
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Busting the myth of spontaneous formation of H2O2 at the air–water interface: contributions of the liquid–solid interface and dissolved oxygen exposed†
Muzzamil Ahmad
Eatoo
 ab and
Himanshu
Mishra
*abc
ab and
Himanshu
Mishra
*abc
aEnvironmental Science and Engineering (EnSE) Program, Biological and Environmental Science and Engineering (BESE) Division, King Abdullah University of Science and Technology (KAUST), 23955-6900, Thuwal, Kingdom of Saudi Arabia
bWater Desalination and Reuse Center (WDRC), King Abdullah University of Science and Technology (KAUST), 23955-6900, Thuwal, Kingdom of Saudi Arabia. E-mail: Himanshu.Mishra@kaust.edu.sa
cCenter for Desert Agriculture (CDA), King Abdullah University of Science and Technology (KAUST), 23955-6900, Thuwal, Kingdom of Saudi Arabia
First published on 24th January 2024
Abstract
Recent reports on the spontaneous formation of hydrogen peroxide (H2O2) at the air–water and solid–water interfaces challenge our current understanding of aquatic chemistry and have ramifications on atmosphere chemistry models, surface science, and green chemistry. Suggested mechanisms underlying this chemical transformation include ultrahigh instantaneous electric fields at the air–water interface and the oxidation of water and reduction of the solid at the solid–water interface. Here, we revisit this curious problem with NMR spectroscopy (with an H2O2 detection limit ≥50 nM) and pay special attention to the effects of nebulizing gas, dissolved oxygen content, and the solid–water interface on this chemical transformation in condensed and sprayed water microdroplets. Experiments reveal that the reduction of dissolved oxygen at the solid–water interface predominantly contributes to the H2O2 formation (not the oxidation of hydroxyl ions at the air–water interface or the oxidation of water at the solid–water interface). We find that the H2O2 formation is accompanied by the consumption (i.e., reduction) of dissolved oxygen and the oxidation of the solid surface, i.e., in the absence of dissolved oxygen, the formation of H2O2(aq) is not observed within the detection limit of ≥50 nM. Remarkably, the tendency of the solids investigated in this work towards forming H2O2 in water followed the same order as their positions in the classic Galvanic series. These findings bust the prevailing myths surrounding H2O2 formation due to the air–water interface, the ultrahigh electric fields therein, or the micro-scale of droplets. The hitherto unrealized role of the oxidation of the solid surface due to dissolved oxygen in the formation of H2O2 is exposed. These findings are especially relevant to corrosion science, surface science, and electrochemistry, among others.
Introduction
Hydrogen peroxide (H2O2) is an industrial chemical with a wide range of applications, such as disinfection,1 chemical synthesis,2 rocket propulsion,3 and wastewater treatment.4 The current production of H2O2 at scale relies on the anthraquinone cycling process, requiring significant energy and generating waste,5 necessitating sustainable alternatives. Recent reports on the laboratory-scale production of H2O2via electrochemical oxygen reduction are promising.6–8 However, electrochemical methods are not devoid of shortcomings; the process is complex and has the risk of spontaneous combustion and explosion. Multiple side reactions may further restrict the scalability of the process.In this context, sensational reports on the spontaneous formation of H2O2 at the aerial interface of water microdroplets seem enticing.9–13 Specifically, about 30 μM H2O2 was found in water microdroplets with a diameter of ≤20 μm sprayed via pressurized gas,9 and ≤115 μM H2O2 was discovered in condensed water microdroplets on common substrates in the relative humidity range of 40% to 70%.10 The presence of ultrahigh electric fields on microdroplet surfaces has been speculated to be the underlying cause.9,10 Moreover, studies have noted the implications for atmospheric chemistry,14 human health and bactericidal applications,13 green chemistry,9,14 and the seasonality of diseases due to the Goldilocks effect.12,15 We introduce the investigation of this chemical transformation by noting that the examination of water's interfaces is notorious for artifacts arising from contamination, incorrect interpretations of experiments, and the lack of encompassing multiscale computational models.16–38 Experience in microdroplet chemistry has demonstrated the need to stress-test the conclusion via multiple experimental techniques.
The prospect of aerosolized water microdroplets producing H2O2 is appealing due to its greenness and potential ease of application.13,39 While some experiments,40–43 computer simulations,44,45 and a Gedankenexperiment46 have given credence to the claim of spontaneous H2O2 formation at the air–water interface, others have disagreed.47–54 We commenced the investigation in 2019 by comparing the various commercially available assays for H2O2(aq). Compared to the potassium titanium oxalate assay (PTO; detection limit ≥10 μM), used in the original reports,9,10 the hydrogen peroxide assay kit (HPAK) affords a 40-times lower detection limit (≥250 nM). Equipped with HPAK, we employed a glovebox to assess H2O2(aq) concentrations in condensed water microdroplets generated in an N2(g) environment by gently heating water (50–70 °C). This experiment revealed that the H2O2 concentrations in the bulk water and the condensates were indistinguishable.47 We also discovered that if the condensates were produced via ultrasonic humidifiers, about 1 μM H2O2(aq) was produced in the water reservoir, the mist, and the condensates.47 This result was rationalized on the basis of the cavitating bubbles formed under ultrasonic acoustic pressure in bulk water, which is known to produce OH˙ radicals.49,55–58 However, why Zare & co-workers found ≤115 μM H2O2 in their experiments remained unclear.
From 2020 to 2021, we broadened the investigation to include microdroplets produced by pneumatic sprays. This device, like the one in the original report,9 facilitated the gas flow speeds of 100 to 1000 ms−1, breaking up water droplets to form sprays. In all these studies, (i) sprayed water microdroplets were collected in glass bottles, and (ii) condensed microdroplets were formed on SiO2/Si wafers. We discovered that the ppm-level of spontaneous H2O2 formation (1 ppm = 29.4 μM H2O2) occurred only in the presence of ozone (O3(g)).48
In their latest report, Zare & co-workers59 repeated the spray experiments in a controlled (ozone-free) gas environment. They employed 1H-NMR to quantify H2O2(aq) at a 40 nM resolution following the protocol of Bax et al. (Bruker 600 MHz Avance III, noncryogenic probe, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans with 0.1 s acquisition time).41,60 When the water was injected through copper tubing in the flow rate range of 25 to 150 μL min−1via a pressurized N2 at 100 psi (6.8 atm), the H2O2(aq) concentration in the sprays ranged from 1.5 to 0.3 μM (95–99% reduction from the original report9). They also found that, for a fixed liquid flow rate, as the nebulizing gas (fixed line pressure) was changed from (i) N2 to (ii) N2 + O2 (2%) to (iii) N2 + O2 (21%) to (iv) O2 (100%), the H2O2(aq) concentration increased from (i) 0.49 ± 0.05 μM to (ii) 0.69 ± 0.05 μM to (iii) 1.12 ± 0.02 μM to (iv) 2.00 ± 0.05 μM, respectively.59 Based on these observations, they contend that their original claims were correct (i.e., the microdroplet air–water interface spontaneously produces H2O2).
000 scans with 0.1 s acquisition time).41,60 When the water was injected through copper tubing in the flow rate range of 25 to 150 μL min−1via a pressurized N2 at 100 psi (6.8 atm), the H2O2(aq) concentration in the sprays ranged from 1.5 to 0.3 μM (95–99% reduction from the original report9). They also found that, for a fixed liquid flow rate, as the nebulizing gas (fixed line pressure) was changed from (i) N2 to (ii) N2 + O2 (2%) to (iii) N2 + O2 (21%) to (iv) O2 (100%), the H2O2(aq) concentration increased from (i) 0.49 ± 0.05 μM to (ii) 0.69 ± 0.05 μM to (iii) 1.12 ± 0.02 μM to (iv) 2.00 ± 0.05 μM, respectively.59 Based on these observations, they contend that their original claims were correct (i.e., the microdroplet air–water interface spontaneously produces H2O2).
Even if we assume that the latest claim is valid, the previous reports,9,10 which applied the PTO assay (H2O2 detection limit ≥10 μM), could not have detected the 0.30 to 2.00 μM H2O2(aq) concentrations (i.e., they were reporting artifacts of the ambient ozone gas48). This admission may help explain the latest59 and previous9 reports by Zare & co-workers of contradictory trends in the H2O2(aq) concentrations in water microdroplets when the concentration of dissolved oxygen (O2(aq)) is increased (explained in concluding remarks).
Lastly, two new experimental reports have surfaced in which H2O2 formation is also observed at the silica–water interface when (i) liquid water passes through a polydimethylsiloxane microfluidic chip placed on glass,11 and (ii) water vapor passes through a packed bed of SiO2 nanoparticles.15 Regarding the mechanism, the authors stated, “In fact, our proposed mechanism is built around the hypothesis that the overlap between the electron clouds of the water molecule and the solid surface during the contact will lead to the generation of H2O+ and OH˙”.11 They also presented an example: “Then, the electron may transfer from the water molecule to the surface of SiO2, which is the so-called contact electrification”.15
In this contribution, we investigate whether the skin of water (the air–water interface) is so unstable that airborne microdroplets can spontaneously produce H2O2, or whether another process is occurring. We investigate the origins of approx. 1 μM H2O2 in condensed and sprayed water microdroplets via1H-NMR, to answer the following interrelated fundamental questions:
(1) Is H2O2 formation in water microdroplets influenced by the nature of the nebulizing gas (viz., N2 or O2)?
(2) Would the H2O2 concentration in condensates collected in an inert gaseous environment be the same or different if the solid surface composition is varied (e.g., a SiO2/Si wafer or stainless steel)?
(3) Is the ‘micro’ scale of droplets necessary for the spontaneous formation of H2O2 at aqueous interfaces? What would the result be if pellets of a solid material (e.g., aluminum or mild steel) were immersed in bulk water, or if a film of water was sandwiched between two solid surfaces to eliminate the air–water interface from the picture?
(4) What is the role of dissolved oxygen (in water) in this chemical transformation? If the dissolved oxygen were removed from the water and sprayed, would H2O2 still form?
(5) During the spontaneous H2O2 formation at the solid–water interface, do water molecules transfer electrons to the solid and therefore reduce it?11,15
(6) Which aqueous interface would produce more H2O2 for a fixed area: the air–water interface or the solid–water interface (solid refers to common materials, such as glass, steel, etc.)?
Results
In a controlled gaseous environment (N2(g), unless specified otherwise) afforded by a clean glovebox, we collected water microdroplets formed via pneumatic sprays (Fig. S1 and Section S1 present the details†) or by condensing the vapor generated by gently heating water (60 °C) onto cold surfaces. First, we probed the effects of the nebulizing gas (N2 or O2) on the H2O2(aq) concentration. We applied the flow rates suggested by Zare & co-workers59 to maximize the H2O2 formation: a water flow rate of 25 μL min−1 through a 0.10 mm-wide silica capillary, nebulizing N2(g) gas at 100 psi shearing through an outer concentric tube of 0.43 mm in diameter (Fig. 1a & S1†). The spray was collected in a custom-built glass container, as described previously48 (Fig. S1†). The quantification of H2O2(aq) relied on 1H-NMR via the remarkable protocol of Bax et al.,60 which was also followed by Zare & co-workers.41,59 We employed a Bruker 950 MHz Avance Neo NMR spectrometer equipped with a 5 mm Z-axis gradient TCI cryoprobe at 275 K. During each measurement, a 6 ms Gaussian 90° pulse was applied to selectively excite the protons of H2O2, followed by a 53 ms acquisition time corresponding to 1024 detection points with a spectral width of 9615 Hz. Over 50000 scans were collected with a recycle delay of 1 ms between the scans. With this technique, we observed H2O2(aq) down to approx. 50 nM detection limit (see Fig. S2† for a representative calibration plot).
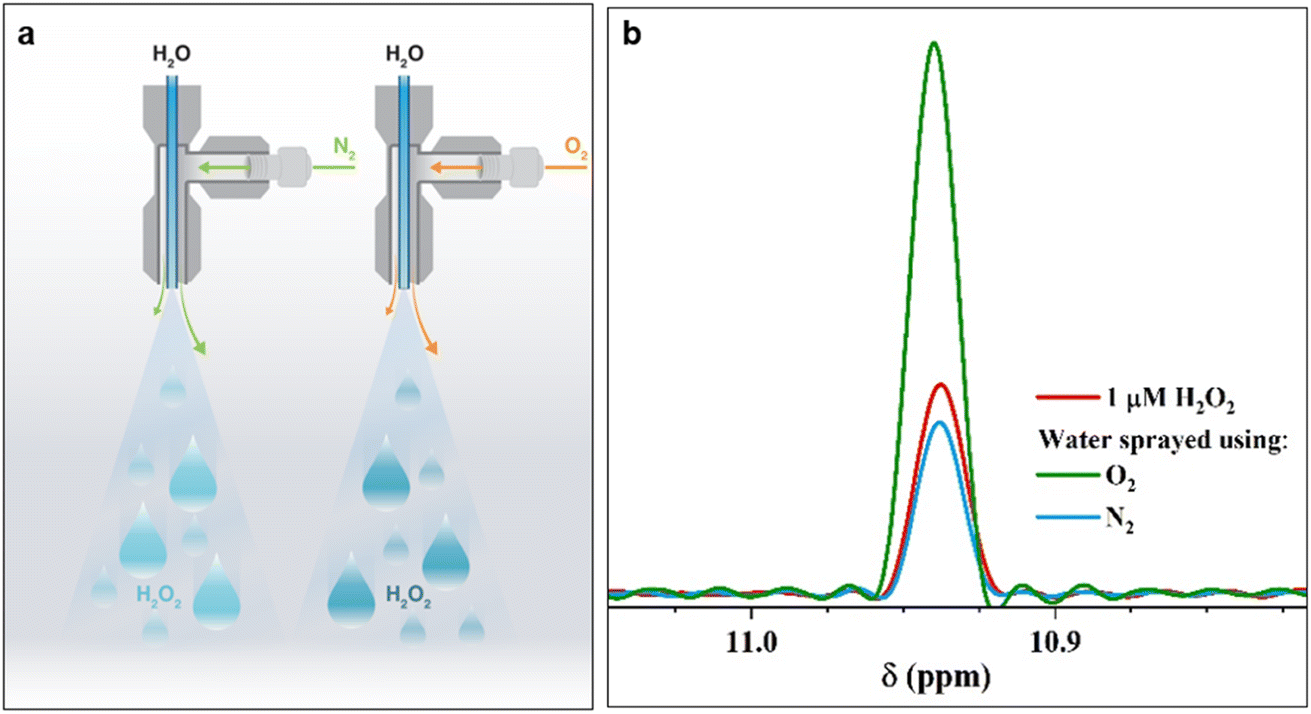 | ||
| Fig. 1 Pneumatic spraying of water into microdroplets produces H2O2. (a) Illustration of the experimental setup: as the nebulizing gas changes from N2(g) to O2(g), the H2O2(aq) concentration increases from 1.0 ± 0.2 μM to 3.0 ± 0.2 μM. (b) Representative 1H-NMR spectra of the reference standard, 1 μM H2O2 solution (red); 1H-NMR spectra of H2O2(aq) in water microdroplets nebulized by high pressure (100 psi) N2 (blue) and O2 (green) gases at an injection rate of 25 μL min−1. A description of our experimental set up is presented in ESI Section S1† along with photographs (Fig. S1†). | ||
The 1H-NMR results confirmed the presence of 1.0 ± 0.2 μM H2O2 in water microdroplets sprayed using N2 gas (Fig. 1). Next, when we switched the nebulizing gas to O2, keeping the water flow the same, the H2O2 concentration increased to 3.0 ± 0.2 μM (Fig. 1). Two crucial questions arose, which we address next: (i) If this phenomenon is driven by an ultrahigh instantaneous electric field at the air–water interface, then why does the nebulizing gas influence it; (ii) Could the solid–water interface drive this chemical transformation?
To answer these questions, we compared the amount of H2O2(aq) formed in water microdroplets condensed onto a variety of smooth and flat substrates: SiO2/Si wafers, polished titanium, polished stainless steel (SS304), polished mild steel, silicon surfaces (obtained by the reactive ion etching of SiO2/Si wafers), polished copper (Cu), polished magnesium alloy (Mg alloy, AZ31B), and polished aluminum (Al) (see Methods for details). Mechanical polishing was performed using emery paper with a grit size ranging from 400 to 1500 to remove the native oxide layer. While the size distribution of the microdroplets formed on these substrates did not vary significantly because of their superhydrophilic nature, a dramatic difference in the amount of H2O2(aq) occurred in the condensates, depending on the nature of the substrate (Fig. 2). For instance, as we replaced the SiO2/Si wafer substrate with a Mg alloy (AZ31B), the H2O2 concentration rose from 0.4 ± 0.2 μM to 68 ± 5 μM (Fig. 2). In this four year-long investigation of this phenomenon, predominantly with null results, this was the first time we observed the formation of ppm-level H2O2 (aq) in water microdroplets in the absence of O3(g).
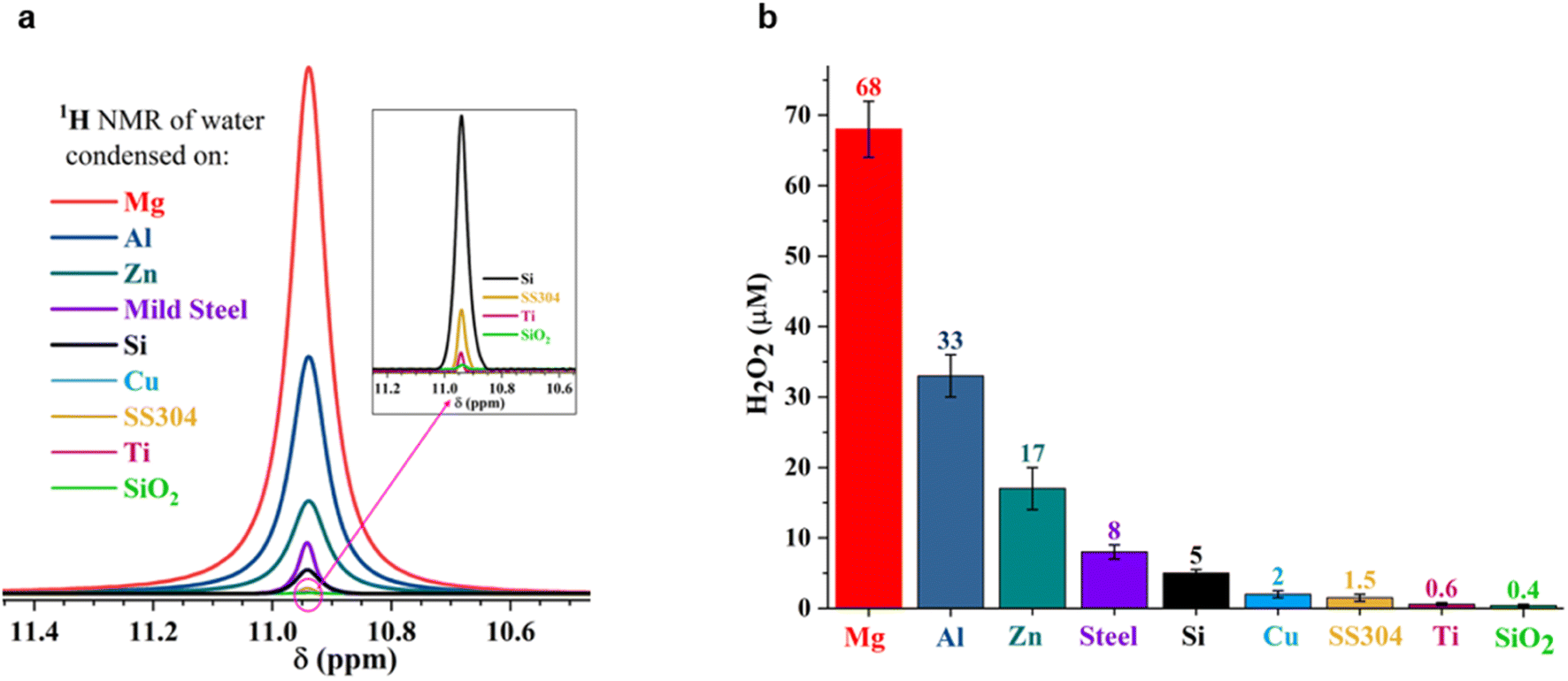 | ||
| Fig. 2 Concentration of H2O2(aq) formed in condensed water microdroplets varies with the nature of the solid substrate. (a) Selective-excitation 1H-NMR spectra for H2O2 quantification following the protocols developed by Bax et al.60 (b) Measured H2O2(aq) concentrations in condensates collected from the various substrates varied by over two orders of magnitude, confirmed via1H-NMR and HPAK. The air–water interfacial area of the microdroplets was not too dissimilar in these scenarios; thus, the solid–water interface drives the formation of H2O2. | ||
Notably, the H2O2 concentrations reported in Fig. 2 were those obtained on freshly prepared surfaces (i.e., without a native oxide layer). Over time, as the condensation experiments were repeated on the same surface, the extent of the H2O2(aq) formation decreased, underscoring the importance of the solid–water interface. For instance, the H2O2 produced on a freshly prepared Al surface was around 30 to 35 μM, which decreased to 7 to 8 μM in the second cycle and to 4 to 5 μM in the third cycle (each separated by 10 min).
Having identified that microdroplets placed on common materials, such as (polished) aluminum, produce ppm-level H2O2, we assessed the importance of the droplet size in this chemical transformation. We formed a 1:1 volumetric solution of deionized (DI) water with the HPAK reaction mixture and placed a macroscopic 1000 μL droplet (a base diameter of 12000 μm) onto an Al plate. Within a few seconds, we observed a sharp blue fluorescence – proof of the formation of H2O2(aq) – with an unambiguous gradient emanating from the Al–water interface (Fig. 3 and ESI Movie 1†). Judging by the fluorescence intensity, the local concentration of H2O2(aq) at the solid–liquid interface is at the ppm level (i.e., the air–water interface produced no fluorescence visible to the naked eye).
Building on this experiment, we reduced the air–water interfacial area from this three-phase system by (i) layering 1 ml of fresh DI-water–HPAK 1:1 mixture between two 20 × 20 cm2 Al plates and (ii) immersing freshly polished Mg pellets (6.2 cm2) in 5 ml of bulk DI water–HPAK 1:1 mixture (Fig. 4c and d). In all these scenarios, we discovered that the solid–water interface was the site for the spontaneous H2O2 formation, and the air–water interface had a negligible effect, if any. This observation is based on the distinct color gradient at the solid–water interface. After these experiments, we collected the water samples (films or bulk) and took 1H-NMR measurements (Table S1†). The H2O2 concentration produced by a sessile water droplet (∼1 ml) placed on a freshly polished Al plate and the scenarios (i) and (ii) listed above after 1 minute was 1.4 ± 0.5 μM, 39 ± 6 μM, and 2.5 ± 0.6 μM, respectively. A systematic study of the effects of the solid–water surface area and the effect of time is underway. These results unambiguously establish that the spontaneous H2O2 production in water does not necessitate microscopic droplets or the air–water interface, and it can occur even in bulk water when specific solid materials are introduced.
Next, we probed the surfaces before and after contact with water via X-ray photoelectron spectroscopy (XPS), which revealed that the spontaneous H2O2 formation at the water-solid surface was accompanied by substrate oxidation. For instance, on contact with water (condensed or both), metallic Al (Al0) was oxidized to Al3+, and semiconductor silicon (Si0) was oxidized to the Si4+ oxidation state (Fig. 5a–c). The water microdroplets spread and merged during each cycle, covering the entire (superhydrophilic) solid surface by the end of each cycle. This finding contradicts the claims that during H2O2 formation at the solid–water interface, OH− ions are oxidized to OH˙ (or H2O molecules are oxidized to H2O+),11,15 because, if this were true, the solid surface would be getting reduced, which is not the case. An in depth characterization of oxidation products is beyond the scope of this study and will be explored in the future.
 | ||
| Fig. 3 Time-dependent formation of H2O2(aq) in a macroscopic droplet of a 1:1 mixture of water and HPAK reaction mixture on an Al plate (see ESI Movie S1†). Within seconds, H2O2(aq) formation at the Al–water interface is evident, proving that the size of the droplet and air–water interface do not matter. The solid–water interface drives this chemical transformation. The Al surface is superhydrophilic, and water spreads on it as a film. Therefore, the Al plate was placed vertically on a polystyrene sheet and formed a 1 ml droplet resting on the Al edge. The 1:1 mixture of water and HPAK reaction on polystyrene did not yield the faintest blue fluorescence visible to the naked eye. | ||
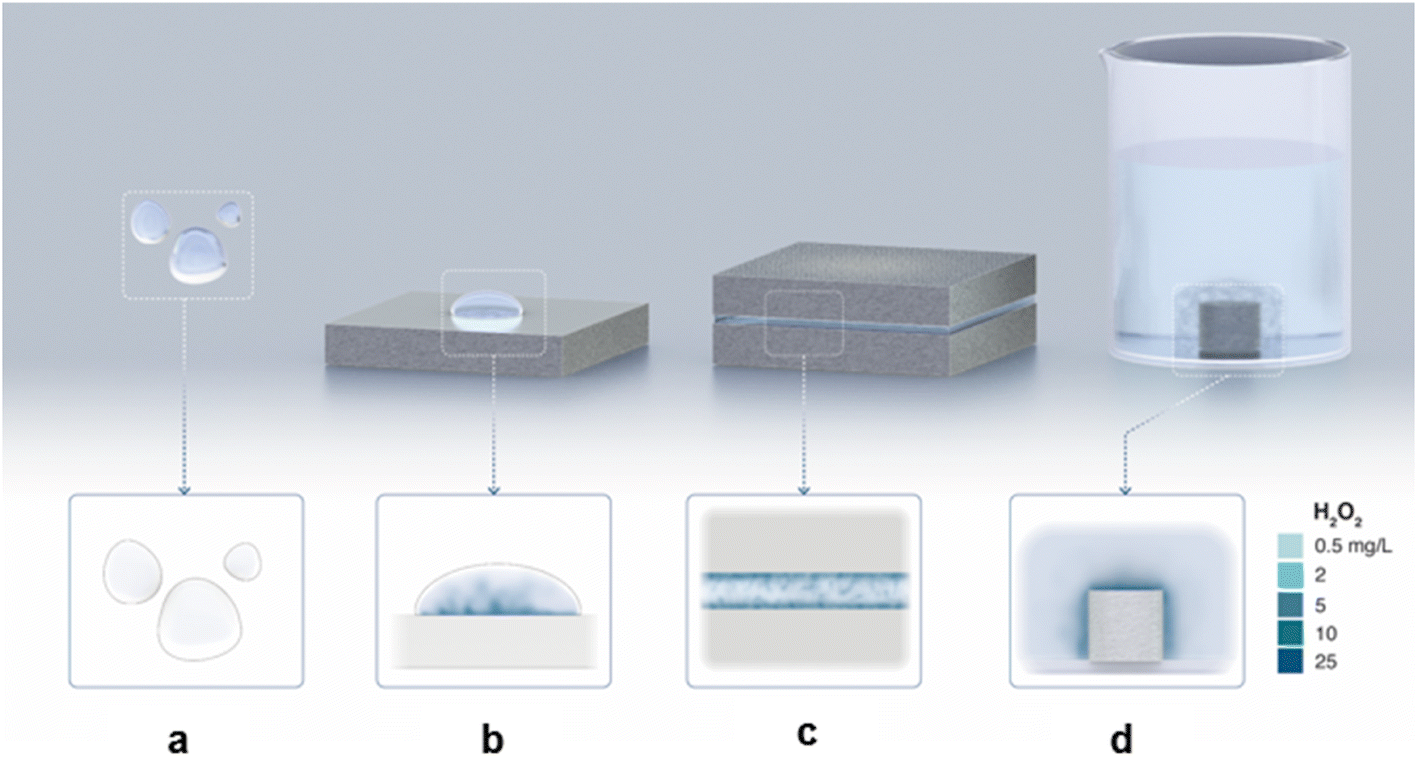 | ||
| Fig. 4 Illustration capturing the experimental observations: (a) No H2O2 is formed in the water microdroplets suspended in the air (i.e., without any contact with a solid). (b) H2O2 formation in water microdroplets on a solid substrate. (c) H2O2 formation in a water film between two solids. (d) H2O2 formation in bulk water at the solid–water interface. In the latter two cases, the air–water interface was practically eliminated. See text above and Table S1† for experimental details. | ||
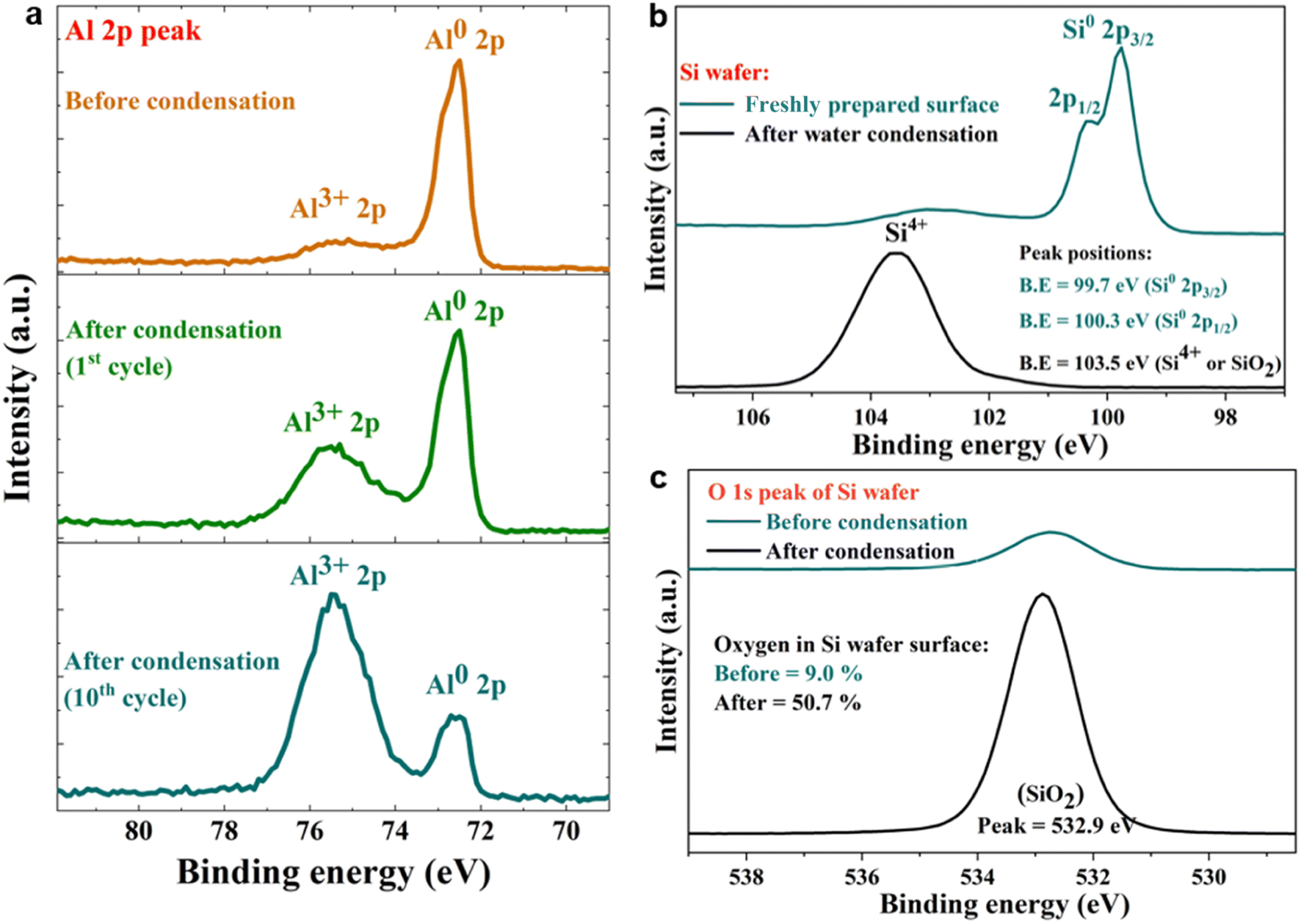 | ||
| Fig. 5 Representative XPS analysis of freshly polished metallic surfaces before and after contact with water. (a) High-resolution spectra of Al 2p before the condensation of water reveal a dominant metallic Al0 peak at 72.6 eV. After condensation cycles, the Al0 peak shrinks while the oxidized Al3+ peak (at 75.4 eV) increases. (b) High-resolution spectra of Si 2p before the water condensation cycle reveal a Si0 peak at 99.7 eV. After a water condensation cycle, the Si0 peak shrinks, and the Si4+ peak at 103.5 eV increases. (c) High-resolution XPS spectra of the O 1s peak on a freshly etched Si surface before and after a water condensation cycle. The surface oxygen composition increases from 9.0% to 50.7% after a condensation cycle due to the formation of the oxide layer. | ||
Following the XPS study, we noticed that the H2O2 formation was accompanied by surface oxidation. Therefore, we got curious whether it was due to the reduction of the dissolved oxygen.61 To examine this, we first removed dissolved O2 from the water by heating it in an autoclave to its boiling point, followed by N2(g) bubbling for 45 minutes and then sealing it inside an N2-purged container (methods). This treatment reduced the O2(aq) concentration to <0.01 mg L−1. Microdroplets of O2-free water were formed via pneumatic spraying using N2(g) in an N2 environment and collected in glass containers (following the same protocol). The H2O2(aq) concentration was compared with that in the microdroplets formed with water containing dissolved O2(g) (Fig. 6a). Remarkably, in the absence of O2(aq), no H2O2(aq) was observed via 1H-NMR (detection limit ≥50 nM; Fig. 6b). Next, we tested the effects of dissolved O2(g) on the formation of H2O2(aq) in bulk water by adding metallic pellets (Mg or Al). We tested the following three scenarios:
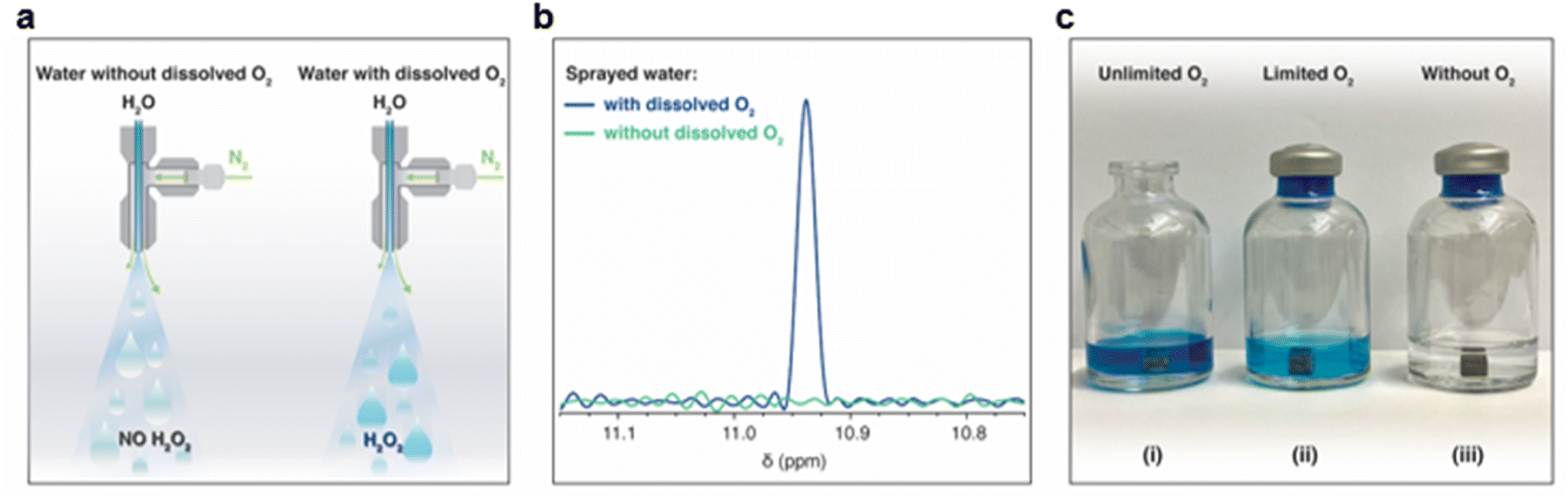 | ||
| Fig. 6 Role of the dissolved oxygen (O2) in water in the formation of H2O2 in microdroplets and bulk forms. (a) An illustration of the experiments in which water containing dissolved O2 and deoxygenated water was sprayed to form microdroplets. The microdroplets were collected in a glass vial, and H2O2(aq) was quantified. (b) Within a detection limit of 50 nM, 1H-NMR revealed that no H2O2 formed in the deoxygenated water, whereas H2O2(aq) was readily detected in the presence of dissolved O2. The air–water interface was common in both scenarios; thus, these experiments prove that the H2O2(aq) formation happens at the solid–water interface and dissolved O2(aq) is the limiting factor. (c) In another experiment, we prepared a 1:1 mixture of the HPAK reaction mixture with deoxygenated water and for the control case prepared the mixture with water containing dissolved O2(aq). Next, H2O2 formation in the following three scenarios was investigated: (i) an Mg pellet was added to the 1:1 mixture saturated with the ambient O2(g) and the vial was exposed to the ambient air (i.e., unlimited oxygen case); (ii) an Mg pellet was added to the 1:1 mixture saturated with dissolved oxygen O2(g), and the vial was sealed (i.e., the limited oxygen case); and (iii) a pellet was added to the 1:1 mixture without dissolved O2(g), and the vial was sealed (i.e., the without O2 case). In the absence of dissolved O2(aq), no H2O2(aq) formed within the detection limit of 0.25 μM, whereas it appeared readily in the presence of O2(aq), demonstrating that this chemical transformation occurs at the solid–water interface and that dissolved O2(aq) is a reactant. Therefore, H2O2 formation is not a property of the air–water interface or dependent on the size of the droplets. (Scale bar: the diameter of the pellet is 1 cm). | ||
(i) An Mg pellet was added to water saturated with the ambient O2(g), and the vial was left open in an O3-free ambient environment.
(ii) An Mg pellet was added to water saturated with ambient oxygen O2(g) and then sealed.
(iii) An Mg pellet was added to O2-free water in an N2(g) environment and sealed.
The vial open to the ambient air had significantly higher H2O2(aq) than that in the sealed vial containing water saturated with dissolved O2(g). Thus, the formation of H2O2 at the solid–water interface consumes dissolved O2(g) (i.e., it is the limiting factor). We also characterized the consumption of the dissolved O2(g) before and after adding the pellets and found that it decreased over time (Fig. S4†). Notably, in the absence of dissolved O2(g), we did not observe H2O2(aq) within the detection limit of 50 nM (Fig. 6b). These results unambiguously establish (i) the importance of dissolved O2(g) in this chemical transformation and that (ii) the air–water interface of microdroplets is incapable of forming H2O2.
Discussion
We draw together the results of this study and previous scientific reports to discuss the mechanisms underlying the formation of H2O2 in interfacial water (Fig. 7). The first finding is that the amount of H2O2(aq) formed in water condensates (or sprayed microdroplets) depends only on the nature of the surface on which it is collected (Fig. 2 and 3). In other words, the air–water interface or the size of the microdroplets has no bearing on the H2O2(aq) formation (Fig. 3, 4d, and S5†). For instance, as the air–water interface is reduced (or eliminated) from the picture, via layering water films between solid plates (Fig. 4c) (or by introducing solid pellets into bulk water) the formation of H2O2(aq) remains unaffected (Fig. 4c, d and S5†). The second crucial finding is that if the dissolved O2 is removed from the water, there is no evidence for H2O2(aq) formation within the detection limit (Fig. 6a–c). This observation contradicts the suggested mechanism for H2O2(aq) formation due to the charge transfer between positively (H3O+ rich) and negatively charged (OH− rich) microdroplets.42,46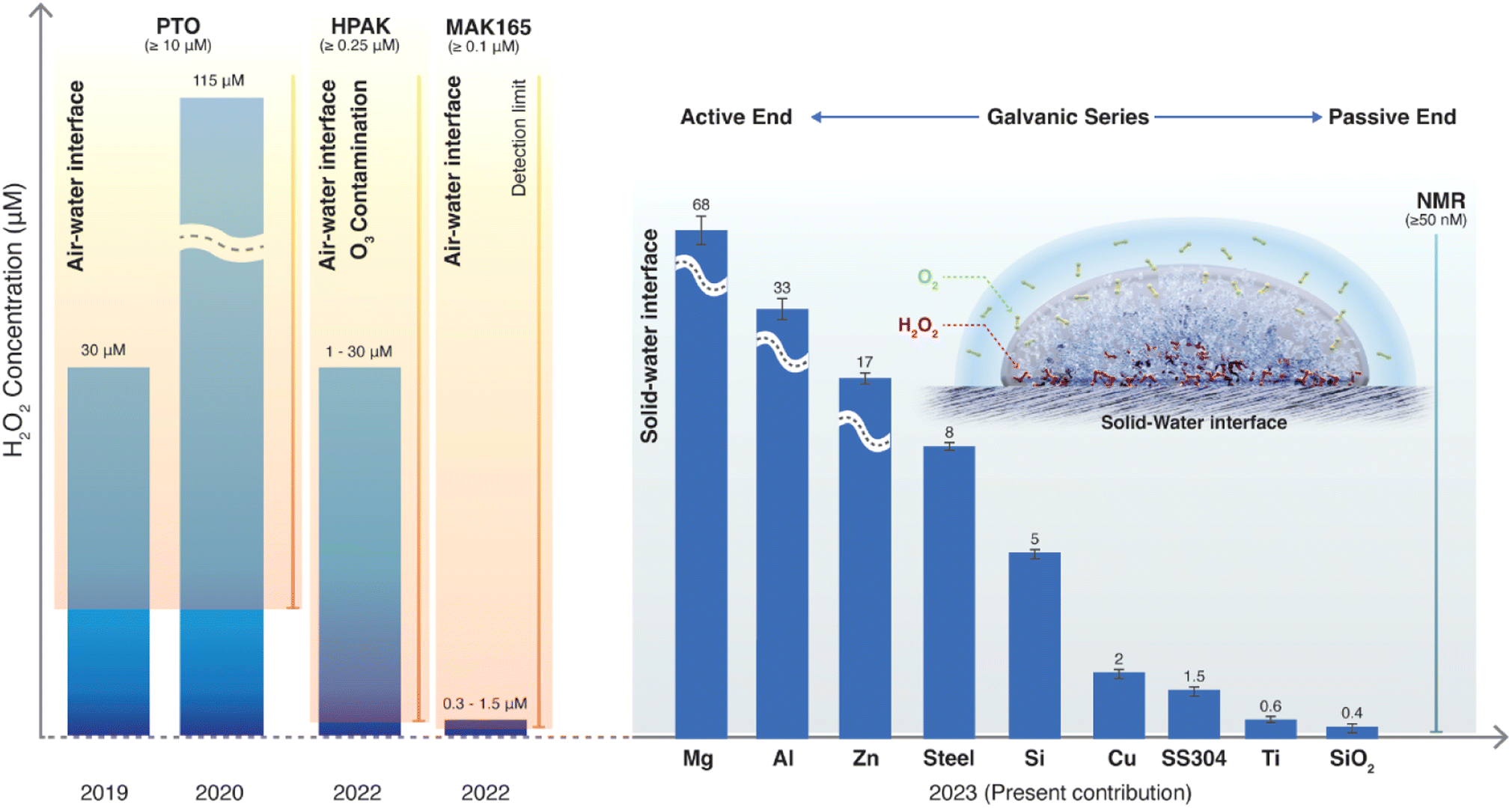 | ||
| Fig. 7 Current understanding of the spontaneous H2O2 formation in water microdroplets since its first report in 2019. Initial papers by Zare & coworkers used the PTO assay (detection limit ≥10 μM) and reported 30 and 110 μM H2O2(aq) in sprays and condensates, respectively.9,10 In 2022, it was revealed that ambient O3(g) could cause severe artifacts in these microdroplet experiments using HPAK (detection limit ≥0.25 μM).48 In 2023, using 1H-NMR (detection limit ≥0.04 μM), it was contended that in an O3-free environment, the air–water interface still produces H2O2 (∼1 μM).59 In the present contribution using 1H-NMR, we reveal that the solid–water interface is the site for H2O2(aq) formation, and the air–water interface does not contribute to H2O2 formation (quantified within the detection limit of ≥0.05 μM). If dissolved O2 is removed from the water, H2O2(aq) is not observed within the detection limit. Next, depending on the nature of the substrate that water contacts (as microdroplets or a film or as bulk water), the amount of H2O2(aq) formed follows the classic Galvanic series.62 | ||
Next, the XPS results demonstrate that the formation of H2O2(aq) is accompanied by the oxidation of the solid surface and the reduction of dissolved O2 (Fig. 5). Fig. S4† illustrates how the absolute concentration of O2(aq) decreases during H2O2 formation. When we evaluated the various commercially available materials used in this study in terms of their ability to form H2O2(aq) in (air-equilibrated) water, the trend followed the Galvanic series: Mg > Al > Zn > mild steel > Si > Cu > stainless steel (SS304) > Ti > SiO2/Si wafer (Fig. 7). Therefore, these findings refute the previous speculations regarding the oxidation of OH− ions to OH˙ (or the oxidation of H2O to H2O+) and the reduction of the solid surface during the H2O2 formation at the solid–water interface.11,15
We postulate that the initiation of this chemistry involves the reduction of dissolved O2(aq) by the solid surface; that is, the surface transfers two electrons into interfacial O2(aq), transforming it into a highly reactive peroxide dianion (O2˙2−).63 We anticipate this anion species to hover near the solid–water interface due to electrostatic attraction. Next, the anion reacts with interfacial water molecules to form H2O2 and hydroxide ions61,63 (the reactions below capture this logic):
| O2 + 2e− → O2˙2− | (1) |
| O2˙2− + 2H2O → H2O2 + 2OH− | (2) |
Reaction (2) indicates that the H2O2(aq) formation is accompanied by pH enhancement. The pH of the condensate collected on the Mg plate was around 7.9 ± 0.2, whereas the pH of the water reservoir used for gentle heating was 5.6 ± 0.1. The H2O2(aq) formation rate is the highest when the surface is free of native oxide and slows down as the oxide layer grows, which was also noticed in our experiments. Similar reaction schemes have been proposed recently, and in some of them, O2 is the byproduct.40 If this were true, then the H2O2(aq) formation due to the addition of a metal pellet (Mg or Al) to bulk water would be the same whether the dissolved O2 content was (i) unlimited (Fig. 6ci), (ii) limited (Fig. 6cii) or (ii) nil (Fig. 6ciii), but that is not the case. An in-depth investigation of the reaction intermediates (Reactions (1) and (2)) and the contribution of metal pellets on water-splitting reactions and the water pH is underway.
Conclusion
These findings put to rest several myths surrounding the spontaneous formation of H2O2 at the air–water interface, including the instantaneous ultrahigh electric fields, the “microscale” of droplets,9,10,12–14,40,41,44,45,59 and arguments based on charge transfer between microdroplets.42,46 For water containing dissolved O2, which is commonplace in environmental and applied scenarios, the solid–water interface is the site where O2(aq) reduces and forms H2O2(aq). One can therefore expect trace level (<0.5 μM) H2O2 to be produced at (clean) glass–water interfaces in laboratories routinely that, depending on the surface-to-volume ratio, may impact ultrasensitive investigation of aqueous interfaces and engineering processes such as semiconductor device fabrication. Notably, the ability of a solid to drive this chemical transformation depends on its position in the Galvanic series. For example, Mg and Al have low oxidation resistance; therefore, they form higher H2O2(aq), and, in contrast, Ti and stainless steel have high oxidation resistance, forming a lower amount of H2O2(aq). Next, our XPS experiments have revealed that the solid surface gets oxidized during the formation of H2O2, which refutes the suggestion that during the formation of H2O2 at the solid–water interface, water molecules transfer electrons to the solid, reducing it.11,15Crucially, in the absence of dissolved O2 in water, H2O2(aq) was not observed in pneumatic sprays or in bulk water containing pellets of Mg or Al (down to the 50 nM detection limit). This demonstrates that (i) the air–water interface of sprayed microdroplets and the putative (instantaneous) ultrahigh electric field therein are not capable of spontaneously forming H2O2 (Fig. 6), and (ii) the presence of dissolved O2 is a required condition for the solid–water interface to form H2O2. Therefore, we submit that the latest claims59,64 of the formation of ≤3 μM H2O2(aq) in water microdroplets (containing dissolved O2) suffered from artifacts arising due to the unavoidable physical contact of water with solid surfaces (e.g., during sample preparation, collection, and analysis), and evaporative concentration.48
When water microdroplets were formed by nebulizing with O2(g), it increased the O2(aq) concentration, promoting the formation of H2O2(aq) at the solid–water interface. Conversely, if the water is devoid of O2(g), H2O2 is not formed spontaneously at the solid–water interface. In that scenario, the air–water interface can contribute to the H2O2 formation in the following two ways: (i) transfer O2(g) to be reduced at the solid–water interface to form H2O2(aq) (see the inset in Fig. 7); or (ii) transfer O3(g) to oxidize water to form H2O2(aq) without the necessity of the solid–water interface. We hope these findings will advance the current knowledge of aquatic chemistry and prove relevant to corrosion science, electrochemistry, and soil chemistry.
Materials and methods
Chemicals
Deionized water was obtained from a Milli-Q Advantage 10 set-up (18.2 MΩ cm resistivity). This study used commercially available 30% hydrogen peroxide (H2O2) solution (Sigma-Aldrich CAS no. 7722-84-1) and deuterium oxide ((D2O, Catalog no. 3000007892).Spraying microdroplets
Inside a glove box with a controlled N2(g) atmosphere to prevent ambient contamination, the water was injected using a stainless steel capillary tube with an inner diameter of 100 μm using a syringe pump (PHD Ultra, Harvard Apparatus). Ultra-pure N2/O2 was pushed through a coaxial stainless steel sheath with an inner diameter of 430 μm to nebulize the water stream (Fig. S1†). The liquid water flow rate was 25 μL min−1, and approximately 2 ml of microdroplet volume was collected in clean glass vials for further analysis.Deoxygenation of water
Water was heated in an autoclave to its boiling point, followed by cooling via N2(g) bubbling for 45 min, lowering the temperature to about 40 °C. An O2 sensor (WTW Multi 3320) measured the dissolved O2 concentration in water with a detection limit of 0.01 mg L−1. After this treatment, we could not observe a signal for the O2(aq), meaning it was below the detection limit. Next, the water was quickly transferred to a glove box filled with N2 gas, where glass bottles were filled and sealed. The water sealed in an N2(g) filled glove box was autoclaved at 121 °C for 10 min to remove organic contamination on the vials.Substrates for condensation
Silicon (SiO2/Si) wafers of about 300 μm thickness, 10 cm in diameter, and 2 μm-thick thermally grown oxide were purchased from Silicon Valley Microelectronics (Catalog #SV010, p-type and 100 orientation). Fresh Si surfaces were prepared by etching the SiO2 layer via reactive ion etching (using C4F8 and O2(g) for 5 mi) inside the KAUST cleanroom.65 Directly afterward, condensation experiments were performed on the etched surfaces, and water samples were collected in clean glass vials for 1H-NMR analysis. Next, the following commercially available plates comprising metals or metallic alloys were used: Mg alloy (AZ31B, Thermo Scientific, Catalog No. AA14066RF), Al (Fisher Scientific, Catalog no. AA42124RF), mild steel, stainless steel (SS304), Zn (Thermo Scientific, Catalog No. AA11914FI), Cu (Thermo Scientific Chemicals, Catalog no. AA43822KS), and Ti (ASTM B 265 Trinity Brand Industries INC part #6T-5). The native oxide on the metal plates was removed via mechanical polishing using SiC emery papers with a grit size of 400 to 1500, followed by cleaning with pressurized N2 gas. For the condensation experiments, DI water was heated to 60 °C inside a closed chamber to produce the water vapor. Water microdroplets formed on cooled substrates (placed directly on ice) were collected using a low-pressure N2 gas stream and transferred to NMR tubes and glass vials for further analysis.Hydrogen peroxide assay kit (HPAK) assay
The H2O2 concentration of the condensed water was quantified using the hydrogen peroxide assay kit (Fluorometric-Near Infrared, Catalog # ab138886). It contains a unique AbIR peroxidase indicator that produces fluorescence independent of the solution pH in the range of 4 to 10. Its maximum excitation wavelength is 647 nm, and the maximum emission is 674 nm. The horseradish peroxidase enzyme catalyzes the reaction between H2O2 and the indicator and enhances the fluorescence signal. This action facilitates the linear detection range from 30 nM to 10 μM. The calibration curve (Fig. S3†) was realized by adding 50 μL of an H2O2 standard solution from a concentration of 50 nM to 10 μM into 50 μL of the H2O2 reaction mixture using a black 96-well microtiter-plate, and the SpectraMax M3 microplate reader (Molecular Devices LLC). The analysis software was SoftMax Pro 7. The water microdroplets were analyzed similarly by mixing 50 μL of each sample with the H2O2 reaction mixture, obtaining the respective concentration from the calibration curve.NMR spectroscopy analysis and sample preparation
No chemical was added to adjust the pH of the samples to avoid contamination. In each case, 10 μL of D2O was added to 490 μL analyte in regular 5 mm quartz NMR tubes for testing. All NMR measurements were conducted on a Bruker 950 MHz Avance Neo NMR spectrometer equipped with a 5 mm Z-axis gradient TCI cryoprobe at the temperature of 275 K. During the measurement, a 6 ms Gaussian 90° pulse was applied to selectively excite the protons of H2O2, followed by a 53 ms acquisition corresponding to 1024 detecting points with a spectral width of 9615 Hz. Over 50000 scans were collected with a recycle delay of 1 ms between scans. The NMR data were analyzed using TopSpin 4.2.0 software.
XPS measurements
A Kratos Axis Supra instrument equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV) operating at a power of 75 W under UHV conditions in the range of 10−9 mbar was used to obtain the data. All spectra were recorded in hybrid mode using magnetic and electrostatic lenses and an aperture slot of 300 × 700 μm. The high-resolution spectra were acquired at a fixed analyzer pass energy of 20 eV. The adventitious carbon (C 1s) peak at 284.5 eV was used as a reference for calibrating all peaks.Data availability
All data needed to evaluate the conclusions in the paper are present in the paper or the ESI.†Author contributions
HM conceived the research plan and oversaw its execution. ME designed and performed the condensation and spray experiments inside the glovebox and collected the 1H-NMR and HPAK data. ME and HM analyzed the data and wrote the manuscript together.Conflicts of interest
The authors declare no competing interests.Acknowledgements
HM acknowledges KAUST for funding (Grant No. BAS/1/1070-01-01). The coauthors thank Dr Adair Gallo and Ms Nayara Musskopf for building a robust experimental setup in HM's laboratory equipped with pneumatic sprays, a glovebox, an ozone meter, and so on. The coauthors are indebted to KAUST's Dr Xianrong Guo, Dr Christian Canlas, Prof. Lukasz Jaremko, and Dr Spyridon Gourdoupis for teaching them how to use 1H-NMR and giving them their precious slots on the Bruker 950 MHz NMR spectrometer for over three months to support this research. The coauthors thank Mr Heno Hwang, scientific illustrator at KAUST, for preparing illustrations in Fig. 1, 4, 6, and 7; Mr Amin Haider, Dr Sankara Arunachalam, Dr Hari Anand Rao, and Dr Nimer Wehbe from KAUST for assistance with Fig. S1,† the SiO2 etching work, water deaeration, and XPS results, respectively. The coauthors dedicate this paper to Prof. Rudy Marcus' centennial celebrations and thank him and Prof. Richard Saykally (UC Berkeley), Prof. Harry Gray (Caltech), Prof. Bill Goddard (Caltech), Prof. Paul Cremer (Penn State University), and Dr Adair Gallo (Terraxy LLC) for productive discussions.References
- J. A. Otter, S. Yezli, F. Barbut and T. M. Perl, in Decontamination in Hospitals and Healthcare, ed. J. Walker, Woodhead Publishing, 2nd edn, 2020, pp. 323–369, DOI:10.1016/B978-0-08-102565-9.00015-7.
- L. Kurti and B. Czakó, Strategic Applications of Named Reactions in Organic Synthesis, Academic Press, Illustrated edition, 2005 Search PubMed.
- W. Kopacz, A. Okninski, A. Kasztankiewicz, P. Nowakowski, G. Rarata and P. Maksimowski, Hydrogen peroxide–A promising oxidizer for rocket propulsion and its application in solid rocket propellants, FirePhysChem, 2022, 2, 56–66 CrossRef.
- M. Ksibi, Chemical oxidation with hydrogen peroxide for domestic wastewater treatment, Chem. Eng. J., 2006, 119, 161–165 CrossRef CAS.
- R. Hans-Joachim and P. Georg, Production of hydrogen peroxide, US Pat., US2158525A, 1939 Search PubMed.
- A. T. Murray, S. Voskian, M. Schreier, T. A. Hatton and Y. Surendranath, Electrosynthesis of Hydrogen Peroxide by Phase-Transfer Catalysis, Joule, 2019, 3, 2942–2954 CrossRef CAS.
- X. Zhang, X. Zhao, P. Zhu, Z. Adler, Z.-Y. Wu, Y. Liu and H. Wang, Electrochemical oxygen reduction to hydrogen peroxide at practical rates in strong acidic media, Nat. Commun., 2022, 13, 2880 CrossRef CAS PubMed.
- K. Wang, J. Huang, H. Chen, Y. Wang and S. Song, Recent advances in electrochemical 2e oxygen reduction reaction for on-site hydrogen peroxide production and beyond, Chem. Commun., 2020, 56, 12109–12121 RSC.
- J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Spontaneous generation of hydrogen peroxide from aqueous microdroplets, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19294–19298 CrossRef CAS PubMed.
- J. K. Lee, H. S. Han, S. Chaikasetsin, D. P. Marron, R. M. Waymouth, F. B. Prinz and R. N. Zare, Condensing water vapor to droplets generates hydrogen peroxide, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30934–30941 CrossRef CAS PubMed.
- B. Chen, Y. Xia, R. He, H. Sang, W. Zhang, J. Li, L. Chen, P. Wang, S. Guo, Y. Yin, L. Hu, M. Song, Y. Liang, Y. Wang, G. Jiang and R. N. Zare, Water–solid contact electrification causes hydrogen peroxide production from hydroxyl radical recombination in sprayed microdroplets, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2209056119 CrossRef CAS PubMed.
- M. T. Dulay, C. A. Huerta-Aguilar, C. F. Chamberlayne, R. N. Zare, A. Davidse and S. Vukovic, Effect of relative humidity on hydrogen peroxide production in water droplets, QRB Discov., 2021, 2, e8 CrossRef PubMed.
- M. T. Dulay, J. K. Lee, A. C. Mody, R. Narasimhan, D. M. Monack and R. N. Zare, Spraying Small Water Droplets Acts as a Bacteriocide, QRB Discov., 2020, 1, e3 CrossRef PubMed.
- C. Zhu and J. S. Francisco, Production of hydrogen peroxide enabled by microdroplets, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19222–19224 CrossRef CAS PubMed.
- Y. Xia, J. Li, Y. Zhang, Y. Yin, B. Chen, Y. Liang, G. Jiang and R. N. Zare, Contact between water vapor and silicate surface causes abiotic formation of reactive oxygen species in an anoxic atmosphere, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2302014120 CrossRef CAS PubMed.
- H. Mishra, S. Enami, R. J. Nielsen, L. A. Stewart, M. R. Hoffmann, W. A. Goddard and A. J. Colussi, Bronsted basicity of the air-water interface, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18679–18683 CrossRef CAS PubMed.
- R. J. Saykally, Air/water interface: Two sides of the acid-base story, Nat. Chem., 2013, 5, 82–84 CrossRef CAS PubMed.
- A. Gallo, A. S. F. Farinha, M. Dinis, A.-H. Emwas, A. Santana, R. J. Nielsen, W. A. Goddard and H. Mishra, The chemical reactions in electrosprays of water do not always correspond to those at the pristine air–water interface, Chem. Sci., 2019, 10, 2566–2577 RSC.
- A. J. Colussi and S. Enami, Comment on “The chemical reactions in electrosprays of water do not always correspond to those at the pristine air–water interface” by A. Gallo Jr, A. S. F. Farinha, M. Dinis, A.-H. Emwas, A. Santana, R. J. Nielsen, W. A. Goddard III and H. Mishra, Chem. Sci., 2019, 10, 2566, 10.1039/C9SC00991D.
- A. Gallo, A. S. F. Farinha, A.-H. Emwas, A. Santana, R. J. Nielsen, W. A. Goddard and H. Mishra, Reply to the ‘Comment on “The chemical reactions in electrosprays of water do not always correspond to those at the pristine air–water interface”’ by A. J. Colussi and S. Enami, 2019, 10, DOI: 10.1039/c9sc00991d, Chem. Sci., 2019, 10, 8253–8255, 10.1039/C9SC02702E.
- J. Nauruzbayeva, Z. Sun, A. Gallo, M. Ibrahim, J. C. Santamarina and H. Mishra, Electrification at water–hydrophobe interfaces, Nat. Commun., 2020, 11, 5285 CrossRef CAS PubMed.
- Y. Uematsu, D. J. Bonthuis and R. R. Netz, Charged Surface-Active Impurities at Nanomolar Concentration Induce Jones-Ray Effect, J. Phys. Chem. Lett., 2018, 9, 189–193 CrossRef CAS PubMed.
- S. J. Byrnes, P. L. Geissler and Y. R. Shen, Ambiguities in surface nonlinear spectroscopy calculations, Chem. Phys. Lett., 2011, 516, 115–124 CrossRef CAS.
- N. Agmon, H. J. Bakker, R. K. Campen, R. H. Henchman, P. Pohl, S. Roke, M. Thämer and A. Hassanali, Protons and Hydroxide Ions in Aqueous Systems, Chem. Rev., 2016, 116, 7642–7672 CrossRef CAS PubMed.
- M. F. Ruiz-Lopez, J. S. Francisco, M. T. C. Martins-Costa and J. M. Anglada, Molecular reactions at aqueous interfaces, Nat. Rev. Chem, 2020, 4, 459–475 CrossRef CAS PubMed.
- M. I. Jacobs, R. D. Davis, R. J. Rapf and K. R. Wilson, Studying Chemistry in Micro-compartments by Separating Droplet Generation from Ionization, J. Am. Soc. Mass Spectrom., 2019, 30, 339–343 CrossRef CAS PubMed.
- G. Rovelli, M. I. Jacobs, M. D. Willis, R. J. Rapf, A. M. Prophet and K. R. Wilson, A critical analysis of electrospray techniques for the determination of accelerated rates and mechanisms of chemical reactions in droplets, Chem. Sci., 2020, 11, 13026–13043 RSC.
- S. Pullanchery, S. Kulik, B. Rehl, A. Hassanali and S. Roke, Charge transfer across C–H⋅⋅⋅O hydrogen bonds stabilizes oil droplets in water, Science, 2021, 374, 1366–1370 CrossRef CAS PubMed.
- H. Wei, E. P. Vejerano, W. Leng, Q. Huang, M. R. Willner, L. C. Marr and P. J. Vikesland, Aerosol microdroplets exhibit a stable pH gradient, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7272–7277 CrossRef CAS PubMed.
- A. J. Colussi, Can the pH at the air/water interface be different from the pH of bulk water?, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7887 CrossRef CAS PubMed.
- M. Li, Y. Kan, H. Su, U. Pöschl, S. H. Parekh, M. Bonn and Y. Cheng, Spatial homogeneity of pH in aerosol microdroplets, Chem, 2023, 9, 1036–1046 CAS.
- K. Roger and B. Cabane, Why Are Hydrophobic/Water Interfaces Negatively Charged?, Angew. Chem., Int. Ed., 2012, 51, 5625–5628 CrossRef CAS PubMed.
- K. Roger and B. Cabane, Uncontaminated Hydrophobic/Water Interfaces Are Uncharged: A Reply, Angew. Chem., Int. Ed., 2012, 51, 12943–12945 CrossRef CAS.
- K. C. Jena, R. Scheu and S. Roke, Surface Impurities Are Not Responsible For the Charge on the Oil/Water Interface: A Comment, Angew. Chem., Int. Ed., 2012, 51, 12938–12940 CrossRef CAS PubMed.
- J. K. Beattie and A. Gray-Weale, Oil/Water Interface Charged by Hydroxide Ions and Deprotonated Fatty Acids: A Comment, Angew. Chem., Int. Ed., 2012, 51, 12941–12942 CrossRef CAS PubMed.
- D. Ben-Amotz, Electric buzz in a glass of pure water, Science, 2022, 376, 800–801 CrossRef CAS PubMed.
- E. Poli, K. H. Jong and A. Hassanali, Charge transfer as a ubiquitous mechanism in determining the negative charge at hydrophobic interfaces, Nat. Commun., 2020, 11, 901 CrossRef CAS PubMed.
- S. Enami, H. Mishra, M. R. Hoffmann and A. J. Colussi, Protonation and Oligomerization of Gaseous Isoprene on Mildly Acidic Surfaces: Implications for Atmospheric Chemistry, J. Phys. Chem. A, 2012, 116, 6027–6032 CrossRef CAS PubMed.
- J. A. Otter, S. Yezli, F. Barbut and T. M. Perl, An overview of automated room disinfection systems: When to use them and how to choose them, Decontamination in Hospitals and Healthcare, 2020, pp. 323–369, DOI:10.1016/B978-0-08-102565-9.00015-7.
- K. Li, Y. Guo, S. A. Nizkorodov, Y. Rudich, M. Angelaki, X. Wang, T. An, S. Perrier and C. George, Spontaneous dark formation of OH radicals at the interface of aqueous atmospheric droplets, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220228120 CrossRef CAS PubMed.
- T. Kakeshpour, B. Metaferia, R. N. Zare and A. Bax, Quantitative detection of hydrogen peroxide in rain, air, exhaled breath, and biological fluids by NMR spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2121542119 CrossRef CAS PubMed.
- S. Lin, L. N. Y. Cao, Z. Tang and Z. L. Wang, Size-dependent charge transfer between water microdroplets, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2307977120 CrossRef CAS PubMed.
- H. Xiong, J. K. Lee, R. N. Zare and W. Min, Strong Electric Field Observed at the Interface of Aqueous Microdroplets, J. Phys. Chem. Lett., 2020, 11, 7423–7428 CrossRef CAS PubMed.
- H. Hao, I. Leven and T. Head-Gordon, Can electric fields drive chemistry for an aqueous microdroplet?, Nat. Commun., 2022, 13, 280 CrossRef CAS PubMed.
- J. P. Heindel, H. Hao, R. A. LaCour and T. Head-Gordon, Spontaneous formation of hydrogen peroxide in water microdroplets, J. Phys. Chem. Lett., 2022, 13, 10035–10041 CrossRef CAS PubMed.
- A. J. Colussi, Mechanism of Hydrogen Peroxide Formation on Sprayed Water Microdroplets, J. Am. Chem. Soc., 2023, 145, 16315–16317 CrossRef CAS PubMed.
- N. H. Musskopf, A. Gallo, P. Zhang, J. Petry and H. Mishra, The Air–Water Interface of Water Microdroplets Formed by Ultrasonication or Condensation Does Not Produce H2O2, J. Phys. Chem. Lett., 2021, 12, 11422–11429 CrossRef CAS PubMed.
- A. Gallo Jr, N. H. Musskopf, X. Liu, Z. Yang, J. Petry, P. Zhang, S. Thoroddsen, H. Im and H. Mishra, On the formation of hydrogen peroxide in water microdroplets, Chem. Sci., 2022, 13, 2574–2583 RSC.
- D. Nguyen and S. C. Nguyen, Revisiting the Effect of the Air–Water Interface of Ultrasonically Atomized Water Microdroplets on H2O2 Formation, J. Phys. Chem. B, 2022, 126, 3180–3185 CrossRef CAS PubMed.
- D. Nguyen, P. Lyu and S. C. Nguyen, Experimental and Thermodynamic Viewpoints on Claims of a Spontaneous H2O2 Formation at the Air–Water Interface, J. Phys. Chem. B, 2023, 127, 2323–2330 CrossRef CAS PubMed.
- M. T. Martins-Costa and M. F. Ruiz-López, Probing solvation electrostatics at the air–water interface, Theor. Chem. Acc., 2023, 142, 29 Search PubMed.
- M. Peplow, Claims of water turning into hydrogen peroxide spark debate, Chem. Eng. News, 2022 Search PubMed , https://cen.acs.org/research-integrity/reproducibility/Claims-water-turning-hydrogen-peroxide/100/web/2022/05.
- T. Cozens, Study casts doubt on water microdroplets’ ability to spontaneously produce hydrogen peroxide, Chem. World, 2022 Search PubMed , https://www.chemistryworld.com/news/study-casts-doubt-on-water-microdroplets-ability-to-spontaneously-produce-hydrogen-peroxide/4015169.article.
- R. Trager, Water surprise: microdroplets have potential to produce hydrogen peroxide, Chem. World, 2019 Search PubMed , https://www.chemistryworld.com/news/water-surprise-microdroplets-have-potential-to-produce-h2o2/3010950.article#commentsJump.
- P. Riesz, D. Berdahl and C. L. Christman, Free radical generation by ultrasound in aqueous and nonaqueous solutions, Environ. Health Perspect., 1985, 64, 233–252 CrossRef CAS PubMed.
- X. Fang, G. Mark and C. von Sonntag, OH radical formation by ultrasound in aqueous solutions Part I: the chemistry underlying the terephthalate dosimeter, Ultrason. Sonochem., 1996, 3, 57–63 CrossRef CAS.
- M. Hoffmann, I. Hua and R. Hoechemer, Application of Ultrasonic Irradiation for the Degradation of Chemical Contaminants in Water, Ultrason. Sonochem., 1996, 3, 168–172 CrossRef.
- J. H. Bang and K. S. Suslick, Applications of Ultrasound to the Synthesis of Nanostructured Materials, Adv. Mater., 2010, 22, 1039–1059 CrossRef CAS PubMed.
- M. A. Mehrgardi, M. Mofidfar and R. N. Zare, Sprayed Water Microdroplets Are Able to Generate Hydrogen Peroxide Spontaneously, J. Am. Chem. Soc., 2022, 144, 7606–7609 CrossRef CAS PubMed.
- T. Kakeshpour and A. Bax, NMR characterization of H2O2 hydrogen exchange, J. Magn. Reson., 2021, 333, 107092 CrossRef CAS PubMed.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Superoxide Ion: Generation and Chemical Implications, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2nd edn, 2001 Search PubMed.
- I. Ivanovic-Burmazovic, Catalytic dismutation vs. reversible binding of superoxide, Adv. Inorg. Chem., 2008, 60, 59–100 CrossRef CAS.
- K. Zhou, H. Su, J. Gao, H. Li, S. Liu, X. Yi, Z. Zhang and W. Wang, Deciphering the Kinetics of Spontaneous Generation of H2O2 in Individual Water Microdroplets, J. Am. Chem. Soc., 2024 DOI:10.1021/jacs.3c09864.
- S. Arunachalam, E. M. Domingues, R. Das, J. Nauruzbayeva, U. Buttner, A. Syed and H. Mishra, Rendering SiO2/Si Surfaces Omniphobic by Carving Gas-Entrapping Microtextures Comprising Reentrant and Doubly Reentrant Cavities or Pillars, J. Vis. Exp., 2020, e60403, DOI:10.3791/60403.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06534k |
| This journal is © The Royal Society of Chemistry 2024 |