
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual catalytic strategy by non-heme Fe(II)/2-oxoglutarate-dependent aspartyl hydroxylase AspH†
Anandhu
Krishnan
 a,
Sodiq O.
Waheed
a,
Ann
Varghese
a,
Fathima Hameed
Cherilakkudy
a,
Christopher J.
Schofield
b and
Tatyana G.
Karabencheva-Christova
*a
a,
Sodiq O.
Waheed
a,
Ann
Varghese
a,
Fathima Hameed
Cherilakkudy
a,
Christopher J.
Schofield
b and
Tatyana G.
Karabencheva-Christova
*a
aDepartment of Chemistry, Michigan Technological University, Houghton, MI 49931, USA. E-mail: tatyanak@mtu.edu
bChemistry Research Laboratory, Department of Chemistry and the Ineos Oxford Institute for Antimicrobial Research, University of Oxford, OX1 3TA, Oxford, UK
First published on 5th February 2024
Abstract
Biocatalytic C–H oxidation reactions are of important synthetic utility, provide a sustainable route for selective synthesis of important organic molecules, and are an integral part of fundamental cell processes. The multidomain non-heme Fe(II)/2-oxoglutarate (2OG) dependent oxygenase AspH catalyzes stereoselective (3R)-hydroxylation of aspartyl- and asparaginyl-residues. Unusually, compared to other 2OG hydroxylases, crystallography has shown that AspH lacks the carboxylate residue of the characteristic two-His-one-Asp/Glu Fe-binding triad. Instead, AspH has a water molecule that coordinates Fe(II) in the coordination position usually occupied by the Asp/Glu carboxylate. Molecular dynamics (MD) and quantum mechanics/molecular mechanics (QM/MM) studies reveal that the iron coordinating water is stabilized by hydrogen bonding with a second coordination sphere (SCS) carboxylate residue Asp721, an arrangement that helps maintain the six coordinated Fe(II) distorted octahedral coordination geometry and enable catalysis. AspH catalysis follows a dioxygen activation-hydrogen atom transfer (HAT)-rebound hydroxylation mechanism, unusually exhibiting higher activation energy for rebound hydroxylation than for HAT, indicating that the rebound step may be rate-limiting. The HAT step, along with substrate positioning modulated by the non-covalent interactions with SCS residues (Arg688, Arg686, Lys666, Asp721, and Gln664), are essential in determining stereoselectivity, which likely proceeds with retention of configuration. The tetratricopeptide repeat (TPR) domain of AspH influences substrate binding and manifests dynamic motions during catalysis, an observation of interest with respect to other 2OG oxygenases with TPR domains. The results provide unique insights into how non-heme Fe(II) oxygenases can effectively catalyze stereoselective hydroxylation using only two enzyme-derived Fe-ligating residues, potentially guiding enzyme engineering for stereoselective biocatalysis, thus advancing the development of non-heme Fe(II) based biomimetic C–H oxidation catalysts, and supporting the proposal that the 2OG oxygenase superfamily may be larger than once perceived.
Introduction
Enzyme-catalyzed hydroxylation of C–H bonds of proteins is a post-translational modification that in animals is involved in many important processes.1 Hydroxylation of specific proline- and lysine-residues in procollagen, for example, as catalyzed by prolyl- and lysine-hydroxylases is essential for the function and stability of collagen, the most abundant protein in metazoans.2,3 Hydroxylated amino acid residues are present in numerous biologically active natural products,4–7 and such hydroxylations provide valuable starting points for further chemical modifications.8 Hydroxylations of aspartyl and asparaginyl residues, as catalyzed by the aspartate/asparagine-β-hydroxylase (AspH) and factor-inhibiting hypoxia-inducing factor (FIH), are proposed to play crucial roles in regulating hypoxia,9,10 calcium signaling,11,12 and blood coagulation.13–15 AspH catalyzes the post-translational C3 hydroxylation of specific aspartate and asparagine residues in the epidermal growth factor-like domains (EGFDs), which perform vital biological functions,16,17 including intercellular signaling,18–21 calcium binding,22–25 and extracellular matrix formation.26–28Direct and selective functionalization of C–H bonds is an efficient and powerful tool for synthesizing complex molecules.29–31 However, achieving regio- and stereoselective C–H bond oxidation poses significant challenges due to the high bond dissociation energy and the presence of multiple C–H bonds within the molecules.32,33 Consequently, the pursuit of environmentally friendly and cost-effective synthetic methods for C–H oxidation reactions has gained substantial attention.34–36 Enzymes provide a sustainable solution, as both natural and engineered enzymes selectively and efficiently mediate diverse C–H oxidation reactions.37–43 Notably, heme- and non-heme iron-containing enzymes catalyze direct C–H oxidations with remarkable stereo- and regio-selectivity8,39,44,45 thus offering valuable insights for applying redesigned enzymes as synthetic utilities.46–49 In contrast to small molecule catalysts, enzymes can be continuously optimized, redesigned and repurposed for performing specific C–H reactions with industrial importance.50–52 Residues in the Second Coordination Sphere (SCS) and Remote Areas (RA) became increasingly important tools for modulating and improving enzyme activity and product selectivity.53–59
Non-heme Fe(II) and 2-oxoglutarate (2OG) dependent oxygenases are gaining prominence as industrial biocatalysts, owing to their scalability and versatility.60 AspH (Fig. 1) is a non-heme Fe(II)/2OG hydroxylase, that catalyzes the stereoselective (3R)-hydroxylation of aspartyl- and asparaginyl-residues in EGFDs.15,61–64 AspH is essential for normal human biology and has assigned roles in placental implantation and fetal growth.16,65,66 AspH variants, such as R735Q/W, R688Q cause the ophthalmological condition-the Traboulsi syndrome67–69 and the G434V variant causes vesicoureteral reflux (VUR).70 The AspH gene is over-expressed in many cancer cells, in a manner proposed to promote cell migration and reduce the life expectancy of cancer patients; AspH is thus a promising target for anticancer therapy.71–74 AspH is also hypoxically upregulated, and its kinetic parameters suggest it has potential as a hypoxia sensing enzyme, as known for some other 2OG oxygenases, including FIH.75 Unveiling the catalytic strategy of AspH thus offers potential biotechnological applications, such as developing AspH-selective inhibitors that do not affect other Fe(II)/2OG enzymes and repurposing the enzyme for stereoselective C–H activation reactions. Such applications could be relevant, for example, for the efficient and sustainable synthesis of β-hydroxy acids/amides, which are structural motifs found in drugs, and biologically active molecules.76
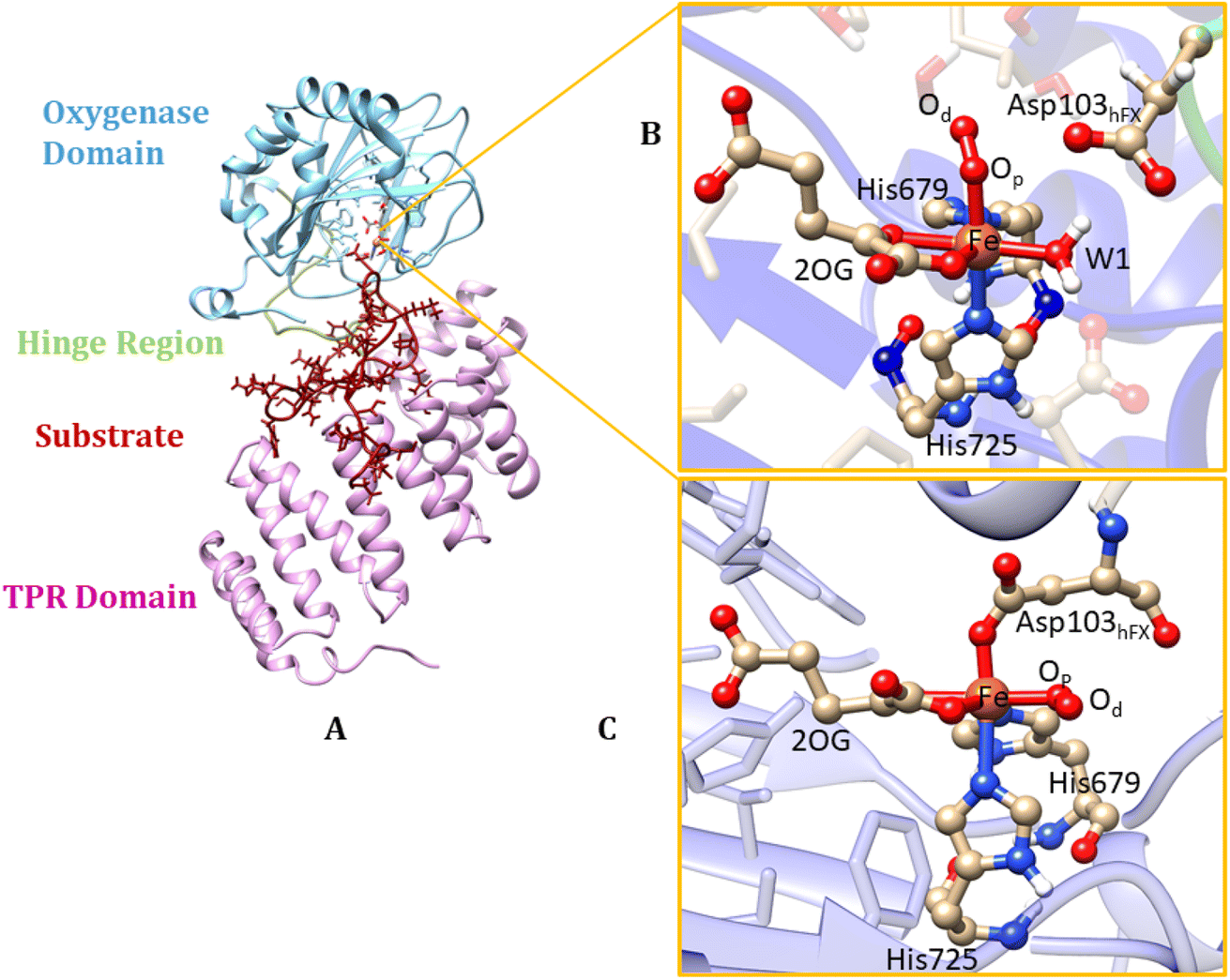 | ||
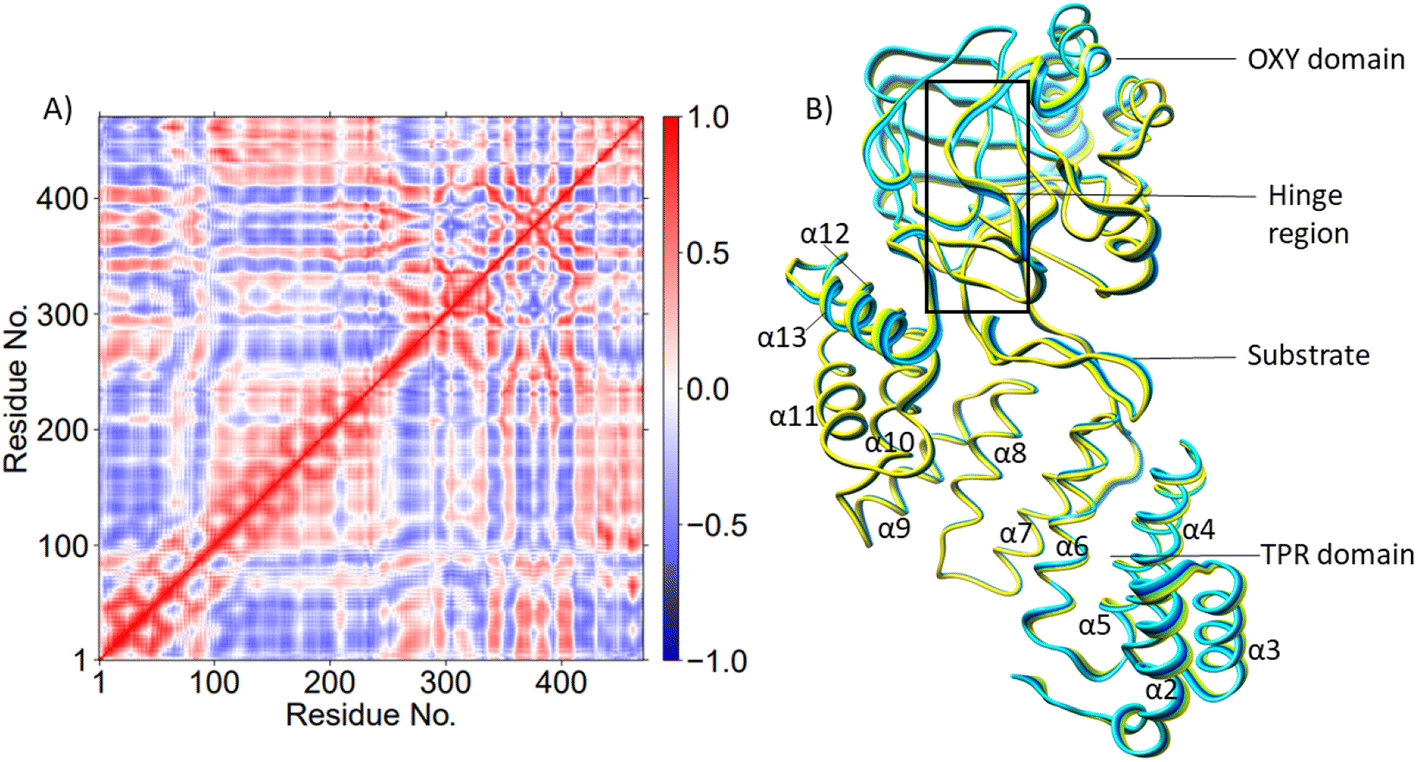
| Fig. 1 Views from an AspH X-ray crystal structure (PDB ID: 5JZ8). (A) displays the OXY domain in blue, the TPR domain in pink, the hinge region in green, and the substrate in red. (B) QM/MM optimized structure with Fe(III) coordinated to the histidines (His679 & His725), 2OG, O2, water (W1) (mode A). In mode A the Asp103hFX does not coordinate to the iron; instead, W1 coordinates the iron. (C) Mode B of the Fe(III)-superoxo complex, where the Asp103hFX coordinates the iron displacing a water molecule. | ||
In their resting state, most 2OG-dependent oxygenases contain a high-spin (HS) Fe(II) center in their active sites coordinated by a highly conserved structural motif, that is, the imidazole rings from two histidine-residues and one carboxylate from an aspartate- or glutamate-residue, sometimes called a ‘facial triad’.77–80 By contrast, AspH contains only two histidine Fe-ligands, and no carboxylate that coordinates the iron.62,81 This arrangement is similar to the Fe(II)/2OG dependent halogenases, such as CytC3 (ref. 82) and SyrB2,83,84 and the catalytically active D201G (but not D201A) variant of factor inhibiting HIF (FIH).85 At the Fe(II) center of AspH, the normal carboxylate ligand is replaced by a coordinating water molecule (W1), the ligation of which is stabilized by a hydrogen bond with the SCS residue Asp721.62 Multiple studies have evidenced the key role of the facial triad in catalysis by 2OG oxygenases;86–88 however, how AspH catalysis proceeds with a dyad comprising only two histidine ligands, without the normal carboxylate ligand is unknown.
AspH is a multidomain protein (Fig. 1A) with a tetratricopeptide repeat (TPR) domain,89 a hinge region, and an oxygenase (OXY) domain. The OXY domain contains a core double-stranded beta-helix (DSBH) motif, which is flanked by several alpha-helices,62 in a manner characteristic of 2OG oxygenases.90,91 TPR domain has a functional role in substrate recognition. It is also present in other 2OG oxygenases such as collagen prolyl hydroxylases (CPH), some JmjC histone demethylases (KDM), e.g. ubiquitously transcribed X (UTX) and Y (UTY), and the leprecans.92–95 The hinge region of AspH connects the OXY domain with the TPR domain.62 Since substrate binding involves both the OXY and TPR domains, inter-domain dynamics and motions are expected to contribute to AspH catalysis.96–100
AspH hydroxylates EGFD substrates with a non-canonical disulfide pattern with disulfide links between Cys 1-2, 3-4, and 5-6, rather than the often observed canonical Cys 1-3, 2-4, 5-6 disulfide pattern.62,64 The EGFD substrate of AspH considered in the present study was the N-terminal EGFD of human coagulation factor X (hFX), which has 39 amino acid residues (86–124), with a disulfide linkage between Cys101hFX and Cys110hFX.62 X-ray crystallographic studies have revealed two different aspartyl-residue substrate binding modes at the active site of the AspH enzyme-substrate (ES) complex. In the first binding mode (binding mode A, Fig. 1B), which most closely corresponds to the productive substrate binding in Fe(II)/2OG enzymes, Asp103hFX (the residue undergoing hydroxylation) of the EGFD hFX is not coordinated to the Fe(II). In the second binding mode B (Fig. 1C), which is likely an unproductive one, the Asp103hFX side chain carboxylate directly coordinates to the active site metal ion, trans to His725, displacing the coordinated water molecule.62
The mechanisms of 2OG oxygenases have been studied experimentally and computationally (Scheme 1).80,101–123 The catalytic cycle is initiated by the co-substrate 2OG, then substrate binding to the Fe(II) center, followed by O2 binding producing a Fe(III)-superoxo anion-radical (Fe(III)-OO˙−) intermediate.106,124 The latter reacts via a succinyl peroxide intermediate to give a ferryl (Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) intermediate125–130 with the formation of CO2 and Fe-linked succinate. During hydroxylation, a substrate C–H hydrogen is abstracted by the Fe(IV)O intermediate, via σ or π-channel electron transfer.131 The resulting Fe(III)–OH intermediate undergoes rebound hydroxylation with the substrate radical, forming an alcohol and regenerating Fe(II). SCS and long-range (LR) interacting residues and correlated motions have been demonstrated to play important roles in the catalysis by non-heme Fe(II)/2OG enzymes controlling substrate binding and the catalytic mechanism.57,58,132–139
O) intermediate125–130 with the formation of CO2 and Fe-linked succinate. During hydroxylation, a substrate C–H hydrogen is abstracted by the Fe(IV)O intermediate, via σ or π-channel electron transfer.131 The resulting Fe(III)–OH intermediate undergoes rebound hydroxylation with the substrate radical, forming an alcohol and regenerating Fe(II). SCS and long-range (LR) interacting residues and correlated motions have been demonstrated to play important roles in the catalysis by non-heme Fe(II)/2OG enzymes controlling substrate binding and the catalytic mechanism.57,58,132–139
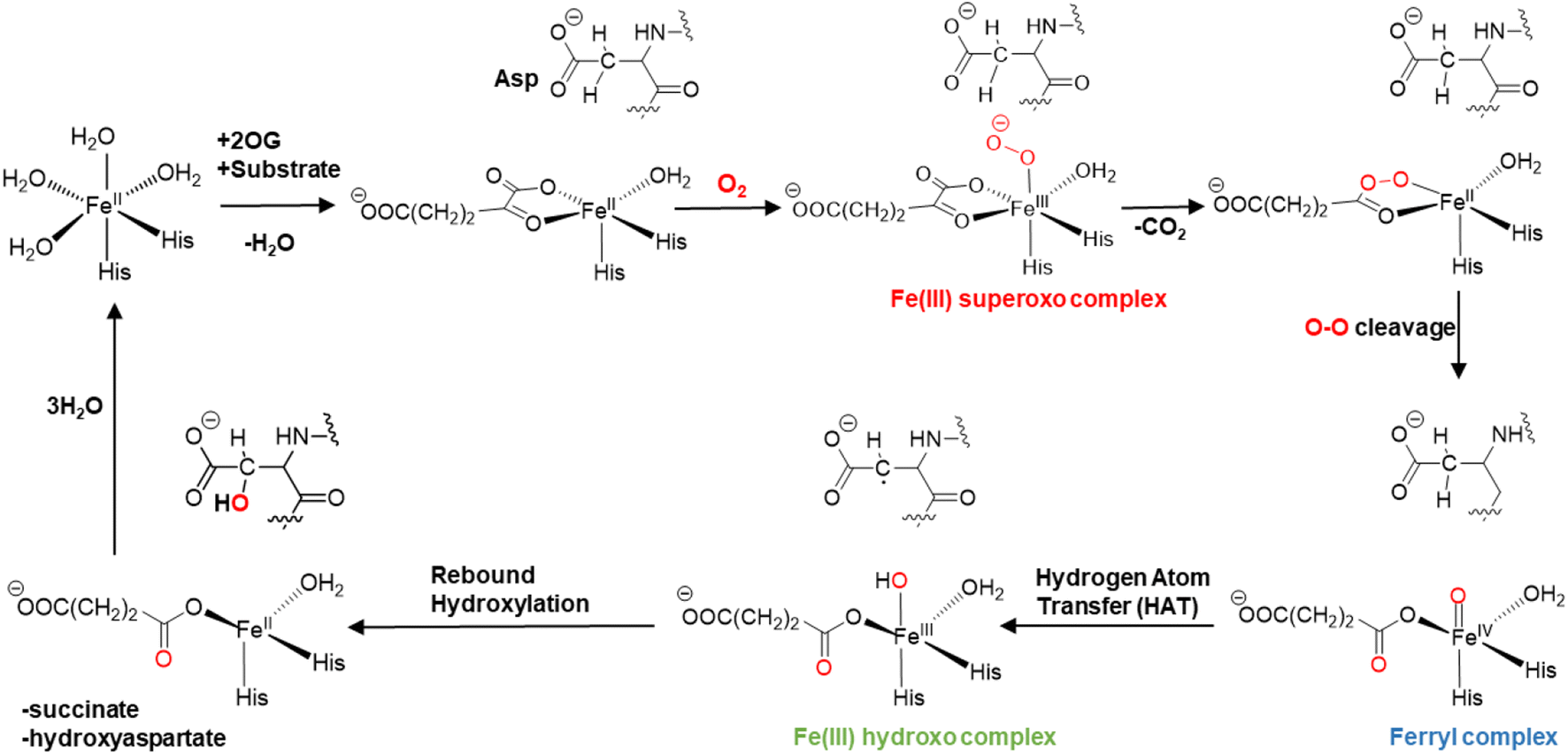 | ||
| Scheme 1 Proposed catalytic cycle for AspH catalyzed hydroxylation. | ||
Although the mechanisms of 2OG oxygenases have been extensively studied, how AspH performs catalysis without the presence of the highly conserved facial triad has remained unknown. However, this is of great interest from the perspective of biocatalysis and for the development of selective inhibitors of AspH, as a cancer target.71,73 Furthermore, the atomistic origins of the stereoselectivity of AspH for both aspartyl and asparaginyl- C–H hydroxylation are not understood. The role of the SCS and LR interacting residues in AspH catalysis, the complex dynamics of the EGFD substrate, and the interdomain motions of the AspH OXY and TPR domains are also unexplored, which could potentially guide in enzyme redesign.
To investigate the nature of catalysis by AspH, we performed molecular dynamics (MD) and quantum mechanics/molecular mechanics (QM/MM) studies. The results reveal how the protein dynamics, SCS, and LR interactions modulate the catalytic process in AspH and enable stereoselectivity without the canonical facial triad.
Computational methods
System preparation
A crystal structure of the human AspH ES complex with EGFD of hFX protein (PDB code: 5JZ8)62 was used as the initial structure for the calculations. Missing residues of the substrate (Asp86-Gln98, Glu117-Phe124) were added using Modeller.140,141 Gaussview 6.0 was used to modify N-oxalglycine (NOG) (used for crystallization) to 2OG and to replace the active site Mn with Fe; Mn was used as an inert Fe substitute in the crystal studies. Propka142 was used to determine the protonation states of the ionizable side chains, except for the Fe coordinating histidine residues and the Cys101hFX, Cys110hFX, Cys641, and Cys648 residues involved in disulfide bonds, were assigned after examining the local environment.The active site parameters were prepared with Metal Center Parameter Builder (MCPB.py)143 in Amber18.144 Hydrogen atoms were added to the protein using the leap module in Amber, and Cl− counter ions were used to neutralize the system. Force constants for bonds and angles were obtained using the Seminario method,145 while the point charge parameters for electrostatic potential were obtained using the RESP charge fitting (ChgModB) method.146 Parameters for 2OG were generated using the Antechamber147 module of Amber18. The system was then solvated using the Transferable Intermolecular Potential 3-Point (TIP3P)148 water molecules in a box with a minimum of 10 Å between the protein surface and the box boundary. The geometry of nonheme iron systems was successfully reproduced in previous studies using the above-mentioned methods.100,121,149 The parameters for the Fe(IV)O complex were generated following a similar procedure, where monodentate succinate replaced the bidentate 2OG. The parameters for the D721A, R688A, K666A mutants were generated using the wild-type (WT) parameters by manual substitution of mutant residue.
Molecular dynamics simulations
After solvating the system, a two-step minimization using MM was conducted. In the first minimization step, only solvent water molecules and Cl− counter ions were optimized; the solute (protein) was restrained using a 500 kcal mol−1 Å−2 harmonic potential. In the second step of the minimization, the restrain was removed from the solute, and all the atoms (solute + solvent) were optimized. Both minimizations were conducted for 5000 steps of steepest descent energy minimization followed by 5000 steps of conjugate gradient energy minimization.After the two minimizations, the system was heated from 0 to 300 K for 50 picoseconds (ps) in a canonical (NVT) ensemble using a Langevin thermostat.150 During heating, the solute molecules were restrained with a harmonic potential of 50 kcal mol−1 Å−2. After heating, the systems were kept at a constant 300 K in an isothermal-isobaric (NPT) ensemble for 1 ns with a restrain of 5 kcal mol−1 Å−2 placed on the solute molecules while the pressure of 1 bar was maintained. The productive MD calculations were conducted using the GPU version of Amber18 for a total of 1 µs at 1 bar with a pressure coupling of 2 ps. All the simulations were performed using the FF14SB151 force field using periodic boundary conditions. A Berendsen barostat152 was used to maintain the pressure, and the SHAKE algorithm153 was used to constrain bonds involving hydrogen atoms. Long-range electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method154 with a direct space and vdW cut-off of 10 Å.
Root mean square deviation (RMSD), Root mean square fluctuations (RMSF), electrostatic interactions, and hydrogen bond analysis were done using the CPPTRAJ module from Ambertools utilities.155 VMD156 and Chimera157 were used to analyze MD trajectories. Dynamic cross-correlation analysis (DCCA) and principal component analysis (PCA) were performed using Bio3D.158
QM/MM calculations
All water molecules greater than 12 Å from the protein were stripped from the structures. The QM region for the Fe(III)-OO˙− intermediate includes Fe(III), O2, 2OG, the imidazole groups of histidines His679 and His725, the coordinated water, and the substrate Asp103hFX (Fig. 2). For the Fe(IV)O complex, O2 was replaced with an oxo group and 2OG with succinate. The MM region was defined as all other protein atoms within 8 Å of the QM region and was described using Amber FF14SB forcefield. The positions of atoms beyond 8 Å from the QM region were fixed. We considered long-range electrostatic interactions for the QM/MM calculations using the electrostatic embedding scheme,159 accounting for the polarizing effect of the MM region on the QM region via incorporation of the MM point charges in the Hamiltonian. The van der Waals interactions between MM and QM atoms are calculated using the Lennard-Jones potential. The QM/MM calculations were conducted using the Chemshell160 package combining Turbomole161 (QM region) and DL_POLY162 (MM region). Hydrogen link atoms were used to cap the bonds spanning the boundary between the QM and MM regions using a charge shift model.163–165 QM/MM geometry optimizations were performed using the Density Functional Theory (DFT) with the unrestricted B3LYP functional (UB3LYP) and def2-SVP basis set (termed B1) for all atoms defined in the QM part. A relaxed potential energy scan along the reaction coordinate in steps of 0.1 Å was performed from the optimized reactant complex (RC) to obtain transition states (TS) and intermediates using DL-find optimizer.166 The highest energy points (TSs) were optimized without any constraints using the dimer method167 in Chemshell. Single-point energy calculations were also conducted with the larger basis set, def2-TZVP (B2) at the DFT UB3LYP level, to improve the calculated energy of each geometry. Frequency calculations were conducted for each optimized geometry to confirm all minima and TSs. Zero-point energy (ZPE) corrections obtained from frequency calculations were then added to the results of the single-point energy calculations to obtain energies reported as B3 (B2+ZPE). The empirical dispersion corrections were included in single-point energy calculations using Grimme's dispersion correction (D3)168,169 with Becke-Johnson damping (D3-BJ).170
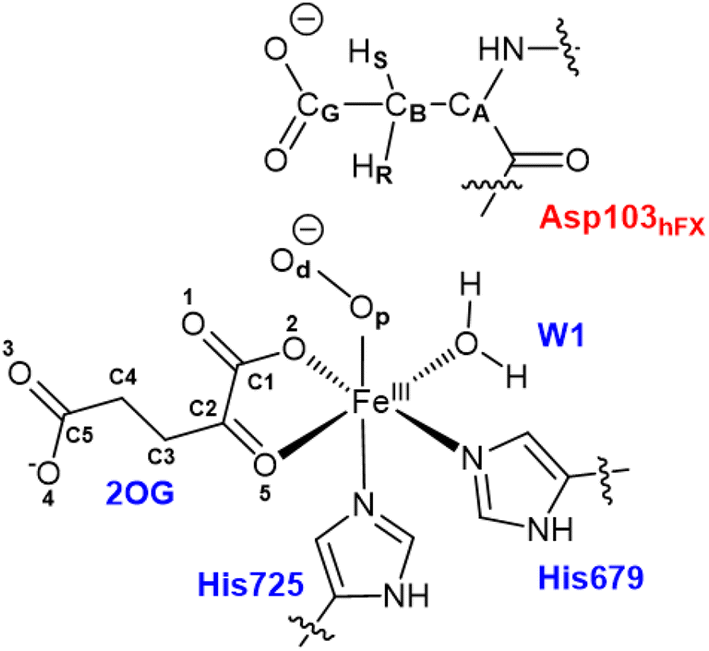 | ||
| Fig. 2 QM region used for the QM/MM calculations of the AspH reaction mechanism includes Fe, 2OG (superoxo), O2 (superoxo), succinate (ferryl), oxo (ferryl), side chain imidazole rings of Fe-ligating histidines (His679 and His725), W1 (mode A), Asp103hFX. | ||
Spin Natural Orbital (SNO) analysis171 was performed to analyze the molecular orbitals involved in the reaction path. TITAN code was used for electric field calculations.172 Energy decomposition analysis (EDA)173–175 was performed on optimized RCs and TSs to obtain total energy contributions/interaction energy (ΔE) (van der Waals + electrostatic) of individual residues. The differences in the interaction energies between TS and RC structures (ΔΔE) were calculated to investigate the contributions of individual residues on TS stabilization.
To evaluate the role of N-CA-CB-CG dihedral angle on stereoselectivity, an aspartate molecule with peptide linkages was optimized at the gas phase using DFT UB3LYP/def2SVP level, and the QM energy scan was performed at the same level of theory using N-CA-CB-CG dihedral as scan coordinate, using Gaussian 16 software.176
QM/MM MD simulations
QM/MM MD simulation was performed with CP2K version 6.1, combining QUICKSTEPS (QM part) and FIST (MM part).177,178 An equilibrated structure from the Fe(IV)O classical MD simulation was used as the initial structure for QM/MM MD. The QM region used was the same as that used for the QM/MM calculations, and the remaining part of the system was treated as MM region. A real-space multigrid method was used to account for the electrostatic coupling between the QM and MM regions.179,180 The DFT-D3 level of theory with B3LYP functional and the Gaussian and plane-waves method (GPW) with dual basis sets was used for the QM part.178 The Gaussian double zeta valence polarized (DZVP) basis set181 was used to describe the wave function, and the auxiliary plane-wave basis set expanded with a cut-off of 360 rydberg (Ry) and Goedecker–Teter–Hutter (GTH) potential was utilized to describe the electron density.182 The auxiliary density matrix method (ADMM) was used to accelerate the Hartree–Fock exchange calculations.183 The hydrogen link atom method was employed to complete the valences of the bonds at the QM-MM boundary.163–165 The QM/MM MD simulation was performed in an NVT ensemble with a time step of 0.5 femtoseconds (fs) and for a total time of 5 ps.
Results and discussion
Structure and dynamics of the AspH Fe(III)-OO˙− ES complex for dioxygen activation
Conformational dynamics underlies the enzymes structure–function relationships by facilitating the substrate binding, catalysis and product release.184–194 To investigate the effect of the substrate binding mode on AspH catalysis, we performed MD simulations of the reactive AspH-Fe(III)-OO˙− complex in the two crystallographically observed substrate binding modes (modes A and B, Fig. 1B and C). RMSD, RMSF, and distance plots for both binding modes are provided in Fig. S1–S4 (ESI (ESI)).† In both substrate binding modes A and B for the AspH- Fe(III)-OO˙− complex, the iron is coordinated by the co-substrate 2OG, two histidine residues (His725 and His679), O2, and a water molecule (mode A) or the substrate (mode B). 2OG coordinates the iron in a bidentate fashion and is additionally stabilized by a network of interactions.62 The network includes (i) a stable salt bridge between the 2OG C5 carboxylate and the Arg735 guanidium group and (ii) hydrogen bonding interactions of 2OG C1 carboxylate oxygens (O1 and O2) with the His690 imidazole –NH (O1) and an Arg688 guanidium group –NH (O2) as observed in the crystal structure, conserved in both binding modes A and B (Fig. S5†).The combined active site and substrate stabilizing interactions described here are consistent with those observed in the crystal structure; however, some interactions facilitating productive substrate orientation, as reported in the crystal structure, were weak throughout the AspH Fe(III)-OO˙− MD. These include the Lys102hFX–Asp616 salt bridge and the network of hydrogen-bonding interactions between residues-Gln627-Asp103hFX, Glu617-Asp103hFX, Glu615-Leu619, Arg686-Lys102hFX (See ESI for additional details†).
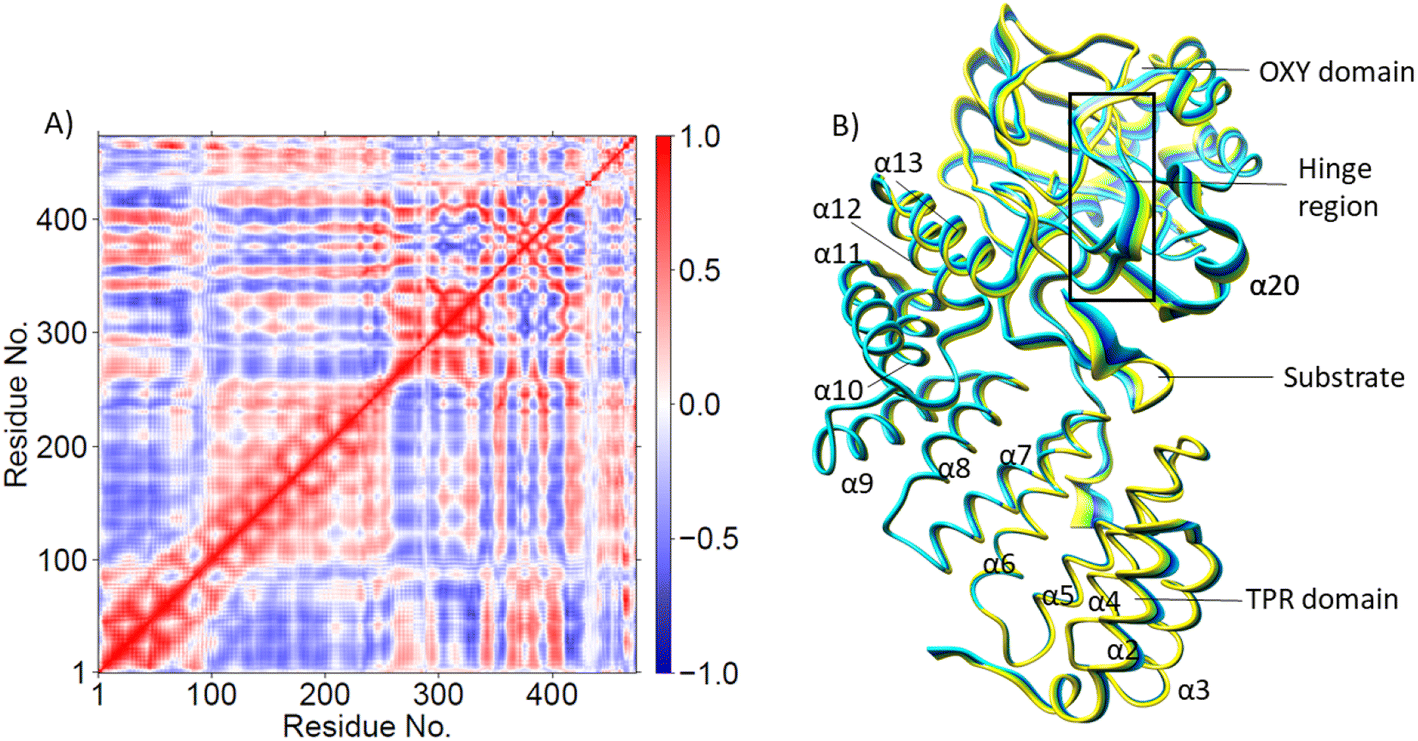 | ||
| Fig. 3 (A) Dynamic Cross Correlation Analysis (DCCA) and (B) Principal Component Analysis (PCA) of Cα atoms in the AspH Fe(III)-OO˙− complex (binding mode A). Residues 1-429 are AspH protein residues; 430-Fe, 431-O2, 432-2OG, 433-W1. Residues 434–472 are EGFD substrate residues; 451-Asp103hFX. The motion of the residues in PCA (part B) is indicated by the color change from yellow to blue. | ||
PCA (Fig. 3B) shows that the α2–α5 TPR helices move towards the substrate, possibly assisting in substrate positioning, while α7–α13, the hinge region, and α20 of the OXY domain move away from the substrate. The OXY domain shows movement towards the active site, potentially stabilizing the productive ES complex. The secondary structure of AspH, including the α helices and β sheets is provided in Fig. S14.†
Understanding the conformational dynamics of the ES complex provides vitally important information about key interactions in the SCS that might be catalytically important, as well as enlightens potentially intriguing LR interactions with RA residues that might be utilized for modulating, optimizing and repurposing the enzyme catalytic functions. The results align with earlier studies that demonstrated the role of dynamics for substrate binding, catalysis and as a valuable tool for enzyme engineering together with other computational and experimental approaches.99,136,137,197–203
O intermediate is formed via peroxosuccinic O–O cleavage which requires 2.0 kcal mol−1 activation energy, and the Fe(IV)O is stabilized by −46.6 kcal mol−1 (Fig. S15†). Thus, the decarboxylation step is the rate-limiting during the dioxygen activation process, in accord with the previous studies on 2OG oxygenases.80,121,122,208–211 Fig. S16† displays the geometric parameters and spin densities of stationary points involved in dioxygen activation via binding mode A (See ESI for additional details†).
QM/MM calculations for the reaction of dioxygen activation starting from binding mode B give a very high barrier of 75.2 kcal mol−1 for the superoxide nucleophilic attack on C2 and the formed peroxosuccinate intermediate is exothermic to RC1′ (Fig. S17†). The reason for such high energy is that the Asp103hFX coordination to the Fe(III) center keeps the O2˙− trans to the C2 of 2OG, thus requiring a significant structural rearrangement for the nucleophilic attack by superoxide onto C2 of 2OG, rendering oxidative decarboxylation highly unfavourable for binding mode B. The geometric parameters for dioxygen activation in binding mode B are given in Fig. S18.†
In summary, reaction path calculations starting from binding modes A and B confirm that the substrate Asp103hFX carboxylate should not be coordinated to the Fe(III) center for productive dioxygen activation reaction in agreement with proposed mechanisms for most 2OG oxygenases.100,106,108,121,123,127,138,208,212,213
Structure and dynamics of the ES complex for the stereospecific Asp hydroxylation
Dioxygen activation via substrate binding mode A leads to the formation of the AspH Fe(IV)O-EGFD complex (RC2 (Scheme 2)/IM3 (Scheme S1†)), which is the intermediate necessary for Asp103hFX hydroxylation. Fig. S19–S22† display the RMSD, RMSF, key distances, and angle plots for Fe(IV)O intermediate. MD simulations revealed that most active site interactions remained similar in the Fe(III)-OO˙− and Fe(IV)O intermediates (Fig. S7–S12†). Key differences include the stable hydrogen bonding of Arg688 guanidium NH with W1 and the Ser668 side chain hydroxyl with Fe(IV)-linked succinate C4 carboxylate in the Fe(IV)O complex, which were less prominent in the Fe(III)-OO˙− complex (Fig. S23†). Further, the Arg735:2OG C5 carboxylate salt bridge observed in Fe(III)-OO˙− complex is disrupted upon succinate formation in Fe(IV)O complex, possibly due to a shorter carbon length in succinate. Instead, a stable solvent-mediated bridging hydrogen bond is observed between Arg735 side chain guanidium NH and the C4 carboxylate of succinate in the Fe(IV)O complex. A stable solvent-mediated (W2) bridging hydrogen bond between the ferryl Op and Asp721 side chain carboxylate helps orientate the Fe(IV)O oxygen towards the pro-R hydrogen of Asp103hFX CB (Fig. S24†). Overall, most active site interactions, particularly those involved in substrate positioning in the Fe(III)-OO˙− simulation, also appear stable in the Fe(IV)O simulation, indicating tight substrate binding.
 | ||
| Scheme 2 Proposed mechanism of AspH catalyzed substrate hydroxylation. | ||
DCCA analysis revealed that most correlated motions in the AspH Fe(IV)O and Fe(III)-OO˙− complexes are similar (Fig. 4A). In the AspH Fe(IV)O complex, Asp721 showed a positive correlation with that of α19 of the OXY domain and the loop residues connecting α19 with βI of DSBH, and anti-correlation with the motions of helices α4 and α6 of the TPR domain. The 2OG/succinate stabilizing residues Arg735 and Ser668 display a strong positive correlation with TPR helices α1-α4. Substrate stabilizing residue Arg688 and 2OG stabilizing residue Arg735 exhibited strong anti-correlation with the motions of TPR helices α6 and α7 in the Fe(IV)O complex, not observed in the Fe(III)–O–O˙− complex. Thus, the LR interactions of helices α6 and α7 with the 2OG, and substrate stabilizing residues likely play a crucial role in AspH catalysis. Interestingly, kidney dysfunction in humans is caused by a mutation of AspH Gly434 residue, which forms the loop connecting α6 and α7. These observations highlight the roles of the TPR domain not only in substrate binding, but also in dynamic motions during catalysis, potentially in a similar manner as with other 2OG oxygenases such as collagen prolyl hydroxylases.92
 | ||
| Fig. 4 (A) Dynamic Cross Correlation Analysis (DCCA) and (B) Principal Component Analysis (PCA) of Cα atoms in the AspH Fe(IV)O complex. Residues 1-429 are AspH protein residues; 430-Fe, 431-Op, 432-succinate, 433-W1. Residues 434-472 are EGFD substrate residues; 451-Asp103hFX. The motion of the residues in PCA (part B) is indicated by the color change from yellow to blue. | ||
PCA analysis also reveals that the AspH Fe(IV)O complex is less flexible than the Fe(III)-OO˙− complex (Fig. 4B), arising from the stronger substrate and active site stabilizing interactions observed in the Fe(IV)O simulation. The TPR domain displayed movement towards the substrate and the OXY domain, potentially contributing to the compactness of the Fe(IV)O complex. The limited flexibility and motions of helices α2-α13 and the hinge region may contribute to a more rigid OXY domain and tight binding of the substrate, as evidenced by their correlated movements with substrate stabilizing residues. Overall, the PCA depicts a more rigid and contract Fe(IV)O complex than the Fe(III)-OO˙− in a manner likely favoring catalysis.
To examine the dynamic behavior of the Fe-coordinated water molecule during the catalytic reaction, we performed a QM/MM MD simulation of the Fe(IV)O intermediate. This type of simulation combines exploration of dynamics/flexibility and changes in electronic structure (such as bond breaking and creation). The simulation shows that the distance between the Fe and the coordinated water molecule remains very stable (an average Fe–W1 distance of 2.12 Å, Fig. S25A†). This indicates that W1 remains firmly coordinated to Fe and does not diffuse away. To further elaborate on the strength of the coordination of W1, we performed a QM/MM potential energy surface (PES) study of the dissociation of W1 from the iron center (Fig. S25B†). The PES scan indicates that the dissociation of W1 is very unfavored leading to a continuous increase of the energy, further indicating that there is no spontaneous water dissociation and formation of a four-coordinate (4C) ferryl complex and that such a process would require high activation energy. We also evaluated if a second solvent water molecule could displace W1 from Fe (Fig. S25C†); however, the high barrier observed for this process suggests such a ligand exchange may not be feasible in the Fe(IV)O intermediate state (Fig. S25C†). Therefore, the coordination of W1 to Fe is essential to form the catalytically productive Fe(IV)O intermediate and to enable the C–H activation.
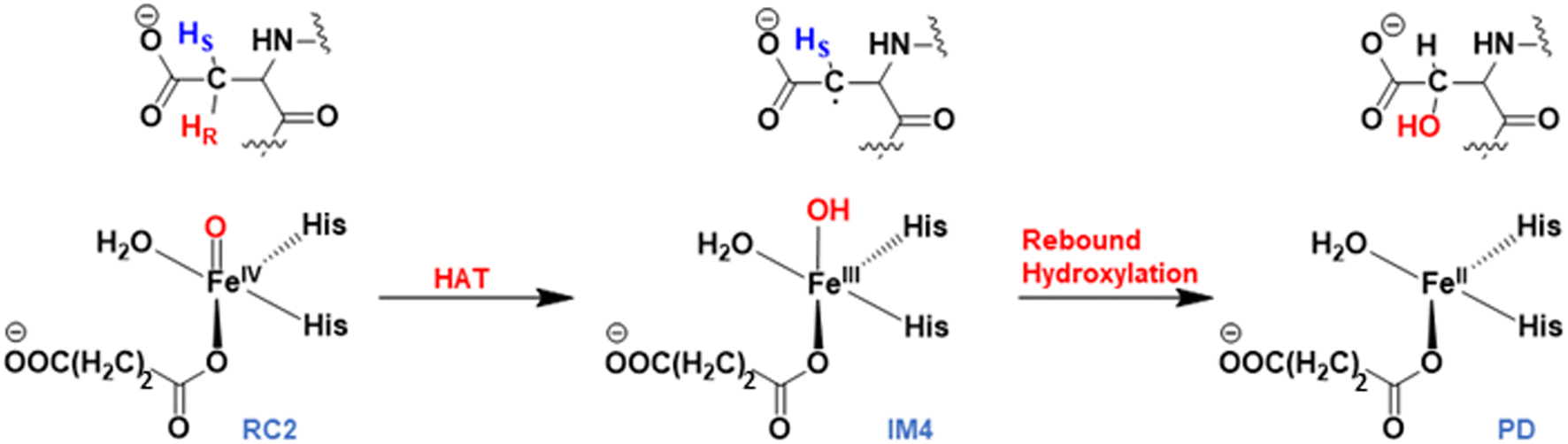
Mechanism of the stereospecific Asp hydroxylation
AspH substrate hydroxylation from the Fe(IV)O complex proceeds in two steps: (1) hydrogen atom transfer (HAT) from the β-carbon (CB) of Asp103hFX by the Fe(IV)O group, and subsequent (2) rebound hydroxylation producing 3-hydroxy-L-aspartate (Scheme 2). The energy profile for HAT and rebound hydroxylation steps is given in Fig. 5. We optimized the Fe(IV)O intermediate in triplet and septet states using the same level of theory used for the quintet calculations (Fig. 5). The results show that the quintet state of the Fe(IV)O intermediate is energetically more favorable compared to triplet and septet states, agreeing with prior studies on Fe(II)/2OG oxygenases.80,122,129,207,214,215 During HAT, a single electron transfers from the substrate pro-R C–H bond to the Fe(IV)O moiety, resulting in CB–HR bond cleavage and alkyl radical (IM4) formation. At the same time, Fe(IV) is reduced to Fe(III)–OH. The activation barrier for pro-R HAT is 20.8 kcal mol−1 (at B3 level). The electron transfer can occur either through a π or σ channel.131,204,215 The σ channel involves a transfer of an α electron (electron with a magnetic spin quantum number (ms) of +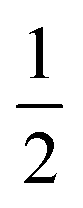 from the substrate σC–H orbital to the Fe σ*z2 orbital, leaving a β electron (electron with a ms = −
from the substrate σC–H orbital to the Fe σ*z2 orbital, leaving a β electron (electron with a ms = −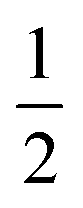 ) on the substrate radical. The head-on overlap of the two σ orbitals results in a more linear Fe–O–H arrangement with an approximate bond angle larger than 120° and closer to 180°.204 Conversely, in the π channel, a β electron from the substrate transfers into the antibonding metal π*xz/yz orbital through angular overlap resulting in a Fe–O–H bond angle approaching 120°.204 SNO analysis (Fig. 6) of the HAT TSs shows that C–H α electron from the substrate transfers to the Fe(IV)O σ*Z2 orbital, and a β electron remain at the carbon radical, suggesting a σ channel mechanism (Fig. 7) in AspH. In TSproR, the Fe–O bond elongates to 1.77 Å (from 1.64 Å in RC2), and the CB–HR bond elongates to 1.34 Å (from 1.11 Å in RC2), while the Op–HR distance shortens to 1.32 Å from 3.18 Å (RC2) (Fig. 8). In IM4, Fe is antiferromagnetically coupled to the substrate radical, as indicated by the spin densities of 4.22 (Fe), 0.27 (Op), and −0.35 (CB) (Fig. 8).
) on the substrate radical. The head-on overlap of the two σ orbitals results in a more linear Fe–O–H arrangement with an approximate bond angle larger than 120° and closer to 180°.204 Conversely, in the π channel, a β electron from the substrate transfers into the antibonding metal π*xz/yz orbital through angular overlap resulting in a Fe–O–H bond angle approaching 120°.204 SNO analysis (Fig. 6) of the HAT TSs shows that C–H α electron from the substrate transfers to the Fe(IV)O σ*Z2 orbital, and a β electron remain at the carbon radical, suggesting a σ channel mechanism (Fig. 7) in AspH. In TSproR, the Fe–O bond elongates to 1.77 Å (from 1.64 Å in RC2), and the CB–HR bond elongates to 1.34 Å (from 1.11 Å in RC2), while the Op–HR distance shortens to 1.32 Å from 3.18 Å (RC2) (Fig. 8). In IM4, Fe is antiferromagnetically coupled to the substrate radical, as indicated by the spin densities of 4.22 (Fe), 0.27 (Op), and −0.35 (CB) (Fig. 8).
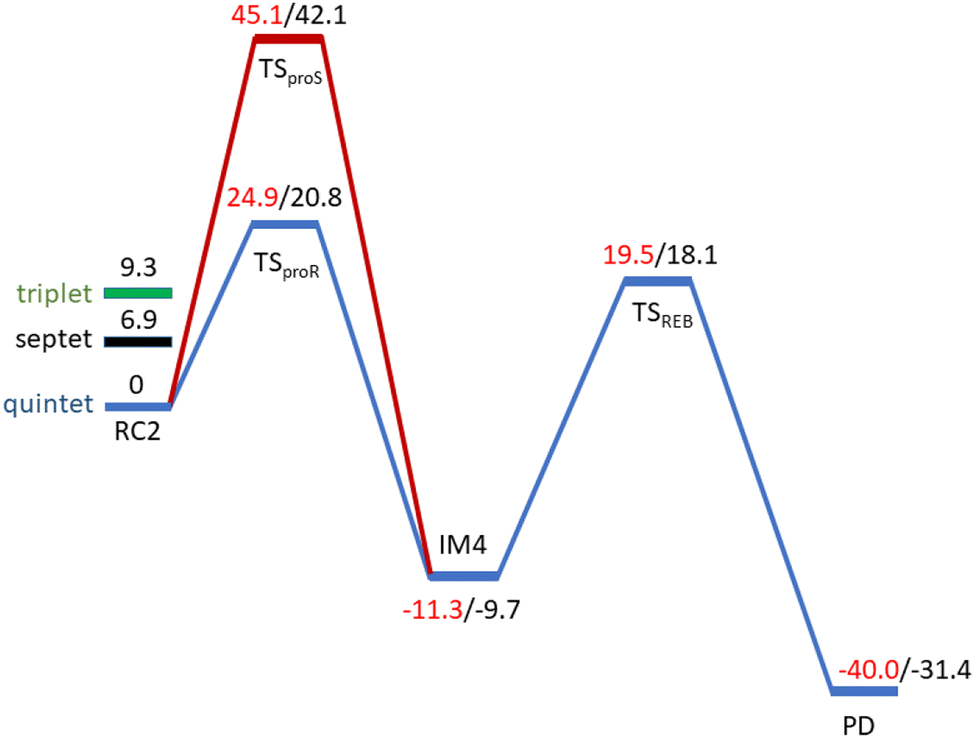 | ||
| Fig. 5 Energy profile for the pro-R (blue) and pro-S (red) HAT and rebound hydroxylation steps calculated at UB3LYP/def2-TZVP (B2) (red) and B2 +ZPE (B3) (black). | ||
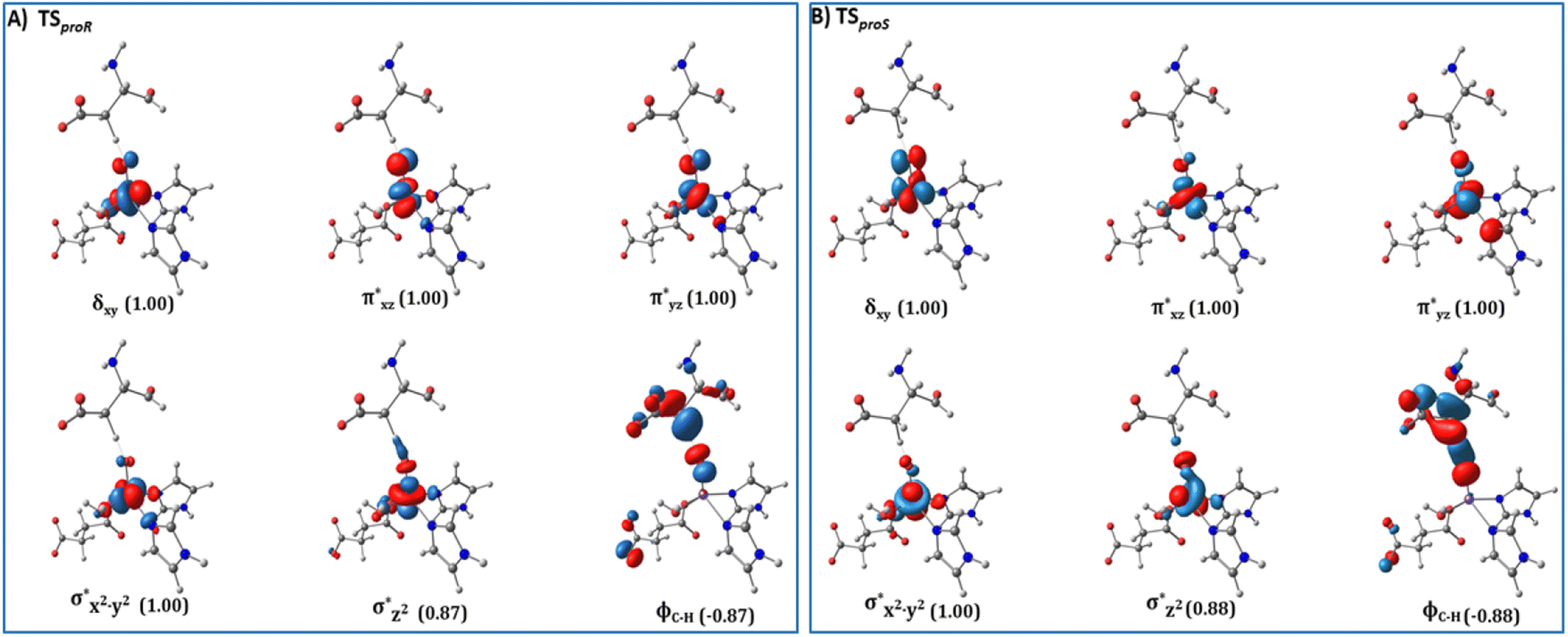 | ||
| Fig. 6 Spin Natural Orbitals (SNO) with corresponding electron occupancies (in brackets) for the HAT TSs: (A) TSproR and (B) TSproS. | ||
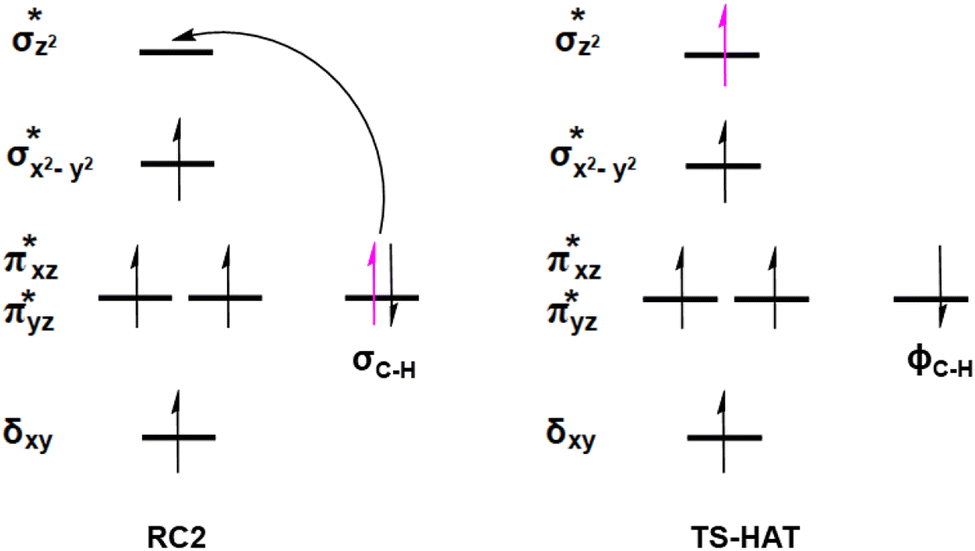 | ||
| Fig. 7 Electron transfer and orbital occupancy during the HAT step. | ||
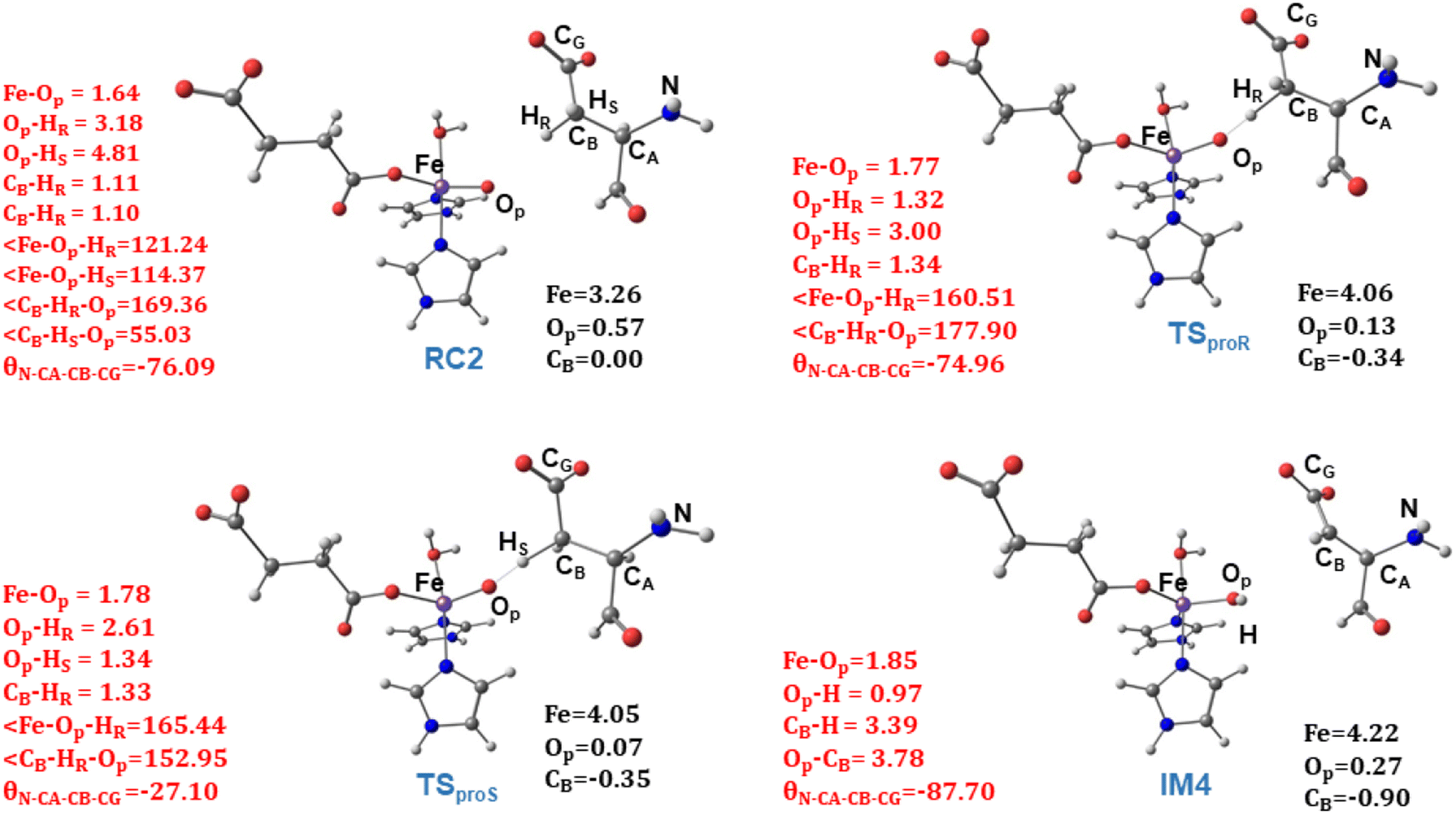 | ||
| Fig. 8 Geometric parameters (red) and spin densities (black) of optimized stationary points in HAT step. | ||
How do SCS residues sterically stabilize the HAT TS
Electrostatic interactions of SCS residues, Lys666, Arg686, and Arg688, help stabilize the side chain carboxylate of Asp103hFX in the HAT TS (Fig. 9). Additionally, hydrogen bonding between the Asp103hFX side chain carboxylate and the Gln664 side chain NH; the solvent-mediated bridging hydrogen bonding interactions of residues Asp721 and W1; the bridging hydrogen bonding interaction of water molecule (W2) with Asp721, the oxo moiety, and the Gly104hFX backbone NH group of the substrate, are likely involved in productive substrate positioning and TS stability (Fig. 9). Solvent-mediated hydrogen bond interactions with the Fe(IV)O moiety have been observed in other 2OG oxygenases, such as AlkB216 and TET2.138 This interaction may contribute to the observed stereoselectivity in AspH by aligning the FeO moiety towards the pro-R hydrogen of Asp103hFX.
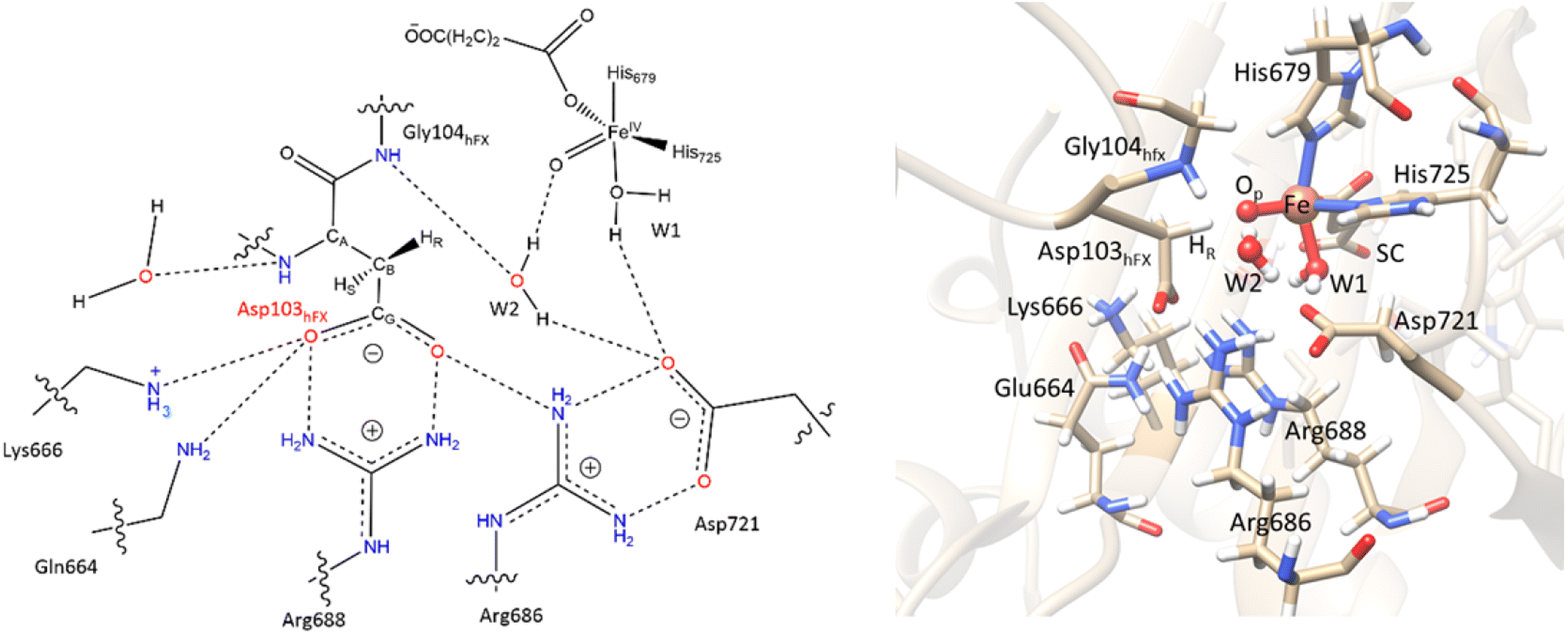 | ||
| Fig. 9 SCS interactions restricting the rotation of Asp103hFX N-CA-CB-CG dihedral and positioning pro-R H atom towards the Fe(IV)O moiety. | ||
How the flexibility of the RC influences the HAT reaction?
To explore how the conformational flexibility of the RC influences the HAT step, we performed QM/MM calculations using five different MD snapshots of the Fe(IV)O intermediate. Tables S1 and S2† show geometric parameters and spin densities variation for the different HAT TSs. The Fe–Op, CB–HR and O–HR bond distances range from 1.76–1.79 Å, 1.34–1.47 Å, and 1.24–1.32 Å respectively, while the Fe–O–HR angle varies from 156.59°–171.91° in different HAT TSs. The calculated Fe–Op–HR angle values, the Asp103hFX CB spin densities in the different pro-R HAT TS structures (−0.37 to −0.29), and SNO analysis indicate that the σ-channel mechanism is exclusively used by AspH. The bond angles and spin densities for HAT TSs corroborate with that of studied 2OG oxygenases such as AlkB/AlkBH2,113,121 TET2,138,200,217 TauD,139,218,219 KDM4A,123 KDM6B,220 KDM7B (PHF8)122 that follow σ-channel mechanism. The calculated electronic energy barriers for the HAT step varied from 20.8 to 25.8 kcal mol−1 (Table 1 and Fig. S26†) with a Boltzmann weighted average of 21.5 kcal mol−1 (B3 level). The computed barriers with dispersion corrections (D3-BJ) for representative snapshots (snapshot1 and snapshot 4) show barriers 19.4 kcal mol−1 and 23.4 kcal mol−1, respectively at the B3 level (Table S3†). The HAT barriers observed in experimental and computational studies on other Fe(II)/2OG oxygenases like prolyl-4-hydroxylase (20.7 kcal mol−1),114,221 TauD (23.2 kcal mol−1),222 histone demethylases (20.2–25.2 kcal mol−1),122,123,220,223 and DNA-demethylases (15.5–26.7 kcal mol−1),121,138,214,224,225 align with the barriers we have calculated for AspH.
| AspH-WT | Activation barrier | Reaction energy | |||||
|---|---|---|---|---|---|---|---|
| B1 (kcal mol−1) | B2 (kcal mol−1) | B3 (kcal mol−1) | B1 (kcal mol−1) | B2 (kcal mol−1) | B3 (kcal mol−1) | ||
| Snapshot 1 | HATproR | 22.0 | 24.9 | 20.8 | −9.3 | −11.3 | −9.7 |
| HATproS | 42.3 | 45.1 | 42.1 | −9.3 | −11.4 | −9.9 | |
| Snapshot 2 | HATproR | 22.2 | 25.7 | 21.1 | −10.0 | −11.6 | −9.8 |
| HATproS | 41.1 | 46.1 | 42.4 | −9.9 | −11.5 | −9.9 | |
| Snapshot 3 | HATproR | 24.4 | 26.9 | 25.8 | −5.8 | −7.6 | −2.9 |
| HATproS | 42.8 | 46.3 | 44.6 | −5.5 | −7.9 | −7.1 | |
| Snapshot 4 | HATproR | 26.7 | 30.2 | 25.4 | −2.7 | −4.5 | −3.9 |
| HATproS | 45.2 | 49.0 | 44.9 | −2.7 | −4.6 | −3.8 | |
| Snapshot 5 | HATproR | 26.2 | 28.8 | 25.3 | −4.0 | −7.2 | −2.2 |
| HATproS | 41.3 | 44.8 | 42.2 | −4.0 | −7.2 | −2.2 | |
Notably, the HAT barriers and reaction energies show a positive correlation with the strength of the hydrogen bonding between W2 and the Op and a negative correlation between that of W1 and Asp721 (Table S4 and Fig. S27–S30†). In the snapshots where W2–Op hydrogen bond is stronger, a higher stability for IM4 and a lower HAT barrier are observed. Therefore, the presence of the carboxylate residue-Asp721 in the SCS is vital in HAT TS stabilization and modulating the HAT rate. All the five snapshots revealed HAT to be exothermic with reaction energies in the range −9.8 to −2.2 kcal mol−1 at B3 level. The Fe(III)–OH hydroxyl moiety in IM4 is stabilized by hydrogen bonding with W2. The EDA (Fig. S31†) manifests that IM4 is stabilized by the SCS residues Arg688, Lys666, and Asp721 (Fig. S32†), by energy contributions of −1.5, −1.3, and −4.7 kcal mol−1, respectively.
Protein control on the stereoselectivity in the HAT
To investigate the origin of stereoselectivity, we performed QM/MM reaction path calculations for the pro-S HAT using the same starting snapshots used for the Fe(IV)O intermediate RC2 as for the pro-R HAT reaction (Fig. 5). Unlike the pro-R HAT, we found that the pro-S HAT is unfeasible due to a relatively high activation barrier of 42.1 kcal mol−1. We observed that the activation barrier for pro-S HAT is consistently high across all five snapshots, with energy differences between pro-R and pro-S HATs ranging from 15.1–20.3 kcal mol−1 at the B1 level and 16.9–21.3 kcal mol−1 at the B3 level (Table 1). A superimposed structure of TSproR and TSproS is given in (Fig. S33†). The dihedral angle (N-CA-CB-CG) is a key structural determinant between the TSproR and TSproS geometries, with the former having a dihedral angle of −74.96° and the latter having a dihedral angle of −27.10°, compared to −76.09 in RC2 for snapshot 1. N-CA-CB-CG dihedral angle varies from −83.33 to −71.40° in TSproR, −29.71° to −16.55° in TSproS, and −76.77° to −72.89° in RC2 (Table S2†). We further performed a QM energy scan to investigate the role of this dihedral angle and the stability of the geometry of the substrate Asp-residue (Fig. S34†). The scan revealed that a change of the N-CA-CB-CG dihedral from a pro-R TS value of −74.96° to a pro-S TS value of −27.10° requires a barrier of ∼9 kcal mol−1, calculated at the UB3LYP-def2SVP level, corresponding to a QM/MM energy difference of 15.1–20.3 kcal mol−1 (at B1 level), suggests that the increased stability (between 6.1 and 11.3 kcal mol−1) of the pro-R compared to the pro-S orientation arises from the interactions with the AspH protein environment that restraints the substrate to favor pro-R HAT. The Asp103hFX side chain carboxylate interacts with Arg686, Arg688, and Lys666 via electrostatic interactions and Gln664 side chain amine via hydrogen bond. In addition, the Gly104hFX-Asp721 solvent-mediated hydrogen bond and the hydrogen bonding interaction of the Asp103hFX backbone amide with solvent water promotes pro-R HAT (Fig. 9). The substantial geometric changes required to reach TSproS from RC2 result in a high activation barrier, thereby disfavoring pro-S HAT. Interestingly, the pro-R and pro-S HAT reactions lead to the same intermediate (IM4), with an N-CA-CB-CG dihedral angle of −87.70°, still preferring the pro-R product. The pro-S configuration of IM4 is also destabilized by repulsive interactions between hydrogen atoms on Asp103hFX CB and CA atoms. Thus, the overall stereoselectivity of the AspH hydroxylation reaction is determined by the HAT step.
Substrate positioning is a critical factor in determining stereoselectivity.139 We further performed EDA on both pro-R and pro-S HAT TSs to investigate the role of SCS residues in determining stereoselectivity. We calculated the differential contributions between each TS (pro-R and pro-S) and the RC2 and then calculated the difference between the two differential contributions (ΔE (TSproR − RC2) − ΔE (TSproS − RC2)). A positive Δ (ΔΔE) value indicates destabilizing, while a negative value indicates stabilizing effect on the TSproR (Fig. S35†). We found that the SCS residues Gln664, Arg686 and Arg688 influence stereoselectivity by showing stronger energetic contributions towards stabilizing the pro-R TS compared to the pro-S TS via hydrogen bonding (Gln664) and electrostatic (Arg686 and Arg688) interactions (Fig. S36†). Comparing TSproR and TSproS structures, we observed that the positioning of the Arg688/Arg686/Lys666 sidechain-Asp103hFX carboxylate salt bridges and Gln664 hydrogen bond influences the Asp103hFX carboxylate position and the orientation of the pro-R hydrogen with respect to the Fe(IV)O moiety to enable stereoselective reaction (Fig. 9). The revealed significant differential catalytic contributions by SCS residues towards stabilizing the pro-R and pro-S TS offer opportunities for engineering novel enzymes or catalysts for stereoselective C–H activation reactions. Furthermore, DCCA analysis of the ferryl complex suggests that the motions of TPR domain helices α2-α7 and α12-α14, as well as OXY domain helices α16, α18, and α19 all involved in substrate binding, show correlations with SCS residues involved in TSproR and TSproS stabilization, indicating that these residues could also influence the stereochemical outcome of the reaction via LR interactions (Fig. S37–S40†), thus providing an intriguing new background for long-range allosteric modulation of AspH activity and stereoselectivity.
Reaction selectivity in enzymatic catalysis can be influenced by electrostatic preorganization, that is the orientation of enzyme's internal electric fields (IEFs).226,227 To evaluate the effect of IEF on stereoselectivity in AspH, the net electric field along the FeO axis was calculated for both pro-R and pro-S HAT (Table S5†). In all five snapshots, the net IEF along the FeO axis was more negative for TSproS than the corresponding TSproR, indicating the role of EF in stereoselectivity and modulating the EF along the FeO direction could be a strategy to alter the stereoselectivity in AspH.
Rebound hydroxylation
At the final stage of the reaction mechanism, substrate hydroxylation proceeds through a radical rebound mechanism, where the hydroxyl group from the Fe(III)–OH intermediate (IM4) transfers to the substrate methylene radical generated after HAT. Subsequently, Fe(III) reduces to Fe(II), and the hydroxylated product forms. Rebound hydroxylation shows a 27.8 kcal mol−1 barrier relative to the stable intermediate IM4 (RC3 in Fig. S41†), corresponding to the lowest energy HAT pathway (snapshot 1) at the B3 level. All five QM/MM snapshots revealed a similar high activation barrier for the rebound step (Table 2 and Fig. S41†) with a Boltzmann weighted average of 26.4 kcal mol−1 at the B3 level of theory, suggesting this step may be rate-limiting in AspH instead of the HAT step which is the most common rate-limiting step in Fe(II)/2OG enzymes.113,114,121,122,138,175,228 Calculations with dispersion corrections (D3-BJ) show a decrease in the B3-level rebound barriers to 25.5 and 23.4 kcal mol−1 for representative snapshots (snapshot 1 and snapshot 4), respectively (Table S6†). Both the HAT and rebound reactions require changes in the torsional angle N-CA-CB-CG of the substrate Asp103hFX (from the respective RCs to the TSs). However, this change is much larger for the rebound step and respectively requires larger energy. In the HAT, during the transition from RC2 to TSproR (snapshot 1), the torsional angle changes from −76.09 to −74.96 degrees (Table S2†). In contrast, for the rebound step, the torsion angle changes from −87.70 (in the IM4) to −79.55 degrees (in the TSREB) (Table S8†). A similar trend is observed across five different snapshots (see Tables S2 and S8†). Such a significant torsional adjustment disturbs the salt bridge and hydrogen bonding interactions that tightly bind the Asp103hFX side chain carboxylate and contribute to the higher energy barrier observed for the rebound step compared to the HAT. Previous studies by Shaik et al. describe a case of rebound hydroxylation as rate limiting in CYP450 catalyzed reaction of dopamine formation.229 Similarly, Lu et al. reported a higher barrier for rebound step than for HAT in the TET2-catalyzed conversion of 5-methylcytosine to 5-hydroxymethylcytosine.224 Furthermore, in nonheme Fe(II)/2OG halogenases, which lack the facial triad carboxylate, a relatively high barrier was observed for the rebound hydroxylation compared to halogen atom transfer which is important in distinguishing reaction selectivity between hydroxylation and halogenation.202,230–232 The finding that the rebound reaction was predicted to be the rate-limiting step in other heme/non-heme enzymes with conventional coordination suggests that other factors than the water coordination might be responsible for the high energy barrier of the rebound hydroxylation, however further studies would be necessary to comprehensively explore this issue.| AspH-WT | Activation barrier | Reaction energy | ||||
|---|---|---|---|---|---|---|
| B1 (kcal mol−1) | B2 (kcal mol−1) | B3 (kcal mol−1) | B1 (kcal mol−1) | B2 (kcal mol−1) | B3 (kcal mol−1) | |
| Snapshot 1 | 26.6 | 30.8 | 27.8 | −41.2 | −40.0 | −31.4 |
| Snapshot 2 | 28.2 | 33.3 | 30.7 | −35.3 | −32.4 | −24.1 |
| Snapshot 3 | 24.8 | 30.2 | 26.9 | −40.5 | −36.1 | −24.7 |
| Snapshot 4 | 22.2 | 28.7 | 25.8 | −35.3 | −30.4 | −22.3 |
| Snapshot 5 | 26.3 | 32.5 | 28.1 | −34.7 | −32.2 | −21.6 |
TSREB represents the TS for rebound hydroxylation (Fig. 10), wherein the Fe–Op distance elongates to 2.20 Å, and the Op–CB distance reduces to 2.23 Å from 1.85 Å and 3.78 Å respectively, in IM4. The N-CA-CB-CG dihedral decreases to −79.55 (TSREB) from −87.70 (IM4), and the hydroxyl Op to CB (Asp103hFX) distance decreases to 2.27 Å from 3.78 Å. Tables S7 and S8† show the spin densities and geometric parameters of all stationary points involved in the rebound step. The hydroxyl moiety in TSREB is stabilized by the solvent-mediated (W2) bridging hydrogen bond with Asp721. As the hydroxyl radical leaves the Fe center, W2 coordinates to the vacant position to generate a stable Fe(II) complex. The hydroxylation product is exothermic by −31.4 kcal mol−1 (Fig. 5), which compares favorably well with previous studies on other non-heme Fe(II)/2OG oxygenases.114,120–123,138,219,230,233
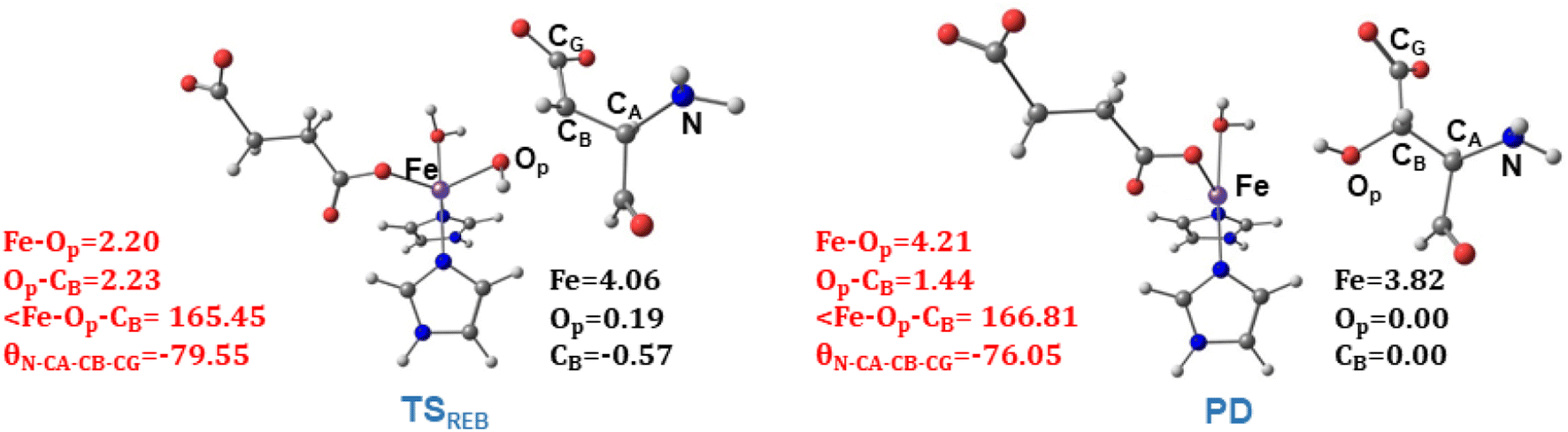 | ||
| Fig. 10 Geometric parameters (red) and spin densities (black) of optimized stationary points in rebound hydroxylation step. | ||
EDA shows TSREB is stabilized by residues Asp721 (−1.4 kcal mol−1), Lys666 (−0.9 kcal mol−1), and Asp741 (−1.9 kcal mol−1), and destabilized by Arg688 (3.5 kcal mol−1) and Arg686 (0.8 kcal mol−1) (Fig. S42–S44†) compared to IM4.
Effect of the D721A mutation on substrate binding and hydroxylation
To explore the importance of Asp721, we performed MD with D721A, which manifests substantially reduced catalytic activity.195 MD for D721A shows an overall increase in the average distance between Fe(IV)O Op and the substrate CB of 7.03 Å compared to 4.23 Å to the AspH-WT. The increased distance between the Fe(IV)O oxygen and the substrate leads to enhanced flexibility of the ES complex (Fig. S45–S47 and Table S9†), disrupting the ES complex stabilizing salt bridges. PCA shows that Fe(IV)O complex for D721A has motions opposite to that of the WT (Fig. S48A†). Specifically, the hinge region and the TPR domain helices show movement away from the active site, making the OXY domain in the D721A mutant less compact. The α1-α5 and α10-α13 helices in the TPR domain move away from the substrate in the D721A mutant, consequently increasing the distance between Op and CB. Therefore, Asp721 acts as an SCS alternative for the missing facial triad carboxylate in AspH, stabilizing the active site and for productive substrate binding. A superimposed view of the AspH-WT and D721A Fe(IV)O complexes is shown in Fig. S49 in the ESI.†
The non-canonical water coordination to the non-heme Fe(II) center (instead of carboxylate as in the canonical Fe(II)/2OG enzymes) elevates the role of the interactions with the SCS. The hydrogen bond between the coordinated water molecule and the SCS residue Asp721 plays a key role in the reactivity of AspH compensating for the absence of coordinated carboxylate. Indeed, in agreement with earlier experimental studies which demonstrate that the mutation of D721A sharply decreases the enzyme activity,195 our QM/MM study reveals that this mutation leads to an increased distance of 6.43 Å between the ferryl oxygen (Op) and the Asp103hFx pro-R hydrogen atom (compared to 3.34 Å in the WT), and consequently to much higher energy barrier of 86.1 kcal mol−1 (Fig. S50†). Such SCS carboxylate is unique for AspH and is not present in other Fe(II)/2OG enzymes such as FIH, prolyl hydroxylase, and TauD where mutation of the Fe-coordinated carboxylate inactivates the enzymes.85,88,234 Such SCS carboxylate is also missing in other Fe(II)/2OG enzymes containing canonical facial triads such as PHF8,196 KDM4A,235 KDM2A,236 ALKBH2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 237 and EFE.133
237 and EFE.133
We further performed in silico mutation of the SCS residues Arg688 and Lys666, identified as crucial for TS stabilization and substrate binding, to alanine (A). The subsequent MD and QM/MM studies reveal that these SCS mutations (R688A and K666A) significantly change the productive orientation of the Fe(IV)O ES complex (Fig. S49 and S51†). The mutant forms show an increased distance between the Op and the CB atom of Asp103hFX compared to that in the WT. The average distances between the Op and the CB atom are 5.70 Å and 6.24 Å for R688A and K666A, respectively in comparison to 4.25 Å in the WT. Similarly, the angle between Fe-Op-CB shows average values of 142.6° (R688A) and 147.4° (K666A) compared to 157.7° in the WT. The subsequent QM/MM studies of the mutant forms show that the increased distance significantly enhances the barrier for HAT (Fig. S52†). Our findings highlight these SCS residues' key role in proper substrate orientation, enabling productive catalysis, and providing insights into potential target residues for enzyme redesign and for altering substrate specificity.
Conclusions
In conclusion, our study informs on the chemistry of the catalytic mechanism of the biologically and synthetically important 2OG-dependent dioxygenase AspH. Instead of using the characteristic HXD/EH facial triad, AspH replaces the carboxylate ligand with a Fe-coordinated water molecule, the position of which is controlled by the sidechain of the SCS carboxylate residue Asp721, which is also critical in catalysis. Our findings demonstrate that of the two crystallographically observed substrate binding modes, only the one not coordinating with the iron is catalytically active, as substrate coordination impedes the formation of the essential Fe(IV)O intermediate. The non-catalytically observed substrate carboxylate binding mode may help promote/stabilize iron binding in the absence of an enzyme supported carboxylate ligand. HAT is exothermic and occurs by σ channel mechanism. Atypically, rebound hydroxylation has a higher barrier than HAT, suggesting the rebound step could be the rate-limiting in AspH. HAT step and SCS residues (Arg688, Gln664, Lys666, Arg686, Asp721) enable stereoselective hydroxylation, highlighting the significance of SCS processes in promoting efficient catalysis and controlling the stereoselectivity in AspH. The TPR domain of AspH plays a significant role in EGFD substrate binding. It manifests dynamic motions during the catalysis which are of interest concerning the role of TPR domains in other 2OG oxygenases. The uncovered catalytic strategy of AspH provides novel insights for advancing the synthetic applications of Fe(II)/2OG enzymes for regio- and stereoselective C–H bond functionalizations in biotechnology. The study affirms the roles of dynamics, SCS, and LR interactions as essential factors affecting catalysis and as potentially exciting tools for optimizing and redesigning enzymes for desired industrial applications. Our findings have implications for enzyme redesign, the development of selective AspH inhibitors, and the advancement of regio- and stereoselective biocatalysis catalysts for C–H activation reactions.
Data availability
The data supporting this study's findings are available from the corresponding author upon reasonable request.Author contributions
Conceptualization, T. G. K.-C.; methodology, T. G. K.-C. and A. K.; investigation and formal analysis, A. K. and S. O. W.; writing-original draft, A. K. and T. G. K.-C.; writing-review and editing, A. K., S. O. W., A. V., F. H. C., C. J. S., T. G. K.-C.; visualization, A. K.; funding acquisition, T. G. K.-C.; supervision, T. G. K.-C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TKC acknowledges NIH grant 2R15GM132873-02 for the support. CJS thanks Cancer Research UK (C8717/A18245).References
- G. Zurlo, J. Guo, M. Takada, W. Wei and Q. Zhang, Biochim. Biophys. Acta - Rev. Cancer, 2016, 1866, 208–220 CrossRef CAS PubMed.
- R. A. Berg and D. J. Prockop, Biochem. Biophys. Res. Commun., 1973, 52, 115–120 CrossRef CAS PubMed.
- K. L. Gorres and R. T. Raines, Crit. Rev. Biochem. Mol. Biol., 2010, 45, 106–124 CrossRef CAS PubMed.
- J. B. Hedges and K. S. Ryan, Chem. Rev., 2020, 120, 3161–3209 CrossRef CAS PubMed.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef CAS PubMed.
- R. Ushimaru and I. Abe, ACS Catal., 2023, 13, 1045–1076 CrossRef CAS.
- L.-F. Wu, S. Meng and G.-L. Tang, Biochim. Biophys. Acta, Proteins Proteomics, 2016, 1864, 453–470 CrossRef CAS PubMed.
- C. R. Zwick III and H. Renata, ACS Catal., 2023, 13, 4853–4865 CrossRef CAS.
- N. Masson and P. J. Ratcliffe, J. Cell Sci., 2003, 116, 3041–3049 CrossRef CAS PubMed.
- D. Lando, D. J. Peet, J. J. Gorman, D. A. Whelan, M. L. Whitelaw and R. K. Bruick, Genes Dev., 2002, 16, 1466–1471 CrossRef CAS PubMed.
- A. K. Ohlin, G. Landes, P. Bourdon, C. Oppenheimer, R. Wydro and J. Stenflo, J. Biol. Chem., 1988, 263, 19240–19248 CrossRef CAS PubMed.
- J. M. Greve, A. M. Pinkham and J. A. Cowan, Metallomics, 2021, 13, mfab044 CrossRef PubMed.
- P. Fernlund and J. Stenflo, J. Biol. Chem., 1983, 258, 12509–12512 CrossRef CAS.
- J. Stenflo, A. Lundwall and B. Dahlback, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 368–372 CrossRef CAS PubMed.
- J. Stenflo, E. Holme, S. Lindstedt, N. Chandramouli, L. H. Huang, J. P. Tam and R. B. Merrifield, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 444–447 CrossRef CAS PubMed.
- J. E. Dinchuk, R. J. Focht, J. A. Kelley, N. L. Henderson, N. I. Zolotarjova, R. Wynn, N. T. Neff, J. Link, R. M. Huber, T. C. Burn, M. J. Rupar, M. R. Cunningham, B. H. Selling, J. Ma, A. A. Stern, G. F. Hollis, R. B. Stein and P. A. Friedman, J. Biol. Chem., 2002, 277, 12970–12977 CrossRef CAS.
- C. Loenarz and C. J. Schofield, Trends Biochem. Sci., 2011, 36, 7–18 CrossRef CAS PubMed.
- R. Jorissen, Exp. Cell Res., 2003, 284, 31–53 CrossRef CAS PubMed.
- P. Wee and Z. Wang, Cancers, 2017, 9, 52 CrossRef.
- N. Prenzel, O. M. Fischer, S. Streit, S. Hart and A. Ullrich, Endocr. Relat. Cancer, 2001, 11–31 CAS.
- M. H. H. Schmidt, F. Bicker, I. Nikolic, J. Meister, T. Babuke, S. Picuric, W. Müller-Esterl, K. H. Plate and I. Dikic, Nat. Cell Biol., 2009, 11, 873–880 CrossRef CAS PubMed.
- D. P. Reinhardt, R. N. Ono, H. Notbohm, P. K. Müller, H. P. Bächinger and L. Y. Sakai, J. Biol. Chem., 2000, 275, 12339–12345 CrossRef CAS PubMed.
- R. W. Glanville, R. Q. Qian, D. W. McClure and C. L. Maslen, J. Biol. Chem., 1994, 269, 26630–26634 CrossRef CAS.
- H. Tran, W. J. VanDusen and W. S. Argraves, J. Biol. Chem., 1997, 272, 22600–22606 CrossRef CAS PubMed.
- C. M. Cardy and P. A. Handford, J. Mol. Biol., 1998, 276, 855–860 CrossRef CAS PubMed.
- J. Engel, FEBS Lett., 1989, 251, 1–7 CrossRef CAS PubMed.
- R. Timpl, T. Sasaki, G. Kostka and M.-L. Chu, Nat. Rev. Mol. Cell Biol., 2003, 4, 479–489 CrossRef CAS PubMed.
- F. Deák, R. Wagener, I. Kiss and M. Paulsson, Matrix Biol., 1999, 18, 55–64 CrossRef PubMed.
- K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem, 2021, 5, 522–545 CrossRef CAS PubMed.
- T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS.
- K. Liao, T. C. Pickel, V. Boyarskikh, J. Bacsa, D. G. Musaev and H. M. L. Davies, Nature, 2017, 551, 609–613 CrossRef CAS PubMed.
- T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed.
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC.
- Y.-F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef CAS PubMed.
- S. Chakrabarty, Y. Wang, J. C. Perkins and A. R. H. Narayan, Chem. Soc. Rev., 2020, 49, 8137–8155 RSC.
- J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC.
- R. K. Zhang, X. Huang and F. H. Arnold, Curr. Opin. Chem. Biol., 2019, 49, 67–75 CrossRef CAS PubMed.
- Y. Nakano, K. F. Biegasiewicz and T. K. Hyster, Curr. Opin. Chem. Biol., 2019, 49, 16–24 CrossRef CAS PubMed.
- F. Li, X. Zhang and H. Renata, Curr. Opin. Chem. Biol., 2019, 49, 25–32 CrossRef CAS.
- X. Ren and R. Fasan, Curr. Opin. Green Sustain. Chem., 2021, 31, 100494 CrossRef CAS PubMed.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- S. Kille, F. E. Zilly, J. P. Acevedo and M. T. Reetz, Nat. Chem., 2011, 3, 738–743 CrossRef CAS.
- S. M. Pratter, C. Konstantinovics, C. M. L. Di Giuro, E. Leitner, D. Kumar, S. P. de Visser, G. Grogan and G. D. Straganz, Angew. Chem., Int. Ed., 2013, 52, 9677–9681 CrossRef CAS.
- M. D. Toscano, K. J. Woycechowsky and D. Hilvert, Angew. Chem., Int. Ed., 2007, 46, 3212–3236 CrossRef CAS.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120–1136 CrossRef CAS PubMed.
- K. Chen and F. H. Arnold, Nat. Catal., 2020, 3, 203–213 CrossRef CAS.
- V. Steck, J. N. Kolev, X. Ren and R. Fasan, J. Am. Chem. Soc., 2020, 142, 10343–10357 CrossRef CAS PubMed.
- D. Mondal, H. M. Snodgrass, C. A. Gomez and J. C. Lewis, Chem. Rev., 2023, 123, 10381–10431 CrossRef CAS PubMed.
- N. P. Dunham and F. H. Arnold, ACS Catal., 2020, 10, 12239–12255 CrossRef CAS PubMed.
- J. Lee and N. M. Goodey, Chem. Rev., 2011, 111, 7595–7624 CrossRef CAS PubMed.
- J. C. Lewis, Acc. Chem. Res., 2019, 52, 576–584 CrossRef CAS PubMed.
- C. Van Stappen, Y. Deng, Y. Liu, H. Heidari, J.-X. Wang, Y. Zhou, A. P. Ledray and Y. Lu, Chem. Rev., 2022, 122, 11974–12045 CrossRef CAS PubMed.
- S. T. Stripp, B. R. Duffus, V. Fourmond, C. Léger, S. Leimkühler, S. Hirota, Y. Hu, A. Jasniewski, H. Ogata and M. W. Ribbe, Chem. Rev., 2022, 122, 11900–11973 CrossRef CAS PubMed.
- S. S. Chaturvedi, S. B. Jaber Sathik Rifayee, S. O. Waheed, J. Wildey, C. Warner, C. J. Schofield, T. G. Karabencheva-Christova and C. Z. Christov, JACS Au, 2022, 2, 2169–2186 CrossRef CAS PubMed.
- S. P. Visser, Chem. –Euro. J., 2020, 26, 5308–5327 CrossRef PubMed.
- R. H. Wilson, S. Chatterjee, E. R. Smithwick, J. J. Dalluge and A. Bhagi-Damodaran, ACS Catal., 2022, 12, 10913–10924 CrossRef CAS.
- C. Peters and R. Buller, Catalysts, 2019, 9, 221 CrossRef.
- R. S. Gronke, W. J. VanDusen, V. M. Garsky, J. W. Jacobs, M. K. Sardana, A. M. Stern and P. A. Friedman, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 3609–3613 CrossRef CAS PubMed.
- I. Pfeffer, L. Brewitz, T. Krojer, S. A. Jensen, G. T. Kochan, N. J. Kershaw, K. S. Hewitson, L. A. McNeill, H. Kramer, M. Münzel, R. J. Hopkinson, U. Oppermann, P. A. Handford, M. A. McDonough and C. J. Schofield, Nat. Commun., 2019, 10, 1–16 CrossRef CAS PubMed.
- Md. S. Islam, T. M. Leissing, R. Chowdhury, R. J. Hopkinson and C. J. Schofield, Annu. Rev. Biochem., 2018, 87, 585–620 CrossRef CAS PubMed.
- L. Brewitz, B. C. Onisko and C. J. Schofield, J. Biol. Chem., 2022, 298, 102129 CrossRef CAS PubMed.
- F. Gundogan, A. Bedoya, J. Gilligan, E. Lau, P. Mark, M. E. De Paepe and S. M. De la Monte, Pathol. Res. Pract., 2011, 207, 545–553 CrossRef CAS PubMed.
- F. Gundogan, J. Gilligan, W. Qi, E. Chen, R. Naram and S. M. De La Monte, Placenta, 2015, 36, 523–530 CrossRef CAS PubMed.
- S. Shawaf, B. Noureddin, A. Khouri and E. I. Traboulsi, Ophthalmic Genet., 1995, 16, 163–169 CrossRef CAS PubMed.
- R. Haddad, S. Uwaydat, R. Dakroub and E. I. Traboulsi, Am. J. Med. Genet., 2001, 99, 185–189 CrossRef CAS PubMed.
- N. Patel, A. O. Khan, A. Mansour, J. Y. Mohamed, A. Al-Assiri, R. Haddad, X. Jia, Y. Xiong, A. Mégarbané, E. I. Traboulsi and F. S. Alkuraya, Am. J. Hum. Genet., 2014, 94, 755–759 CrossRef CAS PubMed.
- C. Lei, T. Guo, S. Ding, L. Liao, H. Peng, Z. Tan and H. Luo, Mol. Genet. Genomic Med., 2021, 9, 1–8 Search PubMed.
- M. Kanwal, M. Smahel, M. Olsen, J. Smahelova and R. Tachezy, J. Exp. Clin. Cancer Res., 2020, 39, 163 CrossRef CAS PubMed.
- G. Hou, B. Xu, Y. Bi, C. Wu, B. Ru, B. Sun and X. Bai, Bosn. J. Basic Med. Sci., 2018, 18, 297–304 CrossRef CAS PubMed.
- R. Benelli, D. Costa, L. Mastracci, F. Grillo, M. J. Olsen, P. Barboro, A. Poggi and N. Ferrari, Cancers, 2020, 12, 971 CrossRef CAS PubMed.
- L. Brewitz, A. Tumber, I. Pfeffer, M. A. McDonough and C. J. Schofield, Sci. Rep., 2020, 10, 8650 CrossRef CAS PubMed.
- L. Brewitz, A. Tumber and C. J. Schofield, J. Biol. Chem., 2020, 295, 7826–7838 CrossRef CAS PubMed.
- Y. Mao, W. Zhang, Z. Fu, Y. Liu, L. Chen, X. Lian, D. Zhuo, J. Wu, M. Zheng and C. Liao, Angew. Chem., Int. Ed., 2023, 62, e202305250 CrossRef CAS PubMed.
- L. Que and R. Y. N. Ho, Chem. Rev., 1996, 96, 2607–2624 CrossRef CAS PubMed.
- E. L. Hegg and L. Q. Jr, Eur. J. Biochem., 1997, 250, 625–629 CrossRef CAS PubMed.
- L. Que, Nat. Struct. Mol. Biol., 2000, 7, 182–184 CrossRef CAS PubMed.
- A. Bassan, T. Borowski and P. E. M. Siegbahn, Dalton Trans., 2004, 20, 3153–3162 RSC.
- A. Brasnett, I. Pfeffer, L. Brewitz, R. Chowdhury, Y. Nakashima, A. Tumber, M. A. McDonough and C. J. Schofield, Angew. Chem., Int. Ed., 2021, 60, 14657–14663 CrossRef CAS PubMed.
- M. Ueki, D. P. Galonić, F. H. Vaillancourt, S. Garneau-Tsodikova, E. Yeh, D. A. Vosburg, F. C. Schroeder, H. Osada and C. T. Walsh, Chem. Biol., 2006, 13, 1183–1191 CrossRef CAS PubMed.
- L. C. Blasiak, F. H. Vaillancourt, C. T. Walsh and C. L. Drennan, Nature, 2006, 440, 368–371 CrossRef CAS PubMed.
- L. C. Blasiak and C. L. Drennan, Acc. Chem. Res., 2009, 42, 147–155 CrossRef CAS PubMed.
- K. S. Hewitson, S. L. Holmes, D. Ehrismann, A. P. Hardy, R. Chowdhury, C. J. Schofield and M. A. McDonough, J. Biol. Chem., 2008, 283, 25971–25978 CrossRef CAS PubMed.
- M. L. Neidig, C. D. Brown, K. M. Light, D. G. Fujimori, E. M. Nolan, J. C. Price, E. W. Barr, J. M. Bollinger, C. Krebs, C. T. Walsh and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 14224–14231 CrossRef CAS PubMed.
- S. R. Iyer, V. D. Chaplin, M. J. Knapp and E. I. Solomon, J. Am. Chem. Soc., 2018, 140, 11777–11783 CrossRef CAS PubMed.
- K. L. Gorres, K. H. Pua and R. T. Raines, PLoS One, 2009, 4, e7635 CrossRef PubMed.
- G. L. Blatch and M. Lässle, BioEssays, 1999, 21, 932–939 CrossRef CAS PubMed.
- W. Aik, M. A. McDonough, A. Thalhammer, R. Chowdhury and C. J. Schofield, Curr. Opin. Struct. Biol., 2012, 22, 691–700 CrossRef CAS PubMed.
- I. J. Clifton, M. A. McDonough, D. Ehrismann, N. J. Kershaw, N. Granatino and C. J. Schofield, J. Inorg. Biochem., 2006, 100, 644–669 CrossRef CAS PubMed.
- M. Pekkala, R. Hieta, U. Bergmann, K. I. Kivirikko, R. K. Wierenga and J. Myllyharju, J. Biol. Chem., 2004, 279, 52255–52261 CrossRef CAS PubMed.
- M. K. Koski, R. Hieta, M. Hirsilä, A. Rönkä, J. Myllyharju and R. K. Wierenga, J. Biol. Chem., 2009, 284, 25290–25301 CrossRef CAS PubMed.
- P. J. Pollard, C. Loenarz, D. R. Mole, M. A. McDonough, J. M. Gleadle, C. J. Schofield and P. J. Ratcliffe, Biochem. J., 2008, 416, 387–394 CrossRef CAS PubMed.
- R. Chowdhury, R. Sekirnik, N. C. Brissett, T. Krojer, C. Ho, S. S. Ng, I. J. Clifton, W. Ge, N. J. Kershaw, G. C. Fox, J. R. C. Muniz, M. Vollmar, C. Phillips, E. S. Pilka, K. L. Kavanagh, F. Von Delft, U. Oppermann, M. A. McDonough, A. J. Doherty and C. J. Schofield, Nature, 2014, 510, 422–426 CrossRef CAS PubMed.
- D. Petrović, V. A. Risso, S. C. L. Kamerlin and J. M. Sanchez-Ruiz, J. R. Soc. Interface, 2018, 15, 20180330 CrossRef PubMed.
- B. J. Bahnson, T. D. Colby, J. K. Chin, B. M. Goldstein and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12797–12802 CrossRef CAS PubMed.
- R. B. Leveson-Gower, C. Mayer and G. Roelfes, Nat. Rev. Chem, 2019, 3, 687–705 CrossRef CAS.
- S. O. Waheed, R. Ramanan, S. S. Chaturvedi, J. Ainsley, M. Evison, J. M. Ames, C. J. Schofield, C. Z. Christov and T. G. Karabencheva-Christova, Org. Biomol. Chem., 2019, 17, 2223–2231 RSC.
- S. S. Chaturvedi, R. Ramanan, S. O. Waheed, J. Ainsley, M. Evison, J. M. Ames, C. J. Schofield, T. G. Karabencheva‐Christova and C. Z. Christov, Chem. –Euro. J., 2019, 25, 5422–5426 CrossRef CAS PubMed.
- R. P. Hausinger, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 21–68 CrossRef CAS PubMed.
- S. Martinez and R. P. Hausinger, J. Biol. Chem., 2015, 290, 20702–20711 CrossRef CAS PubMed.
- E. G. Kovaleva and J. D. Lipscomb, Science, 2007, 316, 453–457 CrossRef CAS PubMed.
- P. K. Grzyska, E. H. Appelman, R. P. Hausinger and D. A. Proshlyakov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3982–3987 CrossRef CAS PubMed.
- H. M. Hanauske-Abel and V. Günzler, J. Theor. Biol., 1982, 94, 421–455 CrossRef CAS PubMed.
- E. I. Solomon, S. Goudarzi and K. D. Sutherlin, Biochemistry, 2016, 55, 6363–6374 CrossRef CAS PubMed.
- N. Lehnert and E. I. Solomon, J. Biol. Inorg. Chem., 2003, 8, 294–305 CrossRef CAS PubMed.
- E. I. Solomon, K. M. Light, L. V. Liu, M. Srnec and S. D. Wong, Acc. Chem. Res., 2013, 46, 2725–2739 CrossRef CAS PubMed.
- E. I. Solomon, S. D. Wong, L. V. Liu, A. Decker and M. S. Chow, Curr. Opin. Chem. Biol., 2009, 13, 99–113 CrossRef CAS PubMed.
- T. Borowski, H. Noack, M. Radoń, K. Zych and P. E. M. Siegbahn, J. Am. Chem. Soc., 2010, 132, 12887–12898 CrossRef CAS PubMed.
- S. D. Wong, M. Srnec, M. L. Matthews, L. V. Liu, Y. Kwak, K. Park, C. B. Bell Iii, E. E. Alp, J. Zhao, Y. Yoda, S. Kitao, M. Seto, C. Krebs, J. M. Bollinger and E. I. Solomon, Nature, 2013, 499, 320–323 CrossRef CAS PubMed.
- H. J. Kulik and C. L. Drennan, J. Biol. Chem., 2013, 288, 11233–11241 CrossRef CAS PubMed.
- M. G. Quesne, R. Latifi, L. E. Gonzalez‐Ovalle, D. Kumar and S. P. de Visser, Chem. –Euro. J., 2014, 20, 435–446 CrossRef CAS PubMed.
- A. Timmins, M. Saint-André and S. P. De Visser, J. Am. Chem. Soc., 2017, 139, 9855–9866 CrossRef CAS PubMed.
- K.-B. Cho, H. Hirao, S. Shaik and W. Nam, Chem. Soc. Rev., 2016, 45, 1197–1210 RSC.
- J. Zhang, P. Wu, X. Zhang and B. Wang, ChemBioChem, 2023, 24, e202300119 CrossRef CAS PubMed.
- W. Singh, D. Quinn, T. S. Moody and M. Huang, J. Phys. Chem. B, 2019, 123, 7801–7811 CrossRef CAS PubMed.
- J. Li, H.-J. Liao, Y. Tang, J.-L. Huang, L. Cha, T.-S. Lin, J. L. Lee, I. V. Kurnikov, M. G. Kurnikova, W. Chang, N.-L. Chan and Y. Guo, J. Am. Chem. Soc., 2020, 142, 6268–6284 CrossRef CAS PubMed.
- J. Xue, J. Lu and W. Lai, Phys. Chem. Chem. Phys., 2019, 21, 9957–9968 RSC.
- S. P. de Visser, Chem. Rec., 2018, 18, 1501–1516 CrossRef CAS PubMed.
- S. O. Waheed, R. Ramanan, S. S. Chaturvedi, N. Lehnert, C. J. Schofield, C. Z. Christov and T. G. Karabencheva-Christova, ACS Cent. Sci., 2020, 6, 795–814 CrossRef CAS PubMed.
- S. S. Chaturvedi, R. Ramanan, N. Lehnert, C. J. Schofield, T. G. Karabencheva-Christova and C. Z. Christov, ACS Catal., 2020, 10, 1195–1209 CrossRef CAS PubMed.
- R. Ramanan, S. S. Chaturvedi, N. Lehnert, C. J. Schofield, T. G. Karabencheva-Christova and C. Z. Christov, Chem. Sci., 2020, 11, 9950–9961 RSC.
- M. M. Mbughuni, M. Chakrabarti, J. A. Hayden, E. L. Bominaar, M. P. Hendrich, E. Münck and J. D. Lipscomb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16788–16793 CrossRef CAS PubMed.
- J. C. Price, E. W. Barr, T. E. Glass, C. Krebs and J. M. Bollinger, J. Am. Chem. Soc., 2003, 125, 13008–13009 CrossRef CAS PubMed.
- C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. Martin Bollinger Jr, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed.
- E. I. Solomon, D. E. DeWeese and J. T. Babicz Jr, Biochemistry, 2021, 60, 3497–3506 CrossRef CAS PubMed.
- P. J. Riggs-Gelasco, J. C. Price, R. B. Guyer, J. H. Brehm, E. W. Barr, J. M. Bollinger and C. Krebs, J. Am. Chem. Soc., 2004, 126, 8108–8109 CrossRef CAS PubMed.
- J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger and C. Krebs, Biochemistry, 2003, 42, 7497–7508 CrossRef CAS PubMed.
- M. Srnec, S. D. Wong, M. L. Matthews, C. Krebs, J. M. Bollinger and E. I. Solomon, J. Am. Chem. Soc., 2016, 138, 5110–5122 CrossRef CAS PubMed.
- M. L. Neidig, A. Decker, O. W. Choroba, F. Huang, M. Kavana, G. R. Moran, J. B. Spencer and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12966–12973 CrossRef CAS PubMed.
- P. Rabe, J. J. A. G. Kamps, K. D. Sutherlin, J. D. S. Linyard, P. Aller, C. C. Pham, H. Makita, I. Clifton, M. A. McDonough, T. M. Leissing, D. Shutin, P. A. Lang, A. Butryn, J. Brem, S. Gul, F. D. Fuller, I.-S. Kim, M. H. Cheah, T. Fransson, A. Bhowmick, I. D. Young, L. O'Riordan, A. S. Brewster, I. Pettinati, M. Doyle, Y. Joti, S. Owada, K. Tono, A. Batyuk, M. S. Hunter, R. Alonso-Mori, U. Bergmann, R. L. Owen, N. K. Sauter, T. D. W. Claridge, C. V. Robinson, V. K. Yachandra, J. Yano, J. F. Kern, A. M. Orville and C. J. Schofield, Sci. Adv., 2021, 7, eabh0250 CrossRef CAS PubMed.
- S. Martinez, M. Fellner, C. Q. Herr, A. Ritchie, J. Hu and R. P. Hausinger, J. Am. Chem. Soc., 2017, 139, 11980–11988 CrossRef CAS PubMed.
- L. Hu, Z. Li, J. Cheng, Q. Rao, W. Gong, M. Liu, Y. G. Shi, J. Zhu, P. Wang and Y. Xu, Cell, 2013, 155, 1545–1555 CrossRef CAS PubMed.
- I. Zoi, J. Suarez, D. Antoniou, S. A. Cameron, V. L. Schramm and S. D. Schwartz, J. Am. Chem. Soc., 2016, 138, 3403–3409 CrossRef CAS PubMed.
- S. S. Chaturvedi, R. Ramanan, J. Hu, R. P. Hausinger and C. Z. Christov, ACS Catal., 2021, 11, 1578–1592 CrossRef CAS.
- R. Mehmood, H. W. Qi, A. H. Steeves and H. J. Kulik, ACS Catal., 2019, 9, 4930–4943 CrossRef CAS.
- S. O. Waheed, S. S. Chaturvedi, T. G. Karabencheva-Christova and C. Z. Christov, ACS Catal., 2021, 11, 3877–3890 CrossRef CAS.
- Z. Wojdyla and T. Borowski, Chem. –Euro. J., 2022, 28, e202104106 CrossRef CAS PubMed.
- A. Šali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef PubMed.
- A. Fiser and A. Šali, Methods Enzymol., 2003, 374, 461–491 CAS.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- P. Li and K. M. Merz, J. Chem. Inf. Model., 2016, 56, 599–604 CrossRef CAS PubMed.
- D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York and P. A. Kollman, AMBER 2020, University of California, San Francisco, 2020 Search PubMed.
- J. M. Seminario, Int. J. Quantum Chem., 1996, 60, 1271–1277 CrossRef.
- R. J. Woods and R. Chappelle, J. Mol. Struct., 2000, 527, 149–156 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graph., 2006, 25, 247–260 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- A. Pabis, I. Geronimo, D. M. York and P. Paneth, J. Chem. Theory Comput., 2014, 10, 2246–2254 CrossRef CAS PubMed.
- R. L. Davidchack, T. E. Ouldridge and M. V. Tretyakov, J. Chem. Phys., 2015, 142, 144114 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- F. Bresme, J. Chem. Phys., 2001, 115, 7564–7574 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- M. Deserno and C. Holm, J. Chem. Phys., 1998, 109, 7678–7693 CrossRef CAS.
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- B. J. Grant, A. P. C. Rodrigues, K. M. ElSawy, J. A. McCammon and L. S. D. Caves, Bioinformatics, 2006, 22, 2695–2696 CrossRef CAS PubMed.
- D. Bakowies and W. Thiel, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef CAS.
- S. Metz, J. Kästner, A. A. Sokol, T. W. Keal and P. Sherwood, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 101–110 CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- W. Smith and T. R. Forester, J. Mol. Graph., 1996, 14, 136–141 CrossRef CAS PubMed.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1986, 7, 718–730 CrossRef CAS.
- M. J. Field, P. A. Bash and M. Karplus, J. Comput. Chem., 1990, 11, 700–733 CrossRef CAS.
- P. Sherwood, A. H. De Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, J. Mol. Struct., 2003, 632, 1–28 CrossRef CAS.
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, J. Phys. Chem. A, 2009, 113, 11856–11865 CrossRef PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Sheong, J.-X. Zhang and Z. Lin, J. Comput. Chem., 2019, 40, 1172–1184 CrossRef CAS PubMed.
- T. Stuyver, J. Huang, D. Mallick, D. Danovich and S. Shaik, J. Comput. Chem., 2020, 41, 74–82 CrossRef CAS PubMed.
- S. E. Graham, F. Syeda and G. A. Cisneros, Biochemistry, 2012, 51, 2569–2578 CrossRef CAS PubMed.
- S. W. Dewage and G. A. Cisneros, J. Phys. Chem. B, 2015, 119, 3669–3677 CrossRef CAS PubMed.
- H. Torabifard and G. A. Cisneros, Chem. Sci., 2018, 9, 8433–8445 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, 2016 Search PubMed.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöß, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- A. Laio, J. VandeVondele and U. Rothlisberger, J. Chem. Phys., 2002, 116, 6941–6947 CrossRef CAS.
- T. Laino, F. Mohamed, A. Laio and M. Parrinello, J. Chem. Theory Comput., 2005, 1, 1176–1184 CrossRef CAS PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B, 1996, 54, 1703–1710 CrossRef CAS PubMed.
- M. Guidon, J. Hutter and J. VandeVondele, J. Chem. Theory Comput., 2010, 6, 2348–2364 CrossRef CAS PubMed.
- M. Karplus and J. A. McCammon, Nat. Struct. Mol. Biol., 2002, 9, 646–652 CrossRef CAS PubMed.
- E. Z. Eisenmesser, D. A. Bosco, M. Akke and D. Kern, Science, 2002, 295, 1520–1523 CrossRef CAS PubMed.
- M. Karplus and J. Kuriyan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6679–6685 CrossRef CAS PubMed.
- J. P. Klinman, Acc. Chem. Res., 2015, 48, 449–456 CrossRef CAS PubMed.
- A. G. Palmer, Acc. Chem. Res., 2015, 48, 457–465 CrossRef CAS PubMed.
- A. Kohen, Acc. Chem. Res., 2015, 48, 466–473 CrossRef CAS PubMed.
- G. Bhabha, J. T. Biel and J. S. Fraser, Acc. Chem. Res., 2015, 48, 423–430 CrossRef CAS PubMed.
- M. H. M. Olsson, W. W. Parson and A. Warshel, Chem. Rev., 2006, 106, 1737–1756 CrossRef CAS PubMed.
- S. S. Chaturvedi, R. Ramanan, S. O. Waheed, T. G. Karabencheva-Christova and C. Z. Christov, in Advances in Protein Chemistry and Structural Biology, Elsevier, 2019, vol. 117, pp. 113–125 Search PubMed.
- T. G. Karabencheva-Christova, C. Z. Christov and G. B. Fields, in Advances in Protein Chemistry and Structural Biology, Elsevier, 2017, vol. 109, pp. 1–24 Search PubMed.
- J. Ainsley, A. Lodola, A. J. Mulholland, C. Z. Christov and T. G. Karabencheva-Christova, in Advances in Protein Chemistry and Structural Biology, Elsevier, 2018, vol. 113, pp. 1–32 Search PubMed.
- J. M. Greve, A. M. Pinkham, Z. Thompson and J. A. Cowan, Metallomics, 2021, 13, mfab056 CrossRef PubMed.
- J. R. Horton, A. K. Upadhyay, H. H. Qi, X. Zhang, Y. Shi and X. Cheng, Nat. Struct. Mol. Biol., 2010, 17, 38–43 CrossRef CAS PubMed.
- C. Lemay-St-Denis, N. Doucet and J. N. Pelletier, Protein Eng. Des. Sel., 2022, 35, gzac015 CrossRef PubMed.
- M. C. C. J. C. Ebert, S. L. Dürr, A. A. Houle, G. Lamoureux and J. N. Pelletier, ACS Catal., 2016, 6, 7426–7437 CrossRef CAS.
- L. Alejaldre, C. Lemay-St-Denis, J. N. Pelletier and D. Quaglia, Biochemistry, 2023, 62, 396–409 CrossRef CAS PubMed.
- M. B. Berger, A. R. Walker, E. A. Vázquez-Montelongo and G. A. Cisneros, Phys. Chem. Chem. Phys., 2021, 23, 22227–22240 RSC.
- K. D. Dubey and S. Shaik, Acc. Chem. Res., 2019, 52, 389–399 CrossRef PubMed.
- J. Huang, C. Li, B. Wang, D. A. Sharon, W. Wu and S. Shaik, ACS Catal., 2016, 6, 2694–2704 CrossRef CAS.
- S. O. Waheed, A. Varghese, I. DiCastri, B. Kaski, C. LaRouche, G. B. Fields and T. G. Karabencheva‐Christova, ChemPhysChem, 2023, 24, e202200649 CrossRef CAS PubMed.
- S. Shaik, H. Chen and D. Janardanan, Nat. Chem., 2011, 3, 19–27 CrossRef CAS PubMed.
- B. Wang, E. M. Johnston, P. Li, S. Shaik, G. J. Davies, P. H. Walton and C. Rovira, ACS Catal., 2018, 8, 1346–1351 CrossRef CAS.
- S. Ye and F. Neese, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1228–1233 CrossRef CAS PubMed.
- S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS PubMed.
- S. Ye, C. Riplinger, A. Hansen, C. Krebs, J. M. Bollinger and F. Neese, Chem.–Euro. J., 2012, 18, 6555–6567 CrossRef CAS PubMed.
- L. M. Mirica, K. P. McCusker, J. W. Munos, H. Liu and J. P. Klinman, J. Am. Chem. Soc., 2008, 130, 8122–8123 CrossRef CAS PubMed.
- I. A. Topol, A. V. Nemukhin, K. Salnikow, R. E. Cachau, Y. G. Abashkin, K. S. Kasprzak and S. K. Burt, J. Phys. Chem. A, 2006, 110, 4223–4228 CrossRef CAS PubMed.
- J. A. Hangasky, H. Gandhi, M. A. Valliere, N. E. Ostrom and M. J. Knapp, Biochemistry, 2014, 53, 8077–8084 CrossRef CAS PubMed.
- S. Goudarzi, S. R. Iyer, J. T. Babicz, J. J. Yan, G. H. J. Peters, H. E. M. Christensen, B. Hedman, K. O. Hodgson and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5152–5159 CrossRef CAS PubMed.
- J. Zhou, M. Gunsior, B. O. Bachmann, C. A. Townsend and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 13539–13540 CrossRef CAS.
- H. Liu, J. Llano and J. W. Gauld, J. Phys. Chem. B, 2009, 113, 4887–4898 CrossRef CAS PubMed.
- C. Geng, S. Ye and F. Neese, Angew. Chem., Int. Ed., 2010, 49, 5717–5720 CrossRef CAS PubMed.
- D. Fang, R. L. Lord and G. A. Cisneros, J. Phys. Chem. B, 2013, 117, 6410–6420 CrossRef CAS PubMed.
- S. O. Waheed, A. Varghese, S. S. Chaturvedi, T. G. Karabencheva-Christova and C. Z. Christov, ACS Catal., 2022, 12, 5327–5344 CrossRef CAS PubMed.
- S. P. De Visser, J. Am. Chem. Soc., 2006, 128, 9813–9824 CrossRef CAS PubMed.
- D. Usharani, D. Janardanan and S. Shaik, J. Am. Chem. Soc., 2011, 133, 176–179 CrossRef CAS PubMed.
- S. B. J. S. Rifayee, S. S. Chaturvedi, C. Warner, J. Wildey, W. White, M. Thompson, C. J. Schofield and C. Z. Christov, Chem. –Euro. J., 2023, 29, e202301305 CrossRef CAS PubMed.
- L. M. Hoffart, E. W. Barr, R. B. Guyer, J. M. Bollinger and C. Krebs, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14738–14743 CrossRef CAS PubMed.
- D. W. Kastner, A. Nandy, R. Mehmood and H. J. Kulik, ACS Catal., 2023, 13, 2489–2501 CrossRef CAS.
- L. J. Walport, R. J. Hopkinson, R. Chowdhury, R. Schiller, W. Ge, A. Kawamura and C. J. Schofield, Nat. Commun., 2016, 7, 11974 CrossRef CAS PubMed.
- J. Lu, L. Hu, J. Cheng, D. Fang, C. Wang, K. Yu, H. Jiang, Q. Cui, Y. Xu and C. Luo, Phys. Chem. Chem. Phys., 2016, 18, 4728–4738 RSC.
- F. Chen, K. Bian, Q. Tang, B. I. Fedeles, V. Singh, Z. T. Humulock, J. M. Essigmann and D. Li, Chem. Res. Toxicol., 2017, 30, 1102–1110 Search PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- W. Peng, S. Yan, X. Zhang, L. Liao, J. Zhang, S. Shaik and B. Wang, J. Am. Chem. Soc., 2022, 144, 20484–20494 CrossRef CAS PubMed.
- B. Wang, Z. Cao, D. A. Sharon and S. Shaik, ACS Catal., 2015, 5, 7077–7090 CrossRef CAS.
- P. Schyman, W. Lai, H. Chen, Y. Wang and S. Shaik, J. Am. Chem. Soc., 2011, 133, 7977–7984 CrossRef CAS PubMed.
- H. J. Kulik, L. C. Blasiak, N. Marzari and C. L. Drennan, J. Am. Chem. Soc., 2009, 131, 14426–14433 CrossRef CAS PubMed.
- M. Srnec and E. I. Solomon, J. Am. Chem. Soc., 2017, 139, 2396–2407 CrossRef CAS PubMed.
- A. Timmins, N. J. Fowler, J. Warwicker, G. D. Straganz and S. P. De Visser, Front. Chem., 2018, 6, 513 CrossRef CAS PubMed.
- E. I. Solomon, A. Decker and N. Lehnert, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3589–3594 CrossRef CAS PubMed.
- P. K. Grzyska, T. A. Müller, M. G. Campbell and R. P. Hausinger, J. Inorg. Biochem., 2007, 101, 797–808 CrossRef CAS PubMed.
- S. S. Ng, K. L. Kavanagh, M. A. McDonough, D. Butler, E. S. Pilka, B. M. R. Lienard, J. E. Bray, P. Savitsky, O. Gileadi, F. Von Delft, N. R. Rose, J. Offer, J. C. Scheinost, T. Borowski, M. Sundstrom, C. J. Schofield and U. Oppermann, Nature, 2007, 448, 87–91 CrossRef CAS PubMed.
- Z. Cheng, P. Cheung, A. J. Kuo, E. T. Yukl, C. M. Wilmot, O. Gozani and D. J. Patel, Genes Dev., 2014, 28, 1758–1771 CrossRef CAS PubMed.
- C. Yi, B. Chen, B. Qi, W. Zhang, G. Jia, L. Zhang, C. J. Li, A. R. Dinner, C.-G. Yang and C. He, Nat. Struct. Mol. Biol., 2012, 19, 671–676 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05974j |
| This journal is © The Royal Society of Chemistry 2024 |