
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient C–N coupling for urea electrosynthesis on defective Co3O4 with dual-functional sites†
Pengsong
Li
ab,
Qinggong
Zhu
 *ab,
Jiyuan
Liu
ab,
Tianbin
Wu
ab,
Xinning
Song
ab,
Qinglei
Meng
ab,
Xinchen
Kang
ab,
Xiaofu
Sun
ab and
Buxing
Han
*abc
*ab,
Jiyuan
Liu
ab,
Tianbin
Wu
ab,
Xinning
Song
ab,
Qinglei
Meng
ab,
Xinchen
Kang
ab,
Xiaofu
Sun
ab and
Buxing
Han
*abc
aBeijing National Laboratory for Molecular Sciences, CAS Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Center for Carbon Neutral Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: qgzhu@iccas.ac.cn; hanbx@iccas.ac.cn
bSchool of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
First published on 18th January 2024
Abstract
Urea electrosynthesis under ambient conditions is emerging as a promising alternative to conventional synthetic protocols. However, the weak binding of reactants/intermediates on the catalyst surface induces multiple competing pathways, hindering efficient urea production. Herein, we report the synthesis of defective Co3O4 catalysts that integrate dual-functional sites for urea production from CO2 and nitrite. Regulating the reactant adsorption capacity on defective Co3O4 catalysts can efficiently control the competing reaction pathways. The urea yield rate of 3361 mg h−1 gcat−1 was achieved with a corresponding faradaic efficiency (FE) of 26.3% and 100% carbon selectivity at a potential of −0.7 V vs. the reversible hydrogen electrode. Both experimental and theoretical investigations reveal that the introduction of oxygen vacancies efficiently triggers the formation of well-matched adsorption/activation sites, optimizing the adsorption of reactants/intermediates while decreasing the C–N coupling reaction energy. This work offers new insights into the development of dual-functional catalysts based on non-noble transition metal oxides with oxygen vacancies, enabling the efficient electrosynthesis of essential C–N fine chemicals.
Introduction
Urea is widely used as a fertilizer, as well as a chemical raw material for medicines and pesticides. However, traditional industrial urea synthesis, relying on the reaction of CO2 and NH3, involves harsh conditions and leaves a significant carbon footprint,1,2 necessitating the pursuit of greener, more energy-efficient, and cost-effective alternatives.3,4 Electrochemical urea synthesis from N-integrated electrocatalytic CO2 reduction is gradually emerging as a promising alternative to reform the conventional urea industry.5–7 Most strikingly, emerging electrocatalytic co-reduction of N2 and CO2 in an aqueous solution has been demonstrated as a feasible method for urea synthesis.8–12 However, the inherent chemical inertness of N2, such as high triple bond dissociation energy (941 kJ mol−1) and low solubility (∼0.02 v/v, 1 atm, 25 °C), poses a considerable limitation on urea yield rates.Alternatively, N-integrated electrochemical CO2 reduction can be performed with nitrate/nitrite, which is a widely existing contaminant in the wastewater and poses severe risks to both public health and the environment. In particular, using nitrite as a N-containing reactant for urea electrosynthesis is highly desirable due to its merits of superior solubility (e.g. KNO2, 2.81 g mL−1, 0 °C) and lower N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond dissociation energy (204 kJ mol−1), which could potentially result in higher performance for urea synthesis.13 Some pioneer works have shown that urea can be synthesized from electrochemically coupling CO2 and nitrate/nitrite.14–20 For example, the In(OH)3–S catalyst could promote co-electrolysis of CO2 and nitrate to urea with an average yield rate of 533.1 mg h−1 gcat−1 and a Faradaic efficiency (FE) of 53.4% at the potential of −0.6 V vs. the reversible hydrogen electrode (RHE).21 Over a Cu1–CeO2 catalyst, the yield rate of urea could reach 52.84 mmol h−1 gcat−1 at −1.6 V vs. RHE.22 Low-valence Cuδ+-doped anatase TiO2 was employed as an electrocatalyst for the co-electrolysis of CO2 and nitrite ions to urea with a yield rate of 20.8 μmol h−1 at a low potential of −0.4 V vs. RHE.23 In another study, a Te-doped Pd nanocrystal was utilized to enhance the electrochemical urea production by coupling CO2 and nitrite reduction with nearly 12.2% FE and 88.7% N atom efficiency.24 Despite substantial progress having been made in urea synthesis, there still remains a significant challenge in improving urea FE and yield rate. This is because the parallel CO2 reduction reaction, nitrate/nitrite reduction reaction and inescapable hydrogen evolution reaction (HER) strongly compete with the desirable C–N coupling reaction, leading to low selectivity of the urea product. The primary obstacle lies in the suppression of selective hydrogenation of intermediate species to unwanted by-products and the hindrance of the unfavorable HER to facilitate C–N coupling for urea production.
O bond dissociation energy (204 kJ mol−1), which could potentially result in higher performance for urea synthesis.13 Some pioneer works have shown that urea can be synthesized from electrochemically coupling CO2 and nitrate/nitrite.14–20 For example, the In(OH)3–S catalyst could promote co-electrolysis of CO2 and nitrate to urea with an average yield rate of 533.1 mg h−1 gcat−1 and a Faradaic efficiency (FE) of 53.4% at the potential of −0.6 V vs. the reversible hydrogen electrode (RHE).21 Over a Cu1–CeO2 catalyst, the yield rate of urea could reach 52.84 mmol h−1 gcat−1 at −1.6 V vs. RHE.22 Low-valence Cuδ+-doped anatase TiO2 was employed as an electrocatalyst for the co-electrolysis of CO2 and nitrite ions to urea with a yield rate of 20.8 μmol h−1 at a low potential of −0.4 V vs. RHE.23 In another study, a Te-doped Pd nanocrystal was utilized to enhance the electrochemical urea production by coupling CO2 and nitrite reduction with nearly 12.2% FE and 88.7% N atom efficiency.24 Despite substantial progress having been made in urea synthesis, there still remains a significant challenge in improving urea FE and yield rate. This is because the parallel CO2 reduction reaction, nitrate/nitrite reduction reaction and inescapable hydrogen evolution reaction (HER) strongly compete with the desirable C–N coupling reaction, leading to low selectivity of the urea product. The primary obstacle lies in the suppression of selective hydrogenation of intermediate species to unwanted by-products and the hindrance of the unfavorable HER to facilitate C–N coupling for urea production.
To address this, we proposed a two-step thermal annealing strategy to synthesize defective Co3O4 catalysts, integrating dual-functional sites to favor urea electrosynthesis. We discovered that the presence of oxygen vacancies (Vo) could construct well-matched adsorption/activation sites, which strengthened the adsorption of CO2 and NO2− reactants. The modulated local environment on Co atoms near the oxygen vacancies, serving as the dual-functional sites, not only strengthened the adsorption of intermediates but also decreased the C–N coupling resistance in urea synthesis. Notably, when using defective Co3O4 obtained under suitable treatment conditions as an electrocatalyst, an impressive urea yield rate of 3361 mg h−1 gcat−1 was achieved with a corresponding FE of 26.3% and 100% carbon selectivity at the potential of −0.7 V vs. RHE.
Results and discussion
We used a two-step thermal annealing strategy to synthesize defective Co3O4 catalysts with tunable Vo concentrations. Briefly, Co(NO3)2·6H2O and NaCl were first mixed in an agate mortar, followed by adding NaOH solution for sufficient grinding and solvent removal. The resulting solid solution was then directly pyrolysed under an Ar atmosphere to get the pristine Co3O4, in which the Vo concentrations could be tuned by thermal treatment under a reducing atmosphere (H2/Ar) for different times (0.5, 1.0 and 1.5 h). The as-synthesized catalysts were denoted as Co3O4-0.5, Co3O4-1.0, and Co3O4-1.5, respectively. We commenced our characterization using scanning electron microscopy (SEM). As shown in Fig. 1a and b and S1,† both the pristine and reduced thermally treated Co3O4 had nanosized and octahedral structures, indicating that the reducing thermal annealing process did not affect the initial morphology. The morphological and structural information was further studied by transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM). Typically, the TEM images in Fig. 1c and d demonstrated that Co3O4-1.0 had a rougher surface than pristine Co3O4, which could be attributed to the formation of high Vo concentration on the catalyst surface. The HRTEM images in Fig. 1e and f showed that the lattice fringe of the Co3O4 nanoparticle was 0.24 nm, which is ascribed to the lattice plane distance of the (311) plane of Co3O4. The main difference was that the pristine Co3O4 featured a flatter crystal surface (Fig. 1e) than Co3O4-1.0 (Fig. 1f). The normalized Co K-edge X-ray absorption near edge structure (XANES) spectra (Fig. 1g) revealed a slightly reduced average valence state of the Co species in Co3O4-1.0, indicating that Vo can manipulate the electronic state of the nearby Co atoms. Additionally, the Co K-edge extended X-ray absorption fine structure (EXAFS) spectra of pristine Co3O4 and Co3O4-1.0 demonstrated three distinct peaks (Fig. 1h), which were identified as Co–O, tetrahedral Co–Co, and octahedral Co–Co bonds, respectively. Comparably, the intensity of the Co–O bond in Co3O4-1.0 was lower than that in the initial Co3O4, suggesting a reduced oxygen coordination number for the Co atoms in Co3O4-1.0. This reduction is attributed to the introduction of additional Vo in Co3O4-1.0.25,26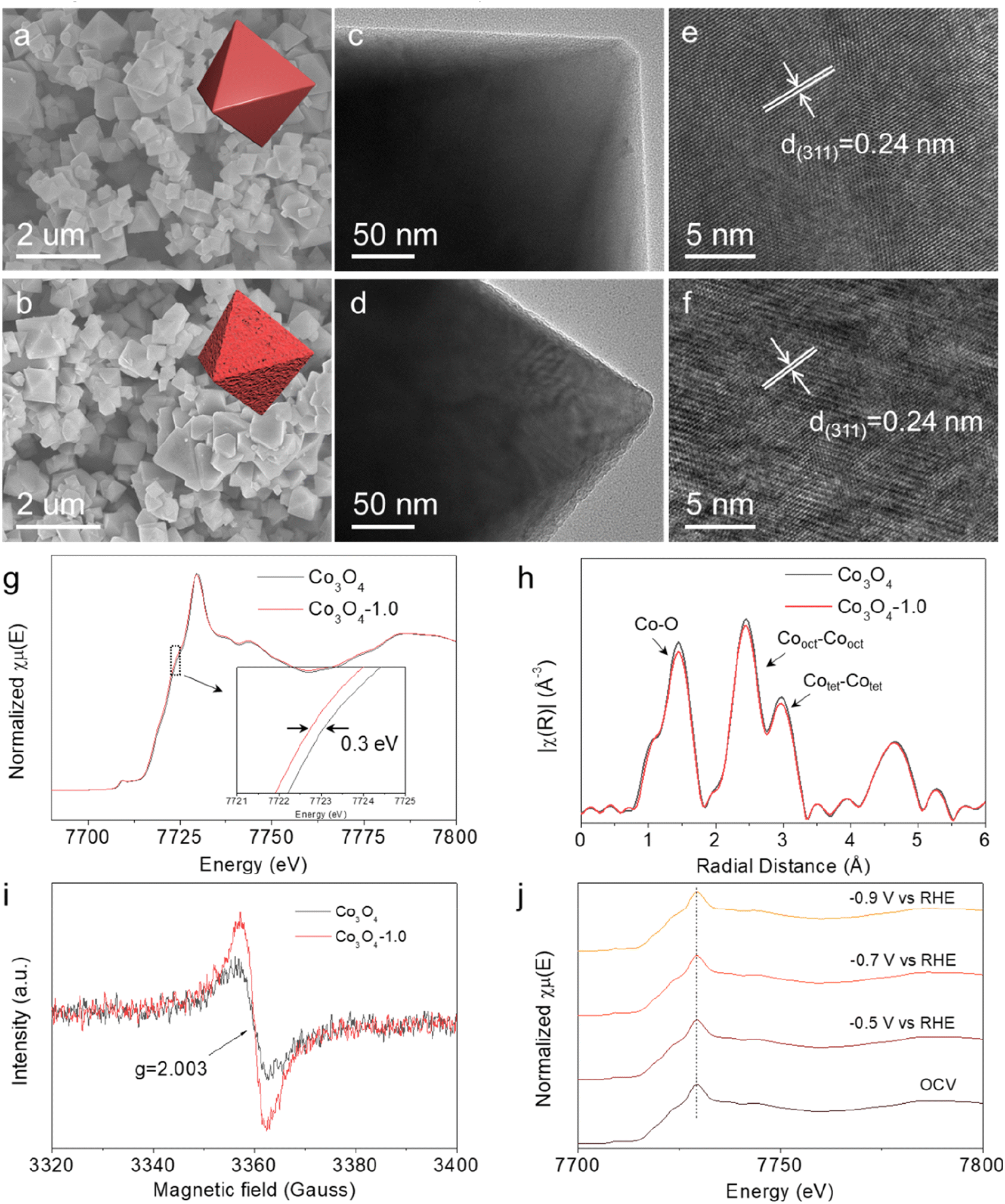 | ||
| Fig. 1 SEM images of (a) pristine Co3O4 and (b) Co3O4-1.0. TEM images of (c) pristine Co3O4 and (d) Co3O4-1.0. HRTEM images of (e) pristine Co3O4 and (f) Co3O4-1.0. (g) XANES and (h) Fourier-transformed EXAFS spectra of the Co K-edge over pristine Co3O4 and Co3O4-1.0. (i) EPR spectra of pristine Co3O4 and Co3O4-1.0. (j) Operando XANES under the electrochemical conditions of Co K-edge over Co3O4-1.0. OCV stands for open-circuit voltage. | ||
The X-ray diffraction (XRD) patterns of the catalysts are shown in Fig. S2.† In detail, the peaks located at 31, 37, 44, 59 and 65° can be indexed to the (220), (311), (400), (511) and (440) crystalline planes of Co3O4 (JCPDS: PDF#43-1003), respectively, confirming the successful synthesis of Co3O4. The electron paramagnetic resonance (EPR) spectra were recorded to identify Vo. The electron deficient oxygen species exhibited an EPR signal at g = 2.003 (Fig. 1i and S3†).27,28 The intensity increased with increasing annealing time, revealing a higher Vo concentration in the Co3O4-1.0 catalyst than in the pristine Co3O4 catalyst. Furthermore, X-ray photoelectron spectroscopy (XPS) measurement analysis was performed to investigate the chemical states and compositions of the catalysts. The XPS spectra of O 1s (Fig. S4†) verified that the lattice oxygen (O1) and oxygen vacancies (O2) existed in all catalysts.29,30 The O2/O1 ratio means the ratio of O2 and O1 area in the O 1s spectra, and followed the sequence of pristine Co3O4 < Co3O4-0.5 < Co3O4-1.0 < Co3O4-1.5, suggesting the presence of more Vo species in the Co3O4-1.5 catalyst, which was consistent with the EPR results. Fig. S5† displayed the impact of Vo on the electronic structure of Co3O4. The peaks at around 780 eV (Co 2p3/2) and 795 eV (Co 2p1/2) retained the characteristic feature of Co species.25 Apparently, the Co2+/Co3+ ratio increased from 0.85 to 1.54 with increasing Vo concentration, suggesting a decrease in Co valence state on the Co3O4 surface after thermal treatment under the reducing atmosphere. Interestingly, we found a positive linear correlation between the ratio of Co2+/Co3+ and the ratio of O2/O1 (Fig. S6†), indicating that the Vo level in the catalysts was directly correlated with the electron cloud density of Co atoms, which is critical for the electrocatalytic activity.
The structural stability of electrocatalyst is a crucial indicator when assessing its performance. Therefore, we first carried out operando X-ray absorption spectroscopy (XAS) to investigate the structural and valence state changes of Co element under the electrochemical reaction conditions. In the operando XAS measurement, the potential was decreased from open-circuit voltage (OCV) to −0.5 V, −0.7 V, and −0.9 V vs. RHE. Fig. 1j and S7† demonstrate that there was no energy shift on the Co K-edge in the XANES spectra as the applied potential decreased. Similarly, no changes were observed in the coordination environment of Co atoms in the EXAFS spectra (Fig. S8†). These results collectively suggest that the local structure and electronic state of Co species in Co3O4-1.0 remained stable during the electrochemical reaction process.
The electrocatalytic activity of the as-prepared catalysts for application in urea synthesis was evaluated in an H-type cell. Cyclic voltammetry (CV) was carried out to preliminarily assess the performance of the Co3O4-1.0 in Ar or CO2 saturated 0.2 M KHCO3 + 0.02 M KNO2 electrolyte. As shown in Fig. 2a, in the presence of CO2, Co3O4-1.0 exhibited larger current density responses during the forward scan from −0.2 V to −0.8 V vs. RHE, resulting from simultaneous electroreduction of CO2 and NO2−. Subsequently, chronoamperometry tests were conducted at applied potentials ranging from −0.5 to −0.9 V vs. RHE to analyze the FE and the yield of products. The yield of urea was quantified using the well-established urease decomposition and indophenol blue spectrophotometric method (Fig. S9 and S10†).15,25,31 The performances of urea electrosynthesis at different applied potentials are displayed in Fig. 2b and S11.† It can be found that Co3O4-1.0 mainly yielded H2, NH3, and urea with a combined FE of around 100%. With increasing applied potential, the current density and the FE of urea are increased. At the potential of −0.7 V vs. RHE, the FE of urea could reach 26.3% with the current density of 34.1 mA cm−2 over the Co3O4-1.0 catalyst. When the potential was beyond −0.7 V vs. RHE, the FE of urea decreased, mainly due to the highly competitive HER. It is noteworthy that no other reduction products from CO2 were detected across the potential range, indicating that the catalyst demonstrated a 100% carbon selectivity for C–N coupling. Fig. S12–S14† show the FE of urea and current density of comparative catalysts (pristine Co3O4, Co3O4-0.5, and Co3O4-1.5) within the potential range from −0.5 V to −0.9 V vs. RHE. This indicates that the product distributions of NH3, urea, and H2 were correlated with the Vo concentration in the catalysts, in which Co3O4-1.0 with an appropriate Vo concentration exhibited the best performance for urea formation among all the catalysts. At the optimized condition, the urea yield rate over the Co3O4-1.0 catalyst could reach 3361 mg h−1 gcat−1 (Fig. 2c), which was nearly 2.3 times that of pristine Co3O4. For the pristine Co3O4 catalyst, the FE of urea was only 13.7% with a limited urea yield rate of 1450 mg h−1 gcat−1. Comparison with the state-of-the-art catalysts for urea synthesis is presented in Fig. 2d and Table S1.† It shows that Co3O4-1.0 was highly efficient as an electrocatalyst for C–N coupling to produce urea and it could reach high FE and yield rate at a low reduction potential.
 | ||
| Fig. 2 (a) CV curves of Co3O4-1.0 at the scan rate of 50 mV s−1 in Ar or CO2 saturated electrolyte. (b) The FE of major products and total current densities over the Co3O4-1.0 electrocatalyst in CO2 saturated 0.2 M KHCO3 + 0.02 M KNO2 electrolyte. (c) The urea yield rate vs. the ratio of O2/O1 at the potential of −0.7 V (vs. RHE). (d) Comparison of the FE (%) of urea and yield rate (mg h−1 gcat−1) of urea of Co3O4-1.0 with those of state-of-the-art catalysts for application in urea synthesis. (e) Stability tests of Co3O4-1.0 at the potential of −0.7 V (vs. RHE). (f) 1H NMR spectra obtained by using K14NO2 and K15NO2 as the reactants with CO2 and a standard 14N-urea sample. | ||
Remarkably, in the ten consecutive cycles at −0.7 V vs. RHE, the Co3O4-1.0 achieved long-term stability with no obvious decays in current density, and the FE of urea and NH3 could be held around 25% and 65%, respectively, during the whole electrolysis process (Fig. 2e). Moreover, the XRD, XPS, and TEM measurements showed that the crystal structure, valence states, Vo, and morphology were well preserved after the long-term electrolysis (Fig. S15–S17†), indicating the excellent electrochemical and structural stability of the Co3O4-1.0 catalyst.
To identify the source of urea production, we performed the isotope-labelling experiments in CO2-saturated 0.2 M KHCO3 + 0.02 M K15NO2 electrolytes. Fig. 2f illustrates the 1H NMR spectrum of the electrolyte with K15NO2, displaying the typical double peaks of 15N-urea at 5.48 and 5.71 ppm, while the 1H NMR spectrum of the electrolyte with K14NO2 only exhibits a single peak of 14N-urea at 5.60 ppm. The 1H NMR results confirm that there is urea production in the coupling electrolysis of CO2 and 14NO2− on Co3O4-1.0. Furthermore, we employed the 1H NMR technique (Fig. S18 and S19†) to quantitatively measure the generated urea and concurrently validate the accuracy of the UV-vis method. The obtained results (Fig. S20†) demonstrate a consistent urea concentration measurement between the 1H NMR and UV-vis methods, confirming the accuracy of the employed quantitative method.
Considering the experimental observations above, we think that the high electrocatalytic activity of the Co3O4-1.0 catalyst could be attributed to the generation of abundant active sites in defective Co3O4, which was kinetically favorable for the reaction. To verify the hypothesis, we estimated the electrochemical active surface area (ECSA) of various catalysts through electrochemical double-layer capacitance (Cdl) measurements. Fig. S21† shows the cyclic voltammetry (CV) curves with various scan rates at the non-faradaic region for the catalysts. The linear slopes in Fig. S22† show that the Co3O4-1.0 had a larger ECSA, suggesting that the higher Vo concentration was responsible for the generation of more active sites. After normalizing the urea yield rate to ECSA (Fig. S23†), Co3O4-1.0 still exhibited the largest urea yield rate (1832 mg h−1 gcat−1) at the potential of −0.7 V vs. RHE, which indicates that the Vo concentration regulation could also improve the intrinsic activity of urea production over the catalysts. In addition, the electrochemical impedance spectrum (EIS, Fig. S24†) was recorded to probe the effect of defects on the charge transport kinetics. It showed that charge resistance (Rct) on Co3O4-1.0 was much lower than that on pristine Co3O4, which is favorable for enhancing the reaction rate.
The defect-engineering would serve as a promising strategy for simultaneously enhancing the chemisorption capability of these inert molecules and regulating the electronic structure of the catalyst to construct dual-functional sites. As effective chemisorption of reactants is essential for the initial stage of urea electrosynthesis, we turn to screen the effect of defects on the CO2 chemical adsorption properties. The improved adsorption capacity of CO2 molecules on Vo-enriched Co3O4 can be verified via the CO2 temperature programmed desorption (CO2-TPD) spectra (Fig. 3a) normalized to ECSA. Comparably, all the defective Co3O4 catalysts exhibited stronger binding strength and larger adsorption peaks than the pristine Co3O4. Typically, it was exhibited that CO2 desorption occurred at around 580 °C on the Co3O4-1.0 catalyst, indicating that it had a strong CO2 adsorption site with excellent CO2 adsorption ability. In contrast, the CO2 desorption peak of the pristine Co3O4 catalyst was at around 540 °C, which was significantly weaker than that of the Co3O4-1.0 catalyst. We then correlated the temperature of the CO2 desorption peak with the concentration of Vo (the ratio of O2/O1) to investigate the correlation between Vo and CO2 adsorption capacity (Fig. 3b). This indicates that an increase in the concentration of Vo in the catalyst leads to an increase in the CO2 adsorption capacity. Combining the previous analysis of electrochemical performance over the as-prepared catalysts (Fig. 2), we can conclude that regulating the CO2 adsorption capacity can control the competing reaction pathways (Fig. 3c). Moderating CO2 adsorption is conducive to the adsorption and activation of CO2 molecules to participate in the desirable C–N coupling process with 100% carbon efficiency.
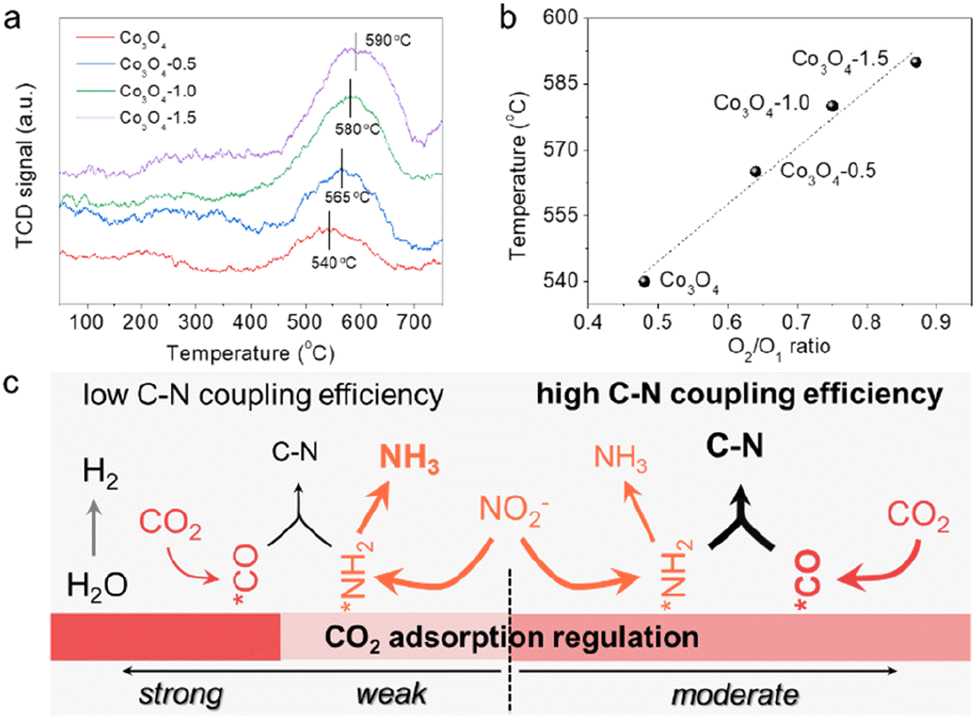 | ||
| Fig. 3 (a) CO2-TPD plots for different catalysts normalized to ECSA. (b) The temperature of CO2 desorption peak vs. the ratio of O2/O1 over different catalysts. (c) Schematic diagram of CO2 adsorption regulation for efficient C–N coupling. | ||
To gain an in-depth understanding of the C–N coupling mechanism, in situ Raman spectroscopy and attenuated total reflection-surface-enhanced Fourier transformed infrared (ATR-FTIR) spectroscopy were carried out. Considering the potential window for urea production, potentials ranging from −0.5 to −0.9 V vs. RHE with 0.1 V intervals were selected for spectral acquisition during the in situ studies. As shown in Fig. 4a, five distinct peaks were observed in Raman spectra that correspond to the characteristic vibration peaks of Co3O4.32 The intensity of the vibration peak did not change obviously at different applied potentials, suggesting that the Co3O4 catalyst is stable during electrolysis. However, no reaction intermediate signal was recorded in the Raman spectra, which could be attributed to the strong Raman signal of Co3O4 that overshadows the faint reaction intermediate signals. Alternatively, some intermediate signals were observed in ATR-FTIR spectra (Fig. 4b). The stretching vibrations observed at around 1700 and 1390 cm−1 are attributed to the formation of *CO and *OCO intermediates, respectively,8,21 indicating the CO2 molecules can adsorb on the surface-active sites of the Co3O4-1.0 catalyst and further be converted into *CO intermediates. The observation of peaks located at around 1675 and 1300 cm−1 corresponded to the bending and wagging modes of the N–H bond, respectively, suggesting the reduction of NO2− in the electrolysis.33 Specifically, a typical peak at around 1445 cm−1 was observed across all tested potentials, demonstrating that the C–N coupling was successfully achieved for urea production.8,21 The enhanced peak intensity of the C–N bond at the potential of −0.7 V vs. RHE was consistent with the aforementioned experimental results, suggesting that the catalyst exhibited higher activity at this potential.
 | ||
| Fig. 4 In situ Raman (a) and ATR-FTIR (b) spectra recorded at different potentials for Co3O4-1.0 in CO2 saturated 0.2 M KHCO3 + 0.02 M KNO2 electrolyte. (c) Schematic structures of Co3O4 and Co3O4-Vo. (d) The PDOS of the Co atoms in Co3O4 and Co3O4-Vo with the d-band center position (Ed) marked by the black line and the Fermi level set as zero. Differential charge densities of (e) Co3O4 and (f) Co3O4-Vo structures. Yellow and blue contours represent electron accumulation and depletion, respectively. (g) Bader charge comparison of the Co atom around the Vo in Co3O4 and Co3O4-Vo. | ||
To strengthen our conclusion, we further conducted density functional theory (DFT) calculations. We built the correlative theoretical models, including Co3O4 and Co3O4-Vo (Fig. 4c and S25†), based on the above experimental characterizations. The partial density of states (PDOS) calculations on the Co atoms in Co3O4 and Co3O4-Vo are shown in Fig. 4d. Notably, the d-band center position of Co atoms in the Co3O4-Vo structure (−1.88 eV, relative to the Fermi level) was found to be higher than that in the Co3O4 structure (−2.00 eV). According to the d-band theory, metal-based catalysts with a higher d-band center (closer to the Fermi level) exhibit stronger interaction between the reaction intermediates and active sites, resulting in an enhanced C–N coupling process. To confirm this assumption, we conducted the simulations on the intermediates involved in CO2 and NO2− adsorption over the Co3O4 and Co3O4-Vo structures. The adsorption structures of *CO2 and *NO2 on Co3O4 (111) and Co3O4-Vo (111) surfaces are shown in Fig. S26 and S27.† Apparently, the adsorption free energy of *CO2 on Co3O4-Vo (−0.31 eV) is lower compared to that on Co3O4 (−0.17 eV), and the adsorption free energy of *NO2 on Co3O4-Vo (−3.39 eV) is also less than that on Co3O4 (−2.88 eV), indicating that the CO2 molecule and NO2− ion are more likely to be adsorbed and activated on the Co3O4-Vo surface. In addition, we pursued theoretical insights into the relationship between electronic structure and the electrocatalytic properties to deeply understand the impact of Vo. To gain further insights into the electronic properties of Co3O4 and Co3O4-Vo, we analyzed the charge transfer within the catalyst structure. As depicted in Fig. 4e and f, we can find that the electrons of Co atoms were consumed in both Co3O4 and Co3O4-Vo, owing to the lower electronegativity of the Co atom compared to the O atom. The detailed Bader charge analysis further revealed that in the Co3O4-Vo structure, the Co4, Co29 and Co44 atoms, which are located in close proximity to the Vo, had Bader charges of 8.41, 8.37 and 8.43e−, respectively. These values are higher than those observed in the Co3O4 structure (Fig. 4g). This indicates that the presence of Vo in the Co3O4 structure leads to the formation of an electron-rich local environment on the Co atoms, which is consistent with the XAS and XPS results. Our computational analysis highlights that the introduction of Vo in the catalysts can effectively regulate the electronic structure of the catalyst, leading to the enhancement of adsorption behaviors of intermediates and electrochemical activity for urea synthesis.
Conclusions
In summary, we achieved urea electrosynthesis by co-reduction of CO2 and nitrite using the Vo-enriched Co3O4 as the electrocatalyst. The as-prepared Co3O4-1.0 catalyst demonstrated an outstanding urea yield rate of 3361 mg h−1 gcat−1 with a corresponding FE of 26.1% and 100% carbon selectivity at the potential of −0.7 V vs. RHE. The remarkable catalytic performance was attributed to the integration of dual-functional sites in defective Co3O4, which can be regulated by the amount of Vo. Experimental measurements and theoretical calculations corroborated the role of Vo in constructing the well-matched adsorption/activation sites, which is beneficial for promoting the chemical adsorption of reactants, redistributing the electronic structure of the catalyst. The modulated local environment on Co atoms further enhanced the adsorption of intermediates and decreased the C–N coupling resistance in urea synthesis. Our study demonstrates a route for highly efficient urea synthesis, and the methodology can be used for designing other electrocatalysts for the C–N coupling process.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
P. S. L., Q. G. Z., and B. X. H. proposed the project, designed the experiments, and wrote the manuscript. P. S. L. performed all the experiments. P. S. L., J. Y. L., T. B. W., X. N. S., Q. L. M., X. C. K., and X. F. S. performed the analysis of experimental data. Q. G. Z. and B. X. H. co-supervised the whole project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the National Natural Science Foundation of China ( 22102192, 22279146, 21972146, 22033009,22293015, 22022307 and 22121002), the National Key Research and Development Program of China (2023YFA1507440), CAS Project for Young Scientists in Basic Research (Grant No. YSBR-050), Photon Science Center for Carbon Neutrality, China Postdoctoral Science Foundation (BX20200336 and 2020M680680), and S&T Program of Hebei (B2021208074). We also acknowledge the 1W1B beamline station of Beijing Synchrotron Radiation Facility and BL14W1 beamline station of Shanghai Synchrotron Radiation Facility.References
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem, 2022, 6, 303–319 CrossRef CAS PubMed.
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- R. Xia, S. Overa and F. Jiao, JACS Au, 2022, 2, 1054–1070 CrossRef CAS PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- M. Jiang, M. Zhu, M. Wang, Y. He, X. Luo, C. Wu, L. Zhang and Z. Jin, ACS Nano, 2023, 17, 3209–3224 CrossRef CAS PubMed.
- X. Peng, L. Zeng, D. Wang, Z. Liu, Y. Li, Z. Li, B. Yang, L. Lei, L. Dai and Y. Hou, Chem. Soc. Rev., 2023, 52, 2193–2237 RSC.
- C. Chen, N. He and S. Wang, Small Science, 2021, 1, 2100070 CrossRef CAS.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Xu, R. Liu, T. Zhao, J. Zhang, Z. Ren, Z. Liu, C. Streb, H. He, C. Yang, S. Zhang and G. Zhang, Energy Environ. Sci., 2021, 14, 6605–6615 RSC.
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef CAS.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Shi, S. Li, X. Wang and G. Zhang, Chem. Sci., 2021, 12, 6048–6058 RSC.
- Z. Mei, Y. Zhou, W. Lv, S. Tong, X. Yang, L. Chen and N. Zhang, ACS Sustain. Chem. Eng., 2022, 10, 12477–12496 CrossRef CAS.
- M. Qiu, X. Zhu, S. Bo, K. Cheng, N. He, K. Gu, D. Song, C. Chen, X. Wei, D. Wang, Y. Liu, S. Li, X. Tu, Y. Li, Q. Liu, C. Li and S. Wang, CCS Chem., 2023, 1–11 CAS.
- M. Sun, G. Wu, J. Jiang, Y. Yang, A. Du, L. Dai, X. Mao and Q. Qin, Angew. Chem., Int. Ed., 2023, 62, e202301957 CrossRef CAS PubMed.
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Nat. Commun., 2022, 13, 5337 CrossRef CAS PubMed.
- S. Zhang, J. Geng, Z. Zhao, M. Jin, W. Li, Y. Ye, K. Li, G. Wang, Y. Zhang, H. Yin, H. Zhang and H. Zhao, EES Catal., 2023, 1, 45–53 RSC.
- H. Wang, Y. Jiang, S. Li, F. Gou, X. Liu, Y. Jiang, W. Luo, W. Shen, R. He and M. Li, Appl. Catal. B Environ., 2022, 318, 121819 CrossRef CAS.
- J. Qin, N. Liu, L. Chen, K. Wu, Q. Zhao, B. Liu and Z. Ye, ACS Sustain. Chem. Eng., 2022, 10, 15869–15875 CrossRef CAS.
- N. Meng, X. Ma, C. Wang, Y. Wang, R. Yang, J. Shao, Y. Huang, Y. Xu, B. Zhang and Y. Yu, ACS Nano, 2022, 16, 9095–9104 CrossRef CAS PubMed.
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustain., 2021, 4, 868–876 CrossRef.
- X. Wei, Y. Liu, X. Zhu, S. Bo, L. Xiao, C. Chen, T. T. T. Nga, Y. He, M. Qiu, C. Xie, D. Wang, Q. Liu, F. Dong, C.-L. Dong, X.-Z. Fu and S. Wang, Adv. Mater., 2023, 35, 2300020 CrossRef CAS PubMed.
- N. Cao, Y. Quan, A. Guan, C. Yang, Y. Ji, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2020, 577, 109–114 CrossRef CAS PubMed.
- Y. Feng, H. Yang, Y. Zhang, X. Huang, L. Li, T. Cheng and Q. Shao, Nano Lett., 2020, 20, 8282–8289 CrossRef CAS PubMed.
- Y. Zhu, J. Wang, T. Koketsu, M. Kroschel, J.-M. Chen, S.-Y. Hsu, G. Henkelman, Z. Hu, P. Strasser and J. Ma, Nat. Commun., 2022, 13, 7754 CrossRef CAS PubMed.
- Z. Cai, Y. Bi, E. Hu, W. Liu, N. Dwarica, Y. Tian, X. Li, Y. Kuang, Y. Li, X.-Q. Yang, H. Wang and X. Sun, Adv. Energy Mater., 2018, 8, 1701694 CrossRef.
- A. Martínez-Arias, J. C. Conesa and J. Soria, Res. Chem. Intermed., 2007, 33, 775–791 CrossRef.
- R. Schmitt, A. Nenning, O. Kraynis, R. Korobko, A. I. Frenkel, I. Lubomirsky, S. M. Haile and J. L. M. Rupp, Chem. Soc. Rev., 2020, 49, 554–592 RSC.
- J. H. Kim, Y. J. Jang, J. H. Kim, J.-W. Jang, S. H. Choi and J. S. Lee, Nanoscale, 2015, 7, 19144–19151 RSC.
- H. Tan, B. Tang, Y. Lu, Q. Ji, L. Lv, H. Duan, N. Li, Y. Wang, S. Feng, Z. Li, C. Wang, F. Hu, Z. Sun and W. Yan, Nat. Commun., 2022, 13, 2024 CrossRef CAS PubMed.
- L. Pan, J. Wang, F. Lu, Q. Liu, Y. Gao, Y. Wang, J. Jiang, C. Sun, J. Wang and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216835 CrossRef CAS PubMed.
- Y. Wang, X. Wei, X. Hu, W. Zhou and Y. Zhao, Catal. Lett., 2019, 149, 1026–1036 CrossRef CAS.
- J. Geng, S. Ji, M. Jin, C. Zhang, M. Xu, G. Wang, C. Liang and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202210958 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06579k |
| This journal is © The Royal Society of Chemistry 2024 |