
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioorthogonal chemistry of polyoxometalates – challenges and prospects
Stanislav K.
Petrovskii
 a,
Elena V.
Grachova
b and
Kirill Yu.
Monakhov
*a
a,
Elena V.
Grachova
b and
Kirill Yu.
Monakhov
*a
aLeibniz Institute of Surface Engineering (IOM), Permoserstr. 15, Leipzig 04318, Germany. E-mail: kirill.monakhov@iom-leipzig.de
bInstitute of Chemistry, St Petersburg University, Universitetskii pr. 26, St. Petersburg 198504, Russia
First published on 26th February 2024
Abstract
Bioorthogonal chemistry has enabled scientists to carry out controlled chemical processes in high yields in vivo while minimizing hazardous effects. Its extension to the field of polyoxometalates (POMs) could open up new possibilities and new applications in molecular electronics, sensing and catalysis, including inside living cells. However, this comes with many challenges that need to be addressed to effectively implement and exploit bioorthogonal reactions in the chemistry of POMs. In particular, how to protect POMs from the biological environment but make their reactivity selective towards specific bioorthogonal tags (and thereby reduce their toxicity), as well as which bioorthogonal chemistry protocols are suitable for POMs and how reactions can be carried out are questions that we are exploring herein. This perspective conceptualizes and discusses advances in the supramolecular chemistry of POMs, their click chemistry, and POM-based surface engineering to develop innovative bioorthogonal approaches tailored to POMs and to improve POM biological tolerance.
1 Introduction
Over the past decades, chemistry has been significantly enriched by new revolutionary approaches that force researchers to take a fresh look and completely rethink molecular design and the creation of new materials. The breakthrough discoveries of click chemistry by Meldal1 and Sharpless2 and the further development of bioorthogonal chemistry by Bertozzi3 redefined the limits of what was possible and gave chemists an extremely powerful and versatile set of tools for the construction of molecular assemblies.4,5 Many transformations that required complex conditions and water–oxygen reaction media were adapted for implementation on biological objects and even in vivo. In essence, bioorthogonal chemistry means high selectivity of reagents towards certain substrates and, therefore, high tolerance to the huge number of functional groups present in the biological environment. Reactions must proceed in high yield even at low reactant concentrations under living cell conditions, making them “reaction flasks” for targeted transformations,6 thus opening many doors for medicine and biotechnology.Hybrid organic–inorganic polyoxometalate (POM) compounds7,8 combine a discrete, nanosized metal–oxo core exhibiting rich redox chemistry and catalytic activity with an organic environment that does not only stabilize these molecular metal oxides and improves their solubility in various media, but can also empower the physicochemical properties to the POM due to synergistic effects. Organic ligation serves as a platform for functionalization and post-functionalization strategies,9–12 providing chemists with the ability to tailor a wide variety of molecular architectures. The latter include numerous conjugates with biological molecules. For example, [MnMo6O24]3− Anderson-type POMs functionalized by two different tris-ligands in an asymmetric way can be incorporated into the backbones of biopolymers both by conjugation with presynthesized peptide chains and by Fmoc solid-phase synthesis as an unique POM bearing unnatural amino acid.13 Symmetric organic–inorganic hybrids based on the same POM unit decorated with bombesin antagonist peptide units are readily assembled through the formation of POM-driven nanoparticles, which are taken up by living cells, while the biological activity of POM is still retained.14 It is noteworthy that in all of these examples, succinimide esters are typically used as labile reagents for amide conjugation and the reactions are therefore intolerant to an aqueous medium.
Despite numerous published reports and reviews15–21 on the use of POMs in biological and medical applications as effective antitumor19 and antibacterial18 agents, chiral bionics based on POMs,22 as well as critical importance of integration of artificial electronic elements into biosystems,23,24 there are still no systematic analyzes and published examples of the implementation of true bioorthogonal processes involving POM building blocks. The development of the concept of bioorthogonal POM chemistry would be extremely useful for POM-based drug targeting (including in situ drug assembly, activation, and modification), cell labeling and in vivo imaging methods, and potential biocompatible positioning of switchable POM units at living neuron interfaces.
Within this perspective, we review critical aspects of implementing bioorthogonal approaches in POM chemistry, including key challenges and possible ways to overcome them. In light of the potential applications of POM-based switchable systems in the field of artificial neural interfaces, biocompatible surface modification approaches have received special attention here.
2 Polyoxometalates as bioorthogonal reagents?
In general, POM typical metals such as V, Mo, and W are quite rare in living organisms and considered as trace elements.25 Molybdenum species are involved in biochemical processes in numerous plants and bacteria. Mo ions are essential for the formation of the FeMo nitrogenase cofactor,26 utilized in ATP-powered nitrogen fixation. Mo ions also play an important role in the function of xanthine dehydrogenase – the enzyme participating in purine metabolism in the human organism.27 Vanadium in its highest oxidation state +5 exhibits significant toxicity and the accumulation of vanadates in tissues leads to oxidative stress.28 Despite this, it is an important trace element for regulating the phosphorylation and oxidation process,29,30 and is used in antidiabetic therapy.31,32 In contrast to highly oxidative VV species, tungstates are less aggressive and can be even used to protect tissues from free radicals by suppressing the oxidative processes through inhibition of xanthine dehydrogenase.33 Tungstate species can induce adipocyte activity and, therefore, serve as promising antiobesity agents.34 The very important factor that determines reactivity and thermodynamic properties of the VV-, MoVI- and WVI-based compounds in solution is their ability to form polyoxoanionic structures, i.e. polyoxometalates (POMs).What's special about POMs? First, common POMs carry a negative charge.35 High charge density along with the considerable polarized M–O bonds promote strong interaction of POMs with a lot of functional groups. Second, because the POM structures are usually built up from transition metal atoms, they are readily involved in redox processes. Most of the POM cores include metal ions in the highest oxidation states (VV, NbV, TaV, MoVI, WVI) and thereby act as oxidizing agents or efficient oxidation catalysts promoting electron transfer processes and generation of oxidative species.36 Finally, inorganic POM structures are not inert towards water media: they are involved in complex equilibria between different species,37,38 that are dependent on pH, ion composition, and temperature. Organic functionalization substantially stabilizes POM structures, expanding their pH range of existence.8
It is clear that the fundamental properties of POMs attracting much attention from scientists working in the fields of catalysis,39 biomedicine,18,19,40,41 or electronics42–45 have their downsides, namely high reactivity and cytotoxicity.46,47 There is no doubt that the properties of POMs literally contradict the formal criteria for bioorthogonal reagents.5 However, does this mean that the very idea of implementing bioorthogonal POM chemistry should be abandoned? No, this doesn't. The idea behind this Perspective is to show what scientists can do to take the concept of bioorthogonal POM chemistry from “fundamentally impossible” to “challenging but possible”.
Key issues we aim to address are two-fold: How can POM and the biological environment be protected from each other? What bioorthogonal chemical transformations are in principle possible for POM-based organic–inorganic hybrids?
The inspiring example of bio-tolerant POMs is the bacterium Azotobacter vinelandii. The cells of this organism not only tolerate POM units, but also use them as compact storage for Mo and W ions. To accumulate these metal ions and protect cells from reactive metal oxide species, this bacterium uses a special molybdenum/tungsten storage protein called Mo/WSto.48–50 The protein scaffold stabilizes POM nanoparticles as a series of unusual architectures, in turn templated by the protein structure, thereby preventing the participation of cytotoxic species in cellular chemistry. This naturally occurring property of POMs is a unique example showing the potential for synthetic POMs to approximate the requirements of bioorthogonal reagents and to build a bridge between the usually barely overlapping concepts of bioinorganic51 and bioorthogonal chemistries.
The specific reactivity and physicochemical properties of POMs that differ dramatically from those of organic and biomolecules, as well as the ability of POMs to encapsulate in surfactant shells, polymers, and biomolecular assemblies could be added up together into a complex key that opens the door to the realization of their bioorthogonal chemistry. In our opinion, the algorithm for implementing this chemistry should include the following steps exploring approaches from various fields of inorganic, organic, and supramolecular chemistry:
(i) synthesis of a hybrid POM precursor including functional groups suitable for bioorthogonal reactions;
(ii) incorporation of hybrid POM molecules into a supramolecular assembly to protect them from the biological environment;
(iii) implementation and adaptation of a bioorthogonal chemical protocol taking into account the properties of reagents containing POM.
The related concept, involving supramolecular protection followed by strain-promoted click conjugation, has recently been demonstrated for the bioorthogonal attachment of magnetic nanoparticles to cell membranes.52 Our Perspective focuses on the challenges and prospects of introducing, implementing, and adapting (post)functionalized POMs9,12,53 to accelerate progress in the development of their bioorthogonal chemistry.
3 Supramolecular approaches to improve biological tolerance
Challenges associated with the non-selective reactivity of POMs towards biological media and, consequently, their poor bioorthogonality, constitute the first task that must be addressed for the successful implementation of bioorthogonal chemistry involving inorganic POMs and POM-based organic–inorganic hybrid compounds. As is often the case, nature has already figured out how to overcome these challenges: the metal storage protein in Azotobacter vinelandii is a brilliant example that POM structures protected by a protein matrix can exist in living cells without damaging them.Incorporating POMs into supramolecular assemblies can be one useful approach15,16,54–56 with far-reaching implications for biological studies of POMs. The idea of protecting certain molecules from others by phase separation between two media with a membrane consisting of amphiphilic subunits is inspired by the structure of living cells. From a fundamental point of view, the formation of such POM-based assemblies will pursue three main goals: (1) protection of POM cores with an inert shell; (2) reducing the chemical potential of the POM-based system, resulting in the formation of less reactive (and therefore more chemically selective) species; (3) fine-tuning of aggregate surface characteristics to modulate interactions with various media, including physiological solutions and cell membranes, thereby improving bioactivity in vitro.57
In this section, we thus outline two different approaches that might be used in biological systems for POMs, namely the formation of POM-surfactant core–shell vesicles and the complexation of POMs with commonly used macrocyclic agents. For each concept, the main patterns and the most illustrative examples are given. Specific attention was paid to the systems with a stimuli-responsive morphology (and thereby controlled reactivity of POMs), as we consider it as one of the possible key factors for targeted POM delivery. Among all cationic surfactants, peptide molecules deserve special attention due to their biocompatibility, as well as structural and functional variability.58,59 Therefore, a special section is paid to supramolecular systems involving POM and peptides.
3.1. Polyoxometalates in core–shell vesicles
POM cores are easily involved in interaction with surfactant molecules, leading to the formation of amphiphilic nanostructures (Fig. 1).15,54,60 These micelles (core–shell structures) and vesicles (hollow layered structures) are supramolecular assemblies that are widely used in biological and medical applications.61,62 In addition, nanoaggregates containing POM vesicles are involved in more complex structures with biological polymers.63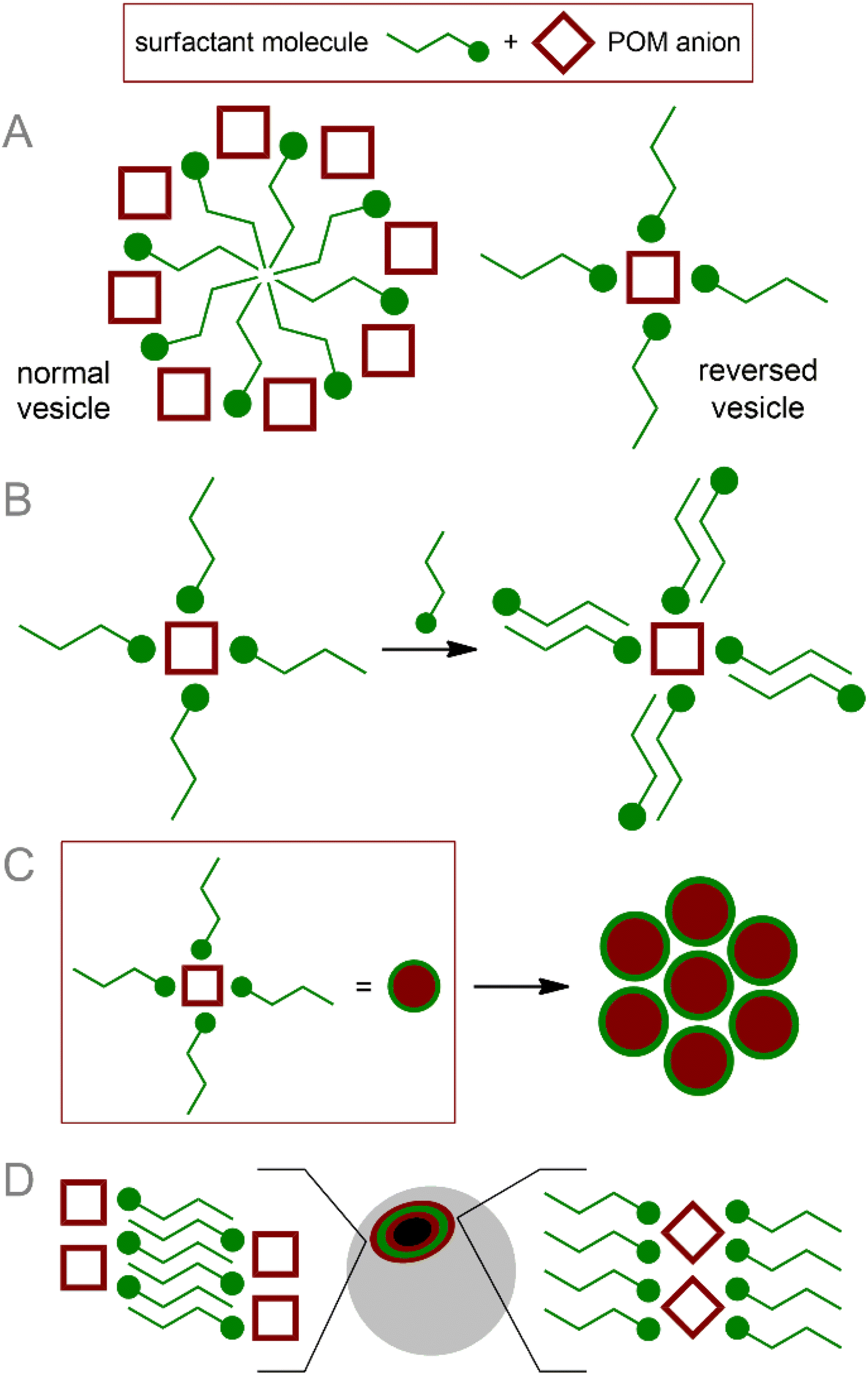 | ||
| Fig. 1 Schematic representation of general POM-surfactant nanoparticle topologies: (A) simple normal and reversed vesicles; (B) concentric multilayer vesicles; (C) “blackberry”-type nanoparticles; (D) hollow membrane normal and reversed vesicles. | ||
There are two general approaches to the formation of POM-based core–shell structures:15 (i) formation of POM–cation pairs, where the cation bears both hydrophilic site and hydrophobic groups providing self-assembling of supramolecular structures, and (ii) covalent modification of POM–organic hybrid platforms by large hydrophobic groups. In both cases, structure formation is determined by non-covalent interactions, which, in turn, are determined by the structural features of the components of the system.64 Thanks to the large structural diversity of POM cores and the rich possibilities of organic ligation and post-functionalization,9 the structure and behavior of supramolecular assemblies can be effectively tuned.54 Since covalent and non-covalent approaches significantly differ in terms of chemical implementation and properties of nanoparticles, we separated them into different sub-sections.
The giant Keplerate-type POM [Mo132O372(CH3CO2)30]42− (Mo132) can be “shielded” by bulky aliphatic tail cations.65–67 The resulting assemblies are capable of forming nanocomposites with electrochemically exploitable graphene, resulting in a material exhibiting synergistically induced supercapacitor properties. Other giant wheel-shaped POMs (Mo154) [MoVI126Mo28VO462H14(H2O)70]0.5·[MoVI124MoV28O457H14(H2O)68]0.5 may participate in the formation of self-assembled vesicles (Mo154)–(TTA)n (TTA = tetradecyltrimethylammonium cation). Moreover, depending on the Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TTA ratio, hydrophobic single-layer or hydrophobic bilayer structures can be formed.68 Notably, giant POMs have a closed metal–oxo cavity and can, therefore, be considered as an encapsulating shell for nanoscale reactor.69
:TTA ratio, hydrophobic single-layer or hydrophobic bilayer structures can be formed.68 Notably, giant POMs have a closed metal–oxo cavity and can, therefore, be considered as an encapsulating shell for nanoscale reactor.69
Smaller POMs generally tend to aggregate into spherical “blackberry” structures. These vesicles, assembled through both strong Coulombic and weak hydrophobic interactions, have been well studied and described in numerous publications.15,54,70–75 Noncovalent encapsulation of POM units into supramolecular assemblies finds application in vivo. For example, POM–organoplatinum hybrids with polyethylene glycol based surfactant nanoparticles demonstrate effective targeted delivery of the [PW11O40(SiC3H6NH2)2Pt(NH3)2Cl2]3− drug agent to living cells.76
The method of POM ligation determines the morphology of the resulting amphiphilic molecule. Monoligated POMs typically present a classic head-to-tail surfactant morphology (Fig. 2A). The most representative examples are Wells–Dawson (WD) POMs conjugated to polystyrene moieties78,79 or long alkyl chains ([P2V3W15O59((OCH2)3CNHCO–C(CH3)2–R)]6−, R = n-C15H31 or polystyrene chain).80 The Lindqvist-type hexamolybdate [Mo6O18(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–(PS)–(PT))]2− (PS and PT are polystyrene and polythiophene polymer chains, respectively) tethered with a polystyrene-polythiophene block copolymer synthesized using CuAAC chemistry,81,82 exhibits flexible aggregation behavior, forming normal or inverted vesicles depending on the solvent. Incorporation of redox-active conjugated aromatics instead of innocent hydrocarbon chains demonstrate a bright possibility of the redox control over hydrophobic properties and, consequently, morphology of self-assembled vesicles. Tuning the structure of hydrophobic chains also makes it possible to control the morphology of nanoparticles, which, in turn, leads to a redox reaction.83 When the POM site is not protected by any shell, such vesicles are very sensitive to ionic composition79 and pH,78 which directly influences effective charge density of the hydrophilic head. These characteristics allow the control of the nanoparticle formation; however, labile behavior can be considered a disadvantage from the point of view of application in a biological environment.
N–(PS)–(PT))]2− (PS and PT are polystyrene and polythiophene polymer chains, respectively) tethered with a polystyrene-polythiophene block copolymer synthesized using CuAAC chemistry,81,82 exhibits flexible aggregation behavior, forming normal or inverted vesicles depending on the solvent. Incorporation of redox-active conjugated aromatics instead of innocent hydrocarbon chains demonstrate a bright possibility of the redox control over hydrophobic properties and, consequently, morphology of self-assembled vesicles. Tuning the structure of hydrophobic chains also makes it possible to control the morphology of nanoparticles, which, in turn, leads to a redox reaction.83 When the POM site is not protected by any shell, such vesicles are very sensitive to ionic composition79 and pH,78 which directly influences effective charge density of the hydrophilic head. These characteristics allow the control of the nanoparticle formation; however, labile behavior can be considered a disadvantage from the point of view of application in a biological environment.
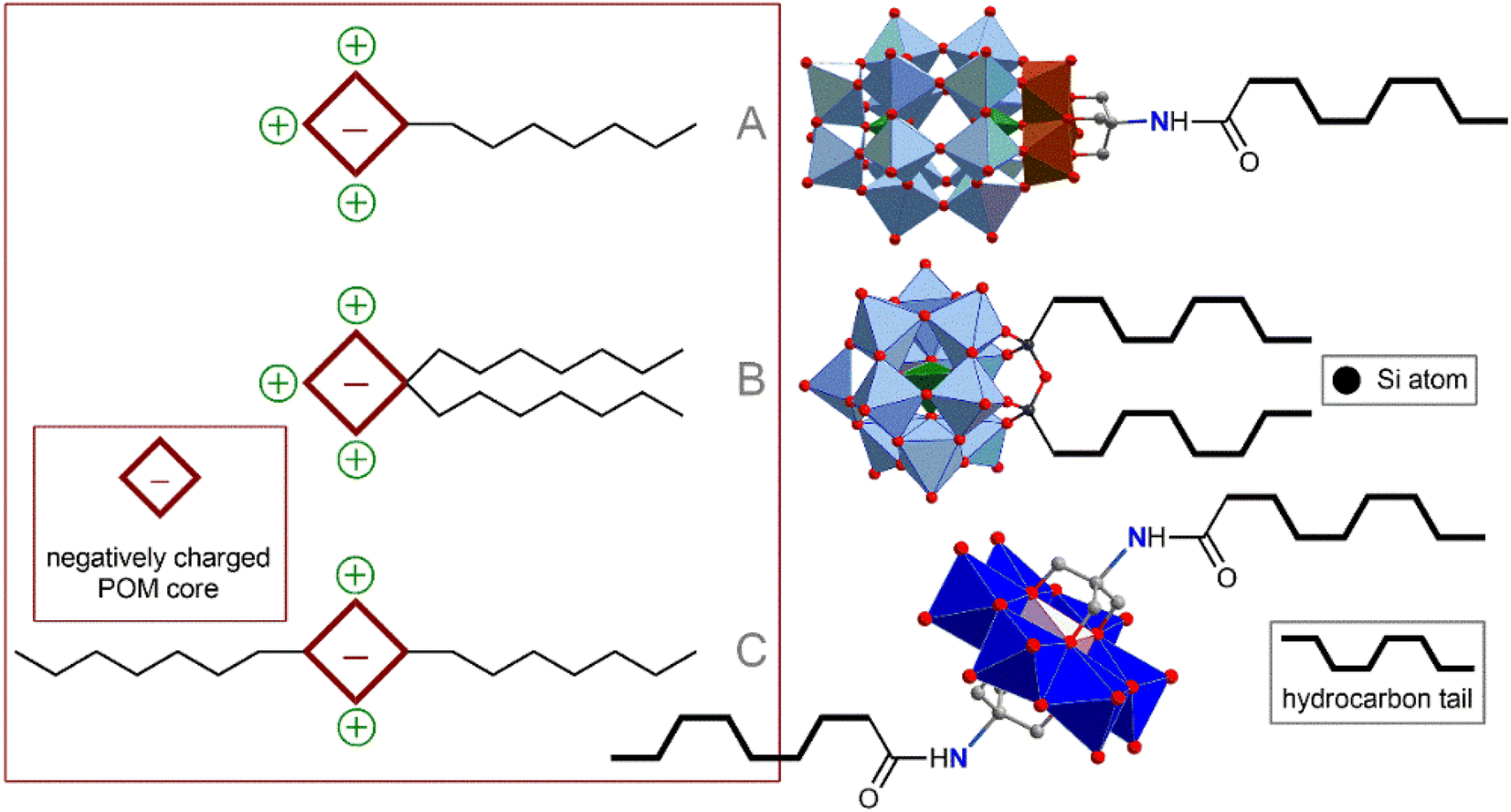 | ||
| Fig. 2 Schematic representation and archetype examples of POM-based surfactants: (A) POM head–hydrophobic tail; (B) POM head–several hydrophobic tails; (C) tail–POM head–tail. | ||
Keggin-type POMs [PW11O39(O(Si–R)2)]3− (R = hydrophobic chain), functionalized on one side with two hydrophobic chains through a siloxane fragment, are very similar in terms of surfactant morphology (Fig. 2B). These surfactants readily organize into micelles in solution and, under some conditions, tend to form self-assembled lyotropic phases.84,85 Larger Preyssler-type POMs [(C16H33)2NCONH(CH2)3SiNaP5W29O110]14− covalently bound to a double-stranded surfactant unit exhibit self-assembly into stable vesicles that are readily taken up cancer cells and demonstrate excellent antitumor activity.57
Symmetrical covalent functionalization of POMs with hydrophobic alkyl chains produces tail–head–tail surfactants (Fig. 2C), which are typically capable of assembling into hollow, inverted vesicular structures. The authors of the work that reported the combination of Anderson-type POMs with two aliphatic chains [MnMo6O18((OCH2)3CNHCO(CH2)14CH3)2]3− indicated that the agglomeration behavior of these surfactants is not as pronounced as for symmetrical POMs.86 The introduction of long-chain dimethyldioctylammonium cations leads to an increase in the supramolecular organization of these POM hybrids, significantly enriching set of available nanostructures.87 Lindqvist-type hexavanadates [V6O13((OCH2)3CNH(CO)CH2C6H4CH2CH2(CF2)nCF3)2]2−, linked to fluorocarbon chains by forming amides, are hybrids that readily form stable bilayer vesicles in solution.88 Fluorocarbon chains exhibit both hydrophobic and lipophobic properties and thus serve as a reliable “shield” for POMs. However, the use of these “forever chemicals” in biochemistry, especially in medicine, is controversial due to possible health concerns.89
The introduction of additional stimuli-responsive elements into the POM-surfactant system makes it possible to control the morphology of assemblies with exposure to some external stimuli. Asymmetric hybrid Anderson-type POM [MnMo6O18((OCH2)3CNHC21H19N2O4)((OCH2)3CNHC16H28O)]3− (SP–POM–C15), bearing spiropyran (SP) and alkyl chain moieties synthesized by subsequent post-functionalization of the diamine precursor, exhibits UV-triggered formation of hollow nanoparticles90 (vesicles in a polar medium and reversed vesicles in a non-polar medium). The mechanism of photoinduced aggregation is based on the photoisomerization of spiropyran into zwitterionic merocyanine (SP → MC). Since MC is a hydrophilic fragment, the molecule acquires the properties of a classic head-to-tail surfactant, easily assembled into two-layer aggregates. The result is of particular interest given that photoisomerization and hence aggregation can be reversed by visible light irradiation promoting MC → SP isomerization, thereby allowing the parent POM molecules to be released in response to stimulus.
3.2. Polyoxometalate-peptide assemblies
Synthetic surfactants can be successfully substituted by biocompatible peptide- or amino acid-derived cations. POM–peptide assemblies are a relatively new but very rapidly developing branch of POM science. The idea of combining the achievements of anionic POMs and cationic peptide chains has already led to numerous excellent discoveries and the creation of materials with outstanding properties.58 In the context of bioorthogonal POM chemistry, the formation of POM–peptide assemblies is attractive from two main points of view: (1) peptide chains are generally biocompatible and stable in the cellular environment; (2) peptides typically carry multiple cationic sites and can, therefore, bind very tightly to the POM core, protecting it from reactive species from solution. Depending on their construction principles, these nanoassemblies can be divided into two classes: electrostatic ionic hybrids and covalently bonded materials.The formed stable nanoparticles were proposed as adaptive containers not only for the POMs and peptides themselves, but also for other guest molecules. The formation of such nanoassemblies was studied using the example of the interaction of highly charged POMs [Eu(SiW10MoO39)2]13− and [EuW10O36]9− with arginine- and lysine-rich peptides.59 This process involved two steps: (1) POM-induced disruption of peptide assembly and formation of primary clusters; (2) self-assembly of these clusters into nanospheres (Fig. 3A). Combining anionic POM cores and peptide chains has helped create photosensors for certain biomolecules.94 Remarkably, the supramolecular design of POM–peptide nanoassemblies can be extended by adding a third component. Zhang et al. reported three-component hierarchical core–shell nanoflowers.95 Uncharged POM Ag3PW12O40 was covered with a polydopamine shell by polymerizing dopamine onto POM, which in turn was modified with antibacterial peptide Nisin. The resulting stable assemblies demonstrated excellent antibiotic activity along with low toxicity towards human cells. It can be assumed that a wide variety of modified peptides can be used to decorate the shell, thereby imparting flexible characteristics to the assemblies.
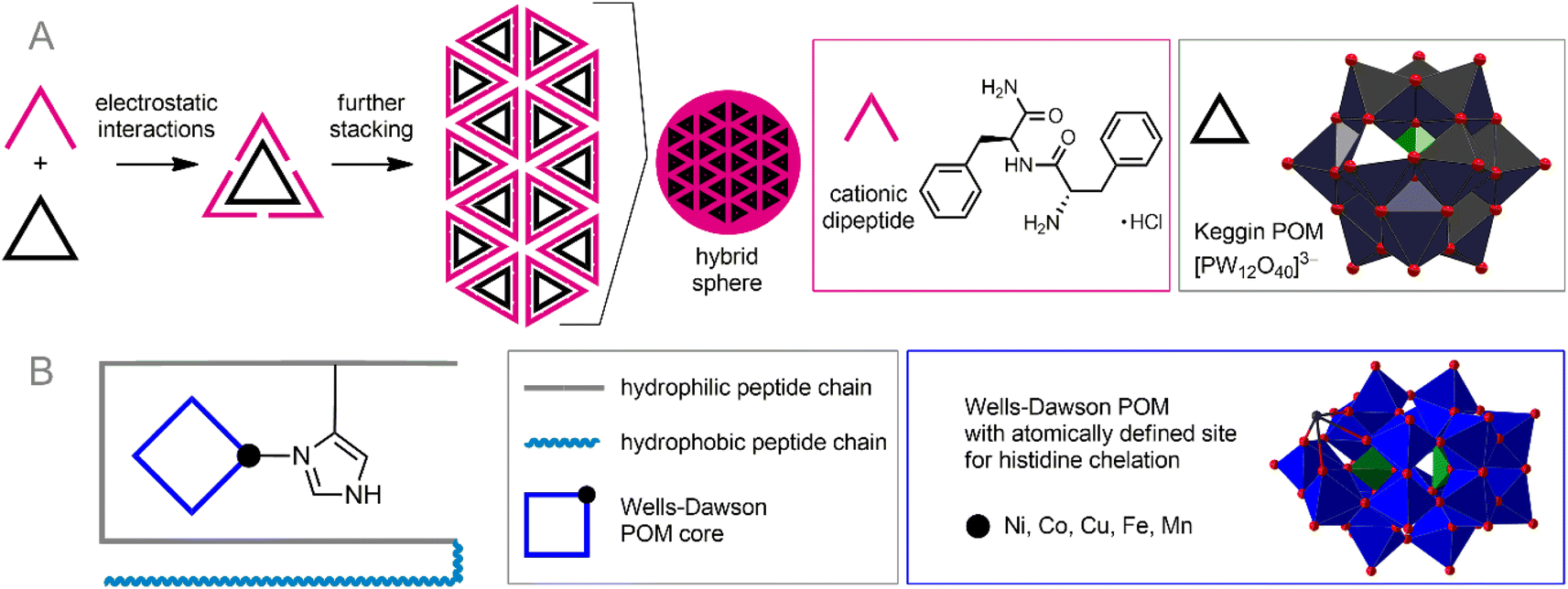 | ||
| Fig. 3 Formation of nanoassemblies of anionic POMs with cationic peptides under the influence of electrostatic forces (A) and the synergetic effect of coordination bonds and electrostatic forces (B). | ||
From the point of view of bioorthogonal chemistry, ideally, supramolecular assemblies containing POMs should be completely inert with respect to biological media, but reactive with respect to certain substrates. Another possible route is to create supramolecular containers that respond to various external stimuli. The morphology of POM–peptide conjugates can be modified, for example, by light irradiation. In Li's work96 a series of inorganic POMs ionically coupled to an azobenzene-modified cationic peptide exhibit light-induced reorganization from water-stable round nanoparticles into transient two-dimensional (2D) assemblies.
Fully inorganic and highly charged POMs such as Anderson–Evans Na6[TeW6O24], Keggin H4[SiW12O40], ZrIV-substituted Wells–Dawson K15H[Zr(α2-P2W17O61)2] or CeIII-substituted Keggin K11[Ce(PW11O39)2] act as linker nodes for self-assembling peptide matrices. The formation of these conjugates is determined by electrostatic97 and symmetry98 factors.
Highly charged inorganic POMs readily promote the self-assembly of short peptide nanofibers99 and vesicle-type nanostructures,100 which exhibit good antibacterial activity. The interaction of Na9[EuW10O36]·32H2O with the human papillomavirus peptide HPV 16 L1-p in solution leads to the formation of stable virus-like particles (VLPs),101 in which the POM core is encapsulated in a peptide shell (virus capsid). Notably, the presence of the charged POM core greatly enhances the stability of these VLPs, and the virus capsid allows them to utilize a biological mechanism to selectively enter cells. Finally, fully biocompatible protein-based liposomal nanocarriers102 were shown to include Ti-modified Keggin-type POM units103 [SiW11TiO40]6−. The resulting aggregates exhibited promising antiviral and antitumor activities.
Heterometallic POMs comprising transition metal ions can efficiently bind peptides through simultaneous coordination bonding and electrostatic interaction. Thus, POMs such as K8[P2NiW17O61] and K8[P2CoW17O61] can specifically bind β-amyloid (Aβ) peptide (Fig. 3B), which is known to be responsible for Alzheimer disease (AD).104 This binding leads to the prevention of the formation of amyloid plaques, inhibiting the toxic effect of Aβ. Interestingly, even Keggin-type homometallic polyoxotungtates, especially the lacunary POM K8[α-SiW11O39] can easily coordinate CuII centers already collected in aggregated Aβ-based structures and form stable ternary POM-CuII-Aβ,105 thereby acting as an analog of heterometallic POM species in the context of AD therapy. Specific supramolecular assembly of transition metal-substituted POMs with peptides may be key to precise targeting of POM-based drugs. Very recently, this was demonstrated by Zhao and co-workers106 using a giant oxocluster {[Ce10Ag6(DMEA)(H2O)27W22O70][B-α-TeW9O33]9}288− (DMEA = N,N-dimethylethanolamine) encapsulated by a brain-targeting peptide (angiopep-2-SH) through Ag–S bonding together with electrostatic interactions. The resulting nanoparticles exhibited superior ability to target glioma.
Covalent modification of proteins by POMs in some special cases can be achieved by combining functional groups with specific reactivity and electrostatic interaction of anionic POM with a charged peptide fragment. This concept was used in recent work of Qu et al.,110 which demonstrated that Wells–Dawson-type POM [P2W17O61SnR]7− (R = thiazolidinethione (TZ)-containing organic unit) selectively binds β-amyloid (Aβ) peptide. This combination, using the site-specific interaction of the TZ functional group with the lysine amino group111 of Aβ together with the strong electrostatic interaction of the POM core with the cationic site of the same peptide, is very strong and effective, leading to inhibition of Aβ aggregation, thus acting as a superior Alzheimer disease treatment. Meanwhile, this example represents an excellent special case of in vivo modification of POMs, which is very close to the concept of true bioorthogonal POM chemistry.
Despite these advances, most of the POM–peptide hybrids known to date belong to the class of non-covalent assemblies. The reason for this is mainly due to the difficulty of synthesizing covalent hybrids. However, some smart approaches could significantly improve their synthetic availability. A very elegant method for producing covalent hybrids was demonstrated by Mitchell, Martín-Rapún and co-workers, who introduced and developed the concept of ‘POMlymers’. These bis-peptide-functionalized Anderson–Evans-type POMs were prepared by ring-opening polymerization of amino acid N-carboxyanhydrides induced by amino-functionalized POM [MnIIIMo6O18((OCH2)3CNH2)2]3−.112 Combination of cationic lysine moieties and hydrophobic benzyloxycarbonyl sites make the material amphiphilic, as a result of which ‘POMlymer’ is able to form stable nanoparticles in solution. The oxidative properties of the parent POM are retained in the covalent hybrid, and the material has been proposed as an efficient skin antiseptic,113 whereas simple ion paring of the same POM with a cationic peptide results in reduced peroxidase-like behavior.
One of the most important properties of biomolecules (including many peptides) is the ability for molecular recognition. This can be a very powerful and selective tool for generating predesigned POM–peptide structures. POMs functionalized with specific functional groups can bind peptides very specifically, forming stable bioconjugates. Thus, Keggin-type POM [γ-SiW10O36]8−, modified with biotin moieties, is easily combined with avidin.97,114 The formation of these conjugates leads to a significant reduction in cytotoxicity and also provides exceptional capabilities for in vivo delivery and tracking of POMs. Bis-biotinylated Anderson–Evans-type POM [MnMo6O18((OCH2)3CNHCOCH2OCOC9H15N2OS)2]3−, equipped with a linker of the required length between the POM core and the pendant biotin group, is capable of forming with streptavidin very stable self-assembled nanoparticles of a regular structure.115 (Fig. 4).
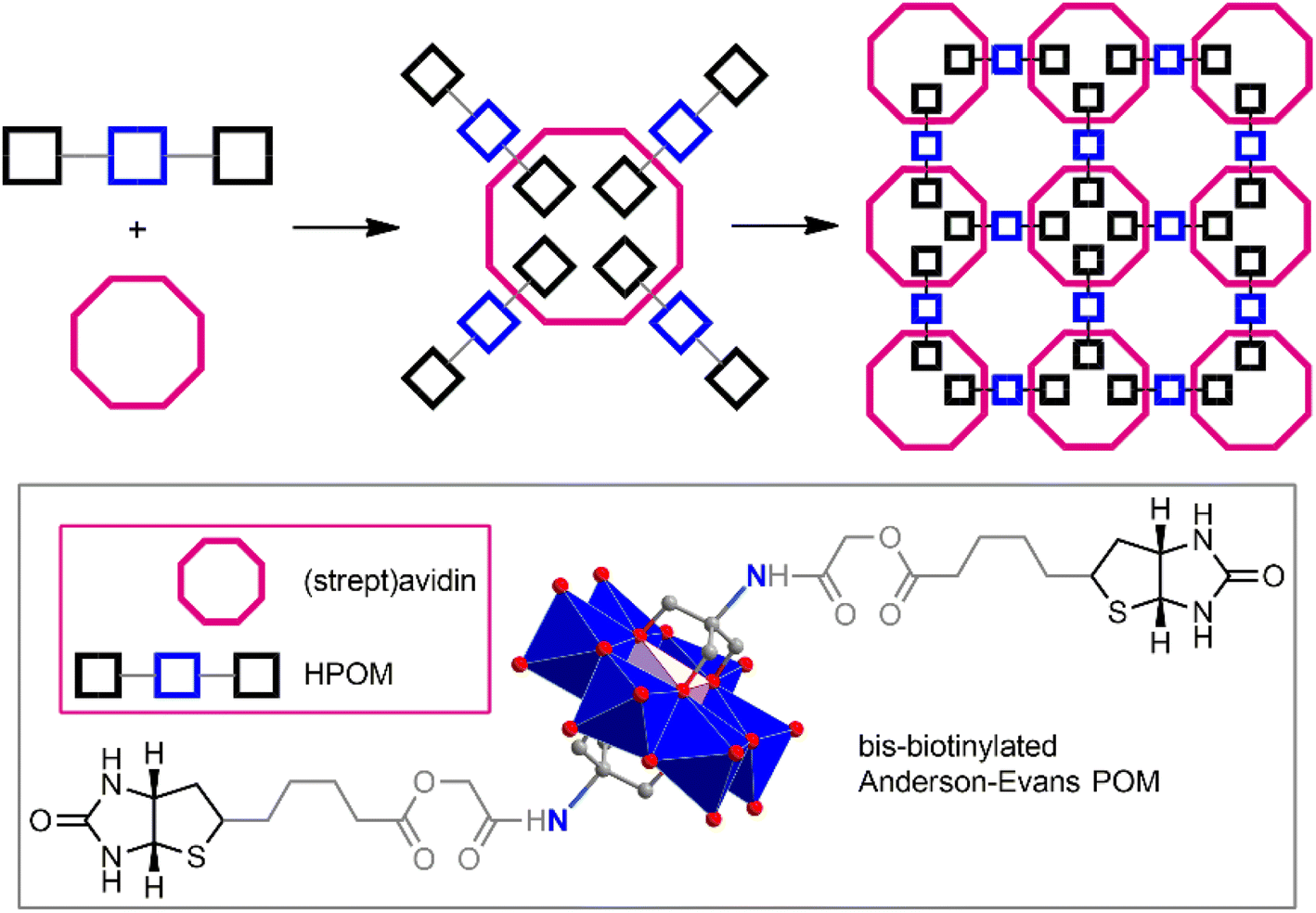 | ||
| Fig. 4 Self-assembly of nanoparticles from bis-biotinylated POM hybrid (HPOM) and streptavidin peptide driven by molecular recognition. | ||
3.3. Host–guest complexes involving polyoxometalates
Supramolecular host–guest complexes that modulate the chemical properties and solubility of target molecules are very widely used for medical and biological applications116,117 and can be employed as an elegant bioorthogonal approach for biorecognition.118 The large structural diversity of POMs, including rich functionalization and post-functionalization chemistry allows one to obtain a wide variety of supramolecular assemblies. POM anions are characterized by a high charge density, as well as nucleophilic oxygen atoms, and can interact with the polar regions of host molecules. On the other hand, functionalization of POMs with organic ligands provides effective hydrophobic interactions.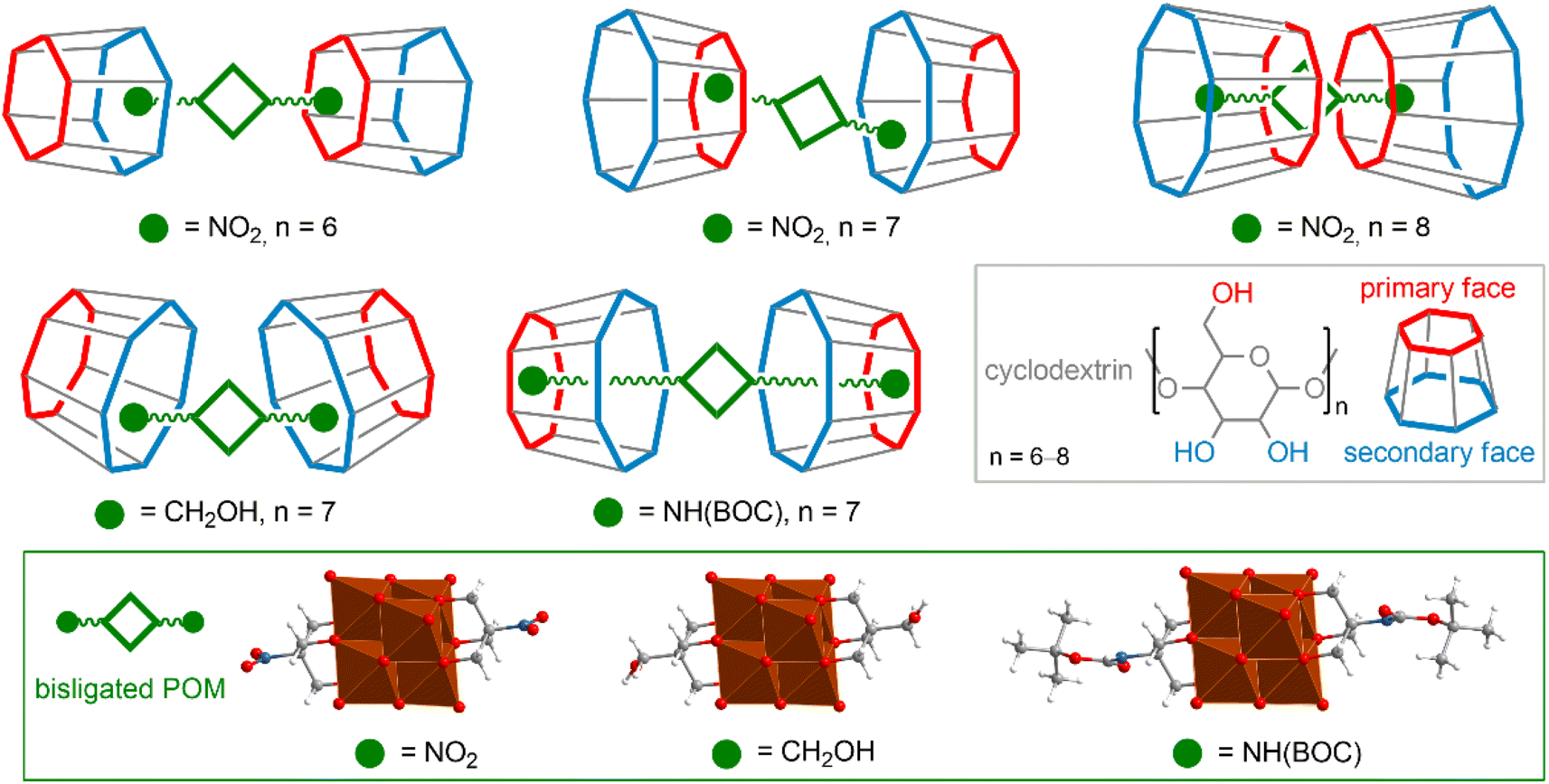 | ||
| Fig. 5 Schematic representation of possible arrangements of (bisligated POM)/CD pairs depending on the organic fragment and the size of the macrocycle. Illustrated using the example of several interacting [V6O13((OCH2)3C–R)2]2− units (R = NO2, CH2OH, NH(BOC)). | ||
Incorporation of completely inorganic POM cores into a suitably sized CD cavity is possible, but typically such complexation is not very robust. It is important to note that the incorporation of POMs into hydrophobic cavities is observed only for clusters with a moderate charge density. Thus, the all-inorganic Lindqvist-type hexamolybdates and hexatungstates [M6O19]2− can be stabilized by γ-CD in aqueous solution,129 whereas the smaller hexavanadate anion with a higher charge density is not stable even in the presence of host CDs.130 Another illustrative example of this concept is the complexation of Keggin-type POMs [XW11M]q− (X = P, Si; M = V or Mo) with γ-cyclodextrin, which is easily reversible by one-electron reduction of the POM core (i.e., increasing its surface charge density).131 Interestingly, giant POMs themselves like [Mo154O462H14(H2O)70]14− can act as host molecules in relation to CD macrocycles, which, in turn, are able to encapsulate smaller POM cores, creating hierarchical structures.132
As mentioned above, organic ligands associated with POM cores easily participate in the formation of host–guest pairs with CD molecules interacting with their hydrophobic cavity.120,133–136 It is noteworthy that in these cases, the interaction of POM with the hydrophobic CD regions is still possible and competes with hydrophobic forces. The interaction energy, as well as the structure of the resulting complex, depend on the components of the POM–organic hybrid.120 Generally, longer and more hydrophobic chains favor hydrophobic pairs, but pairs exhibiting both complexation mechanisms (interactions of the POM core with the hydrophilic site of CD and complexation of the hydrophobic organic tail with the CD cavity) are also common. The variety of patterns of complex POM–CD formation is well illustrated by a series of [V6O13((OCH2)3C–R)2]2− hybrids (Fig. 5), where the nature of the substituent –R and CD size are the factors that determine the energy and structure of the supramolecular assemblies.
Complexation of hybrid POM–organic molecules with CDs can be considered not only as a way to stabilize reagents in various environments, but also opens up very intriguing possibilities for preorganization of reagents in solution,134 chiral transport,135 and modulation of the self-assembly of supramolecular systems.136 Importantly, complexation with macrocyclic agents improves the stability of POM anions not only in solution, but also in the gas phase,137–139 and on surfaces,137 leading to advantages in related analytical protocols and technical applications.
Even less information is available about other macrocycles widely used in medicine, cucurbiturils.147 The Mn-substituted Anderson–Evans-type POMs covalently functionalized with β-cucurbituril (β-CB) moieties148 [MnMo6O18((OCH2)3CNH(CO)(CH2)3(CO)NH–(β-CB))2]3− can interact with surfactant agents of different nature, forming multi-stimuli responsive structured emulsions in two-phase media. The reported result is of particular interest due to the possibility of pre-targeting the host to the guest, which demonstrates a very promising non-classical bioorthogonal approach.118 Cucurbituril macrocycles have an exclusively hydrophobic cavity that can be used as a carrier for various molecules, as well as an ideal nanoreactor for a variety of pre-organized reagents for UV-induced coupling and copper-free alkyne–azide click chemistry.149 Therefore, one can hope that in the near future these systems will prove their value in biocompatible processes as well.
4 Polyoxometalate click chemistry
Among the toolset of methods150 that are generally classified as bioorthogonal chemical transformations, click coupling reactions, especially copper-assisted azide–alkyne cycloadditions (CuAAC), have found a significant place in the field of individual modifications of organic–inorganic POM hybrids.9,12,151–153 However, realizing of bioorthogonal click chemistry using these reagents is a challenging task that faces a number of fundamental difficulties. The section above was devoted to protecting POMs and biological media from each other, however, when the click process is achieved, one has to wonder if the reagents and catalysts in biological media are sufficiently stable in the presence of POMs and if they are tolerant to biological media and living cells. The properties of POMs as efficient oxidation catalysts seem to be the reason why no example of classical Staudinger154 conjugations involving POMs has yet been published. Azide and alkyne functional groups are not easily oxidized, so cycloaddition involving them can be considered the most promising approach. This seems to be valid for the case of catalyst-free processes, however, the addition of copper ions immediately leads to two problems: (i) Cu(I) and especially Cu(II) cations are significantly toxic and, therefore, incompatible with living cells; (ii) oxidation processes involving POMs can lead to deactivation of Cu(I) active sites and often prevent the use of additional reducing agents (such as sodium ascorbate). Thus, the development of biocompatible CuAAC chemistry requires very high activity of Cu catalysts along with stabilization of Cu(I) ions. Remarkable advances that have made CuAAC chemistry, initially nonbioorthogonal,3 suitable for biological applications include the chelation of azidomethylpyridine derivatives as click reagents155–157 and the use of tris(triazolyl)amines as Cu(I) stabilizing ligands.158–160 As a result, the reaction kinetics are dramatically accelerated, and very small amounts of toxic copper compounds are required. Despite the lack of examples of literal implementation of bioorthogonal processes involving POMs, we believe that it is worthwhile to summarize published data on click reactions of hybrid organic–inorganic POM compounds to provide a starting point for developing possible reaction conditions, expanding known reactivities, and exploiting available “click” substrates.4.1. Copper(I) assisted azide–alkyne cycloaddition
Implementing a traditional CuAAC protocol utilizing POM–organic structures as synthons (Fig. 6) requires finding process conditions that do not affect the POM core (i.e., the POM core must act as a chemically and redox innocent unit). In general, the modification processes of POM-based compounds described in the literature can be divided into two approaches: the use of a simple source of Cu(II) together with a reducing agent, usually CuSO4/NaAsc (method A); and direct introduction of “proper” Cu(I) ions in the form of CuI or some soluble complexes (method B). From a “chemistry in a flask” point of view, a combination of factors of catalyst activity and the stability of the reagents and catalyst with respect to reaction media dictates the choice of approach and reaction parameters. From the point of view of bioorthogonal chemistry, cytotoxicity, biocompatibility, and cell membrane permeability are extremely important and must be considered. | ||
| Fig. 6 Click chemistry for (post)functionalization of POMs: CuAAC conjugation of azide-functionalized POM to terminal acetylene (A); CuAAC conjugation of POM bearing a terminal acetylene group to azide (B); SPAAC conjugation of azide-functionalized POM to a 3′-dibenzocyclooctyne-derived building block (C). | ||
Simple and effective for “chemistry in a flask”152 method A is definitely not the method of choice for biological systems due to the high toxicity of copper ions not protected by strong ligands, as well as the oxidation products of ascorbate (Asc).4 Method B uses organic reaction media. In addition, Cu(I) centers complexed by nitrogen ligands or in equilibrium with the poorly soluble CuI solid phase exhibit significantly lower activity and productivity, so reactions require higher Cu loadings and harsh conditions. Moreover, since no reducing agent is used, the reactions must be carried out under an inert atmosphere. Thus, the implementation of typical CuAAC protocols related to both methods A and B could not be considered a method of bioorthogonal chemistry. Nevertheless, the fundamental possibility of CuAAC processes for numerous POM–organic hybrids as synthons and the available solid body of experimental data on this chemistry are of great importance.
The goal of this section is to analyze existing examples of CuAAC reactions involving organic–inorganic POM hybrids, especially in terms of the reaction media, conditions, and the mutual redox tolerance of POM cores, Cu(I) centers, and addition reagents.
Method A, which uses reducing agents in combination with Cu(II) compounds, can generally be used for POMs which oxidation potential is insufficient to accelerate the rapid oxidation of ascorbate induced by POM. The first work published in this area concerned low-oxidizing and stable polyoxotungstate azide and acetylene derivatives of the Keggin-type [PW11O61SnR]4− and Wells–Dawson-type [P2W17O61SnR]7− POMs (R = organic substituent bearing an azido or terminal acetylene moiety)161 coupled with different organic building blocks and even among themselves. The conventional CuAAC protocol involving a CuSO4/NaAsc – acetonitrile/aqueous system was found to work very well for these low-oxidizing POMs and gave good yields of the desired products without reducing the POM core despite the presence of ascorbate. After a short time, a series of works appeared devoted to Wells–Dawson-type polyoxotungstates, which carry two clickable functionalities, namely [α2-P2W17O61(Si–R)2O]6− and [α2-P2W17O61(P(O)–R)2]6−.162–164
Based on Wells–Dawson-type POMs grafted with azide162–164 or terminal acetylene162 functions and utilizing the CuSO4/NaAsc system, a wide variety of hybrid architectures were obtained, including hybrids bearing covalently attached zinc porphyrin moieties, which were proposed as promising molecular materials for light energy harvesting.163 The same approach also gave good results in the click post-functionalization of the Keggin-type POM [SiW11O39(SnCH2CH2CONH(CH2)3N3)]5− with an alkyne-derived ferrocene unit.165 Cronin et al. reported140 that sodium ascorbate in the presence of water readily reduces an Anderson–Evans-type POM [MnMo6O18((OCH2)3CNH(CO)R)2]3−. The same system demonstrates excellent results for modifications of this type of POM hybrids in DMF media.152,166 Thus, it can be concluded that the successful application of method A may be rather sensitive to process conditions due to the dependence of redox potentials on the solvent.167
Direct use of Cu(I) compounds as catalyst precursors without the addition of reducing agents (method B) is usually the method of choice for more oxidative POMs. To stabilize Cu(I) compounds, amines (usually N,N-diisopropylethylamine (DIPEA)) are added to the solution. The reactions are carried out in degassed polar organic solvents (DMF or MeCN). Thus, the aforementioned Anderson–Evans ‘POM–N3’ and ‘POM–acetylene’ MnMo-derivatives can be linked to each other,168 resulting in the formation of a series of monodisperse oligomeric POM hybrids from dimer to pentamer. This demonstrates the great potential of CuAAC chemistry for the controlled assembly of POM structures, even in the case of their vast redox activity and associated chemical reactivity.
The use of the [Cu(MeCN)4]BF4 precatalyst also makes it possible to obtain hybrids of these [MnMo6O24]3− POMs with photoactive moieties BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene).169,170 Related process using Cu(I) diimine complexes was reported for the CuAAC modification of Wells–Dawson-type POMs [α2-P2W17O61(P(O)CH2C6H4–N3)2]6− with fused aromatic pendant groups.145 This process in cyclohexanone requires microwave irradiation, which shows that strong α-diimine ligands provide very effective stabilization of Cu(I) in the presence of oxidative POMs, even under harsh conditions. However, unless the ligands are specifically tuned, the coordination of strong ligands significantly reduces catalyst activity. On the other hand, rational ligand design can not only stabilize Cu(I) in solution, but also modulate the solubility of the active species and significantly increase the click reaction rate. In bioorthogonal chemistry in aqueous solutions, the most promising ligands are water-soluble (and, apparently, promoting the formation of nanoaggregates in solution) chelating derivatives of methyltriazolylamine, such as 3-(4-((bis((1-(tert-butyl)-1H-1,2,3-triazol-4-yl)methyl)amino)methyl)-1H-1,2,3-triazol-1-yl)propane-1-sulfonic acid (BTTES).158,159
Vanadium(V)–oxo clusters are even stronger oxidizing agents, so the use of ascorbate-containing systems, as well as other reducing agents, for their modification is hardly possible, which makes method B the only possible synthetic approach. Therefore, azide-functionalized Wells–Dawson-type POM [P2V3W15O59((OCH2)3NH(CO)CH2–N3)]6− can be successfully clicked with alkynes in the presence of Cu(CH3CN)4]PF6 and DIPEA in MeCN.153 The system CuI/DIPEA in DMF showed good results in the clicking Lindqvist-type hexavanadates [V6O13((OCH2)3CCH2OCH2CCH)2]2− with organic azides,171 [V6O13((OCH2)3CCH2N3)2]2− with various organic acetylenes,151,172 and even with phosphinalkynyl complexes of gold(I) acting as the acetylene component.173,174 These reactions were carried out under oxygen-free conditions at elevated temperatures and lasted about 70 hours. Alternatively, the process can be significantly accelerated using microwave radiation.174 Sometimes method B is the method of choice even for non-oxidative POMs. For example, it was shown175 that in the presence of Cu(I) ions, the [γ-H2SiW10O36Cu2(μ-1,1-N3)2]4− POM can act as an azide component in cycloaddition reactions.
4.2. Other cycloadditions
Copper-free cycloaddition reactions are of great interest for bioorthogonal chemistry due to the absence of toxic copper ions. In addition, challenges related to the redox stability of catalysts, as well as potentially toxic reducing agents and ligands, are avoided.A powerful bioorthogonal, strain-promoted azide–alkyne cycloaddition176 (SPAAC, Fig. 6C) can be successfully implemented to covalently couple [V6O13((OCH2)3CCH2N3)2]2− to ssDNA strands conjugated with 3′-dibenzocyclooctyne tags177 (Fig. 7A). The resulting hybrid is incorporated into a DNA origami structure178 and, although not fully realized in living cells, can be considered, to our knowledge, the only successful implementation of true bioorthogonal POM chemistry to date.
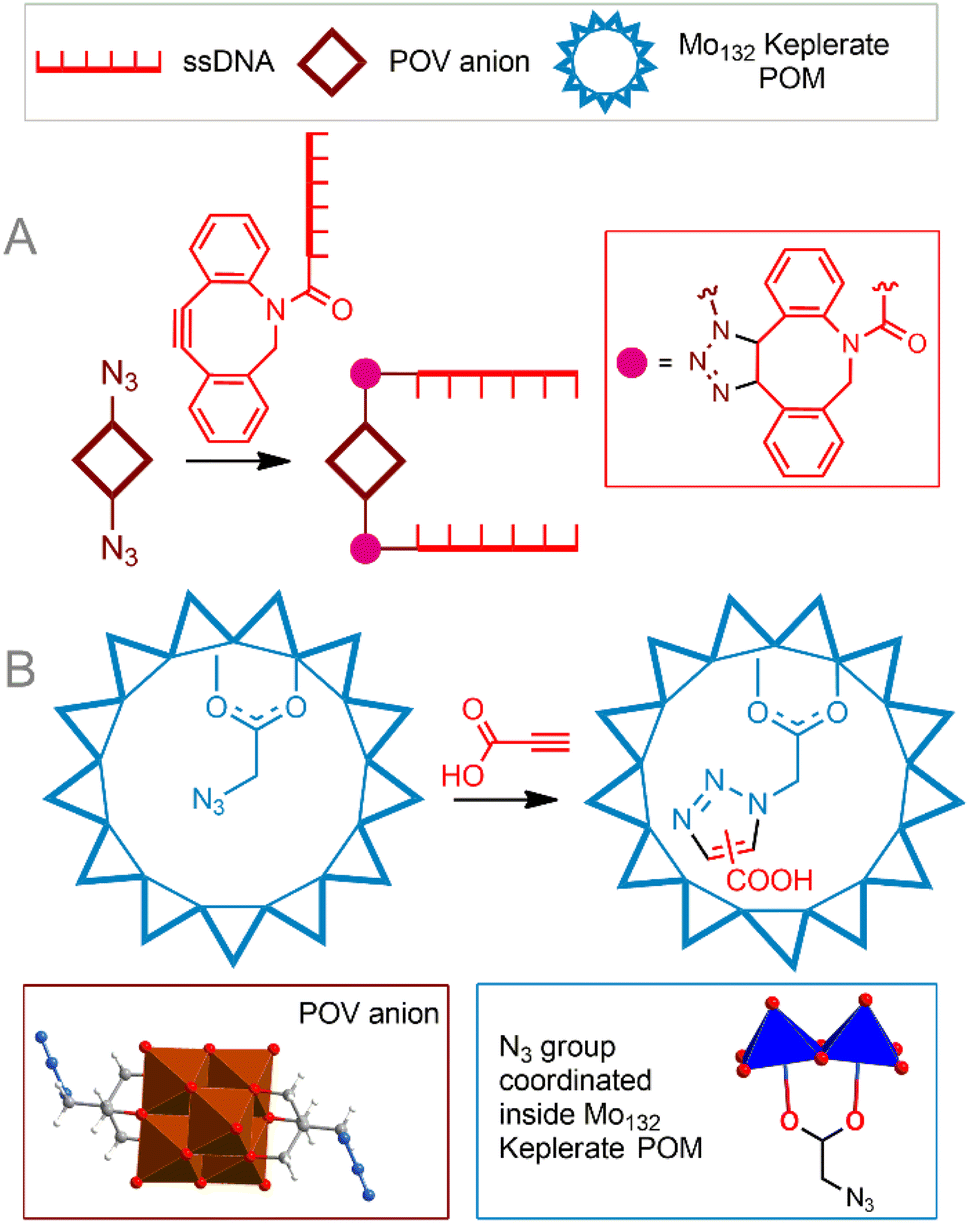 | ||
| Fig. 7 Strain-promoted azide–alkyne cycloaddition to tailor POM–DNA conjugates (A). Cycloaddition reaction in a cavity of the Mo132-nuclearity Keplerate nanoreactor (B). POV is polyoxovanadate. | ||
An interesting example of the copper-free 1,3-dipolar azide–alkyne Huisgen cycloaddition is the reaction process that causes a supramolecular preorganization of the reactants. Kögerler et al.69 demonstrated the catalyst-free cycloaddition of propiolic acid to the azide functionality structurally exposed within the [Mo132O372(AcO)30(H2O)72]42− Keplerate cavity, leading to a mixture of 1,4- and 1,5-triazole products (Fig. 7B). Although the regioselectivity of the process could not compete with classical CuAAC reactions, this approach can be considered a good solution for introducing functional groups inside the POM cavity, since it avoids both the challenge of accessing the transition metal catalyst inside the cavity and its release after the reaction. Given the possibility of supramolecular protection of these giant POMs in solution, they can be viewed as potential biocompatible nanoreactors. The next promising approach is to incorporate catalytically active transition metal sites into the POM structure. Metal centers are capable of activating organic molecules, thereby leading to the functionalization of POM. This concept was demonstrated by Sokolov et al.179 Keggin-type polyoxotungstate modified with a Ru center [PW11O39{RuIII(N3)}]4− was functionalized with a tetrazolyl group by activating acetonitrile and azide ligands in the ruthenium coordination sphere. Reported reaction conditions included high temperatures and organic solvents; the reaction is not selective. It is interesting to note that for the related molybdenum iodide cluster, azide-nitrile cycloadditions exhibited better selectivity.180 It is also important that the introduction of a potentially catalytically active center into the POM core can significantly change its properties and the selectivity of the catalyst. Thus, the Cu(II)–N3 modified POM [γ-H2SiW10O36Cu2(μ-1,1-N3)2]4− did not enter into cycloaddition with acetylenes, but acted as an effective catalyst for the oxidative homocoupling of acetylene.181
An alternative to introducing a transition metal center into the POM structure is to carry out inorganic iClick reactions, which use σ-alkynyl as well as azide complexes of Au,182–186 Ir, Rh187 or Pt188,189 as synthetic analogs of organic acetylenes and azides, respectively. It was shown that Au(I) azide complexes stabilized by phosphine-donor ligands readily react with terminal acetylene groups bound on the Lindqvist-type hexavanadate structure in the absence of a copper catalyst, forming stable [V6O13{(OCH2)3CCH2OCH2(C2N3H)AuP(C6H4OCH3)3}2]2− heterometallic hybrids.173 However, biological aqueous environments with complex ionic compositions pose a major challenge for such processes due to the generally limited stability of metal–C bonds. In addition, although toxic copper is not used, the toxicity of other heavy transition metal ions may be even higher, which also seriously limits the biological application of iClick approach.
5 Bioorthogonal approaches for surface engineering
The developed principles of bioorthogonal chemistry provide a very powerful set of tools not only for in vivo reactions, but also for applications involving the selective reactivity of complex biomolecules,5 including surface modification and other engineering challenges.190,191 The unique properties of redox-active POMs are the reason for their many potential applications, e.g. in electronic42–45 and sensor technologies.192 The remarkable opportunities for precision surface engineering offered by biomolecular self-assembly approaches,193 as well as the prospects for using POM-modified surface assemblies in biological applications, are prompting scientists to pay attention to the bioorthogonality of POMs and the chemical approaches used. The use of biomolecules is of particular importance for the implementation of switchable POM elements in neuromorphic electronics.194In this section, we point out some of the features and key approaches used to create assemblies incorporating various surfaces, biological molecules, and POMs. Some of the examples included here are not directly related to bioorthogonal chemistry and biological entities but demonstrate excellent states of the art and concepts, that are important for surface engineering and can be easily transferred in the bio-related fields.
The interaction of POMs with surfaces is the subject of a lot of research, summarized in numerous reviews.195–199 Surfaces of various natures can be functionalized with POMs (or POM–organic hybrids), which usually leads to a spontaneous arrangement of POM molecules on the surface. To make POM hybrids resistant to desorption on the surface, they are often modified with certain moieties (Fig. 8) that exhibit strong noncovalent interactions with the surface (e.g., condensed aromatic moieties for graphene-like surfaces200,201 or gold-coordinating sulfur atoms202). Covalent functionalization of surfaces by POMs198 through e.g., amine bonds, click chemistry or S–Au bonding also typically results in a random spatial distribution of hybrid POM molecules.174
 | ||
| Fig. 8 Organo-functionalized POMs tethered on the surfaces with covalent or non-covalent interaction (A–D). Incorporation of monoligated POMs into a self-assembled monolayer on the Au surface (E). | ||
Numerous examples demonstrate the high potential of using CuAAC click chemistry to graft Wells–Dawson polyoxotungstates onto polymer chains,203 ethynyl-functionalized polymer resin surfaces,204 and graphene.175 Notably, in the published cases the reactions were not optimized to meet bioorthogonality criteria and typically used high Cu loadings along with toxic amine and organic media. We believe that the approaches discussed in Section 3 can greatly improve their applicability to biological systems.
A useful approach to surface modification with a controlled number of POM units is the incorporation of POM building blocks into a self-assembled monolayer comprising POM–organic hybrids carrying some surface targeting function and molecules forming an auxiliary layer with the same function. This approach, together with the principles of biological chemistry, can be successfully used to create, for example, a superior electrochemical sensor for detecting single nucleotide polymorphisms. The authors of a number of publications described the covalent addition of 7-deaza-7-propargylaminopurines to activated Keggin-type [SiW11O39(Sn(CH2)2CO)]4− and Dawson-type [P2W17O61(Sn(CH2)2CO)]6− POMs via amide formation.205–207 The resulting POM–dideoxynucleotide conjugates could then be incorporated into the DNA chain using Terminator® polymerase.207 The resulting POM-decorated chains were attached to the Au surface using a previously developed approach involving the incorporation of thiolate-functionalized target molecules into thiolate-based self-assembled monolayers (Fig. 8E).208
As mentioned in Section 2.2, anionic POMs show very strong interaction with peptide molecules. The latter can be derivatized by functional groups (which are derived from natural or synthetic amino acids), providing affinity to certain surfaces,209,210 and opening up exciting possibilities in terms of supramolecular and surface chemistry.211 For example, peptide-functionalized acrylic monomers were used to construct a polymer chain that carries multiple peptide residues and thus acts as a “brush” that absorbs POMs.212 Peptides bearing aromatic groups have good affinity for graphene-like surfaces. Thus, diphenylalanine peptide readily forms hybrid nanoparticles encapsulating H3PW12O40 POMs. These nanoparticles were then immobilized on graphene oxide nanosheets, forming a ternary hybrid system.213
The random and not always uniform distribution of grafted functional fragments on the surface leads to potentially low reproducibility of the properties of the composite. This is an important challenge from the point of view of molecular electronics, where the controlled addressing of logic elements is crucial. However, these surface characteristics can be improved by supramolecular self-organization of POMs through special organic ligation.
For example, modification of hydrophilic Wells–Dawson-type {P2V3W15}-POM with a dendritic tether bearing four hydrophobic oligomeric silsesquioxane polyhedra leads to the formation of amphiphilic hybrids. The pre-organized geometry of the latter allows them to be assembled into mesoscale, hexagonal honeycomb lattice of single- or multi-layers214 (Fig. 9A).
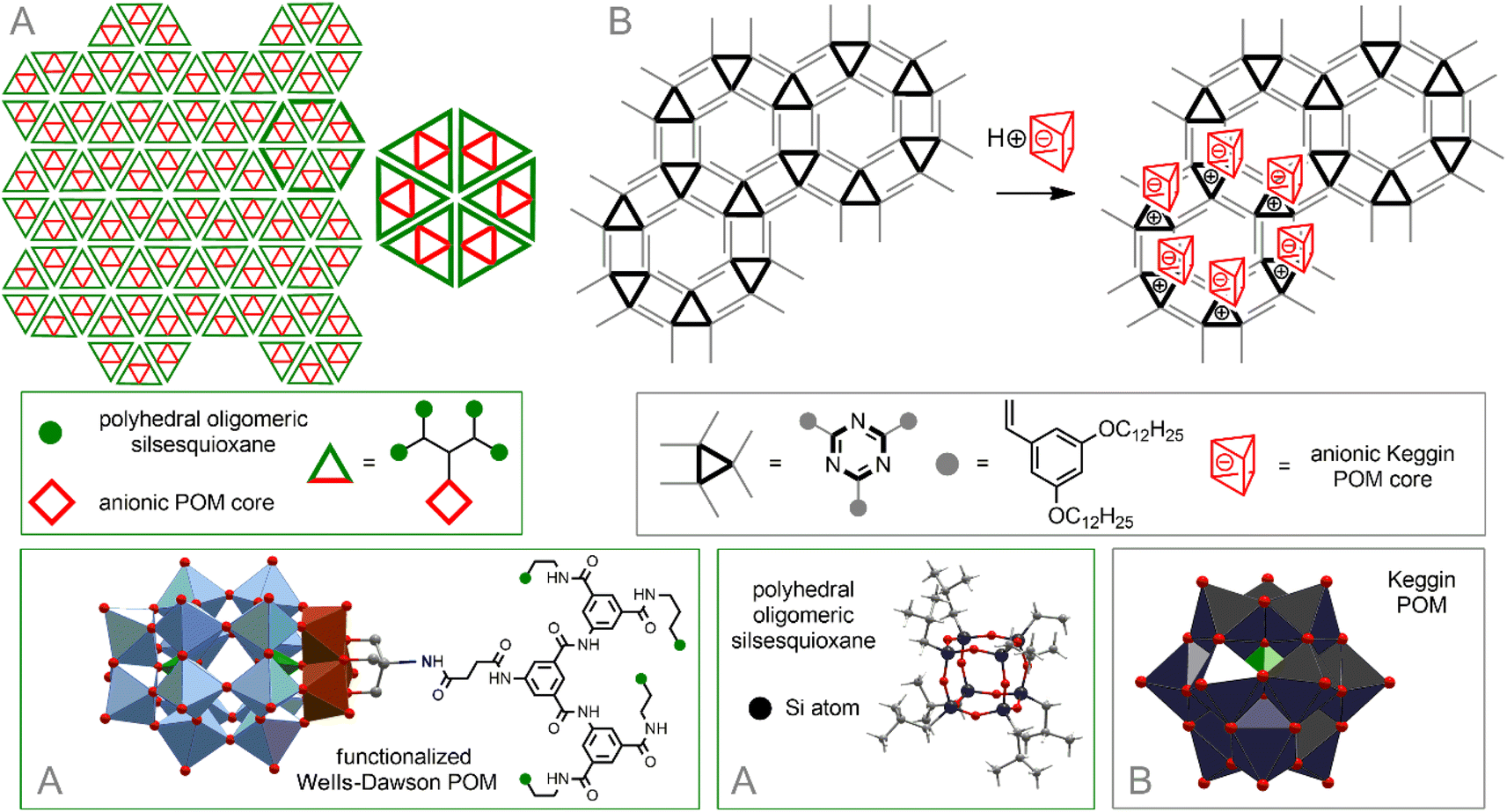 | ||
| Fig. 9 Self-organization of POM–silsesquioxane co-clusters (A). Arrangement of inorganic POM in a hexagonal network formed by triazine-based units (B). | ||
A powerful method for arranging molecules on surfaces is to prefunctionalize the surface with a preorganized template matrix. It was shown that a supramolecular 2D self-assembled network formed as a result of hydrophobic interactions between triazine derivatives with hydrophobic strands serves as an ideal matrix for placing hydrophilic inorganic POMs on the surface of HOPG215 (Fig. 9B). The formation of a well-organized hexagonal network assembled by strong intramolecular H-bonds between melamine and perylene-3,4,9,10-tetracarboxylic acid diimide on the Au surface was also described.216 POM–organic hybrids [PW11O39Ge(p–C6H4–CC–C6H4–NHC(O)(CH2)4(–CH(CH2)2S–S–))]4− augmented with an 1,2-dithiolane group readily enter into supramolecular interactions with this network, simultaneously with the immobilization of hybrid molecules on the surface through Au–S bonds.
The breakthrough discovery217 of DNA folding principles for 2D and 3D molecular (DNA origami) engineering of materials greatly expands the possibilities for programmable assembly of nanostructures. A method that allows controlled design of nanostructures starting from a computer model218 is the subject of numerous extensive reviews,178,193,219–223 demonstrating the virtually unlimited variety of structures with arbitrary shape and precise addressability that can be achieved. The rich structural capabilities as well as the apparent biocompatibility of DNA origami lead to its numerous uses in delicate applications such as gene engineering224 and precision drug delivery.225
The unique folding properties of DNA molecules that make them not only genetic material but also ideal building blocks for bio-nanotechnology include: (i) specific lock-and-key interactions between complementary bases in DNA polymers, resulting in DNA strand pairing and helix packing formation; (ii) ability of DNA strands to cross from one double-stranded DNA helix (dsDNA) to another, forming the rigid junction (Fig. 10A and B); (iii) a targeted and well-controlled structure that can be arbitrarily tuned using well-characterized DNA template synthesis approaches.226
 | ||
| Fig. 10 DNA crossover between helices: helix representation (A); base pairing and schematic line representation showing antiparallel base pairing with 5′ and 3′ ends denoted by squares and triangles, respectively (B); DNA origami structure involving scaffold ssDNA strand (black line) and multiple short staple ssDNA strands (colored lines) (C). | ||
The beauty of the DNA origami method lies in the combination of long single-stranded scaffold DNA (scaffold ssDNA) and short-stranded oligonucleotides (staple ssDNA), which are arranged in a programmed structure by forming cross-over bonding. The combination of these short and long strands, together with the selectivity of crossover formation, results in high yields of desired structures, while rational design of both scaffold and staple DNA sequences is key to precise folding of structures. The formed DNA origami building blocks can further participate in interactions with each other, forming complex predesigned structures (Fig. 10C). Additionally, it is important to note that ssDNA staple strands can be easily modified by incorporating desired functional groups, thereby assembling a biocompatible nanostructure with well-defined positions of these groups. From a surface engineering point of view, this allows, on the one hand, to link the DNA origami assembly to fragments that can be immobilized, e.g., on a metallic surface. On the other hand, the desired functional fragment of interest may also be included. Recently, this approach was successfully implemented177 to construct POM–DNA hybrid structures on the Au(111) surface. For their self-assembling, commercially available ssDNA staple strands modified with thiolate groups (providing immobilization on the Au surface), as well as ssDNA strands serving as complementary pairs for separately synthesized ssDNA hybrids with POM fragments attached via bioorthogonal strain-promoted click reactions were used. Since the POM here is the symmetric azide-augmented hexavanadate [V6O13((OCH2)3CCH2N3)2]2−, the resulting POM–DNA hybrids are symmetric POM(–ssDNA)2 molecules. Thus, the latter can be coordinated to the origami structure with only one or both staple strands of ssDNA (Fig. 11). Remarkably, the hexavanadate units attached to the DNA origami retained their potential-induced multistate switching at room temperature. As a result, the use of bioorthogonal and biocompatible approaches (SPAAC + DNA origami) to fabricate switchable, POM-modified Au materials was successfully demonstrated. However, the statistical distribution of these mono- and double-paired POM fragments is a limiting factor for accurate assembly and precise control of the DNA origami structure, since such factors as the number of bound POM–ssDNA hybrids as well as their surface density are difficult to completely monitor. It can be assumed that the use of monoazide derivatives (monofunctionalized POMs or asymmetric bifunctionalized derivatives) may be beneficial for more controlled assembly.
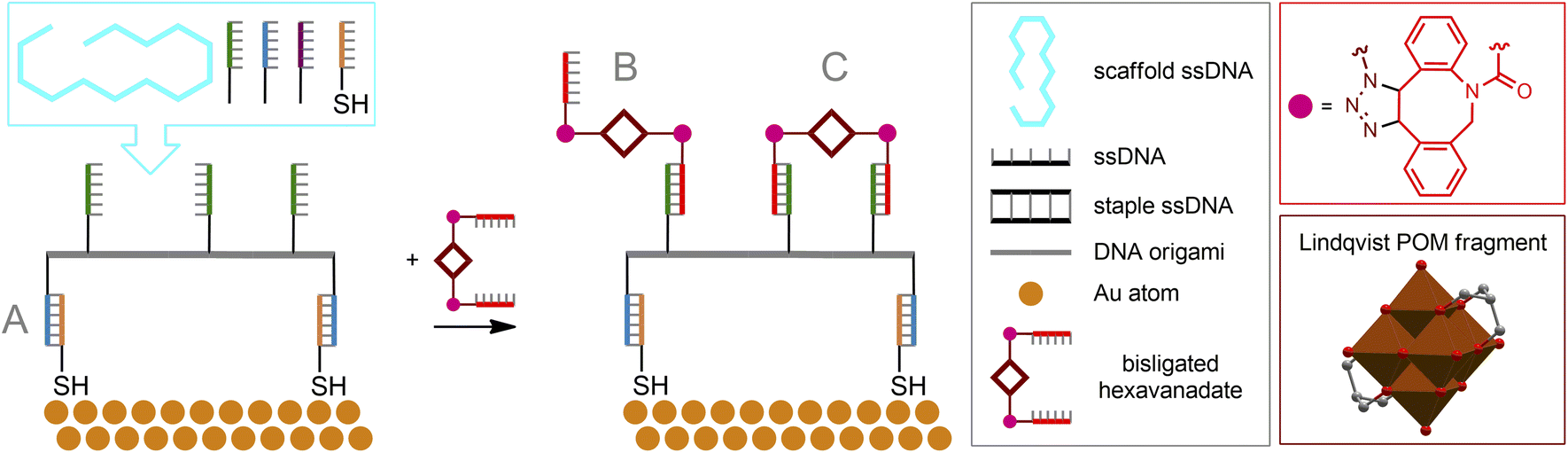 | ||
| Fig. 11 DNA origami structure assembly involving POM–ssDNA building blocks. The preformed DNA origami structure immobilized on Au(111) through thiol groups (A) and conjugated to Lindqvist-type hexavanadate units augmented with ssDNA fragments by pairing complementary nucleotide sequences (B and C). | ||
6 Conclusion and outlook
The programmable structural and electronic properties of POMs have led to numerous advances in their application in catalysis, electronics, sensing, and many other fields. Their potential application in medicine, bio-imaging, and cell labeling, as well as the prospects for integrating POM-based electronics with biological systems, make the idea of realizing powerful bioorthogonal chemistry using the polyoxoanions as molecular substrates/reagents very attractive. However, this idea faces numerous challenges and limitations, resulting in a paucity of reported examples of such chemistry. Strong oxidative behavior and polarizing effect generally lead to non-selective interaction of POMs with biological molecules and the cellular environment. Tailored organic ligation and the use of a supramolecular approach can significantly improve the stability of POM units in aqueous media, resulting in better cell permeability and tolerance to biomolecules. Incorporation of POM anions into the core–shell vesicles or nanoassemblies with peptides is a versatile and efficient way of POM introduction in living cells. This approach is widely used in biomedical studies of POM-based drugs and can be suggested as a key element of bioorthogonal POM chemistry. The rich possibilities of peptide synthesis and modification, as well as advances in the design of POM–peptide conjugates, including examples of the use of the principle of site-specific molecular recognition, have brought POM building blocks significantly closer to bioorthogonal reagents.High reactivity and oxidative potential of POMs require very careful adaptation of the available bioorthogonal chemistry reactions to these substrates. There are numerous examples of the use of click chemistry to create hybrid organic–inorganic POM compounds. Most of them are conventional CuAAC reactions that cannot be considered bioorthogonal processes. Nevertheless, the use of specific substrates and advanced ligand design approaches can make CuAAC processes highly suitable for bioorthogonal chemistry, so the fundamental feasibility of such processes and the synthetic availability of appropriate substrates are of great importance in terms of potential participation in biocompatible processes. Furthermore, the successful examples of fully bioorthogonal SPAAC reactions have also been described for POMs. This click chemistry coupled with a DNA origami approach provides an attractive toolkit for surface engineering, including precisely targeted biological building blocks as well as POMs.
The main goal of the present perspective is to show that, despite the above-mentioned challenges, bioorthogonal POM chemistry is not impossible. Our Perspective cannot be used as a definitive guide and due to the still insufficient amount of experimental data represents only a systematic overview of approaches to implementing bioorthogonal processes for solution chemistry and functional surface engineering with POMs. We believe that the described examples and methods should inspire scientists to develop new efficient protocols for incorporating POMs into bioorthogonal chemical transformations, which will open the door to a variety of biomedical applications over the coming decade.
Author contributions
S. K. P.: conceptualization, investigation, data curation, writing and editing – original draft; E. V. G.: visualization, editing – original draft; K. Y. M.: conceptualization, project supervision, writing and editing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) project number 495326276. S. P. thanks the German-Russian Interdisciplinary Science Center (G-RISC) for travel grant F-2021 b-10_r. E. V. G. appreciates financial support from the Russian Science Foundation, grant 21-13-00052.References
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211–1219 CrossRef CAS.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. Am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Nat. Rev. Methods Primers, 2021, 1, 30 CrossRef CAS PubMed.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS.
- A. Dolbecq, E. Dumas, C. d. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- M. T. Pope and U. Kortz, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2012 Search PubMed.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- D. E. Salazar Marcano, G. Kalandia, M. A. Moussawi, K. Van Hecke and T. N. Parac-Vogt, Chem. Sci., 2023, 14, 5405–5414 RSC.
- P. Gouzerh, R. Villanneau, R. Delmont and A. Proust, Chem.–Eur. J., 2000, 6, 1184–1192 CrossRef.
- G. Izzet, F. Volatron and A. Proust, Chem. Rec., 2017, 17, 250–266 CrossRef CAS PubMed.
- C. Yvon, A. J. Surman, M. Hutin, J. Alex, B. O. Smith, D. L. Long and L. Cronin, Angew. Chem., Int. Ed., 2014, 53, 3336–3341 CrossRef CAS.
- D. Ventura, A. Calderan, C. Honisch, S. Krol, S. Serratì, M. Bonchio, M. Carraro and P. Ruzza, Pept. Sci., 2018, 110, e24047 CrossRef.
- Y. Zhang, J. Wang, Z. Chen, Y. Xie, J. Wu, X. Wang, D. Zang and T. Liu, Mater. Today Commun., 2022, 33, 104991 CrossRef CAS.
- M. Aureliano, S. G. Mitchell and P. Yin, Front. Chem., 2022, 10, 977317 CrossRef.
- N. I. Gumerova and A. Rompel, Inorg. Chem., 2021, 60, 6109–6114 CrossRef CAS PubMed.
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169 RSC.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980–2999 CrossRef CAS PubMed.
- D. Chang, Y. Li, Y. Chen, X. Wang, D. Zang and T. Liu, Nanoscale Adv., 2022, 4, 3689–3706 RSC.
- A. Blazevic and A. Rompel, Coord. Chem. Rev., 2016, 307, 42–64 CrossRef CAS.
- W.-L. Chen, H.-Q. Tan and E.-B. Wang, J. Coord. Chem., 2011, 65, 1–18 CrossRef.
- G. Malliaras and I. McCulloch, Chem. Rev., 2022, 122, 4323–4324 CrossRef CAS PubMed.
- M. Berggren, E. D. Glowacki, D. T. Simon, E. Stavrinidou and K. Tybrandt, Chem. Rev., 2022, 122, 4826–4846 CrossRef CAS PubMed.
- M. Jelikic-Stankov, S. Uskokovic-Markovic, I. Holclajtner-Antunovic, M. Todorovic and P. Djurdjevic, J. Trace Elem. Med. Biol., 2007, 21, 8–16 CrossRef CAS.
- B. M. Hoffman, D. Lukoyanov, Z. Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- C. H. Wang, C. Zhang and X. H. Xing, Bioengineered, 2016, 7, 395–405 CrossRef CAS PubMed.
- S. J. Stohs and D. Bagchi, Free Radical Biol. Med., 1995, 18, 321–336 CrossRef CAS PubMed.
- D. Rehder, Coord. Chem. Rev., 1999, 182, 297–322 CrossRef.
- D. Rehder, Inorganics, 2023, 11(6), 256 CrossRef CAS.
- J. B. Majithiya, R. Balaraman, R. Giridhar and M. R. Yadav, J. Trace Elem. Med. Biol., 2005, 18, 211–217 CrossRef CAS PubMed.
- K. M. Wasan, V. Risovic, V. G. Yuen and J. H. McNeill, J. Trace Elem. Med. Biol., 2006, 19, 251–258 CrossRef CAS.
- S. Pawa and S. Ali, Biochim. Biophys. Acta, Mol. Basis Dis., 2004, 1688, 210–222 CrossRef CAS PubMed.
- M. C. Carmona, M. Amigó, S. Barceló-Batllori, M. Julià, Y. Esteban, S. Moreno and R. Gomis, Int. J. Obes., 2009, 33, 534–540 CrossRef CAS PubMed.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed., 1991, 30, 34–48 CrossRef.
- I. A. Weinstock, R. E. Schreiber and R. Neumann, Chem. Rev., 2018, 118, 2680–2717 CrossRef CAS PubMed.
- N. I. Gumerova and A. Rompel, Sci. Adv., 2023, 9, eadi0814 CrossRef CAS PubMed.
- N. I. Gumerova and A. Rompel, Chem. Soc. Rev., 2020, 49, 7568–7601 RSC.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- X. Kong, G. Wan, B. Li and L. Wu, J. Mater. Chem. B, 2020, 8, 8189–8206 RSC.
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327–358 CrossRef CAS PubMed.
- L. Vila-Nadal, S. G. Mitchell, S. Markov, C. Busche, V. Georgiev, A. Asenov and L. Cronin, Chem.–Eur. J., 2013, 19, 16502–16511 CrossRef CAS.
- C. Busche, L. Vila-Nadal, J. Yan, H. N. Miras, D. L. Long, V. P. Georgiev, A. Asenov, R. H. Pedersen, N. Gadegaard, M. M. Mirza, D. J. Paul, J. M. Poblet and L. Cronin, Nature, 2014, 515, 545–549 CrossRef CAS PubMed.
- N. S. Sterin, N. Basu, M. Cahay, M. N. Satyanarayan, S. S. Mal and P. P. Das, Phys. Status Solidi A, 2020, 217, 2000306 CrossRef.
- M. Moors, J. Warneke, X. López, C. de Graaf, B. Abel and K. Y. Monakhov, Acc. Chem. Res., 2021, 54, 3377–3389 CrossRef CAS.
- L. S. Van Rompuy and T. N. Parac-Vogt, Curr. Opin. Biotechnol., 2019, 58, 92–99 CrossRef CAS PubMed.
- D. E. Salazar Marcano, N. D. Savic, K. Declerck, S. A. M. Abdelhameed and T. N. Parac-Vogt, Chem. Soc. Rev., 2024, 53, 84–136 RSC.
- S. Brunle, M. L. Eisinger, J. Poppe, D. J. Mills, J. D. Langer, J. Vonck and U. Ermler, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 26497–26504 CrossRef.
- B. Kowalewski, J. Poppe, U. Demmer, E. Warkentin, T. Dierks, U. Ermler and K. Schneider, J. Am. Chem. Soc., 2012, 134, 9768–9774 CrossRef CAS.
- J. Schemberg, K. Schneider, U. Demmer, E. Warkentin, A. Muller and U. Ermler, Angew. Chem., Int. Ed., 2007, 46, 2408–2413 CrossRef CAS PubMed.
- S. J. Lippard and J. M. Berg, Principles Of Bioinorganic Chemistry, University Science Books, 1994 Search PubMed.
- J. Idiago-Lopez, E. Moreno-Antolin, M. Eceiza, J. M. Aizpurua, V. Grazu, J. M. de la Fuente and R. M. Fratila, Bioconjugate Chem., 2022, 33, 1620–1633 CrossRef CAS PubMed.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC.
- P. Yin, D. Li and T. Liu, Chem. Soc. Rev., 2012, 41, 7368–7383 RSC.
- M. Stuckart and K. Y. Monakhov, Chem. Sci., 2019, 10, 4364–4376 RSC.
- J. M. Cameron, G. Guillemot, T. Galambos, S. S. Amin, E. Hampson, K. Mall Haidaraly, G. N. Newton and G. Izzet, Chem. Soc. Rev., 2022, 51, 293–328 RSC.
- L. Fu, H. Gao, M. Yan, S. Li, X. Li, Z. Dai and S. Liu, Small, 2015, 11, 2938–2945 CrossRef CAS PubMed.
- H. Soria-Carrera, E. Atrian-Blasco, R. Martin-Rapun and S. G. Mitchell, Chem. Sci., 2022, 14, 10–28 RSC.
- J. Li, J. Wang, Y. Zhao, P. Zhou, J. Carter, Z. Li, T. A. Waigh, J. R. Lu and H. Xu, Coord. Chem. Rev., 2020, 421, 213418 CrossRef CAS.
- S. Polarz, S. Landsmann and A. Klaiber, Angew. Chem., Int. Ed., 2014, 53, 946–954 CrossRef CAS PubMed.
- N. Dib, V. R. Girardi, J. J. Silber, N. M. Correa and R. D. Falcone, Org. Biomol. Chem., 2021, 19, 4969–4977 RSC.
- Y. Lu, G. Allegri and J. Huskens, Mater. Horiz., 2022, 9, 892–907 RSC.
- L. De Matteis, S. G. Mitchell and J. M. de la Fuente, J. Mater. Chem. B, 2014, 2, 7114–7117 RSC.
- Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849–2864 CrossRef CAS.
- D. G. Kurth, P. Lehmann, D. Volkmer, A. Müller and D. Schwahn, J. Chem. Soc., Dalton Trans., 2000, 21, 3989–3998 RSC.
- D. Pakulski, A. Gorczyński, W. Czepa, Z. Liu, L. Ortolani, V. Morandi, V. Patroniak, A. Ciesielski and P. Samorì, Energy Storage Mater., 2019, 17, 186–193 CrossRef.
- S. Polarz, B. Smarsly and M. Antonietti, ChemPhysChem, 2001, 2, 457–461 CrossRef CAS.
- X. Jia, D. Fan, P. Tang, J. Hao and T. Liu, J. Cluster Sci., 2006, 17, 467–478 CrossRef CAS.
- C. Besson, S. Schmitz, K. M. Capella, S. Kopilevich, I. A. Weinstock and P. Kögerler, Dalton Trans., 2012, 41, 9852–9854 RSC.
- H. Li, Y. Yang, Y. Wang, W. Li, L. Bi and L. Wu, Chem. Commun., 2010, 46, 3750–3752 RSC.
- F. Wang, S. Cao, J. Men, N. Lei and R. Wang, Colloids Surf., A, 2020, 601, 125056 CrossRef CAS.
- Y. Yan, B. Li, W. Li, H. Li and L. Wu, Soft Matter, 2009, 5, 4047–4053 RSC.
- Y. F. Song, D. L. Long, C. Ritchie and L. Cronin, Chem. Rec., 2011, 11, 158–171 CrossRef CAS PubMed.
- A. Nisar and X. Wang, Dalton Trans., 2012, 41, 9832–9845 RSC.
- A. Nisar, X. Xu, S. Shen, S. Hu and X. Wang, Adv. Funct. Mater., 2009, 19, 860–865 CrossRef CAS.
- T. Sun, W. Cui, M. Yan, G. Qin, W. Guo, H. Gu, S. Liu and Q. Wu, Adv. Mater., 2016, 28, 7397–7404 CrossRef CAS.
- G. Hu, W. Chang, S. An, B. Qi and Y.-F. Song, Chin. Chem. Lett., 2022, 33, 3968–3972 CrossRef CAS.
- S.-J. Yu, Y.-K. Han and W. Wang, Polymer, 2019, 162, 73–79 CrossRef CAS.
- Y. Xiao, Y. K. Han, N. Xia, M. B. Hu, P. Zheng and W. Wang, Chem.–Eur. J., 2012, 18, 11325–11333 CrossRef CAS PubMed.
- P. Yin, C. P. Pradeep, B. Zhang, F. Y. Li, C. Lydon, M. H. Rosnes, D. Li, E. Bitterlich, L. Xu, L. Cronin and T. Liu, Chem.–Eur. J., 2012, 18, 8157–8162 CrossRef CAS PubMed.
- S. Chakraborty, L. Jin, Y. Li, Y. Liu, T. Dutta, D. M. Zhu, X. Yan, A. Keightley and Z. Peng, Eur. J. Inorg. Chem., 2013, 1799–1807 CrossRef CAS.
- F. Haso, R. Wang, P. Yin, J. Zhou, L. Jin, Z. Peng and T. Liu, Eur. J. Inorg. Chem., 2014, 4589–4592 CrossRef CAS.
- S. She, Z. Huang, P. Yin, A. Bayaguud, H. Jia, Y. Huang, Y. Wei and Y. Wei, Chem.–Eur. J., 2017, 23, 14860–14865 CrossRef CAS PubMed.
- S. Landsmann, M. Wessig, M. Schmid, H. Colfen and S. Polarz, Angew. Chem., Int. Ed., 2012, 51, 5995–5999 CrossRef CAS PubMed.
- S. Landsmann, C. Lizandara-Pueyo and S. Polarz, J. Am. Chem. Soc., 2010, 132, 5315–5321 CrossRef CAS.
- J. Zhang, Y. F. Song, L. Cronin and T. Liu, J. Am. Chem. Soc., 2008, 130, 14408–14409 CrossRef CAS PubMed.
- Y. F. Song, N. McMillan, D. L. Long, J. Thiel, Y. Ding, H. Chen, N. Gadegaard and L. Cronin, Chem.–Eur. J., 2008, 14, 2349–2354 CrossRef CAS PubMed.
- B. Zhang, J. Song, D. Li, L. Hu, C. L. Hill and T. Liu, Langmuir, 2016, 32, 12856–12861 CrossRef CAS PubMed.
- M. G. Evich, M. J. B. Davis, J. P. McCord, B. Acrey, J. A. Awkerman, D. R. U. Knappe, A. B. Lindstrom, T. F. Speth, C. Tebes-Stevens, M. J. Strynar, Z. Wang, E. J. Weber, W. M. Henderson and J. W. Washington, Science, 2022, 375, eabg9065 CrossRef CAS PubMed.
- Y. Chu, A. Saad, P. Yin, J. Wu, O. Oms, A. Dolbecq, P. Mialane and T. Liu, Chem.–Eur. J., 2016, 22, 11756–11762 CrossRef CAS.
- X. Yan, P. Zhu, J. Fei and J. Li, Adv. Mater., 2010, 22, 1283–1287 CrossRef CAS PubMed.
- L. Tong, Z. Wang, C. Xia, Y. Yang, S. Yuan, D. Sun and X. Xin, J. Phys. Chem. B, 2017, 121, 10566–10573 CrossRef CAS PubMed.
- H. Zhang, L. Guo, Z. Xie, X. Xin, D. Sun and S. Yuan, Langmuir, 2016, 32, 13736–13745 CrossRef CAS PubMed.
- P. F. Gao, Y. X. Liu, L. Zhang, S. Zhang, H. W. Li, Y. Wu and L. Wu, J. Colloid Interface Sci., 2018, 514, 407–414 CrossRef CAS PubMed.
- C. Zhang, M. Zhao, H. Zou, X. Zhang, R. Sheng, Y. Zhang, B. Zhang, C. Li and Y. Qi, J. Inorg. Biochem., 2020, 212, 111212 CrossRef CAS PubMed.
- X. Xie, L. Wang, X. Liu, Z. Du, Y. Li, B. Li, L. Wu and W. Li, Chem. Commun., 2020, 56, 1867–1870 RSC.
- L. Vandebroek, H. Noguchi, K. Kamata, J. R. H. Tame, L. Van Meervelt, T. N. Parac-Vogt and A. R. D. Voet, Cryst. Growth Des., 2021, 21, 1307–1313 CrossRef CAS.
- L. Vandebroek, H. Noguchi, K. Kamata, J. R. H. Tame, L. Van Meervelt, T. N. Parac-Vogt and A. R. D. Voet, Chem. Commun., 2020, 56, 11601–11604 RSC.
- J. Li, Z. Chen, M. Zhou, J. Jing, W. Li, Y. Wang, L. Wu, L. Wang, Y. Wang and M. Lee, Angew. Chem., Int. Ed., 2016, 55, 2592–2595 CrossRef CAS PubMed.
- J. Luo, B. Zhang, C. Yvon, M. Hutin, S. Gerislioglu, C. Wesdemiotis, L. Cronin and T. Liu, Eur. J. Inorg. Chem., 2019, 380–386 CrossRef CAS.
- D. Y. Fu, S. Zhang, Z. Qu, X. Yu, Y. Wu and L. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 6137–6145 CrossRef CAS PubMed.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS PubMed.
- X. Wang, F. Li, S. Liu and M. T. Pope, J. Inorg. Biochem., 2005, 99, 452–457 CrossRef CAS PubMed.
- N. Gao, H. Sun, K. Dong, J. Ren, T. Duan, C. Xu and X. Qu, Nat. Commun., 2014, 5, 3422 CrossRef PubMed.
- E. Atrian-Blasco, L. de Cremoux, X. Lin, R. Mitchell-Heggs, L. Sabater, S. Blanchard and C. Hureau, Chem. Commun., 2022, 58, 2367–2370 RSC.
- N. Song, M. Lu, J. Liu, M. Lin, P. Shangguan, J. Wang, B. Shi and J. Zhao, Angew. Chem., Int. Ed., 2024, e202319700 CAS.
- D. Vilona, D. Lachkar, E. Dumont, M. Lelli and E. Lacote, Chem.–Eur. J., 2017, 23, 13323–13327 CrossRef CAS PubMed.
- S. She, N. L. Bell, D. Zheng, J. S. Mathieson, M. D. Castro, D.-L. Long, J. Koehnke and L. Cronin, Chem, 2022, 8, 2734–2748 CAS.
- V. Tagliavini, C. Honisch, S. Serrati, A. Azzariti, M. Bonchio, P. Ruzza and M. Carraro, RSC Adv., 2021, 11, 4952–4957 RSC.
- N. Gao, Z. Liu, H. Zhang, C. Liu, D. Yu, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2022, 61, e202115336 CrossRef CAS PubMed.
- Y. Yasueda, T. Tamura, A. Fujisawa, K. Kuwata, S. Tsukiji, S. Kiyonaka and I. Hamachi, J. Am. Chem. Soc., 2016, 138, 7592–7602 CrossRef CAS PubMed.
- H. Soria-Carrera, I. Franco-Castillo, P. Romero, S. Martin, J. M. de la Fuente, S. G. Mitchell and R. Martín-Rapún, Angew. Chem., Int. Ed., 2021, 60, 3449–3453 CrossRef CAS PubMed.
- H. Soria-Carrera, E. Atrian-Blasco, J. M. de la Fuente, S. G. Mitchell and R. Martin-Rapun, Nanoscale, 2022, 14, 5999–6006 RSC.
- V. A. Zamolo, G. Modugno, E. Lubian, A. Cazzolaro, F. Mancin, L. Giotta, D. Mastrogiacomo, L. Valli, A. Saccani, S. Krol, M. Bonchio and M. Carraro, Front. Chem., 2018, 6, 278 CrossRef.
- D. E. Salazar Marcano, S. Lentink, J. J. Chen, A. V. Anyushin, M. A. Moussawi, J. Bustos, B. Van Meerbeek, M. Nyman and T. N. Parac-Vogt, Small, 2024, e2312009 CrossRef PubMed.
- J. Wankar, N. G. Kotla, S. Gera, S. Rasala, A. Pandit and Y. A. Rochev, Adv. Funct. Mater., 2020, 30, 1909049 CrossRef CAS.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- Y.-L. Ma, S. Yan, X.-J. Xu, H. Cao and R. Wang, Chin. Chem. Lett., 2024, 35, 108645 CrossRef CAS.
- E. M. M. Del Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef CAS.
- I. Fa Bamba, C. Falaise, J. Marrot, P. Atheba, G. Gbassi, D. Landy, W. Shepard, M. Haouas and E. Cadot, Chem.–Eur. J., 2021, 27, 15516–15527 CrossRef CAS PubMed.
- Y. Wu, R. Shi, Y. L. Wu, J. M. Holcroft, Z. Liu, M. Frasconi, M. R. Wasielewski, H. Li and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 4111–4118 CrossRef CAS PubMed.
- M. A. Moussawi, N. Leclerc-Laronze, S. Floquet, P. A. Abramov, M. N. Sokolov, S. Cordier, A. Ponchel, E. Monflier, H. Bricout, D. Landy, M. Haouas, J. Marrot and E. Cadot, J. Am. Chem. Soc., 2017, 139, 12793–12803 CrossRef CAS.
- L. Ni, H. Li, H. Xu, C. Shen, R. Liu, J. Xie, F. Zhang, C. Chen, H. Zhao, T. Zuo and G. Diao, ACS Appl. Mater. Interfaces, 2019, 11, 38708–38718 CrossRef CAS PubMed.
- S. Yao, C. Falaise, N. Leclerc, C. Roch-Marchal, M. Haouas and E. Cadot, Inorg. Chem., 2022, 61, 4193–4203 CrossRef CAS PubMed.
- X. Liu, J. Zhang, Y. Lan, Q. Zheng and W. Xuan, Inorg. Chem. Front., 2022, 9, 6534–6543 RSC.
- S. Khlifi, J. Marrot, M. Haouas, W. E. Shepard, C. Falaise and E. Cadot, J. Am. Chem. Soc., 2022, 144, 4469–4477 CrossRef CAS PubMed.
- L. Ni, J. Gu, X. Jiang, H. Xu, Z. Wu, Y. Wu, Y. Liu, J. Xie, Y. Wei and G. Diao, Angew. Chem., Int. Ed., 2023, 62, e202306528 CrossRef CAS PubMed.
- P. Yang, W. Zhao, A. Shkurenko, Y. Belmabkhout, M. Eddaoudi, X. Dong, H. N. Alshareef and N. M. Khashab, J. Am. Chem. Soc., 2019, 141, 1847–1851 CrossRef CAS PubMed.
- C. Falaise, M. A. Moussawi, S. Floquet, P. A. Abramov, M. N. Sokolov, M. Haouas and E. Cadot, J. Am. Chem. Soc., 2018, 140, 11198–11201 CrossRef CAS PubMed.
- A. C. F. Ribeiro, V. M. M. Lobo, A. J. M. Valente, S. M. N. Simões, A. J. F. N. Sobral, M. L. Ramos and H. D. Burrows, Polyhedron, 2006, 25, 3581–3587 CrossRef CAS.
- S. Yao, C. Falaise, S. Khlifi, N. Leclerc, M. Haouas, D. Landy and E. Cadot, Inorg. Chem., 2021, 60, 7433–7441 CrossRef CAS PubMed.
- M. A. Moussawi, M. Haouas, S. Floquet, W. E. Shepard, P. A. Abramov, M. N. Sokolov, V. P. Fedin, S. Cordier, A. Ponchel, E. Monflier, J. Marrot and E. Cadot, J. Am. Chem. Soc., 2017, 139, 14376–14379 CrossRef CAS PubMed.
- G. Izzet, M. Menand, B. Matt, S. Renaudineau, L. M. Chamoreau, M. Sollogoub and A. Proust, Angew. Chem., Int. Ed., 2012, 51, 487–490 CrossRef CAS PubMed.
- W. Guan, G. Wang, J. Ding, B. Li and L. Wu, Chem. Commun., 2019, 55, 10788–10791 RSC.
- B. Zhang, L. Yue, Y. Wang, Y. Yang and L. Wu, Chem. Commun., 2014, 50, 10823–10826 RSC.
- H. Li, F. Jiang, G. Zhang, B. Li and L. Wu, Dalton Trans., 2019, 48, 5168–5175 RSC.
- F. Yang, M. Moors, D. A. Hoang, S. Schmitz, M. Rohdenburg, H. Knorke, A. Charvat, X.-B. Wang, K. Y. Monakhov and J. Warneke, ACS Appl. Nano Mater., 2022, 5, 14216–14220 CrossRef CAS.
- P. Su, A. J. Smith, J. Warneke and J. Laskin, J. Am. Soc. Mass Spectrom., 2019, 30, 1934–1945 CrossRef CAS PubMed.
- Y. Fan, Y. Zhang, Q. Jia, J. Cao and W. Wu, Mass Spectrom. Lett., 2015, 6, 13–16 CrossRef.
- S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366–386 RSC.
- A. Isik, M. Oguz, A. Kocak and M. Yilmaz, J. Inclusion Phenom. Macrocyclic Chem., 2022, 102, 439–449 CrossRef CAS.
- C. Aronica, G. Chastanet, E. Zueva, S. A. Borshch, J. M. Clemente-Juan and D. Luneau, J. Am. Chem. Soc., 2008, 130, 2365–2371 CrossRef CAS PubMed.
- M. Wang, Y. Guo, G. Zhao, B. Chen and Y. Bi, Chin. Chem. Lett., 2023, 34, 107365 CrossRef CAS.
- M. Zhang, M. Chen, Y. Bi, L. Huang, K. Zhou and Z. Zheng, J. Mater. Chem. A, 2019, 7, 12893–12899 RSC.
- X. Hang, Y. Yu, Z. Wang and Y. Bi, Cryst. Growth Des., 2020, 20, 7934–7940 CrossRef CAS.
- Z. Wang, Y. J. Zhu, B. L. Han, Y. Z. Li, C. H. Tung and D. Sun, Nat. Commun., 2023, 14, 5295 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- Z. Xia, C. G. Lin, Y. Yang, Y. Wang, Z. Wu, Y. F. Song, T. P. Russell and S. Shi, Angew. Chem., Int. Ed., 2022, 61, e202203741 CrossRef CAS PubMed.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, J. Org. Chem., 2002, 48, 3619–3620 CrossRef.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- H. Jia, Q. Li, A. Bayaguud, Y. Huang, S. She, K. Chen and Y. Wei, Dalton Trans., 2018, 47, 577–584 RSC.
- S. Vanhaecht, J. Jacobs, L. Van Meervelt and T. N. Parac-Vogt, Dalton Trans., 2015, 44, 19059–19062 RSC.
- S. Vanhaecht, T. Quanten and T. N. Parac-Vogt, Dalton Trans., 2017, 46, 10215–10219 RSC.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- C. Uttamapinant, A. Tangpeerachaikul, S. Grecian, S. Clarke, U. Singh, P. Slade, K. R. Gee and A. Y. Ting, Angew. Chem., Int. Ed., 2012, 51, 5852–5856 CrossRef CAS PubMed.
- C. Uttamapinant, M. I. Sanchez, D. S. Liu, J. Z. Yao and A. Y. Ting, Nat. Protoc., 2013, 8, 1620–1634 CrossRef PubMed.
- H. Jiang, T. Zheng, A. Lopez-Aguilar, L. Feng, F. Kopp, F. L. Marlow and P. Wu, Bioconjugate Chem., 2014, 25, 698–706 CrossRef CAS PubMed.
- C. Besanceney-Webler, H. Jiang, T. Zheng, L. Feng, D. Soriano del Amo, W. Wang, L. M. Klivansky, F. L. Marlow, Y. Liu and P. Wu, Angew. Chem., Int. Ed., 2011, 50, 8051–8056 CrossRef CAS PubMed.
- D. Soriano Del Amo, W. Wang, H. Jiang, C. Besanceney, A. C. Yan, M. Levy, Y. Liu, F. L. Marlow and P. Wu, J. Am. Chem. Soc., 2010, 132, 16893–16899 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- K. Micoine, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, Org. Lett., 2007, 9, 3981–3984 CrossRef CAS PubMed.
- A. Harriman, K. J. Elliott, M. A. H. Alamiry, L. L. Pieux, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer and F. Odobel, J. Phys. Chem. C, 2009, 113, 5834–5842 CrossRef CAS.
- K. J. Elliott, A. Harriman, L. Le Pleux, Y. Pellegrin, E. Blart, C. R. Mayer and F. Odobel, Phys. Chem. Chem. Phys., 2009, 11, 8767–8773 RSC.
- F. Odobel, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer, K. J. Elliott and A. Harriman, Chem.–Eur. J., 2009, 15, 3130–3138 CrossRef CAS.
- A. M. Debela, M. Ortiz, C. K. Ósullivan, S. Thorimbert and B. Hasenknopf, Polyhedron, 2014, 68, 131–137 CrossRef CAS.
- S. Cetindere, S. T. Clausing, M. Anjass, Y. Luo, S. Kupfer, B. Dietzek and C. Streb, Chem.–Eur. J., 2021, 27, 17181–17187 CrossRef CAS.
- H. Svith, H. Jensen, J. Almstedt, P. Andersson, T. Lundbäck, K. Daasbjerg and M. Jonsson, J. Phys. Chem. A, 2004, 108, 4805–4811 CrossRef CAS.
- A. Macdonell, N. A. B. Johnson, A. J. Surman and L. Cronin, J. Am. Chem. Soc., 2015, 137, 5662–5665 CrossRef CAS PubMed.
- B. Matt, S. Renaudineau, L. M. Chamoreau, C. Afonso, G. Izzet and A. Proust, J. Org. Chem., 2011, 76, 3107–3112 CrossRef CAS PubMed.
- A. Saad, O. Oms, A. Dolbecq, C. Menet, R. Dessapt, H. Serier-Brault, E. Allard, K. Baczko and P. Mialane, Chem. Commun., 2015, 51, 16088–16091 RSC.
- O. Linnenberg, A. Kondinski, C. Stöcker and K. Y. Monakhov, Dalton Trans., 2017, 46, 15636–15640 RSC.
- O. Nachtigall and J. Spandl, Chem.–Eur. J., 2018, 24, 2785–2789 CrossRef CAS PubMed.
- S. K. Petrovskii, V. V. Khistiaeva, A. A. Sizova, V. V. Sizov, A. V. Paderina, I. O. Koshevoy, K. Y. Monakhov and E. V. Grachova, Inorg. Chem., 2020, 59, 16122–16126 CrossRef CAS PubMed.
- S. K. Petrovskii, M. Moors, S. Schmitz, E. V. Grachova and K. Y. Monakhov, Chem. Commun., 2023, 59, 9517–9520 RSC.
- X. Zhang, Y. Li, Y. Li, S. Wang and X. Wang, ACS Appl. Nano Mater., 2019, 2, 6971–6981 CrossRef CAS.
- D. Bauer, S. M. Sarrett, J. S. Lewis and B. M. Zeglis, Nat. Protoc., 2023, 18, 1659–1668 CrossRef CAS PubMed.
- E. Vogelsberg, M. Moors, A. S. Sorokina, D. A. Ryndyk, S. Schmitz, J. S. Freitag, A. V. Subbotina, T. Heine, B. Abel and K. Y. Monakhov, Chem. Mater., 2023, 35, 5447–5457 CrossRef CAS.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- A. A. Mukhacheva, A. L. Gushchin, V. V. Yanshole, P. A. Abramov and M. N. Sokolov, Molecules, 2020, 25, 1859 CrossRef CAS.
- A. Mironova, A. Gushchin, P. Abramov, I. Eltsov, A. Ryadun and M. Sokolov, Polyhedron, 2021, 205, 115282 CrossRef CAS.
- K. Kamata, S. Yamaguchi, M. Kotani, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2008, 47, 2407–2410 CrossRef CAS.
- D. V. Partyka, J. B. Updegraff, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2007, 26, 183–186 CrossRef CAS.
- T. J. Del Castillo, S. Sarkar, K. A. Abboud and A. S. Veige, Dalton Trans., 2011, 40, 8140–8144 RSC.
- A. R. Powers, I. Ghiviriga, K. A. Abboud and A. S. Veige, Dalton Trans., 2015, 44, 14747–14752 RSC.
- X. Yang, S. Wang, I. Ghiviriga, K. A. Abboud and A. S. Veige, Dalton Trans., 2015, 44, 11437–11443 RSC.
- C. C. Beto, C. J. Zeman, Y. Yang, J. D. Bullock, E. D. Holt, A. Q. Kane, T. A. Makal, X. Yang, I. Ghiviriga, K. S. Schanze and A. S. Veige, Inorg. Chem., 2020, 59, 1893–1904 CrossRef CAS PubMed.
- C. C. Beto, X. Yang, A. R. Powers, I. Ghiviriga, K. A. Abboud and A. S. Veige, Polyhedron, 2016, 108, 87–92 CrossRef CAS.
- K. Peng, R. Einsele, P. Irmler, R. F. Winter and U. Schatzschneider, Organometallics, 2020, 39, 1423–1430 CrossRef CAS.
- K. Peng, V. Mawamba, E. Schulz, M. Löhr, C. Hagemann and U. Schatzschneider, Inorg. Chem., 2019, 58, 11508–11521 CrossRef CAS PubMed.
- P. Zhan, T. Wen, Z. G. Wang, Y. He, J. Shi, T. Wang, X. Liu, G. Lu and B. Ding, Angew. Chem., Int. Ed., 2018, 57, 2846–2850 CrossRef CAS.
- Y. X. Chen, G. Triola and H. Waldmann, Acc. Chem. Res., 2011, 44, 762–773 CrossRef CAS PubMed.
- M. I. S. Verissimo, D. V. Evtuguin and M. Gomes, Front. Chem., 2022, 10, 840657 CrossRef CAS PubMed.
- T. Torring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646 RSC.
- J. Li, H. Abbas, D. S. Ang, A. Ali and X. Ju, Nanoscale Horiz., 2023, 8, 1456–1484 RSC.
- E. Rafiee and S. Eavani, RSC Adv., 2016, 6, 46433–46466 RSC.
- S. Herrmann, C. Ritchie and C. Streb, Dalton Trans., 2015, 44, 7092–7104 RSC.
- Y. Ji, L. Huang, J. Hu, C. Streb and Y.-F. Song, Energy Environ. Sci., 2015, 8, 776–789 RSC.
- Y. Zhou, G. Chen, Z. Long and J. Wang, RSC Adv., 2014, 4, 42092–42113 RSC.
- A. S. Cherevan, S. P. Nandan, I. Roger, R. Liu, C. Streb and D. Eder, Adv. Sci., 2020, 7, 1903511 CrossRef CAS.
- I. Werner, J. Griebel, A. Masip-Sanchez, X. Lopez, K. Zaleski, P. Kozlowski, A. Kahnt, M. Boerner, Z. Warneke, J. Warneke and K. Y. Monakhov, Inorg. Chem., 2023, 62, 3761–3775 CrossRef CAS PubMed.
- L. Huang, J. Hu, Y. Ji, C. Streb and Y. F. Song, Chem.–Eur. J., 2015, 21, 18799–18804 CrossRef CAS PubMed.
- O. Linnenberg, M. Moors, A. Notario-Estévez, X. López, C. de Graaf, S. Peter, C. Baeumer, R. Waser and K. Y. Monakhov, J. Am. Chem. Soc., 2018, 140, 16635–16640 CrossRef CAS.
- M. B. Hu, N. Xia, W. Yu, C. Ma, J. Tang, Z. Y. Hou, P. Zheng and W. Wang, Polym. Chem., 2012, 3, 617–620 RSC.
- Y. Xiao, D. Chen, N. Ma, Z. Hou, M. Hu, C. Wang and W. Wang, RSC Adv., 2013, 3, 21544–21551 RSC.
- M. Ortiz, A. M. Debela, M. Svobodova, S. Thorimbert, D. Lesage, R. B. Cole, B. Hasenknopf and C. K. O'Sullivan, Chem.–Eur. J., 2017, 23, 10597–10603 CrossRef CAS PubMed.
- A. M. Debela, M. Ortiz, V. Beni, S. Thorimbert, D. Lesage, R. B. Cole, C. K. O'Sullivan and B. Hasenknopf, Chem.–Eur. J., 2015, 21, 17721–17727 CrossRef CAS PubMed.
- N. Chahin, L. A. Uribe, A. M. Debela, S. Thorimbert, B. Hasenknopf, M. Ortiz, I. Katakis and C. K. O'Sullivan, Biosens. Bioelectron., 2018, 117, 201–206 CrossRef CAS PubMed.
- O. Y. Henry, J. L. Sanchez and C. K. O'Sullivan, Biosens. Bioelectron., 2010, 26, 1500–1506 CrossRef CAS PubMed.
- X. Liu, J. Xu, X. Xie, Z. Ma, T. Zheng, L. Wu, B. Li and W. Li, Chem. Commun., 2020, 56, 11034–11037 RSC.
- X. Li, T. Zheng, X. Liu, Z. Du, X. Xie, B. Li, L. Wu and W. Li, Langmuir, 2019, 35, 4995–5003 CrossRef CAS.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- L. P. Datta, R. Mukherjee, S. Biswas and T. K. Das, Langmuir, 2017, 33, 14195–14208 CrossRef CAS.
- Z. Ma, Y. Qiu, H. Yang, Y. Huang, J. Liu, Y. Lu, C. Zhang and P. Hu, ACS Appl. Mater. Interfaces, 2015, 7, 22036–22045 CrossRef CAS.
- X. S. Hou, G. L. Zhu, L. J. Ren, Z. H. Huang, R. B. Zhang, G. Ungar, L. T. Yan and W. Wang, J. Am. Chem. Soc., 2018, 140, 1805–1811 CrossRef CAS.
- J. Salhi, J. P. Calupitan, M. Mattera, D. Montero, A. Miche, R. Maruchenko, A. Proust, G. Izzet, D. Kreher, I. Arfaoui and F. Volatron, Nanoscale, 2023, 15, 13233–13238 RSC.
- A. Lombana, C. Rinfray, F. Volatron, G. Izzet, N. Battaglini, S. Alves, P. Decorse, P. Lang and A. Proust, J. Phys. Chem. C, 2016, 120, 2837–2845 CrossRef CAS.
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- S. M. Douglas, A. H. Marblestone, S. Teerapittayanon, A. Vazquez, G. M. Church and W. M. Shih, Nucleic Acids Res., 2009, 37, 5001–5006 CrossRef CAS PubMed.
- S. Dey, C. Fan, K. V. Gothelf, J. Li, C. Lin, L. Liu, N. Liu, M. A. D. Nijenhuis, B. Saccà, F. C. Simmel, H. Yan and P. Zhan, Nat. Rev. Methods Primers, 2021, 1, 13 CrossRef CAS.
- N. C. Seeman, Mol. Biotechnol., 2007, 37, 246–257 CrossRef CAS PubMed.
- J. M. Majikes and J. A. Liddle, J. Res. Natl. Inst. Stand., 2021, 126, 126001 CrossRef CAS.
- P. Zhan, A. Peil, Q. Jiang, D. Wang, S. Mousavi, Q. Xiong, Q. Shen, Y. Shang, B. Ding, C. Lin, Y. Ke and N. Liu, Chem. Rev., 2023, 123, 3976–4050 CrossRef CAS PubMed.
- Y. Zhou, J. Dong and Q. Wang, NPG Asia Mater., 2023, 15, 25 CrossRef CAS.
- X. Wu, C. Yang, H. Wang, X. Lu, Y. Shang, Q. Liu, J. Fan, J. Liu and B. Ding, J. Am. Chem. Soc., 2023, 145, 9343–9353 CrossRef CAS PubMed.
- Q. Zhang, Q. Jiang, N. Li, L. Dai, Q. Liu, L. Song, J. Wang, Y. Li, J. Tian, B. Ding and Y. Du, ACS Nano, 2014, 8, 6633–6643 CrossRef CAS PubMed.
- R. K. O'Reilly, A. J. Turberfield and T. R. Wilks, Acc. Chem. Res., 2017, 50, 2496–2509 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |