
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-spin alkylperoxo–iron(III) complex with cis-anionic ligands: implications for the superoxide reductase mechanism†
Tarali
Devi
 ab,
Kuheli
Dutta
a,
Jennifer
Deutscher
a,
Stefan
Mebs
c,
Uwe
Kuhlmann
d,
Michael
Haumann
c,
Beatrice
Cula
a,
Holger
Dau
c,
Peter
Hildebrandt
d and
Kallol
Ray
*a
ab,
Kuheli
Dutta
a,
Jennifer
Deutscher
a,
Stefan
Mebs
c,
Uwe
Kuhlmann
d,
Michael
Haumann
c,
Beatrice
Cula
a,
Holger
Dau
c,
Peter
Hildebrandt
d and
Kallol
Ray
*a
aInstitut für Chemie, Humboldt-Universitat zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany. E-mail: kallol.ray@chemie.hu-berlin.de
bDepartment of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore, Karnataka-560012, India
cDepartment of Physics, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany
dInstitut für Chemie, Technische Universität Berlin, Fakultät II, Straße des 17. Juni 135, 10623 Berlin, Germany
First published on 6th December 2023
Abstract
The N3O macrocycle of the 12-TMCO ligand stabilizes a high spin (S = 5/2) [FeIII(12-TMCO)(OOtBu)Cl]+ (3-Cl) species in the reaction of [FeII(12-TMCO)(OTf)2] (1-(OTf)2) with tert-butylhydroperoxide (tBuOOH) in the presence of tetraethylammonium chloride (NEt4Cl) in acetonitrile at −20 °C. In the absence of NEt4Cl the oxo–iron(IV) complex 2 [FeIV(12-TMCO)(O)(CH3CN)]2+ is formed, which can be further converted to 3-Cl by adding NEt4Cl and tBuOOH. The role of the cis-chloride ligand in the stabilization of the FeIII–OOtBu moiety can be extended to other anions including the thiolate ligand relevant to the enzyme superoxide reductase (SOR). The present study underlines the importance of subtle electronic changes and secondary interactions in the stability of the biologically relevant metal–dioxygen intermediates. It also provides some rationale for the dramatically different outcomes of the chemistry of iron(III)peroxy intermediates formed in the catalytic cycles of SOR (Fe–O cleavage) and cytochrome P450 (O–O bond lysis) in similar N4S coordination environments.
Introduction
Iron(III)–hydroperoxo (FeIII–OOH) and iron(III)–alkylperoxo (FeIII–OOR) species have been identified as key intermediates in heme and non-heme iron enzymes.1–10 In the non-heme enzyme superoxide reductase (SOR), the [FeIII(His)4(Cys)OOH] intermediate undergoes a proton-mediated Fe–O bond cleavage to release H2O2. In contrast, the [FeIII(protoporphyrin-IX)(Cys)OOR(H)] intermediate in the heme protein cytochrome P450 (P-450) undergoes O–O bond cleavage to generate a high-valent iron–oxido species, which is then used in a number of hydrogen atom transfer (HAT) and oxygen atom transfer (OAT) reactions. The different fate of the FeIII–OOR(H) intermediates in an otherwise similar N4S coordination frame has been rationalized based on the different spin states of the FeIII–OOR intermediates in SOR and P-450. The electron donation from a thiolate ligand in SOR is proposed to increase the lifetime of the high-spin (HS) iron(III)–hydroperoxo species to allow its protonation and subsequent release of H2O2. In contrast, the “push” of electron density from the trans-thiolate ligand is believed to assist in the O–O bond cleavage step in the low-spin (LS) iron(III)–hydroperoxo species in P-450 leading to the generation of the iron(IV)oxo porphyrin cation radical species. The speculated role of the spin-state (S = 1/2 vs. 5/2) of the iron(III)–hydroperoxo species in controlling the Fe–O vs. O–O bond cleavage step is, however, challenged in biomimetic studies, where both HS and LS FeIII–OOR(H) species are found to be amenable to O–O bond lysis in the presence of Lewis bases occupying the 6th coordination site of the Fe(III)–OOR species in both cis- and trans- orientations.11–18 Furthermore, recent theoretical19 and experimental20,21 studies have also raised the possibility of the presence of a low-spin intermediate with a weak Fe–O bond in the course of O2 reduction by SOR. Thus, the factors that account for the dramatically different outcomes of the chemistry of SOR versus P-450 in similar N4S coordination environment remain to be determined.In our effort to understand nature's rationale for using similar active sites for apparently opposite functions, we now report the synthesis and characterization of S = 5/2 [FeIII(12-TMCO)(OOR)L′]+ (R = -tBu, -Cumene; L′ = MePhS−, F−, Cl−, Br−, OTf−; 12-TMCO = 4,7,10-trimethyl-1-oxa-4,7,10-triazacyclododecane) and S = 1 [FeIV(12-TMCO)(O)(CH3CN)]2+ complexes supported by the macrocyclic 12-TMCO ligand involving a N3O coordination. In particular, the ligand donation from the cis-anionic ligands like chloride or aryl thiolates and the H-bonding interaction mediated by 12-TMCO are shown to be prerequisites for the stabilization of [FeIII(12-TMCO)(OOR)L′]+. The present study provides a rationale for the presence of a cis- (rather than the speculated trans-) thiolate ligation in SOR and highlights the importance of subtle electronic changes in the stability and reactivity of the biologically relevant metal–dioxygen intermediates.
Results and discussion
Combining the tetradentate 12-TMCO ligand with Fe(OTf)2(CH3CN)2 yielded the iron(II) precursor complex [FeII(12-TMCO)(OTf)2] (1-(OTf)2). The X-ray structure of 1-(OTf)2 (Scheme 1, inset) displays a distorted octahedral geometry with two cis-positions occupied by anionic triflate (–OTf−) ligands. Notably, the smaller ring size of the 12-TMCO ligand forces the iron(II) center out of the equatorial plane involving the three nitrogen atoms of 12-TMCO and an oxygen atom of one of the –OTf− ligands. The etheral oxygen atom of 12-TMCO and the second –OTf ligand occupy the axial binding sites. The macrocyclic ring adopts a conformation in which all of the methyl groups are arranged cis to the axial –OTf− ligand, with iron–ligand bond lengths (Table S1a; for crystallographic information see Table S1b; ESI†) typical of a high-spin (HS) iron(II) complex. The zero-field Mössbauer spectrum of 1-(OTf)2 (Fig. S1†) revealed a single doublet with an isomer shift (δ) of 1.17 mm s−1 and a large quadrupole splitting (ΔEQ) of 3.38 mm s−1, which is also consistent with the presence of HS S = 2 Fe(II) center in 1-(OTf)2. In CH3CN solution, the two –OTf− ligands are replaced by CH3CN to form 1-(CH3CN)2, as evident from 19F-NMR (Fig. S2†) and extended X-ray absorption fine structure (EXAFS) measurements performed on a solution of 1-(OTf)2 in CH3CN (Fig. S3b and c, Table S2†). A chloride-bound complex, 1-Cl was also synthesized combining FeClBF4 with 12-TMCO (Experimental section, ESI).† The formation of 1-Cl was structurally confirmed by X-ray absorption studies (XAS) (Fig. S3†). EXAFS analysis of a solution of 1-Cl in CH3CN, (Fig. S3b, c and Table S2†) revealed close to one Cl scatterer at 2.32 Å and a further shell of three N and one O scatterers at 2.14 and 2.04 Å, respectively (corresponding to the N/O donors of 12-TMCO).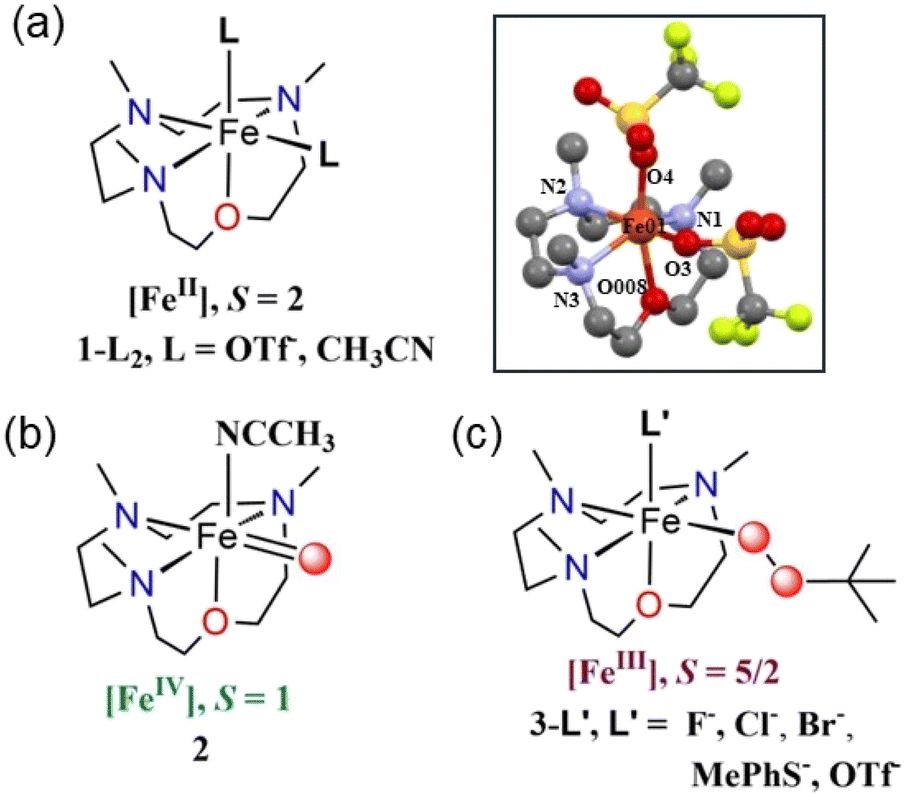 | ||
| Scheme 1 Representative chemical structures of the species (a) 1-L2, (b) 2, and (c) 3-L′. Inset shows the XRD-determined molecular structure of 1-(OTf)2. Hydrogen atoms are removed for clarity. The selected bond distances: Fe–N(1) = 2.215(3) Å, Fe–N(2) = 2.223(3) Å, Fe–N(3) = 2.196(3) Å, Fe–O(008) = 2.167(2) Å, Fe–O(3) = 2.131(2) Å, Fe–O(4) = 2.045(2) Å. See Table S1a† for detailed bond lengths and bond angles. Color code: C: gray; N: blue; O: red; Fe: orange. | ||
The reaction of 1-(CH3CN)2 with excess tBuOOH in CH3CN at −20 °C affords a light green chromophore (2; t1/2 = 1.5 h at −20 °C) with absorption maxima (λmax) centered at 770 nm (ε = 142 M−1 cm−1) and 935 nm (ε = 170 M−1 cm−1). The conversion of 1-(CH3CN)2 to 2 takes place via the intermediate formation of a transient species 3-CH3CN with λmax ≈ 517 nm (Fig. 1a). Further characterizations of 2 were carried out by a variety of spectroscopic methods. The zero-field Mössbauer spectrum of 2 (Fig. 1b; ΔEQ = 1.37 mm s−1, δ = 0.01 mm s−1) is consistent with the presence of an FeIV center in the S = 1 ground-state. Analysis of the EXAFS data of 2 in CH3CN solution (Fig. S3b and c, Table S2†) yields about one oxygen ligand at 1.64 Å (assigned to the Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O unit) aside from the 4 further N/O ligands (at ∼2.05 Å and ∼1.90 Å) of the 12-TMCO group. The Fe K-edge spectrum of 2 (Fig. 2a) reveals a pronounced pre-edge amplitude at ca. 7114.3 eV and a K-edge energy of 7124.3 eV, which are typical for FeIVO complexes.22–27 The resonance Raman (rR) spectrum obtained with 406 nm excitation exhibits a band at 861 cm−1, assigned to the FeO stretching mode [ν(FeO)] (Fig. S4† and 2c). This band disappears upon decay of 2 (data not shown).28 Thus, in the absence of any anionic ligation, the HS FeII center in 1-(CH3CN)2 performs O–O bond homolysis to yield an [FeIV(TMCO)(O)(CH3CN)]2+ complex 2, presumably via the transient formation of [FeIII(TMCO)(OOtBu)(CH3CN)]2+ (3-CH3CN). The homolytic O–O bond cleavage mechanism is further confirmed in the reaction of 1-(CH3CN)2 in CH3CN at −20 °C with cumenehydroperoxide (CumOOH), which is often used as a mechanistic probe.29–33 Complex 2 is obtained in near-stoichiometric yield in the reaction (Fig. S5†) and the gas chromatographic analysis revealed the formation of acetophenone (Fig. S6†) confirming the homolytic O–O bond cleavage mechanism.29–33 No generation of cumylalcohol expected from O–O bond heterolysis was observed. DFT calculations on 2 (Fig. 3a, Table S3a†) predict an S = 1 ground-state with calculated FeO bond distance (1.60 Å), and stretching mode frequency (867 cm−1), in good agreement with experiments (Table S4†).
O unit) aside from the 4 further N/O ligands (at ∼2.05 Å and ∼1.90 Å) of the 12-TMCO group. The Fe K-edge spectrum of 2 (Fig. 2a) reveals a pronounced pre-edge amplitude at ca. 7114.3 eV and a K-edge energy of 7124.3 eV, which are typical for FeIVO complexes.22–27 The resonance Raman (rR) spectrum obtained with 406 nm excitation exhibits a band at 861 cm−1, assigned to the FeO stretching mode [ν(FeO)] (Fig. S4† and 2c). This band disappears upon decay of 2 (data not shown).28 Thus, in the absence of any anionic ligation, the HS FeII center in 1-(CH3CN)2 performs O–O bond homolysis to yield an [FeIV(TMCO)(O)(CH3CN)]2+ complex 2, presumably via the transient formation of [FeIII(TMCO)(OOtBu)(CH3CN)]2+ (3-CH3CN). The homolytic O–O bond cleavage mechanism is further confirmed in the reaction of 1-(CH3CN)2 in CH3CN at −20 °C with cumenehydroperoxide (CumOOH), which is often used as a mechanistic probe.29–33 Complex 2 is obtained in near-stoichiometric yield in the reaction (Fig. S5†) and the gas chromatographic analysis revealed the formation of acetophenone (Fig. S6†) confirming the homolytic O–O bond cleavage mechanism.29–33 No generation of cumylalcohol expected from O–O bond heterolysis was observed. DFT calculations on 2 (Fig. 3a, Table S3a†) predict an S = 1 ground-state with calculated FeO bond distance (1.60 Å), and stretching mode frequency (867 cm−1), in good agreement with experiments (Table S4†).
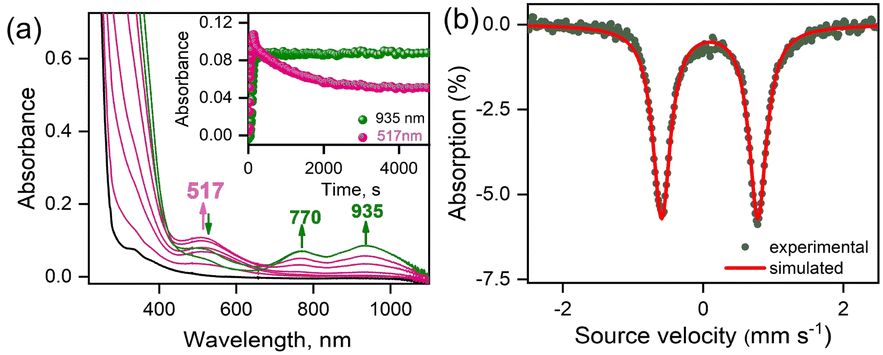 | ||
| Fig. 1 (a) UV-visible spectral changes observed in the formation of 2 (green line) after the oxidation of 1-(CH3CN)2 (0.50 mM, black line) by tBuOOH (20 equiv.) in CH3CN at 253 K. The intermediate spectra are shown in pink. Inset shows the time profiles monitored at 935 nm due to the formation of 2 and at 517 nm for the concomitant formation and decay of the preequilibrium species 3-CH3CN. (b) The zero-field Mössbauer spectrum of 2 with isomer shift, δ = 0.01 mm s−1 and quadrupole splitting, ΔEQ = 1.37 mm s−1 recorded at 4.2 K in CH3CN. | ||
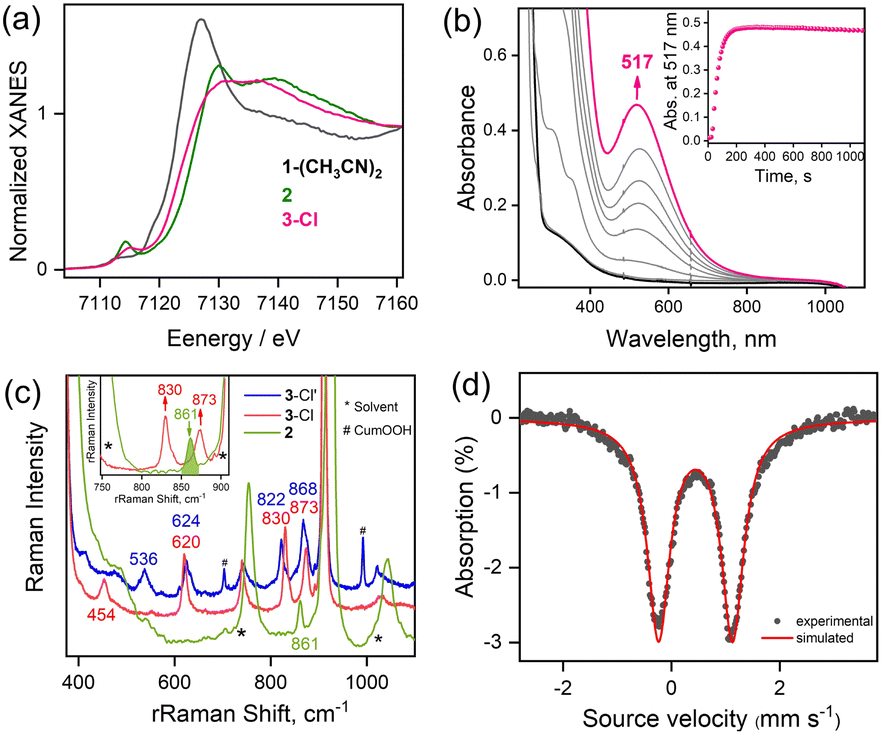 | ||
| Fig. 2 (a) X-ray absorption near edge structure (XANES) spectra of solutions of 1-(CH3CN)2, 2 and 3-Cl in CH3CN. (b) UV-visible spectral changes observed due to the formation of 3-Cl after the reaction of 1-(CH3CN)2 (0.50 mM) with tBuOOH (20 equiv.) in the presence of NEt4Cl (5 equiv.) in CH3CN at 253 K. Inset shows the time profile monitored at 517 nm due to the formation of 3-Cl. (c) rRaman spectra of 3-Cl synthesized with tBuOOH (red) and 3-Cl′ synthesized with CumOOH (blue) upon laser irradiation at 514 nm in CH3CN at −40 °C. The rRaman spectra of 2 synthesized with tBuOOH (green) using a 406 nm laser excitation in CH3CN at −40 °C is shown for comparison. Inset shows the disappearance of rRaman band at 861 cm−1 for 2 (green) upon generation of 3-Cl (red). (d) The zero-field Mössbauer spectrum of 3-Cl with isomer shift, δ = 0.45 mm s−1 and quadrupole splitting, ΔEQ = 1.37 mm s−1 recorded at 4.2 K. | ||
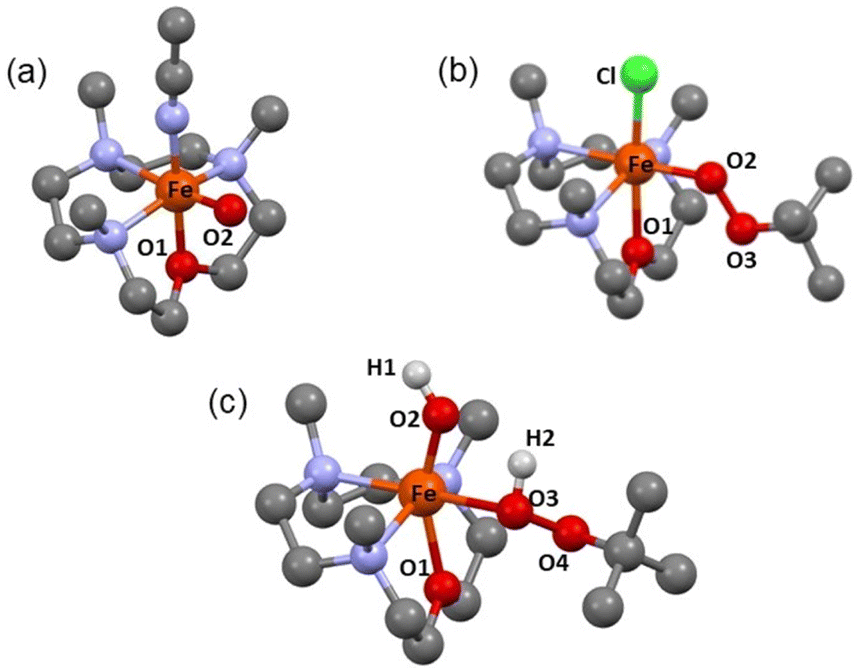 | ||
| Fig. 3 DFT Optimized Structures of (a) 2, (b) 3-Cl and (c) 5. The hydrogens are removed for clarity. See Tables S3a–c† for further information. | ||
Interestingly, the transient 3-CH3CN intermediate involved during the conversion of 1-(CH3CN)2 to 2 can be stabilized in the presence of externally added anionic ligands. For example, the reaction of 1-(CH3CN)2 with excess tBuOOH in CH3CN at −20 °C in the presence of NEt4Cl results in the generation of a burgundy-colored species (3-Cl) with λmax at 517 nm (ε = 880 M−1 cm−1) (Fig. 2b). 3-Cl is also generated in the direct reaction of 1-Cl with tBuOOH at −20 °C in CH3CN (Fig. S7†). Species 3-Cl exhibits an EPR spectrum with signals at g = 9.52, and 4.3 that corresponds to a high-spin iron(III) (S = 5/2) center (Fig. S8†) and a rRaman spectrum with four resonance-enhanced vibrational modes at 536, 620, 830, and 873 cm−1 that arise from coordinated alkylperoxide34 (Fig. 2c and Table S5†). Notably, these bands are significantly shifted in 3-Cl′, involving a bound cumyl peroxide (–OOCum) ligand; 3-Cl′ is generated in the reaction of 1-(CH3CN)2 with excess CumOOH in CH3CN at −20 °C in the presence of NEt4Cl (Fig. 2c). This pattern of rRaman bands has been observed for high-spin alkylperoxoiron(III) intermediates and is distinct from that associated with their low-spin counterparts (Table S5†).12–16,18,19 Further characterization of the Fe-center by zero-field Mössbauer analysis revealed a single doublet with an isomer shift (δ) of 0.45 mm s−1 and a quadrupole splitting ΔEQ = 1.37 mm s−1, consistent with the HS Fe(III) assignment of 3-Cl (Fig. 2d). XAS revealed an Fe K-edge energy of 3-Cl in between the energies of 1-(CH3CN)2 and 2 (Fig. 2a), supporting the Fe(III) assignment of 3-Cl. The EXAFS analysis of 3-Cl in a frozen CH3CN solution (Fig. S3b and c, Table S2†) reveals a Fe–Cl scatterer at 2.26 Å, and three N (at 2.19 Å) and one O (at 2.04 Å) scatterers assignable to the 12-TMCO ligand. Furthermore, two distinct Fe–O shells, at 1.82 Å and 3.15 Å, respectively, confirm the end-on binding mode of –OOtBu in 3-Cl. Thus 3-Cl can be characterized as a high-spin [FeIII(12-TMCO)(OOtBu)(Cl)]+ ion. DFT calculations on 3-Cl (Fig. 3b; Table S3b†) predict the presence of a distorted octahedral (Oh) Fe(III) center in an S = 5/2 ground-state. The three nitrogen donors of the 12-TMCO ligand and the –OOtBu moiety occupy the equatorial positions of the Oh, whereas the etheral oxygen atom and the chloride anion constitute the axial ligands. The calculated metrical parameters are in reasonable agreement with the experimental data (Table S6†). 3-Cl did not exhibit any electrophilic hydrogen atom transfer (HAT) abilities even in the presence of substrates containing weak C–H bonds (for example xanthene and 1,4-cyclohexadiene). Similarly, it was found to be incapable of performing oxygen atom transfer (OAT) to triphenylphosphine. 3-Cl also didn't show any nucleophilic reactivity with substrates like 2-phenylpropionaldehyde.
Parallel reactions using either F−, Br− or aryl thiolate (MePhS−) anionic ligands yield intermediates 3-F, 3-Br, or 3-SPhMe, respectively, with varying absorbance maxima (Fig. 4, Table S7†) depending on the anions used. In particular, the significant blue shift of the absorption band observed from 3-Cl (λmax ≈ 517 nm) to 3-SPhMe (λmax ≈ 460 nm) can be rationalized by the substitution of the axial chloride ligand in 3-Cl with a more Lewis basic thiolate ligand in 3-SPhMe and is consistent with the assignment of the chromophore as an alkylperoxo-to-iron(III) charge-transfer transition.15 Notably, the reaction of 1-(OTf)2 with excess tBuOOH in a non-coordinating solvent like acetone, where the –OTf− ligands stay bound to the Fe(II) center, also affords a deep purple chromophore with λmax = 510 nm corresponding to 3-OTf (Fig. S9†). This is in contrast to the reaction in a coordinating solvent like CH3CN whereby the FeIII–OOtBu species is transient and undergoes spontaneous O–O bond homolysis to yield the oxo–iron(IV) species. Thus, a cis-anionic ligand donation appears to stabilize the HS FeIII–OOtBu unit by retarding the O–O bond lysis step. The decay of 3-Cl and 3-SPhMe have been monitored at temperatures between −20 °C to +25 °C (Fig. S10†); no generation of the oxoiron(IV) species 2 by O–O bond homolysis has been observed.
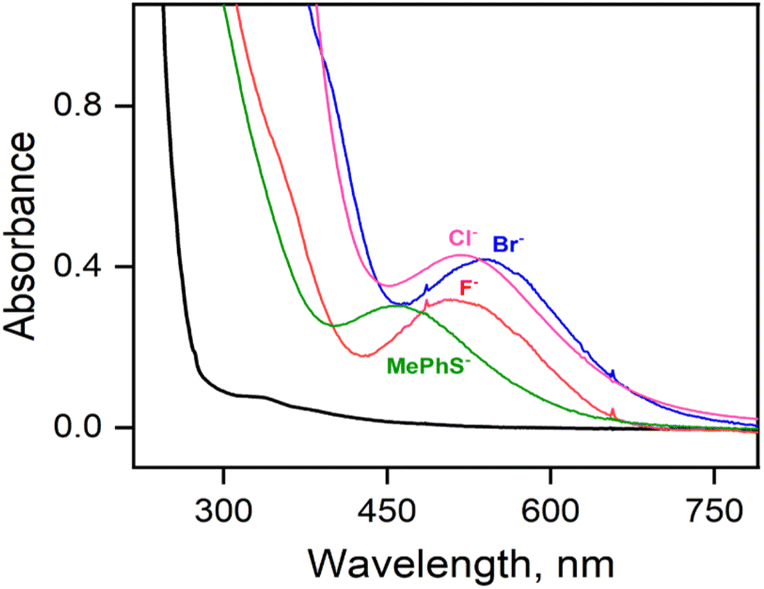 | ||
| Fig. 4 UV-visible spectral changes observed due to the formation of 3-L′ from the reaction of 1-(CH3CN)2 (0.5 mM, black line) with tBuOOH (20 equiv.) in the presence of 5 equiv. sodium toluene-p-thiolate (NaSPhMe, green), tetramethylammonium fluoride (NMe4F, red), tetrabutylammonium bromide (NBu4Br, blue) and tetraethylammonium chloride (NEt4Cl, pink), in CH3CN at −20 °C. | ||
The oxoiron(IV) complex 2 can, however, be converted to the FeIII–OOtBu species in the presence of excess tBuOOH and anionic ligands (Fig. S11†). For example, the addition of NEt4Cl to a CH3CN solution of 2 in the presence of excess tBuOOH at −20 °C leads to the immediate decay of the characteristic absorption bands of 2 at 770 nm and 935 nm, and the concomitant appearance of the band at 517 nm corresponding to 3-Cl. Notably, the time trace of the decay of the 770 nm band showed a pseudo first-order dependence on the concentration of NEt4Cl. In contrast, the rate of generation of 3-Cl was found to be independent of the NEt4Cl concentration. We therefore propose a mechanism where the conversion of 2 to 3-Cl takes place via the intermediate formation of an FeIII–OH species (4), which is generated by the decay of 2 in the presence of NEt4Cl (Scheme S1†). The FeIII–OH moiety, as suggested previously,13,35 then reacts with tBuOOH to yield FeIII–OOtBu, which is stabilized by binding a cis-anionic chloride ligand. In contrast to chloride ligand, which led to a stoichiometric conversion of 2 to 3-Cl, other anions like F− or Br− did not lead to a clean conversion, presumably because of the lower stabilities of 3-Br and 3-F compared to 3-Cl (Fig. S12†).
The N3O macrocycle in 12-TMCO is found to be a prerequisite for the stabilization of FeIVO or FeIII–OOtBu cores in reactions of the (12-TMCO)FeII compounds with tBuOOH. Notably, the corresponding iron(II) compound based on the N4 macrocycle [FeII(12-TMC)(CH3CN)2]2+ (8) (12-TMC = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane)36 yields an Fe2O3 precipitate and free ligand in its reaction with excess tBuOOH. Although, both 1-(CH3CN)2 and [FeII(12-TMC)(CH3CN)2]2+ contain cis-labile binding sites, the different products formed in their reactions with tBuOOH is presumably attributed to the different reactivities of their iron(III)hydroxide complexes, which are plausible intermediates necessary for the generation of the FeIII–OOtBu core.13,35 DFT calculations reveal that the transition state leading to the formation of 2 is preceded by an intermediate [FeIII(12-TMCO)(OH)(HOOtBu)]2+ (5; Scheme 2 and Fig. 3c), where the axial hydroxide donor is involved in hydrogen-bonding interaction with tBuOOH (Fig. S13a†), thereby making tBuOOH binding and the subsequent release of water to form 3-L′ (in the presence of anionic ligands), highly exergonic. Notably, our efforts to optimize the corresponding FeIII(12-TMC)(OH)(HOOtBu)]2+ complex involving the N4 macrocycle resulted in the loss of the tBuOOH fragment in the calculation (Fig. S13b, Table S3d†); so no H-bonding mediated binding of tBuOOH to [FeIII(12-TMC)(OH)]+ is possible, and the latter decays to form Fe2O3 and free ligand.
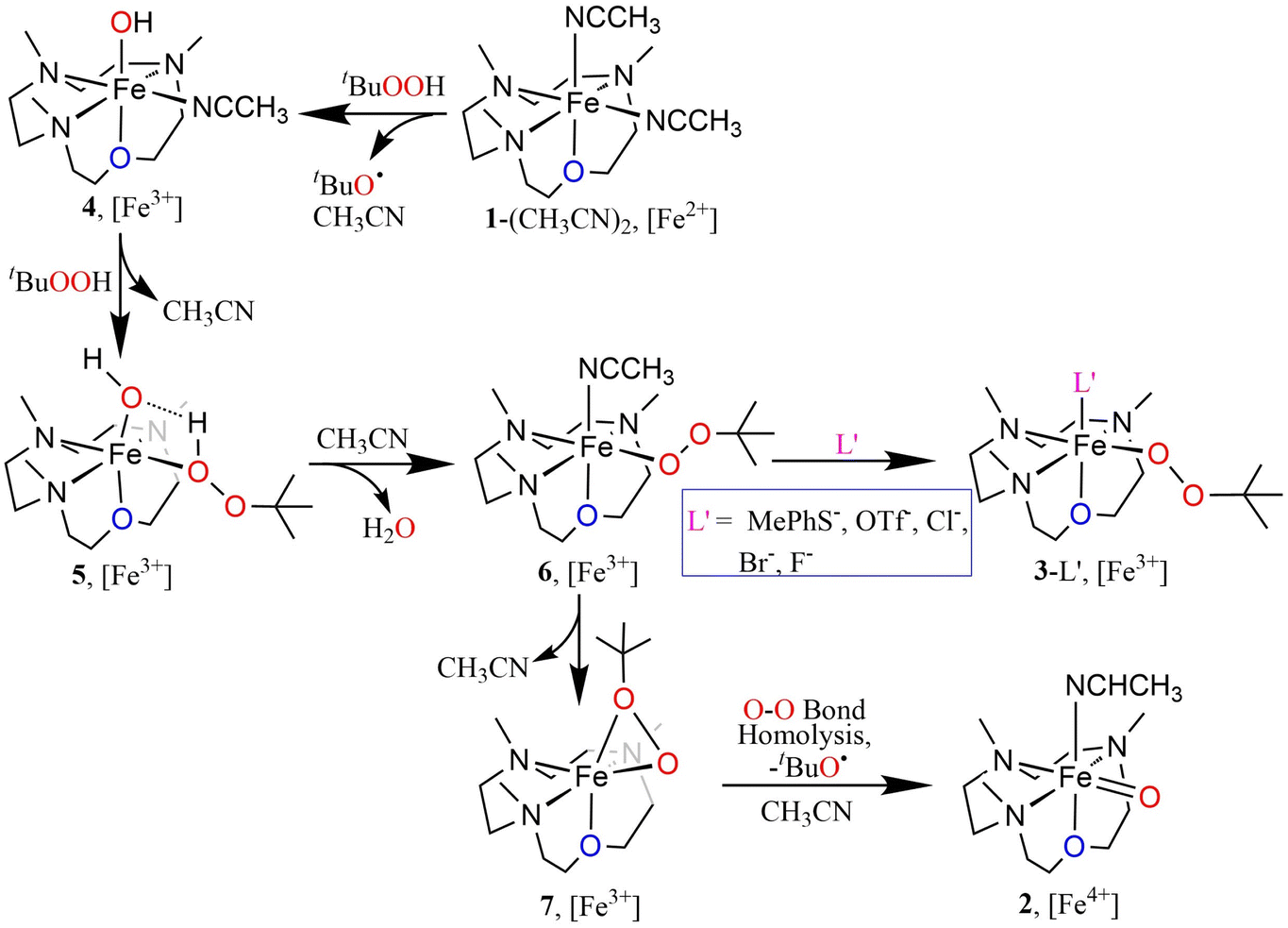 | ||
| Scheme 2 Schematic representation of the proposed reaction mechanism. | ||
Conclusion
In summary, in the present study, we have demonstrated the importance of cis-anionic ligand donations (including the thiolate relevant to SOR) for the stabilization of HS iron(III)–peroxo complexes. These results contrast the previously reported “push” effect11,14 that promotes O–O bond cleavage in HS and LS iron(III) peroxo complexes. The current study provides a basis to discuss the function of the high-spin {FeII(His)4(Cys)} active site of SOR. Although the putative O2-binding site is considered trans- to the axial thiolate ligand,6,8,9 the redox induced structural changes at the level of the peptide bond(s), as previously demonstrated by one of us and others,37,38 may lead to the formation of a HS cis-[FeIII(OOH)(Cys)]+ moiety. The O–O bond cleavage to produce the undesired high-valent oxo–iron species is presumably prevented by the high-spin iron(III) center and the presence of the equatorial thiolate ligand. Furthermore, the importance of secondary interactions in the stabilization of [FeIII–OOtBu] moiety, may also attribute to increasing the lifetime of the peroxo–iron(III) species in SOR to allow its protonation by a second-sphere residue and subsequent release of H2O2. The demonstrated contrasting effects of cis- and trans-anionic ligands on the fate of the HS FeIII–OOtBu moiety may, therefore, provide a rationale for nature's use of axial- and equatorial-thiolate ligations in P-450 and SOR, respectively, for two apparently distinct roles.Data availability
The data underlying this study, which are not included in the main text are available in the ESI.† The source data files can be provided upon request.Author contributions
TD, KD, JD contributed in the experimental work and SM contributed in DFT calculations. UK and PH contributed in rRaman measurements and analysis of the data. MH and HD contributed in carrying out XAS studies. BC recorded the crystallographic data. KR and TD initiated the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Alexander-von-Humboldt (AvH) Postdoctoral Research Fellowship from AvH Research foundation, Germany to T. D. and K. D. was funded by the Einstein Foundation Berlin. We thank the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) for EXC-2008—390540038—“UniSysCat”. We acknowledge the Helmholtz Zentrum Berlin (HZB) for allocating beamtime at beamline KMC-3 of the BESSY synchrotron and I. Zizak (HZB) for experimental support. H. D. and M. H. thank the German Bundesministerium für Bildung und Forschung (BMBF) for funding (Grant 05K22KE1, life-XAS).Notes and references
- L. Que Jr and R. Y. N. Ho, Chem. Rev., 1996, 96, 2607 CrossRef PubMed.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330, 933 CrossRef CAS PubMed.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949 CrossRef CAS PubMed.
- E. G. Kovaleva, M. B. Neibergall, S. Chakrabarty and J. D. Lipscomb, Acc. Chem. Res., 2007, 40, 475 CrossRef CAS PubMed.
- A. P. Yeh, Y. Hu, F. E. Jenney Jr, M. W. W. Adams and D. C. Rees, Biochemistry, 2000, 39, 2499 CrossRef CAS PubMed.
- K. Auclair, P. Moënne-Loccoz and P. R. O. de Montellano, J. Am. Chem. Soc., 2001, 123, 4877 CrossRef CAS PubMed.
- J. P. Emerson, E. D. Coulter, R. S. Phillips and D. M. Kurtz, J. Biol. Chem., 2003, 278, 39662 CrossRef CAS PubMed.
- T. Kitagawa, A. Dey, P. Lugo-Mas, J. B. Benedict, W. Kaminsky, E. I. Solomon and J. A. Kovacs, J. Am. Chem. Soc., 2006, 128, 14448 CrossRef CAS PubMed.
- A. A. A. A. Attia, D. Cioloboc, A. Lupan and R. Silaghi-Dumitrescu, J. Biol. Inorg. Chem., 2013, 18, 95 CrossRef CAS PubMed.
- J. Kaizer, M. Costas and L. Que Jr, Angew. Chem., Int. Ed., 2003, 42, 3671 CrossRef CAS PubMed.
- Y. Zang, J. Kim, Y. Dong, E. C. Wilkinson, E. H. Appelman and L. Que Jr, J. Am. Chem. Soc., 1997, 119, 4197 CrossRef CAS.
- M. R. Bukowski, H. L. Halfen, T. A. van den Berg, J. A. Halfen and L. Que Jr, Angew. Chem., 2005, 117, 590 CrossRef.
- S. Hong, Y.-M. Lee, K.-B. Cho, M. S. Seo, D. Song, J. Yoon, R. Garcia-Serres, M. Clémancey, T. Ogura, W. Shin, J.-M. Latour and W. Nam, Chem. Sci., 2014, 5, 156 RSC.
- F. Namuswe, T. Hayashi, Y. Jiang, G. D. Kasper, A. A. N. Sarjeant, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2010, 132, 157 CrossRef CAS PubMed.
- N. Lehnert, R. Y. N. Ho, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 8271 CrossRef CAS PubMed.
- H. Jeon and S. Hong, Chem. Lett., 2019, 48, 80 CrossRef CAS.
- L. R. Widger, Y. Jiang, A. C. Mc Quilken, T. Yang, M. A. Siegler, H. Matsumura, P. Moënne-Loccoz, D. Kumar, S. P. de Visser and D. P. Goldberg, Dalton Trans., 2014, 43, 7522 RSC.
- T.-C. Yang, R. L. Mc Naughton, M. D. Clay, F. E. Jenney Jr, R. Krishnan, D. M. Kurtz Jr, M. W. W. Adams, M. K. Johnson and B. M. Hoffman, J. Am. Chem. Soc., 2006, 128, 16566 CrossRef CAS PubMed.
- D. Krishnamurthy, G. D. Kasper, F. Namuswe, W. D. Kerber, A. A. N. Sarjeant, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2006, 128, 14222 CrossRef CAS PubMed.
- F. Namuswe, G. D. Kasper, A. A. N. Sarjeant, T. Hayashi, C. M. Krest, M. T. Green, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2008, 130, 14189 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491 CrossRef CAS PubMed.
- R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320 CrossRef CAS PubMed.
- X. Engelmann, I. Monte-Perez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632 CrossRef CAS PubMed.
- M. Puri and L. Que Jr, Acc. Chem. Res., 2015, 48, 2443 CrossRef CAS PubMed.
- J.-U. Rohde, A. Stubna, E. L. Bominaar, E. Münck, W. Nam and L. Que Jr, Inorg. Chem., 2006, 45, 6435 CrossRef CAS PubMed.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13 CrossRef CAS PubMed.
- Due to the non-availability of 18-O labelled tBuOOH and the decay of 2 in presence of 18-O labelled H2O synthesis of 18O labelled 2 was not possible. Notably, complex 2 can also be generated in presence of CumOOH (Fig. S4†) and shows a FeO vibration at 861 cm−1.
- T. Tano, H. Sugimoto, N. Fujieda and S. Itoh, Eur. J. Inorg. Chem., 2012, 4099 CrossRef CAS.
- A. Kunishita, H. Ishimaru, S. Nakashima, T. Ogura and S. Itoh, J. Am. Chem. Soc., 2008, 130, 4244 CrossRef CAS PubMed.
- S. Adachi, S. Nagano, K. Ishimori, Y. Watanabe, I. Morishima, T. Egawa, T. Kitagawa and R. Makino, Biochemistry, 1993, 32, 241 CrossRef CAS PubMed.
- D. V. Avila, C. E. Brown, K. U. Ingold and J. Lusztyk, J. Am. Chem. Soc., 1993, 115, 466 CrossRef CAS.
- E. Baciocchi, M. Bietti, M. Salamone and S. Steenken, J. Org. Chem., 2002, 67, 2266 CrossRef CAS PubMed.
- N. Lehnert, R. Y. N. Ho, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 12802 CrossRef CAS PubMed.
- A. L. Balch, Inorg. Chim. Acta, 1992, 198–200, 297–307 CrossRef CAS.
- W. Zhu, A. Kumar, J. Xiong, M. J. Abernathy, X.-X. Li, M. S. Seo, Y.-M. Lee, R. Sarangi, Y. Guo and W. Nam, J. Am. Chem. Soc., 2023, 145, 4389 CrossRef CAS PubMed.
- C. Berthomieu, F. Dupeyrat, M. Fontecave, A. Verméglio and V. Nivière, Biochemistry, 2002, 41, 10360 CrossRef CAS PubMed.
- M. Horsch, T. Utesch, P. Hildebrandt, M. A. Mroginski and I. Zebger, Phys. Chem. Chem. Phys., 2016, 18, 23053 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; synthesis, characterization, and DFT details. CCDC 2296883. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05603a |
| This journal is © The Royal Society of Chemistry 2024 |