
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic enhancement of electrocatalytic nitroarene hydrogenation over Mo2C@MoS2 heteronanorods with dual active-sites†
Wanling
Zhang
a,
Wenbiao
Zhang
*ab,
Kun
Yu
a,
Jingwen
Tan
a,
Yi
Tang
 b and
Qingsheng
Gao
*a
b and
Qingsheng
Gao
*a
aCollege of Chemistry and Materials Science, Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangzhou 510632, P. R. China. E-mail: wbzhang1994@hotmail.com; tqsgao@jnu.edu.cn
bDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Laboratory of Advanced Materials and Collaborative Innovation Centre of Chemistry for Energy Materials, Fudan University, Shanghai 200433, P. R. China
First published on 6th February 2024
Abstract
Electrocatalytic hydrogenation (ECH) enables the sustainable production of chemicals under ambient conditions, in which catalysts catering for the different chemisorption of reactants/intermediates are desired but still challenging. Here, Mo2C@MoS2 heteronanorods with dual active-sites are developed to accomplish efficient nitroarene ECH according to our theoretical prediction that the binding of atomic H and nitro substrates would be synergistically strengthened on Mo2C–MoS2 interfaces. They afford high faradaic efficiency (>85%), yield (>78%) and selectivity (>99%) for the reduction of 4-nitrostyrene (4-NS) to 4-vinylaniline (4-VA) in neutral electrolytes, outperforming not only the single-component counterparts of Mo2C nanorods and MoS2 nanosheets, but also recently reported noble-metals. Accordingly, in situ Raman spectroscopy combined with electrochemical tests clarifies the rapid ECH of 4-NS on Mo2C–MoS2 interfaces due to the facilitated elementary steps, quickly refreshing active sites for continuous electrocatalysis. Mo2C@MoS2 further confirms efficient and selective ECH toward functional anilines with other well-retained reducible groups in wide substrate scope, underscoring the promise of dual-site engineering for exploring catalysts.
Introduction
Renewable electricity powered chemical processes are burgeoning for the sustainable production of high-value chemicals.1–3 In particular, electrocatalytic hydrogenation (ECH) utilizes electrochemically formed hydrogen H* (* denotes an active site) from protons/water to upgrade various groups (e.g., carbonyl, nitro, alkenyl, etc.) under ambient conditions,4–9 avoiding harsh operations associated with high temperature and H2 pressure adopted in thermocatalysis. For example, the reduction of nitroarenes to anilines is significant for manufacturing medicine, dyes, spices, and explosives.10 Witnessed progress has been made in the thermocatalytic hydrogenation of nitroarenes using H2,11–13 but it is challenged by the trade-off between activity and selectivity due to the difficult control over reaction pathways under heated and pressurized conditions.10,14 ECH, at room temperature and atmospheric pressure, neatly offers a controllable alternative to accommodate such multi-electron transfer processes via varying working potentials or currents.6,15 Following a typical Langmuir–Hinshelwood (L–H) mechanism,16,17 ECH proceeds via the stepwise reactions of adsorbed organics and H* on the catalyst surface, highly dependent on their competitive chemisorption. As H* is dominant, the hydrogen evolution reaction (HER) via its self-combination occurs easily, decreasing the faradaic efficiency (FE) of ECH,18 while the bimolecular coupling of nitroarenes or aldehydes/ketones toward azoxybenzenes or pinanols would exceed the target hydrogenation when the coverage of adsorbed organics is excessively high.19,20 It is necessary to design a catalyst surface that can counterbalance the difference in chemisorption toward efficient hydrogenation.21,22As a noble-metal-free electrocatalyst, MoS2 demonstrates its efficiency in the ECH of aldehydes and nitroarenes;16,23 however, the sluggish H2O activation and discharging to H* (H2O + e− →H* + OH−) under basic/neutral conditions unfortunately limit ECH kinetics. Efforts have been devoted to creating lattice defects and adjusting the phase transition,23,24 which fail to balance the competitive chemisorption of H* and organic substrates due to non-selective enhancement on a single kind of active sites. Constructing heterointerfaces involving dual active sites would be a promising alternative. The combination of MoS2 with noble-metals is capable of accommodating the different elementary steps of multi-electron transfer processes (e.g., N2 and H2O reduction),25–27 but cannot meet the requirement of ECH because of the seriously retarded FEs and increased operation cost after introducing precious elements highly active for the HER.28
Thanks to the relatively more favorable H2O activation and stronger binding with H*,29–31 noble-metal-like molybdenum carbides (MoxC) would be available to construct qualified interfaces on MoS2. Moreover, such two moieties share the same Mo element, enabling the facile construction and proliferative synergy of their interfaces. Here, Mo2C@MoS2 with dual sites was developed to enable the efficient ECH of nitroarenes. As indicated by density functional theory (DFT) calculations, the binding of H* and 4-nitrostyrene (4-NS*) was strengthened on the Mo2C and nearby MoS2 of Mo2C–MoS2 interfaces, respectively, which would promote the surface elementary steps of hydrogenation. As expected, Mo2C@MoS2 heteronanorods consisting of Mo2C axes and MoS2 shells accomplished high FE (>85%), selectivity (>99%) and yield (>78%) for the ECH of 4-NS to 4-vinylaniline (4-VA) in neutral electrolytes at −0.05 to −0.45 V vs. RHE, superior to single-component Mo2C nanorods and MoS2 nanosheets and even recently reported noble-metals. In situ Raman spectroscopy further confirmed the rapid ECH of 4-NS on Mo2C–MoS2 interfaces, quickly refreshing active sites for continuous electrocatalysis. In sharp comparison, the slow turnover due to the absence of either effective nitro activation on Mo2C or rich H* on MoS2 spawned the accumulation of hydrophobic reactants/intermediates and consequently an extra interfacial impedance on electrodes verified by electrochemical impedance spectroscopy (EIS). Moreover, Mo2C@MoS2 afforded efficient and selective ECH toward functional anilines in wide substrate scope, highlighting its efficiency for electrocatalytic refinery.
Experimental section
Chemicals
Ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O, AHM), aniline, thiourea and hydrochloric acid (HCl, AR) were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). 4-Nitrostyrene and Nafion solution (5 wt% in lower aliphatic alcohols and water) were purchased from Sigma-Aldrich. All chemicals were of analytical grade and used as received without further purification. All aqueous solutions were prepared using ultrapure water (>18.2 MΩ).Material synthesis
Physical characterization
X-ray diffraction (XRD) analysis was conducted on a Bruker D8 Advance diffractometer with Cu Kα radiation (λ = 1.54056 Å). Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were conducted on a JEOL JEM-2100F and ZEISS ULTRA 55, respectively. X-ray photoelectron spectroscopy (XPS) was carried out on a Thermo Fisher Scientific (Escalab 250Xi), and the binding energy was calibrated with respect to the C 1s (284.6 eV) of contaminated carbon. Raman spectra were collected on a LabRAM HR Evolution, with an excitation laser wavelength of 532 nm. Nitrogen adsorption–desorption isotherms were used to determine the specific surface areas of electrocatalysts on an automatic gas adsorption analyzer (Quantachrome Autosorb-iQ-MP) by a multipoint Brunauer–Emmett–Teller (BET) method. Electron paramagnetic resonance (EPR) spectra were recorded on a Magnettech ESR500.Electrochemical test
To prepare working electrodes, 10 mg of electrocatalysts and 100 μL of 5 wt% Nafion solution were dispersed in 1.0 mL water–ethanol with a Vwater/Vethanol ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and then the mixture was vigorously sonicated for at least 30 min to get a homogeneous ink. The as-obtained ink (500 μL) was loaded onto pre-treated carbon cloth (1.5 cm × 1.5 cm) carefully and dried naturally in air. The working electrode was directly subjected to the ECH of nitroarenes, which was conducted on a CHI 650E electrochemical workstation (Chenhua Instruments Shanghai Co., Ltd, China), within a gas-tight two-compartment electrochemical cell equipped with a piece of a proton-exchange membrane (Nafion 117) as the separator. A saturated calomel electrode (SCE) and a platinum electrode were used as the reference and counter electrodes, respectively. The cathodic electrolyte (pH 6.8) contained 4-NS (12.5 mmol L−1), LiClO4 (0.1 mol L−1) and methanol/water (1:1), which was degassed using a N2 gas flow for 30 min prior to the ECH and saturated with the same flow-rate N2 during the test. Methanol was used to improve the homogeneous dispersion of the 4-NS phase in the electrolyte. Linear sweep voltammetry (LSV) was conducted in an undivided cell with and without 12.5 mmol L−1 4-NS in 40 mL LiClO4 solutions (pH 6.8).
:1 and then the mixture was vigorously sonicated for at least 30 min to get a homogeneous ink. The as-obtained ink (500 μL) was loaded onto pre-treated carbon cloth (1.5 cm × 1.5 cm) carefully and dried naturally in air. The working electrode was directly subjected to the ECH of nitroarenes, which was conducted on a CHI 650E electrochemical workstation (Chenhua Instruments Shanghai Co., Ltd, China), within a gas-tight two-compartment electrochemical cell equipped with a piece of a proton-exchange membrane (Nafion 117) as the separator. A saturated calomel electrode (SCE) and a platinum electrode were used as the reference and counter electrodes, respectively. The cathodic electrolyte (pH 6.8) contained 4-NS (12.5 mmol L−1), LiClO4 (0.1 mol L−1) and methanol/water (1:1), which was degassed using a N2 gas flow for 30 min prior to the ECH and saturated with the same flow-rate N2 during the test. Methanol was used to improve the homogeneous dispersion of the 4-NS phase in the electrolyte. Linear sweep voltammetry (LSV) was conducted in an undivided cell with and without 12.5 mmol L−1 4-NS in 40 mL LiClO4 solutions (pH 6.8).
To identify the involvement of H* during the ECH, EPR measurement with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the trapping agent was conducted. After the chronoamperometry test at a given constant potential of −0.45 V vs. RHE for 5 min, 0.1 mM DMPO was added into the cathodic electrolyte and stirred for 1 min. Afterwards, the electrolyte was taken out for the EPR test. Meanwhile, t-BuOH (25 wt% in 0.1 M LiClO4) was used to quench hydrogen radicals during chronoamperometry at −0.45 V vs. RHE, in which 4-VA was detected every 60 min. The obviously prohibited yield of 4-VA in comparison with that free-from t-BuOH again proved the involvement of H*.
Product analysis
After reactions, liquid products were collected from the cathode compartment and analyzed via a HPLC (SHIMADZU LC-20AT) equipped with a UV detector at 254 and 370 nm. The column (INERTSUSTAIN C18) was operated at 40 °C and the binary gradient method was adopted at a flow rate of 0.8 mL min−1, which contained H2O and CH3OH (VCH3OH:VH2O = 6:4). The FE (%), yield (%) and selectivity (%) of products were calculated based on the following equations: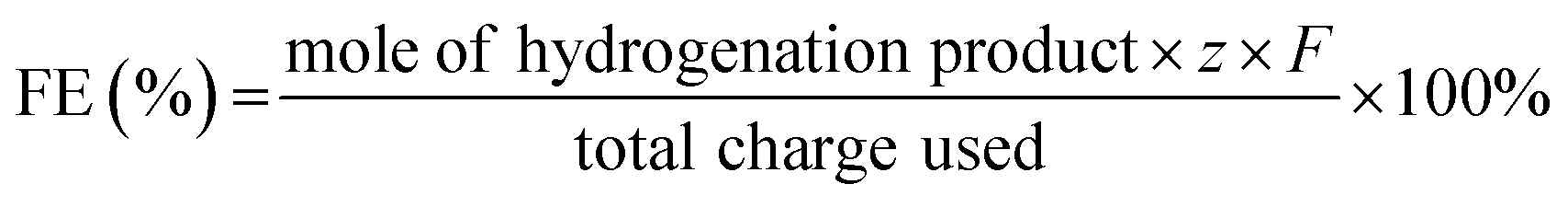
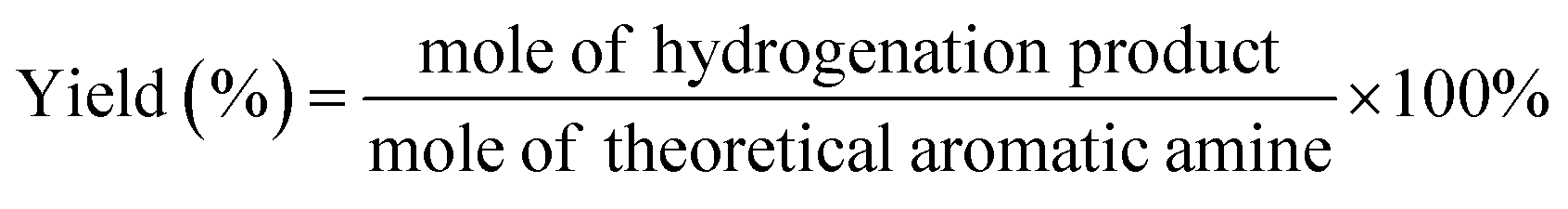
where, z is the number of transferred electrons, which for 4-VA, 4-ethyl-nitrobenzene (4-ENB) and 4-ethylaniline (4-EA) is 6, 2 and 8, respectively. F is the faradaic constant (96
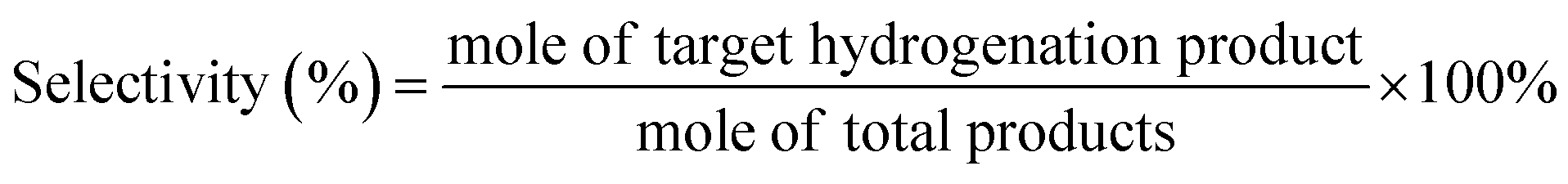 485 C mol−1).
485 C mol−1).
Theoretical calculations
In this work, all the theoretical computations were based on the DFT method, as implemented in the CASTEP package of Materials Studio. For the exchange–correlation functional, we used the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional. The projector augmented wave (PAW) method was applied to depict the interaction between ion and electron, and the cut of energy was set at 400.0 eV. The force and energy cutoff were set at 0.01 eV Å−1 and 10−6 eV for the geometry optimization, respectively. A 5 × 5 × 1 Monkhorst–Pack grid k-point sampling was conducted for the bulk and slab structure optimization. The optimized bulk Mo2C (space group: P63/mmc; a = b = 3.0 Å, c = 4.7 Å) and 2H–MoS2 (space group: P63/mmc; a = b = 3.2 Å, c = 12.3 Å) were selected. Then, we selected a 3 × 3 relaxed rhombus MoS2(002) bilayer on top of the relaxed 4 × 4 Mo2C(001) surface to model Mo2C–MoS2 heterojunctions, in which S atoms were introduced to saturate the coordination of Mo atoms on the Mo2C(001). The adsorption slab would be curved as the (010) of the Mo2C–MoS2 layer and a vacuum layer of 12 Å was used to isolate the slab as the boundary condition, which made the interactions between neighboring slabs negligible according to other previous work.33The binding energy (BE) of an adsorbate was calculated as:
| BE(adsorbate) = E(slab+adsorbate) − E(slab) − E(adsorbate)w |
Results and discussion
DFT calculations were conducted to analyze the varied binding energies of H* (BEH) and 4-NS* (BE4-NS) on MoS2, Mo2C and their interfaces. According to previous reports34,35 and the experimentally evidenced selective hydrogenation of the nitro group in this work, the vertical chemisorption of 4-NS via bonding nitro with Mo sites was reasonably taken into account.36 In further regard to H*, the Mo atom on the Mo2C(101) surface was indicated as the available site because of the obviously stronger BEH (−0.68 eV) than that on a carbon site (+0.19 eV) (Fig. S1, ESI†), which also exceeded those on the Mo (−0.14 eV) and S sites (−0.1 eV) of MoS2. For a reliable comparison, the Mo sites on Mo2C, MoS2 and Mo2C–MoS2 models were further considered in the context of the competitive chemisorption of 4-NS* and H*. As depicted in Fig. 1a, the edge-site rich MoS2(010) presented obviously weaker binding with H* (−0.14 eV) but the stronger one with 4-NS* (−1.39 eV) relative to Mo2C (101) (BEH: −0.68 eV; BE4-NS: −0.48 eV), which suggested the favorable activation of nitro on MoS2 and the rich H* on Mo2C when these sites were combined together. As expected, a Mo2C–MoS2 interface showed the synchronously strengthened BEH (−0.73 eV) on Mo2C and BE4-NS (−2.77 eV) on MoS2 (Fig. 1b), in favor of the ECH via the L-H mechanism. Accordingly, the projected density of states (PDOS) for d bands (Fig. 1c) was visibly broadened near the Fermi level (EF) on Mo2C–MoS2, benefiting the chemisorption of either H* or 4-NS* due to the easy overlap of orbitals. In the chemisorption configurations (Fig. 1d), the electron density distribution of 4-NS* on MoS2 was closer than that on Mo2C, confirming its more favorable activation on the MoS2 moiety. It's thus reasonable to design Mo2C–MoS2 interfaces to boost 4-NS reduction thanks to the capability of counterbalancing the difference between H* and 4-NS* (Fig. 1e). The ECH of 4-NS can produce 4-VA and 4-ENB via selectively hydrogenating nitro and vinyl groups, respectively, both of which would be further reduced to 4-EA. 4-VA is targeted due to its high added-value for chemical synthesis.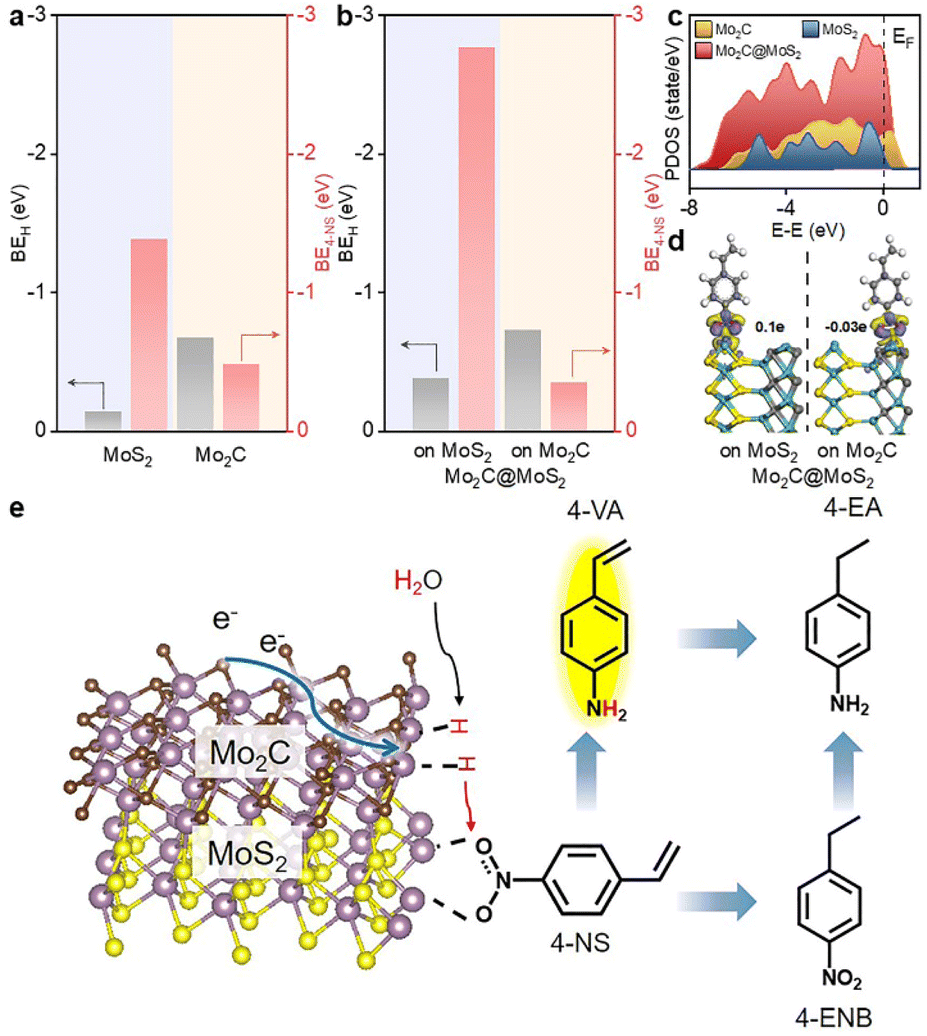 | ||
| Fig. 1 BEH and BE4-NS on (a) Mo2C and MoS2, and (b) Mo2C–MoS2 interfaces. (c) PDOS of Mo2C, Mo2C–MoS2 and MoS2. (d) Electron density difference of Mo2C@MoS2 with 4-NS adsorbed on the Mo2C or MoS2 moiety. (e) Schematic illustration for the boosted ECH of 4-NS on Mo2C–MoS2 interfaces. | ||
Here, the sequent carbonization and sulfidation of Mo3O10(C6H8N)2·2H2O nanowires (Fig. S2, ESI†) was employed to fabricate Mo2C@MoS2 heteronanorods for efficient hydrogenation (Fig. 2a). XRD analysis confirmed the co-presence of Mo2C and MoS2 phases in Mo2C@MoS2 (Fig. 2b), in which the (100), (002) and (101) of Mo2C and the (002), (101) and (110) of MoS2 were observed. Accordingly, it showed the characteristic E12g, A1g and 2LA(M) vibration modes of MoS2, while Mo2C was transparent in Raman spectra (Fig. S3, ESI†). As shown in SEM and TEM (Fig. 2c), Mo2C@MoS2 retained the rod-like morphology of parent Mo2C, but generated nanosheet shells on the surface, resulting in a hierarchical structure that combines the structural merits of 1D Mo2C (Fig. S4, ESI†) and 2D MoS2 (Fig. S5, ESI†). High-resolution TEM (HR-TEM) clearly identified the Mo2C–MoS2 interfaces with visible Mo2C(101) and MoS2(002) lattices (Fig. 2d). Besides, the corresponding elemental mapping confirmed the uniform distribution of Mo, S, and C in Mo2C@MoS2 (Fig. S6, ESI†). Such a hierarchical 1D@2D nanostructure enabled its relatively larger surface area of 39.5 m2 g−1 (Fig. S7, ESI†), as compared to Mo2C (24.9 m2 g−1) and MoS2 (25.0 m2 g−1). Moreover, a series of Mo2C@MoS2 prepared with different feeding ratios had a similar nanostructure (Fig. S8 and S9, ESI†). In addition, the chemical states of elements were analyzed by XPS (Fig. 2e, f and Table S1, ESI†). In the deconvoluted Mo 3d profile, the doublets at 228.3 and 231.4 eV were ascribed to the Mo 3d5/2 and Mo 3d3/2 of Mo2+ due to the presence of Mo2C, and those at 228.7 and 231.8 eV corresponded to Mo4+ of MoS2.37 The S 2p profile also indicated the consistent chemical state of S in Mo2C@MoS2 and MoS2.
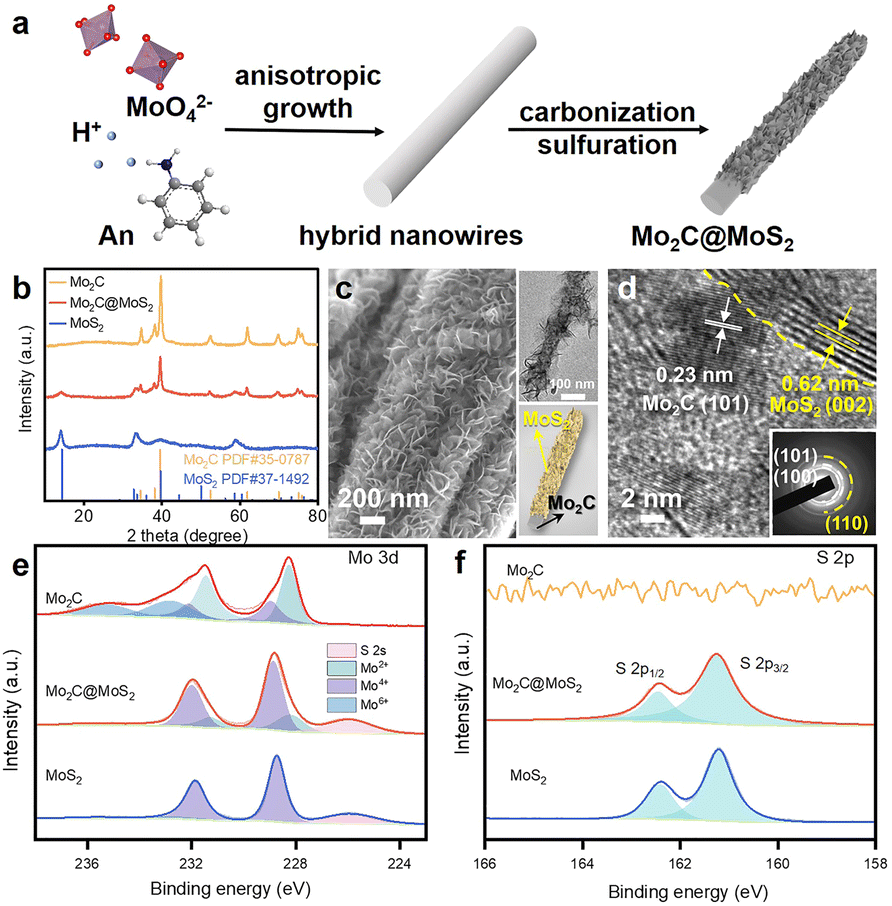 | ||
| Fig. 2 (a) Schematic illustration for the preparation of hierarchical Mo2C@MoS2 nanorods. (b)XRD patterns, (c) SEM and (d) TEM images of Mo2C@MoS2 heteronanorods. (e) Mo 3d, (f) S 2p XPS spectra of Mo2C, Mo2C@MoS2 and MoS2. | ||
The ECH performances of Mo2C, MoS2 and Mo2C@MoS2 were tested in a H-type cell separated by a proton exchange membrane (Nafion 117). Cyclic voltammetry (CV) curves were collected in 0.1 M LiClO4 solution (pH = 6.8) containing 50 wt% methanol to improve the solubility of 4-NS (Fig. S10, ESI†), which exhibited a cathodic reduction peak at −0.29 to −0.50 V vs. RHE, corresponding to the stepwise reduction.38 Then, their polarization curves in 0.1 M LiClO4 with and without 4-NS were compared. The cathodic currents of all three catalysts increased after introducing 4-NS and that of Mo2C@MoS2 was the highest (Fig. 3a), indicative of its outstanding activity for the ECH. Subsequently, chronoamperometry was performed at −0.05 to −0.45 V vs. RHE for 5 h (Fig. 3b). Mo2C@MoS2 maintained an obviously higher FE and yield of 4-VA, as compared to Mo2C and MoS2. The total FEs were lower than 100% associated with the concomitant HER. At −0.45 V vs. RHE, the FE, yield and selectivity of 4-VA were as high as ∼85%, ∼80%, and 99%, respectively (Fig. 3b and S11, ESI†). Furthermore, the electrochemical surface areas (ECSAs) of these electrodes were visualized through calculating the double-layer capacitances (Cdl) according to the proportional relationship (Fig. S12, ESI†) and subsequently used for normalizing the rate of 4-VA production to access the difference in specific activity. The higher specific reaction rate on Mo2C@MoS2 than those on Mo2C and MoS2 indicated the intrinsic superiority associated with Mo2C–MoS2 interfaces (Fig. S13, ESI†). In further comparison with recently reported electrocatalysts including noble-metals (Fig. 3c and Table S2, ESI†), Mo2C@MoS2 performed among the best and featured mild operation with neutral electrolytes, avoiding the use of alkalis that easily trigger by-reactions (e.g., bi-molecular coupling to azoxybenzenes) and equipment corrosion.
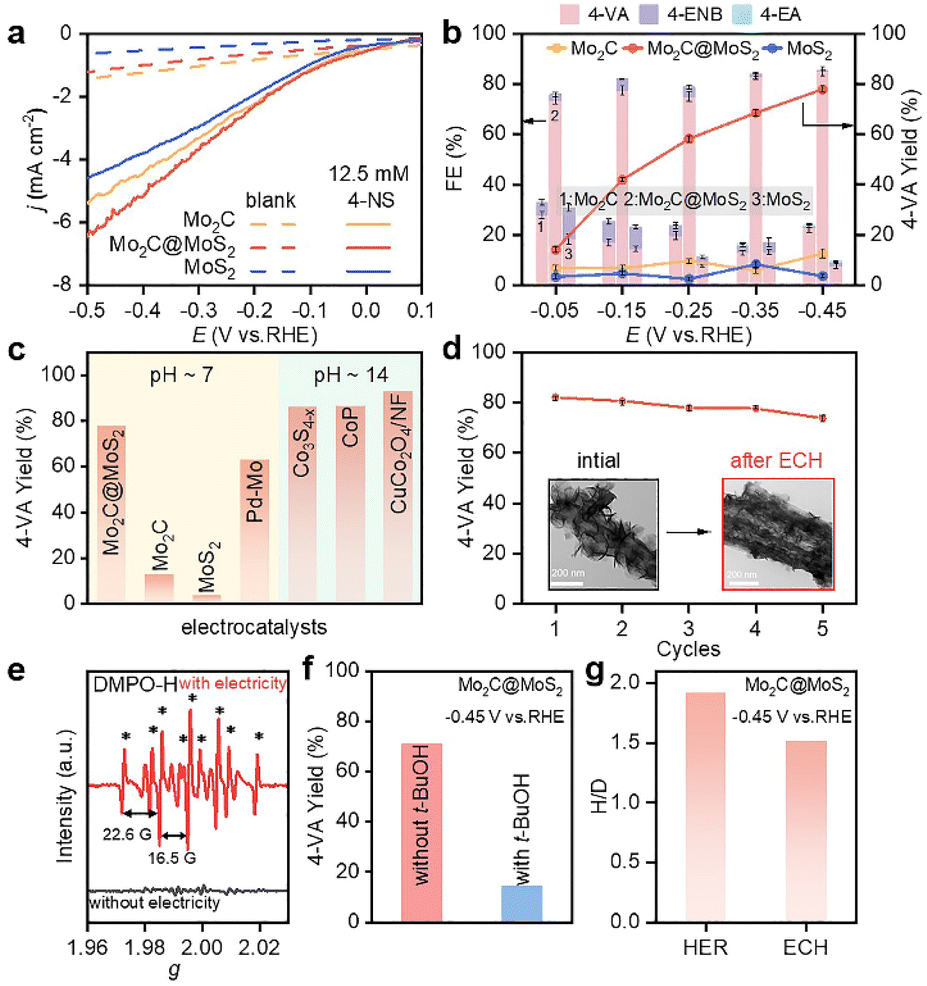 | ||
| Fig. 3 (a) Polarization curves of Mo2C, Mo2C@MoS2 and MoS2 in 0.1 M LiClO4 with and without 12.5 mM 4-NS, and (b) product FEs and yields at −0.05 to −0.45 V vs. RHE with 12.5 mM 4-NS (reaction time: 5 h). (c) Comparison of 4-VA yield of Mo2C@MoS2 with that of state-of-the-art electrocatalysts, including Pd–Mo,39 Co3S4−x,35 CoP,40 and CuCo2O4/NF.41 (d) Cycle-dependent 4-VA yield over Mo2C@MoS2 at −0.45 V. The insets of panel d show the TEM images of Mo2C@MoS2 before and after the test. (e) Quasi in situ EPR trapping of hydrogen radicals over Mo2C@MoS2. (f) Yield of 4-VA with and without t-BuOH as the hydrogen radical scavenger. (g) Kinetic isotopic effect of Mo2C@MoS2 for the HER and ECH. | ||
The time-dependent conversion of 4-NS to 4-VA on Mo2C@MoS2 was in accordance with the visible decrease of 4-NS and the quick emergence of 4-VA in the HPLC chromatogram (Fig. S14–S16, ESI†). The by-product of 4-ENB, derived from the hydrogenation of the vinyl group, was limited to a low level, confirming the highly selective hydrogenation of the nitro group due to the vertical chemisorption. Moreover, Mo2C@MoS2 showed stability in repeated ECH tests (Fig. 3d). The slight decrease of 4-VA yield in each cycle should be ascribed to the inevitable loss of the catalyst in the processes of cleaning. The post-test characterization studies (e.g., TEM, XRD and XPS) identified the negligible change of both hierarchical nanostructures and surface states (insets of Fig. 3d, S17 and S18, ESI†). In addition, the series of Mo2C@MoS2 obtained with the varied feeding ratio showed enhanced activity with the increased amount of thiourea (Fig. S19, ESI†), probably thanks to the enriched interfaces.
In order to detect the active species of ECH, quasi in situ electron paramagnetic resonance (EPR) spectroscopy coupled with electrochemical tests was conducted with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the trapping agent (Fig. 3e). The characteristic EPR signals of DMPO-H adducts (nine peaks, αN = 16.5 G and αH = 22.6 G) were visible at −0.45 V vs. RHE, suggesting that the active H* species was electrochemically generated via water reduction.39 Accordingly, when tert-butanol (t-BuOH) was introduced as a hydrogen radical scavenger, the conversion of 4-NS decreased significantly (Fig. 3f and S20, ESI†), confirming H* as the key intermediate.42 Furthermore, the kinetic isotopic effect (KIE) was tested for both the HER and ECH on Mo2C@MoS2, in terms of the ratio of current densities within H2O and D2O at −0.45 V vs. RHE (Fig. S21, ESI†). Mo2C@MoS2 showed a value of 1.92 for the HER, higher than that for ECH (1.52) (Fig. 3g). It's suggested that the H* is not the only determinant in the ECH. Meanwhile, the productivity of 4-VA on Mo2C@MoS2 presented a positively logarithmical correlation with the initial 4-NS concentration (Fig. S22, ESI†), and the CVs of 4-NS showed a good linear correlation of anodic peak currents with scan rates (Fig. S23, ESI†), which together confirmed the nonnegligible contribution of 4-NS*.16,43 The strengthened 4-NS* adsorption on Mo2C@MoS2 was evidenced by the more significant increase in open-circuit potential (73.1 mV) after injecting 4-NS into the blank electrolyte (Fig. S24, ESI†), as compared with those observed on Mo2C (0.3 mV) and MoS2 (26.5 mV). This experimental result consistent with our theoretical prediction (Fig. 1b) indicated the effective chemisorption and activation of the nitro group. According to the typical L–H mechanism, the conversion of 4-NS to 4-VA requires the co-participation of both 4-NS* and H*, which thereby highly depends on the competitive adsorption of these two substrates. A strong 4-NS* adsorption will inevitably weaken one of the H* and vice versa. As verified by the above experimental and theoretical results, the Mo2C–MoS2 interface is conducive to excellent catalytic performance because of its capability of balancing the competitive adsorption of H* and 4-NS*.
We collected in situ Raman spectra on Mo2C, Mo2C@MoS2 and MoS2 to understand their varied ECH performance (Fig. 4a–c). The initial spectra at 0 min exhibited two major signals at about 1350 and 1600 cm−1 attributing to the carbon matrix of Mo2C and Mo2C@MoS2 (Fig. S25, ESI†). The ECH of 4-NS occurred as reaction time went on, and accordingly new bands appearing at 1100, 1200, 1400, 1450, and 1150 cm−1 could be attributed to the stretching vibrations of C–N, N–O, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and benzene ring and the in-plane bending one of CCH, respectively (Table S3, ESI†).44,45 In detail, the band of C–N (1100 cm−1) was detected after 5–10 min on Mo2C and MoS2, and its progressive enhancement on MoS2 suggested the accumulation of 4-NS due to the strong chemisorption of 4-NS* but insufficient *H for the ECH. Owing to the relatively weaker binding with 4-NS* on Mo2C, the C–N band increased slowly. Besides, the others assigned to CCH, N–O, NO and benzene ring were due to the presence of intermediates, such as phenylhydroxylamine and nitroso benzene. By contrast, there were nearly no signals of 4-NS and intermediates detected on Mo2C@MoS2 before 60 min, confirming the rapid surface elementary steps of ECH involving the co-presented H* and 4-NS* on Mo2C–MoS2 interfaces.
O and benzene ring and the in-plane bending one of CCH, respectively (Table S3, ESI†).44,45 In detail, the band of C–N (1100 cm−1) was detected after 5–10 min on Mo2C and MoS2, and its progressive enhancement on MoS2 suggested the accumulation of 4-NS due to the strong chemisorption of 4-NS* but insufficient *H for the ECH. Owing to the relatively weaker binding with 4-NS* on Mo2C, the C–N band increased slowly. Besides, the others assigned to CCH, N–O, NO and benzene ring were due to the presence of intermediates, such as phenylhydroxylamine and nitroso benzene. By contrast, there were nearly no signals of 4-NS and intermediates detected on Mo2C@MoS2 before 60 min, confirming the rapid surface elementary steps of ECH involving the co-presented H* and 4-NS* on Mo2C–MoS2 interfaces.
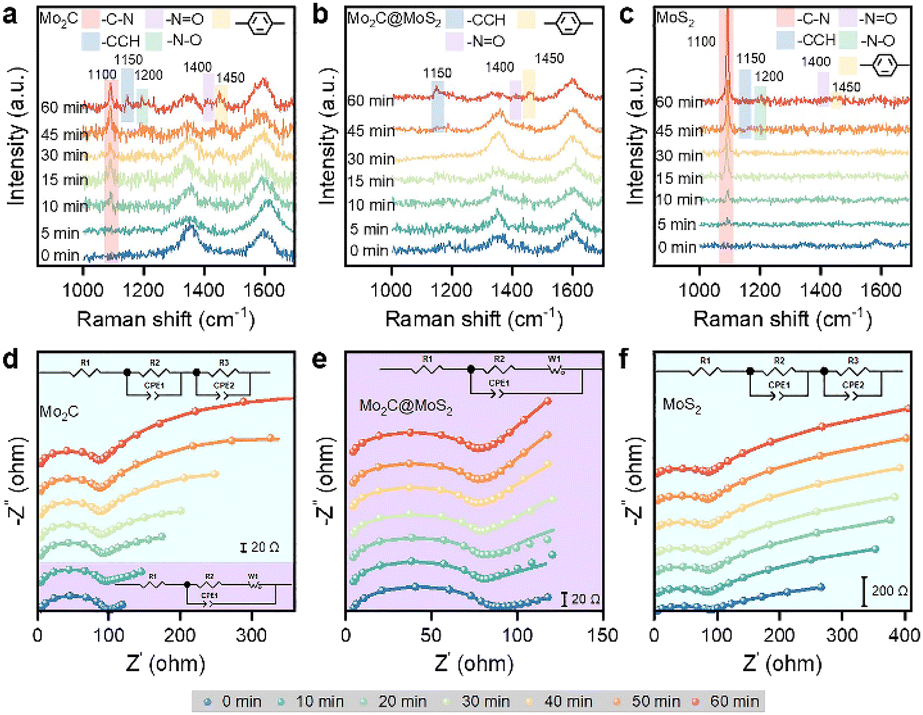 | ||
| Fig. 4 (a–c) In situ Raman spectra and (d–f) Nyquist plots collected on (a and d) Mo2C, (b and e) Mo2C@MoS2, and (c and f) MoS2 at −0.45 V vs. RHE. | ||
Quasi in situ EIS, an efficient technique for identifying reaction interfaces, was performed at intervals during the ECH (Fig. 4d, e and S26, ESI†). The series resistance (Rs) was consistent over Mo2C, Mo2C@MoS2 and MoS2 (Table S4, ESI†), but the configurations at low frequencies were quite different, which were well-fitted with the varied equivalent circuit diagram. It's shown that the kinetic behavior of Mo2C has changed from infinite diffusion to multiple interface diffusion after 20 min and MoS2 kept the feature of multiple interface diffusion. In sharp comparison, Mo2C@MoS2 showed kinetic behavior approximating infinite diffusion, consistent with the result of in situ Raman spectroscopy. In other words, the consistent interfacial charge transfer on Mo2C@MoS2 is ascribed to the quickly refreshed active surface; however, those of Mo2C and MoS2 suffer from an extra interfacial impedance owing to reactant/intermediate accumulation on their surfaces.
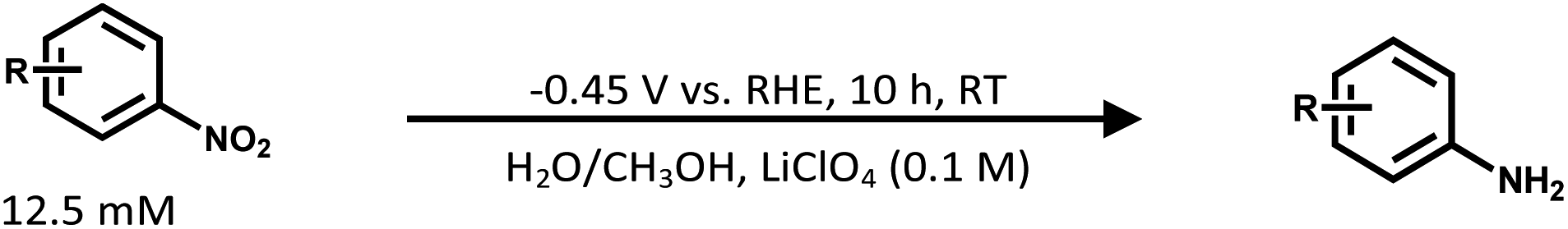
We further examined the efficiency of Mo2C@MoS2 in wide substrate scope (Table 1) and analyzed the products by 1H NMR (Fig. S27–S46, ESI†). For meta (m) and para (p) -halogenated nitrobenzenes (X = I, Br, Cl), Mo2C@MoS2 exhibited excellent performance with high FE (84–99%) and considerable yield (72–96%) of target halogenated anilines, in which the by-products of hydro-dehalogenation and H2 were rarely detected. This demonstrated the extraordinary tolerance for halogen functional groups. Moreover, it achieved the excellent efficacy of ECH for nitroarenes with a methoxy group, keeping the high FE (∼99%) and yield (88–92%) of target anilines. These results verified the promise of Mo2C@MoS2 for the ECH of nitroarenes.
|
|
||
|---|---|---|
| RCl |
FE 90% yield 88% (m) | FE 87% yield 72% (p) |
| RBr |
FE 84% yield 80% (m) | FE 86% yield 82% (p) |
| RI |
FE 99% yield 96% (m) | FE 99% yield 84% (p) |
| ROCH3 |
FE 99% yield 92% (m) | FE 99% yield 88% (p) |
Conclusions
In summary, Mo2C@MoS2 heteronanorods with dual sites were proposed to enable the efficient ECH of nitroarenes on synergized interfaces. As predicated using DFT calculations, the Mo2C–MoS2 interface can strengthen the binding of H* on Mo2C and 4-NS* on nearby MoS2, conducive for the successive hydrogenation toward the corresponding anilines. Mo2C@MoS2 achieved the high FE (>85%), yield (>78%) and selectivity (>99%) of 4-NS to value-added 4-VA, superior to Mo2C and MoS2. Accordingly, in situ Raman spectroscopy and quasi in situ EIS confirmed the facilitated chemisorption and further hydrogenation of 4-NS on Mo2C–MoS2 interfaces owing to the synergistically strengthened binding. Moreover, Mo2C@MoS2 delivered high efficiency for the ECH of nitroarenes to produce functional anilines in wide substrate scope. This work will provide new opportunities to develop high-performance electrocatalysts via the rational engineering of nanostructures and interfaces. Nevertheless, there still existed some challenges for ECH in comparison with thermo-catalysis, such as relatively lower space-time yield and immature reactor design. Drawing lessons from the mature experience of thermal-catalysis will expedite further progress in ECH.Data availability
Data supporting the findings of this study are available within the article ESI.†Author contributions
Wanling Zhang: synthesis, investigations, formal analysis, writing of the original draft, data curation. Wenbiao Zhang: writing-review and editing, DFT calculations, supervision. Kun Yu: experimental analysis, image polish. Jingwen Tan: proofreading of the original draft. Yi Tang: conceptualization, supervision, funding acquisition. Qingsheng Gao: conceptualization, supervision, writing-review and editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFA0209402), National Natural Science Foundation of China (grant No. 22175077), Innovation Team Project in Guangdong Colleges and Universities (2021KCXTD009), Guangzhou Science and Technology Project (202201020071), and Fundamental Research Funds for the Central Universities (No. 21623103).Notes and references
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- G. Papanikolaou, G. Centi, S. Perathoner and P. Lanzafame, ACS Catal., 2022, 12, 2861–2876 CrossRef CAS PubMed.
- C. Tang, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- C. Liu, Y. Wu, B. Zhao and B. Zhang, Acc. Chem. Res., 2023, 56, 1872–1883 CrossRef CAS PubMed.
- C. Han, J. Zenner, J. Johny, N. Kaeffer, A. Bordet and W. Leitner, Nat. Catal., 2022, 5, 1110–1119 CrossRef CAS.
- S. A. Akhade, N. Singh and O. Y. Gutiérrez, et al. , Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS PubMed.
- L. Zhang, T. U. Rao, J. Wang, D. Ren, S. Sirisommboonchai, C. Choi, H. Machida, Z. Huo and K. Norinaga, Fuel Process. Technol., 2022, 226, 107097 CrossRef CAS.
- P. Zhou, L. Li, V. S. S. Mosali, Y. Chen, P. Luan, Q. Gu, D. R. Turner, L. Huang and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202117809 CrossRef CAS PubMed.
- J. T. Kleinhaus, J. Wolf, K. Pellumbi, L. Wickert, S. C. Viswanathan, K. junge Puring, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2023, 52, 7305–7332 RSC.
- J. Song, Z.-F. Huang, L. Pan, K. Li, X. Zhang, L. Wang and J.-J. Zou, Appl. Catal., B, 2018, 227, 386–408 CrossRef CAS.
- H. Jin, P. Li, P. Cui, J. Shi, W. Zhou, X. Yu, W. Song and C. Cao, Nat. Commun., 2022, 13, 723 CrossRef CAS PubMed.
- Y. Ren, Y. Tang and L. Zhang, et al. , Nat. Commun., 2019, 10, 4500 CrossRef PubMed.
- P. Deng, J. Duan and F. Liu, et al. , Angew. Chem., Int. Ed., 2023, 62, e202307853 CrossRef CAS PubMed.
- G. Liu, C. Chen and J. Chen, J. Phys. Chem. C, 2023, 127, 4375–4386 CrossRef CAS.
- R. Xia, D. Tian, S. Kattel, B. Hasa, H. Shin, X. Ma, J. G. Chen and F. Jiao, Nat. Commun., 2021, 12, 1949 CrossRef CAS PubMed.
- J. Tan, J. Shao, Y. Shi, W. Zhang and Q. Gao, ACS Sustain. Chem. Eng., 2022, 10, 13525–13533 CrossRef CAS.
- J. A. Lopez-Ruiz, E. Andrews and S. A. Akhade, et al. , ACS Catal., 2019, 9, 9964–9972 CrossRef CAS.
- J. Tan, W. Zhang, Y. Shu, H. Lu, Y. Tang and Q. Gao, Sci. Bull., 2021, 66, 1003–1012 CrossRef CAS PubMed.
- J. Anibal and B. Xu, ACS Catal., 2020, 10, 11643–11653 CrossRef CAS.
- C. Liu, R. Li and W. Zhou, et al. , ACS Catal., 2021, 11, 8958–8967 CrossRef CAS.
- U. Sanyal, S. F. Yuk and K. Koh, et al. , Angew. Chem., Int. Ed., 2021, 60, 290–296 CrossRef CAS PubMed.
- H. Wu, J. Song, C. Xie, Y. Hu, P. Zhang, G. Yang and B. Han, Chem. Sci., 2019, 10, 1754–1759 RSC.
- S. Huang, B. Gong, Y. Jin, P. H.-L. Sit and J. C.-H. Lam, ACS Catal., 2022, 12, 11340–11354 CrossRef CAS.
- S. Wang, D. Zhang, B. Li, C. Zhang, Z. Du, H. Yin, X. Bi and S. Yang, Adv. Energy Mater., 2018, 8, 1801345 CrossRef.
- J. Liu, Y. Zheng, D. Zhu, A. Vasileff, T. Ling and S.-Z. Qiao, Nanoscale, 2017, 9, 16616–16621 RSC.
- T. Yang, T. T. Song, J. Zhou, S. Wang, D. Chi, L. Shen, M. Yang and Y. P. Feng, Nano Energy, 2020, 68, 104304 CrossRef CAS.
- Z. Shi, X. Zhang and X. Lin, et al. , Nature, 2023, 621, 300–305 CrossRef CAS PubMed.
- K. Pellumbi, L. Wickert and J. T. Kleinhaus, et al. , Chem. Sci., 2022, 13, 12461–12468 RSC.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef PubMed.
- L. Lin, W. Zhou and R. Gao, et al. , Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- X. Fan, C. Liu and M. Wu, et al. , Appl. Catal., B, 2022, 318, 121867 CrossRef CAS.
- H. Lin, Z. Shi, S. He, X. Yu, S. Wang, Q. Gao and Y. Tang, Chem. Sci., 2016, 7, 3399–3405 RSC.
- B. Q. Lv, H. M. Weng and B. B. Fu, et al. , Phys. Rev. X, 2015, 5, 031013 Search PubMed.
- Y. Sun, A. J. Darling, Y. Li, K. Fujisawa, C. F. Holder, H. Liu, M. J. Janik, M. Terrones and R. E. Schaak, Chem. Sci., 2019, 10, 10310–10317 RSC.
- Y. Zhao, C. Liu, C. Wang, X. Chong and B. Zhang, CCS Chem., 2021, 3, 507–515 CrossRef CAS.
- H. Li, Y. Gao, Y. Wu, C. Liu, C. Cheng, F. Chen, Y. Shi and B. Zhang, J. Am. Chem. Soc., 2022, 144, 19456–19465 CrossRef CAS PubMed.
- Y. Shu, L. Zhang, H. Cai, Y. Yang, J. Zeng, D. Ma and Q. Gao, Sens. Actuators, B, 2020, 311, 127863 CrossRef CAS.
- J. M. Mayer, J. Am. Chem. Soc., 2023, 145, 7050–7064 CrossRef CAS PubMed.
- W. Zhang, W. Zhang, J. Tan, D. Pan, Y. Tang and Q. Gao, J. Mater. Chem. A, 2023, 11, 7505–7512 RSC.
- X. Chong, C. Liu, Y. Huang, C. Huang and B. Zhang, Natl. Sci. Rev., 2020, 7, 285–295 CrossRef CAS PubMed.
- S. Wu, X. Huang, H. Zhang, Z. Wei and M. Wang, ACS Catal., 2021, 12, 58–65 CrossRef.
- M. Li, C. Liu, Y. Huang, S. Han and B. Zhang, Chin. J. Catal., 2021, 42, 1983–1991 CrossRef CAS.
- Y. Zheng, Z. Wang, P. Chen, W. Zhang and Q. Gao, ChemSusChem, 2023, 16, e202300180 CrossRef CAS PubMed.
- J. Ma, Z. Wang, T. Majima and G. Zhao, ACS Catal., 2022, 12, 14062–14071 CrossRef CAS.
- X.-M. Zhu, S.-Q. Zhang, X. Zheng and D. L. Phillips, J. Phys. Chem. A, 2005, 109, 3086–3093 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and data for degrader evaluation, EDS mapping, additional XRD, XPS and CVs, and details of NMR spectra. See DOI: https://doi.org/10.1039/d3sc06010a |
| This journal is © The Royal Society of Chemistry 2024 |