghosh and M. Ravikanth, Inorg. Chem., 2012, 51, 6700–6709 CrossRef CAS PubMed.
- C.-H. Chuang, C.-K. Ou, S.-T. Liu, A. Kumar, W.-M. Ching, P.-C. Chiang, M. A. C. dela Rosa and C.-H. Hung, Inorg. Chem., 2011, 50, 11947–11957 CrossRef CAS PubMed.
- C. Brückner, Acc. Chem. Res., 2016, 49, 1080–1092 CrossRef PubMed.
- Y. Ning, G. Q. Jin and J. L. Zhang, Acc. Chem. Res., 2019, 52, 2620–2633 CrossRef CAS PubMed.
- Y. Li, M. Zhou, L. Xu, B. Zhou, Y. Rao, H. Nie, T. Gu, J. Zhou, X. Liang, B. Yin, W. Zhu, A. Osuka and J. Song, Org. Lett., 2020, 22, 6001–6005 CrossRef CAS PubMed.
- Q. C. Chen, M. Soll, A. Mizrahi, I. Saltsman, N. Fridman, M. Saphier and Z. Gross, Angew. Chem., Int. Ed., 2018, 57, 1006–1010 CrossRef CAS PubMed.
- M. G. P. M. S. Neves, R. M. Martins, A. C. Tomé, A. J. D. Silvestre, A. M. S. Silva, V. Félix, J. A. S. Cavaleiro and M. G. B. Drew, Chem. Commun., 1999, 385–386 RSC.
- T. Sarma, G. Kim, S. Sen, W.-Y. Cha, Z. Duan, M. D. Moore, V. M. Lynch, Z. Zhang, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 12111–12119 CrossRef CAS PubMed.
- S. Mori and A. Osuka, J. Am. Chem. Soc., 2005, 127, 8030–8031 CrossRef CAS PubMed.
- T. Nédellec, B. Boitrel and S. Le Gac, J. Am. Chem. Soc., 2023, 145, 27067–27079 CrossRef PubMed.
- S. Mori, S. Shimizu, R. Taniguchi and A. Osuka, Inorg. Chem., 2005, 44, 4127–4129 CrossRef CAS PubMed.
- T. Tanaka, T. Sugita, S. Tokuji, S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 6619–6621 CrossRef CAS PubMed.
- M. Inoue and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 9488–9491 CrossRef CAS PubMed.
- F. Luo, L. Liu, H. Wu, L. Xu, Y. Rao, M. Zhou, A. Osuka and J. Song, Nat. Commun., 2023, 14, 5028 CrossRef CAS PubMed.
- S. Mori and A. Osuka, Inorg. Chem., 2008, 47, 3937–3939 CrossRef CAS PubMed.
- K. Shimomura, H. Kai, Y. Nakamura, Y. Hong, S. Mori, K. Miki, K. Ohe, Y. Notsuka, Y. Yamaoka, M. Ishida, D. Kim and H. Furuta, J. Am. Chem. Soc., 2020, 142, 4429–4437 CrossRef CAS PubMed.
- A. Srinivasan, T. Ishizuka, A. Osuka and H. Furuta, J. Am. Chem. Soc., 2003, 125, 878–879 CrossRef CAS PubMed.
- K. Yamasumi, K. Nishimura, Y. Hisamune, Y. Nagae, T. Uchiyama, K. Kamitani, T. Hirai, M. Nishibori, S. Mori, S. Karasawa, T. Kato, K. Furukawa, M. Ishida and H. Furuta, Chem.–Eur. J., 2017, 23, 15322–15326 CrossRef CAS.
- T. K. Chandrashekar and S. Venkatraman, Acc. Chem. Res., 2003, 36, 676–691 CrossRef CAS PubMed.
- A. Alka, V. S. Shetti and M. Ravikanth, Coord. Chem. Rev., 2019, 401, 213063 CrossRef CAS.
- S. Gokulnath, K. Yamaguchi, M. Toganoh, S. Mori, H. Uno and H. Furuta, Angew. Chem., Int. Ed., 2011, 50, 2302–2306 CrossRef CAS PubMed.
- Y. Wang, H. Kai, M. Ishida, S. Gokulnath, S. Mori, T. Murayama, A. Muranaka, M. Uchiyama, Y. Yasutake, S. Fukatsu, Y. Notsuka, Y. Yamaoka, M. Hanafusa, M. Yoshizawa, G. Kim, D. Kim and H. Furuta, J. Am. Chem. Soc., 2020, 142, 6807–6813 CrossRef CAS PubMed.
- H. Shu, M. Guo, M. Wang, M. Zhou, B. Zhou, L. Xu, Y. Rao, B. Yin, A. Osuka and J. Song, Angew. Chem., Int. Ed., 2022, 61, e202209594 CrossRef CAS PubMed.
- Y. Liu, D. Xie, Y. Rao, L. Xu, M. Zhou, A. Osuka and J. Song, Angew. Chem., Int. Ed., 2022, 61, e202206601 CrossRef CAS PubMed.
- L. Liu, Z. Hu, F. Zhang, Y. Liu, L. Xu, M. Zhou, T. Tanaka, A. Osuka and J. Song, Nat. Commun., 2020, 11, 6206 CrossRef CAS PubMed.
- Y. Rao, L. Xu, M. Zhou, B. Yin, A. Osuka and J. Song, Angew. Chem., Int. Ed., 2022, 61, e202206899 CrossRef CAS PubMed.
- Y. Xu, B. Zhu, Q. Li, F. Sha, G. Baryshnikov, L. He, Y. Feng, J. Tang, Y. Wei, C. Li, X. Wu, H. Ågren and Y. Xie, Org. Lett., 2023, 25, 1793–1798 CrossRef CAS PubMed.
- T. Yoneda and A. Osuka, Chem.–Eur. J., 2013, 19, 7314–7318 CrossRef CAS PubMed.
- S. Gokulnath, K. Nishimura, M. Toganoh, S. Mori and H. Furuta, Angew. Chem., Int. Ed., 2013, 52, 6940–6943 CrossRef CAS PubMed.
- Q. Li, M. Ishida, H. Kai, T. Gu, C. Li, X. Li, G. Baryshnikov, X. Liang, W. Zhu, H. Ågren, H. Furuta and Y. Xie, Angew. Chem., Int. Ed., 2019, 58, 5925–5929 CrossRef CAS PubMed.
- Y. Du, B. Zhu, Q. Li, G. Baryshnikov, C. Wei, Y. Lin, G. Su, C. Li, H. Ågren and Y. Xie, Org. Lett., 2020, 22, 9648–9652 CrossRef CAS PubMed.
- G. Su, Q. Li, M. Ishida, C. Li, F. Sha, X.-Y. Wu, L. Wang, G. Baryshnikov, D. Li, H. Ågren, H. Furuta and Y. Xie, Angew. Chem., Int. Ed., 2020, 59, 1537–1541 CrossRef CAS PubMed.
- Q. Li, M. Ishida, Y. Wang, C. Li, G. Baryshnikov, B. Zhu, F. Sha, X. Wu, H. Ågren, H. Furuta and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202212174 CrossRef CAS PubMed.
- W. Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC.
- J. W. Buchler, J. Porphyrins Phthalocyanines, 2000, 04, 337–339 CrossRef CAS.
- T. D. Lash, Chem.–Asian J., 2014, 9, 682–705 CrossRef CAS PubMed.
- C. Wu, X. Li, M. Shao, J. Kan, G. Wang, Y. Geng and Y.-B. Dong, Chin. Chem. Lett., 2022, 33, 4559–4562 CrossRef CAS.
- Z. Liang, H. Guo, H. Lei and R. Cao, Chin. Chem. Lett., 2022, 33, 3999–4002 CrossRef CAS.
- B. Babu, J. Mack and T. Nyokong, Dalton Trans., 2023, 52, 5000–5018 RSC.
- C. Liu, K. Liu, C. Wang, H. Liu, H. Wang, H. Su, X. Li, B. Chen and J. Jiang, Nat. Commun., 2020, 11, 1047 CrossRef CAS PubMed.
- T. Sakurai, Y. Hiraoka, H. Tanaka, Y. Miyake, N. Fukui and H. Shinokubo, Angew. Chem., Int. Ed., 2023, 62, e202300437 CrossRef CAS PubMed.
- G. I. Vargas-Zúñiga and J. L. Sessler, Coord. Chem. Rev., 2017, 345, 281–296 CrossRef PubMed.
- J. Luo, Z. Xie, J. Zou, X. Wu, X. Gong, C. Li and Y. Xie, Chin. Chem. Lett., 2022, 33, 4313–4316 CrossRef CAS.
- J. Zou, Y. Wang, G. Baryshnikov, J. Luo, X. Wang, H. Ågren, C. Li and Y. Xie, ACS Appl. Mater. Interfaces, 2022, 14, 33274–33284 CrossRef CAS PubMed.
- C. Li, K. Zhang, M. Ishida, Q. Li, K. Shimomura, G. Baryshnikov, X. Li, M. Savage, X.-Y. Wu, S. Yang, H. Furuta and Y. Xie, Chem. Sci., 2020, 11, 2790–2795 RSC.
- A. Nakai, T. Yoneda, T. Tanaka and A. Osuka, Chem. Commun., 2021, 57, 3034–3037 RSC.
- D. Comandella, M. Werheid, F.-D. Kopinke and K. Mackenzie, Chem. Eng. J., 2017, 319, 21–30 CrossRef CAS.
- J. Zhu, J. Wood, K. Deplanche, I. Mikheenko and L. E. Macaskie, Appl. Catal., B, 2016, 199, 108–122 CrossRef CAS.
- J. Y. Shin, H. Furuta, K. Yoza, S. Igarashi and A. Osuka, J. Am. Chem. Soc., 2001, 123, 7190–7191 CrossRef CAS PubMed.
- H. Matsumoto, T. Okujima, S. Mori, A. C. C. Bacilla, M. Takase, H. Uno and N. Kobayashi, Org. Lett., 2021, 23, 3442–3446 CrossRef CAS PubMed.
- Y. Xu, B. Zhu, Q. Li, G. Baryshnikov, M. Zhou, C. Li, F. Sha, X. Wu, H. Ågren, J. Song and Y. Xie, Org. Lett., 2021, 23, 8307–8311 CrossRef CAS PubMed.
- X.-J. Zhu, S.-T. Fu, W.-K. Wong, J.-P. Guo and W.-Y. Wong, Angew. Chem., Int. Ed., 2006, 45, 3150–3154 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2277039–2277043. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05473j |
| This journal is © The Royal Society of Chemistry 2024 |