
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico MS/MS prediction for peptidoglycan profiling uncovers novel anti-inflammatory peptidoglycan fragments of the gut microbiota†
Jeric Mun Chung
Kwan
 ab,
Yaquan
Liang
a,
Evan Wei Long
Ng
a,
Ekaterina
Sviriaeva
b,
Chenyu
Li
a,
Yilin
Zhao
a,
Xiao-Lin
Zhang
a,
Xue-Wei
Liu
a,
Sunny H.
Wong
b and
Yuan
Qiao
*a
ab,
Yaquan
Liang
a,
Evan Wei Long
Ng
a,
Ekaterina
Sviriaeva
b,
Chenyu
Li
a,
Yilin
Zhao
a,
Xiao-Lin
Zhang
a,
Xue-Wei
Liu
a,
Sunny H.
Wong
b and
Yuan
Qiao
*a
aSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore. E-mail: yuan.qiao@ntu.edu.sg
bLee Kong Chian School of Medicine, Nanyang Technological University, 11 Mandalay Road, 308232, Singapore
First published on 5th January 2024
Abstract
Peptidoglycan is an essential exoskeletal polymer across all bacteria. Gut microbiota-derived peptidoglycan fragments (PGNs) are increasingly recognized as key effector molecules that impact host biology. However, the current peptidoglycan analysis workflow relies on laborious manual identification from tandem mass spectrometry (MS/MS) data, impeding the discovery of novel bioactive PGNs in the gut microbiota. In this work, we built a computational tool PGN_MS2 that reliably simulates MS/MS spectra of PGNs and integrated it into the user-defined MS library of in silico PGN search space, facilitating automated PGN identification. Empowered by PGN_MS2, we comprehensively profiled gut bacterial peptidoglycan composition. Strikingly, the probiotic Bifidobacterium spp. manifests an abundant amount of the 1,6-anhydro-MurNAc moiety that is distinct from Gram-positive bacteria. In addition to biochemical characterization of three putative lytic transglycosylases (LTs) that are responsible for anhydro-PGN production in Bifidobacterium, we established that these 1,6-anhydro-PGNs exhibit potent anti-inflammatory activity in vitro, offering novel insights into Bifidobacterium-derived PGNs as molecular signals in gut microbiota-host crosstalk.
Introduction
All bacteria possess a peptidoglycan layer. As an essential exoskeletal polymer that surrounds the bacterial cytoplasmic membrane, peptidoglycan protects bacterial cells against internal turgor pressure and also serves as a scaffold for other cell surface proteins and polymers.1 Apart from a structural role, bacterial peptidoglycan also participates in diverse intra- and inter-kingdom signalling.2,3 Soluble peptidoglycan fragments, also known as PGNs or muropeptides, are continuously generated by bacteria during growth and released into the milieu, exerting a broad-range impact on different organisms.4 In the context of the human gut microbiota, trillions of resident bacteria produce a multitude of PGNs in the gut niche,5 which can disseminate into host systemic circulation under steady-state conditions,6 influencing host biology including autoimmunity, brain development, appetite, and body temperature, as well as efficacies of cancer immunotherapy.7–10 Remarkably, subtle structural changes in PGNs can significantly alter their biological activities in hosts.11 Thus, profiling peptidoglycan compositions and characteristics in gut bacteria is of paramount importance to facilitate studies of gut microbiota-derived PGNs in hosts.While the chemical makeup of peptidoglycan polymers is largely conserved, the exact compositions and structural modifications of peptidoglycan are highly variable across bacteria and under different environmental conditions (Fig. 1).1,12,13 In general, the ‘glycan’ component of peptidoglycan consists of alternating units of N-acetylglucosamine (GlcNAc, or herein NAG) and N-acetylmuramic acid (MurNAc, or herein NAM) linked via β-1,4-glycosidic bonds; the ‘peptido’ portion refers to the short stem pentapeptide connected onto the lactoyl group of each NAM, which has the common sequence L-Ala1-γ-D-Glu/isoGln2-AA3-D-Ala4-D-Ala5, with AA3 being either L-Lys attached to a lateral bridge peptide (that is specific to each bacterial species) or a non-proteogenic diamino acid such as meso-diaminopimelic acid (mDAP) (Fig. 1A). These stem peptides on adjacent glycan strands can form 3–4 or 3–3 crosslinks through iso-peptide bonds, thereby strengthening the peptidoglycan layer (Fig. 1C). Furthermore, a great deal of structural diversity in peptidoglycan comes from the cell wall remodeling process, where bacterial enzymes catalyze specific reactions at distinct positions in peptidoglycan to generate new structural moieties, such as modifications of the glycan backbone, trimming of pentapeptides to shorter stems, and incorporation of non-canonical D-amino acids (NCDAA) into the stem peptide (Fig. 1A and B).14 While most insights on peptidoglycan structural diversity were gained from analyses of model bacterial organisms, our knowledge of the scope and variety of peptidoglycan in the gut microbiota is still in its infancy. Recognizing the biological significance of peptidoglycan modifications, we seek to develop a robust and automated workflow to characterize peptidoglycan compositions and structural features in any bacteria of interest, especially those in the gut microbiota.
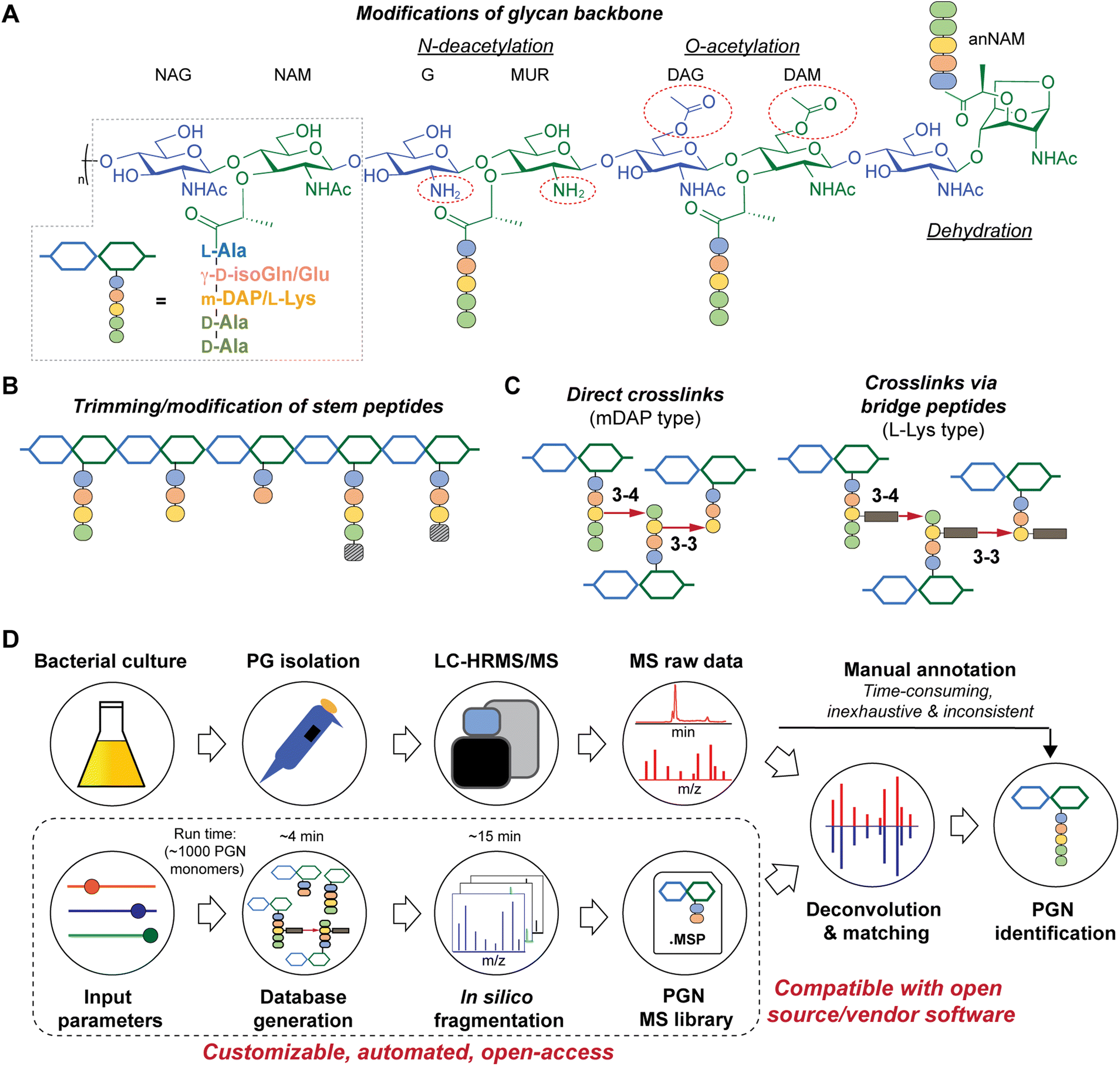 | ||
| Fig. 1 Schematic representations of bacterial peptidoglycan compositions (A–C) and our in silico peptidoglycan fragment (PGN) library analysis pipeline (D). (A) Peptidoglycan is composed of repeating muropeptide units, i.e., N-acetylglucosamine (NAG)-N-acetylmuramic acid (NAM) disaccharides with a stem peptide. Various modifications can be present on the disaccharide backbone (right). Glu: glutamate and isoGln: iso-glutamine; mDAP: meso-diaminopimelic acid. (B) Remodelling of peptidoglycan may include trimming of stem peptides and/or incorporation of non-canonical D-amino acids (striped box). (C) Stem peptides can be crosslinked via direct crosslinks (left, for mDAP-type PGN) or indirect crosslinks, attached through a species-specific bridge peptide (right, for L-Lys type PGN). (D) Manual analysis of MS/MS spectra for structural determination is a bottleneck in bacterial PGN analysis. PGN_MS2 (bottom box) creates a PGN database that includes in silico predicted MS/MS spectra. The resulting spectral library (.msp) is open-access and compatible with mass spectra analysis software for automated deconvolution and analysis of PGNs using m/z, isotopic pattern, and spectral similarity from the LC-MS/MS raw data. | ||
There are significant gaps in the current workflow of bacterial peptidoglycan analysis, with the widely adopted experimental procedure developed >30 years ago.15 Briefly, the peptidoglycan polymer (i.e., sacculi) isolated from bacteria is digested with a muramidase (e.g., lysozyme) that hydrolyzes the NAM-β-1,4-NAG linkages along the peptidoglycan backbone, generating soluble PGNs that are disaccharide-containing muropeptides in nature.16 The collection of these soluble PGNs is then subjected to high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) analysis for structural characterization and profiling (Fig. 1D, top row). Improvements in HPLC-MS/MS instrumentation such as higher resolution and faster scanning rate have improved the quality of acquired data; however, analyzing raw MS data to elucidate PGN structures remains a painstaking manual task, where one needs to come up with the potential structures of PGNs (i.e., search space, which can be as large as >6000 structures on ChemDraw)17,18 and look for matches of the expected m/z values in the acquired LC-MS dataset. Such manual annotations of MS data are considerably time-consuming, laborious, and inconsistent, remaining as an undesirable bottleneck for robust and comprehensive peptidoglycan analysis with higher throughput.19,20 This may deter the discovery of novel structural features of peptidoglycan, especially in the gut microbiota, where the scope of peptidoglycan diversity has not been much explored.
Towards these challenges, we present a novel and customizable PGN database integrated with in silico MS/MS spectra to enable automated MS/MS deconvolution for PGN identification (Fig. 1D, bottom row). The spectral library (.msp format) encompasses the in silico predicted MS/MS fragmentation for each PGN in the dataset, which is compatible with open-access and vendor software for automated matching and scoring of the experimental MS/MS peaks, thus streamlining PGN analysis with unmatched confidence and throughput. Applying this automated PGN analysis pipeline, we profiled the peptidoglycan compositions of five different gut bacteria. Intriguingly, an unusually high abundance of anhydro-PGNs (i.e., PGNs containing a 1,6-anhydro-muramyl moiety, anNAM) (Fig. 1A, far right) was found in Bifidobacterium, the common probiotic bacteria that confer anti-inflammatory effects in hosts.21,22 We further demonstrated that MltG and RfpB homologs in Bifidobacterium possess robust lytic transglycosylase (LT) activity towards distinct peptidoglycan substrates to generate anhydro-PGN moieties. Importantly, we established that these anhydro-PGNs of Bifidobacterium exhibit novel anti-inflammatory effects in vitro, which opens up exciting opportunities for postbiotic development.
Results
Generation of a customizable PGN MS1 database
To streamline the PGN searching process, we envisioned a method to automatically generate a PGN MS1 database with user-defined parameters. The basic muropeptide scaffold of PGNs (upon muramidase digestion in the sample preparation) features a (NAG)(NAM) disaccharide with a stem peptide, where distinct structural modifications are possible at each position.1 To build the PGN database, the user, through a graphical user interface, conveniently selects the possible range of modifications on the (NAG)(NAM) backbone, including O-acetylation, de-N-acetylation, or anNAM termini, followed by selecting the possible amino acid identities at each stem peptide position. Next, additional structural modifications can be included, such as Braun's lipoprotein attachment, substitution of the terminal amino acid with lactate, endopeptidase-cleaved products, and reduction of muramyl termini. Lastly, the user can select the amount and types of PGN polymerization, either through peptide crosslinks or glycosidic bonds. All parameters can be adjusted. Next, PGN molecules are constructed in silico with RDKit,23 and the database is saved as an Excel worksheet (.xlsx). Each PGN is assigned a unique descriptive name (Fig. S1†). With the graphical user interface, no coding experience from the user is required to build the PGN database (Fig. S2 and SI1†).Apart from its descriptive name, the PGN database (.xlsx) also includes chemical descriptors for individual PGNs, e.g., chemical formula, adducts m/z, clogP, InChIKey, SMILES, and PGN-specific descriptors, e.g., the degree of acetylation, degree of amidation, and stem peptide length, thereby facilitating subsequent PGN categorization and comparative analysis (Fig. 2, right). Accompanying the PGN database, an image output that summarizes user-defined parameters is automatically generated for convenient referencing (Fig. 2, left). For a typical database of 3000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 PGNs, it takes ∼1 min per 1000 PGN to generate when run on a computer with a 2.60 GHz processor and 16 GB RAM. To reduce analysis time, PGN_MS2 includes various ways to skip illogical/unreasonable PGN polymers (Table S1†).
000 PGNs, it takes ∼1 min per 1000 PGN to generate when run on a computer with a 2.60 GHz processor and 16 GB RAM. To reduce analysis time, PGN_MS2 includes various ways to skip illogical/unreasonable PGN polymers (Table S1†).
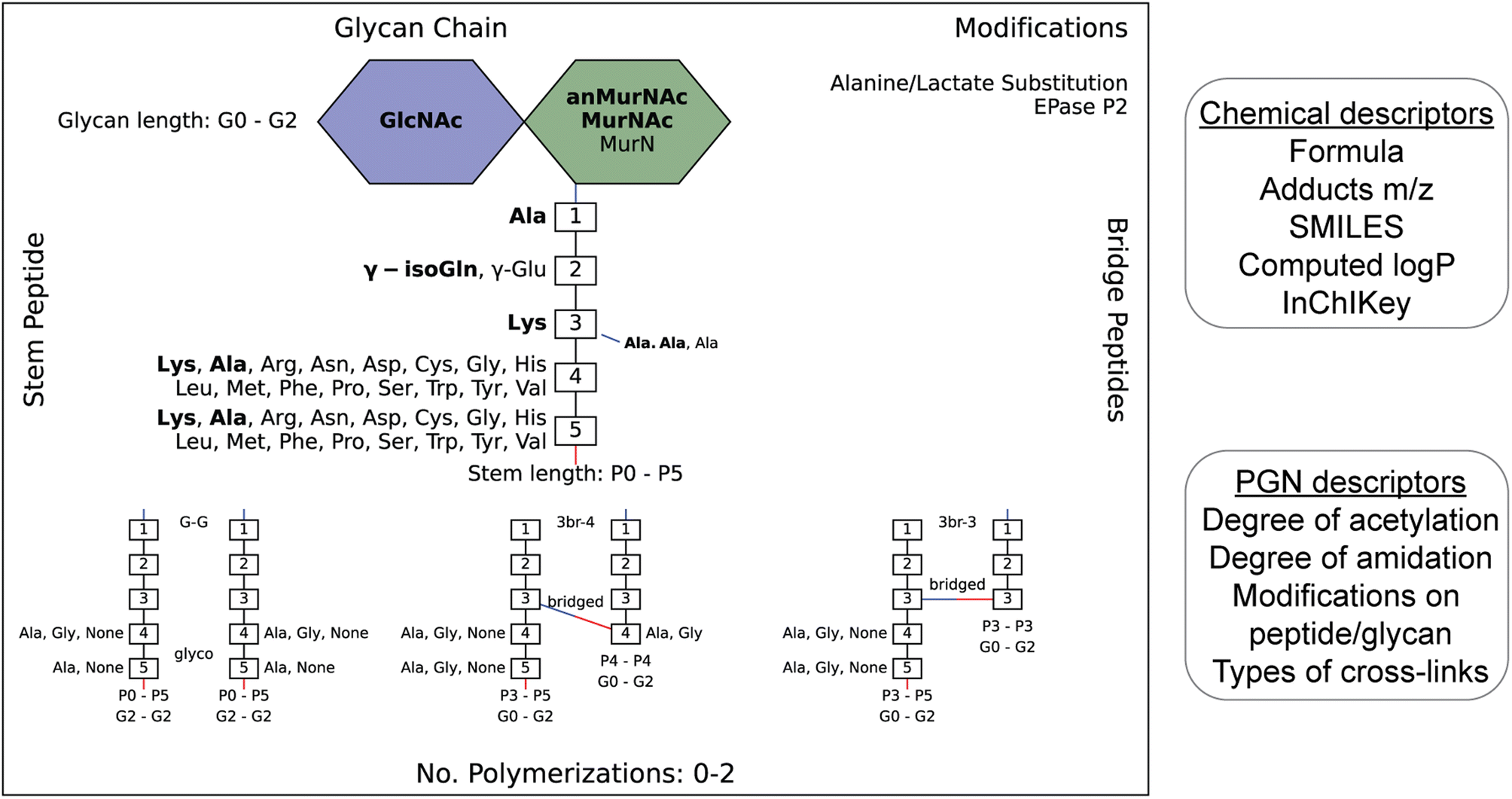 | ||
| Fig. 2 Example of an image output that summarizes the diversity of PGNs present in the user-defined in silico PGN library (left). The glucosamines (blue), muramic acids (green), and amino acids (numbered white boxes) used to construct PGNs are listed. Bridge peptide sequences are indicated to the right of the connecting amino acid with a connecting line. Its color indicates the type of connection (red: through COOH; blue: NH2.) The user-defined canonical components are bolded. Glycan lengths (0–2 glycans), peptide lengths (0–5 amino acids), and polymerizations (0 to 2) are indicated. Peptidoglycan modifications are listed in the top-right corner. The requirement for each polymerization is shown at the bottom. For instance, G–G polymerization is only formed between PGNs with glycan length 2, peptide lengths 0–5, and with either Ala, Gly or no amino acid in positions 4/5 on both the acceptor and donor. N- and C-peptide termini are colored blue and red, respectively. The PGN library is saved as an Excel file (.xlsx) which contains their chemical and PGN-specific descriptors (right). | ||
Development of PGN_MS2 for in silico MS/MS prediction
Although PGNs can be identified by their m/z values alone (MS1 identification), additional analysis by tandem mass spectrometry (MS/MS) is necessary to resolve structural isomers of PGNs with mass coincidences. In the fields of metabolomics and proteomics, compound identification is routinely performed by matching and scoring experimental MS/MS spectra against a reference library of actual MS/MS spectra of standard compounds and/or in silico simulated MS/MS spectra for compounds whose experimental data are not available.24–27 Given the limited availability of empirically collected MS/MS data, in silico MS/MS prediction can greatly improve compound identification.28 However, due to the unique sugar and amino acid compositions present in PGNs, existing MS/MS simulation tools in metabolomics and proteomics are not well-suited for PGN identification.19 Toward the automated PGN analysis workflow, we next sought to augment the PGN MS1 database with in silico predicted MS/MS spectra.To derive in silico PGN MS/MS spectra, we first studied the ESI-MS/MS spectra of known PGNs. Recent studies by Tan et al. and Anderson et al. reported the experimental MS/MS spectra for selected PGNs from E. coli, S. aureus, and P. aeruginosa, providing a suitable starting point for our evaluation.17,29 In addition, we also acquired experimental LC-HRMS/MS data for several major PGNs with known structures from E. faecalis and L. plantarum. Notably, these spectra were acquired using different MS instruments, namely, Orbitrap Exploris 120 (our study), LCQ Fleet (Tan et al.), and Q-TOF (Anderson et al.), enabling us to derive common ESI-MS/MS fragmentation rules for most PGNs. We recognized that the PGN precursor ions frequently undergo B/Z-type glycan fragmentation (nomenclature according to Domon and Costello30) and b/y-type peptide fragmentation, with multiple b/y cleavages to yield lighter ions (Fig. 3A). Additionally, the lactoyl bond connecting the glycan and peptide in PGNs also fragments readily, with the peptide fragment ion henceforth named L (Fig. 3A). Furthermore, isomeric PGNs that contain stem peptides such as Aqm and Aem(NH2) with differing amidation positions can be easily distinguished by their MS/MS patterns (Fig. S3†). The y2 peptide fragments (i.e. qm or em(NH2), m/z: 319.1619) undergo further e1/e2 or q1/q2 fragmentations due to prominent neutral losses at the N-terminus.31 For instance, em(NH2) yields 301.1465 (e1) and 256.1280 (e2) fragments, whereas qm gives rise to signature MS/MS peaks of 302.1347 (q1) and 257.1103 (q2); with q2 fragments showing higher relative intensities (Fig. S3A and B†). These abundant MS/MS features are useful to distinguish PGNs that bear e or q in the stem peptides, as in the case of L. plantarum (Fig. S3†). Upon evaluating the experimental MS/MS spectra for ∼30 PGNs, we found that most of the fragmentation peaks can be explained by 19 fragmentation reactions or a combination thereof (Fig. 3A).
 | ||
| Fig. 3 The design and construction of in silico PGN fragmenter, PGN_MS2. (A) Empirical analysis of PGN MS/MS spectra reveals the possible types of fragmentation reactions, which are encoded using PGN_MS2. Fragmentation of the glycan backbone (B/Y/C/Z) follows Domon and Costello's nomenclature; fragmentation of the stem peptide is denoted as bn and yn, where n indicates the position of the peptide bond, with the peptide bond nearest to the glycan backbone denoted as “1”; fragmentation of the bridge peptide is indicated with a quotation mark (’). In addition, the γ-Glu-containing PGNs yield e1/e2 fragments due to the neutral loss of H2O and COOH + NH3, respectively; similarly, γ-isoGln-containing PGNs generate q1/q2 fragments by neutral loss of NH3 and CONH2 + NH3, respectively. Furthermore, further fragmentation of GlcNAc/MurNAc (Glc-1, Glc-2, Glc-3, and Mur-1) and neutral loss of H2O or NH3 are also included as fragmentation reactions. (B) In silico MS/MS spectrum generation schematic. The fragmentation reactions for each PGN are encoded as SMARTS, where the m/z and relative intensity are calculated for each product ion. Each product ion undergoes further fragmentation (2 repeats) to create the in silico spectrum. (C) Comparison of experimental MS/MS spectra (top, red) vs. in silico predicted spectra (bottom, blue) for canonical PGN in E. coli (left) and S. aureus (right). Matched peaks are coloured black, and the cosine similarity scores between the spectra are shown in the top right corner. | ||
Based on these common fragmentation reactions, we developed PGN_MS2, an in silico MS/MS prediction tool for PGNs. As shown in Fig. 3B, we encoded each fragmentation as a chemical reaction in SMARTS and simulated it with RDKit.23 Each parental PGN ion (generation-0) is fragmented via all possible 19 reactions to form generation-1 product ions, which are further fragmented to yield generation-2 and generation-3 product ions sequentially. Fragmentation is discontinued after no new product ions are generated. For every fragmentation, the m/z value and relative intensity for each fragment are calculated. Relative intensity is estimated based on an empirically derived formula (that accounts for the number of peptide bonds or mass ratio of the precursor and product ions) together with a fragmentation-specific adjustment factor. Finally, the assembly of possible fragment ions affords the in silico predicted MS/MS spectra. To account for the different precursor adducts (i.e., [M + H]+, [M + 2H]2+, and [M + 3H]3+), separate MS/MS spectra are created for each adduct, whereby fragment ions with m/z greater than that of the precursor ion are removed. In sum, our PGN library integrates the predicted MS/MS spectra of all PGNs in the database as a NIST format text file (.msp, Fig. S2C†).
Reliable PGN identification with in silico MS/MS prediction
To test the accuracy and reliability of MS/MS prediction by PGN_MS2, we first compared the experimental spectra of a panel of distinct PGNs from different bacteria with their respective predicted spectra by calculating the cosine spectral similarity scores.32 As expected, PGN_MS2 consistently afforded high similarity scores of 0.7–0.8 for most PGNs, which significantly outperformed other spectral prediction tools such as CFM-ID and ms2pip (Fig. S4A–C, Table S2†),26,27 showcasing the specialized applications of PGN_MS2 for PGN analysis. In addition, we validated PGN_MS2 by demonstrating its ability to predict MS/MS spectra of synthetic PGN standards (Fig. S4D†).Next, we confirmed that the in silico predicted spectra by PGN_MS2 match well with the MS/MS spectra acquired using either an Orbitrap spectrometer via higher-energy C-trap dissociation (HCD)-based fragmentation or a Q-TOF instrument via collisional dissociation (CID)-based fragmentation (Fig. S5A–D†).33 In addition, to benchmark our PGN_MS2 with the available PGN dataset, we also evaluated the experimental data of P. aeruginosa PGNs deposited by Anderson et al., which was collected using a Q-TOF mass spectrometer.17 Consistently, using PGN_MS2 and MS-DIAL, we readily confirmed 54 PGNs that were manually identified in the previous work (62, with MS/MS) (Fig. S5E–F†). Taken together, our observations demonstrate the robustness and reliability of PGN_MS2 in simulating ESI-MS/MS spectra of PGNs for structural determination.
To further investigate if PGN_MS2 could indeed aid accurate assignment of PGNs among closely related structural isomers, we challenged it to identify the canonical E. coli or S. aureus PGN, (NAG)(NAM)-AemA and (NAG)(NAM)-AqKAA[3-NH2-GGGGG] respectively, from a set of four intentionally generated mock PGNs with identical molecular formulae (Fig. S6†). Satisfactorily, we correctly assigned the two PGN structures, since they both emerged as the top hits with the highest spectral similarity scores compared to other possible isomers, albeit by a small margin (Fig. S6†). Based on our analysis, we noted that although the top matched in silico PGN usually represents the accurate structure, other criteria such as the presence or absence of certain signature MS/MS fragments are particularly useful for PGN determination too. For instance, fragments containing the intact mDAP–mDAP bond (i.e., m/z: 617.2777, 746.3203, and 889.3785) are observed in the MS/MS spectra of the 3–3 but not 3–4 crosslinked PGNs in E. coli, allowing convenient distinction between the two isomers (Fig. 4C and S7†). Therefore, it is prudent to check for these signature fragments for PGN identification. To assist with this, PGN_MS2 also annotates the chemical structures of each fragment in the predicted MS/MS spectra as SMILES (Fig. S2D†).
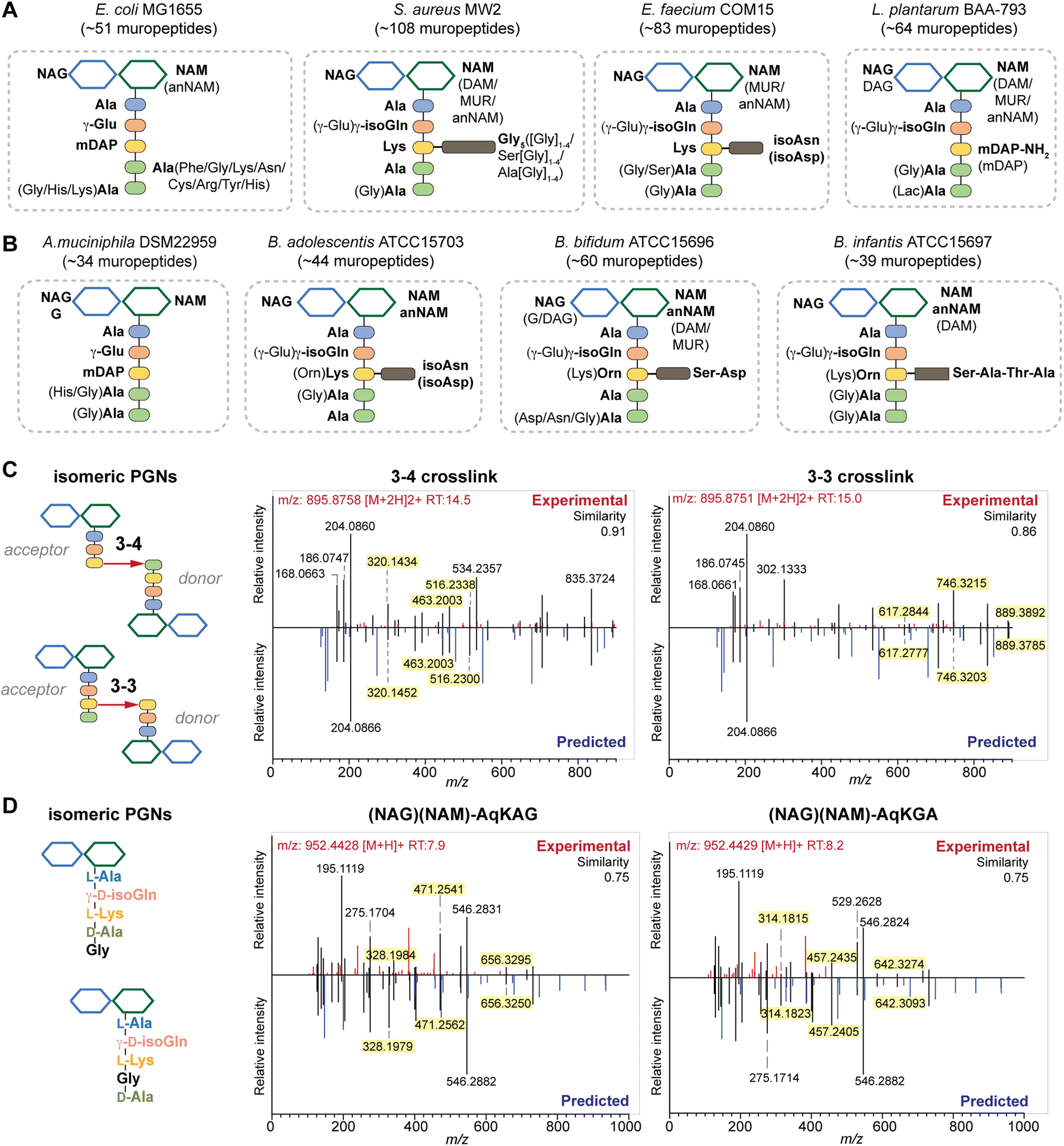 | ||
| Fig. 4 Summary of peptidoglycan compositions in the model (A) and gut bacteria (B) with the canonical makeup shown in bold and variable components not bolded. For instance, the canonical makeup in E. coli is (NAG)(NAM)-AemAA and the fifth amino acid, Ala, can be substituted with His, Gly, or Lys. The total number of PGNs identified in each species of bacteria is listed. Compositions for E. faecalis and F. nucleatum are shown in Fig. S8G† instead. PGN_MS2 enables distinctions between isomeric PGNs by matching experimental spectra against in silico predicted MS/MS patterns for: (C) tetrapeptide-tripeptide dimers with either 3–4 or 3–3 crosslinks in E. coli; (D) monomeric PGNs that incorporate Gly at either the 4th or 5th position in E. faecium. The key fragments that are essential for resolving the respective isomers are highlighted in yellow. Fig. S6, S7, S9, and S10† showcase additional examples of differentiating isomeric PGNs by PGN_MS2. | ||
Validation of the MS/MS-integrated workflow for model bacterial PGN profiling
Upon demonstrating the reliability of PGN_MS2 for identifying individual PGN molecules, we then sought to evaluate its potential application for profiling bacterial peptidoglycan compositions. We first constructed PGN libraries customized for different model bacteria, including E. coli, S. aureus, E. faecium, and L. plantarum. We next acquired experimental LC-HRMS/MS data of PGNs from these bacteria. NaBH4 reduction was omitted to prevent potential acid hydrolysis during the addition of phosphoric acid and preserve the natural structure of PGN. Next, we utilized open-source software MS-DIAL for automated data analysis by importing the respective in silico MS/MS libraries as spectral references for PGN identification.34 In general, our findings are consistent with previous knowledge of PGN compositions in these bacteria,16,35–43 validating our MS/MS-integrated PGN MS library for automated PGN profiling. We summarized the canonical PGN monomeric makeup (Fig. 4A and S8G†) and listed the detailed PGN compositions in these bacteria (Tables S3–S8†). Below we highlight the discovery of several PGN structural features that exemplify the virtue of the in silico MS/MS spectral library.Amidation of stem peptides is a unique feature in PGNs of Gram-positive bacteria (Fig. 5B).1 For instance, the canonical monomeric PGNs in E. faecium and L. plantarum each contain two possible amidated residues in the stem peptides, q and isoAsn, q and m(NH2), respectively (Fig. 4A). Although most PGNs in both bacteria are amidated at both positions, substantial amounts of singly amidated PGNs are also observed, which require MS/MS analysis to determine the exact amidation position in the isomeric PGNs (Fig. S3†). In addition, some L. plantarum PGNs have D-lactate instead of D-Ala at the stem peptide's terminus,43 which further complicates identification. The three structural isomers, (NAG)(NAM)-Aem(NH2)AA, (NAG)(NAM)-AqmAA, and (NAG)(NAM)-Aqm(NH2)ALac have identical m/z values that are indistinguishable solely based on MS1 analysis and require in-depth MS/MS evaluation. With our approach, the in silico predicted MS/MS spectra by PGN_MS2 revealed signature fragments for each of the three PGN isomers, which significantly improved the confidence and throughput of MS/MS identification (Fig. S9†). For instance, the experimental spectra of (NAG)(NAM)-Aqm(NH2)ALac showed the best match to the in silico spectra for this particular isomer and contained all key fragments, allowing us to easily assign the correct structure (Fig. S9†). Moreover, with our MS/MS-integrated analysis pipeline, we also uncovered that amidation at the second residue (q) of the stem peptide is more prominent than that at the side chain (β-Asp) in E. faecium, whereas similar amidation rates were observed for both q and m(NH2) in PGNs of L. plantarum (Fig. S8A†).43 Recognizing that bacterial peptidoglycan amidations are associated with increased levels of crosslinking and also implicate antibiotic resistance,44–49 we anticipate that our workflow for the facile analysis of such amidated PGNs will facilitate the development of novel antimicrobials targeting bacterial peptidoglycan amidations.
 | ||
| Fig. 5 Summary of peptidoglycan features in model and gut bacteria. (A) Varying lengths of stem peptide in monomeric PGNs across bacteria. (B) Amidation rate in stem peptides across bacteria. (C) Frequency of NCDAA incorporation in stem peptides across bacteria. (D) Amount of anNAM termini in bacteria. Bifidobacterium spp. showcase a high abundance of anNAM that differs from that of typical Gram-positive bacteria. All statistics indicate the relative muropeptide composition (in %) except for (B), where the amidation rate is instead defined as the number of amidated residues (γ-D-isoGln/β-D-isoAsn/mDAP(NH2)) per muropeptide. L. plantarum, E. faecium, and B. adolescentis feature two amidated amino acids, and the values shown are the combined rates for both. The data represent the average of three to four biological replicates with error bars representing standard deviations. Additional profiling analysis can be found in Fig. S8A–F.† | ||
Peptidoglycan crosslinking via stem peptides confers strength and resistance to certain antibiotics and stress conditions. For instance, E. coli typically manifests 3–4 crosslinking but significantly increases 3–3 crosslinking under stress conditions.50,51 The 3–4 and 3–3 crosslinked tripeptide-tetrapeptide dimeric PGNs are structural isomers that differ only in the isopeptide bond position, which were easily distinguished using our MS/MS-integrated PGN analysis workflow (Fig. 4C and S7†). Interestingly, across all bacteria, we also detected tetra-saccharide PGN dimers that are isomeric to the crosslinked dimers (Fig. S10†). Although such tetra-saccharide motifs are possible products of incomplete muramidase digestion during sample preparation, additional rounds of enzymatic digestion could not fully eliminate them.52 Compared to the crosslinked PGN dimers, these tetra-saccharide PGNs generally yielded fewer MS/MS fragments with lower relative intensity for B-type fragments and higher intensity for L-type fragments (Fig. S10†), which is consistent with the presence of only one terminal GlcNAc and two free-stem peptides in these structures. The ability to easily identify such tetra-saccharide PGNs in our workflow may provide the impetus to investigate their physiological relevance in bacteria.
Comprehensive and automated PGN profiling in gut bacteria
Encouraged by the proof-of-concept studies in model bacteria, we next set out to comprehensively profile the PGNs in a panel of human gut bacteria: Bifidobacterium adolescentis, Bifidobacterium bifidum, Bifidobacterium infantis, Fusobacterium nucleatum and Akkermansia muciniphila. Among them, A. muciniphila and Bifidobacterium spp. are commensal species that help maintain the gut microbiota balance and reduce inflammation, whereas F. nucleatum is associated with colorectal and other cancers.21,22,53–55 Notably, except for a recent study that analyzed PGNs in A. muciniphila using LC-MS,56 our knowledge of Bifidobacterium and F. nucleatum PGNs is only from early studies in the 1970s.57–60 To address their potential biological functions in the host, there is an imperative need to perform in-depth PGN profiling of these gut bacteria.We first elucidated the canonical PGN makeup in the respective gut bacteria (Fig. 4B). A. muciniphila possesses mDAP-type PGNs,56 similar to most other Gram-negative bacteria. However, F. nucleatum PGNs exclusively feature the non-proteinogenic lanthionine at the third position of the stem peptide, whose structure closely resembles that of mDAP.58,59 On the other hand, Gram-positive Bifidobacterium spp. possess either L-Lys or L-Orn as the third residue that is further appended with distinct bridge peptides (Fig. 4B).57 Surprisingly, we found that whereas the L-Lys containing PGNs are only minor constituents in B. bifidum and B. infantis (2.7 and 3.6% respectively, Fig. S8B†), they are the major constituents in B. adolescentis (62.3%, Fig. S8B†). This could imply that MurE, the ligase that incorporates the third amino acid residue in soluble peptidoglycan precursors, exhibits unique substrate tolerances amongst different species of Bifidobacterium. Furthermore, B. adolescentis PGNs also sport an identical bridge peptide (i.e., β-Asp/β-isoAsn) as those in E. faecium and L. lactis,42,61,62 which are constructed by the sequential enzymatic activities of the D-aspartate ligase, Aslfm, and the asparagine synthase, AsnH.61–63 Consistently, B. adolescentis encodes homologs of both enzymes (Table S14†).
Evaluating the lengths of stem peptides in PGNs across different bacteria, we found that PGNs in F. nucleatum, B. infantis, and S. aureus predominantly possess penta- and tetra-peptides, whereas B. adolescentis, L. plantarum, and E. faecium showcase variable PGNs with shorter stems ranging from one to four amino acids, which are likely products of enzymatic cleavages by DD-carboxypeptidases, LD-endopeptidases or DL-endopeptidases during PG maturation in bacteria (Fig. 5A).14 Recent studies have revealed that SagA-like DL-endopeptidases secreted by commensal gut bacteria such as E. faecium and Lactobacillus generate bioactive PGN motifs that regulate host gut homeostasis.42,64,65 Interestingly, both B. bifidum and B. adolescentis have a significant proportion of PGNs with dipeptide stems (∼15%) (Fig. 5A), suggesting the activities of SagA-like enzymes in these two Bifidobacterium that could be potentially relevant to their anti-inflammatory effects.
In all bacteria, NCDAAs are commonly found in the stem peptides of PGNs, substituting D-Ala in the fourth or fifth position (Fig. 4A and B and 5B).12 PGNs from A. muciniphila and F. nucleatum mostly contain basic NCDAAs such as His, Arg, Asn, or Lys at the fifth position of the stem peptides (Fig. 4B and S8G†), which could be incorporated by transpeptidases and/or Ddl in these bacteria.66 Notably, E. coli possesses the greatest diversity of NCDAAs in PGNs, including Phe, Tyr, Gly, Lys, Cys, Arg, etc., whereas other bacteria, B. adolescentis, B. infantis, L. plantarum, and S. aureus appear to solely utilize Gly as the non-canonical amino acid in the PGN stem peptides (Fig. 4A and B). Empowered by in silico MS/MS spectral references, we readily distinguished PGN isomers with Gly at either the fourth or fifth position of the pentapeptide stem in E. faecium (Fig. 4D). NCDAAs in peptidoglycan confer bacterial resistance against hydrolases of rival bacterial species, which are consistently found at elevated levels in bacteria under stress conditions.12,67 Our work reveals the widespread presence of NCDAAs in bacterial PGNs under steady-state conditions compared to what was previously appreciated.
Besides stem peptide motifs, we also profiled structural features on the (NAG)(NAM) backbone in PGNs across bacteria, including O-acetylation (i.e., DAG and DAM) or de-N-acetylation (i.e., G and MUR) (Fig. 4A and B).68 Modifications to acetylation in peptidoglycan may help bacteria evade lytic enzymes such as lysozyme.69 With our MS/MS-integrated analysis workflow, we could readily determine if acetylation/de-acetylation occurs on the NAG or NAM residue in disaccharide PGNs. Such alterations only account for a minor extent (<5%) in PGNs of B. bifidum and L. plantarum; hence no significant changes in the overall acetylation rate of PGNs were observed for most bacteria (Fig. S8E and F†). One remarkable exception is A. muciniphila that showcases 43% de-N-acetylation of NAG (Fig. S8E and F†), which is in good agreement with the recent analysis by Garcia-Vello et al. (∼40%).56 Notably, these de-N-acetylated PGNs are still potent agonists to both NOD1 and NOD2 immune sensors;56 thus, it remains to be determined if such de-N-acetylated motifs exhibit any distinct functions in the host.
Next, 1,6-anhydroMurNAc (anNAM) termini are unique features that mark the end of the peptidoglycan strands in Gram-negative bacteria.20 Correspondingly, anhydro-PGNs constituted 4–5% of total peptidoglycan composition in Gram-negative bacteria, E. coli and F. nucleatum, but are nearly undetectable in model Gram-positive bacteria and A. muciniphila (Fig. 5D).56 Surprisingly, we found that all three Bifidobacterium spp. contain a remarkably high abundance of anhydro-PGNs, which is unusual for Gram-positive bacteria (Fig. 5D and S11†). For instance, the anNAM-containing PGNs comprise nearly 40% of total PGNs in B. adolescentis (Fig. 5D). The exceedingly high amounts of anhydro-PGNs in Bifidobacterium suggest the presence of active lytic transglycosylases (LTs) in catalyzing the non-hydrolytic cleavage of the peptidoglycan backbone, which are elusive in Gram-positive bacteria.52,70,71 We next set out to establish putative LTs in Bifidobacterium responsible for anhydro-PGN formation.
Identification and characterization of putative LTs in Bifidobacterium
To identify putative LTs in Bifidobacterium, we searched for homologous proteins containing the catalytic domains of known LTs in other species (i.e. E. coli MltA-G and Slt70).72 Interestingly, all three Bifidobacterium species encode proteins (BaMltG, BbMltG and BiMltG) that possess the catalytic domain, IPR003770, of MltG, consistent with the broad conservation of MltG across bacteria (Fig. S12A and B†).73 Protein sequence alignment with ClustalOmega revealed that BaMltG, BbMltG, and BiMltG are ∼55–57% similar to one another and are 23–27% similar to E. coli MltG, B. subtilis MltG, and S. pneumoniae MpgA, whose biochemical activities have been characterized (Fig. S13A†).52,73,74 While MltG in Gram-negative bacteria represents the sole inner membrane-bound LT that is responsible for the cleavage of nascent PG strands and generates 1,6-anhydro-MurNAc termini, the MltG homolog in S. pneumoniae, MpgA acts as a muramidase instead.52 Notably, the identity of a single amino acid in the active site of MltG serves as a key determinant for the corresponding enzymatic activity, with Asp for LTs and Asn for muramidases.52 We found that Bifidobacterium MltG harbors an Asp at this position, implying its potential role as an LT (Fig. S13A†). For biochemical characterization of Bifidobacterium MltG, we cloned, overexpressed, and purified the respective MltG lacking the N-terminal transmembrane domain (Fig. 6B). Initial attempts at incubating the recombinant MltG protein with bacterial sacculi did not yield any products, which was in agreement with previous findings that mature sacculi are poor/not suitable substrates for MltG.52,73,74 We next sought to evaluate the activity of Bifidobacterium MltG with nascent peptidoglycan as a substrate, generated from in situ Lipid II polymerization with SgtB.52,74 Since it is challenging to isolate native Lipid II molecules from large-scale cultures of Bifidobacterium, it was substituted with E. faecalis Lipid II instead,75 as its structure resembles that of Bifidobacterium Lipid II. We added Bifidobacterium MltG and SgtB to the Lipid II substrate, followed by mutanolysin to release soluble muropeptides for LC-MS analysis. As shown in Fig. 6A and B, the addition of Bifidobacterium MltG indeed led to a significant increase in anhydro-PGN products, indicating its robust LT activity in vitro (Fig. 6B and S14†). As a negative control, we showed that mutating the catalytic residue Asp to Ala in Bifidobacterium MltG completely abolishes the observed LT activity in vitro (Fig. S15†). In addition, we showed that Lipid II is not a substrate for Bifidobacterium MltG, as no LT products were detected in the absence of SgtB (Fig. S15†).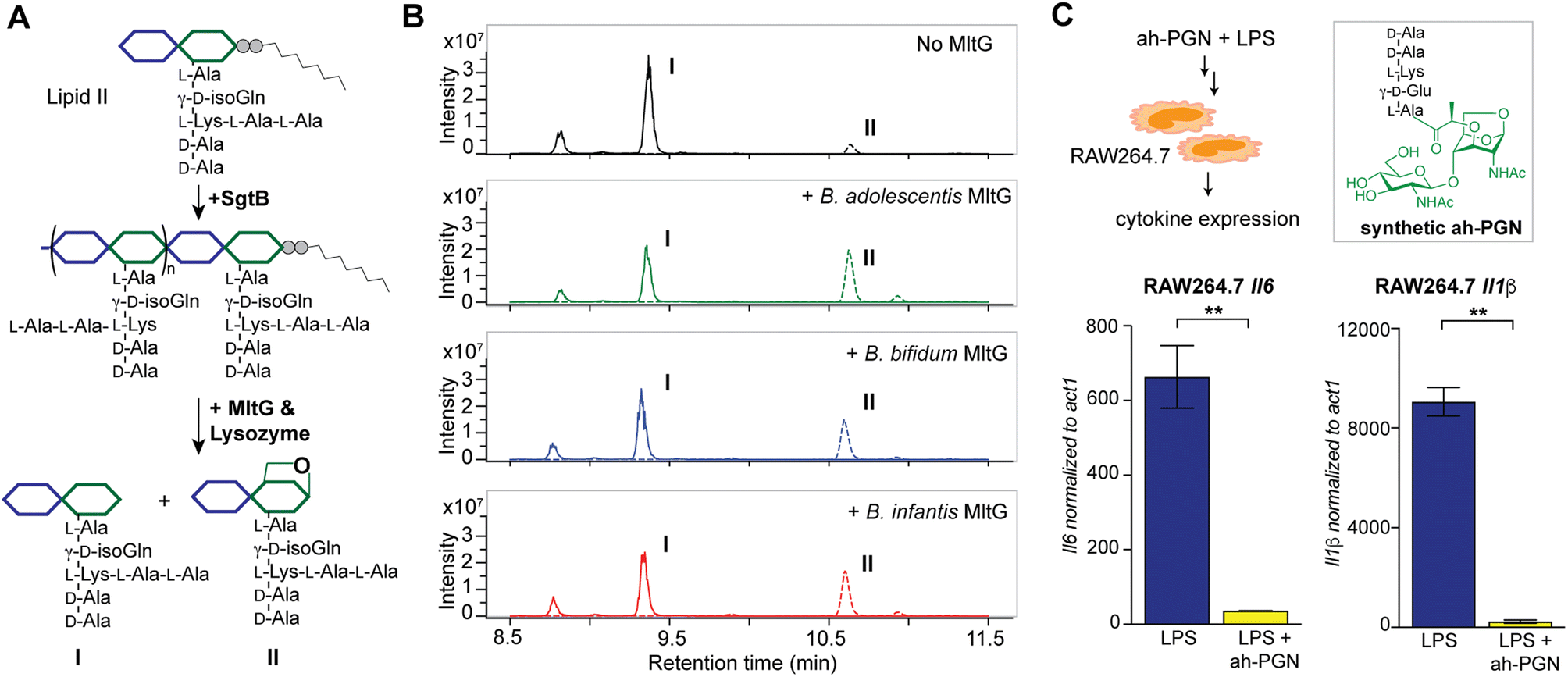 | ||
| Fig. 6 Bifidobacterium anhydro-PGNs from the cleavage of lytic transglycosylases (LTs) exhibit potent anti-inflammatory effects in vitro. (A) Biochemical reconstitution of recombinant Bifidobacterium MltG with nascent peptidoglycans as substrates. Lipid II was extracted from E. faecalis. (B) LC-MS chromatograms of the muropeptide products indicate the formation of anhydro-PGNs, II. Extracted ion chromatograms (EICs) for the [M + 2H]2+ adduct are shown. Additional control experiments are shown in Fig. S15.† (C) Pre-treatment of synthetic anhydro-PGN (ah-PGN), (NAG)(NAM)-AeKAA, significantly suppresses LPS-induced inflammatory responses in murine macrophage RAW264.7 cells. The synthetic ah-PGN mimics the natural anhydro-PGNs found in B. adolescentis. | ||
Apart from the well-characterized LTs in Gram-negative bacteria, certain Gram-positive bacteria that undergo dormancy also encode a large family of cell wall lytic enzymes that are known as resuscitation-promoting factors (Rpfs), some of which are LTs.76,77 Since Bifidobacterium can also enter a viable but non-culturable (VBNC) state similar to dormancy,78,79 we explored if Bifidobacterium could possess any Rpfs with LT activity. Using sequence similarity searching by BLAST, we identified two candidate proteins (RpfB-FL and RpfB-Truncated) in Bifidobacterium containing the lysozyme-like domain (IPR023346) that show weak homology to M. tuberculosis and S. coelicolor RpfB (Fig. S12A and C†). Interestingly, both the full-length and truncated RpfB proteins of B. adolescentis display dual LT and amidase activities with bacterial sacculi in vitro (Fig. S16†), indicating their possible involvement in sacculi remodeling. Taken together, our results established three bona fide LTs (MltG, RpfB-FL length, and RpfB-Truncated) in Bifidobacterium that may act in concert contributing to the high abundance of anNAM in Bifidobacterium peptidoglycan.
Bifidobacterium anhydro-PGNs exhibit potent anti-inflammatory activity in vitro
Intrigued by the predominant anhydro-PGNs in Bifidobacterium spp., we hypothesize that the remarkable anti-inflammatory functions of Bifidobacterium spp. as probiotics may be attributed to these unique anhydro-PGN molecules. Although most bacterial PGNs belong to pathogen-associated molecular patterns (PAMPs) that are agonists of mammalian NOD immune sensors to trigger downstream proinflammatory responses,80 these anhydro-PGN motifs in Bifidobacterium spp. lack critical structural features for both NOD1 and NOD2 activations. Specifically, Bifidobacterium PGNs harbor an L-Lys or L-Orn instead of mDAP in the stem peptide, rendering them non-agnostic to NOD1.81,82 Moreover, these PGNs with 1,6-anhydro-MurNAc termini effectively evade NOD2 recognition, which strictly senses the reducing-end anomeric configuration of MurNAc in PGNs.83,84 As expected, we demonstrated that crude PGNs of B. adolescentis exhibit significantly reduced capacity in activating NOD signaling pathways in cell-based reporter assays compared to PGNs of other Gram-positive and Gram-negative bacteria including S. aureus, E. faecalis, and E. coli (Fig. S17†), highlighting the distinct characteristics of PGNs from probiotic Bifidobacterium spp. To further explore the potential anti-inflammatory effects of these anhydro-PGNs, we used an in vitro immunological assay, where we pre-treated murine macrophage RAW246.7 cells with a synthetic anhydro-PGN before the addition of LPS, followed by gene expression analysis by RT-qPCR (Fig. 6C and S17†). To our surprise, the presence of the anhydro-PGN effectively suppressed the expressions of several key proinflammatory cytokines including tnfa, il1b, and il6 in RAW246.7 induced by LPS, highlighting the potent anti-inflammatory properties of Bifidobacterium anhydro-PGNs in vitro.Discussion
With access to our open-access and customizable MS/MS-integrated PGN library, it is now possible to automate the PGN analysis workflow. We demonstrated the use of an open-source program, MS-DIAL for data processing,34 which easily performs the searching and scoring of the experimental data against our in silico PGN spectral reference, rendering the entire PGN identification process more accurate and robust than ever.Firstly, our in silico PGN MS1 database, which centers around the (NAG)(NAM)-containing disaccharide muropeptide as the core PGN structure, is customizable with user-defined parameters to accommodate diverse structural modifications and polymerizations/crosslinking in PGNs. Currently, our algorithms support most known PGN modifications as built-in selections, including O-acetylation, de-N-acetylation, NCDAA incorporation, 3–3/3–4 crosslinking, etc.; additional structural features can be conveniently incorporated to expand the search space for identification of novel PGNs in the gut microbiota.
Secondly, for each PGN molecule in the MS1 database, an in silico predicted MS/MS pattern is automatically generated by PGN_MS2. The collection of these simulated MS/MS spectra affords a comprehensive in silico PGN spectral library that enables automated analysis. In contrast to PGFinder,35 a PGN analysis pipeline based solely on MS1 values, our PGN MS library integrates in silico MS/MS spectral prediction, marking a significant advance for accurate and robust PGN identification. Similar to the iterative searching strategy in PGFinder,35 we also recommend users specify selective parameters to build the in silico PGN polymer pool focusing on the major canonical features in the PGN monomers, to reduce the number of possible polymers created. As a novel feature of our PGN library, the PGN_MS2 tool also outputs an image summarizing the diversity of PGNs with their respective nomenclatures and chemical- and PGN-specific properties.
Notably, our PGN_MS2 represents a dedicated in silico MS/MS spectral prediction algorithm for PGNs, whose unique sugar and non-proteogenic amino acids defy reliable predictions by existing tools developed for small molecules or proteins. For instance, the proteomics analysis software Byonic has been previously used for PGN analysis,19 which takes a peptide-centric approach such that common PGN structures are viewed as variable modifications of the stem peptide. As a result, one needs to manually annotate the masses of various moieties, such as anhydro- and de-acetylation of the disaccharide backbone, non-proteogenic and amidated amino acids for PGN search and analysis. In contrast, PGN_MS2 is specialized to predict MS/MS spectra for PGN chemotypes, where the user simply selects the desired structural features of PGNs without needing to calculate and input their respective masses, rendering the analysis process user-friendly and flexible to accommodate novel modifications.
To validate the reliability of our workflow, we compared the cosine similarity scores between the in silico predicted spectra of PGNs and the authentic spectra of several PGN motifs. Remarkably, PGN_MS2 consistently outperformed other spectral simulation software packages in metabolomics and proteomics. Moreover, the PGN_MS2 predicted spectra matched well with the fragmentation data acquired using different instruments (i.e., Orbitrap and Q-TOF), showcasing the congruity of the in silico PGN fragmentation rules. We further demonstrated the facile and accurate assignment of closely related PGN isomers via automated spectral matching and scoring. However, we also noted that the experimental MS/MS spectra of low abundant analytes tend to have lower quality, which led to the top predicted PGN structures having very close similarity scores. In these cases, manual inspections are needed to ensure accurate structural assignment. To facilitate such manual analysis, our PGN_MS2 records the precursor, fragmentation type, and chemical structure of all fragment peaks generated (Fig. S2D†).
During the preparation of our manuscript, Hsu et al. reported a high-throughput automated muropeptide analysis (HAMA) framework that generates in silico MS/MS fragments for PGN analysis.85 However, we note several key distinctions between our workflow and HAMA. First, for in silico prediction of MS/MS patterns, HAMA focuses on fragmentation of the stem peptide, solely generating the b- and y-ions of stem peptides without any fragmentation of the sugar moieties in PGNs. Secondly, HAMA restricts the types of PGN modifications to <6 (including those on sugar motifs and peptide aminations etc.) to avoid mass coincidences. On the other hand, our PGN_MS2 is developed especially for simulating MS/MS patterns of soluble muropeptide chemotypes, whose fragmentation rules were derived from empirical analysis that include both sugar and peptide moieties in PGNs, showcasing superior matches to actual MS/MS data from HCD and CID fragmentations. As a result, our workflow accommodates much more diverse PGNs in the database and accurately distinguishes structural isomers by MS/MS matching. Notably, HAMA is reportedly unable to differentiate the 3–4 and 3–3 crosslinks in dimeric PGNs and hence can only consider 3–4 crosslinks currently. In contrast, with our PGN workflow, the 3–4 and 3–3 crosslinked PGN isomers can be facilely identified with signature fragments from the in silico MS/MS patterns (Fig. 4C and S7†). Moreover, our PGN_MS2 also includes specific fragmentations pertinent to the isoGln/Glu (q1/q2 and e1/e2), exhibiting its unique power in determining the amidation positions on isomeric PGNs (Fig. S3 and S9†). Furthermore, PGN_MS2 creates a PGN MS library that is compatible with various vendors or open-source MS analysis software, offering users flexibility in choosing their preferred platforms for data analysis. To promote open-access research in the PGN field, PGN_MS2 itself is open source (https://github.com/jerickwan/PGN_MS2), where users can download directly to use or modify the code to increase the scope of fragmentations and the identities of amino acids and/or glycan motifs, etc. Recognizing the lack of PGNs in the existing metabolomics databank, we also uploaded the annotated MS/MS spectra of PGNs across different bacteria to the metabolomic data repository MoNA (https://mona.fiehnlab.ucdavis.edu/spectra/browse?query=exists(tags.text:%27QiaoLab_PGN%27)).
Aided by PGN_MS2, we uncovered that Bifidobacterium spp. features a large abundance of anNAM termini in peptidoglycan, which are non-hydrolytic cleavage products of LT enzymes.72 By homology searching, we identified and biochemically characterized three enzymes as LTs in Bifidobacterium, namely, MltG, RpfB-FL, and RpfB-Truncated, respectively. Interestingly, MltG strictly requires nascent peptidoglycan strands as substrates for non-hydrolytic cleavage, whereas RpfBs robustly use mature sacculi to produce anhydro-NAM termini. The complementary substrate preferences of these LTs may account for the remarkably high amount of anhydro-PGNs. Importantly, Bifidobacterium spp. are well-known probiotics that confer beneficial effects on hosts such as reducing LPS-induced inflammation in vitro and in vivo.21,22 We demonstrated that pre-treatment with anhydro-PGN effectively suppressed LPS-induced proinflammatory cytokine expression in murine macrophages in vitro. As Bifidobacterium anhydro-PGNs are non-agnostic to canonical NOD1 and NOD2 immune receptors,81–84 the underlying mechanisms of their anti-inflammatory roles are yet to be elucidated. We are currently working to genetically manipulate putative LTs in Bifidobacterium spp. to evaluate the anti-inflammatory activities of the mutants in vivo, which may lead to improved probiotics.
Conclusions
In summary, we established a novel and robust PGN_MS2 tool to facilitate automated PGN identification and analysis, addressing the key bottleneck in the current analysis workflow. Empowered by PGN_MS2, we characterized the peptidoglycan composition of various gut bacteria species. We discovered an abundance of anhydro-PGNs (i.e., LT products) in Bifidobacterium spp., which is unusual for Gram-positive bacteria, and further biochemically characterized three putative LTs in Bifidobacterium. Lastly, we established that Bifidobacterium anhydro-PGNs exhibit anti-inflammatory activity in vitro, offering insights into novel bioactive PGNs in gut microbiota-host crosstalk.Data availability
PGN_MS2 is available on GitHub (https://github.com/jerickwan/PGN_MS2). Annotated MS/MS spectra of muropeptides have been deposited on MoNA (https://mona.fiehnlab.ucdavis.edu/spectra/browse?query=exists(tags.text:%27QiaoLab_PGN%27)).Author contributions
J. M. C. K. and Y. Q. designed the research; J. M. C. K. performed coding and developed the PGN analysis pipeline; J. M. C. K. and E. W. L. N. performed biochemical characterization of Bifidobacterium LTs; Y. L. performed the in vitro assays with RAW264.7 and HEK-Blue reporter cells; Y. L., C. L., and Y. Z. contributed to LC-MS method development; E. K. and S. W. provided anaerobic cultures of the gut bacteria; X. Z. and X. L. provided synthetic ah-PGN for testing; J. M. C. K., Y. L. and Y. Q. analyzed the data; S. W. and Y. Q. supervised the work; J. M. C. K. and Y. Q. wrote the paper with inputs from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge members of the Qiao lab for critical reading of the manuscript. J. M. C. K.’s PhD candidature is supported by the Nanyang Presidential Graduate Scholarship. This work was supported by the National Research Foundation (NRF) Singapore, NRF-NRFF12-2020-0006, NTU-Start-up grant, and MOE AcRF Tier 1, RG3/22 to Y. Q.Notes and references
- W. Vollmer, D. Blanot and M. A. De Pedro, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS PubMed.
- O. Irazoki, S. B. Hernandez and F. Cava, Front. Microbiol., 2019, 10, 500 CrossRef PubMed.
- J. Dworkin, Annu. Rev. Microbiol., 2014, 68, 137–154 CrossRef CAS PubMed.
- R. Wheeler, G. Chevalier, G. Eberl and I. Gomperts Boneca, Cell. Microbiol., 2014, 16, 1014–1023 CrossRef CAS PubMed.
- J. D. Laman, B. A. ’t Hart, C. Power and R. Dziarski, Trends Mol. Med., 2020, 26, 670–682 CrossRef CAS PubMed.
- R. Wheeler, P. A. D. Bastos, O. Disson, A. Rifflet, I. Gabanyi, J. Spielbauer, M. Bérard, M. Lecuit and I. G. Boneca, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2209936120 CrossRef CAS PubMed.
- M. E. Griffin, J. Espinosa, J. L. Becker, J. D. Luo, T. S. Carroll, J. K. Jha, G. R. Fanger and H. C. Hang, Science, 2021, 373, 1040–1046 CrossRef CAS PubMed.
- Z. Huang, J. Wang, X. Xu, H. Wang, Y. Qiao, W. C. Chu, S. Xu, L. Chai, F. Cottier, N. Pavelka, M. Oosting, L. A. B. Joosten, M. Netea, C. Y. L. Ng, K. P. Leong, P. Kundu, K. P. Lam, S. Pettersson and Y. Wang, Nat. Microbiol., 2019, 4, 766–773 CrossRef CAS PubMed.
- T. Arentsen, Y. Qian, S. Gkotzis, T. Femenia, T. Wang, K. Udekwu, H. Forssberg and R. Diaz Heijtz, Mol. Psychiatry, 2017, 22, 257–266 CrossRef CAS PubMed.
- I. Gabanyi, G. Lepousez, R. Wheeler, A. Vieites-Prado, A. Nissant, S. Wagner, C. Moigneu, S. Dulauroy, S. Hicham, B. Polomack, F. Verny, P. Rosenstiel, N. Renier, I. G. Boneca, G. Eberl and P.-M. Lledo, Science, 2022, 376, eabj3986 CrossRef CAS PubMed.
- K. L. Bersch, K. E. DeMeester, R. Zagani, S. Chen, K. A. Wodzanowski, S. Liu, S. Mashayekh, H.-C. Reinecker and C. L. Grimes, ACS Cent. Sci., 2021, 7, 688–696 CrossRef CAS PubMed.
- P. Horcajo, M. A. de Pedro and F. Cava, Microb. Drug Resist., 2012, 18, 306–313 CrossRef CAS PubMed.
- A. J. F. Egan, J. Errington and W. Vollmer, Nat. Rev. Microbiol., 2020, 18, 446–460 CrossRef CAS PubMed.
- J. M. C. Kwan and Y. Qiao, ChemBioChem, 2023, 24, e202200693 CrossRef CAS PubMed.
- B. Glauner, Anal. Biochem., 1988, 172, 451–464 CrossRef CAS PubMed.
- D. Kühner, M. Stahl, D. D. Demircioglu and U. Bertsche, Sci. Rep., 2014, 4, 1–7 Search PubMed.
- E. M. Anderson, D. Sychantha, D. Brewer, A. J. Clarke, J. Geddes-McAlister and C. M. Khursigara, J. Biol. Chem., 2020, 295, 504–516 CrossRef CAS PubMed.
- E. M. Anderson, N. Shaji Saji, A. C. Anderson, D. Brewer, A. J. Clarke and C. M. Khursigara, mSystems, 2022, 7(3), e00156 CrossRef PubMed.
- M. Bern, R. Beniston and S. Mesnage, Anal. Bioanal. Chem., 2017, 409, 551–560 CrossRef CAS PubMed.
- S. Porfírio, R. W. Carlson and P. Azadi, Trends Microbiol., 2019, 27, 607–622 CrossRef PubMed.
- S. Singh, R. Bhatia, P. Khare, S. Sharma, S. Rajarammohan, M. Bishnoi, S. K. Bhadada, S. S. Sharma, J. Kaur and K. K. Kondepudi, Sci. Rep., 2020, 10, 18597 CrossRef CAS PubMed.
- S. Sun, L. Luo, W. Liang, Q. Yin, J. Guo, A. M. Rush, Z. Lv, Q. Liang, M. A. Fischbach, J. L. Sonnenburg, D. Dodd, M. M. Davis and F. Wang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 27509–27515 CrossRef CAS.
- G. Landrum, RDKit: Open-source cheminformatics, https://www.rdkit.org, accessed 20 May 2022 Search PubMed.
- C. Ruttkies, E. L. Schymanski, S. Wolf, J. Hollender and S. Neumann, J. Cheminf., 2016, 8, 1–16 Search PubMed.
- T. Kind, K.-H. H. Liu, D. Y. Lee, B. Defelice, J. K. Meissen and O. Fiehn, Nat. Methods, 2013, 10, 755–758 CrossRef CAS PubMed.
- F. Wang, J. Liigand, S. Tian, D. Arndt, R. Greiner and D. S. Wishart, Anal. Chem., 2021, 93, 11692–11700 CrossRef CAS PubMed.
- S. Degroeve, D. Maddelein and L. Martens, Nucleic Acids Res., 2015, 43, W326–W330 CrossRef CAS PubMed.
- T. Kind, H. Tsugawa, T. Cajka, Y. Ma, Z. Lai, S. S. Mehta, G. Wohlgemuth, D. K. Barupal, M. R. Showalter, M. Arita and O. Fiehn, Mass Spectrom. Rev., 2018, 37, 513–532 CrossRef CAS PubMed.
- C. T. Tan, X. Xu, Y. Qiao and Y. Wang, Nat. Commun., 2021, 12, 2560 CrossRef CAS PubMed.
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS.
- P. Zhang, W. Chan, I. L. Ang, R. Wei, M. M. T. Lam, K. M. K. Lei and T. C. W. Poon, Sci. Rep., 2019, 9, 6453 CrossRef PubMed.
- S. E. Stein and D. R. Scott, J. Am. Soc. Mass Spectrom., 1994, 5, 859–866 CrossRef CAS PubMed.
- D. Szabó, G. Schlosser, K. Vékey, L. Drahos and Á. Révész, J. Mass Spectrom., 2021, 56, e4693 CrossRef PubMed.
- H. Tsugawa, K. Ikeda, M. Takahashi, A. Satoh, Y. Mori, H. Uchino, N. Okahashi, Y. Yamada, I. Tada, P. Bonini, Y. Higashi, Y. Okazaki, Z. Zhou, Z. J. Zhu, J. Koelmel, T. Cajka, O. Fiehn, K. Saito, M. Arita and M. Arita, Nat. Biotechnol., 2020, 38, 1159–1163 CrossRef CAS PubMed.
- A. V. Patel, R. D. Turner, A. Rifflet, A. E. Acosta-Martin, A. Nichols, M. M. Awad, D. Lyras, I. G. Boneca, M. Bern, M. O. Collins and S. Mesnage, eLife, 2021, 10, 1–4 Search PubMed.
- B. Glauner, J. V. Holtje and U. Schwarz, J. Biol. Chem., 1988, 263, 10088–10095 CrossRef CAS PubMed.
- B. L. M. De Jonge, Y. S. Chang, D. Gage and A. Tomasz, J. Biol. Chem., 1992, 267, 11248–11254 CrossRef CAS PubMed.
- S. Willing, E. Dyer, O. Schneewind and D. Missiakas, J. Biol. Chem., 2020, 295, 13664–13676 CrossRef CAS.
- J. D. Chang, A. G. Wallace, E. E. Foster and S. J. Kim, Biochemistry, 2018, 57, 1274–1283 CrossRef CAS PubMed.
- S. Magnet, A. Arbeloa, J. L. Mainardi, J. E. Hugonnet, M. Fourgeaud, L. Dubost, A. Marie, V. Delfosse, C. Mayer, L. B. Rice and M. Arthur, J. Biol. Chem., 2007, 282, 13151–13159 CrossRef CAS PubMed.
- J. D. Chang, E. E. Foster, A. G. Wallace and S. J. Kim, Sci. Rep., 2017, 7, 1–8 CrossRef PubMed.
- B. Kim, Y.-C. Wang, C. W. Hespen, J. Espinosa, J. Salje, K. J. Rangan, D. A. Oren, J. Y. Kang, V. A. Pedicord and H. C. Hang, eLife, 2019, 8, e45343 CrossRef PubMed.
- E. Bernard, T. Rolain, P. Courtin, A. Guillot, P. Langella, P. Hols and M.-P. Chapot-Chartier, J. Biol. Chem., 2011, 286, 23950–23958 CrossRef CAS PubMed.
- A. J. Apostolos, S. E. Pidgeon and M. M. Pires, ACS Chem. Biol., 2020, 15, 1261–1267 CrossRef CAS PubMed.
- F. Ngadjeua, E. Braud, S. Saidjalolov, L. Iannazzo, D. Schnappinger, S. Ehrt, J. E. Hugonnet, D. Mengin-Lecreulx, D. Patin, M. Ethève-Quelquejeu, M. Fonvielle and M. Arthur, Chem.–Eur. J., 2018, 24, 5743–5747 CrossRef CAS PubMed.
- A. Zapun, J. Philippe, K. A. Abrahams, L. Signor, D. I. Roper, E. Breukink and T. Vernet, ACS Chem. Biol., 2013, 8, 2688–2696 CrossRef CAS PubMed.
- S. E. Pidgeon, A. J. Apostolos, J. M. Nelson, M. Shaku, B. Rimal, M. N. Islam, D. C. Crick, S. J. Kim, M. S. Pavelka, B. D. Kana and M. M. Pires, ACS Chem. Biol., 2019, 14, 2185–2196 CAS.
- A. J. Apostolos, J. M. Nelson, J. R. A. Silva, J. Lameira, A. M. Achimovich, A. Gahlmann, C. N. Alves and M. M. Pires, ACS Chem. Biol., 2020, 15, 2966–2975 CrossRef CAS PubMed.
- A. M. Strandén, M. Roos and B. Berger-Bächi, Microb. Drug Resist., 1996, 2, 201–207 CrossRef.
- J.-E. Hugonnet, D. Mengin-Lecreulx, A. Monton, T. den Blaauwen, E. Carbonnelle, C. Veckerlé, Y. V. Brun, M. van Nieuwenhze, C. Bouchier, K. Tu, L. B. Rice and M. Arthur, eLife, 2016, 5, e19469 CrossRef PubMed.
- N. Morè, A. M. Martorana, J. Biboy, C. Otten, M. Winkle, C. K. G. Serrano, A. Montón Silva, L. Atkinson, H. Yau, E. Breukink, T. den Blaauwen, W. Vollmer and A. Polissi, mBio, 2019, 10(1), e02729 CrossRef PubMed.
- A. Taguchi, J. E. Page, H.-C. T. Tsui, M. E. Winkler and S. Walker, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2103740118 CrossRef CAS PubMed.
- S. H. Wong and J. Yu, Nat. Rev. Gastroenterol. Hepatol., 2019, 16(11), 690–704 CrossRef CAS PubMed.
- P. D. Cani and W. M. de Vos, Front. Microbiol., 2017, 8, 1–8 Search PubMed.
- J. Chen, X. Chen and C. L. Ho, Front. Bioeng. Biotechnol., 2021, 9, 770248 CrossRef PubMed.
- P. Garcia-Vello, H. L. P. Tytgat, J. Gray, J. Elzinga, F. Di Lorenzo, J. Biboy, D. Vollmer, C. De Castro, W. Vollmer, W. M. de Vos and A. Molinaro, Glycobiology, 2022, 1–8 Search PubMed.
- O. Kandler, Int. J. Syst. Bacteriol., 1970, 20, 491–507 CrossRef CAS.
- E. N. Vasstrand, T. Hofstad, C. Endresen and H. B. Jensen, Infect. Immun., 1979, 25, 775–780 CrossRef CAS PubMed.
- K. Kato, T. Umemoto, H. Sagawa and S. Kotani, Curr. Microbiol., 1979, 3, 147–151 CrossRef CAS.
- E. N. Vasstrand, Infect. Immun., 1981, 33, 75–82 CrossRef CAS PubMed.
- S. Bellais, M. Arthur, L. Dubost, J.-E. Hugonnet, L. Gutmann, J. van Heijenoort, R. Legrand, J.-P. Brouard, L. Rice and J.-L. Mainardi, J. Biol. Chem., 2006, 281, 11586–11594 CrossRef CAS PubMed.
- P. Veiga, S. Piquet, A. Maisons, S. Furlan, P. Courtin, M. P. Chapot-Chartier and S. Kulakauskas, Mol. Microbiol., 2006, 62, 1713–1724 CrossRef CAS PubMed.
- P. Veiga, M. Erkelenz, E. Bernard, P. Courtin, S. Kulakauskas and M. P. Chapot-Chartier, J. Bacteriol., 2009, 191, 3752–3757 CrossRef CAS PubMed.
- J. Gao, X. Zhao, S. Hu, Z. Huang, M. Hu, S. Jin, B. Lu, K. Sun, Z. Wang, J. Fu, R. K. Weersma, X. He and H. Zhou, Cell Host Microbe, 2022, 30, 1435–1449.e9 CrossRef CAS PubMed.
- J. Gao, L. Wang, J. Jiang, Q. Xu, N. Zeng, B. Lu, P. Yuan, K. Sun, H. Zhou and X. He, Nat. Commun., 2023, 14, 3338 CrossRef CAS PubMed.
- F. Cava, M. A. De Pedro, H. Lam, B. M. Davis and M. K. Waldor, EMBO J., 2011, 30, 3442–3453 CrossRef CAS PubMed.
- N.-H. Le, K. Peters, A. Espaillat, J. R. Sheldon, J. Gray, G. Di Venanzio, J. Lopez, B. Djahanschiri, E. A. Mueller, S. W. Hennon, P. A. Levin, I. Ebersberger, E. P. Skaar, F. Cava, W. Vollmer and M. F. Feldman, Sci. Adv., 2020, 6, eabb5614 CrossRef CAS PubMed.
- W. Vollmer, FEMS Microbiol. Rev., 2008, 32, 287–306 CrossRef CAS PubMed.
- K. M. Davis and J. N. Weiser, Infect. Immun., 2011, 79, 562–570 CrossRef CAS PubMed.
- H. C. T. Tsui, J. J. Zheng, A. N. Magallon, J. D. Ryan, R. Yunck, B. E. Rued, T. G. Bernhardt and M. E. Winkler, Mol. Microbiol., 2016, 100, 1039–1065 CrossRef CAS PubMed.
- M. R. Stapleton, M. J. Horsburgh, E. J. Hayhurst, L. Wright, I. M. Jonsson, A. Tarkowski, J. F. Kokai-Kun, J. J. Mond and S. J. Foster, J. Bacteriol., 2007, 189, 7316–7325 CrossRef CAS PubMed.
- D. A. Dik, D. R. Marous, J. F. Fisher and S. Mobashery, Crit. Rev. Biochem. Mol. Biol., 2017, 52, 503–542 CrossRef PubMed.
- R. Yunck, H. Cho and T. G. Bernhardt, Mol. Microbiol., 2016, 99, 700–718 CrossRef CAS PubMed.
- J. Sassine, M. Pazos, E. Breukink and W. Vollmer, Cell Surf., 2021, 7, 100053 CrossRef CAS PubMed.
- M. A. Welsh, A. Taguchi, K. Schaefer, D. Van Tyne, F. Lebreton, M. S. Gilmore, D. Kahne and S. Walker, J. Am. Chem. Soc., 2017, 139, 17727 CrossRef CAS PubMed.
- V. D. Nikitushkin, G. R. Demina, M. O. Shleeva, S. V. Guryanova, A. Ruggiero, R. Berisio and A. S. Kaprelyants, FEBS J., 2015, 282, 2500–2511 CrossRef CAS PubMed.
- D. L. Sexton, R. J. St-Onge, H. J. Haiser, M. R. Yousef, L. Brady, C. Gao, J. Leonard and M. A. Elliot, J. Bacteriol., 2015, 197, 848–860 CrossRef PubMed.
- S. J. Lahtinen, M. Gueimonde, A. C. Ouwehand, J. P. Reinikainen and S. J. Salminen, Appl. Environ. Microbiol., 2005, 71, 1662–1663 CrossRef CAS PubMed.
- A. R. Ortiz Camargo, O. van Mastrigt, R. S. Bongers, K. Ben-Amor, J. Knol, T. Abee and E. J. Smid, Microbiol. Spectr., 2023, 11, e0256822 CrossRef PubMed.
- A. J. Wolf and D. M. Underhill, Nat. Rev. Immunol., 2017, 18(4), 243–254 CrossRef.
- S. E. Girardin, I. G. Boneca, L. A. M. Carneiro, A. Antignac, M. Jéhanno, J. Viala, K. Tedin, M. K. Taha, A. Labigne, U. Zähringer, A. J. Coyle, P. S. DiStefano, J. Bertin, P. J. Sansonetti and D. J. Philpott, Science, 2003, 300, 1584–1587 CrossRef CAS PubMed.
- Y. Fujimoto, A. R. Pradipta, N. Inohara and K. Fukase, Nat. Prod. Rep., 2012, 29, 568–579 RSC.
- S. E. Girardin, I. G. Boneca, J. Viala, M. Chamaillard, A. Labigne, G. Thomas, D. J. Philpott and P. J. Sansonetti, J. Biol. Chem., 2003, 278, 8869–8872 CrossRef CAS PubMed.
- N. Inohara, Y. Ogura, A. Fontalba, O. Gutierrez, F. Pons, J. Crespo, K. Fukase, S. Inamura, S. Kusumoto, M. Hashimoto, S. J. Foster, A. P. Moran, J. L. Fernandez-Luna and G. Nuñez, J. Biol. Chem., 2003, 278, 5509–5512 CrossRef CAS PubMed.
- H. Ya-Chen, S. Pin-Rui, H. Lin-Jie, C. Kum-Yi, C. Chun-hsien and H. Cheng-Chih, eLife, 2023, 12, 88491 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05819k |
| This journal is © The Royal Society of Chemistry 2024 |