
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evidence for Suzuki–Miyaura cross-couplings catalyzed by ligated Pd3-clusters: from cradle to grave†
Neda
Jeddi
a,
Neil W. J.
Scott
 a,
Theo
Tanner
a,
Simon K.
Beaumont
*b and
Ian J. S.
Fairlamb
*a
a,
Theo
Tanner
a,
Simon K.
Beaumont
*b and
Ian J. S.
Fairlamb
*a
aDepartment of Chemistry, University of York, York, YO20 5DD, UK. E-mail: ian.fairlamb@york.ac.uk
bDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: simon.beaumont@durham.ac.uk
First published on 15th January 2024
Abstract
Pdn clusters offer unique selectivity and exploitable reactivity in catalysis. Understanding the behavior of Pdn clusters is thus critical for catalysis, applied synthetic organic chemistry and greener outcomes for precious Pd. The Pd3 cluster, [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3][Cl] (denoted as Pd3Cl2), which exhibits distinctive reactivity, was synthesized and immobilized on a phosphine-functionalized polystyrene resin (denoted as immob-Pd3Cl2). The resultant material served as a tool to study closely the role of Pd3 clusters in a prototypical Suzuki–Miyaura cross-coupling of 4-fluoro-1-bromobenzene and 4-methoxyphenyl boronic acid at varying low Pd ppm concentrations (24, 45, and 68 ppm). Advanced heterogeneity tests such as Hg poisoning and the three-phase test showed that leached mononuclear or nanoparticulate Pd are unlikely to be the major active catalyst species under the reaction conditions tested. EXAFS/XANES analysis from (pre)catalyst and filtered catalysts during and after catalysis has shown the intactness of the triangular structure of the Pd3X2 cluster, with exchange of chloride (X) by bromide during catalytic turnover of bromoarene substrate. This finding is further corroborated by treatment of immob-Pd3Cl2 after catalyzing the Suzuki–Miyaura reaction with excess PPh3, which releases the cluster from the polymer support and so permits direct observation of [Pd3(μ-Br)(μ-PPh2)2(PPh3)3]+ ions by ESI-MS. No evidence is seen for a proposed intermediate in which the bridging halogen on the Pd3 motif is replaced by an aryl group from the organoboronic acid, i.e. formed by a transmetallation-first process. Our findings taken together indicate that the ‘Pd3X2’ motif is an active catalyst species, which is stabilized by being immobilized, providing a more robust Pd3 cluster catalyst system. Non-immobilized Pd3Cl2 is less stable, as is followed by stepwise XAFS of the non-immobilized Pd3Cl2, which gradually changes to a species consistent with ‘Pdx(PPh3)y’ type material. Our findings have far-reaching future implications for Pd3 cluster involvement in catalysis, showing that immobilization of Pd3 cluster species offers advantages for rigorous mechanistic examination and applied chemistries.
Introduction
Suzuki–Miyaura cross-coupling (SMCC) reactions are ubiquitous in applied chemical synthesis laboratories around the world, forming a keystone in the synthesis of an eclectic array of high value organic products.1 Palladium is the metal catalyst of choice for many of these transformations, although to be sustainable this precious metal ought to be used responsibly, i.e. used at low catalyst loadings, and be efficiently recovered and recycled, particularly in large scale chemical processes.2 Advances in the design of well-defined ligand systems (e.g. the Buchwald-type ligands such as XPhos, SPhos and many others) have positively contributed towards applied SMCC reaction processes.3 On the other hand, multinuclear Pd species, from small Pdn clusters to Pd nanoparticles, offer unique reactivity and activity in SMCC reactions, as well as other cross-coupling chemistries.4 Immobilized-Pd5 or the use of supported Pd nanoparticles allow for more effective6 catalyst recycling, particularly when used in conjunction with magnetic co-additives such as iron.7 Moving towards 2030, one can argue that simply improving the activity and recyclability of Pd catalysts is not enough. However, if one can alter reaction outcomes of cross-coupling reactions based on unique reactivity of a reproducible, well-defined multinuclear Pd species, then that could be a highly exploitable tool in synthetic chemistry.8 In this context, the identification of Pd3 clusters of the type [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X (X can be a variety of coordinating or non-coordinating anions; structures originally discovered and characterized by Coulson and Dixon)9 as competent, highly active and selective catalysts for cross-coupling reactions has prompted several research groups to exploit their distinctive and unique behavior.4d,10 SMCC reactions with di- and tetra-nuclear Pd complexes have also been reported, although with less detailed mechanistic evaluation.11 A guiding example with Pd3 is the cross-coupling of 2,4-dibromopyridine, where typical mononuclear Pd species containing phosphine ligands exhibit C2 site-selectivity.12 On the other hand, [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X species (X = Cl or OAc) exhibit C4 site-selectivity.10b Indeed, higher order stabilized Pd nanoparticles exhibit similar behavior.13 Although the mechanism for this site-selectivity switch is not fully known, the ability to program either reaction outcome by the choice of catalyst provides synthetic chemists with a powerful tool for wider applications.In 2017, Li et al. reported the reactivity and behavior of [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X species in SMCC reactions, suggesting it to be a highly active catalyst for the cross-coupling of an array of coupling partners, including aryl chlorides.4d The latter point is remarkable, as clearly the aggregated cyclic Pd3 cluster increases the reactivity of this Pd-phosphorus based catalyst system. Moreover, the authors suggested that there was an inversion of the steps in SMCC reactions catalyzed by [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X species (X = SbF6). Despite the mechanistic complexity of typical SMCC reactions, i.e. the difficulty in elucidating Pd nuclearity and speciation (both Pd and B), the scientific community broadly agrees with a sequence of oxidative addition, transmetallation and reductive elimination steps. However, a transmetallation-first step was proposed for the action of [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X species, which is followed by oxidative addition (σ-bond metathesis suggested) and reductive elimination at the intact Pd3 cluster center. Thus it would appear that [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X operates in a mechanistically distinct manner to typical mononuclear Pd catalysts, particularly Pd0(PPh3)n catalyst systems. Indeed, Fairlamb et al. demonstrated the normally expected sequence is only observed for Pd0(PPh3)n where n > 3 in reactions of 2-bromopyridine10a or 2,4-dibromopyridine10b with aryl boronic acids. When n < 3 higher order Pdn clusters play a key role.
Despite the work conducted on [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X catalyst species, there is limited information available on the pathways for catalyst activation, understanding productive catalyst turnover and potential pathways for catalyst deactivation, i.e. from cradle to grave. Indeed, apart from our preliminary kinetic studies,10a there is very little reported on the speciation and kinetic behavior of [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X species in catalytic cross-coupling reactions. Preliminary X-ray Absorption Spectroscopy (XAS) data on the behavior of [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X species reported by Li et al. show changes,4d however the EXAFS data were not fitted to structures proposed making speciation assignments difficult. This prior work, in addition to Pd3Cl2's distinct selectivity and high activity in cross couplings, prompted us to conduct further XAS studies and mechanistic experiments on the Pd3 cluster system.
Herein, we report our findings on the behavior of a phosphine-supported (immobilized) [Pd3(μ-Cl)(μ-PPh2)2(PPh3)2(PPh2)-Ar-supported]Cl species in SMCC reactions. Various immobilized systems have been applied to SMCC reactions and are reviewed elsewhere,14 however, we have focused on a supported catalyst system to facilitate our structural study of the intact Pd3 cluster species in a manner that [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X cannot alone. We have further conducted studies using [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]Cl as the catalyst system, so that there is a degree of comparability enabling broader conclusions to be reached. In a wider context, the combination of catalyst immobilization strategies and in solution structural characterization (XAS) presented in this study provides a robust approach for discriminating reaction participants and evaluating Pd speciation in SMCC reactions. In doing so we signpost how this approach can be invaluable for furthering our understanding of this important class of reactions, while recognizing the complexity of the catalyst speciation (summarized in Fig. 1).
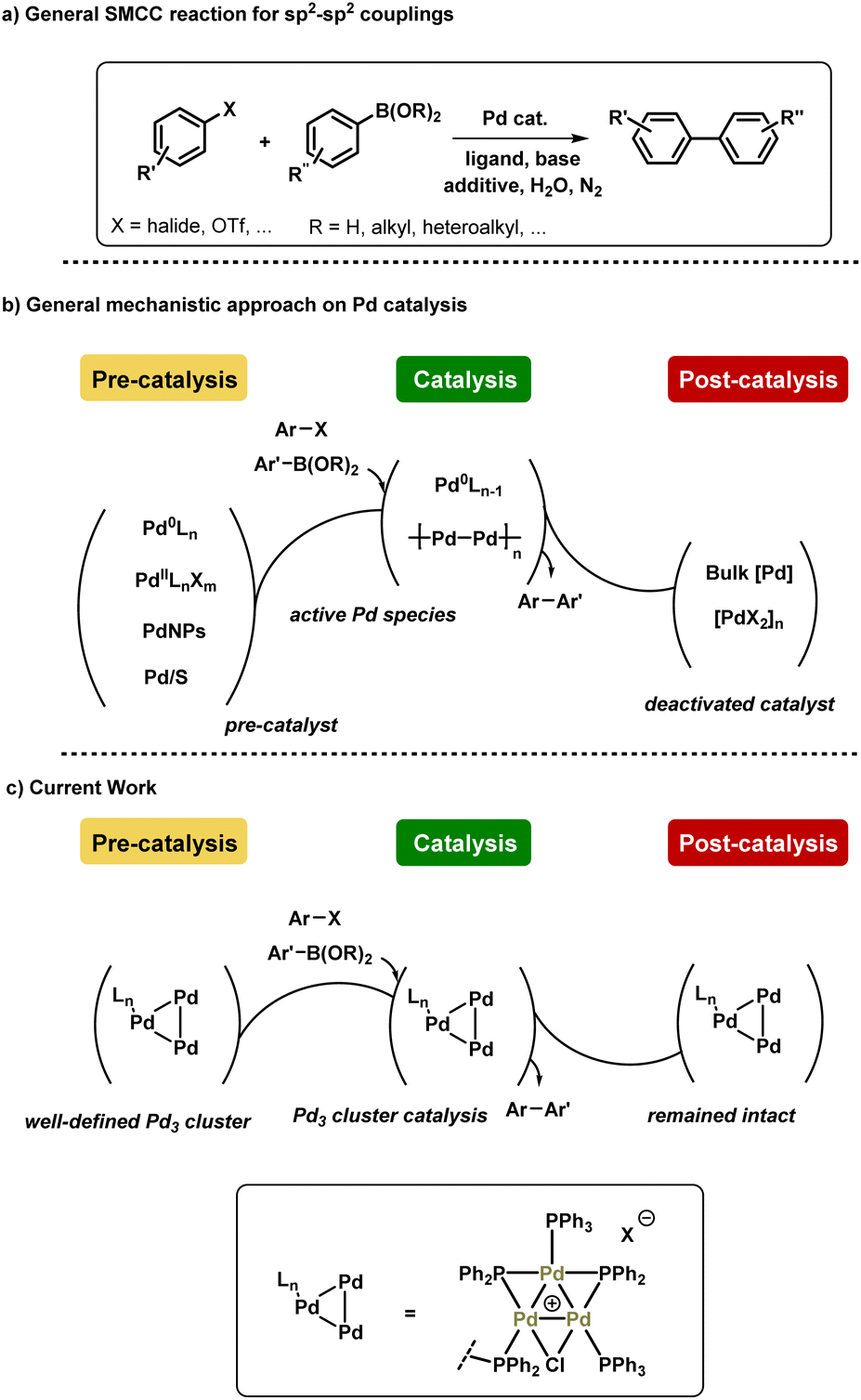 | ||
| Fig. 1 (a) General Pd-catalyzed SMCC reaction scheme, (b) the mechanistic approach on Pd catalyst speciation during and after catalysis, (c) and current work showing the higher order Pd3 catalyst robustness during and after catalysis. | ||
Results and discussion
Synthesis and characterization of immobilized-Pd3Cl23
The synthesis of [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]Cl 1 (referred to as Pd3Cl21) can be accomplished by the reaction of trans-PdCl2(PPh3)2 with H2 in aniline at 90 °C.10a The immobilization of 1 used of an (aminomethyl)polystyrene resin (∼1.5 mmol g−1 amine loading, 200–400 mesh), which was treated with a linkable N-succinimidyl 3-(diphenylphosphino)-propionate phosphine ligand to afford 2, which was then directly reacted with Pd3Cl21, giving the immobilized-Pd3Cl23 (referred to as immob-Pd3Cl23) (Scheme 1). The non-polar polystyrene resin was hypothesized to provide a suitable environment in which to interact with the Pd3 cluster, since the PPh3/PPh2 groups give it a non-polar hydrophobic chemical environment, particularly above and below the plane of the triangular Pd3 structural motif. Furthermore, we anticipated that these interactions protect the Pd3 cluster from the harshness of the basic environment induced by the use of n-Bu4NOH.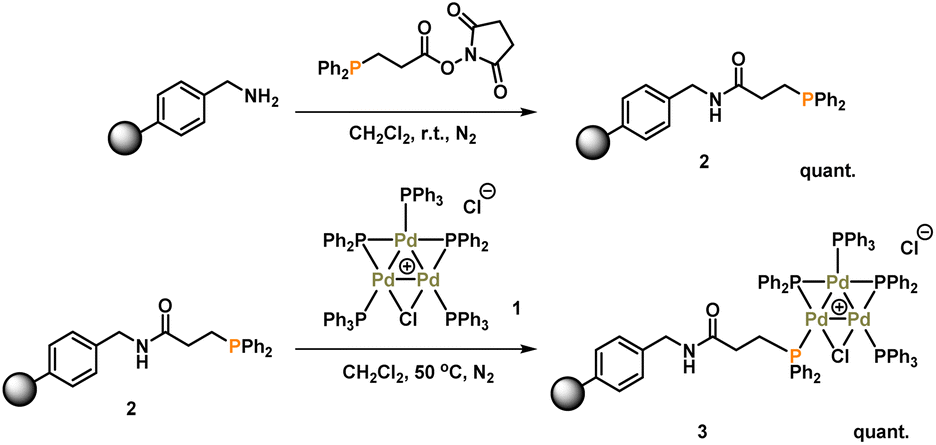 | ||
| Scheme 1 Synthesis of immobilized-Pd3Cl2 cluster 3. | ||
The connectivity of the Pd3Cl2 cluster motif during the immobilization process was confirmed by a solid-state 31P NMR experiment (Fig. 2a). The 31P NMR spectra of Pd3Cl21 and immob-Pd3Cl23 confirm the presence of the triangular Pd3 motif anchored with functionalized (aminomethyl)polystyrene resin; similar 31P chemical shifts are seen for the phosphorus chemical environments. For Pd3Cl21 we observed a broad peak with some fine structure at ca. δ −7 to 43 ppm, which possesses spinning side bands at δ −44 and 80 ppm. These signals are attributed to the distal and proximal phosphines ligated to Pd. A similar but broadened 31P NMR spectrum is seen for immob-Pd3Cl23 with the same main feature around −7 to 43 ppm, which given the sensitivity of 31P NMR points to the presence of the same phosphorous environments. The phosphine functionalized resin prior to addition of the Pd cluster is quite different with a singlet centered at −13 ppm with two side bands (Fig. 2a). None of this signal at −13 ppm remains in immob-Pd3Cl2, showing that all the resin bound phosphine is interacting with the Pd cluster. As expected, free PPh3 and excess Pd3Cl2 are observed in the solution phase by 1H and 31P NMR after the immobilization step (Fig. S3 and S4†).
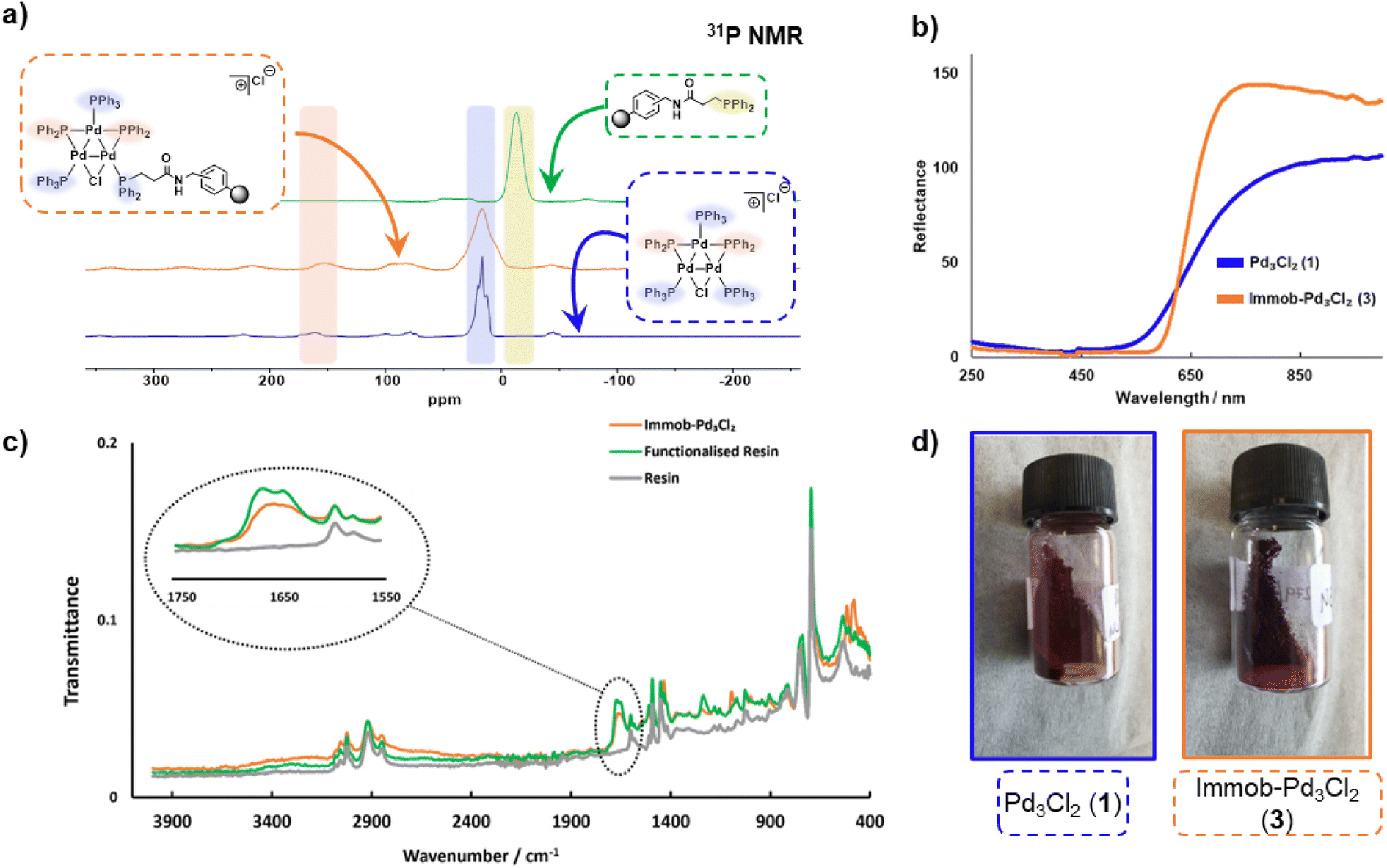 | ||
| Fig. 2 (a) Solid-state 31P NMR spectral data for Pd3Cl21, the functionalized resin and the immob-Pd3Cl23 (at 10 kHz); (b) reflectance UV-vis spectra of Pd3Cl21 and immob-Pd3Cl23; (c) overlay ATR-IR spectral of (aminomethyl) polystyrene resin (blue), phosphine-functionalized resin 2 (green) and immob-Pd3Cl23 (orange); (d) material appearance of 1 (dark red) and 3 (dark purple). | ||
A similar, but broader peak is seen for immob-Pd3Cl23, along with spinning side bands. The 31P NMR chemical shifts of the coordinating terminal PPh3 ligands share similarities with those seen in solution for Pd3Cl21. The signals for the two bridging PPh2 environments in Pd3Cl21 are distinctive, centered at δ 162, with spinning side bands at δ 220 and 100 (note: there is further evidence of extended anisotropy at wider chemical shifts within this spectral data). Importantly, these chemical shifts closely match the signals observed for immob-Pd3Cl23, noting the broader signals, which we associate with the subtly different pore environments of the resin support, and the substituted possibility of phosphine being distal or proximal to chloride. However, there is no evidence to show which phosphine (i.e. distal or proximal position to chloride) undergoes substitution or how many substitutions occur within the cluster. Importantly, the reflectance UV-vis spectrum of immob-Pd3Cl23 (red-shifted) shares similarity with Pd3Cl21 (Fig. 2b), confirming the structure of the triangular Pd3 motif following immobilization. There is a physical difference in the color of these materials – immob-Pd3Cl23 is dark purple whereas Pd3Cl21 is of a dark red appearance (Fig. 2d). Attenuated total reflectance infrared spectroscopic analysis of immob-Pd3Cl23 revealed the presence of the vibrational peaks associated with the amide carbonyl group, that overlay the amide group in the spectrum of the phosphine-functionalized resin 2 between 1640–1680 cm−1 (Fig. 2c). This implies the preservation of the amide linkage in immob-Pd3Cl23 and that the phosphine remained tethered to the polystyrene during the Pd3 immobilization reaction. These are not present in the stating resin material (no amide carbonyl). The retention of the amide linkage is important as it confirms the tethered phosphine was not released from the polystyrene anchor such that it was able to act as a new solution phase ligand and that any exchange really was the result of interaction of Pd3Cl21 with the resin bound phosphine 2.
Summarizing the initial characterization of the immob-Pd3Cl23, 31P SSNMR provides evidence of the Pd3 motif remaining, and no significant other P containing species being present, UV-vis data show a lack of change to the metal–ligand interactions and ATR-IR confirms, via presence of the carbonyl, that the linker unit is intact.
Suzuki–Miyaura cross-coupling (SMCC) catalysis
The reactivity of immob-Pd3Cl23 was examined in a benchmark SMCC reaction of 4-fluoro-bromobenzene 4 and 4-methoxyphenyl boronic acid 5 under optimized reaction conditions (Table 1 and Fig. 3). The conditions were identified from our earlier study on the regioselective SMCC reaction of 2,4-dibromopyridine with aryl boronic acids, which revealed the unusual C4 site-selectivity for Pd3Cl21 and Pd(OAc)2/≤2PPh3 precatalyst systems.10b As an aside, the same atypical C4 site selectivity is observed with immob-Pd3Cl23 as catalyst, demonstrating the generalizability of immob-Pd3Cl23 as a direct analogue for non-tethered Pd3Cl21 in SMCC catalysis (Table S4†). In the present mechanistic study, however, we chose to focus on the simpler reaction of 4 with 5 to avoid possible regioselectivity complications. The base, n-Bu4NOH, is employed in a THF/H2O solvent mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). While it is a highly basic, mixed solvent system, there is one single phase, where the organoboron species in solution is [ArB(OH)3]−[n-Bu4N]+. Where there is a question about Pd speciation, it is imperative to limit speciation in respect to boron, which makes the conditions advantageous for studying Pd catalyst speciation.
:1, v/v). While it is a highly basic, mixed solvent system, there is one single phase, where the organoboron species in solution is [ArB(OH)3]−[n-Bu4N]+. Where there is a question about Pd speciation, it is imperative to limit speciation in respect to boron, which makes the conditions advantageous for studying Pd catalyst speciation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Pd content (ppm)a | T (°C) | Time (min) | Conv.b (%) |
| a Pd ppm (moles) is three times catalyst ppm (moles) for the Pd3 cluster; the Pd concentration of 68 ppm (moles) equates to a 0.34 mol% catalyst loading with respect to 4 the [ArBr]. b Product 6 conversion (19F NMR; note product conversions mirrored in 1H NMR spectral data) of crude reaction sample with internal standards, 1,3,5-trimethoxybenzene and 4,4′-difluorobenzophenone. c Conversion to 6 obtained at room temperature. | |||||
| 1 | Supported Pd3Cl2 | 68 | 40 | 15 | 99 |
| 2 | Supported Pd3Cl2 | 45 | 40 | 18 | 97 |
| 3 | Supported Pd3Cl2 | 24 | 40 | 28 | 97 |
| 4 | Pd3Cl2 | 68 | 23 | 5 | 95c |
| 5 | Pd3(OAc)6/6PPh3 | 68 | 40 | 4 | 82 |
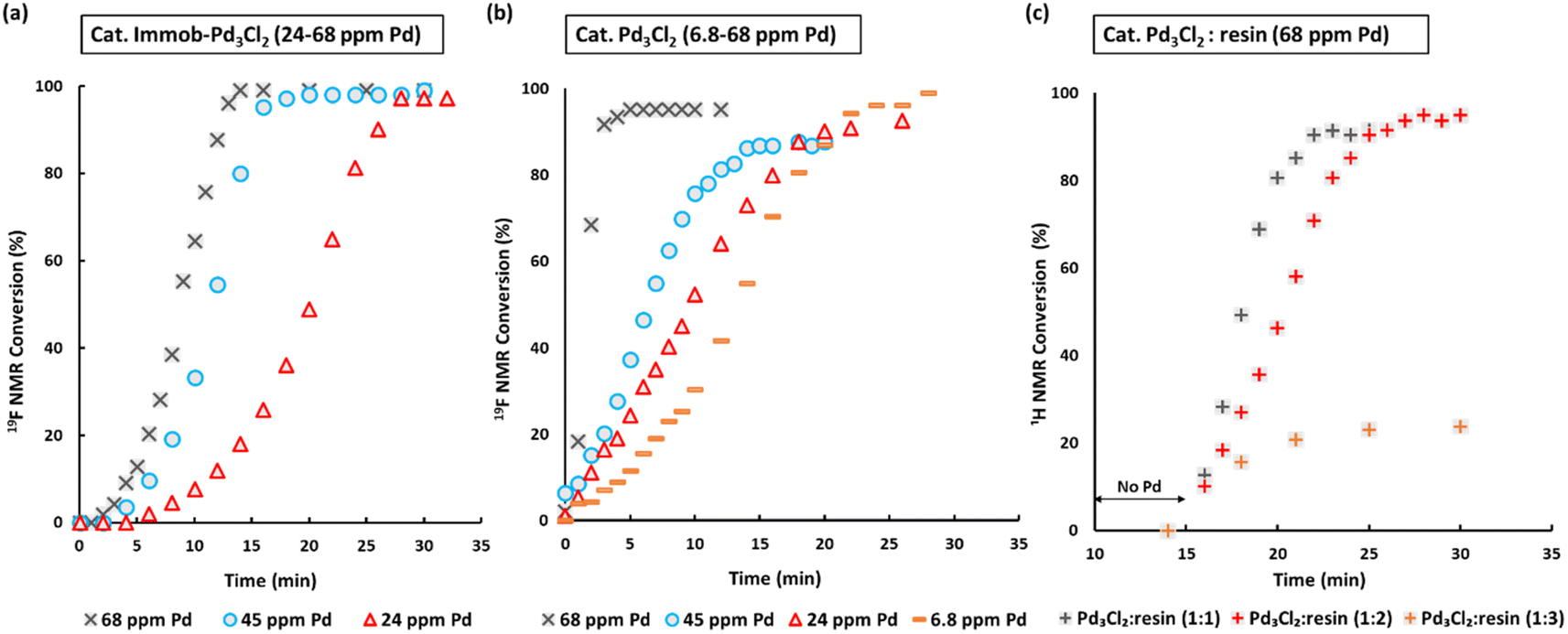 | ||
| Fig. 3 SMCC reaction kinetic profiles using immob-Pd3Cl23 and Pd3Cl21 using different catalyst loadings (monitored by 1H and 19F NMR; 1,3,5-trimethoxybenzene and 4,4′-difluorobenzophenone as internal standards): (a) immob-Pd3Cl23; (b) Pd3Cl21; (c) Pd3Cl21 with exogenous phosphine resin support 2 (in a ratio of 1:1, to 1:2 and 1:3), with the reaction triggered by addition of Pd3Cl2 at t = 15 minutes. Note: the Pd concentration of 68 ppm (moles) equates to a 0.34 mol% catalyst loading with respect to 4 [the ArBr]. Details concerning sampling, NMR analysis and reproducibility are included in the ESI.† | ||
We selected to employ 68 ppm (by moles) of Pd (note: total ppm Pd is three times catalyst ppm; this Pd concentration equates to a 0.34 mol% catalyst loading with respect to 4). The SMCC reaction catalyzed by immob-Pd3Cl23 reaches completion within 15 minutes to give cross-coupled product 6 (entry 1, Table 1). Lowering the Pd catalyst loadings to 0.22 mol% (immob-Pd3Cl23, 45 ppm by moles Pd) and 0.12 mol% (immob-Pd3Cl23, 24 ppm by moles Pd) led to extended reaction times (18 and 28 minutes, respectively) (entries 2 and 3, Table 1).
Remarkably, under comparable conditions using Pd3Cl21, the reaction proceeded to completion within 5 minutes (entry 4, Table 1). We anticipated a higher rate of catalysis for Pd3Cl21 since the accessibility to the “Pd3Cl2” core structure in immob-Pd3Cl23 would be subject to greater substrate diffusion limitations. Nevertheless, the high rate of this reaction is remarkable, which supports the claims made about high reactivity using aryl cross-coupling partners (including aryl chlorides) made by Li and co-workers in their independent study.4d By way of comparison, the “Pd(OAc)2/2PPh3” pre-catalyst system, known to invoke Pd3 cluster catalysis in SMCC and Kumada cross-coupling reactions,10b also effectively catalyzed the reaction (entry 5, Table 1).
The SMCC reaction progress was monitored using 1H/19F NMR spectroscopic analysis, enabling kinetic profiles to be obtained. We examined both immob-Pd3Cl23 and Pd3Cl21, at different catalyst loadings. Interestingly, sigmoidal kinetic profiles were seen for reactions catalyzed by both immob-Pd3Cl23 and Pd3Cl21, suggesting that a pre-catalyst activation step is necessary. The induction period extends to slightly longer times on reducing the catalyst loading, which is longer for the immob-Pd3Cl23 system than for Pd3Cl21, presumably because of diffusion issues for the former catalyst. For the Pd3Cl21 catalyst, one needs to go down to very low catalyst loadings (6.8 ppm Pd, 0.034 mol%) to reveal the full extent of the induction period.
Given the high catalytic activity associated with Pd3Cl21 we hypothesized that the influence of the exogenous phosphine resin support 2 also merited assessment. The kinetic profiles in Fig. 3c for the reaction catalyzed by Pd3Cl21: exogenous phosphine resin 2 in a 1:1, 1:2 and 1:3 ratio revealed that the activity is reduced versus Pd3Cl21 alone and further reduced with increasing equivalents of resin (Fig. 3c). The model SMCC reaction was also recharged with both reactants and additives at t = 15 min (based on when the reaction ends as judged from the kinetic curve shown in Fig. 3) to assess catalyst reactivity and catalyst deactivation. 1H NMR analysis of samples taken at the end of each run revealed the temporal reactivity of 3 (six runs) with enduring good conversions to cross-coupled product (see Fig. 4). The result of this experiment suggests that the active catalyst can re-enter into the catalytic cycle in the presence of fresh reactants.
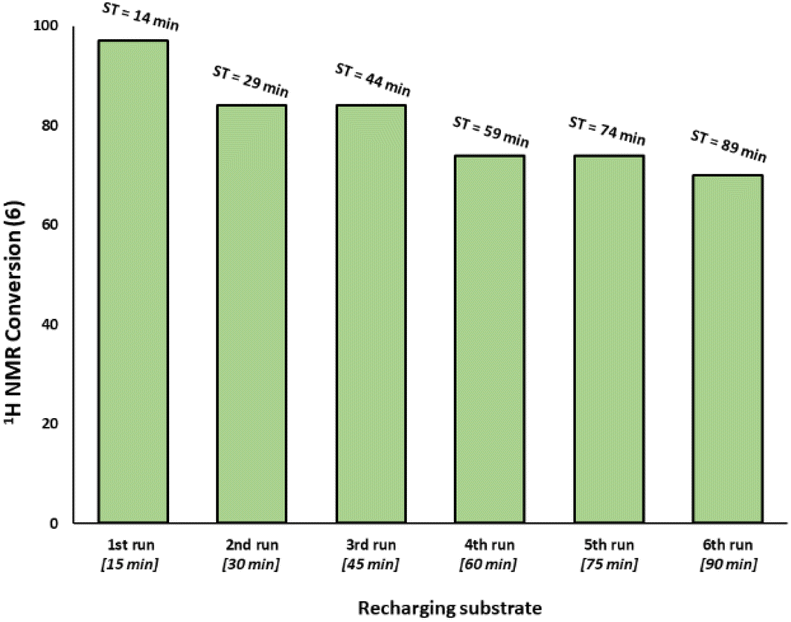 | ||
| Fig. 4 Recharging SMCC reaction promoted by immob-Pd3Cl23 (1 mol%, 68 ppm Pd) with fresh batch of substrates and base at the end of catalysis (taken to be at 15 minutes intervals based on earlier reaction profiles in Fig. 3) and monitoring the reaction progress up to six runs (ST = sampling time during the overall experiment). The reaction was analyzed by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. | ||
Probing the heterogeneous behavior in the SMCC reactions
To assess whether the SMCC reaction occurs heterogeneously, or through a Pd leaching mechanism from the polymer resin, experiments were conducted to gauge the heterogeneity/homogeneity of the processes involving the immob-Pd3Cl23 catalyst system. The mercury-drop test has been one of the common tools for examining heterogeneous catalyst behavior, where a positive test can result in poisoning of any aggregated Pd0 present (inferred as being the active catalyst species). The alternative outcome is that mercury has no effect, indicating a role for soluble molecular catalyst species (likely of low metal nuclearity).15 However, the applicability of the test for reactions involving different molecular complexes of Pd,16 Pt17 and Rh18 has been recently questioned, due to direct reactions of mercury with homogenous metal species such as palladacycles,19 or a high dependence of the test outcomes to the operational conditions (e.g. stirring rate/size of the reaction vessel).20 The validity of the mercury poisoning test for M0/NHC or MII/NHC (M = Pd, Pt) complexes was brought into question by Ananikov et al.20,21 Certain mononuclear PdIILn complexes react with metallic Hg through an oxidative-reductive transmetalation process to form HgIILn and HgxPdy species, whereas Pd0Ln complexes decompose under exposure to metallic mercury.20 Thus, the issue is that a poisoned catalytic reaction system could have (potentially) involved mononuclear PdLn complexes, bringing about an incorrect interpretation when poisoning does occur. Conversely, if a catalyst system is unperturbed by mercury addition, where the operational conditions are carefully selected to ensure the test can poison reactivity (see ESI†), the attribution of activity to aggregated Pd(0) may still be reasonably ruled out. That is, if the test is conducted correctly to ensure mercury contacts the entire system in adequate excess, an outcome of unperturbed reactivity in the presence of Hg is sufficient to exclude Pd(0) particles, which would suffer from Hg poisoning (while the converse test outcome is inconclusive for the reasons above).With all these studies in mind, we conducted a mercury poisoning test for the model SMCC reaction catalyzed by immob-Pd3Cl23 (68 ppm, 0.34 mol%), which reached 50% product conversion after 8 minutes (see Fig. 2a). Addition of metallic mercury (300 equiv. with fast agitation on a standard magnetic stirrer hot plate – see Section 2.8 in the ESI†) at t = 8 minutes had no effect on the reaction rate or product conversion, reaching completion within 15 minutes (Fig. 5). Interestingly, in a control reaction we determined that Pd3Cl21 does react with metallic mercury, but only on longer timescales (see Fig. S21†), leading to decomposition. Our results, taken together show that: (a) Pd nanoparticles (Pd0 species) are not operative in SMCC reactions catalyzed by immob-Pd3Cl23; (b) the Pd3Cl2 cluster motif is likely protected from metallic Hg in immob-Pd3Cl23, under the reaction conditions tested.
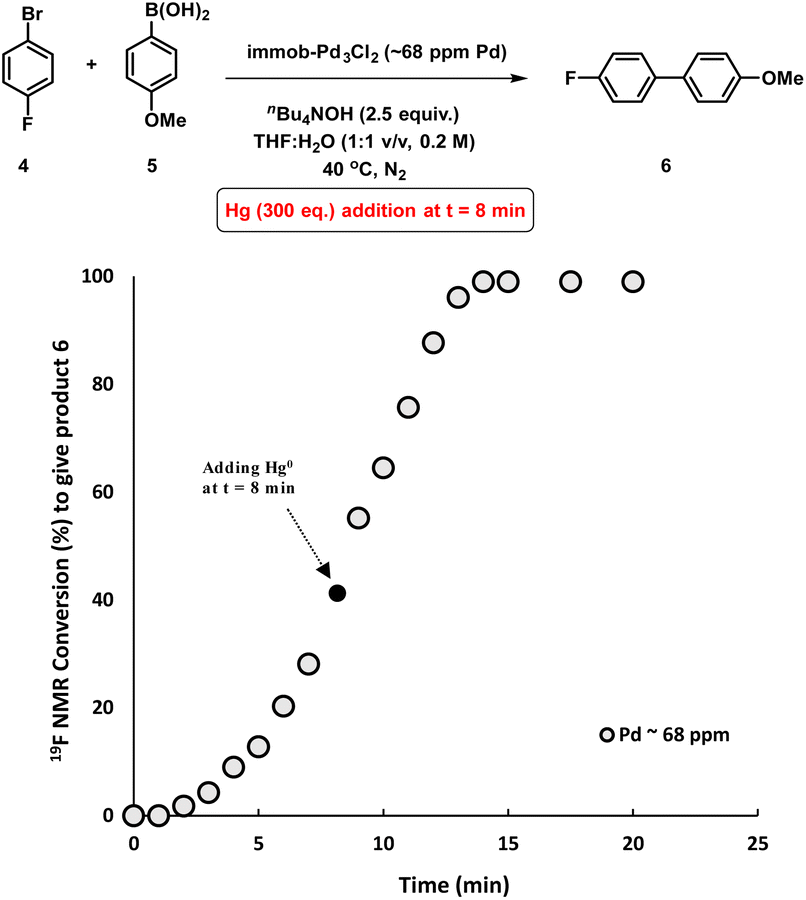 | ||
| Fig. 5 Kinetic profile of the model SMCC reaction obtained when excess metallic Hg (300 eq.) added at t = 8 min and reaction progress monitored by 19F NMR analysis. The Pd concentration of 68 ppm (moles) equates to a 0.34 mol% catalyst loading with respect to 4 [the ArBr]. | ||
In another experiment, the immob-Pd3Cl23 catalyst was separated from the model SMCC reaction after 4 minutes to monitor the reaction progress. The analysis showed that the reaction proceeds although at a significantly slower rate (see Fig. S22†). While the result could suggest the leaching of soluble Pd0 species into the solution (note: not supported by the Hg drop test), some of the supported Pd catalyst particles evaded removal by filtration, even using syringe filters (with 0.22-micron pore size). Moreover, the presence of small particles were observed in the reaction flask post-filtration. Nevertheless, the removal of the majority of immob-Pd3Cl23 showed reaction retardation. Such retardation (taken in isolation) on filtration of the solid species can't explicitly exclude the possibility of leached Pd nanoparticles or leached molecular species becoming bound to the filter, but the much-decreased rate on filtration (given the difficulty of excluding all the solid material) is consistent with the attribution of reactivity drop to the removed solid immob-Pd3Cl23.
The three-phase test, devised by Rebek and Gavina,22 was used to test for potentially reactive soluble catalytic Pd species that detach from a heterogenous (pre)catalyst and leach into the solution (being the active catalyst species). This is typically done by immobilizing one of the reactants and probing its reaction with another soluble coupling partner, in the presence of the immobilized catalyst.23 The diffusion of two solids or solid-bound reaction components should be inhibited versus diffusion of soluble component(s). For this experiment, we prepared a polystyrene-bound aryl iodide 7. SMCC reactions of 7 with 5 were examined using either immob-Pd3Cl23 or soluble Pd3Cl21, under otherwise identical conditions (Table 2).
|
|
|||
|---|---|---|---|
| Catalyst | Conversiona,b,c to 8 (%) | ||
| 1 hour | 5 hours | 24 hours | |
| a Conversion monitored by 1H NMR (consumption of 5). b <3% cross-coupled byproduct formed as result of amide hydrolysis. c The Pd concentration of 68 ppm (moles) equates to a 0.34 mol% catalyst loading with respect to 7 [the ArI]. | |||
| Pd3Cl2 | 1 | 8 | 28 |
| Immob-Pd3Cl2 | 1 | 1 | 1 |
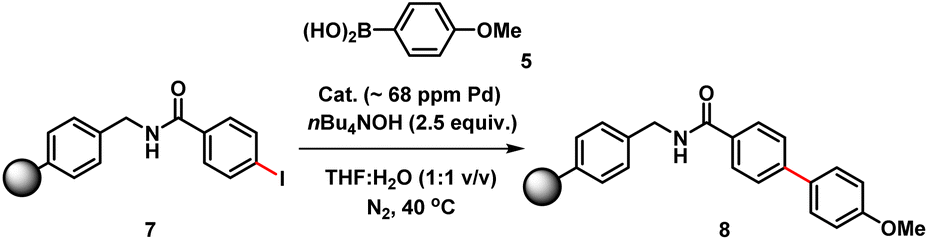
The immob-Pd3Cl23 catalyst was effectively inactive in the three-phase test (less than 1% conversion to cross-coupled product 8 after 24 h). Immobilization of the aryl halide restricts the accessibility of the immob-Pd3Cl23 catalyst; thus, any catalytic reaction cannot proceed under the employed reaction conditions. The outcome confirms that leached soluble Pd species are not playing a significant role under the reaction conditions tested. The equivalent reaction using Pd3Cl21 as the catalyst was sluggish, as might be expected given the introduction of a diffusion constraint on one species involved, and the possibility the amide linker in immobilized derivative 7 might be considered inhibitory to the aryl halide (28% product conversion noted after 24 h).24 It should be noted that in an effort to keep the reactions more similar in timescale we have used the more reactive aryl iodide for these experiments (weak C–I bond, facile oxidative addition), which, while not perfect, removes the risk of speciation differences over longer timescales of reaction.
We therefore also conducted a control to determine that immob-Pd3Cl23 was able to effectively cross-couple 4-iodo-N-methylbenzenamide 9 with p-methoxy-phenyl boronic acid 5 (gave 86% conversion to 10 after 1 h) (Scheme 2). It was important to confirm this, as it shows similar reactivity to that of our normal test reaction between 4 and 5, but with a similar inhibitory group/aryl iodide combination to that in immobilized derivative 7. Table 2 three-phase test experiment for the SMCC reaction of an immobilized aryl iodide 7 with 4-methoxyphenyl boronic acid 5 to give immobilized product 8.
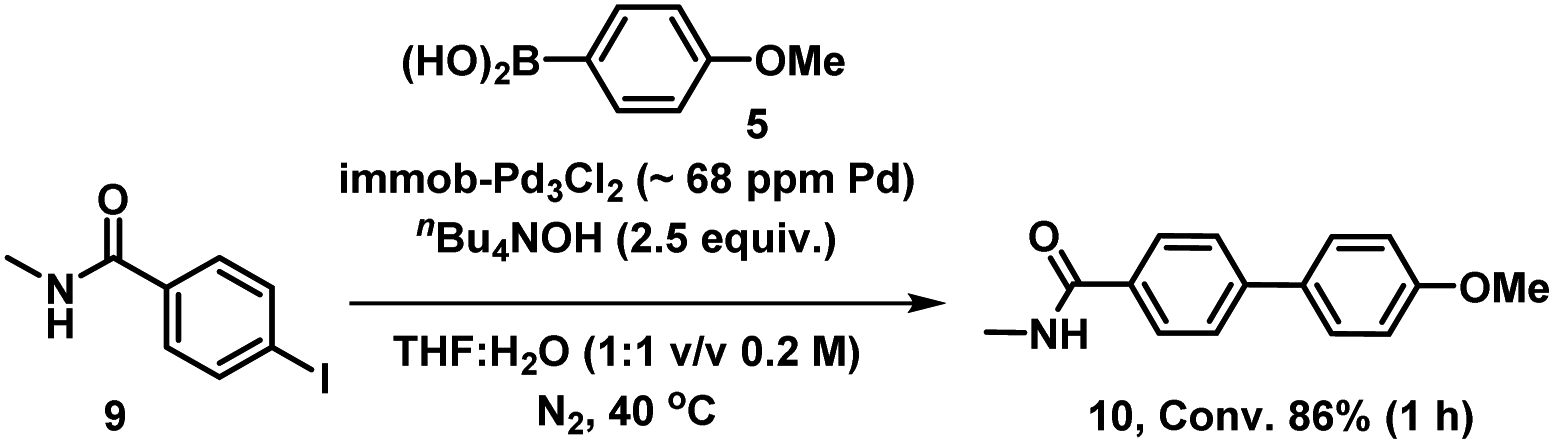 | ||
| Scheme 2 Control SMCC of 4-iodo-N-methylbenzeneamide 9 with 4-methoxyphenyl boronic acid 5, catalyzed by immob-Pd3Cl23. | ||
The three-phase test results cannot rule out the potential release of a “Pd3Cl2” species (or derivative) cluster from the support into the solution following a reaction with substrate(s), which could be subsequently redeposited on to the support. Supported Pd catalysts are known to exhibit release-redeposition (catch) mechanisms in catalysis.25 There is no evidence for loss of the “Pd3Cl2” species from immob-Pd3Cl23. Indeed, the only way to trigger release is by the reaction of exogenous (excess) PPh3 with immob-Pd3Cl23 in CH2Cl2, which occurs instantaneously at room temperature to deliver Pd3Cl21 into solution; confirmed by 31P NMR and ESI-MS (+ve mode) (Fig. 6a).
 | ||
| Fig. 6 (a) Reaction of immob-Pd3Cl23 with 5 equiv. PPh3 in CH2Cl2 at 22 °C; 31P NMR spectrum was recorded upon dissolution (within 10 min). ESI-MS (+ve mode) confirms pseudo-molecular ion at m/z 1509, with the correct isotopic distribution pattern centered at m/z 1511 (bottom right); (b) ESI-MS (+ve mode) analysis of the solution after reaction of excess PPh3 with post-reaction immob-Pd3Cl23 cluster which was filtered off 10 minutes into Suzuki–Miyaura catalytic turnover and thoroughly washed, showing both [Pd3Cl]+ and [Pd3Br]+ ions shown, with their correct isotopic distribution patterns. | ||
This ability to trigger release of the Pd3 cluster from the resin support affords an additional test as to whether the Pd3 cluster remains intact on the resin during reaction. An SMCC reaction of 4-fluoro-bromobenzene 4 and 4-methoxyphenyl boronic acid 5 was again performed under the normal reaction conditions with 68 ppm Pd, but stopped after 10 minutes by filtration (where Fig. 3 shows ∼60% conversion, but catalytically relevant species are still present). The filtered resin was then washed thoroughly and reacted with excess PPh3 to trigger release of the cluster into clean solvent (details given in the ESI†). ESI-MS of the released Pd3 shows not only the initial [Pd3Cl]+ ion, but also [Pd3Br]+ (Fig. 6b), pointing to the resin bound species having been involved in a process that swaps out the bridging halide.
The results taken together suggest that the induction period observed from the cross-coupling catalysis is not a consequence of leached mononuclear or nanoparticulate Pd catalyst formation. At 10 minutes where >50% conversion has occurred ICP showed only leaching of 0.58% of the initial Pd into the liquid phase for a sample filtered to remove the solid (and some of the polymer bound material undoubtedly evades the filter, so this is a worst-case scenario). This is further corroborated by the quantification of the XAS step-edge height for the pre-reaction immob-Pd3Cl23 and ‘during reaction’ samples—proportional to concentration—implying the solid contained the same Pd amount, within error, in both cases. In addition to these tests performed to exclude catalysis by leached molecular or nanoparticle species, the overall rates of reaction imply a minor component would have to exhibit far greater performance than those seen for SMCC reactions at these temperatures ∼40 °C (see Table S11;† it should be noted that typical rates of “ppb Pd” SMCCs or catalysis by “impurity Pd” are generally identified only at significantly higher temperatures, e.g. 110 °C),26a in keeping with relating Pd-catalyzed cross-coupling reactions where ppm-Pd descriptors have been rigorously examined.26b
A possible explanation for the induction period is the interplay of planar aromatic [Pd3X]+ species with structurally distorted non-aromatic [Pd3X/X'] species, both of which are plausible species characterized by single crystal XRD methods.10a The possible importance of d-orbital aromatic stability metal aromaticity27 in rendering, for example the [Pd3(μ-SR)3(PR3)3]+Y− cluster complexes as being oxygen and moisture-stable,28 has been previously highlighted,29 and its potential relevance to Pd3 cross-coupling discussed.30 Here the postulated change from more stable aromatic Pd3Cl2 to the structural-distorted bromide bridged cluster being accompanied by an induction and rate increase is consistent with this being an important effect in these systems. The primary point, however, is that the immob-Pd3Cl23 is a viable tool to study the component mediating Pdn cluster catalysis.
Studies by X-ray absorption spectroscopy (XAS: EXAFS/XANES)
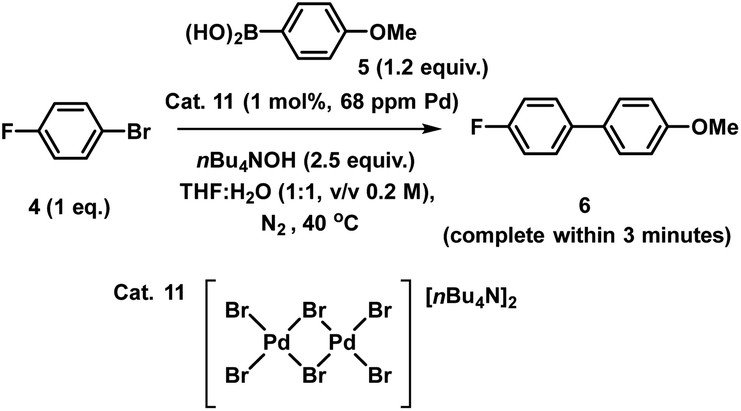
XAS has been used widely to study the physical form of Pd catalysts,31 particularly immobilized Pd catalyst systems25b,32 and well-defined and/or evolving Pd nanoparticle species.33 The pros of the approach are: (1) X-ray absorption is unambiguously specific to a given element, in this case Pd; (2) the penetrating nature of X-rays makes it possible to obtain measurements in complex matrices without perturbing the system, here we do not remove from the solvent for analysis; (3) the spectral information obtained is rich, with information about oxidation state, co-ordination environment and geometries being possible to obtain with appropriate modelling; and (4) it is inherently quantitative with normalized spectra of different species allowing their relative contributions to a mixture to be ascribed. The cons of the XAS approach are: (1) it is an averaging method so small components can be overlooked (but this is true of many techniques); and (2) high intensity beam damage to the Pd samples is possible (reassuringly in the present case excellent agreement is seen for the as prepared immob-Pd3Cl23; fitting models are based on expected structure). The reference standards selected in our study include combinations of bromide, phosphines and nBu4N+ species, the latter from the base used in reaction, and these have largely not been reported previously. PdBr2 is consistent with spectra reported in the literature.34To explore the identity of the catalytic Pd species during the SMCC reactions, and to identify the fate of the catalyst after reaction completion, samples of reactions during and after catalysis were analyzed using Pd K-edge extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES) techniques. For preparation of the samples, the catalyst particles were separated from the reaction mixture by cannula filtration (paper filter), after 5 minutes (during productive catalyst turnover, i.e. catalyst activated) and also after reaction completed. We further examined the concentrated filtrate of the reaction solution after separation of the supported catalyst system, which was evaporated to dryness both after 5 minutes and after reaction completion (Fig. 7). Comparison of these samples allows us to understand whether Pd3Cl2 clusters remained intact and potentially give information about any leached Pd species present.
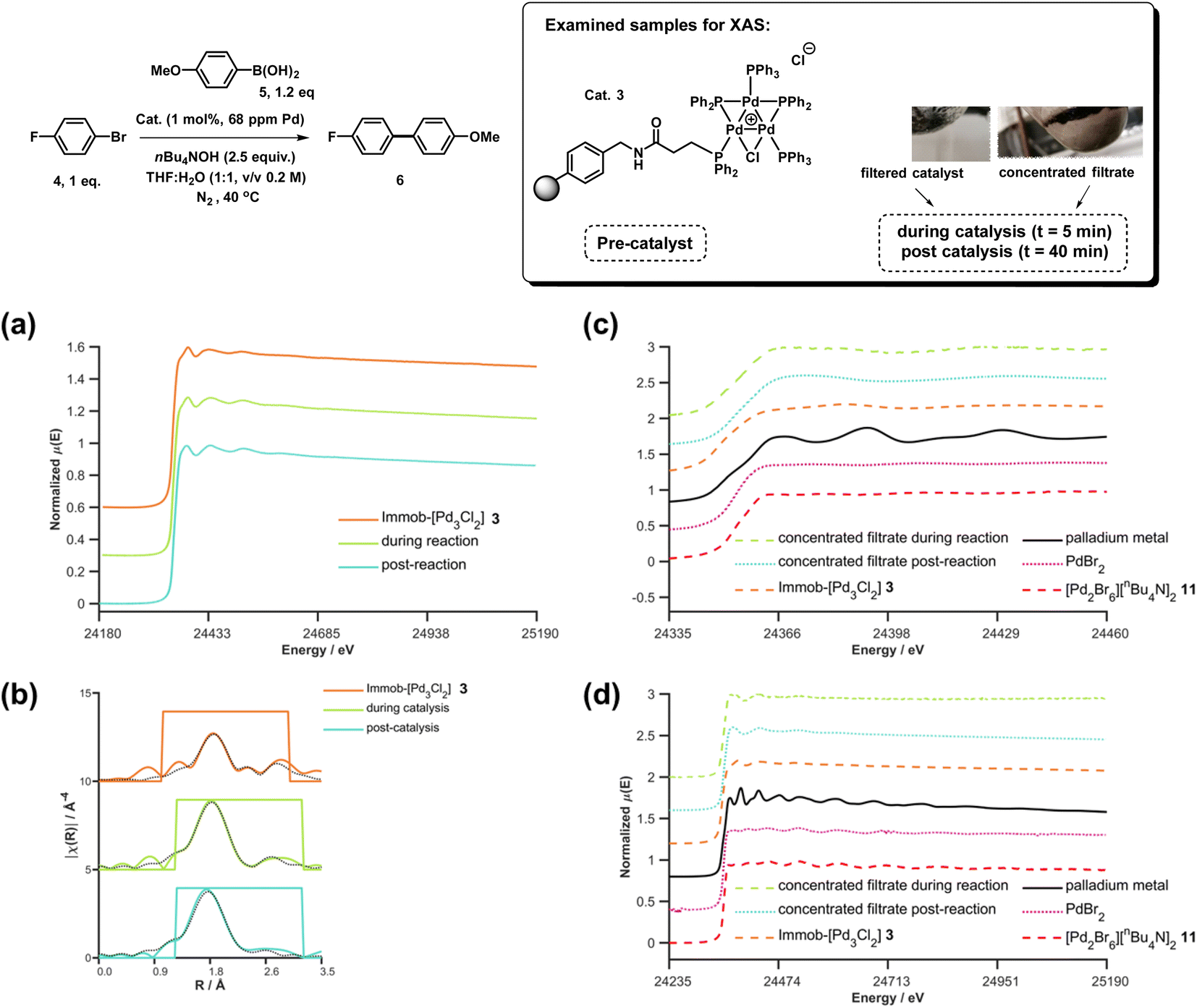 | ||
| Fig. 7 Pd K-edge XANES spectra of the immobilized-[Pd3Cl2] cluster 3 as reference (orange), during reaction (green, reaction stopped at 5 min) and post-reaction (cyan) (a); Pd K-edge R-space EXAFS data and fits of the immobilized-[Pd3Cl2] cluster 3 before reaction, during reaction and post-reaction, solid colored lines denote the data, dotted black lines the fits, with the fitting window indicated in grey (full details given in the ESI†) (b); Pd K-edge XAS spectra (XANES (c) and extended (d)) of the concentrated filtrate during reaction and post-reaction, compared to immobilized-[Pd3Cl2] 3, and PdBr2 and [Pd2Br6][nBu4N]2 as references compounds. Spectra offset for clarity, the lowest spectrum correctly placed on the y-axis in each case, with XANES spectra all normalized between 0 and 1 and R-space EXAFS data offset by 5 units per spectrum. | ||
The Pd K-edge XAS spectra of the filtered catalyst, both during and after reaction in both the XANES region (Fig. 7a) and based on fits of the EXAFS data to the Pd3 motif (Fig. 7b), are in excellent agreement with the data obtained for immob-Pd3Cl23 before reaction (fitting parameters and details are given in the ESI†). The invariant XANES suggest no marked change in either oxidation state or local co-ordination environment, and the fact that the Pd3 motif (with Pd, P and halogen nearest neighbors) is preserved well in the fitting of the EXAFS data strongly points to the fact that the structure of solid-supported Pd3 clusters remains similar during the catalysis and even after catalysis. As was highlighted in Fig. 1, this situation afforded by our immobilization strategy is an unusual and valuable occurrence in mechanistic studies of Pd cross-coupling catalysts, where deactivated Pd species are typically all that remains upon post-mortem analysis. The structure of immob-Pd3Cl23 therefore remains a realistic model for the species bound to the polymer throughout the reaction. Note: XAS and the use of nearest neighbor fits cannot strictly distinguish species 1 and 3, but the difference in reactivity between them, three-phase test and fact that this species is extracted for analysis by filtration of the polymer all imply this is likely to be 3 rather than 1.
Another significant finding from EXAFS analysis was revealed by comparing the goodness of fit for bridging –Br, –Cl and –Ar (a carbon atom in the 1st co-ordination shell, which scatters less than Cl) for each sample. As expected in the pre-reaction catalyst, Cl provides a suitable fit. For the post-reaction samples placing Br in the bridging position between two palladiums (average co-ordination number of Pd by Br = 0.67) provides a distinctly better fit, and in neither the during reaction or post-reaction sample will placing a carbon in the bridging position, representing a bridging aryl, provide an acceptable fit (see ESI†). The better fit for the bromide-bridged Pd3 cluster suggested a substitution of chloride by bromide in the structure of the Pd3 cluster. This is supported by the reaction clusters released from the resin post-filtration after 10 minutes of reaction in Fig. 6b being a mixture of Cl and Br bridged Pd3 clusters (also seen by Li et al. using ESI-MS with both Br and I containing aryl halide reactants).4d The precise steps by which the bromide-containing Pd3 cluster forms likely involve reactions with substrates (depicted in Scheme 3). Repetition of the reaction kinetic profile for immob-Pd3Cl23, but in the presence of a large excess of Br− (1 equiv. nBu4NBr with respect to aryl halide) showed no significant change in kinetics, suggesting this incorporation must occur reactively (see ESI†).
 | ||
| Scheme 3 The halide exchange of bridging chloride by bromide sourcing from the substrate (in immob-Pd3Cl23). The best fit of the EXAFS spectra being obtained for Br-bridged Pd3 cluster indicates that the exchange probably occurred during catalyst activation and remained intact during catalyst deactivation in the model SMCC reaction. Note that the EXAFS analysis cannot preclude formation of non-aromatic Pd3Br2 or Pd3BrOH species. | ||
Plots of the filtrate samples (where polymer resin containing immob-Pd3Cl23 was filtered out and the solvent removed to dryness) obtained during and after catalysis against a variety of reference samples in both XANES (Fig. 7c) and EXAFS (Fig. 7d) regions show no match to the spectral features for palladium metal, Pd3 clusters, nor other reference samples, including PdBr2 and [Pd2Br6][n-Bu4N]211. It should be noted at this point that only very subtle differences are seen experimentally (Pd7nm–Pdfoil)35 or predicted theoretically as a function of metallic palladium nanoparticle size (Pd13-atom–Pdfoil),36 and so the Pd foil standard is a suitable reference for metallic Pd particles of any size. Attempts to use mass spectrometry to identify the Pd species in the filtrate were unsuccessful, either due to other species present swamping the signal or there being a poorly defined mixture of end product species. The nature of this species is discussed later, but at this point we can confidently rule out this leached Pd being present as Pd3 clusters, which would readily be seen by HRMS, metallic palladium particles (readily seen in XAS) or the other reference samples considered.
The reference sample [Pd2Br6][n-Bu4N]211 is reported to be catalytically active in cross-coupling reactions and we were therefore careful to consider its possible formation from the Pd3X2 catalyst system.25b,37 Here, complex 11 was prepared through treatment of n-Bu4NBr and PdBr2 in THF and H2O (1:1) under inert atmosphere at room temperature and was characterized by far infrared, elemental analysis and X-ray single crystal diffraction (Fig. 8).
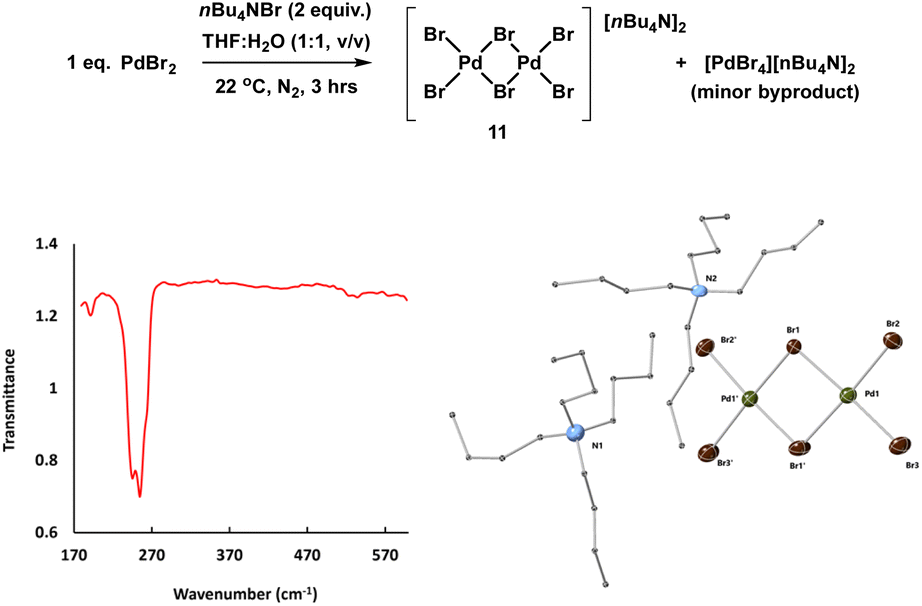 | ||
Fig. 8 Synthesis and key characterization for 11: far IR spectrum (left) showing characteristic bridged Pd–Br stretch bands at 244.9 and 254.6 cm−1 and X-ray single crystal structure of [Pd2Br6][nBu4N]211 (right), thermal ellipsoids set at 30% probability and hydrogen atoms removed from both structures for clarity. Selected bond distances for 11 (Å): Pd1–Br1 = 2.445(2), Pd1–Pd2 = 2.407(2), selected bond angles (°): Br1–Pd1–Br2: 91.114(8), 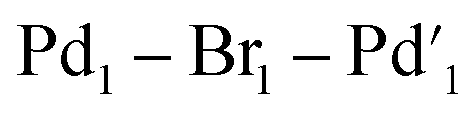 : 93.547(7), Br2–Pd1–Br3: 91.893(9). : 93.547(7), Br2–Pd1–Br3: 91.893(9). | ||
The pre-catalyst 11 showed very high reactivity in forming cross-coupled product 6. The result of this experiment supports the XAS outcome that leached Pd species (which were relatively inactive) do not share any similarity with 11. It is also notable that even if such species were present at low concentration (below those that we could detect), they would not be capable of promoting the cross-coupling reaction at this temperature at the rate seen by the present highly reactive immob-Pd3Cl23 catalytic system.
The catalytic activity of 11 was examined in the model SMCC reaction of 4 and 5 (using 0.1 mol%, 68 ppm Pd), under our working reaction conditions, showing quantitative conversion to 6 within 3 minutes (Scheme 4), cf. 15 minutes for immob-Pd3Cl23 (Table 1).
 | ||
| Scheme 4 Model SMCC reaction of 4 and aryl 5 under the optimized reaction conditions, using [Pd2Br6][nBu4N]211 (0.1 mol%, 68 ppm Pd) as catalyst, giving complete conversion to 6 within 3 minutes. The crude reaction mixture was analyzed by both 1H and 19F NMR analysis using 1,3,5-trimethoxybenzene and 4,4′-difluorobenzophenone as internal standards. | ||
:1 v/v solvent after n-Bu4NOH base added; after aryl boronic acid added; after aryl bromide added) an aliquot of the reaction mixture containing all components was rapidly quenched to LN2 temperatures in a Nalgene cryovial for XAS analysis. The resulting sequence of XANES spectra are shown in Fig. 9a. The initial spectrum of Pd3Cl21 is an excellent fit to the expected first shell co-ordination of Pd in the cluster (Fig. 9b).
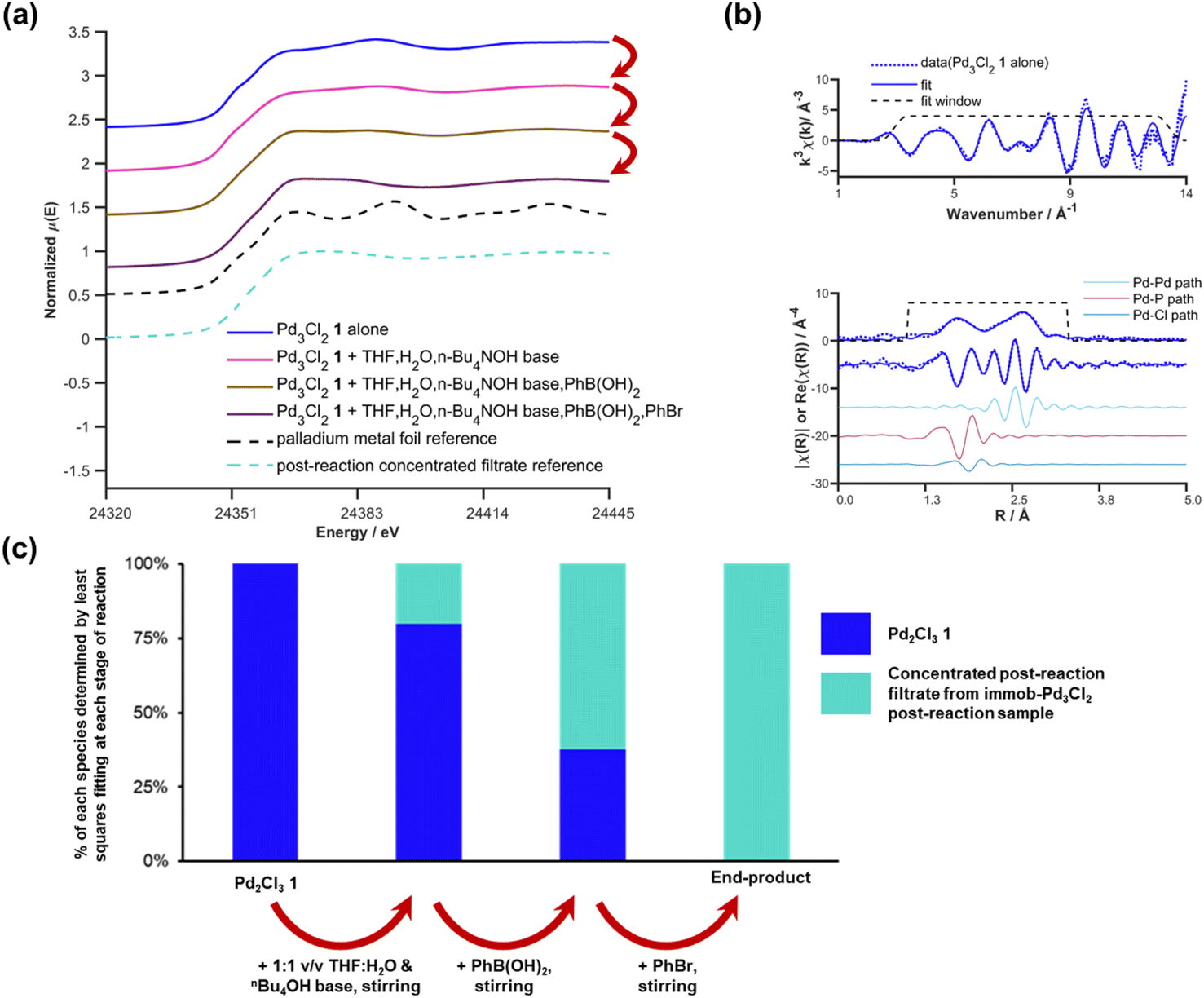 | ||
| Fig. 9 Pd K-edge XANES spectra of the Pd3Cl2 cluster 1 alone (blue), and after stepwise addition of H2O/THF and nBu4NOH base (pink), phenylboronic acid (brown), and phenyl bromide (platinate purple) (a); Pd K-edge k-space and R-space EXAFS data and fits of the initial Pd3Cl2 cluster 1, dashed blue lines denote the data, solid blue lines show the fit, with the fitting window shown above and the contributing paths from Pd–Pd, Pd–P and Pd–Cl shown below (b); plot of the contributions determined by least squares fitting the spectra shown in (a) for the stepwise reaction of the initial Pd3Cl2 cluster 1 and the concentrated post reaction filtrate from the immob-Pd3Cl2 experiments as explained in the text (c). Spectra offset for clarity, the lowest XANES spectrum is correctly placed on the y-axis with all XANES spectra normalized between 0 and 1 and EXAFS oscillations centered around y = 0 prior to any offset. | ||
However, it is immediately clear from the XANES that even combining with THF/H2O/base begins to alter the Pd speciation. Further time and/or reagent addition caused further changes in the spectra (it is difficult to discriminate which, as both time and addition of reagents took place). The changes in the spectra mirror the visual observation that there was an immediate change to a black color upon adding the aqueous n-Bu4NOH base to a THF solution of Pd3Cl21. While our first thoughts turned to precipitation of Pd black, no distinct large Pd particles were visible by eye from this point throughout the subsequent reagent addition steps performed (during which the appearance of the reaction solution remained unchanged). The impression of there being a single liquid phase can be misleading if the Pd is nanoparticulate in nature and does not precipitate. However, as will be seen shortly the conclusion that no Pd metal is being formed (nanoparticulate or otherwise) is supported by further analysis of the XAS data.
Qualitatively the XANES spectra through the reaction steps appear to become progressively more similar to the filtrate during reaction from the immob-Pd3Cl23, while bearing little similarity to Pd metal (spectra both shown for reference in Fig. 9a). It is important to note that the above work on immob-Pd3Cl23 showed that the leached species seen in the concentrated post-reaction filtrate were inactive for the test SMCC reaction versus either Pd3Cl21 or immob-Pd3Cl23.
A more quantitative analysis of the XANES data in Fig. 9a and k-space EXAFS data can be achieved with principal component analysis (PCA) and least squares fitting. PCA of the XANES series in Fig. 9a shows significance above the noise in the first two components (Fig. S34,† the edge region is slightly present in the third component, but noise at the edge where a large change is present leads to artificial variation because of slight energy misalignments). A scree plot (Fig. S34b†) also has an elbow after the first two points, pointing to only two principal components that contribute significantly. Similar analysis using an approach used elsewhere34 on the k-space EXAFS spectrum region between k = 0 and k = 11.5 is again largely comprised of only two components that are significantly above the noise level of the data (Fig. S36†). It should be noted that the sample after adding base, but no other reagents, is slightly less well fitted in the EXAFS region by only two components, which may hint at transitional species being present, but there is insufficient data to infer more than this being a possibility.
Given the visual similarity between the post-reaction filtrate sample (leached from immob-Pd3Cl23) to the later stages of the reaction of Pd3Cl21, a two-component linear combination fit was undertaken for each spectrum in the sequence in Fig. 9a. This produced a good fit in the XANES region of the data (Fig. S40†), with the corresponding quantities of Pd3Cl21 and the filtrate-like species being shown in Fig. 9c. This pleasingly shows a similarity between the chemical fate of the homogenous and albeit more stable immobilized Pd3Cl2 catalysts, supporting the idea that the immobilized system can facilitate broader studies of Pd3Cl21. Given the above inactivity of the leached species in the filtrate that appears over time and the fast kinetics of the reaction, we have no evidence for other species besides Pd3Cl21 being involved in the reaction process.
While the precise chemical identity of the species from the filtrate is somewhat unimportant if the species is ascribed as inactive, understanding the fate of Pd3Cl21 is still of interest to understand the reactions that bring about its formation. As noted earlier the XANES spectra of the species in the filtrate did not fit visually with spectra of any common standards, such as PdBr2, Pd metal, or [Pd2Br6]2−. While a visual similarity in the spectra is a good first check, in the ESI† we consider a more quantitative approach using principal component analysis, which has been employed in XANES studies for some time (Section S2.16†).38 This speculatively points to some form of Pdx(PPh3)y-type species being the end fate of Pd in this system.
Finally, the stepwise approach used in this experiment allows us to re-examine the mechanistically surprising claim of Li and co-workers that an aryl-bridge Pd3 cluster forms as a stable transmetallation intermediate that can be observed by XAS.4d The details of the experiments conducted previously are unclear, but the concentrations used in transmission XAS appear to have been around 0.01 wt% Pd from the details given in their publication, lower than is typically suitable for transmission experiments and likely produce noisy data. The claim concerning transmetallation is on the basis of reduced amplitude at low values of k (<5 Å), but this is extremely susceptible to the background functions and the energy spectra from which the k-space data were obtained were not given. The R-space data presented by Li and coworkers seem distinctly different from those we have consistently obtained for the cluster (homogenous or immobilized) with weaker than expected amplitudes for the Pd–Pd scatter (which dominates the region 2.5–3 Å), and an absence of the Pd–Cl scatter contribution (easier to identify when fitting the real part of the data than by inspection of the magnitude R-space plots). Repeating the stepwise experiment with the 1:1 v/v 2-propanol/H2O solvent mixture they report and using K2CO3 base as they did (which is only moderately soluble, although can form methoxide with alcohols39) we were able to see the Pd3Cl21 cluster intact at each step of the process, with only a halogen exchange on adding bromobenzene (Fig. S36–S40†). Crucially in the sample after all other reagents were added except bromobenzene the spectrum is a significantly better fit to chloride than carbon (aryl) in the bridging position. In short, obtaining higher quality data at the low concentrations of Pd typically used in SMCC reactions by taking advantage of modern fluorescence detection capabilities (B18 Diamond Light Source) and fitting this using a summation of theoretically calculated scattering paths (ARTEMIS40) we saw no evidence with either the immobilized cluster or attempts to reproduce their experiment using Pd3Cl21 for the proposed [Pd3Ar]+ intermediate. The other evidence they presented for this was based on ESI-MS of CH2Cl2 solutions of the reagents.4d In our hands we found that the base (K2CO3) is insoluble in this solvent/it is not the medium in which the reactions were conducted (note: limited experimental details given in the original publication). It also appears the signal associated with [Pd3Ar]+ is still <20% of the Pd3Cl21 cluster signal, and so even if present in the same amount, XAS being an average technique the Cl bridge would still yield a better fit (and have dominated their data also). It is also noteworthy that using some routes to prepare the Pd3Cl21 cluster, we have sometimes seen the aryl bridged Pd3 cluster as a synthetic biproduct (rather than reactive intermediate – Fig. S27†), which is a possible alternative origin for such species. We have repeated the synthetic route to 1 described by Li and co-workers (yield = 2%) and have not been able to obtain evidence showing the intermediacy of [Pd3Ar]+. One final point is a further potential for confusion in their study arises from the fact that the phenyl group in the PhB(OH)2 substrate used is the same as that found in the PPh3/PPh2 ligands in Pd3Cl21, avoided in the present study through the use of the methoxy-functionalized boronic acid 5. In short, the current study does not provide any evidence to support the existence of the aryl-bridged intermediate suggested by Li and co-workers.4d However, additional work is required to fully investigate this hypothesis and establish if such a species is kinetically relevant, using a series of organoboron compounds to probe the transmetalation step first hypothesis.
Conclusions
In conclusion, we studied ‘Pd3Cl2’ catalytic behavior in a typical SMCC reaction by tethering it to a resin support to afford a valuable tool for mechanistic studies. Experimental heterogeneity tests, such as Hg drop test and three-phase test, showed no active Pd leached from the immobilized Pd catalyst within the reaction's time scale. Analysis of XAS data from the filtered catalyst during and after reaction showed the triangular Pd3 cluster motif intact, with only exchange of chloride for bromide in the bridging position during the substrate turnover. XAS of the filtrate after catalyst removal showed non-active Pd species, with no match to a series of reference samples such as PdBr2, [Pd2Br6][n-Bu4N]2, and Pd foil. XAS analysis of non-tethered (non-mobilized) Pd3Cl21 catalyst in the same SMCC reaction, conducted stepwise, indicates the Pd speciation at the later stages of the reaction is like that in the reaction filtrate when immob-Pd3Cl23 was used as catalyst. Using principal component analysis and target transformation from a range of potential standards along with the available EXAFS data, the identity of the inactive Pd species in the filtrate sample from reaction of immob-Pd3Cl2 or later stage reaction of Pd3Cl2 was suggested to be Pdx(PPh3)y, exhibiting a blend of Pd–Pd and Pd–P interactions. This study provides the first experimentally compelling insight30 into the speciation of Pd involving the catalytic behavior of Pd3 type clusters stabilized by phosphorus containing ligands within cross-coupling reactions. Finally, during the proof stage for our manuscript we became aware of a study reported by Scott et al. involving the temperature-inducated activation of the Coulson-cluster on a carbon support. The results are complementary to those described here, and provide insight into how changes in the ‘Pd3Cl2’ cluster can occur at significantly higher temperatures (between 150 and 250 °C).41Abbreviations
| EXAFS | Extended X-ray absorption fine structure |
| LN2 | Liquid N2 |
| PCA | Principal component analysis |
| SMCC | Suzuki–Miyaura cross-coupling |
| THF | Tetrahydrofuran |
| XANES | X-ray absorption near edge structure |
| XAS | X-ray absorption spectroscopy |
Data availability
A dataset containing raw experimental data has been deposited to Research Data York, which can be accessed through the following link (https://pure.york.ac.uk/portal/en/datasets/evidence-for-suzukimiyaura-cross-couplings-catalyzed-by-ligated-p).Author contributions
I. J. S. F. conceived the project and plans, with input from N. J., concerning the design, synthesis and application of the immobilization Pd3 cluster species. I. J. S. F. secured the research funding to support N. J. and N. W. J. S., S. K. B. led the synchrotron research work; S. K. B. and I. J. S. F. proposed experiments for and secured access to diamond facilities. S. K. B. assisted with the interpretation of catalytic data and design of appropriate control experiments, with input from N. J., N. W. J. S. and I. J. S. F., T. T. carried out the single crystal X-ray diffraction analysis. N. J., N. W. J. S., S. K. B. and I. J. S. F. executed the majority of synchrotron experiments at diamond (supported by diamond/UK Catalysis Hub teams – see acknowledgements). N. J. and S. K. B. conducted the XANES/EXAFS analysis, the interpretation of which was supported by discussion with I. J. S. F. N. J. wrote an initial draft manuscript, with input from all authors. The final version of the paper was written jointly between I. J. S. F. and S. K. B., with contributions and confirmation provided by all co-authors. Much of the synthetic and catalytic work was conducted by N. J. which forms an integral part of her PhD thesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Tony Wild for PhD studentship funding (platinum scholarship) to support N. J., and the RSC Summer Bursary scheme (reference E21-3331237928) for specifically supporting the collaboration between Durham University and the University of York. We are grateful to the Royal Society for an Industry Fellowship to I. J. S. F., and to June Callison and Martin Wilding (EPSRC UK Catalysis Hub BAG) and Giannantonio Cibin, Veronica Celorrio and Diego Gianolio (STFC Diamond Light Source) for invaluable practical assistance with XAS measurements.Notes and references
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) A. Suzuki, Acc. Chem. Res., 1982, 15, 178–184 CrossRef CAS; (c) A. B. Pagett and G. C. Lloyd-Jones, Suzuki–Miyaura Cross-Coupling, in Organic Reactions, pp. 547–620 Search PubMed; (d) J. Almond-Thynne, D. C. Blakemore, D. C. Pryde and A. C. Spivey, Chem. Sci., 2017, 8, 40–62 RSC; (e) M. F. Lipton, M. A. Mauragis, M. T. Maloney, M. F. Veley, D. W. VanderBor, J. J. Newby, R. B. Appell and E. D. Daugs, Org. Process Res. Dev., 2003, 7, 385–392 CrossRef CAS.
- (a) I. J. S. Fairlamb, Pd-catalysed Cross-couplings for the Pharmaceutical Sector and a Move to Cutting-edge C–H Bond Functionalization: Is Palladium Simply Too Precious?, in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, The Royal Society of Chemistry, 2016, pp. 129–139, ch. 11 Search PubMed; (b) S. McCarthy, A. Lee Wei Jie, D. C. Braddock, A. Serpe and J. D. Wilton-Ely, Molecules, 2021, 26, 5217 CrossRef CAS PubMed; (c) S. McCarthy, D. C. Braddock and J. D. Wilton-Ely, Coord. Chem. Rev., 2021, 442, 213925 CrossRef CAS; (d) T. Ma, Z. Li, R. Zhao, J. Song, Y. Tian, S. Ding and G. Zhu, ACS Appl. Nano Mater., 2022, 5, 15115–15122 CrossRef CAS; (e) K. S. Song, T. Ashirov, S. N. Talapaneni, A. H. Clark, A. V. Yakimov, M. Nachtegaal, C. Copéret and A. Coskun, Chem, 2022, 8, 2043–2059 CrossRef CAS.
- (a) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed; (b) S. H. Newman-Stonebraker, S. R. Smith, J. E. Borowski, E. Peters, T. Gensch, H. C. Johnson, M. S. Sigman and A. G. Doyle, Science, 2021, 374, 301–308 CrossRef CAS PubMed; (c) R. A. Altman and S. L. Buchwald, Nat. Protoc., 2007, 2, 3115–3121 CrossRef CAS PubMed; (d) J. P. Norman, N. G. Larson and S. R. Neufeldt, ACS Catal., 2022, 12, 8822–8828 CrossRef CAS PubMed; (e) T. Gensch, S. R. Smith, T. J. Colacot, Y. N. Timsina, G. Xu, B. W. Glasspoole and M. S. Sigman, ACS Catal., 2022, 12, 7773–7780 CrossRef CAS.
- (a) A. Monfredini, V. Santacroce, L. Marchiò, R. Maggi, F. Bigi, G. Maestri and M. Malacria, ACS Sustain. Chem. Eng., 2017, 5, 8205–8212 CrossRef CAS; (b) M. Lanzi, T. Cañeque, L. Marchiò, R. Maggi, F. Bigi, M. Malacria and G. Maestri, ACS Catal., 2018, 8, 144–147 CrossRef CAS; (c) C. Fricke, G. J. Sherborne, I. Funes-Ardoiz, E. Senol, S. Guven and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 17788–17795 CrossRef CAS PubMed; (d) F. Fu, J. Xiang, H. Cheng, L. Cheng, H. Chong, S. Wang, P. Li, S. Wei, M. Zhu and Y. Li, ACS Catal., 2017, 7, 1860–1867 CrossRef CAS; (e) C. J. Diehl, T. Scattolin, U. Englert and F. Schoenebeck, Angew. Chem., 2019, 131, 217–221 CrossRef; (f) K. M. Appleby, E. Dzotsi, N. W. J. Scott, G. Dexin, N. Jeddi, A. C. Whitwood, N. E. Pridmore, S. Hart, S. B. Duckett and I. J. S. Fairlamb, Organometallics, 2021, 40, 3560–3570 CrossRef CAS.
- (a) M. Wilson, R. Kore, R. Fraser, S. K. Beaumont, R. Srivastava and J. Badyal, Colloids Surf., A, 2017, 520, 788–795 CrossRef CAS; (b) S.-B. Jang, Tetrahedron Lett., 1997, 38, 1793–1796 CrossRef CAS.
- N. Nikbin, M. Ladlow and S. V. Ley, Org. Process Res. Dev., 2007, 11, 458–462 CrossRef CAS.
- Z. Zhu, Z. Wang, Y. Jian, H. Sun, G. Zhang, J. M. Lynam, C. R. McElroy, T. J. Burden, R. L. Inight, I. J. S. Fairlamb, W. Zhang and Z. Gao, Green Chem., 2021, 23, 920–926 RSC.
- M. V. Polynski and V. P. Ananikov, ACS Catal., 2019, 9, 3991–4005 CrossRef CAS.
- (a) D. R. Coulson, Chem. Commun., 1968, 1530–1531 RSC; (b) J. S. Cartwright, K. R. Dixon and A. D. Rattray, Inorg. Chem., 1980, 19, 1120–1124 CrossRef.
- (a) N. W. J. Scott, M. J. Ford, C. Schotes, R. R. Parker, A. C. Whitwood and I. J. S. Fairlamb, Chem. Sci., 2019, 10, 7898–7906 RSC; (b) N. W. J. Scott, M. J. Ford, N. Jeddi, A. Eyles, L. Simon, A. C. Whitwood, T. Tanner, C. E. Willans and I. J. S. Fairlamb, J. Am. Chem. Soc., 2021, 143, 9682–9693 CrossRef CAS PubMed; (c) Y. Yun, H. Sheng, J. Yu, L. Bao, Y. Du, F. Xu, H. Yu, P. Li and M. Zhu, Adv. Synth. Catal., 2018, 360, 4731–4743 CrossRef CAS; (d) J. Campos, A. Nova, E. L. Kolychev and S. Aldridge, Chem.–Eur. J., 2017, 23, 12655–12667 CrossRef CAS PubMed.
- H. Qian, T. Zhang, L. Song, S. Yu, Q. Yuan, L. Sun, D. Zhang, Z. Yin and Y. Dai, Eur. J. Org Chem., 2017, 1337–1342 CrossRef CAS.
- (a) C. Sicre, J.-L. Alonso-Gómez and M. M. Cid, Tetrahedron, 2006, 62, 11063–11072 CrossRef CAS; (b) C. Y. Legault, Y. Garcia, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 12664–12665 CrossRef CAS PubMed.
- (a) D.-T. D. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450–7453 CrossRef CAS PubMed; (b) M. Yang, J. Chen, C. He, X. Hu, Y. Ding, Y. Kuang, J. Liu and Q. Huang, J. Org. Chem., 2020, 85, 6498–6508 CrossRef CAS PubMed.
- (a) M. Opanasenko, P. Stepnicka and J. Cejka, RSC Adv., 2014, 4, 65137–65162 RSC; (b) M. Pagliaro, V. Pandarus, R. Ciriminna, F. Beland and P. D. Cara, ChemCatChem, 2012, 4, 432–445 CrossRef CAS; (c) Y. Uozumi, Recent progress in polymeric palladium catalysts for organic synthesis, in Immobilized Catalysts: Solid Phases, Immobilization and Applications, ed. A. Kirschning, vol. 242, 2004, pp. 77–112 Search PubMed.
- (a) K. C. Campbell and J. S. Hislop, J. Catal., 1969, 13, 12–19 CrossRef CAS; (b) J. A. Steckel, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 115412 CrossRef; (c) D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 855–859 CrossRef CAS; (d) P. Foley, R. DiCosimo and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 6713–6725 CrossRef CAS.
- N. G. Ol'ga, M. V. Livantsov, Y. K. Grishin, M. M. Ilyin Jr, K. A. Kochetkov, A. V. Churakov, L. G. Kuz'mina, V. N. Khrustalev and V. V. Dunina, J. Organomet. Chem., 2013, 737, 59–63 CrossRef.
- J. Stein, L. Lewis, Y. Gao and R. Scott, J. Am. Chem. Soc., 1999, 121, 3693–3703 CrossRef CAS.
- E. Bayram, J. C. Linehan, J. L. Fulton, J. A. Roberts, N. K. Szymczak, T. D. Smurthwaite, S. Ozkar, M. Balasubramanian and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 18889–18902 CrossRef CAS PubMed.
- O. N. Gorunova, I. M. Novitskiy, Y. K. Grishin, I. P. Gloriozov, V. A. Roznyatovsky, V. N. Khrustalev, K. A. Kochetkov and V. V. Dunina, Organometallics, 2018, 37, 2842–2858 CrossRef CAS.
- V. M. Chernyshev, A. V. Astakhov, I. E. Chikunov, R. V. Tyurin, D. B. Eremin, G. S. Ranny, V. N. Khrustalev and V. P. Ananikov, ACS Catal., 2019, 9, 2984–2995 CrossRef CAS.
- In a control reaction we found that Hg does react with non-immobilized Pd3Cl21 to give uncharacterizable species. Given that catalysis continues, it is unlikely we are seeing any appreciable leaching into solution for immobilized Pd3Cl23.
- J. Rebek and F. Gavina, J. Am. Chem. Soc., 1975, 97, 1591–1592 CrossRef CAS.
- (a) M. Hori, D. J. Gravert, P. Wentworth Jr and K. D. Janda, Bioorg. Med. Chem. Lett., 1998, 8, 2363–2368 CrossRef CAS PubMed; (b) I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 10139–10140 CrossRef CAS PubMed; (c) R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed.
- (a) P. Albers, J. Pietsch and S. F. Parker, J. Mol. Catal. A: Chem., 2001, 173, 275–286 CrossRef CAS; (b) P. Marcazzan, B. O. Patrick and B. R. James, Organometallics, 2003, 22, 1177–1179 CrossRef CAS; (c) A. Meißner, E. Alberico, H.-J. Drexler, W. Baumann and D. Heller, Catal. Sci. Technol., 2014, 4, 3409–3425 RSC.
- (a) C. Yue, Q. Xing, P. Sun, Z. Zhao, H. Lv and F. Li, Nat. Commun., 2021, 12, 1–14 CrossRef PubMed; (b) S. Reimann, J. Stötzel, R. Frahm, W. Kleist, J.-D. Grunwaldt and A. Baiker, J. Am. Chem. Soc., 2011, 133, 3921–3930 CrossRef CAS PubMed; (c) F. Zhao, K. Murakami, M. Shirai and M. Arai, J. Catal., 2000, 194, 479–483 CrossRef CAS; (d) P. A. Albinana, J. El Haskouri, M. D. Marcos, F. Estevan, P. Amorós, M. Á. Úbeda and F. Pérez-Pla, J. Catal., 2018, 367, 283–295 CrossRef; (e) C. Gnad, A. Abram, A. Urstöger, F. Weigl, M. Schuster and K. Köhler, ACS Catal., 2020, 10, 6030–6041 CrossRef CAS.
- (a) Z. Novák, R. Adamik, J. T. Csenki, F. Béke, R. Gavaldik, B. Varga, B. Nagy, Z. May, J. Daru, Z. Gonda and G. L. Tolnai, Nat. Catal., 2021, 4, 991–993 CrossRef; (b) C. S. Horbaczewskyj and I. J. S. Fairlamb, Org. Process Res. Dev., 2022, 26, 2240–2269 CrossRef CAS PubMed.
- J. M. Mercero, A. I. Boldyrev, G. Merino and J. M. Ugalde, Chem. Soc. Rev., 2015, 44, 6519–6534 RSC.
- Y. Wang, A. Monfredini, P.-A. Deyris, F. Blanchard, E. Derat, G. Maestri and M. Malacria, Chem. Sci., 2017, 8, 7394–7402 RSC.
- F. Bigi, G. Cera, R. Maggi, Y. Wang, M. Malacria and G. Maestri, J. Phys. Chem. A, 2021, 125, 10035–10043 CrossRef CAS PubMed.
- N. Jeddi, N. W. J. Scott and I. J. S. Fairlamb, ACS Catal., 2022, 12, 11615–11638 CrossRef CAS.
- (a) C. Lv, H. Cheng, W. He, M. I. A. Shah, C. Xu, X. Meng, L. Jiao, S. Wei, J. Li and L. Liu, Nano Res., 2016, 9, 2544–2550 CrossRef CAS; (b) M. Tromp, J. R. Sietsma, J. A. van Bokhoven, G. P. van Strijdonck, R. J. van Haaren, A. M. van der Eerden, P. W. N. M. van Leeuwen and D. C. Koningsberger, Chem. Commun., 2003, 128–129 RSC.
- N. Yuan, V. Pascanu, Z. Huang, A. Valiente, N. Heidenreich, S. Leubner, A. K. Inge, J. Gaar, N. Stock and I. Persson, J. Am. Chem. Soc., 2018, 140, 8206–8217 CrossRef CAS PubMed.
- (a) P. J. Ellis, I. J. S. Fairlamb, S. F. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820–1824 CrossRef CAS PubMed; (b) A. F. Lee, P. J. Ellis, I. J. S. Fairlamb and K. Wilson, Dalton Trans., 2010, 39, 10473–10482 RSC; (c) B. D. Briggs, N. M. Bedford, S. Seifert, H. Koerner, H. Ramezani-Dakhel, H. Heinz, R. R. Naik, A. I. Frenkel and M. R. Knecht, Chem. Sci., 2015, 6, 6413–6419 RSC.
- M. Tromp, J. A. van Bokhoven, G. P. F. van Strijdonck, P. W. N. M. van Leeuwen, D. C. Koningsberger and D. E. Ramaker, J. Am. Chem. Soc., 2005, 127, 777–779 CrossRef CAS PubMed.
- C.-M. Lin, T.-L. Hung, Y.-H. Huang, K.-T. Wu, M.-T. Tang, C.-H. Lee, C. T. Chen and Y. Y. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125426 CrossRef.
- J. Nilsson, P.-A. Carlsson and M. Skoglundh, Top. Catal., 2017, 60, 283–288 CrossRef CAS.
- S. R. Wasserman, P. G. Allen, D. K. Shuh, J. J. Bucher and N. M. Edelstein, J. Synchrotron Radiat., 1999, 6, 284–286 CrossRef CAS PubMed.
- A. Manceau, M. Marcus and T. Lenoir, J. Synchrotron Radiat., 2014, 21, 1140–1147 CrossRef CAS PubMed.
- A. Y. Platonov, A. N. Evdokimov, A. V. Kurzin and H. D. Maiyorova, J. Chem. Eng. Data, 2002, 47, 1175–1176 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- S. Singh, K. O. Sulaiman, Mahwar and R. W. J. Scott, Catal. Sci. Technol., 2023 10.1039/d3cy01316b.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, compound characterization, catalysis results and XAFS data. Single crystal XRD data for [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]SbF6. CCDC 2281111. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06447f |
| This journal is © The Royal Society of Chemistry 2024 |