
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRGB tri-luminescence in organic–inorganic zirconium halide perovskites†
Chuying
Wang
a,
Wen
Meng
a,
Guigen
Luo
a,
Guangyong
Xu
a,
Min
Peng
a,
Bin
Xu
a,
Shuming
Nie
 b and
Zhengtao
Deng
*a
b and
Zhengtao
Deng
*a
aCollege of Engineering and Applied Sciences, State Key Laboratory of Analytical Chemistry for Life Science, National Laboratory of Micro-structures, Nanjing University, Nanjing, Jiangsu 210023, P. R. China. E-mail: dengz@nju.edu.cn
bDepartments of Bioengineering, Chemistry, Electrical and Computer Engineering, and Materials Science and Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
First published on 15th January 2024
Abstract
Materials with two or more fluorescence features under different excitation sources have great potential in optical applications, but luminous materials with three emission characteristics have been largely undeveloped. Here, we report a novel zero-dimensional (0D) organic–inorganic hybrid ((C2H5)4N)2ZrCl6 perovskite with multiple emissions. The zirconium-based perovskite exhibits a red emission around 620 nm, a green emission at 527 nm, and a blue emission around 500 nm. The red and green emissions come from self-trapped excitons (STEs) and the d–d transitions of Zr(IV), respectively, which are caused by distortion of the [ZrCl6]2− octahedra. The blue emission is caused by thermally activated delayed fluorescence (TADF), which is similar to that of Cs2ZrCl6. The absolute photoluminescence quantum yield (PLQY) of the red and blue double emission is up to 83% and the PLQY of the green emission is 27%. With different combinations of ((C2H5)4N)2ZrCl6 samples, we achieve a variety of applications, including a two-color luminescent anti-counterfeiting device, a white light-emitting diode (WLED) with a color rendering index (CRI) of 95 and information encryption with different excitations. We also synthesize other hybrid zirconium perovskites with tri-luminescence through a similar method. Our work provides a potential set of excitation-dependent luminescent materials and is expected to expand the basic research and practical applications of multi-luminescence materials.
Introduction
Materials with multi-excitonic emission within a single structure have attracted much attention due to their properties in optoelectronic applications,1 such as imaging,2 multi-excitation anti-counterfeiting3 and white light emitting diodes (WLED).4–10 For instance, multi-excitonic emission, originating from multiple types of excitons within one phosphor, achieves broadband emission for WLEDs with a high colour rendering index (CRI). Broadband emission through multi-excitonic emission within a single structure can be obtained in two main ways. One approach is to achieve multi-luminescence through structure, which has been applied in various lead-free halides.11–14 By applying (TBA)2Cu2I4 as a single phosphor, a WLED with a CRI of 89 and low correlated color temperature (CCT) of 4516 K was fabricated.11 The other way is by adding a luminescent dopant to the host.15,16 In particular, Bi3+, Sb3+, and Te4+ can be doped into A2BX6 perovskite structures, providing additional luminescence energy levels.3,17–22 Among such materials, Cs2ZrCl6:1.5%Sb has been used in a stable single-component WLED with a record half-lifetime of 2003 h.19 Co-doped Cs2HfCl6:Bi3+/Te4+ microcrystals show warm-white emission and were applied to a WLED with a CRI of 75.4 and CIE colour coordinate of (0.370, 0.393).23As a typical group of A2BX6 perovskites, Zr-based halide zero-dimensional (0D) perovskites have been researched as both hosts and luminescence centers for lighting applications.17–21,24–30 Emission from Zr-based perovskites mainly arises via three pathways. The first is through thermally activated delayed fluorescence (TADF). TADF was observed in Cs2ZrCl6, in which the photoluminescence and lifetime exhibited a temperature dependence.25 Typically, Cs2ZrCl6 exhibits a broad blue emission centered at 450 nm with a large Stokes shift of up to 200 nm. The large Stokes shift and broadband emission suggest that the photoluminescence (PL) may originate from host self-trapped excitons (STEs) caused by the distortion of the [ZrCl6]2− octahedron. Thus, the emission is also called STE emission.25,28–31 As the blue fluorescence exhibit thermal activation delayed nature, we call the broad blue emission through the first pathway TADF to distinguish it. The second pathway is through extrinsic STE emission, also exhibiting a broadband emission, which has been reported in research on Hf-based 0D perovskites.32–34 Recently, another emission mode was attributed to the spin-allowed d–d transitions of Zr4+ in the green emission region.35 The d–d transition has only been reported for 0D organic–inorganic hybrid Zr-based perovskites, which is possibly due to the distortion between the [ZrCl6]2− octahedra, similar to [MnX6]4− complexes.36 Thus, organic–inorganic hybrid Zr-based perovskites with distorted structures are potential multiple-emission materials. Recently, organic–inorganic Zr-based perovskites have been explored as potential materials for X-ray imaging and anticounterfeiting with different dopants.37 However, the multiple emissions in zirconium perovskites are aroused by other dopant ions, such as Sb3+,21 Bi3+ and Te4+.17,33 Controllable synthesis and the multi-emission nature of zirconium perovskites still require further research.
Herein, we apply organic cations to synthesize 0D organic–inorganic hybrid ((C2H5)4N)2ZrCl6 perovskite samples. The ((C2H5)4N)2ZrCl6 samples exhibit tri-luminescence: blue emission around 500 nm and red emission at 620 nm through STE emission and green emission at 527 nm through the d–d transitions of Zr(IV). The ((C2H5)4N)2ZrCl6 crystallizes in the monoclinic phase with distorted [ZrCl6]2− octahedra, leading to STE emission and d–d transitions in the samples. Compared with the inorganic Cs2ZrCl6, the organic–inorganic hybrid ((C2H5)4N)2ZrCl6 exhibits multiple emissions, which enables different luminescence applications without introducing other rare-earth materials. By controlling the reaction conditions, the emission behaviour of ((C2H5)4N)2ZrCl6 can be adjusted. Various applications, such as anti-counterfeiting, WLEDs and information encryption, are achieved through different combinations of the ((C2H5)4N)2ZrCl6 samples in this work.
Results and discussion
The ((C2H5)4N)2ZrCl6 samples exhibit excitation-dependent multiple emissions, as shown in Fig. 1. The absorption spectrum of ((C2H5)4N)2ZrCl6 single crystals exhibits a sharp peak at 261 nm, with shoulders around 310 nm and 380 nm (Fig. 1a), suggesting the existence of multiple excited states. The absorption spectra of ((C2H5)4N)2ZrCl6 powder and single-crystal samples show similar peak positions, but the relative intensity of each peak is different. Compared with the overall excitation in Fig. S1† and Fig. 1c, the overall excitation spectra and absorption spectra both exhibit a strong peak at 261 nm. The excitation at 261 nm is mainly linked to the blue emission at 500 nm, meaning that the absorption at 261 nm can effectively excite the blue emission part. Thus, the absorption at 261 nm is related to the blue emission part. Similarly, the second shoulder around 310 nm is related to both the green and red emission parts. The third shoulder around 380 nm is related to the red emission part.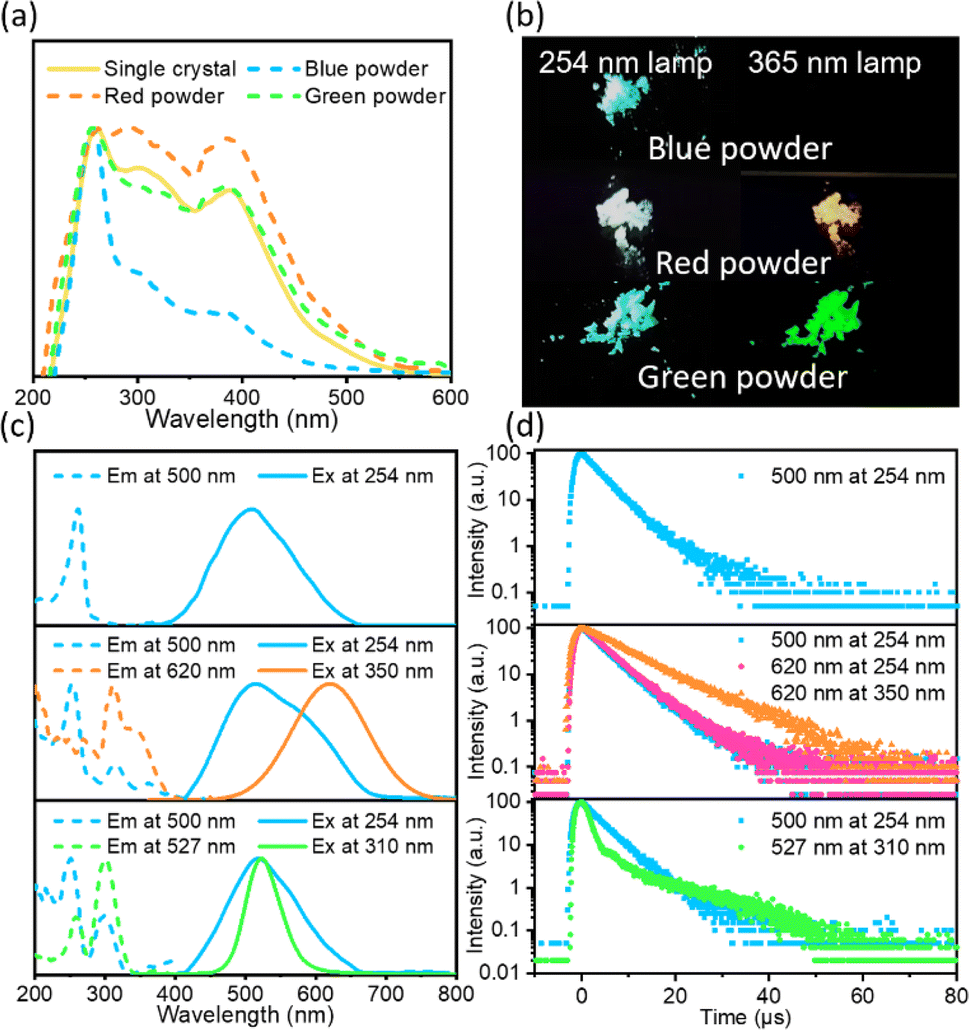 | ||
| Fig. 1 Optical properties of ((C2H5)4N)2ZrCl6 samples. (a) Absorption spectra of ((C2H5)4N)2ZrCl6 single crystal, blue powder, red powder and green powder. (b) Photographs, (c) photoluminescence (PL) emission spectra and photoluminescence excitation (PLE) spectra and (d) time-resolved photoluminescence spectra of ((C2H5)4N)2ZrCl6 powder samples. Samples only exhibiting obvious blue emission are defined as blue powder; samples exhibiting white and orange-red emission are defined as red powder; samples exhibiting blue and green emission are defined as green powder. | ||
The emission behaviour of the ((C2H5)4N)2ZrCl6 powders can be adjusted through the reaction conditions. Here, we define the main three groups of powder samples in Fig. 1b and Fig. S2:†: a sample with blue emission under a 254 nm lamp and almost no emission under a 365 nm lamp is defined as blue powder; a sample with white emission under a 254 nm lamp and orange-red emission under a 365 nm lamp is defined as red powder; a sample with blue emission under a 254 nm lamp and green emission under a 365 nm lamp is defined as green powder. As shown in Fig. 1c, the blue powder exhibits an emission peak around 500 nm with a wide full width at half maximum (FWHM) of 131 nm under irradiation at 254 nm. This broad emission band with a large Stokes shift of 239 nm has a strong excitation band at around 260 nm, which is similar to previous results for Cs2ZrCl6.25,29 For the blue emission from blue powder, the PL decay spectrum exhibits a PL decay lifetime of 3.9 μs at 300 K (Fig. 1d and Table S1†). With the temperature decreasing to 273 K and 77 K, the average PL decay lifetime of the ((C2H5)4N)2ZrCl6 blue powder increases to 4.9 μs and 146.7 μs, respectively (Fig. S3 and Table S1†). The changes in lifetime involve reverse intersystem crossing (RISC) from long-lived triplet excited states back to short-lived singlet excited states, validating a TADF nature according to previous research on the blue emission of [ZrCl6]2− perovskites.25,29,30
The red powder exhibits a wide emission under a 254 nm lamp. The wide emission originates from a blue emission around 500 nm and a red emission around 620 nm (Fig. S4†). With decreasing excitation energy, the blue emission gradually disappears and the sample exhibits a single emission peak at 620 nm with a FWHM of 124 nm (Fig. 1c). Interestingly, the blue emission of the blue powder can only be effectively excited in the range of 220∼280 nm, while the blue emission of the red powder can be excited at lower energy. The blue emission state in the red powder sample is related to the red emission state. For the blue emission band, the PL decay spectrum exhibits an average lifetime of 5.5 μs (τ1 = 4.6 μs, 96%; τ2 = 28.1 μs, 4%) for excitation under 254 nm light (Fig. 1d and Table S1†) owing to the TADF state. For the red powder, the FWHM of the red emission band decreased from 124 nm to 120 nm with the temperature decreasing from 300 K to 273 K (Fig. S5†). The PL position shifts slightly from 620 nm at 300 K to 619 nm at 273 K. This phenomenon is similar to the STE emission of ((CH3)4N)2ZrCl6 in previous research.35 In addition, the PL decay spectrum exhibits a lifetime of 4.9 μs to 8.7 μs with excitation from 254 nm to 350 nm (Fig. S6 and Table S1†). The extension of the lifetime is attributed to STE emission showing an excitation-energy-dependent property.38 Based on these results, the red emission is attributed to STEs.
As displayed in Fig. 1c, the green powder exhibits a broad blue emission under irradiation by a 254 nm lamp and a narrow green emission under a 365 nm lamp (Fig. S7†). The narrow green emission peak centred at 527 nm with a FWHM of 47 nm (Fig. 1c) has not been reported for zirconium halide complexes yet. For the blue emission, the PL decay spectrum exhibits an average lifetime of 4.7 μs for excitation under 254 nm light (Fig. 1d and Table S1†) owing to a TADF state. For the green emission band, the PL decay spectrum exhibits a shorter lifetime τ1 = 1.6 μs (71%) and a longer lifetime τ2 = 19.7 μs (29%) for excitation under 310 nm light. The shorter lifetime is owing to non-radiative recombination and the longer lifetime is ascribed to the d–d transitions of Zr(IV).35 The blue and green emission bands of the green powder both exhibit strong excitation around 250 nm and 300 nm. The similar excitation spectra may originate from the overlap in the blue and green emission. Thus, a three-dimensional excitation–emission matrix (EEM) fluorescence spectrum is necessary.
For better understanding of the multi-emissive behaviour of the ((C2H5)4N)2ZrCl6 powders, the EEM fluorescence spectra of the red and green powders are displayed in Fig. 2. Different from the narrower excitation range (220–280 nm) of the blue emission of the blue powder, the blue emission of the red powder can be excited at lower energy, with a wavelength of 340 nm (Fig. 2a and c). The red sample exhibits double-peak emission around 500 nm and 620 nm with excitation from 250 to 340 nm. With the excitation energy decreasing below that at 340 nm, the red sample exhibits a single broad emission peak around 620 nm. The emission peak position did not shift with the change of the excitation wavelength, which means that the red emission is not generated by a defect state. The emission at 620 nm, with a large Stokes shift of 270 nm and broad FWHM of 124 nm, is attributed to STE emission due to lattice distortions.35,39 As shown in Fig. 2a and c, the blue emission of the red sample dominates the luminescence under 254 nm light. The lifetime of the blue emission of the red powder is similar to that of the blue and green powder (Table S1†). Under excitation at 310 nm and 320 nm, the red sample exhibits a double peak at 500 nm and 620 nm. Both the shorter lifetime τ1 and the longer lifetime τ2 decrease, and the ratio of the longer τ2 increases. Under excitation at 340 nm, the blue luminescence part is relatively weak compared with the red part. Both τ1 and τ2 exhibit a decreasing trend. Under 350 nm light, the red powder only exhibits a single emission peak at 620 nm. The decrease in τ1 and τ2 for the blue emission accompanied with the appearance of the red emission suggests energy transfer between the two states. The absolute photoluminescence quantum yield (PLQY) of the double-peak emission is up to 83% at 270 nm and for the single red emission it is up to 14% at 340 nm (Fig. 2c).
Unlike the relation between the blue and red emissions of the red powder sample, the blue and green emissions of the green powder are relatively independent. Although the PLE spectra of the blue and green emissions are similar (Fig. 1c), the EEM spectra in Fig. 2b and d imply that the two emission groups belong to two independent pathways. The blue emission part for the green powder can only be excited with energy higher than that at 280 nm, similar to the blue powder. Besides the blue emission, a new emission at 527 nm appears with excitation from 265 nm to 340 nm (Fig. S8†). The absolute PLQY of the blue emission is up to 57% at 250 nm and for the green emission it is up to 27% at 295 nm (Fig. 2f). The green emission under a 365 nm lamp is stimulated by the broad emission of the source (Fig. S7†). The broad peak enables us to observe the green emission at 527 nm, which has not yet been discovered in previous research on Zr-based halide 0D perovskites. As the driving voltage increases from 3 V to 12 V, the PL peak positions for both the green and red powders show no obvious shift. The green powder exhibits luminescence at 527 nm, while the red powder exhibits a double peak around 500 nm and 620 nm (Fig. S9 and S10†). The PL intensity of both samples shows a linear dependence on the normalized excitation power. This phenomenon may exclude the possibility that the additional luminescence peak originates from permanent defects.12,40,41 In addition, the red emission observed for other hybrid Zr-based perovskites has been identified as STE emission in previous research.35
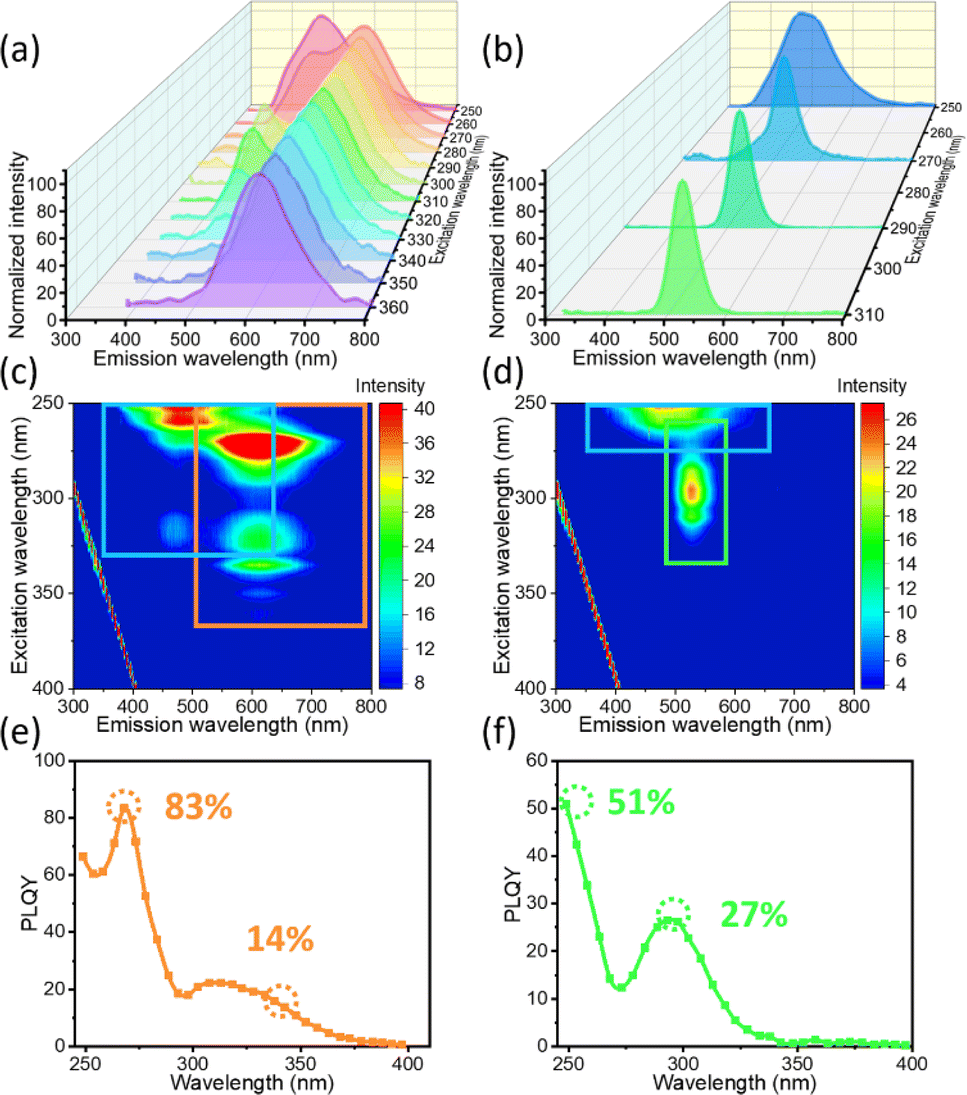 | ||
| Fig. 2 Luminescence properties of red and green ((C2H5)4N)2ZrCl6 powder samples. PL emission spectra under different excitations for (a) red and (b) green ((C2H5)4N)2ZrCl6 powders. Three-dimensional excitation-emission matrix (EEM) fluorescence spectra for (c) red and (d) green ((C2H5)4N)2ZrCl6 powders. PLQY under various excitation wavelengths for (e) red and (f) green powders. | ||
((C2H5)4N)2ZrCl6 single crystals are grown via the evaporation method. It is worth noting that the absorption spectra (Fig. 1a) show that both the powder and the single crystals show multiple excited states with different relative intensities. By slowly evaporating the precursor solution, a single crystal is obtained, which is 4 mm × 3 mm × 1 mm in size, and transparent under room lighting (Fig. 4a). Scanning electron microscopy (SEM) imaging and energy dispersive X-ray spectroscopy (EDS) mappings show that the distribution shapes of the Cl, Zr and N elements are consistent with the single-crystal profile, indicating uniformity of the final product (Fig. S11 and S12†). Single-crystal X-ray diffraction (SCXRD) measurements showed that the ((C2H5)4N)2ZrCl6 single crystal adopted the monoclinic space group C2/c (no. 15). The crystal structure consists of two tetraethylammonium cations and a zirconium hexachloride(II) anion complex (Fig. 3a). In the ((C2H5)4N)2ZrCl6 crystals, each Zr4+ is coordinated with six Cl− ions to form [ZrCl6]2− octahedra, and each adjacent octahedron exhibits a different spatial orientation (Fig. S13†). The Zr–Cl1, Zr–Cl2 and Zr–Cl3 lengths are 2.457, 2.463 and 2.474 Å, respectively. The Cl1–Zr–Cl2, Cl1–Zr–Cl3 and Cl2–Zr–Cl3 angles in the octahedron are 90.55°, 90.39° and 89.50°, respectively (Fig. 3b). It is obvious that the [ZrCl6]2− octahedron is slightly deformed. Furthermore, the torsion between two neighbouring [ZrCl6]2− octahedra, evaluated through the longest Cl–Zr–Cl bond, is 14.49°, as displayed in Fig. 3c. In 0D systems, the STE property mainly depends on the distortion in a single octahedron.38,39,42 In a centrosymmetric octahedron, the d–d transition is forbidden by the Laporte selection rule, making the luminescence very weak.43 In previous research on Mn(II) complexes in the octahedral crystal field, the distortion between adjacent octahedra breaks the symmetry, leading to observable luminescence.36,44,45 Thus, the distortion of [ZrCl6]2− octahedra in the structure also enables the d–d transitions of Zr(IV). Therefore, structural distortion of [ZrCl6]2− octahedra leads to the multiple emission of the ((C2H5)4N)2ZrCl6 samples.
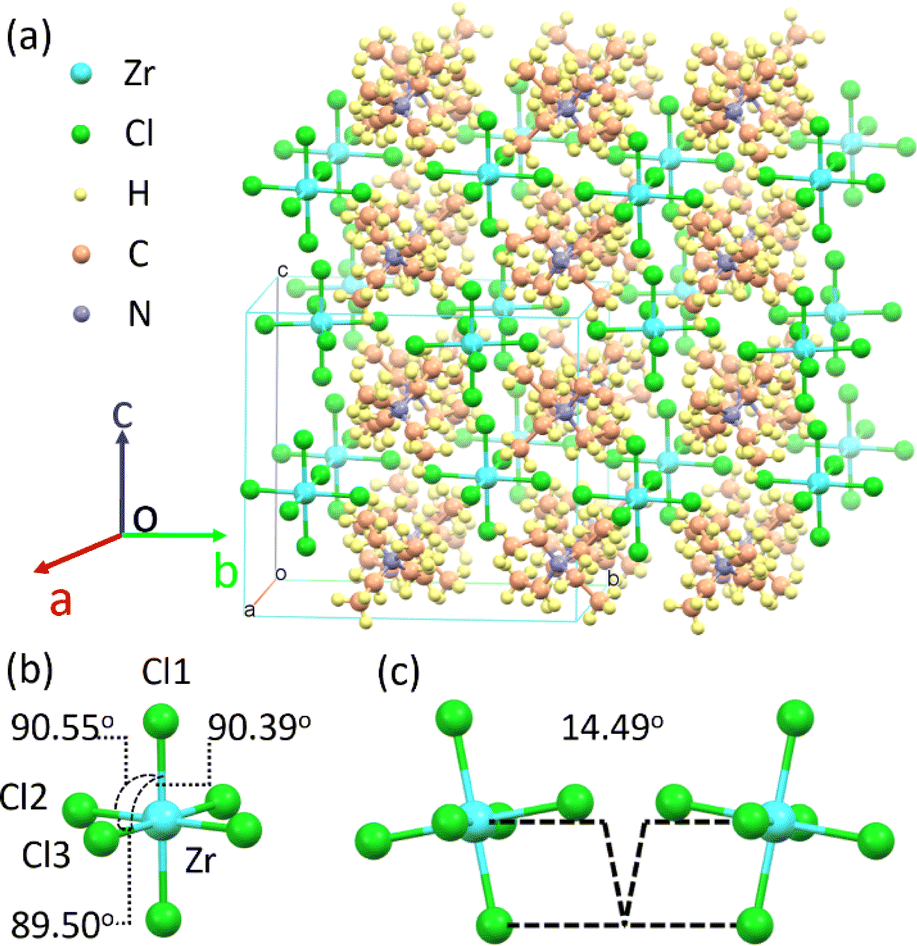 | ||
| Fig. 3 Crystal structure of ((C2H5)4N)2ZrCl6. (a) Detailed view of the crystal of ((C2H5)4N)2ZrCl6. (b) Detailed view of a [ZrCl6]2− octahedron. (c) Detailed view of adjacent [ZrCl6]2− octahedra of the ((C2H5)4N)2ZrCl6 sample. | ||
As displayed in Fig. 4a & b, ((C2H5)4N)2ZrCl6 single crystals exhibit blue luminescence under a 254 nm lamp, green luminescence under a 310 nm UV chip and weak red luminescence under 375 nm irradiation. The blue emission exhibits a peak around 500 nm and the weak red emission exhibits a peak around 620 nm. The ((C2H5)4N)2ZrCl6 single crystal exhibits yellow luminescence under a 365 nm lamp (Fig. S14†). In addition, ((C2H5)4N)2ZrCl6 single crystals exhibit weak broadband emission at lower excitation energy (Fig. S15†). We infer that energy transfer between the two states occurs in the single crystals. Unlike the green powder samples, the green luminescence of the single crystal exhibits split peaks centred at 498, 527, 550, and 576 nm (Fig. 4d), which are derived from the d–d transitions of Zr(IV).35,46 Emission through the d–d transitions of Zr(IV) has only been reported for 0D organic–inorganic hybrid Zr-based perovskites, and is due to the distortion of the [ZrCl6]2− octahedra, similar to [MnX6]4− complexes.36 Although the green powder exhibits a single emission peak under a 365 nm lamp, its peak shape fits well with the split peaks of the ((C2H5)4N)2ZrCl6 single crystal (Fig. 4d). We posit that the narrow green emission of the green powder is attributed to the d–d transitions of Zr(IV), which will be elucidated later. In addition, the XRD patterns of both the single crystal and powder samples match well with the simulated structure, as displayed in Fig. S16.† Briefly, there are three emission pathways for((C2H5)4N)2ZrCl6: TADF emission around 500 nm, STE emission around 620 nm and d–d transitions of Zr(IV) due to lattice distortion, as shown in Fig. 4e. Energy transfer occurs between the two STE emission states.
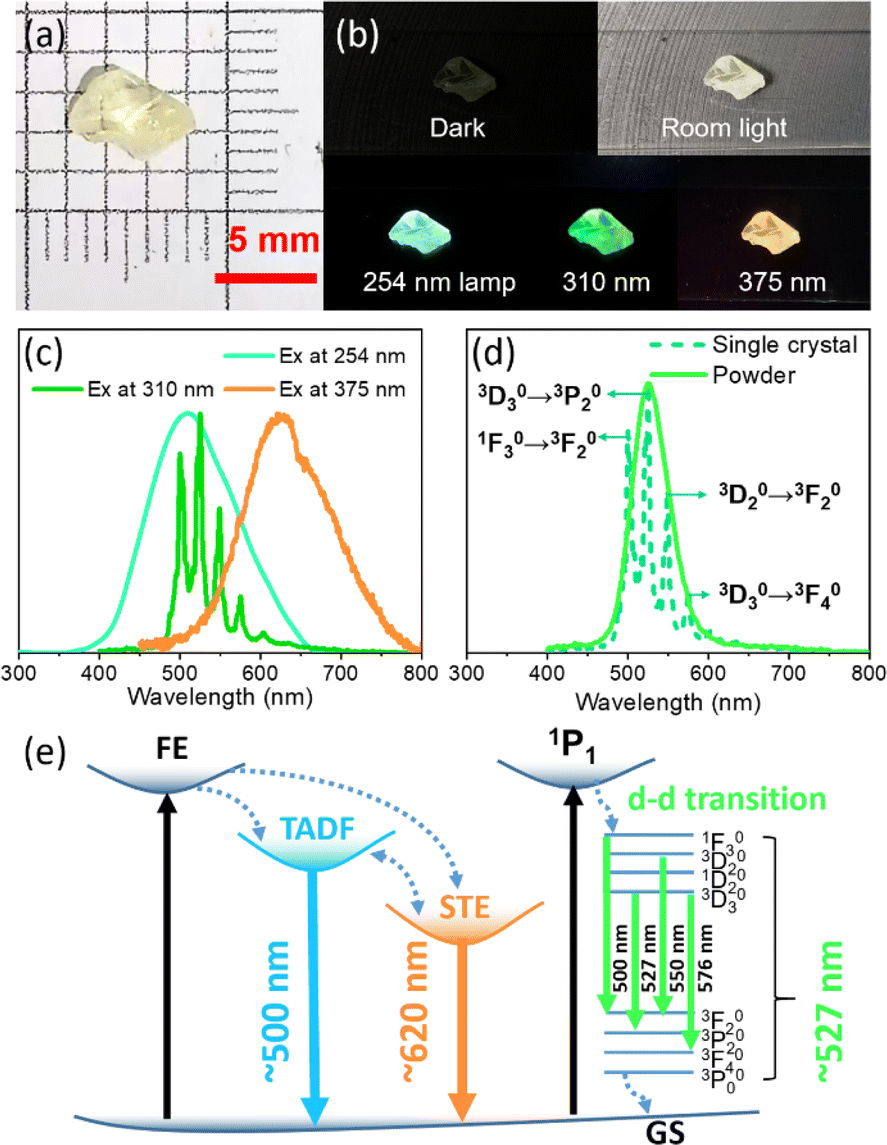 | ||
| Fig. 4 Luminescence and structural properties of ((C2H5)4N)2ZrCl6. Photographic images under (a) room lighting and (b) UV irradiation. (c) PL emission spectra of the ((C2H5)4N)2ZrCl6 single crystal under different excitations. (d) PL spectra of the single crystal and green powder under excitation at 310 nm. (e) Energy transfer diagram of FE, TADF, STE and d–d transition states. | ||
It is worth noting that all the ((C2H5)4N)2ZrCl6 samples, including different powder samples and single crystals, exhibit multiple excited states, but their relative intensities are different (Fig. 1a). As shown in Fig. S17,† the blue powder exhibits octahedral-shaped granules with sizes of ∼30 μm while the red and green powders exhibit smaller-sized granules. For the narrow green emission at 527 nm, we attribute it to the small-sized zirconium perovskite granules, which has not been reported in previous research on organic–inorganic hybrid zirconium perovskite single crystals.37 To confirm this hypothesis, we applied a top-down method to grind the ((C2H5)4N)2ZrCl6 single crystal to a powder with different luminescent properties. As shown in Fig. S18,† ((C2H5)4N)2ZrCl6 powder only exhibits blue emission under a 254 nm lamp after being ground once, while the ((C2H5)4N)2ZrCl6 powder ground twice to a smaller size exhibits blue emission under a 254 nm lamp and green emission under a 365 nm lamp. The green emission peak is located at 527 nm (Fig. S19†). We further apply ball milling to ((C2H5)4N)2ZrCl6 single crystals to fully pulverize the samples. As shown in Fig. S20,† the ((C2H5)4N)2ZrCl6 powders exhibit uniform blue and green emission under 254 nm and 365 nm lamps, respectively. The blue and green emissions are from the same areas (Fig. S21†). Therefore, we assume that the narrow green emission of the green powder is attributed to the d–d transitions of Zr(IV) and is obtained by controlling the grain size of ((C2H5)4N)2ZrCl6 powder.
For the red emission from the STE states, we find that the emission behaviour can be adjusted by humidity or water. After being stored at a relative humidity (RH) of 100% for one day, some of the green powder turns into red powder, forming yellow luminescence (Fig. S22†). By applying the powder on a 310 nm UV chip, blue, green and red luminescence are achieved simultaneously (Fig. S23†). This phenomenon is more obvious in another 0D organic–inorganic hybrid (C13H30N)2ZrCl6 perovskite sample. (C13H30N)2ZrCl6 exhibits similar multiple emissions around 510 nm, 660 nm and 527 nm (Fig. S24†). The red emission part attributed to STE gradually increases with increasing numbers of washes with water, as shown in Fig. S25.† The CIE chromaticity coordinate of the (C13H30N)2ZrCl6 sample changes from (0.38, 0.47) to (0.44, 0.43) after being washed twice.
Both the blue and green powders exhibit high stability in high-temperature and dry storage environments. As shown in Fig. S26,† after being stored in an oven at 80 °C for 60 days, the PLQY of the blue powder decreased slightly from 90% to 89% under excitation at 250 nm. The PLQY of the green powder remains at 27% under 300 nm irradiation. Compared with the blue and green powders, the red powder is less stable. After being stored in an oven for 60 days, the PLQY of the red powder decreases from 83% to 53% at 270 nm, from 22% to 12% at 315 nm and from 14% to 7% at 340 nm. Therefore, we think the red emission is related to humidity or water. However, the changes in structure need further study in future research.
The organic–inorganic hybrid ((C2H5)4N)2ZrCl6 perovskite powders exhibit multiple emissions and the emission tendency can be adjusted by the reaction conditions. Utilizing the unique luminescence properties, we explore various applications with the ((C2H5)4N)2ZrCl6 powders. First, the ((C2H5)4N)2ZrCl6 powders with multiple luminescence wavelengths under different excitation energies in one matrix, such as the red and green powder samples, are suitable for anti-counterfeiting security. Traditional anti-counterfeiting fluorescent materials exhibit monochromatic emission under different excitations, which make them easily counterfeited.47 However, simply mixing phosphors with different emissions may lead to inhomogeneous dispersion of the materials.48 Therefore, it is necessary to develop a single-matrix material exhibiting multiple emissions, which would be difficult to replicate and have a higher level of anti-counterfeiting security.21,27,49Fig. 5a shows that the ((C2H5)4N)2ZrCl6 green powder emits blue and green luminescence under different UV lamps, exhibiting great potential for application in the anti-counterfeiting field. Compared with previous 0D Zr-based perovskite anti-counterfeiting applications,3,21,27 the ((C2H5)4N)2ZrCl6 powders do not require the introduction of other rare-earth materials.
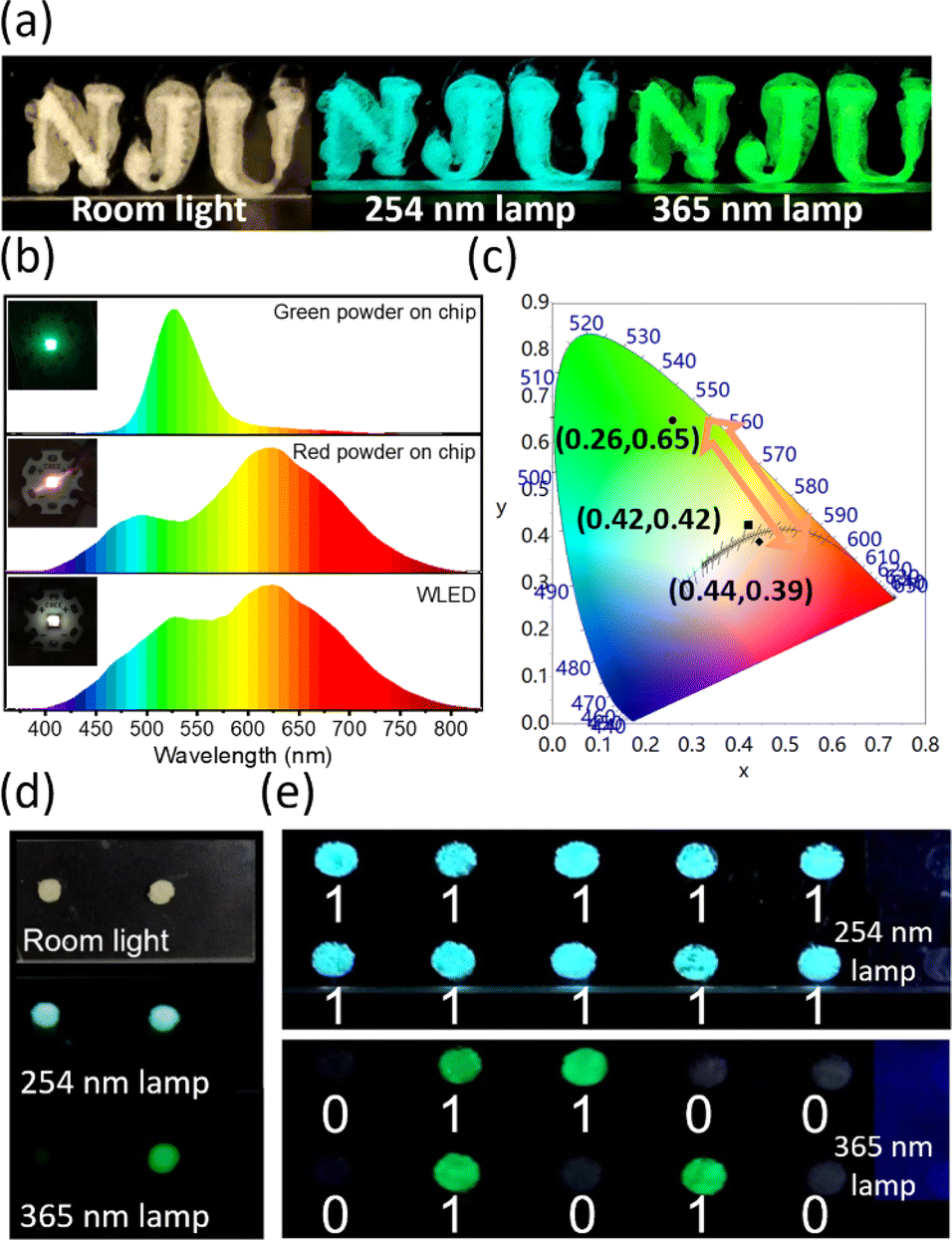 | ||
| Fig. 5 Applications of ((C2H5)4N)2ZrCl6 samples. (a) Photographs of an anti-counterfeiting pattern using the ((C2H5)4N)2ZrCl6 powder sample under different lights. (b) Emission spectra and (c) CIE chromaticity diagram of green, red and white LEDs through green, red and mixed samples on a UV chip (310 nm). The insets are the photographs of the corresponding LEDs. (d) Photographs of the green and blue powders under different irradiations. (e) Photographs of pixelated patterns of the green and blue powders under different irradiation conditions. | ||
Besides anti-counterfeiting, the ((C2H5)4N)2ZrCl6 powders can further be applied to UV-pumped WLEDs. The LEDs are fabricated by coating the ((C2H5)4N)2ZrCl6 powders on a UV chip (310 nm). Fig. 5b shows the spectra of the LEDs through different ((C2H5)4N)2ZrCl6 powders. The CIE chromaticity coordinates of the green LED and red LED are (0.26, 0.65) and (0.44, 0.39), respectively, at a driving voltage of 3 V. The CRI of the red LED reaches as high as 89 with a CCT of 2747 K. From the spectra, it is noted that the emission of the green LED can cover the absence of emission of the red LED in the range of 500 nm to 600 nm via excitation at 310 nm. Therefore, by mixing the green and red ((C2H5)4N)2ZrCl6 powders on a UV chip, a WLED is produced that can achieve a high CRI of up to 95 and low CCT of 3441 K with a CIE chromaticity coordinate of (0.42, 0.42) in the 1931 CIE colour space.
Finally, the different excitation-dependent emission performance of the blue and green ((C2H5)4N)2ZrCl6 powders provides an opportunity for information encryption applications. The blue and green powders exhibit similar luminescence under a 254 nm lamp but different luminescence under a 365 nm lamp (Fig. 5d and e and S27†). Thus, the blue and green powders are chosen to be selectively deposited into a pixelated hole pattern (ϕ = 2 mm, d = 5 mm) in a PMMA plate (Fig. S28†). As shown in Fig. 5d and S28,† the blue and green powders both exhibit a pale yellow colour under room lighting and do not exhibit luminescence under lower energy excitation with a wavelength of 375 nm, which makes them hard to distinguish. The pattern composed of the blue and green powders showed uniformly blue luminescence under 254 nm excitation, expressing signals in the pixel holes (Fig. 5g). However, on excitation under a 365 nm UV lamp, cryptographic information emerged as green emission due to the excitation-dependent emission of the green powder (Video S1†). Different luminescent behaviours in each pixel hole can be decoded as binary data of “0” or “1” for digital information.
Besides light conversion applications, ((C2H5)4N)2ZrCl6 can also be applied to X-ray detection. The radioluminescence (RL) spectrum of the blue ((C2H5)4N)2ZrCl6 samples is carried acquired under X-ray irradiation with a tube voltage of 100 kV and tube current of 100 mA. The emission peak is around 500 nm, which is similar to the emission peak under a 254 nm lamp (Fig. S29†).
Conclusions
In summary, we synthesized ((C2H5)4N)2ZrCl6 samples with multiple emissions. The ((C2H5)4N)2ZrCl6 crystallized in the monoclinic phase with distorted [ZrCl6]2− octahedra, leading to multi-mode luminescence. The blue, red and green luminescence are attributed to STE emission and the d–d transitions of Zr(IV). The three emission modes exist simultaneously in both the single crystal and powder samples. The relative intensity of the three luminescence colours can be adjusted in experiments to form three major types of powder sample. The blue powder sample mainly exhibits emission at 500 nm with an excitation energy higher than that at 280 nm. The red powder sample exhibits blue emission at 500 nm and a red emission peak at 620 nm. Energy transfer occurs between the two STE emissions, which is observed for the red powder sample. The green sample exhibits blue emission at 500 nm and a narrow green emission peak at 527 nm with a FWHM of 47 nm. Various optical applications are achieved through different combinations of the three kinds of powders. An anti-counterfeiting application with two chromatic emissions is achieved using the green ((C2H5)4N)2ZrCl6 powder under 254 nm and 365 nm UV lamps. A WLED with a CIE chromaticity coordinate of (0.42, 0.42), CCT of 3441 K and CRI of up to 95 is obtained by applying green and red ((C2H5)4N)2ZrCl6 powders on a 310 nm UV chip. The application of information encryption is achieved by depositing blue and green powders in a pixelated hole pattern in a PMMA plate. Besides the ((C2H5)4N)2ZrCl6 samples, (C13H30N)2ZrCl6 with a different organic cation also exhibits multiple emissions. We believe the unique tri-luminescent ((C2H5)4N)2ZrCl6 developed in this study could pave the way towards multi-emitting lead-free perovskites and is expected to expand the application of multi-luminescence materials in lighting, anti-counterfeiting and encryption fields.Experimental
Materials
Hydrochloric acid (HCl, Shanghai Lingfeng Chemical Reagent Co., Ltd, 36.0–38.0 wt%), zirconium tetrachloride (ZrCl4, Aladdin, 98%), tetraethylammonium chloride ((C2H5)4NCl, HEOWNS, 98%), methyltributylammonium chloride (C13H30NCl, HEOWNS, 98%), isopropanol (IPA, Aladdin, AR), toluene (99.9%, Sinopharm Chemical Reagent Co., Ltd), methyl acetate (≥99.0%, Macklin), polystyrene (PS, Aladdin) and deionized water were used in this work. All chemicals were used as received without any further purification.Synthesis of the ((C2H5)4N)2ZrCl6 powder
In a typical synthesis of the ((C2H5)4N)2ZrCl6 powder (nominal concentrations in moles), 4 mmol ZrCl4 (2 mmol ZrCl4 for the green powder) were dissolved in 8 mL HCl and 4 mmol (C2H5)4NCl were dissolved in 8 mL HCl. Subsequently, 1 mL of the (C2H5)4NCl solution was injected into 1 mL of the ZrCl4 solution under vigorous stirring; then a white precipitate appeared. The crude mixture was centrifuged at 6000 rpm for 10 minutes and the precipitate was re-dispersed in 0.5 mL HCl, then dried in a tube in an oven at 80 °C to obtain the blue ((C2H5)4N)2ZrCl6 powder. For the green ((C2H5)4N)2ZrCl6 powder and the red ((C2H5)4N)2ZrCl6 powder, the precipitates were dried on a glass substrate at 80 °C and 50 °C, respectively. The green powder can also be obtained by grinding ((C2H5)4N)2ZrCl6 single crystals in a mortar and pestle.Synthesis of the ((C2H5)4N)2ZrCl6 single crystals
0.4 mmol (C2H5)4NCl was dissolved in 1.5 mL DI water, then 1.6 mL ZrCl4 solution (0.25 M) was injected into the water solution to form a clear solution. Subsequently, the clear solution was sealed, with a small hole, then placed in an oven and kept at 80 °C. Transparent crystals precipitated at the bottom of the sample vial after 1 week. Finally, the crystals were washed with IPA and dried at 80 °C.Fabrication of LEDs
The as-synthesized green and red ((C2H5)4N)2ZrCl6 powders were mixed with 25% polystyrene (PS) toluene ink at room temperature in air, and the ink was dropped onto a UV-LED chip (310 nm).Characterization details
Single-crystal X-ray diffraction (XRD) data were collected using an Apex DUO with Mo-Kα radiation (λ = 0.71073 Å) at room temperature. The structures were resolved and refined using direct methods with SHELXL and Olex2. X-ray diffraction (XRD) measurements were carried out using a Rigaku Ultima III X-ray diffractometer equipped with Cu Kα radiation (λ = 1.541841 Å). Ultraviolet and visible (UV-vis) absorption spectra were measured using a Shimadzu UV-3600 Plus spectrophotometer at room temperature. PL spectra were measured using a Horiba PTI QuantaMaster 400. Time-resolved photoluminescence spectra were detected using an Edinburgh Instruments FLS980 at room temperature. The absolute fluorescence quantum yields were measured using a Horiba PTI QuantaMaster 400 steady-state fluorescence system with an integrating sphere. Scanning electron microscopy (SEM) and X-ray spectroscopy (EDS) were performed on a JEOL JSM-7800F at 15 kV. Fluorescence microscopy images were captured using an Axio Vert A1 FL-LED at room temperature.Data availability
More experimental or data associated with this article is available on request from the authors.Author contributions
Z. D. supervised the project. C. W. conceived the study, synthesized the perovskites and performed steady-state absorption and PL spectral measurements and time-resolved photoluminescence spectra (TCSPC) measurements. W. M. and G. L. fabricated the LED devices, and G. X., M. P. and B. X. performed characterization of the LED devices. All authors discussed the results and assisted during manuscript preparation. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22075129).Notes and references
- K. M. McCall, V. Morad, B. M. Benin and M. V. Kovalenko, Efficient Lone-Pair-Driven Luminescence: Structure–Property Relationships in Emissive 5s2 Metal Halides, ACS Mater. Lett., 2020, 2, 1218–1232 CrossRef CAS PubMed.
- J. Sun, W. Zheng, P. Huang, M. Zhang, W. Zhang, Z. Deng, S. Yu, M. Jin and X. Chen, Efficient Near-Infrared Luminescence in Lanthanide-Doped Vacancy-Ordered Double Perovskite Cs2ZrCl6 Phosphors via Te4+ Sensitization, Angew. Chem., Int. Ed., 2022, 61, e202201993 CrossRef CAS PubMed.
- M. Cao, Z. Li, X. Zhao and X. Gong, Achieving Ultrahigh Efficiency Vacancy-Ordered Double Perovskite Microcrystals via Ionic Liquids, Small, 2022, 18, 2204198 CrossRef CAS PubMed.
- M. Li and Z. Xia, Recent progress of zero-dimensional luminescent metal halides, Chem. Soc. Rev., 2021, 50, 2626–2662 RSC.
- S. Sun, M. Lu, X. Gao, Z. Shi, X. Bai, W. W. Yu and Y. Zhang, 0D Perovskites: Unique Properties, Synthesis, and Their Applications, Adv. Sci., 2021, 8, 2102689 CrossRef PubMed.
- C. Zhou, L.-J. Xu, S. Lee, H. Lin and B. Ma, Recent Advances in Luminescent Zero-Dimensional Organic Metal Halide Hybrids, Adv. Opt. Mater., 2021, 9, 2001766 CrossRef CAS.
- H. Peng, X. He, Q. Wei, Y. Tian, W. Lin, S. Yao and B. Zou, Realizing High-Efficiency Yellow Emission of Organic Antimony Halides via Rational Structural Design, ACS Appl. Mater. Interfaces, 2022, 14, 45611–45620 CrossRef CAS PubMed.
- R. L. Zhang, X. Xu, X. Mao, Z. Y. Wang, P. Y. Wang, Y. Yang, J. S. Chen, R. F. Lu, W. Q. Deng and K. L. Han, Excitation-Dependent Emission in All-Inorganic Lead-Free Cs2ScCl5 center dot H2O Perovskite Crystals, Laser Photonics Rev., 2022, 16, 2100689 CrossRef CAS.
- J. C. Li, Y. Y. Sheng, G. Q. Tong, H. W. Zhu, X. Y. Tao, C. Y. Wu, Y. J. Chang, Z. Y. Tang, J. T. Yang, S. B. Zhang and Y. Jiang, Anti-Solvent Synthesis of Three-Color Indium-Based Halide Perovskite Microplate/Microcrystal Phosphors for High Color Rendering WLEDs, Adv. Opt. Mater., 2023, 11, 2300100 CrossRef CAS.
- S. Das Adhikari, C. Echeverría-Arrondo, R. S. Sánchez, V. S. Chirvony, J. P. Martínez-Pastor, S. Agouram, V. Muñoz-Sanjosé and I. Mora-Seró, White light emission from lead-free mixed-cation doped Cs2SnCl6 nanocrystals, Nanoscale, 2022, 14, 1468–1479 RSC.
- W. Meng, C. Wang, Y. Li, G. Hu, S. Sui, G. Xu, M. Peng and Z. Deng, Synthesis of Efficient and Stable Tetrabutylammonium Copper Halides with Dual Emissions for Warm White Light-Emitting Diodes, Chem.–Eur. J., 2023, 29, e202202675 CrossRef CAS PubMed.
- J.-H. Wei, J.-F. Liao, X.-D. Wang, L. Zhou, Y. Jiang and D.-B. Kuang, All-Inorganic Lead-Free Heterometallic Cs4MnBi2Cl12 Perovskite Single Crystal with Highly Efficient Orange Emission, Matter, 2020, 3, 892–903 CrossRef.
- Z. Li, Y. Li, P. Liang, T. Zhou, L. Wang and R.-J. Xie, Dual-Band Luminescent Lead-Free Antimony Chloride Halides with Near-Unity Photoluminescence Quantum Efficiency, Chem. Mater., 2019, 31, 9363–9371 CrossRef CAS.
- M. Li, J. Zhou, G. Zhou, M. S. Molokeev, J. Zhao, V. Morad, M. V. Kovalenko and Z. Xia, Hybrid Metal Halides with Multiple Photoluminescence Centers, Angew. Chem., Int. Ed., 2019, 58, 18670–18675 CrossRef CAS PubMed.
- J. Chen, Y. Guo, B. Chen, W. Zheng and F. Wang, Ultrafast and Multicolor Luminescence Switching in a Lanthanide-Based Hydrochromic Perovskite, J. Am. Chem. Soc., 2022, 144, 22295–22301 CrossRef CAS PubMed.
- Y. Guo, J. Chen, B. Chen, W. Zheng, X. Zhang, H. Suo, F. Chun, X. Wei and F. Wang, Sequential thermochromic switching in zero-dimensional Cs2ZnCl4 metal halides, Mater. Today Phys., 2023, 35, 101111 CrossRef CAS.
- T. Chang, Q. Wei, R. Zeng, S. Cao, J. Zhao and B. Zou, Efficient Energy Transfer in Te4+-Doped Cs2ZrCl6 Vacancy-Ordered Perovskites and Ultrahigh Moisture Stability via A-Site Rb-Alloying Strategy, J. Phys. Chem. Lett., 2021, 12, 1829–1837 CrossRef CAS PubMed.
- B. Chen, Y. Guo, Y. Wang, Z. Liu, Q. Wei, S. Wang, A. L. Rogach, G. Xing, P. Shi and F. Wang, Multiexcitonic Emission in Zero-Dimensional Cs2ZrCl6:Sb3+ Perovskite Crystals, J. Am. Chem. Soc., 2021, 143, 17599–17606 CrossRef CAS PubMed.
- F. Zhang, X. Chen, X. Qi, W. Liang, M. Wang, Z. Ma, X. Ji, D. Yang, M. Jia, D. Wu, X. J. Li, Y. Zhang, Z. Shi and C.-X. Shan, Regulating the Singlet and Triplet Emission of Sb3+ Ions to Achieve Single-Component White-Light Emitter with Record High Color-Rendering Index and Stability, Nano Lett., 2022, 22, 5046–5054 CrossRef CAS PubMed.
- F. Zhang, Z. Chen, Z. Liu, M. Jia, X. Chen, D. Wu, X. Li and Z. Shi, Highly stable vacancy-ordered double perovskite Rb2ZrCl6 with broadband emission for down-conversion white light-emitting diodes, J. Lumin., 2022, 251, 119150 CrossRef CAS.
- Z. Li, Q. Li, M. Cao, Z. Rao, X. Shi, L. Zhou, X. Zhao and X. Gong, Multimodal Luminescent Low-Dimension Cs2ZrCl6:xSb3+ Crystals for White Light-Emitting Diodes and Information Encryption, Langmuir, 2023, 39, 3792–3799 CrossRef CAS PubMed.
- X. Yun, J. Nie, H. Wang, H. Hu, H. Zhong, D. Xu, Y. Shi and H. Li, Multiexcitonic Emission of Organic–Inorganic (C4H12N)2ZrCl6:Sb3+ Perovskites across the Full Visible Region for Anticounterfeiting Applications, ACS Appl. Nano Mater., 2023, 6, 19581–19587 CrossRef CAS.
- W. Yang, P. Dang, G. Zhang, H. Lian, G. Li and J. Lin, Tunable Dual Emission in Bi3+/Te4+-Doped Cs2HfCl6 Double Perovskites for White Light-Emitting Diode Applications, Inorg. Chem., 2022, 61, 5903–5911 CrossRef CAS PubMed.
- A. Abfalterer, J. Shamsi, D. J. Kubicki, C. N. Savory, J. Xiao, G. Divitini, W. Li, S. Macpherson, K. Gałkowski, J. L. MacManus-Driscoll, D. O. Scanlon and S. D. Stranks, Colloidal Synthesis and Optical Properties of Perovskite-Inspired Cesium Zirconium Halide Nanocrystals, ACS Mater. Lett., 2020, 2, 1644–1652 CrossRef CAS PubMed.
- S. Liu, B. Yang, J. Chen, D. Wei, D. Zheng, Q. Kong, W. Deng and K. Han, Efficient Thermally Activated Delayed Fluorescence from All-Inorganic Cesium Zirconium Halide Perovskite Nanocrystals, Angew. Chem., Int. Ed., 2020, 59, 21925–21929 CrossRef CAS PubMed.
- P. Cheng, D. Zheng, L. Feng, Y. Liu, J. Liu, J. Li, Y. Yang, G. Wang and K. Han, Doped all-inorganic cesium zirconium halide perovskites with high-efficiency and tunable emission, J. Energy Chem., 2022, 65, 600–604 CrossRef CAS.
- X. Shi, Z. Li, M. Cao, Z. Rao, X. Zhao and X. Gong, Fast HCl-free Synthesis of Lead-free Rb2ZrCl6:xSb3+ Perovskites, Inorg. Chem., 2022, 61, 14095–14101 CrossRef CAS PubMed.
- S.-Y. Yao, H. Li, M. Zhou, T.-C. Wang, X. Yu, Y.-S. Xu, J.-H. Yi, J.-B. Qiu, J. Yu and X.-H. Xu, Visualization of X-rays with an Ultralow Detection Limit via Zero-Dimensional Perovskite Scintillators, ACS Appl. Mater. Interfaces, 2022, 14, 56957–56962 CrossRef CAS PubMed.
- F. Zhang, Y. Zhou, Z. Chen, M. Wang, Z. Ma, X. Chen, M. Jia, D. Wu, J. Xiao, X. Li, Y. Zhang, Z. Shi and C. Shan, Thermally Activated Delayed Fluorescence Zirconium-Based Perovskites for Large-Area and Ultraflexible X-ray Scintillator Screens, Adv. Mater., 2022, 34, e2204801 CrossRef PubMed.
- C. Fang, J. Yang, G. Zhou, Z. Zhang, Y. Mao, X. Yun, L. Liu, D. Xu, X. Li and J. Zhou, Energy transfer from self-trapped excitons to rare earth ions in Cs2ZrCl6 perovskite variants, J. Mater. Chem. C, 2023, 11, 1095–1102 RSC.
- Y. He, S. Liu, Z. Yao, Q. Zhao, P. Chabera, K. Zheng, B. Yang, T. Pullerits and J. Chen, Nature of Self-Trapped Exciton Emission in Zero-Dimensional Cs2ZrCl6 Perovskite Nanocrystals, J. Phys. Chem. Lett., 2023, 14, 7665–7671 CrossRef CAS PubMed.
- R. Liu, W. Zhang, W. Liu and G. Li, Synthesis of a Bi3+-Doped Cs2HfCl6 Double Perovskite with Highly Efficient Blue Light Emission at Room Temperature, Inorg. Chem., 2021, 60, 10451–10458 CrossRef CAS PubMed.
- H. Wan, F. Jia, F. Dinic, M. Imran, B. Rehl, Y. Liu, W. Paritmongkol, P. Xia, Y.-K. Wang, Y. Liu, S. Wang, Q. Lyu, G. F. Cotella, P. Chun, O. Voznyy, S. Hoogland and E. H. Sargent, Enhanced Blue Emission in Rb2HfCl6 Double Perovskite via Bi3+ Doping and Cs+ Alloying, Chem. Mater., 2023, 35, 948–953 CrossRef CAS.
- M. Adhikari, N. Shrivastava, S. T. McClain, C. M. Adhikari, B. Guzelturk, R. Khanal, B. Gautam and Z. Luo, Luminescence from Self-Trapped Excitons and Energy Transfers in Vacancy-Ordered Hexagonal Halide Perovskite Cs2HfF6 Doped with Rare Earths for Radiation Detection, Adv. Opt. Mater., 2022, 10, 2201374 CrossRef CAS.
- G. Dai, Z. Ma, Y. Qiu, Z. Li, X. Fu, H. Jiang and Z. Ma, Excitation-Dependent Luminescence of 0D ((CH3)4N)2ZrCl6 across the Full Visible Region, J. Phys. Chem. Lett., 2022, 13, 7553–7560 CrossRef CAS PubMed.
- S. Yan, W. Tian, H. Chen, K. Tang, T. Lin, G. Zhong, L. Qiu, X. Pan and W. Wang, Synthesis of 0D Manganese-Based Organic–Inorganic Hybrid Perovskite and Its Application in Lead-Free Red Light-Emitting Diode, Adv. Funct. Mater., 2021, 31, 2100855 CrossRef CAS.
- Z. X. Wang, J. A. Lai, Q. Huang, D. F. Wu, F. Qi, N. Zhang, Y. Y. Pu, C. Q. Tian, W. W. Chen, Y. F. Liu, P. He, K. An, X. S. Tang, F. Wang, Y. Liu and G. Q. Han, Ultrahigh PLQY Lead-Free Organic-Inorganic Hybrid Zirconium-Based Perovskites in Anticounterfeiting Applications, Adv. Opt. Mater., 2023, 11, 2300399 CrossRef CAS.
- Y. Wada, U. Lemmer, E. O. Göbel, M. Yamashita and K. Toriumi, Time-resolved luminescence study of self-trapped-exciton relaxation in quasi-one-dimensionalhalogen-bridged mixed-valence metal complexes, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 8276–8282 CrossRef CAS PubMed.
- S. Li, J. Luo, J. Liu and J. Tang, Self-Trapped Excitons in All-Inorganic Halide Perovskites: Fundamentals, Status, and Potential Applications, J. Phys. Chem. Lett., 2019, 10, 1999–2007 CrossRef CAS PubMed.
- F. Zhang, Y. Zhou, Z. Chen, X. Niu, H. Wang, M. Jia, J. Xiao, X. Chen, D. Wu, X. Li, Z. Shi and C. Shan, Large-Area X-Ray Scintillator Screen Based on Cesium Hafnium Chloride Microcrystals Films with High Sensitivity and Stability, Laser Photonics Rev., 2023, 17, 2200848 CrossRef CAS.
- W. Gao, M. Leng, Z. Hu, J. Li, D. Li, H. Liu, L. Gao, G. Niu and J. Tang, Reversible luminescent humidity chromism of organic–inorganic hybrid PEA2MnBr4 single crystals, Dalton Trans., 2020, 49, 5662–5668 RSC.
- S. Tomimoto, H. Nansei, S. Saito, T. Suemoto, J. Takeda and S. Kurita, Femtosecond Dynamics of the Exciton Self-Trapping Process in a Quasi-One-Dimensional Halogen-Bridged Platinum Complex, Phys. Rev. Lett., 1998, 81, 417 CrossRef CAS.
- V. Morad, I. Cherniukh, L. Pöttschacher, Y. Shynkarenko, S. Yakunin and M. V. Kovalenko, Manganese(II) in Tetrahedral Halide Environment: Factors Governing Bright Green Luminescence, Chem. Mater., 2019, 31, 10161–10169 CrossRef CAS PubMed.
- X.-H. Lv, W.-Q. Liao, P.-F. Li, Z.-X. Wang, C.-Y. Mao and Y. Zhang, Dielectric and photoluminescence properties of a layered perovskite-type organic–inorganic hybrid phase transition compound: NH3(CH2)5NH3MnCl4, J. Mater. Chem. C, 2016, 4, 1881–1885 RSC.
- W. Li, Z. Zhou, C. Wang, Y. Li, S. Kurosawa, G. Ren, X. OuYang and Y. Wu, Red-Emitting Organic–Inorganic Hybrid Manganese(II) Halides for X-Ray Imaging, Adv. Sens. Res., 2023, 2, 2200083 CrossRef.
- J. Reader and N. Acquista, Spectrum and energy levels of four-times ionized zirconium (Zr v), J. Opt. Soc. Am., 1979, 69, 239–253 CrossRef CAS.
- B. K. Gupta, D. Haranath, S. Saini, V. N. Singh and V. Shanker, Synthesis and characterization of ultra-fine Y2O3:Eu3+ nanophosphors for luminescent security ink applications, Nanotechnology, 2010, 21, 055607 CrossRef PubMed.
- W. Tian, J. Zhang, J. Yu, J. Wu, J. Zhang, J. He and F. Wang, Phototunable Full-Color Emission of Cellulose-Based Dynamic Fluorescent Materials, Adv. Funct. Mater., 2018, 28, 1703548 CrossRef.
- T. Si, Q. Zhu, J. Xiahou, X. Sun and J.-G. Li, Regulating Mn2+/Mn4+ Activators in ZnGa2O4 via Mg2+/Ge4+ Doping to Generate Multimode Luminescence for Advanced Anti-Counterfeiting, ACS Appl. Electron. Mater., 2021, 3, 2005–2016 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photographs, PL spectra, UV-vis absorption spectra, XRD patterns, SEM images, EDS mapping and crystal structure of hybrid Zr-based perovskite samples. See DOI: https://doi.org/10.1039/d3sc06178g |
| This journal is © The Royal Society of Chemistry 2024 |