
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile fabrication of Ni, Fe-doped δ-MnO2 derived from Prussian blue analogues as an efficient catalyst for stable Li–CO2 batteries†
Xiaoyang
Chen‡
a,
Jian
Chen‡
 a,
Yun
Qiao
*a,
Yun
Gao
b,
Siwei
Fan
a,
Yijie
Liu
a,
Li
Li
a,
Yang
Liu
*a and
Shulei
Chou
*b
a,
Yun
Qiao
*a,
Yun
Gao
b,
Siwei
Fan
a,
Yijie
Liu
a,
Li
Li
a,
Yang
Liu
*a and
Shulei
Chou
*b
aSchool of Environmental and Chemical Engineering, Shanghai University, Shanghai 200444, China. E-mail: yunqiao@shu.edu.cn; liuy986@163.com
bInstitute for Carbon Neutralization, College of Chemistry and Materials, Engineering, Wenzhou University, Zhejiang 325035, China. E-mail: chou@wzu.edu.cn
First published on 3rd January 2024
Abstract
Rechargeable Li–CO2 batteries are regarded as an ideal new-generation energy storage system, owing to their high energy density and extraordinary CO2 capture capability. Developing a suitable cathode to improve the electrochemical performance of Li–CO2 batteries has always been a research hotspot. Herein, Ni–Fe-δ-MnO2 nano-flower composites are designed and synthesized by in situ etching a Ni–Fe PBA precursor as the cathode for Li–CO2 batteries. Ni–Fe-δ-MnO2 nanoflowers composed of ultra-thin nanosheets possess considerable surface spaces, which can not only provide abundant catalytic active sites, but also facilitate the nucleation of discharge products and promote the CO2 reduction reaction. On the one hand, the introduction of Ni and Fe elements can improve the electrical conductivity of δ-MnO2. On the other hand, the synergistic catalytic effect between Ni, Fe elements and δ-MnO2 will greatly enhance the cycling performance and reduce the overpotential of Li–CO2 batteries. Consequently, the Li–CO2 battery based on the Ni–Fe-δ-MnO2 cathode shows a high discharge capacity of 8287 mA h g−1 and can stabilize over 100 cycles at a current density of 100 mA g−1. The work offers a promising guideline to design efficient manganese-based catalysts for Li–CO2 batteries.
Introduction
Nowadays, the over-dependence on traditional fossil fuels and the continuous release of greenhouse gases (CO2) have led to serious environmental problems and hindered the sustainable development of the world.1,2 To reduce carbon emissions or repurpose CO2, various chemical processes have been adopted to convert CO2 into value-added carbon compounds, such as methanol, organic materials and plastics.3–7 However, the conventional CO2 reduction system usually shows less energy conversion efficiency than the ideal value, due to the complex multi-electron reaction on the surface of the electrode.8–10 Therefore, direct electrochemical reduction of CO2 through a new energy storage device is an ideal strategy. Currently, metal–CO2 batteries, especially Li–CO2 batteries, show great potential in novel electrochemical CO2 reduction energy storage systems, due to their high energy density and reversible CO2 drive.11,12 Unfortunately, the development and exploration of Li–CO2 batteries are still in the early stage, and their practical application is restricted by large polarization, poor cycle stability and low coulombic efficiency.13–15Typically, the widely recognized electrochemical reaction of Li–CO2 batteries is confirmed to be 4Li + 3CO2 = 2Li2CO3 + C (E = 2.80 V vs. Li/Li+).16,17 However, it is found that Li2CO3, the discharge product of Li–CO2 batteries, is a kind of insulator with a wide band gap, and its decomposition kinetics is slow. As a result, Li2CO3, which is difficult to decompose, gradually accumulates on the surface of the cathode during cycling, inducing the coverage on the active sites of the catalyst and thus causing the cell deactivation.18,19 Therefore, the study of cathodes with high catalytic activity to achieve reversible decomposition of Li2CO3 is a key strategy to improve the performance of Li–CO2 batteries. Noble metal catalysts have been shown to promote the reversible formation and decomposition of Li2CO3 at lower potentials, greatly improving the electrochemical performance of Li–CO2 batteries.20–24 Nevertheless, their large-scale application is limited due to the high price and scarce reserves of noble metal catalysts.
Transition metal catalysts are considered as good substitutes for noble metal catalysts because of their excellent catalytic activity and relatively low cost.25–27 Numerous studies have proved that MnO2 catalyst can effectively promote the CO2 reduction reaction (CO2RR) and CO2 evolution reaction (CO2ER) processes.28 In previous studies, Lei et al. reported that the α-MnO2/CNT electrode could provide sufficient active sites for Li2CO3 nucleation and CO2 capture, and the cathode structure could be tuned.29 Similarly, Wang et al. successfully synthesized the CNT@MnO2 (Birnessite δ-MnO2) cathode; the as-assembled Li–CO2 batteries exhibited low overpotential and excellent cycle stability with the synergistic effect of δ-MnO2 and CNTs.30 However, further improvement of MnO2 catalytic activity is restricted by its poor conductivity and limited active sites. Previous results have shown that the electrical conductivity of MnO2 can be improved by modifying its band level and electronic structure through the introduction of atomic doping. Based on these inspirations, Peng et al. first applied Co-doped α-MnO2 nanowires for Li–CO2 batteries, achieving a low overpotential of 0.73 V and long cycle performance (500 cycles at 100 mA g−1). They attributed the excellent electrochemical performance to the high conductivity of Co-α-MnO2 nanowires, enhanced specific surface area, and synergistic catalysis of doped Co atoms.31 However, its irregular pore size and low active specific surface area lead to non-uniform deposition of Li2CO3 and low discharge capacity, which will limit its practical application.
Herein, Ni, Fe co-doped δ-MnO2 (Ni–Fe-δ-MnO2) nanoflowers as a cathode for Li–CO2 batteries were synthesized by in situ etching the Ni–Fe PBA precursors via a sacrificing-template strategy. Ni–Fe-δ-MnO2 nanoflowers can significantly promote the CO2RR and CO2ER processes and improve the cycle stability of Li–CO2 batteries. These excellent electrochemical properties are attributed to the unique nanoflower structure, abundant exposed surfaces, and a large number of oxygen vacancy defects of Ni–Fe-δ-MnO2. Furthermore, the introduction of Ni and Fe elements can not only improve the conductivity of δ-MnO2, but also significantly promote the reversible formation and decomposition of Li2CO3. Therefore, this work will provide an ideal guidance for the design of efficient Li–CO2 battery cathodes in the future.
Results and discussion
Highly crystalline three-dimensional nanocubes with a uniform diameter of 50 nm were synthesized by the co-precipitation method, and are denoted as Ni–Fe-PBA (Fig. S1a and b†). As shown in Fig. 1, nanoflowers with a diameter of 170 nm are grown vertically and uniformly on the Ni–Fe-PBA template after the hydrothermal process, denoted as the Ni–Fe-δ-MnO2 precursor (Fig. S1c and d†). Subsequently, Ni–Fe-δ-MnO2 was obtained by calcining the Ni–Fe-δ-MnO2 precursor in an Ar atmosphere at 300 °C for 2 h. The X-ray diffraction (XRD) characteristic peaks of Ni–Fe-δ-MnO2 at 12.5°, 25.2°, 37.3°, and 65.6° can be well attributed to the (001), (002), (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1), and (020) crystal faces of δ-MnO2 (JCPDS no. 80-1098), as shown in Fig. 2a. Scanning electron microscopy (Fig. 2b and c) revealed that the morphology of Ni–Fe-δ-MnO2 is well maintained compared with that of the Fe-δ-MnO2 precursor, which means that these nanoflowers possess superior stability. Interestingly, the nanoflower structure exposes a large surface, which can provide a large space and rich active sites for the deposition and decomposition of discharge products. Meanwhile, no residues of other PBA-derived precursors are observed in the SEM images, which further indicates that Ni–Fe PBA has been completely etched during the hydrothermal process. Transmission electron microscopy (TEM) was performed to further demonstrate the cubic nanoflower morphology of Ni–Fe-δ-MnO2 (Fig. 2d). It is worth noting that the ultra-thin nanosheets tend to bend and fold, and their thickness can be determined by measuring the folded area. The high-resolution transmission electron microscopy (HRTEM) image displays a lattice spacing of 0.67 nm, corresponding to the (001) plane of Ni–Fe-δ-MnO2 (Fig. 2e). Furthermore, the thickness of the nanosheet is about 2.7 nm, corresponding to Ni–Fe-δ-MnO2 (001) crystal surfaces.32 Additionally, considerable micropores can be seen in Fig. 2f, which can provide more nucleation regions and oxygen vacancies, conducive to electron transport and nucleation of Li2CO3. The high-angle annular dark field-scanning transmission microscope (HAADF-STEM) image of Ni–Fe-δ-MnO2 in Fig. 2g and the corresponding energy dispersive spectrometer (EDS) elemental mapping in Fig. 2h–l clearly prove the presence of Ni, Fe, Mn, and O elements in the catalyst, which are evenly distributed in Ni–Fe-δ-MnO2 nanoflowers. It is worth noting that the presence of K element was also detected in the EDS spectrum, which was also verified by inductively coupled plasma mass spectrometry (ICP-MS) (Table S1†). This indicates that Ni–Fe-δ-MnO2 contains a certain amount of K+. The presence of K+ can not only improve the structural stability and charge balance of MnO2, but also promote the octahedral effect of MnO6 and the diffusion rate of Li+, which will further enhance the catalytic activities of the CO2RR and CO2ER.33 The contents of Ni and Fe in Ni–Fe-δ-MnO2 are 7.57 and 4.22 wt%, respectively, which means that Ni and Fe with a mass ratio of 2
1), and (020) crystal faces of δ-MnO2 (JCPDS no. 80-1098), as shown in Fig. 2a. Scanning electron microscopy (Fig. 2b and c) revealed that the morphology of Ni–Fe-δ-MnO2 is well maintained compared with that of the Fe-δ-MnO2 precursor, which means that these nanoflowers possess superior stability. Interestingly, the nanoflower structure exposes a large surface, which can provide a large space and rich active sites for the deposition and decomposition of discharge products. Meanwhile, no residues of other PBA-derived precursors are observed in the SEM images, which further indicates that Ni–Fe PBA has been completely etched during the hydrothermal process. Transmission electron microscopy (TEM) was performed to further demonstrate the cubic nanoflower morphology of Ni–Fe-δ-MnO2 (Fig. 2d). It is worth noting that the ultra-thin nanosheets tend to bend and fold, and their thickness can be determined by measuring the folded area. The high-resolution transmission electron microscopy (HRTEM) image displays a lattice spacing of 0.67 nm, corresponding to the (001) plane of Ni–Fe-δ-MnO2 (Fig. 2e). Furthermore, the thickness of the nanosheet is about 2.7 nm, corresponding to Ni–Fe-δ-MnO2 (001) crystal surfaces.32 Additionally, considerable micropores can be seen in Fig. 2f, which can provide more nucleation regions and oxygen vacancies, conducive to electron transport and nucleation of Li2CO3. The high-angle annular dark field-scanning transmission microscope (HAADF-STEM) image of Ni–Fe-δ-MnO2 in Fig. 2g and the corresponding energy dispersive spectrometer (EDS) elemental mapping in Fig. 2h–l clearly prove the presence of Ni, Fe, Mn, and O elements in the catalyst, which are evenly distributed in Ni–Fe-δ-MnO2 nanoflowers. It is worth noting that the presence of K element was also detected in the EDS spectrum, which was also verified by inductively coupled plasma mass spectrometry (ICP-MS) (Table S1†). This indicates that Ni–Fe-δ-MnO2 contains a certain amount of K+. The presence of K+ can not only improve the structural stability and charge balance of MnO2, but also promote the octahedral effect of MnO6 and the diffusion rate of Li+, which will further enhance the catalytic activities of the CO2RR and CO2ER.33 The contents of Ni and Fe in Ni–Fe-δ-MnO2 are 7.57 and 4.22 wt%, respectively, which means that Ni and Fe with a mass ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 occupy the surroundings of the MnO6 octahedron. Thus, more oxygen vacancies are formed and can enhance the CO2RR.
:1 occupy the surroundings of the MnO6 octahedron. Thus, more oxygen vacancies are formed and can enhance the CO2RR.
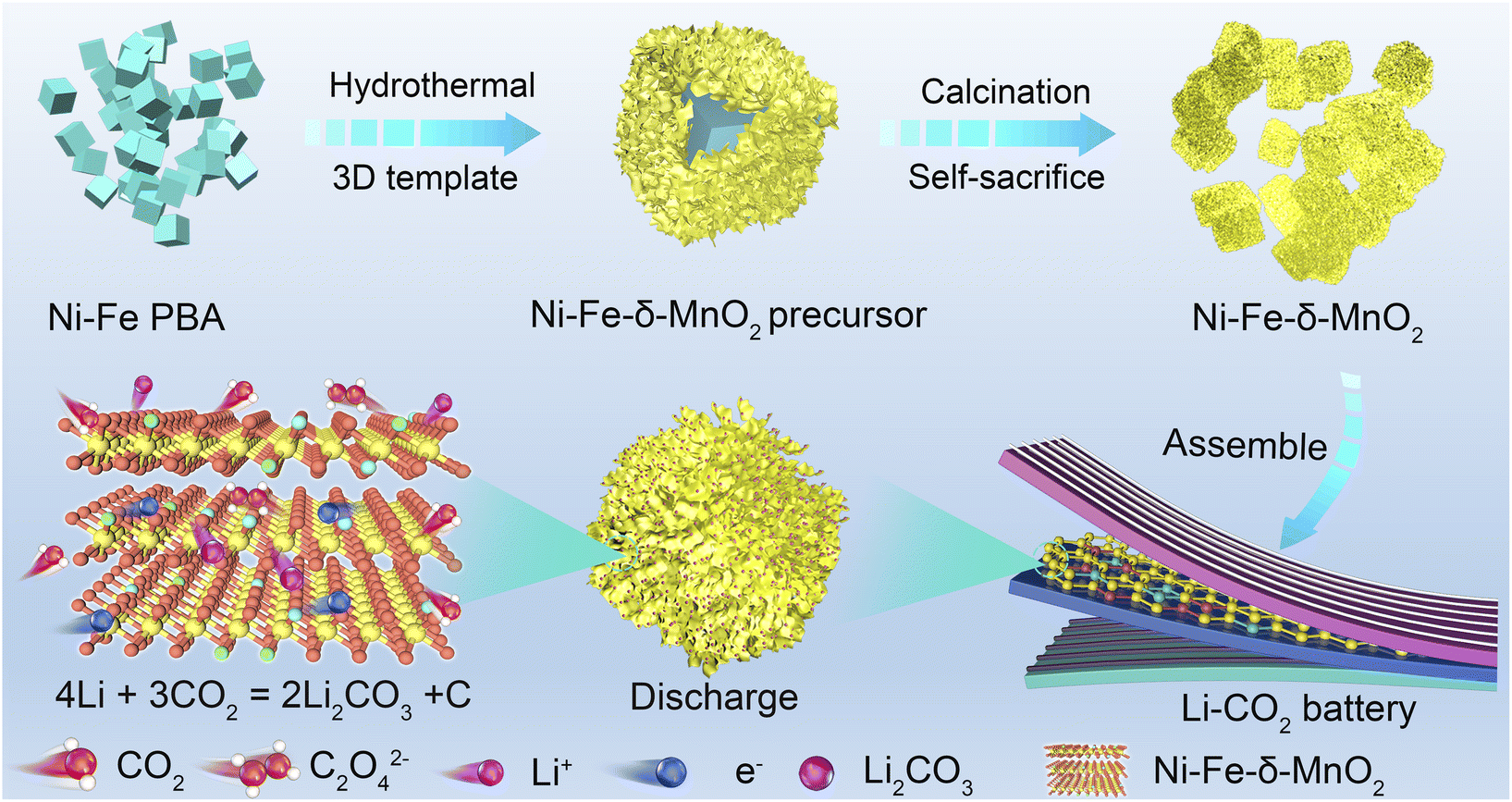 | ||
| Fig. 1 Schematic illustration for the fabrication of the Ni–Fe-δ-MnO2 cathode. | ||
 | ||
| Fig. 2 (a) XRD patterns of δ-MnO2, Ni–Fe-δ-MnO2 precursor and Ni–Fe-δ-MnO2, (b and c) SEM, (d) TEM, (e and f) HRTEM, (g) HAADF-STEM images and corresponding EDS elemental mapping results of (h) Ni, (i) Fe, (j) Mn, (k) O and (l) K elements for Ni–Fe-δ-MnO2. | ||
X-Ray photoelectron spectroscopy (XPS) was used to detect the valence state and electronic structure of Ni–Fe-δ-MnO2, Ni–Fe-δ-MnO2 precursor and δ-MnO2. The XPS full spectra show obvious characteristic peaks of Fe, Ni, Mn, O and K elements (Fig. 3a), which are consistent with the EDS analysis results. As shown in Fig. 3b, the high-resolution Mn 2p XPS spectra exhibit three peaks, which can be divided into Mn4+ from MnO2 at 642.2 eV, and the Mn3+ peaks at 654.0 and 645.4 eV due to the oxygen defects induced by Mn4+ transformation. The high-resolution O 1s XPS spectra of Ni–Fe-δ-MnO2 were deconvoluted into three spin-orbital peaks at 529.46, 530.03, and 531.32 eV, corresponding to the lattice oxygen (O1), oxygen vacancy (O2), and hydroxyl or adsorbed water molecules (O3) of Mn–O bonds (Fig. 3c). Ni–Fe-δ-MnO2 has the highest O2 ratio (45.97%), which further proves abundant oxygen vacancy defects in its structure.34,35 As shown in Fig. 3d, the main peak of Ni 2p3/2 and its satellites at 854.9 and 861.4 eV, and the main peak of Ni 2p1/2 and its satellites at 872.2 and 879.3 eV are attributed to nickel in its oxides.36 Similarly, the Fe 2p spectra (Fig. 3e) can be deconvoluted into five peaks at 710.4/723.5 eV (Fe3+), 712.8/725.8 eV (Fe2+) and 769.2 eV (satellite peaks).37 The result can further confirm the successful synthesis of Ni–Fe-δ-MnO2. As shown in Fig. 3d and e, both Ni 2p and Fe 2p peaks of Ni–Fe-δ-MnO2 move to the lower binding energy in contrast to that of the Ni–Fe-δ-MnO2 precursor, which can increase the density of the surrounding electron cloud. This phenomenon can be attributed to the co-coordination between Ni/Fe and O atoms in the center of Mn. In this case, this structure can inhibit Jahn–Teller distortion, stabilize the MnO2 structure and expose more active sites. In addition, increasing the electron density of the metal center can enhance the adsorption between the metal center and the intermediate and change the coordination around MnO2 to form more oxygen vacancies. Electron paramagnetic resonance (EPR) spectroscopy was employed to detect the unpaired electrons in Ni–Fe-δ-MnO2. The EPR signal for Ni–Fe-δ-MnO2 is dramatically enhanced in contrast to that of δ-MnO2 and Ni–Fe-δ-MnO2 precursor, which suggests its enhanced oxygen vacancy density (Fig. 3f).38 Interestingly, the co-doping of Ni and Fe makes Ni–Fe-δ-MnO2 expose more active sites and enhance the oxygen vacancy density through the coordination of adjacent O atoms around Mn, and thus promotes the activities of the CO2RR and CO2ER. Brunauer–Emmett–Teller (BET) analysis was applied to investigate the specific surface area of the as-prepared Ni–Fe-δ-MnO2. The nitrogen adsorption and desorption curve (Fig. S2a†) shows that its specific surface area is up to 154.99 m2 g−1 for Ni–Fe-δ-MnO2, meaning that the catalyst can expose considerable active sites and provide sufficient space for the CO2RR process. The pore size distribution results reveal that Ni–Fe-δ-MnO2 possesses a large number of mesoporous structures with pore size in the range of 5.0–33.0 nm (Fig. S2b†). These mesoporous structures can accommodate more deposition of discharge products, enhance the diffusion of CO2 and electrolyte, and expose more active sites. Based on the above analysis, we can conclude that Ni–Fe-δ-MnO2 nanoflowers are successfully prepared. The presence of Ni and Fe elements can improve the electrical conductivity of δ-MnO2. Moreover, their synergistic effect with Mn atoms will further improve the catalytic activity of the cathode. In addition, the large surface area and abundant oxygen vacancy defects for Ni–Fe-δ-MnO2 can greatly enhance the reactivity of the CO2RR and CO2ER. Therefore, Ni–Fe-δ-MnO2 nanoflowers are expected to be an ideal cathode for Li–CO2 batteries.
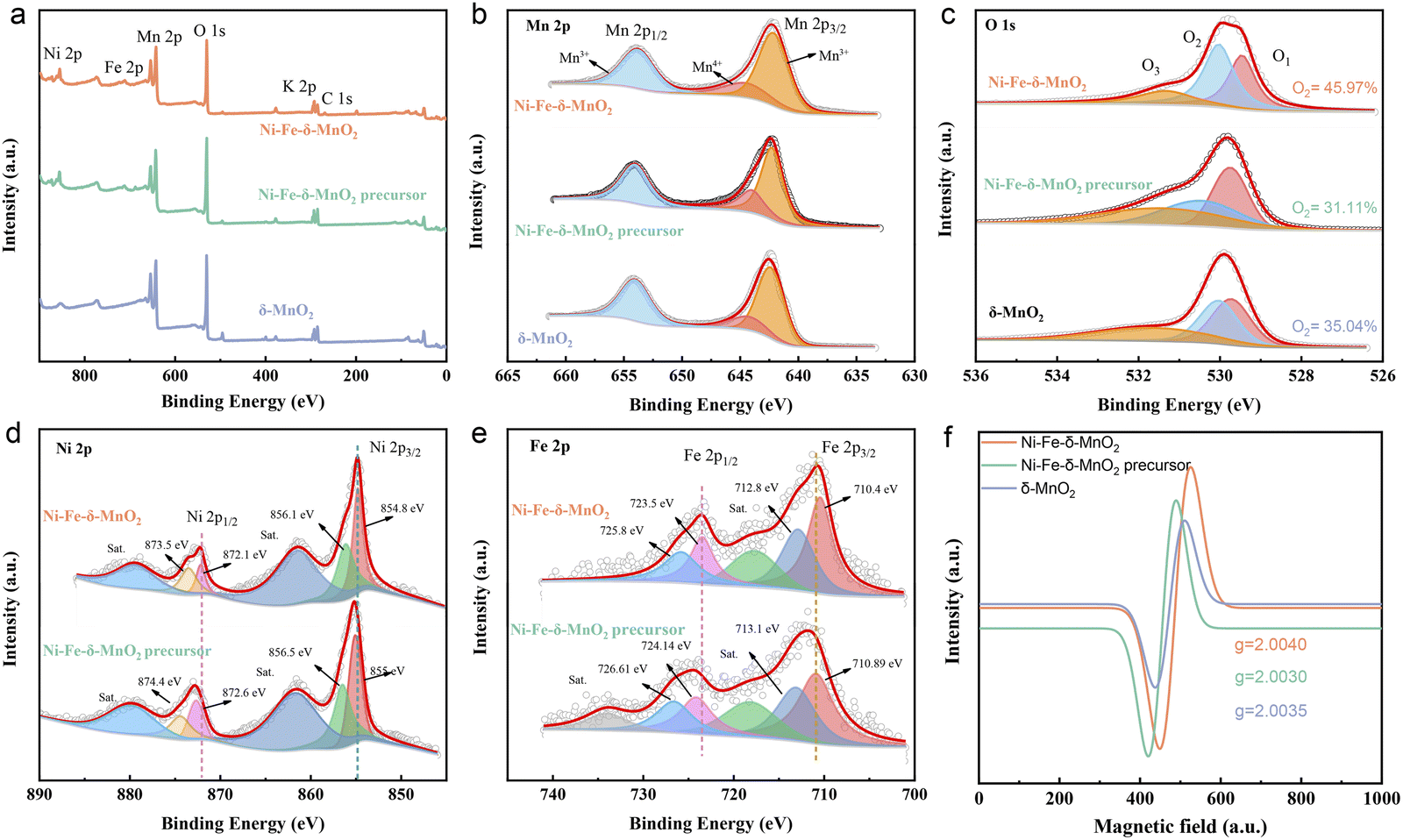 | ||
| Fig. 3 (a) The survey XPS spectra, high-resolution XPS spectra of (b) Mn 2p, (c) O 1s of Ni–Fe-δ-MnO2, Ni–Fe-δ-MnO2 precursor and δ-MnO2. High-resolution XPS spectra of (d) Ni 2p and (e) Fe 2p of Ni–Fe-δ-MnO2 and Ni–Fe-δ-MnO2 precursor. (f) EPR curves of Ni–Fe-δ-MnO2, Ni–Fe-δ-MnO2 precursor and δ-MnO2. | ||
The electrochemical performance of the Ni–Fe-δ-MnO2 cathode in Li–CO2 batteries was systematically evaluated. As shown in Fig. S3a,† the Ni–Fe-δ-MnO2 cathode shows more obvious reduction and oxidation peaks in a CO2 atmosphere than in an Ar atmosphere. In addition, Fig. S3b† reveals that the discharge capacity of the Ni–Fe-δ-MnO2 cathode is 8287 mA h g−1, which is 2.1 times that of the δ-MnO2 cathode (3923 mA h g−1), suggesting that the Ni–Fe-δ-MnO2 cathode possesses a relatively better catalytic activity for the CO2RR. In order to investigate the stability of the Li–CO2 battery based on the Ni–Fe-δ-MnO2 cathode, charge–discharge tests were conducted at a current density of 100 mA g−1 with a cutoff capacity of 1000 mA h g−1. Fig. 4a and b display the charge and discharge cycles of Ni–Fe-δ-MnO2 and δ-MnO2 cathodes. The Li–CO2 battery with Ni–Fe-δ-MnO2 as the cathode can stabilize for 100 cycles, and its overpotential drops to 1.32 V (Fig. 4a). The charge terminal voltage remains below 4.2 V despite a slight increase after 100 cycles. As a contrast, we also investigated the cycle performance of δ-MnO2 under the same test conditions, as shown in Fig. 4b. It is obvious that δ-MnO2 exhibits extremely poor cycle stability. After 60 cycles, the voltage of its charging platform begins to gradually decrease. Unfortunately, both the terminal charge and discharge voltages reach the cut-off voltage (2.0 and 4.5 V) after 90 cycles. By comparing the discharge and charge terminal voltages of δ-MnO2 and Ni–Fe-δ-MnO2 (Fig. 4c), it can be clearly seen that the Ni–Fe-δ-MnO2 exhibits much better cycle stability than δ-MnO2. In addition, the overpotential of the Ni–Fe-δ-MnO2 cathode is always lower than that of δ-MnO2. Furthermore, we compared the overpotentials of the two cathodes at the 10th cycle (Fig. 4d). Compared with δ-MnO2, the overpotential of the Ni–Fe-δ-MnO2 is decreased by 0.23 V, further indicating that the introduction of Ni and Fe elements can significantly improve the catalytic activity of δ-MnO2.
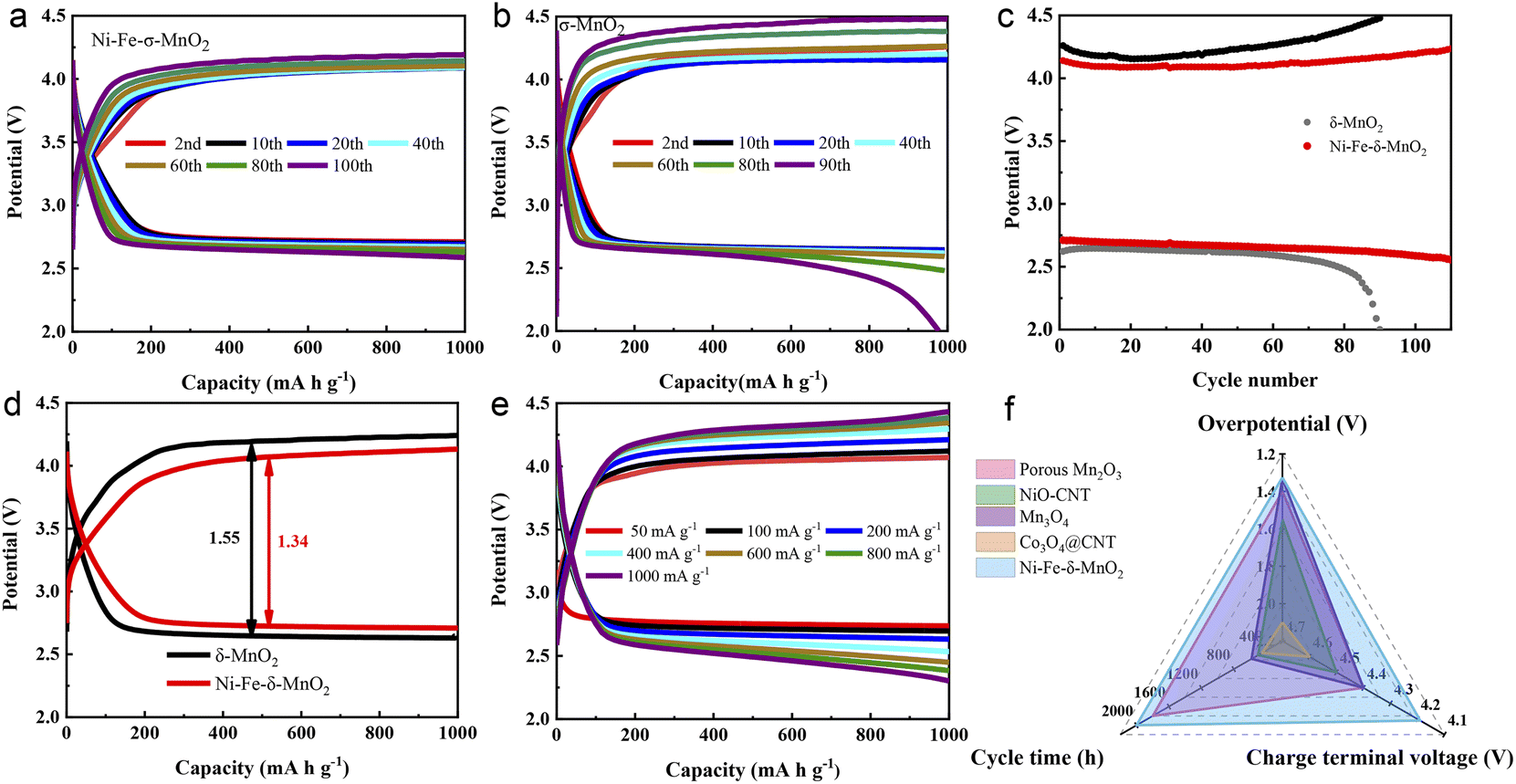 | ||
| Fig. 4 The discharge and charge curves of (a) Ni–Fe-δ-MnO2 and (b) δ-MnO2 at 100 mA g−1 with a limited capacity of 1000 mA g−1. (c) The discharge and charge terminal voltages of Ni–Fe-δ-MnO2 and δ-MnO2 upon cycling, (d) the overpotential comparison for the two cathodes at the 10th cycle. (e) The discharge and charge profiles of the Ni–Fe-δ-MnO2 cathode at different current densities. (f) Comparison of the electrochemical properties of the Ni–Fe-δ-MnO2 cathode and manganese based or other transition metal oxides. | ||
Fig. 4e exhibits the charge–discharge curves of the Li–CO2 battery with Ni–Fe-δ-MnO2 as the cathode at different current densities. When the current density increases from 400 to 1000 mA g−1, the charge terminal voltage increases by only 0.13 V. The charge terminal voltage of Ni–Fe-δ-MnO2 is only 4.43 V (<4.5 V) even at the current density of 1000 mA g−1, indicating that the Ni–Fe-δ-MnO2 electrode can still maintain excellent CO2ER activity even at a higher current density. Furthermore, the electrochemical performance of the Ni–Fe-δ-MnO2 cathode was compared with that of other Mn-based and related transition metal catalysts. The radar diagram in Fig. 4f demonstrates that the Ni–Fe-δ-MnO2 cathode endows obvious advantages in terms of cycle time, overpotential and charge terminal voltage.39–42 These results indicate that Ni–Fe-δ-MnO2 can improve the catalytic activities of the CO2RR and CO2ER, and thus significantly enhance the cycle stability of Li–CO2 batteries.
In order to further explore the mechanism of the excellent electrochemical performance of the Ni–Fe-δ-MnO2 cathode in a Li–CO2 battery, SEM, XPS, in situ Raman and Fourier transform infrared (FTIR) spectroscopies were performed to systematically characterize the formation and decomposition of discharge products during the cycling processes. Fig. S6† displays the morphologies and structures of the Ni–Fe-δ-MnO2 cathode at the discharged and recharged states. As shown in Fig. S6a,† discharge products with a flake morphology densely cover on the surface of the entire electrode after the first discharge process. Fortunately, the electrode returns to its pristine morphology, and the carbon nanotubes wrapped around its surface are also exposed after recharging (Fig. S6b†), indicating the reversible formation and decomposition of the discharge products. This result proves that the Ni–Fe-δ-MnO2 electrode possesses excellent reversibility for the formation and decomposition of discharge products. In order to further verify the composition of the discharge products and the reversibility of the Ni–Fe-δ-MnO2 electrode, the XPS spectrum was recorded. As shown in Fig. 5a, the high-resolution C 1s spectra can be deconvoluted into four distinct characteristic peaks at 284.96, 286.78, 290.45 and 293.53 eV after discharge, wherein the characteristic peak at 290.45 eV corresponds to the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of Li2CO3, indicating the existence of Li2CO3 as the discharge product. Importantly, this characteristic peak almost completely disappears after the subsequent recharging (Fig. 5b), demonstrating the reversibility of the discharge product. Fig. 5c shows the FTIR spectra of the pristine, discharged and recharged Ni–Fe-δ-MnO2 cathodes. The discharged electrode exhibits three new peaks at 865, 1408 and 1476 cm−1, compared with the pristine electrode, which are consistent with the standard spectrum of commercial Li2CO3.43 Similarly, the characteristic peaks of Li2CO3 almost completely disappear in the subsequent recharging process, which is consistent with the XPS results. Moreover, we further investigated the electrochemical behaviour of the discharge product Li2CO3 in the whole charge and discharge processes by in situ Raman spectroscopy. As shown in Fig. 5d, the characteristic peaks of MnO2 (633 cm−1), D band (1343 cm−1) and G band (1574 cm−1) of carbon can be observed in the Raman spectra at the initial discharge stage. As the discharge progresses, the peak intensities of Li2CO3 (1081 cm−1), D and G bands continuously increase, which can be attributed to the formation of discharge products (Li2CO3 and C).44,45 In addition, the ID/IG ratio gradually decreases with the continuous discharge process, indicating that the C product possesses a high degree of graphitization. Noteworthily, higher graphitization is conducive to electron transfer and decomposition of discharge products, as the gradual weakening and eventual disappearance of Li2CO3 and C in the in situ Raman spectra during the subsequent charging process proves the complete decomposition of discharge products.46 The corresponding 2D contour map in Fig. 5e further intuitively reflects this trend of evolution. These results fully demonstrate the reversible formation and decomposition of Li2CO3 during the cycling processes and the excellent catalytic activity of the Ni–Fe-δ-MnO2 electrode for the CO2RR and CO2ER processes.47 Besides, we can also notice that the characteristic peak of MnO2 gradually disappears during the discharge stage, which may be due to the coverage of Li2CO3 on the surface of the Ni–Fe-δ-MnO2 electrode, which affects the detected signal of MnO2. It is worth noting that the characteristic peak of MnO2 appears with the gradual disappearance of Li2CO3, which is exactly opposite to the evolution trend of Li2CO3 after recharging. Furthermore, the existence of the MnO2 signal after charging also indicates the stability of the Ni–Fe-δ-MnO2 electrode. Based on the above results, we can conclude that Ni–Fe-δ-MnO2 can promote not only the transport of electrons and Li+via the four-electron interaction but also the nucleation and reversible decomposition of Li2CO3, which could improve its catalytic performance.
O bond of Li2CO3, indicating the existence of Li2CO3 as the discharge product. Importantly, this characteristic peak almost completely disappears after the subsequent recharging (Fig. 5b), demonstrating the reversibility of the discharge product. Fig. 5c shows the FTIR spectra of the pristine, discharged and recharged Ni–Fe-δ-MnO2 cathodes. The discharged electrode exhibits three new peaks at 865, 1408 and 1476 cm−1, compared with the pristine electrode, which are consistent with the standard spectrum of commercial Li2CO3.43 Similarly, the characteristic peaks of Li2CO3 almost completely disappear in the subsequent recharging process, which is consistent with the XPS results. Moreover, we further investigated the electrochemical behaviour of the discharge product Li2CO3 in the whole charge and discharge processes by in situ Raman spectroscopy. As shown in Fig. 5d, the characteristic peaks of MnO2 (633 cm−1), D band (1343 cm−1) and G band (1574 cm−1) of carbon can be observed in the Raman spectra at the initial discharge stage. As the discharge progresses, the peak intensities of Li2CO3 (1081 cm−1), D and G bands continuously increase, which can be attributed to the formation of discharge products (Li2CO3 and C).44,45 In addition, the ID/IG ratio gradually decreases with the continuous discharge process, indicating that the C product possesses a high degree of graphitization. Noteworthily, higher graphitization is conducive to electron transfer and decomposition of discharge products, as the gradual weakening and eventual disappearance of Li2CO3 and C in the in situ Raman spectra during the subsequent charging process proves the complete decomposition of discharge products.46 The corresponding 2D contour map in Fig. 5e further intuitively reflects this trend of evolution. These results fully demonstrate the reversible formation and decomposition of Li2CO3 during the cycling processes and the excellent catalytic activity of the Ni–Fe-δ-MnO2 electrode for the CO2RR and CO2ER processes.47 Besides, we can also notice that the characteristic peak of MnO2 gradually disappears during the discharge stage, which may be due to the coverage of Li2CO3 on the surface of the Ni–Fe-δ-MnO2 electrode, which affects the detected signal of MnO2. It is worth noting that the characteristic peak of MnO2 appears with the gradual disappearance of Li2CO3, which is exactly opposite to the evolution trend of Li2CO3 after recharging. Furthermore, the existence of the MnO2 signal after charging also indicates the stability of the Ni–Fe-δ-MnO2 electrode. Based on the above results, we can conclude that Ni–Fe-δ-MnO2 can promote not only the transport of electrons and Li+via the four-electron interaction but also the nucleation and reversible decomposition of Li2CO3, which could improve its catalytic performance.
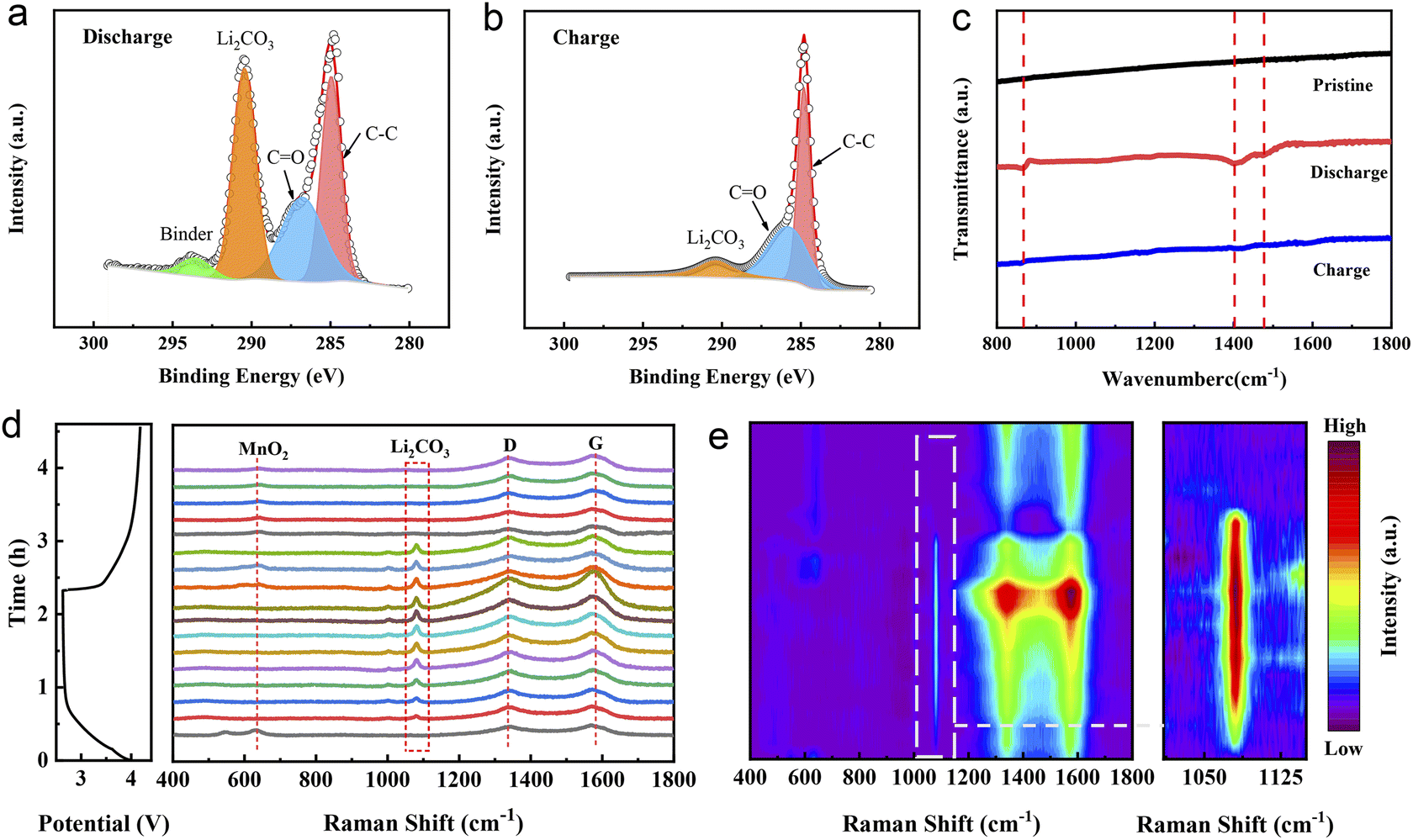 | ||
| Fig. 5 The ex situ and in situ characterizations of the Ni–Fe-δ-MnO2 cathode in Li–CO2 batteries. XPS spectra of C 1s after (a) discharge and (b) charge processes. (c) FTIR spectra of the pristine, discharged and charged cathodes. (d) In situ Raman spectra and (e) the corresponding 2D contour plots of the Ni–Fe-δ-MnO2 cathode. | ||
Conclusions
In summary, Ni–Fe-doped δ-MnO2 nanoflowers (Ni–Fe-δ-MnO2) are designed and synthesized by etching a Ni–Fe-δ-MnO2 precursor using the sacrificial template method. The Ni–Fe-δ-MnO2 nanoflowers composed of ultra-thin nanosheets possess considerable surface spaces, which can not only provide abundant catalytic active sites, but also facilitate the nucleation of discharge products and promote the CO2 reduction reaction. In addition, the introduction of Ni and Fe elements could improve the electrical conductivity of δ-MnO2. It is worth noting that their synergistic catalytic effect with δ-MnO2 could greatly enhance the cycling performance and reduce the overpotential of the Li–CO2 battery. Due to these advantages, the Li–CO2 battery with the Ni–Fe-δ-MnO2 cathode delivers the highest discharge capacity of 8287 mA h g−1 at the current density of 100 mA g−1, which is 2.1 times that of the bare δ-MnO2. Additionally, the Li–CO2 battery based on the Ni–Fe-δ-MnO2 cathode can achieve a low overpotential of 1.32 V and outstanding cycle performance of 100 cycles. This work provides a reference direction for the design of highly efficient multi-atom co-catalysts in the future.Data availability
Synthetic procedures, experimental details, and characterization data are available in the ESI.†Author contributions
Y. Q., Y. L. and S. L. C. proposed the concept and supervised the work; X. Y. C. and J. C. designed the experiments and wrote the paper; S. W. F., Y. G., and Y. J. L. contributed to the discussion and provided suggestions. Y. J. L. helped to analyze the data; L. L. helped to summarize the data. All authors have discussed the results, drafted the manuscript and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X.-Y. C. and J. C. contributed equally to this work. This work was financially supported by the National Natural Science Foundation of China (No. 51971124 and 52171217). The Innovative Research Team of High-level Local Universities in Shanghai is gratefully acknowledged for their financial support.Notes and references
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. de Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2019, 11, 1167 CrossRef CAS.
- K. S. Lackner, Science, 2003, 300, 1677 CrossRef CAS.
- S. A. Theofanidis, A. N. Antzaras and A. A. Lemonidou, Curr. Opin. Chem. Eng., 2023, 39, 100902 CrossRef.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609 CrossRef CAS.
- U. Ulmer, T. Dingle, P. N. Duchesne, R. H. Morris, A. Tavasoli, T. Wood and G. A. Ozin, Nat. Commun., 2019, 10, 3169 CrossRef PubMed.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952 CrossRef CAS.
- R. Zhao, P. Ding, P. Wei, L. Zhang, Q. Liu, Y. Luo, T. Li, S. Lu, X. Shi, S. Gao, A. M. Asiri, Z. Wang and X. Sun, Adv. Funct. Mater., 2021, 31, 2009449 CrossRef CAS.
- D.-H. Guan, X.-X. Wang, M.-L. Li, F. Li, L.-J. Zheng, X.-L. Huang and J.-J. Xu, Angew. Chem., Int. Ed., 2020, 59, 19518 CrossRef CAS.
- X. Sun, X. Mu, W. Zheng, L. Wang, S. Yang, C. Sheng, H. Pan, W. Li, C.-H. Li, P. He and H. Zhou, Nat. Commun., 2023, 14, 536 CrossRef CAS.
- T. Jian, W. Ma, C. Xu, H. Liu and J. Wang, eScience, 2023, 3, 100114 CrossRef.
- Y.-F. Wang, G.-J. Ji, L.-N. Song, X.-X. Wang and J.-J. Xu, ACS Energy Lett., 2023, 8, 1026 CrossRef CAS.
- Y. Qiao, S. Xu, Y. Liu, J. Dai, H. Xie, Y. Yao, X. Mu, C. Chen, D. J. Kline, E. M. Hitz, B. Liu, J. Song, P. He, M. R. Zachariah and L. Hu, Energy Environ. Sci., 2019, 12, 1100 RSC.
- J. Chen, X.-Y. Chen, Y. Liu, Y. Qiao, S.-Y. Guan, L. Li and S.-L. Chou, Energy Environ. Sci., 2023, 7, 130 Search PubMed.
- C. Yang, K. Guo, D. Yuan, J. Cheng and B. Wang, J. Am. Chem. Soc., 2020, 142, 6983 CrossRef CAS.
- X. Ma, W. Zhao, Q. Deng, X. Fu, L. Wu, W. Yan and Y. Yang, J. Power Sources, 2022, 535, 231446 CrossRef CAS.
- D.-H. Guan, X.-X. Wang, F. Li, L.-J. Zheng, M.-L. Li, H.-F. Wang and J.-J. Xu, ACS Nano, 2022, 16, 12364 CrossRef CAS.
- X.-X. Wang, D.-H. Guan, F. Li, M.-L. Li, L.-J. Zheng and J.-J. Xu, Small, 2021, 17, 2100642 CrossRef CAS PubMed.
- B. Chen, D. Wang, J. Tan, Y. Liu, M. Jiao, B. Liu, N. Zhao, X. Zou, G. Zhou and H.-M. Cheng, J. Am. Chem. Soc., 2022, 144, 3106 CrossRef CAS PubMed.
- S. Yang, P. He and H. Zhou, Energy Environ. Sci., 2016, 9, 1650 RSC.
- Z. Guo, J. Li, H. Qi, X. Sun, H. Li, A. G. Tamirat, J. Liu, Y. Wang and L. Wang, Small, 2019, 15, e1803246 CrossRef.
- C. Wang, Q. Zhang, X. Zhang, X.-G. Wang, Z. Xie and Z. Zhou, Small, 2018, 14, e1800641 CrossRef.
- S. Yang, Y. Qiao, P. He, Y. Liu, Z. Cheng, J.-j. Zhu and H. Zhou, Energy Environ. Sci., 2017, 10, 972 RSC.
- H. Zhao, D. Li, H. Li, A. G. Tamirat, X. Song, Z. Zhang, Y. Wang, Z. Guo, L. Wang and S. Feng, Electrochim. Acta, 2019, 299, 592 CrossRef CAS.
- S. Thoka, C.-J. Chen, A. Jena, F.-M. Wang, X.-C. Wang, H. Chang, S.-F. Hu and R.-S. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 17353 CrossRef CAS PubMed.
- A. Hu, C. Shu, C. Xu, R. Liang, J. Li, R. Zheng, M. Li and J. Long, J. Mater. Chem. A, 2019, 7, 21605 RSC.
- X.-X. Wang, G.-J. Ji, P. She, F. Li, Q.-C. Liu, H.-F. Wang and J.-J. Xu, Chem. Eng. J., 2021, 426, 131101 CrossRef CAS.
- X. Chen, J. Chen, Y. Liu, Y. Liu, Y. Gao, S. Fan, X. He, X. Liu, C. Shen, Y. Jiang, L. Li, Y. Qiao and S. Chou, ACS Appl. Mater. Interfaces, 2023, 15, 28106 CrossRef CAS.
- D. Lei, S. Ma, Y. Lu, Q. Liu and Z. Li, J. Electron. Mater., 2019, 48, 4653–4659 CrossRef CAS.
- Q. Liu, Z. Hu, L. Li, W. Li, C. Zou, H. Jin, S. Wang and S.-L. Chou, ACS Appl. Mater. Interfaces, 2021, 13, 16585 CrossRef CAS PubMed.
- B. Ge, Y. Sun, J. Guo, X. Yan, C. Fernandez and Q. Peng, Small, 2019, 15, e1902220 CrossRef PubMed.
- Y. Mao, C. Tang, Z. Tang, J. Xie, Z. Chen, J. Tu, G. Cao and X. Zhao, Energy Storage Mater., 2019, 18, 405 CrossRef.
- Z. Tang, M. Yuan, H. Zhu, G. Zeng, J. Liu, J. Duan and Z. Chen, Front. Chem., 2021, 9, 670612 CrossRef CAS.
- Q. Deng, Y. Yang, S. Qu, W. Wang, Y. Zhang and X. Ma, Energy Storage Mater., 2021, 42, 484 CrossRef.
- J. Xia, Y. Zhou, J. Zhang, T. Lu, W. Gong, D. Zhang, X. Wang and J. Di, Small, 2023, 19, 2301906 CrossRef CAS PubMed.
- X. Guo, T. Zheng, G. Ji, N. Hu, C. Xu and Y. Zhang, J. Mater. Chem. A, 2018, 6, 10243 RSC.
- M. Al-Dossary, A. A. Ismail, J. L. G. Fierro, H. Bouzid and S. A. Al-Sayari, Appl. Catal., B, 2015, 165, 651 CrossRef CAS.
- J. Chen, X. Qi, C. Liu, J. Zeng and T. Liang, ACS Appl. Mater. Interfaces, 2020, 12, 51418 CrossRef CAS.
- W. Ma, S. Lu, X. Lei, X. Liu and Y. Ding, J. Mater. Chem. A, 2018, 6, 20829 RSC.
- X. Zhang, C. Wang, H. Li, X.-G. Wang, Y.-N. Chen, Z. Xie and Z. Zhou, J. Mater. Chem. A, 2018, 6, 2792 RSC.
- L. Liu, L. Zhang, K. Wang, H. Wu, H. Mao, L. Li, Z. Sun, S. Lu, D. Zhang, W. Yu and S. Ding, ACS Appl. Mater. Interfaces, 2020, 12, 33846 CrossRef CAS.
- S. Thoka, C.-J. Chen, A. Jena, F.-M. Wang, X.-C. Wang, H. Chang, S.-F. Hu and R.-S. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 17353 CrossRef CAS PubMed.
- Y. Qiao, J. Wu, J. Zhao, Q. Li, P. Zhang, C. Hao, X. Liu, S. Yang and Y. Liu, Energy Storage Mater., 2020, 27, 133 CrossRef.
- D. Na, H. Jeong, J. Baek, H. Yu, S.-M. Lee, C.-R. Lee, H.-K. Seo, J.-K. Kim and I. Seo, Electrochim. Acta, 2022, 419, 140408 CrossRef CAS.
- A. Bharti, G. Manna, P. Saha, G. Achutarao and A. J. Bhattacharyya, J. Phys. Chem. Lett., 2022, 13, 7380 CrossRef CAS PubMed.
- W. Ma, S. Lu, X. Lei, X. Liu and Y. Ding, J. Mater. Chem. A, 2018, 6, 20829 RSC.
- H. Liao, X. Guo, Y. Hou, H. Liang, Z. Zhou and H. Yang, Small, 2020, 16, e1905223 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05794a |
| ‡ X.-Y. C and. J. C. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |