
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective nickel-catalyzed Mizoroki–Heck cyclizations of amide electrophiles†
Ana S.
Bulger‡
 ,
Daniel J.
Nasrallah‡
,
Arismel
Tena Meza‡
and
Neil K.
Garg
*
,
Daniel J.
Nasrallah‡
,
Arismel
Tena Meza‡
and
Neil K.
Garg
*
Department of Chemistry and Biochemistry, University of California at Los Angeles, Los Angeles, California 90095, USA. E-mail: neilgarg@chem.ucla.edu
First published on 8th January 2024
Abstract
Amide cross-couplings that rely on C–N bond activation by transition metal catalysts have emerged as valuable synthetic tools. Despite numerous discoveries in this field, no catalytic asymmetric variants have been disclosed to date. Herein, we demonstrate the first such transformation, which is the Mizoroki–Heck cyclization of amide substrates using asymmetric nickel catalysis. This proof-of-concept study provides an entryway to complex enantioenriched polycyclic scaffolds and advances the field of amide C–N bond activation chemistry.
Introduction
Metal-catalyzed cross-coupling reactions are essential tools for synthetic chemists.1 Our laboratory and others have been interested in investigating unconventional cross-coupling partners to discover new fundamental reactivity, enable chemoselective reactions, and advance the field by expanding the synthetic toolbox.2 One class of unconventional cross-coupling handles, acyl electrophiles3 (Fig. 1A, 2) are underexplored compared to traditional electrophiles, such as aryl halides and pseudohalides. Of the known acyl electrophiles, thioesters (2a) have been investigated the most in conventional asymmetric transition-metal catalyzed cross-couplings (i.e., 2a → 1).4 Anhydrides (2b) and acid chlorides (2c) have also been used in asymmetric catalysis in the context of reductive or photochemical cross-couplings (2b–c → 1).5 Recently, the far less reactive acyl electrophiles, esters6 (2d) and amides7 (2e), have been investigated as electrophiles in C–C and C–heteroatom bond-forming reactions. Of note, no catalytic asymmetric examples have been reported using these species as electrophiles (i.e., 2d or 2e → 3).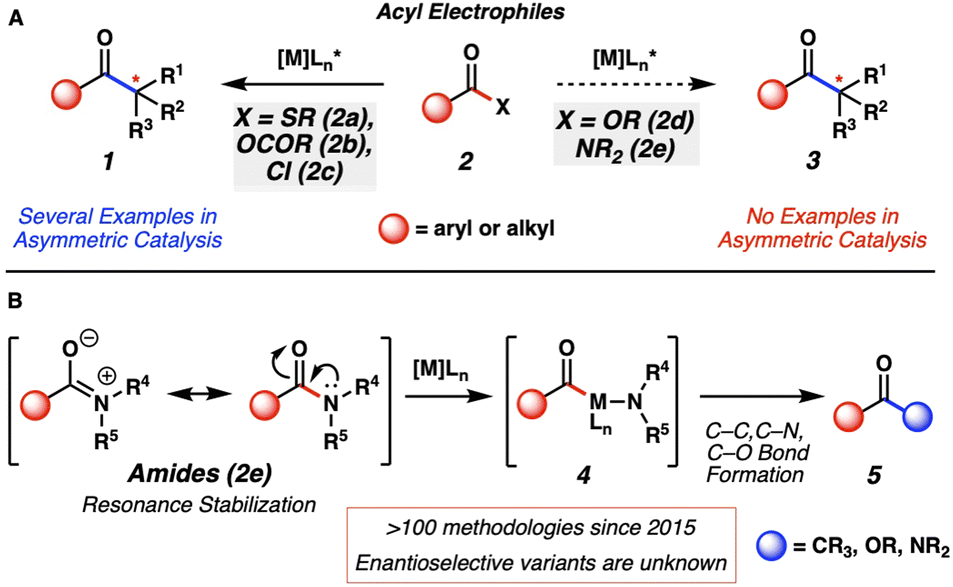 | ||
| Fig. 1 (A) Asymmetric metal-catalyzed reactions of acyl electrophiles and current limitations. (B) Current state-of-the-art of amide cross-coupling reactions. | ||
Amides (Fig. 1B, 2e) are particularly attractive as synthetic building blocks. They are known for their pronounced stability under various reaction conditions, including hydrolysis and redox processes. Amides can also be used as directing groups in other useful processes, such as aryl C–H bond functionalization.8 Thus, amides have the potential to be employed in synthetic sequences, then activated late-stage to access a variety of other functional groups. The pronounced stability of amides is largely attributed to resonance delocalization of the nitrogen lone pair.9 This leads to the high kinetic barrier associated with breaking the amide C–N bond and has led to the historical avoidance of amides as versatile synthetic handles.10 Thus, our laboratory and others11 have been interested in expanding known reactivity of these historically underutilized functional handles under mild conditions. Our specific interests pertain to cross-coupling reactions of amides using transition-metal catalysis, which led to our first report of an amide cross-coupling, published in 2015.7a Subsequently, amides have become versatile electrophiles in Pd- and Ni-catalyzed cross-coupling reactions.
Mechanistically, these reactions are typically thought to proceed by oxidative addition to the amide C–N bond to generate acyl metal species 4.7a Subsequent interception by a nucleophile gives rise to products 5. Using this platform, amides can now be readily converted to functional groups such as ketones, carboxylic acids, esters, and other amides, through relatively simple net substitution processes. We questioned if the use of a chiral ligand on the metal could provide an opportunity to access enantioenriched products. Despite there being more than a hundred amide cross-couplings involving C–N bond activation reported since 2015,12 asymmetric variants that utilize a chiral metal catalyst have not been discovered. Such processes would provide a new avenue in amide bond activation chemistry to allow access to enantioenriched molecules from simple amide precursors.
We envisioned that an asymmetric Mizoroki–Heck cyclization would be a suitable platform to access enantioenriched products from amide electrophiles.13 Our reaction design, inspired by prior racemic Heck-type reactions of amide electrophiles discovered by our laboratory7j and Stanley's,7l and other asymmetric examples involving acyl-metal intermediates14,15 is shown in Fig. 2. Representative amide substrate 6 bearing a tethered olefin would undergo oxidative addition using a metal catalyst to form acyl metal species 7. Subsequent olefin coordination and migratory insertion (the enantiodetermining step) would furnish intermediate 8. β-Hydride elimination would then provide the desired product 9. Importantly, 9 would bear a stereodefined quaternary center, which is notable as quaternary stereocenters remain challenging to access and are highly sought-after motifs.16
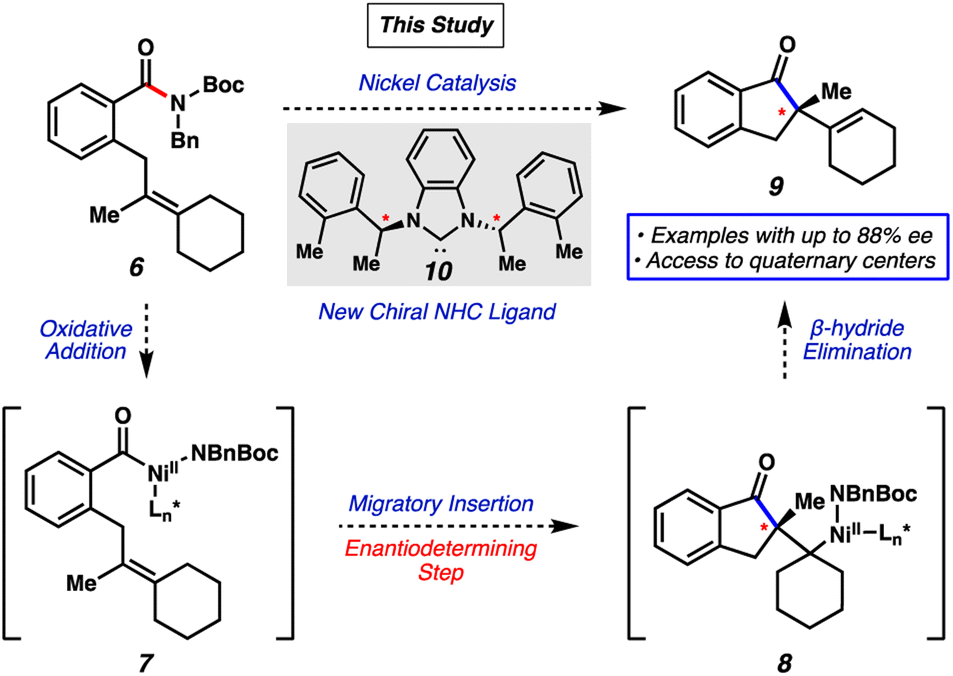 | ||
| Fig. 2 Overview of the reaction design and proposed mechanism. | ||
Herein, we demonstrate the success of this approach, which provides the first asymmetric transition-metal-catalyzed reaction involving amide C–N bond cleavage. The transformation is enabled by the design of a new chiral N-heterocyclic carbene (NHC)17 ligand 10 (Fig. 2) and the use of amine additives, and ultimately delivers select products containing quaternary centers in up to 88% enantiomeric excess (ee). Although currently limited in scope, this proof-of-concept study allows access to several highly complex, enantioenriched scaffolds. Moreover, this study demonstrates the ability to use catalytic amide C–N bond cleavage as an approach to achieve enantioselective synthesis.
Results and discussion
Our studies commenced by attempting the asymmetric Mizoroki–Heck cyclization using nickel catalysis (Fig. 3). Prior studies involving amide substrates with tethered olefins (e.g., 11) showed that a highly nucleophilic metal (i.e., nickel18), accompanied by strongly electron-donating NHC ligands was required to activate the sterically encumbered and electron-rich amide C–N bond. A small library of new and known chiral NHC salts was accessible by a combination of acquisition from colleagues in academia (Fig. 3 top panel; see acknowledgements) and chemical synthesis (Fig. 3 bottom panel). Substrate 11 was utilized for initial studies and select results are shown in Fig. 3, with further results available in the ESI.†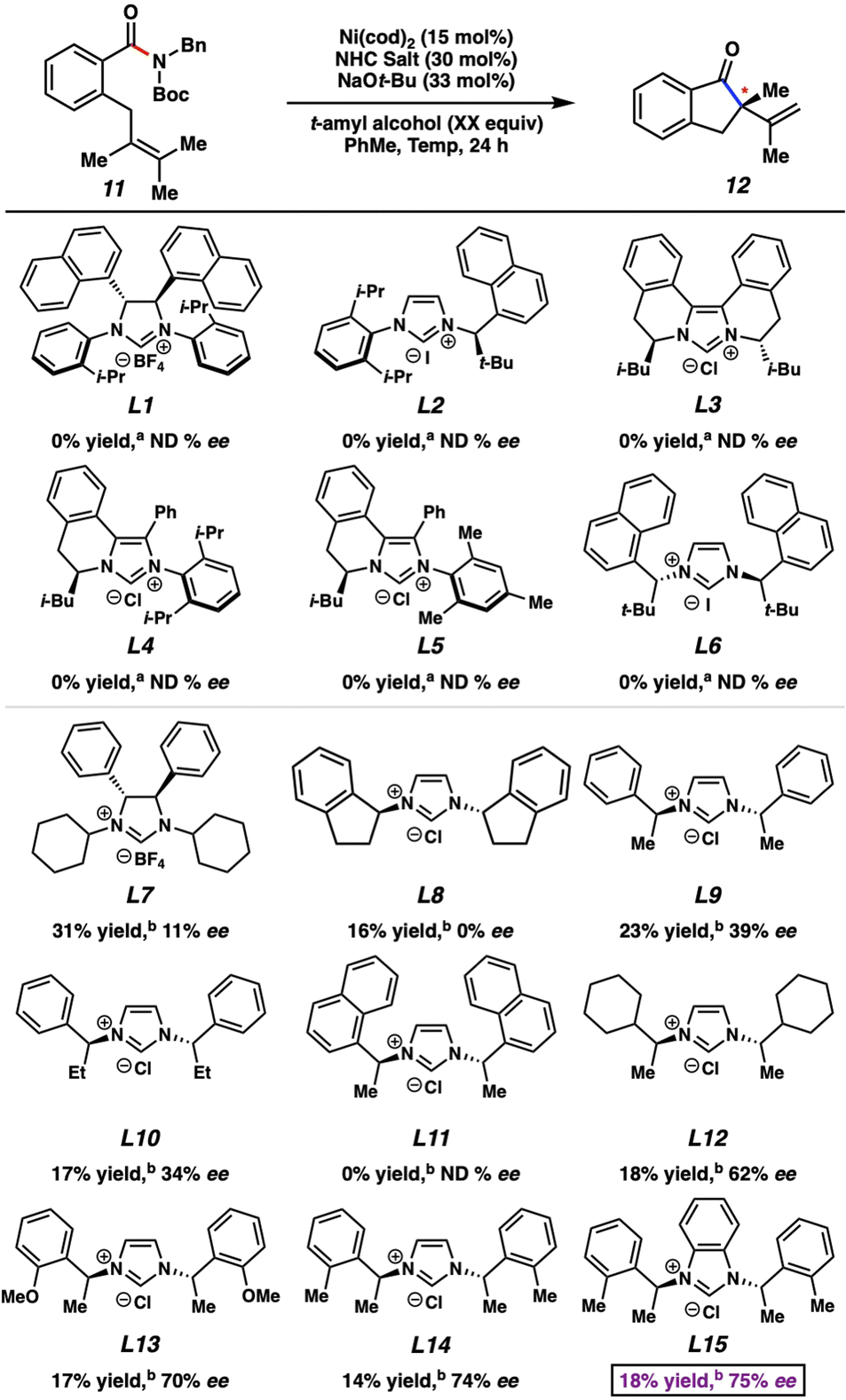 | ||
| Fig. 3 Survey of chiral NHC salts with in situ free-basing. aConditions: Ni(cod)2 (15 mol%), NHC Salt (30 mol%), NaOt-Bu (33 mol%), PhMe (0.5 M), 100 °C; 24 h in a sealed vial. bConditions: Ni(cod)2 (15 mol%), NHC Salt (30 mol%), NaOt-Bu (33 mol%), t-amyl alcohol (3.0 equiv.), PhMe (0.5 M), 60 °C; 24 h in a sealed vial. a,bYields determined by 1H NMR using hexamethylbenzene as an external standard. ND is defined as not determined. | ||
Known chiral NHC salts L1–L6 were tested using conditions developed for the racemic transformation7j to effect the desired conversion of amide 11 to indanone 12. Despite the fact that these ligand salts have been employed in asymmetric reactions such as Ni-catalyzed annulations,19 Pd-catalyzed C–H activation,20 Cu-catalyzed allylic alkylations21 and conjugate borylations,22 they were found to be ineffective, even at elevated reaction temperatures. We attribute this lack of reactivity to the high degree of steric hindrance and structural rigidity of these chiral NHC ligands, which could render oxidative addition challenging. In order to circumvent this issue, we examined less sterically bulky ligand salts to potentially enable oxidative addition, while still permitting selectivity in the enantiodetermining step. Using L7–L9 we saw more promising results. Of these, L9, which has acyclic chiral centers in close proximity to the bound metal, gave the best combination of yield and enantioenrichment of 12 (23% yield with 39% ee). Whereas using a ligand salt with ethyl groups in place of methyl groups (i.e., L10) led to minimal change, more pronounced effects were seen by altering the phenyl ring on L9. For example, the use of L11, which contains bulky naphthyl groups, led to no reaction, whereas the use of L12–L14 gave product 12 with higher enantioselectivity. Finally, the use of L15, the benzimidazolium derivative of L14, led to the formation of 12 in 18% yield with 75% ee. Importantly, the two most promising NHC salts, L14 and L15, were not known prior to our investigations, and NHC salts L9, L12, and L13 had not been used previously in nickel catalysis.
To better understand the influence of different olefin substitution patterns on enantioselectivity, we tested amide substrate 6 using the most promising ligands from our aforementioned studies of substrate 11 (Fig. 4). Specifically, amide 6 bears a more sterically congested olefin with the incorporation of an additional ring. Although the use of L7 led to poor enantioselectivity, more promising results were observed using ligand salts L9 and L12–L15. The use of L15 produced the best combination of yield and enantiomeric excess, furnishing 9 in 32% yield and 88% ee. Although beyond the scope of our present study, these ligands may prove valuable in other applications, such as in the asymmetric reactions of other electrophiles that contain strong bonds.
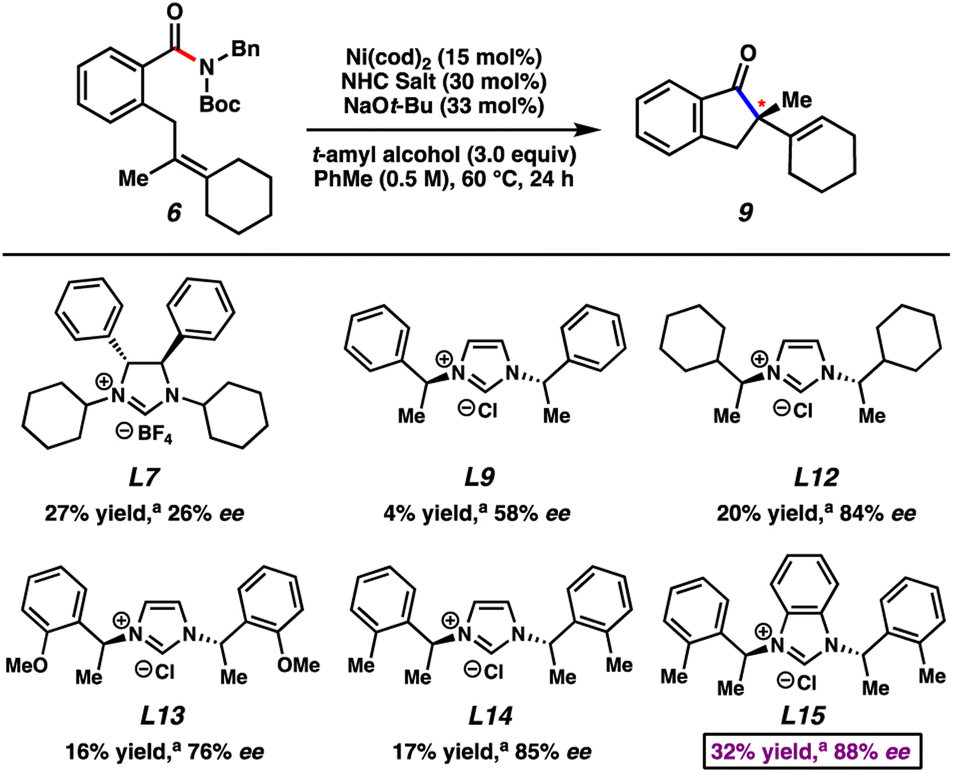 | ||
| Fig. 4 Results using select chiral NHC ligand salts with amide 6. aYields determined by 1H NMR using hexamethylbenzene as an external standard. | ||
We attempted an initial optimization, focusing on increasing the yield of the product, using substrate 6 with L15. Gratifyingly, this new chiral NHC salt, L15, can be readily synthesized from commercially available building blocks on gram-scale and is bench-stable, rendering it an attractive choice for practical applications. Attempts to optimize the reaction by varying the stoichiometry of NaOt-Bu and the identity of the base did not lead to significant increase in yield (see ESI† for details). Exhaustive optimization with L15 involving other reaction parameters, such as variation of amide N-substituents, solvents, and stoichiometric loading of reagents did not lead to significant catalytic turnover. Thus, in order to reduce variables in the reaction and mitigate potential unwanted base-mediated side pathways (see ESI† for details), we sought to generate the free carbene of L15 and subsequently add it as a discrete reagent (Fig. 5). Accordingly, this would avoid the use of a base in the catalytic transformation. To our delight, carbene 10 could be generated in solution upon treatment of L15 with KOt-Bu, analogous to a procedure previously reported by Cramer.23 The identification of 10 was made using 13C NMR, as 10 displays a diagnostic signal at 225.0 ppm. Carbene 10 was found to be stable under inert conditions in toluene for multiple weeks without any detectable dimerization.
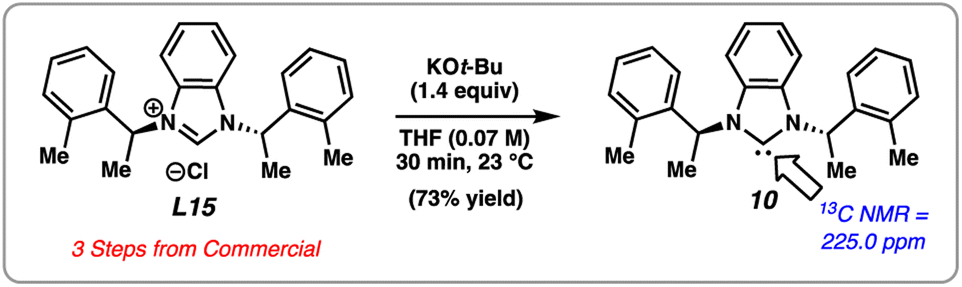 | ||
| Fig. 5 Generation of free carbene 10. | ||
Thorough variation of reaction parameters (e.g., variation of N-substitution,24 temperature, solvent, catalyst loading) was performed in the absence of an additive, but did not lead to improved outcomes (see ESI† for details). We turned to examining additives, with inspiration from the literature.25 Additionally, the improvement seen in employing t-amyl alcohol (13) as an additive (Fig. 6, entry 2) compared to the lower yield observed in the absence of additive (entry 1) signified that an additive could be important in increasing the conversion of the reaction. A selection of results are shown in Fig. 6, with further results available in the ESI.† Whereas the use of other alcohols (e.g., 14) was ultimately found to give comparable results, amine additives were generally found to be most impactful, with varying results.26 For example, when primary amines 15 and 16 were evaluated, α-tertiary amine 16 was found to improve the yield of the reaction to 42%, whereas α-primary amine 15 inhibited product formation, giving only 4% yield of 9. Moreover, the use of amines 17–19, which represent acyclic secondary, tertiary, and diamines, respectively, did not lead to superior yields of 9. Cyclic amines were also evaluated. Although the use of piperidine (20) as an additive was not very promising (entry 9), the employment of morpholine (21) led to complete consumption of substrate 6 and 42% yield of indanone 9 (entry 10). The majority of the remaining mass balance was comprised of an undesired morpholine transamidation side-product. As such, the equivalents of morpholine were reduced from three to one. Using these optimized conditions gave indanone 9 in 55% yield with 88% ee (entry 11). It is hypothesized that the additive may have an effect on either the rate of β-hydride elimination, potentially due to reduced steric bulk around the metal, or that it could facilitate regeneration of the Ni(0) catalyst. However, future mechanistic studies would be necessary to support either conclusion as to the effect of the amine additive on the turnover-limiting step of this reaction.27,28 Nonetheless, the findings with regard to ligand optimization and additive evaluation should provide insight into effective reaction conditions for the activation of strong-bond electrophiles in asymmetric catalytic processes.
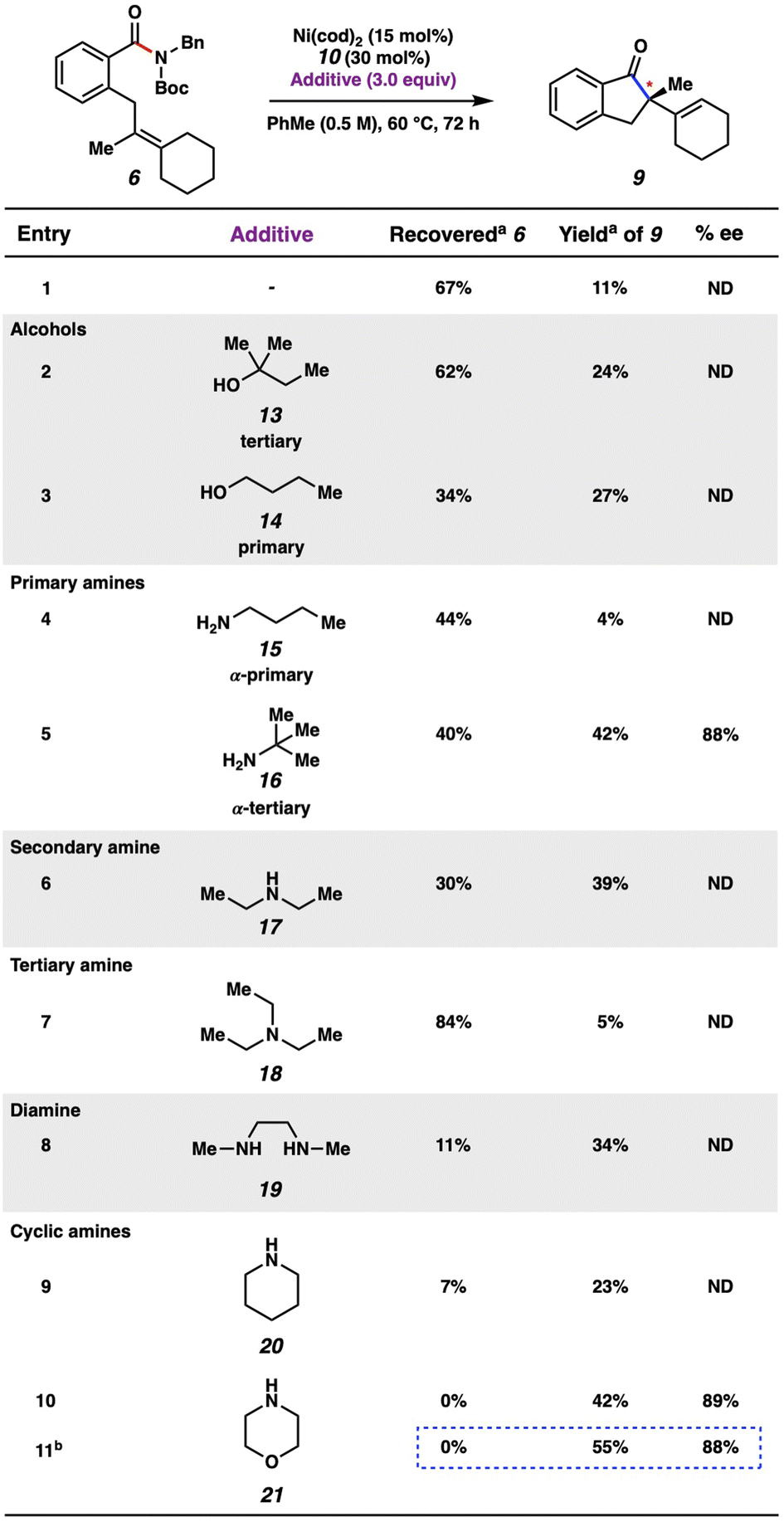 | ||
| Fig. 6 Survey of alcohol and amine additives in the transformation. aConversions and yields determined by 1H NMR using hexamethylbenzene as an external standard. bMorpholine (1.0 equiv.). | ||
Several substrates bearing different olefin substitution patterns were evaluated in the transformation (Fig. 7). Cyclohexylidene substrate 6 underwent the desired enantioselective transformation to deliver 9 in 54% isolated yield and 88% ee. Additionally, we demonstrated the tolerance of heterocycles through the employment of amides 22 and 24, which gave rise to products 23 and 25, respectively, with good enantioenrichment, albeit in modest yields. Amide 26, bearing a sterically bulky geminal dimethyl moiety, gave rise to spirocycle 27 in good yield and moderate enantiomeric excess (80% yield with 54% ee). Additionally, an acyclic olefin (entry 5) could be tolerated in the methodology, giving rise to indanone 12 in 41% yield with 78% ee. Although the scope of the transformation is currently limited, these examples demonstrate the potential for amides to serve as acyl electrophiles in enantioselective transformations with tolerance of heterocycles and tri- or tetra-substituted olefins.
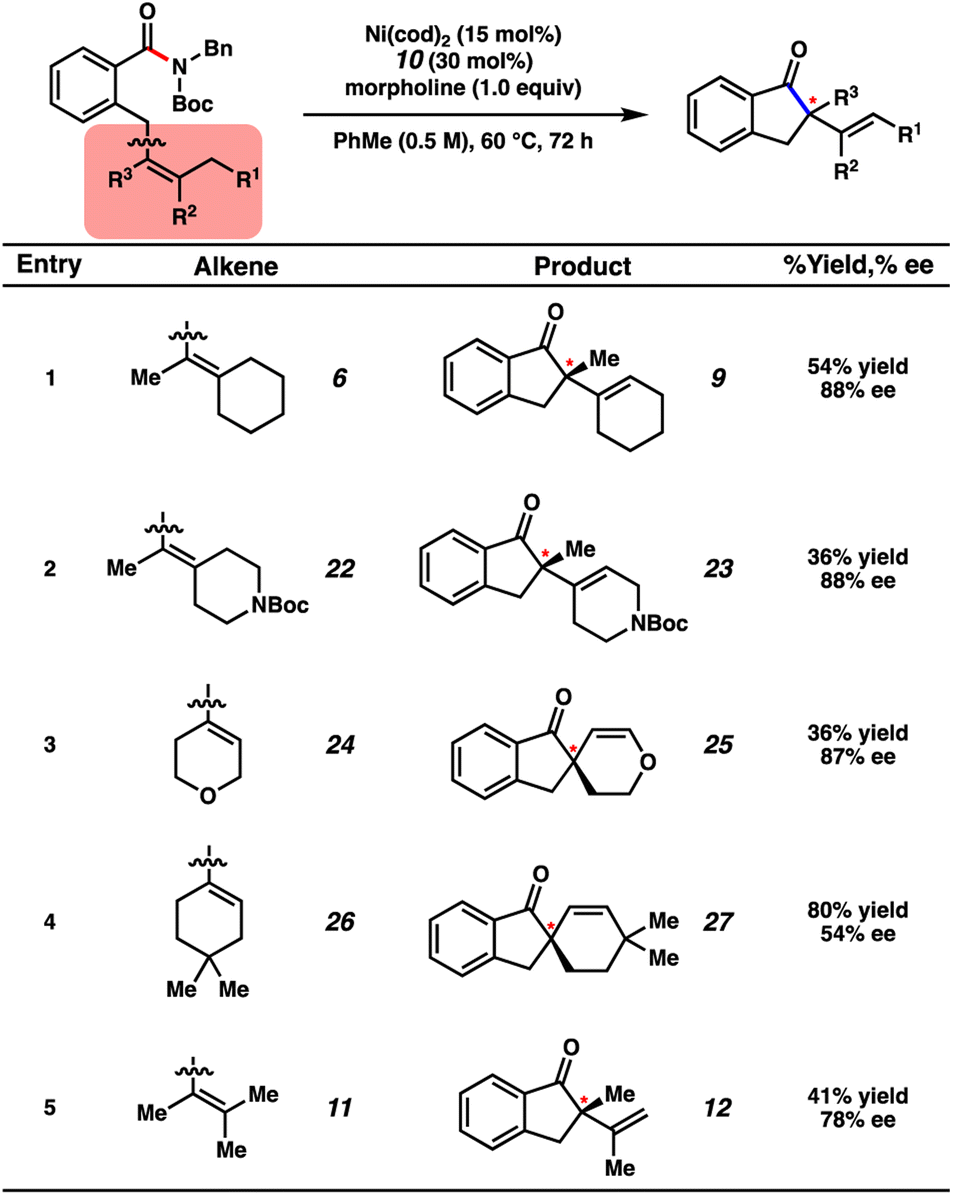 | ||
| Fig. 7 Examples of methodology. Yields shown reflect the average of two isolation experiments. | ||
The Mizoroki–Heck cyclization provides access to complex quaternary center-containing products that are well poised to undergo further synthetic elaboration. As shown in Fig. 8, we demonstrate this virtue through the elaboration of cyclization product 9 to several scaffolds, including stereochemically complex products. One key effort involved condensation of indanone 9 with N-brosyl hydrazine to give brosyl hydrazone 28. This adduct could be crystallized, which enabled the assignment of absolute stereochemistry through single crystal X-ray analysis. The absolute configuration at the quaternary stereocenter (in 28 and other derivatives of 9) is as depicted throughout Fig. 8.
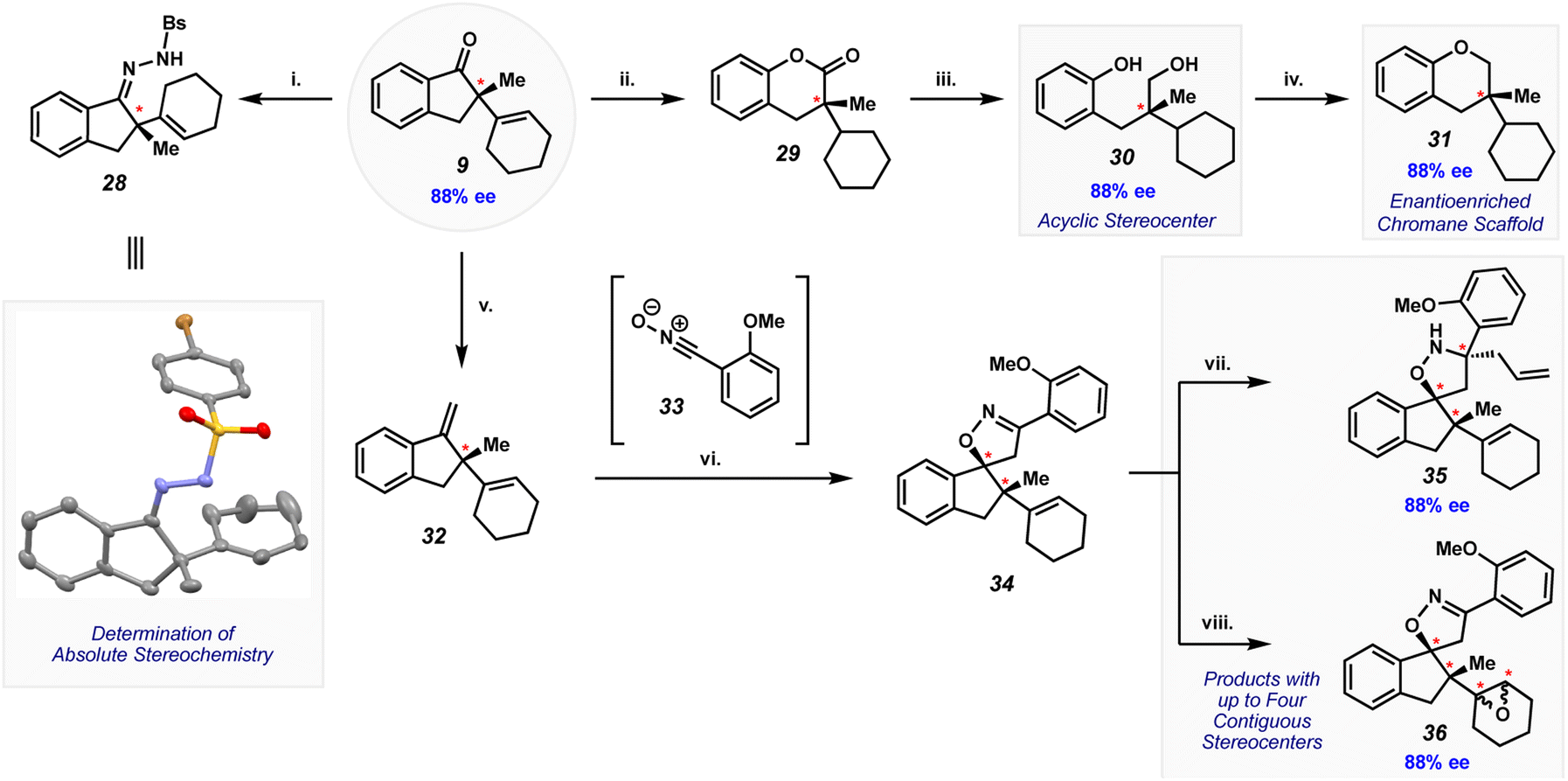 | ||
Fig. 8 Elaboration of (+)-9 to enantioenriched compounds. (i) N-Brosyl hydrazine (5.0 equiv.), acetyl chloride (10 equiv.), EtOH (0.07 M), 0 → 60 °C; 72 h, 47% yield. (ii) (a) Fe(acac)3 (40 mol%), PhSH (40 mol%), PhSiH3 (4.0 equiv.), EtOH (0.1 M), 23 °C, 25 h. (b) TBAF (25 equiv.), 16 h, 87% yield over two steps (c) urea hydrogen peroxide (43 equiv.), TFA (19 equiv.), BF3·OEt3 (73 equiv.), CH2Cl2 (0.1 M), 0 → 23 °C, 16 h, 60% yield, 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 rr. (iii) LiAlH4 (1.5 equiv.), Et2O (0.03 M), 0 → 23 °C, 0.5 h, quant. yield. (iv) PPh3 (1.5 equiv.), DBAD (1.5 equiv.), CH2Cl2 (0.07 M), 10 °C, 3 h, quant. yield. (v) PPh3MeBr (2.4 equiv.), n-BuLi (1.7 equiv.), THF (0.2 M), 23 °C, 2 h, 97% yield. (vi) 2-(Methoxy)benzohydroximinoyl chloride (2.0 equiv.), NEt3 (2.0 equiv.), MeCN (0.1 M), 23 °C, 25 h, 79% yield, 5.3:1 dr. (vii) BF3·OEt2 (3.0 equiv.), allylMgCl (15 equiv.), toluene (0.05 M), −78 °C, 2 h, 63% yield, 1.6:1 dr. (viii) m-CPBA (1.5 equiv.), NaHCO3 (3.0 equiv.), CH2Cl2 (0.07 M), 0 °C, 2 h, 83% yield, 2.2:1 dr. :1 rr. (iii) LiAlH4 (1.5 equiv.), Et2O (0.03 M), 0 → 23 °C, 0.5 h, quant. yield. (iv) PPh3 (1.5 equiv.), DBAD (1.5 equiv.), CH2Cl2 (0.07 M), 10 °C, 3 h, quant. yield. (v) PPh3MeBr (2.4 equiv.), n-BuLi (1.7 equiv.), THF (0.2 M), 23 °C, 2 h, 97% yield. (vi) 2-(Methoxy)benzohydroximinoyl chloride (2.0 equiv.), NEt3 (2.0 equiv.), MeCN (0.1 M), 23 °C, 25 h, 79% yield, 5.3:1 dr. (vii) BF3·OEt2 (3.0 equiv.), allylMgCl (15 equiv.), toluene (0.05 M), −78 °C, 2 h, 63% yield, 1.6:1 dr. (viii) m-CPBA (1.5 equiv.), NaHCO3 (3.0 equiv.), CH2Cl2 (0.07 M), 0 °C, 2 h, 83% yield, 2.2:1 dr. | ||
Compounds 29–31 are additional derivatives of indanone 9 that each bear a defined quaternary stereocenter (Fig. 8). Although the scaffolds for these derivatives depart from that of 9, each are readily accessible through the depicted sequence. Lactone 29 was obtained by reduction of the olefin using hydrogen-atom transfer (HAT) conditions, followed by Baeyer–Villiger oxidation. Using urea hydrogen peroxide, trifluoroacetic acid (TFA), and boron trifluoride etherate (BF3·OEt2), the product arising from the migration of the phenyl group was observed as the major regioisomer. Subsequent reduction of lactone 29 using lithium aluminum hydride led to ring cleavage, providing diol 30 in quantitative yield. Lastly, diol 30 underwent an intramolecular Mitsunobu reaction to form chromane 31, a common motif in bioactive molecules and pharmaceuticals.29 The elaboration of 9 to enantioenriched products 29–31 is reminiscent of the skeletal editing paradigm,30 which has recently received significant interest from the synthetic community.
As reflected in the remaining sequence shown in Fig. 8, we sought to elaborate indanone 9 to enantioenriched products that bear additional rings and multiple stereocenters. Wittig homologation of 9 provided olefin 32 in nearly quantitative yield. A (3 + 2) cycloaddition of 32 with in situ-generated nitrile oxide 33 delivered spiroheterocycle 34. The reaction proceeds with high chemoselectivity for the more reactive styrenyl olefin in 32. Moreover, the cycloaddition proceeds diastereoselectively (5.3:1 dr), with a preference for the isomer shown, presumably by approach of the 1,3-dipole from the less sterically encumbered face of the styrenyl olefin. Finally, spiroheterocycle 34 could be elaborated further to isoxazolidine 35 and epoxides 36. The former was obtained by 1,2-addition of allyl magnesium chloride, while the latter was accessed by epoxidation of the trisubstituted cyclic olefin. In addition to retaining the quaternary stereocenter generated in the Mizoroki–Heck cyclization, products 35 and 36 contain multiple stereocenters, as well as multiple heteroatoms, rings, and functional handles for further manipulation. The results shown in Fig. 8 underscore the notion that a range of highly complex, enantioenriched scaffolds can now be accessed from amide precursors, ultimately stemming from our methodology involving amide C–N bond activation and asymmetric catalysis.
Conclusions
We have demonstrated that amides can serve as cross-coupling partners in the asymmetric transition-metal-catalyzed Mizoroki–Heck cyclization. This is notable, as more than a hundred amide cross-couplings involving C–N bond cleavage by a transition metal catalyst have been reported since 2015, but none are asymmetric. Our transformation was enabled by the development of a new NHC ligand 10, as well as extensive reaction optimization. Through cyclization and subsequent synthetic elaborations, we show that a variety of enantioenriched polycyclic products containing difficult-to-access quaternary centers can be made, including complex compounds with up to four stereogenic centers (i.e., 36). Future studies aim to expand the scope of this reaction and develop new methods that rely on the use of amides in C–N bond cleavage reactions using asymmetric transition metal catalysis. Our studies challenge the conventional understanding of how amides can be used in chemical synthesis and, more broadly, should encourage the study of other non-traditional electrophiles in asymmetric transformations by expanding the library of, and providing knowledge on, the types of ligands that could be employed in such transformations.Data availability
Full details on the synthesis and characterization of compounds are accessible in the ESI.†Author contributions
A. S. B., D. J. N., and A. T. M. designed and performed experiments and analyzed experimental data. All authors contributed to the preparation of the manuscript and participated in discussions. N. K. G. directed the investigations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NIH-NIGMS (R35 GM139593 for N. K. G. and Supplement for A. T. M., and F31 GM149161 for A. T. M), and the Trueblood Family (for N. K. G.), as well as the NSF (DGE-2034835 for A. S. B.), and the Foote Family (for A. S. B. and A. T. M.). We thank Dr Saeed Khan (UCLA) for X-ray analysis. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804), the National Center for Research Resources (S10RR025631), and the NIH Office of Research Infrastructure Programs (S10OD028644). The authors acknowledge Professor Kendall Houk (UCLA) and Pan-Pan Chen (UCLA) for helpful discussions. The authors are also grateful to Dr Sophie Racine (UCLA) for experimental assistance and Professor Nicolai Cramer (EPFL) for gifting chiral NHC salts.Notes and references
- (a) C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (b) J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed; (c) L. Jiang and S. L. Buchwald, in Metal-Catalyzed Cross- Coupling Reactions, 2nd edn, ed. A. Meijere and F. Diederich, Wiley VCH, Weinheim, 2004, pp. 699−760 Search PubMed; (d) J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed; (e) E. Negishi, Bull. Chem. Soc. Jpn., 2007, 80, 233–257 CrossRef CAS; (f) H. C. Shen, in Application of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, ed. M. L. Crawley and B. M. Trost, Wiley, Hoboken, 2012, pp. 25−96 Search PubMed; (g) L. C. Campeau and N. Hazari, Organometallics, 2019, 38, 3–35 CrossRef CAS PubMed; (h) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed; (i) E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- For select reviews on C–O, C–N and C–F bond cleaving reactions, see: (a) T. B. Boit, A. S. Bulger, J. E. Dander and N. K. Garg, ACS Catal., 2020, 10, 12109–12126 CrossRef CAS PubMed; (b) S. Q. Zhang and X. Hong, Acc. Chem. Res., 2021, 54, 2158–2171 CrossRef CAS PubMed; (c) B. A. Baviskar, P. V. Ajmire, D. S. Chumbhale, M. S. Khan, V. G. Kuchake, M. Singupuram and P. R. Laddha, Sustainable Chem. Pharm., 2023, 32, 100953 CrossRef CAS; (d) X. Zeng and X. Huang, Adv. Anal. Chem., 2022, 12, 68–75 CrossRef; (e) M. Tian and M. Liu, Pure Appl. Chem., 2021, 93, 799–810 CrossRef CAS; (f) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC; (g) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717–1726 CrossRef CAS PubMed; (h) F. Liu, H. J. Jiang, Y. Zhou and Z. Shi, Chin. J. Chem., 2020, 38, 855–863 CrossRef CAS; (i) T. Zhou and M. Szostak, Catal. Sci. Technol., 2020, 10, 5702–5739 RSC; (j) Z. Qiu and C. J. Li, Chem. Rev., 2020, 120, 10454–10515 CrossRef CAS PubMed; (k) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (l) L. Fu, Q. Chen and Y. Nishihara, Chem. Rec., 2021, 21, 3394–3410 CrossRef CAS PubMed; (m) J. Becica and D. C. Leitch, Synlett, 2021, 32, 641–646 CrossRef CAS.
- For select reviews on acyl electrophiles see: (a) J. Buchspies and M. Szostak, Catal., 2019, 9, 53 Search PubMed; (b) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC; (c) T. Zhou and M. Szostak, Catal. Sci. Technol., 2020, 10, 5702–5739 RSC; (d) L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef PubMed; (e) J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413–1423 CrossRef CAS PubMed; (f) G. Li, S. Ma and M. Szostak, Trends Chem., 2020, 2, 914–928 CrossRef CAS.
- For this context, “conventional” is used to refer to cross-couplings employing a nucleophile and electrophile. For select studies on asymmetric transition-metal-catalyzed reactions using thioester electrophiles, see: (a) F. Banchini, B. Leroux, E. Le Gall, M. Presset, O. Jackowski, F. Chemla and A. Perez-Luna, Chem.–Eur. J., 2023, e202301084 CrossRef CAS PubMed; (b) R. Oost, A. Misale and N. Maulide, Angew. Chem., Int. Ed., 2016, 55, 4587–4590 CrossRef CAS PubMed; (c) M. Liu, X. Wang, Z. Guo, H. Li, W. Huang, H. Xu and H.-X. Dai, Org. Lett., 2021, 23, 6299–6304 CrossRef CAS PubMed.
- For select studies on asymmetric transition-metal-catalyzed reductive or photochemical cross-coupling reactions using anhydride or acid chloride electrophiles, see: (a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442–7445 CrossRef CAS PubMed; (b) H. Ji, D. Lin, L. Tai, X. Li, Y. Shi, Q. Han and L.-A. Chen, J. Am. Chem. Soc., 2022, 144, 23019–23029 CrossRef CAS PubMed; (c) E. Gandolfo, X. Tang, S. R. Roy and P. Melchiorre, Angew. Chem., 2019, 131, 17010–17014 CrossRef.
- For select recent work on transition-metal-catalyzed ester C–O bond activation and cross-couplings, see: (a) T. Yamamoto, J. Ishizu, T. Kohara, S. Komiya and A. Yamamoto, J. Am. Chem. Soc., 1980, 102, 3758–3764 CrossRef CAS; (b) K. Amaike, K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 13573–13576 CrossRef CAS PubMed; (c) L. Meng, Y. Kamada, K. Muto, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2013, 52, 10048–10051 CrossRef CAS PubMed; (d) K. Muto, J. Yamaguchi, D. G. Musaev and K. Itami, Nat. Commun., 2015, 6, 7508 CrossRef PubMed; (e) K. Muto, R. Hatakeyama, K. Itami and J. Yamaguchi, Org. Lett., 2016, 18, 5106–5109 CrossRef CAS PubMed; (f) L. J. Gooßen and J. Paetzold, Angew. Chem., Int. Ed., 2002, 41, 1237–1241 CrossRef; (g) L. J. Gooßen and J. Paetzold, Angew. Chem., Int. Ed., 2004, 43, 1095–1098 CrossRef PubMed; (h) N. Chatani, H. Tatamidani, Y. Ie, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 4849–4850 CrossRef CAS PubMed; (i) H. Tatamidani, K. Yokota, F. Kakiuchi and N. Chatani, J. Org. Chem., 2004, 69, 5615–5621 CrossRef CAS PubMed; (j) H. Tatamidani, F. Kakiuchi and N. Chatani, Org. Lett., 2004, 6, 3597–3599 CrossRef CAS PubMed; (k) N. A. LaBerge and J. A. Love, Eur. J. Org Chem., 2015, 2015, 5546–5553 CrossRef CAS; (l) T. Ben Halima, J. K. Vandavasi, M. Shkoor and S. G. Newman, ACS Catal., 2017, 7, 2176–2180 CrossRef CAS; (m) A. Kruckenberg, H. Wadepohl and L. H. Gade, Organometallics, 2013, 32, 5153–5170 CrossRef CAS; (n) H. Yue, L. Guo, H.-H. Liao, Y. Cai, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 4282–4285 CrossRef CAS PubMed; (o) H. Yu, L. Guo, S.-C. Lee, X. Liu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 3972–3976 CrossRef PubMed; (p) T. Ben Halima, W. Zhang, I. Yalaoui, X. Hong, Y. Yang, K. N. Houk and S. G. Newman, J. Am. Chem. Soc., 2017, 139, 1311–1318 CrossRef CAS PubMed; (q) L. Hie, N. Fine Nathel, X. Hong, Y.-F. Yang, K. N. Houk and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 2810–2814 CrossRef CAS PubMed; (r) T. Ben Halima, J. Masson-Makdissi and S. G. Newman, Angew. Chem., Int. Ed., 2018, 57, 12925–12929 CrossRef CAS PubMed; (s) Y.-L. Zheng and S. G. Newman, ACS Catal., 2019, 9, 4426–4433 CrossRef CAS; (t) C. W. Cheung, M. L. Ploeger and X. Hu, Nat. Commun., 2017, 8, 14878 CrossRef PubMed; (u) H. K. Banovetz, K. L. Vickerman, C. M. David, M. Alkan and L. M. Stanley, Org. Lett., 2021, 23, 3507–3521 CrossRef CAS PubMed; (v) Y.-L. Zheng, P.-P. Xie, O. Daneshfar, K. N. Houk, X. Hong and S. G. Newman, Angew. Chem., 2021, 133, 13588–13595 CrossRef; (w) S. Yang, T. Zhou, A. Poater, L. Cavallo, S. P. Nolan and M. Szostak, Catal. Sci. Technol., 2021, 11, 3189–3197 RSC.
- For recent articles on nickel-catalyzed reactions proceeding through amide C–N bond cleavage, see: (a) L. Hie, N. Fine Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79–83 CrossRef CAS PubMed; (b) N. A. Weires, E. L. Baker and N. K. Garg, Nat. Chem., 2016, 8, 75–79 CrossRef CAS PubMed; (c) E. L. Baker, M. M. Yamano, Y. Zhou, S. M. Anthony and N. K. Garg, Nat. Commun., 2016, 7, 11554 CrossRef PubMed; (d) B. J. Simmons, N. A. Weires, J. E. Dander and N. K. Garg, ACS Catal., 2016, 6, 3176–3179 CrossRef CAS PubMed; (e) S. Shi and M. Szostak, Org. Lett., 2016, 18, 5872–5875 CrossRef CAS PubMed; (f) S. Shi and M. Szostak, Chem.–Eur. J., 2016, 22, 10420–10424 CrossRef CAS PubMed; (g) L. Hie, E. L. Baker, S. M. Anthony, J.-N. Desrosiers, C. Senanayake and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 15129–15132 CrossRef CAS PubMed; (h) A. Dey, S. Sasmal, K. Seth, G. K. Lahiri and D. Maiti, ACS Catal., 2017, 7, 433–437 CrossRef CAS; (i) S. Ni, W. Zhang, H. Mei, J. Han and Y. Pan, Org. Lett., 2017, 19, 2536–2539 CrossRef CAS PubMed; (j) J. M. Medina, J. Moreno, S. Racine, S. Du and N. K. Garg, Angew. Chem., Int. Ed., 2017, 56, 6567–6571 CrossRef CAS PubMed; (k) X. Li and G. Zou, Chem. Commun., 2015, 51, 5089–5092 RSC; (l) J. A. Walker, K. L. Vickerman, J. N. Humke and L. M. Stanley, J. Am. Chem. Soc., 2017, 139, 10228–10231 CrossRef CAS PubMed; (m) J. Hu, M. Wang, X. Pu and Z. Shi, Nat. Commun., 2017, 8, 14993 CrossRef PubMed; (n) N. A. Weires, D. D. Caspi and N. K. Garg, ACS Catal., 2017, 7, 4381–4385 CrossRef CAS PubMed; (o) S. Shi and M. Szostak, Synthesis, 2017, 3602–3608 CAS; (p) J. E. Dander, E. L. Baker and N. K. Garg, Chem. Sci., 2017, 8, 6433–6438 RSC; (q) P.-Q. Huang and H. Chen, Chem. Commun., 2017, 53, 12584–12587 RSC; (r) T. Deguchi, H.-L. Xin, H. Morimoto and T. Ohshima, ACS Catal., 2017, 7, 3157–3161 CrossRef CAS; (s) J. E. Dander, M. Giroud, S. Racine, E. R. Darzi, O. Alvizo, D. Entwistle and N. K. Garg, Commun. Chem., 2019, 2, 82 CrossRef PubMed; (t) M. M. Mehta, T. B. Boit, J. E. Dander and N. K. Garg, Org. Lett., 2020, 22, 1–5 CrossRef CAS PubMed; (u) R. R. Knapp, A. S. Bulger and N. K. Garg, Org. Lett., 2020, 22, 2833–2837 CrossRef CAS PubMed; (v) T. B. Boit, M. M. Mehta, J. Kim, E. L. Baker and N. K. Garg, Angew. Chem., Int. Ed., 2021, 60, 2472–2477 CrossRef CAS PubMed; (w) J. Buchspies, M. Rahman and M. Szostak, Molecules, 2021, 26, 188 CrossRef CAS PubMed.
- For a select example, see: G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed.
- (a) The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, ed. A. Greenberg, C. M. Breneman, J. F. Liebman, Wiley, Hoboken, vol. 1, 2003, pp. 33−43 Search PubMed; (b) L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205–211 CrossRef CAS PubMed.
- For examples of traditional amide C–N bond cleavage reactions, see: (a) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS; (b) S. Balasubramaniam and I. S. Aidhen, Synthesis, 2008, 2008, 3707–3738 CrossRef.
- For select reviews on mild amide activation strategies, see: (a) G. Meng, S. Shi and M. Szostak, Synlett, 2016, 27, 2530–2540 CrossRef CAS; (b) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC; (c) D. Kaiser, A. Bauer, M. Lemmerer and N. Maulide, Chem. Soc. Rev., 2018, 47, 7899–7925 RSC.
- This number represents a conservative estimate of the array of metal-catalyzed cross-couplings developed for amide electrophiles, demonstrated by citations of our first paper that was published in 2015.
- Initial attempts towards an intermolecular Mizoroki–Heck reaction proved unsuccessful, but will be the subject of future investigation.
- T. Hayashi, J. Tang and K. Kato, Org. Lett., 1999, 1, 1487–1489 CrossRef CAS.
- P. Fan, Y. Lan, C. Zhang and C. Wang, J. Am. Chem. Soc., 2020, 142, 2180–2186 CrossRef CAS PubMed.
- (a) J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591–4597 CrossRef CAS; (b) C. Li, S. S. Ragab, G. Liu and W. Tang, Nat. Prod. Rep., 2020, 37, 276–292 RSC.
- N-heterocyclic carbene ligands were of particular interest for this investigation, as opposed to other ligand classes, as they are highly electron-rich and have uniquely enabled similar non-asymmetric catalytic amide activation reactions (see T. Droge and F. Glorius, Angew. Chem., Int. Ed. 2010, 49, 6940–6952. for a review on NHC ligands).
- V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- (a) J. S. E. Ahlin and N. Cramer, Org. Lett., 2016, 18, 3242–3245 CrossRef CAS PubMed; (b) J. S. E. Ahlin, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 13229–13233 CrossRef CAS PubMed.
- M. Nakanishi, D. Katayev, C. Besnard and E. P. Kündig, Angew. Chem., Int. Ed., 2011, 50, 7438–7441 CrossRef CAS PubMed.
- H. Seo, D. Hirsch-Weil, K. A. Abboud and S. Hong, J. Org. Chem., 2008, 73, 1983–1986 CrossRef CAS PubMed.
- D. Hirsch-Weil, K. A. Abboud and S. Hong, Chem. Commun., 2010, 46, 7525–7527 RSC.
- J. S. E. Ahlin, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 13229–13233 CrossRef CAS PubMed.
- A variety of other N-substituents were employed in the transformation, including: N-Ph-N-Boc, N-Ph-N-Ts, N-Ph-N-Ms, N-Bn-N-CO2Et, and N-Me-N-Ph, but either led to trace amounts or no observed formation of the desired product.
- Although the exact impact of the additive on the mechanism is currently under investigation, alcohol additives have been shown to be enabling in nickel-catalyzed cross-couplings: see ref. 7j and M. R. Harris, L. E. Hanna, M. A. Greene, C. E. Moore and E. R. Jarvo, J. Am. Chem. Soc., 2013, 135, 3303–3306 CrossRef CAS PubMed . Amine additives have also been proposed to activate low-valent nickel species toward oxidative addition in photoredox nickel catalysis: S. Gizbertz, S. Reischauer and B. Pieber, Nat. Catal., 2020, 3, 611–620 CrossRef.
- The use of enantioenriched proline derivatives was also investigated; however, these attempts either resulted in low yields of the product or comparable levels of enantioselectivity.
- Although the turnover limiting step of the reaction has not yet been elucidated, our data from ligand optimization indicate that the β-hydride elimination step or regeneration of Ni(0) could be slow. This is supported by other proposed nickel catalysis mechanisms in the literature (see ref. 18 for more explanation on the properties of nickel as a catalyst).
- During our optimization campaign with the morpholine additive, we also found empirically that a 1:2 ratio of Ni(cod)2: chiral ligand 10 was necessary to achieve the desired transformation. Although the reason for this ratio has not been unambiguously established, similar findings have been seen previously. In one study, it has been suggested based on computations that two NHC ligands could be implicated in the catalyst resting state (see ref. 7a). Alternatively, an additional NHC ligand could be enabling for reductive elimination.
- H. C. Shen, Tetrahedron, 2009, 65, 3931–3952 CrossRef CAS.
- J. Jurczyk, J. Woo, S. F. Kim, B. D. Dherange, R. Sarpong and M. D. Levin, Nat. Synth., 2022, 1, 352–364 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2271421. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05797f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |