
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the carbonate issue through the regulation of mass transport and charge transfer in mild acid†
Zhongshuo
Zhang
a,
Qian
Lu
ac,
Jiping
Sun
ab,
Guangchao
Li
ab,
Weixing
Wu
a,
Zhanyou
Xu
a,
Liangpang
Xu
a and
Ying
Wang
 *a
*a
aDepartment of Chemistry, The Chinese University of Hong Kong, Hong Kong, S. A. R., China. E-mail: ying.b.wang@cuhk.edu.hk
bSchool of Metallurgy and Environment, Central South University, Changsha, P. R. China
cJiangsu Collaborative Innovation Center of Atmospheric Environment and Equipment Technology, Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, UNIST-NUIST Environment and Energy Jointed Lab, School of Environmental Science and Technology, Nanjing University of Information Science and Technology, Nanjing, P. R. China
First published on 17th January 2024
Abstract
The electrochemical CO2 reduction reaction (CO2RR) triggered by renewable electricity provides a promising route to produce chemical feedstocks and fuels with low-carbon footprints. The intrinsic challenge for the current CO2RR electrolyzer is the carbonate issue arising from the reaction between hydroxide and CO2. Acid CO2RR electrolyzers, in principle, can effectively solve the carbonate formation, but it remains inevitable practically. In this work, we thoroughly investigated the electrode processes of the CO2RR on the benchmark Ag catalyst in mild acid. The root of the carbonate issue arises from the imbalanced supply–consumption rate of protons—the electron transfer vs. mass transport. Regulating the hydrodynamics substantially reduces the proton diffusion length by 80%, increasing the single-pass carbon utilization efficiency of CO2-to-CO to 44% at −100 mA cm−2. The fundamental difference between mass transport and electron transfer on the spatial and temporal scale still leads to unavoidable carbonate formation. Future work to design intrinsically active catalysts in strong acid or metal-cation-free media is critical to solving the carbonate issue.
Introduction
Electrocatalytic conversion of CO2 offers an attractive route for producing chemicals and fuels with low carbon emissions. Over the past few decades, substantial progress has been made in advancing low temperature (<100 °C) electrochemical CO2 reduction reaction (CO2RR). Most current state-of-the-art CO2RR electrolyzers operate under alkaline conditions, where the ultra-low proton concentration and extra energy barrier for water dissociation suppress the competitive hydrogen evolution reaction (HER).1–4 Appreciable activity and selectivity have been achieved at industrially relevant current densities (>100 mA cm−2).5,6 However, operating under alkaline conditions suffers from the “carbonate issue”—the fast acid–base reaction between hydroxide and CO2 to form electrochemically inactive carbonate.2,7,8 The alkaline electrolyte consumes a significant amount of the input CO2, turning the electrolyte into a near-neutral solution, producing high overpotentials for both the cathode and anode reactions, and giving rise to considerable penalties on carbon utilization and energy efficiency.9,10Performing efficient and selective CO2RR in strong acid (pH = 0) can solve the carbonate issue, but it is challenging due to the lack of intrinsically active electrocatalysts.11 Mildly acidic conditions, such as pH 2 and above, are commonly adopted.12–14 Such conditions enable a suppressed HER at the alkaline electrode interface and minimize the carbonate issue benefiting from the bulk acidic environment.12 Recent work showed nearly 100% faradaic efficiency (FE) towards CO at −250 mA cm−2,14 and 89% FE towards multi-carbon products at −500 mA cm−2 in pH 2 electrolyte.12 However, the alkaline interface and the high concentration of metal cations lead to unavoidable salt precipitation during long-term operation.15,16 The reported stability of acidic electrolyzers at current density > −200 mA cm−2 lies between 1 and 30 h, with salt precipitation as the primary cause of performance decay.11,12,17–19
For CO2RR electrolyzers in mild acid, the carbonate formation results from the locally imbalanced consumption-supply rate of protons, which is determined by electron transfer and mass transport. These are spatially and temporally different in scale—electron transfer happens near the electrode surface (ca. 10–20 Å) while mass transport occurs at the scale of sub-μm and above.20 For the flow cell typically used, the mass transport of the proton from bulk to the surface is decided by the diffusion in the stagnant liquid layer at the surface and the fluid flow in the bulk electrolyte.21 The diffusion layer at the electrode surface always exists regardless of the hydrodynamics, but the flow rate decides the thickness.20 A reduced diffusion length for protons can be expected at a fast flow rate, which is beneficial to create an acidic interface for suppressed carbonate formation. Under pH 2 conditions, 11 s is required to deliver 1 μmol cm—2 of H+ (the flux for 100 mA cm—2) for a 100 μm diffusion layer thickness, while the value is reduced to 0.11 s for a 1 μm diffusion layer.20 However, a low pH may hinder the reactivity of the CO2RR as protons and CO2 compete for the active sites.11 The ideal scenario is the mass transport rate of protons is sufficient to address the bicarbonate/carbonate at the interface while not influencing the CO2RR activity. In reality, achieving such a design depends on the relationship between mass transport and electron transfer—the carbon–hydrogen–electron (CHE) relationship.
In this work, we elucidate the electrode process of the CO2RR under mild acidic conditions to investigate the mass transport–electron transfer relation. As a scaffold, we choose Ag, where the electrode reactions are relatively more straightforward than Cu. Multiple electrode reactions, the hydrogen evolution reaction (HER) and the CO2RR, are coupled together, deviating the local environment from the bulk solution. Through online differential electrochemical mass spectrometry (DEMS) and scanning electrochemical microscopy (SECM), we found that the CO2RR occurs after local protons depleted at pH 11—unavoidable carbonate formation at the interface. Increasing the flow rate from 13 standard cubic centimetres per minute (sccm) to 63 sccm can reduce the proton diffusion length from 182 μm to 37 μm, around 80% reduction. This, in return, alleviates the carbonate issue—the single-pass carbon utilization efficiency of CO2-to-CO reaches 44% at a current density of −100 mA cm−2. Although regulating mass transport effectively relieves the carbonate issue, carbonate formation is still unavoidable due to the fundamental differences in the scale of rate for mass transport and electron transfer.
Results and discussion
We first seek to understand the electrode processes for the CO2RR on the benchmark Ag catalyst under mild acidic conditions by using online differential electrochemical mass spectrometry (DEMS) at a sampling time interval of 1 s (ESI Schematic 1†) in the flow cell. The gaseous products are ionized and detected in DEMS, which generates ionic current (II). The magnitude of II is proportional to the generation rate of each product (detailed in the ESI†).22 No obvious change in the crystal orientation, electronic structure and morphology of Ag was observed after electrolysis (Fig. S1†), which demonstrates the robustness of the Ag catalyst. The ionic current of H2 (IHI2) was observed at −3.8 to −6.4 mA cm—2, while no signal related to CO (ICOI) was shown (Fig. 1a). ICOI appeared at −7.6 mA cm—2, followed by a continuous increase to reach a plateau. This is accompanied by a constant decay of the IHI2. In the initial 680 seconds, the potential decay exhibited no difference under Ar and CO2 saturated conditions at a constant current density of −7.6 mA cm—2 (Fig. 1b). In this region, H2 was the only species detected in the DEMS, indicating the dominant HER (blue and red dash-dotted lines in Fig. 1c). This potential decay can be attributed to the gradual depletion of protons during constant electrolysis. A sharp drop in potential was observed after 680 s regardless of the gas fed (Fig. 1b), and CO began to appear in CO2-saturated solution concurrently. The enhanced CO signal was accompanied by a suppressed H2, which is likely associated with the reduced proton availability due to the formation of a thick diffusion layer. With limited protons around, the reduction of other species was initiated, that is CO2 in CO2 saturated solution and H2O in Ar saturated solution. The HER is significantly impeded by the increased CO production as the IHI2 falls almost to zero from 680 s to 900 s (red dash-dotted line in Fig. 1c), suggesting that the CO2RR overtakes the HER in this region.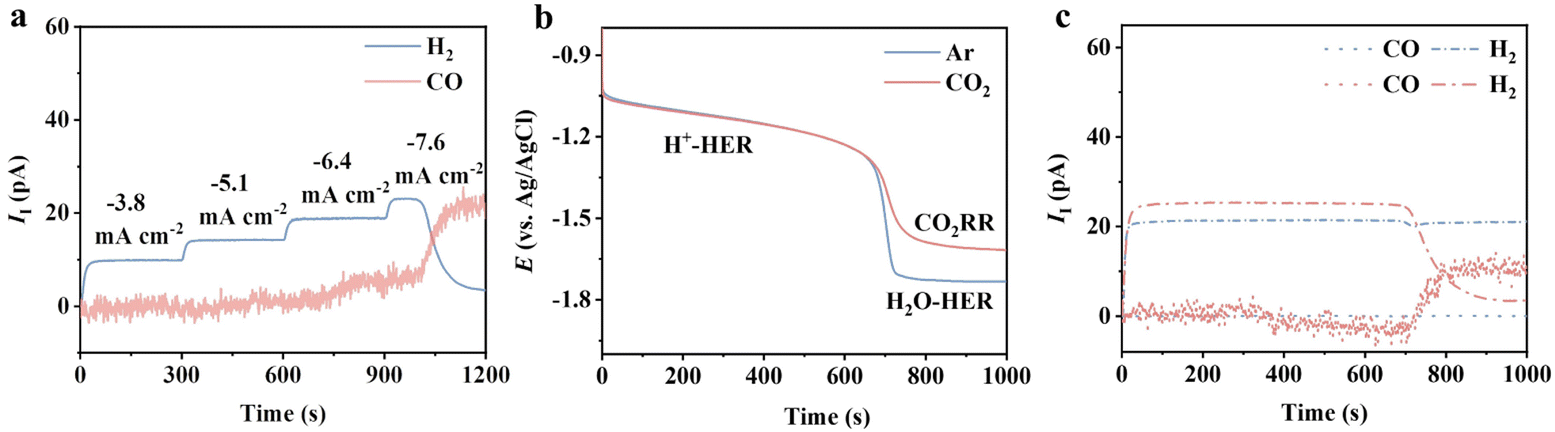 | ||
| Fig. 1 (a) Ionic currents of H2 and CO at applied current densities from −3.8 to −7.6 mA cm−2 on Ag. (b) Chronopotentiometry at −7.6 mA cm−2 under Ar (blue solid line) and CO2 (red solid line). (c) Ionic currents of H2 (dash-dotted line) and CO (dotted line) under Ar (blue) and CO2 flow (red) during the chronopotentiometry at −7.6 mA cm−2. The electrode is an Ag GDE (detailed in the ESI†). The electrolytes used are 0.5 M K2SO4 (pH = 1.5 adjusted with H2SO4). The H+-HER and H2O-HER refer to the HER with two different proton sources, protons and water. | ||
Such behaviours indicate the occurrence of multiple electrode processes caused by the proton-coupled electron transfer (PCET) nature of the HER and the CO2RR.3 No differences were observed for the CO production at different pH at the standard hydrogen electrode scale, indicating a pH-independent manner (Fig. S3†). The HER is a bit more complicated under mild acidic conditions—the production of H2 is pH-dependent at low current density while switching to pH-independent at high current density.23 This is due to the different proton sources, protons for the former and water for the latter, involved in the HER in these two regions.24 Considering all the above, we propose that the dominant reaction in the first 680 s in Fig. 1b is the proton-HER under both Ar and CO2-saturated conditions. The depleted proton concentration and the increased diffusion layer thickness lead to the slow potential decay in the first 680 s in Fig. 1b. Once the surface concentration of the proton reaches a threshold value, the CO2RR starts to kick off, and the proton source of the HER switches to water, evidenced by the appearance of ICOI. As CO2 has a stronger binding affinity to the Ag surface, the CO2RR dominates the HER in this region, agreeing with the disappearance of the hydrogen signal in DEMS (Fig. 1c).25 Thus, it is reasonable to propose eqn (1)–(3) for the charge transfer reactions for the CO2RR in mild acid.
| 2H+ + 2e− → H2 | (1) |
| H2O + 2e− → H2 + 2OH− | (2) |
| CO2 + H2O + 2e− → CO + 2OH− | (3) |
The threshold value of the proton concentration for the commencement of the CO2RR is of fundamental interest. Such a value enables us to evaluate the rate difference for eqn (1) and (3), and serves as an index to design the operation conditions in the flow reactor. We employed a more sensitive technique, scanning electrochemical microscopy (SECM), to deepen the understanding of the local chemical environment.
The experiments were conducted in a stationary cell due to the technical requirement of SECM. The temporary switch of the electrolyte to monoacid (KClO4 + HClO4) was for facile local pH quantification.26 The Ag substrate was biased for 400 s to reach a steady state at a series of current densities from −2.5 mA cm−2 to −5.5 mA cm−2. A 10 μm Pt microelectrode was positioned at 50 μm above the substrate electrode to detect CO production (Fig. 2a) and local pH (Fig. 2b) via electrooxidation and limiting current measurement, respectively (detailed in the ESI†). A broad peak at −0.2 V to 0.85 V for hydrogen oxidation was observed for current densities below −4.5 mA cm−2 (Fig. 2c), indicating the absence of the CO2RR.27 Benefiting from the high sensitivity of SECM, the occurrence of the CO2RR was observed at −5.5 mA cm−2, evidenced by the sharp oxidation peaks at 0.62 V and 0.65 V for CO oxidation.27 The local pH at 50 μm barely changes with the bulk value for current densities from −2.5 to −4.5 mA cm−2 (blue dot in Fig. 2d). The local pH increases to 3.5 at −5.5 mA cm−2 within 50 s. We note that the experimentally measured pH is at a distance of 50 μm, which deviates from the local conditions.
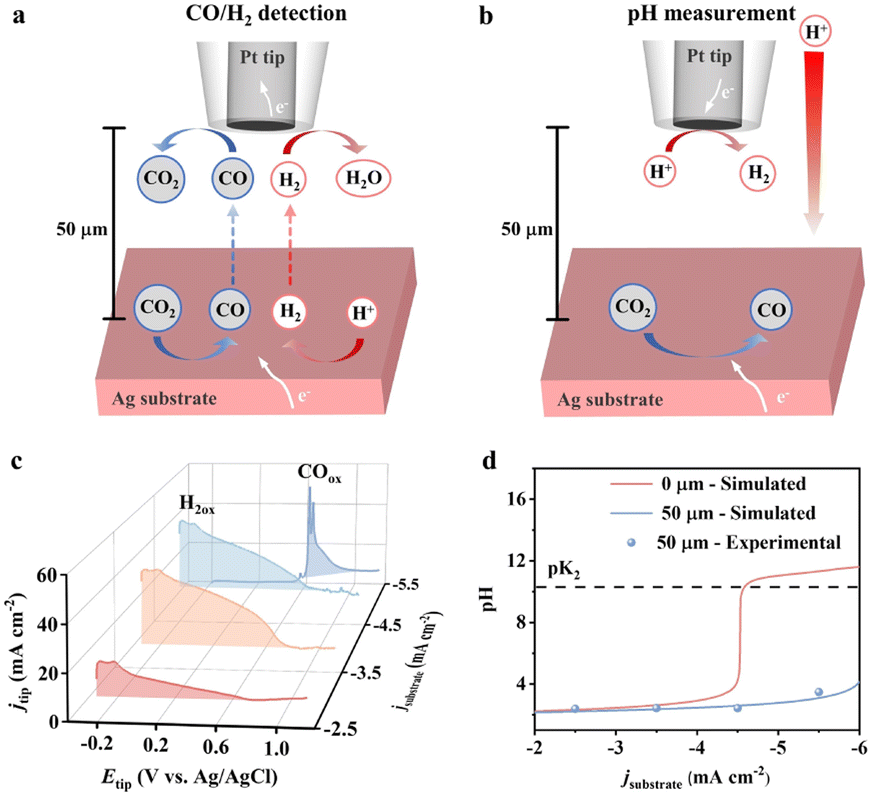 | ||
| Fig. 2 (a) Schematic of CO and H2 detection by SECM. (b) Schematic of pH measurement by SECM. (c) Tip voltammetry at different substrate current densities. (d) Experimental (50 μm) and simulated (50 μm and 0 μm) local pH at different applied current densities. The electrolyte is 0.1 M KClO4 solution (pH = 2 adjusted with HClO4). The substrate is a planar Ag electrode and the tip is a 10 μm Pt microelectrode. | ||
We, therefore, employed multiphysics modelling to explore the pH change at the electrode interface. An electrode and an adjacent diffusion layer with experimentally determined thickness were simulated. The electrochemical processes, homogeneous reactions and species transport were incorporated to solve for the local pH (detailed in the ESI†). The simulation results (blue line) at a distance of 50 μm agree very well with the experiments (blue dot). A noticeable pH change is observed at the electrode surface (0 μm) based on the simulation (red line). The surface pH sharply increases to above 10.3, where carbonate is the primary form, at a current density larger than −4.5 mA cm−2. This also explains the observed CO2RR at −5.5 mA cm−2. The results imply that an alkaline surface is needed for appreciable CO2RR, arising from the significant difference in the intrinsic reaction rate of the HER-H+ (eqn (1)) and CO2RR-H2O (eqn (3)). However, around 70% of CO2 is converted to the electrochemical inactive carbonate at this pH (Fig. S6†).
The above results show that the carbonate issue is barely avoidable under mild acidic conditions. The next question is if we can suppress the carbonate formation while maintaining the reactivity of the CO2RR. The apparent reaction rate of the CO2RR is a combination of intrinsic activity, which is a few orders of magnitude slower than the HER-H+, and the mass transport—both CO2 and H+. Increasing the concentration of CO2 enhances the overall reaction rate, as indicated by the rate law.20 The proton concentration, as discussed previously, suppresses the carbonate issue.
Due to the much larger transport resistance of CO2 in liquid than gas, constructing a local gas channel to shorten the diffusion layer of CO2 is necessary to enhance the local concentration.28–30 As shown in Fig. 3a and b, the depletion of CO2 concentration is much slower in the presence of gas channels. We introduce the hydrophobic polymer polytetrafluoroethylene (PTFE) in the catalyst layer for local gas channels. The surface hydrophobicity increases when increasing the mass ratio of PTFE (Fig. 3c). The limiting current density of CO showed a volcano shape with the amount of PTFE introduced, peaking at 30% PTFE for a limiting current density of −640 mA cm−2 (Fig. 3d). The adverse effect on the limiting current density at high PTFE content (50% and 70%) is related to the insufficient water availability for the CO2RR.31 Salt precipitation was confirmed under such conditions when we switched the electrolyte to less soluble Li salt—a dramatic decay of IICO after 15 min (Fig. 3e and f).
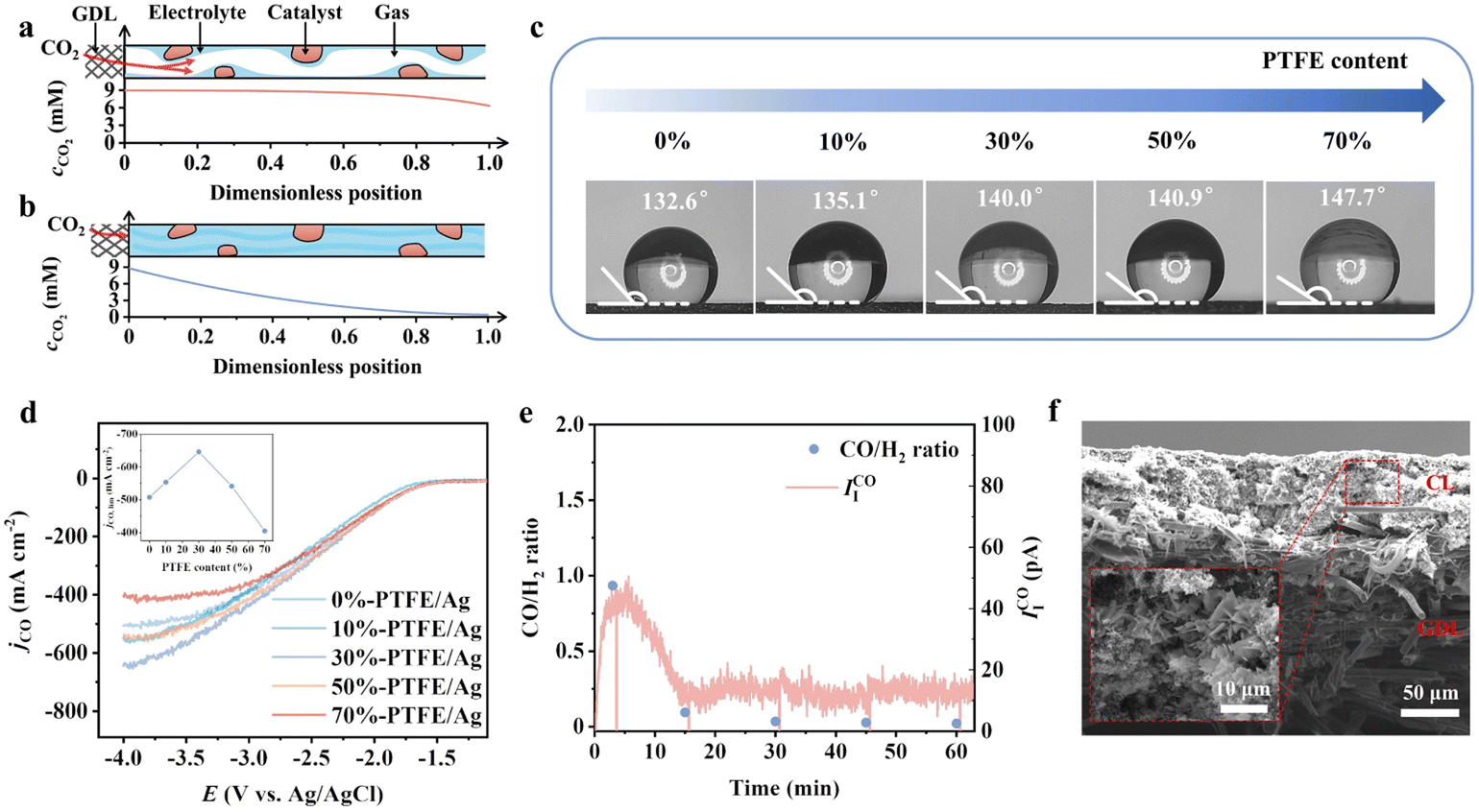 | ||
| Fig. 3 The schematic representation and simulated CO2 concentration profile in a partially wetted GDE (a) and a fully wetted GDE (b) at −500 mA cm−2. (c) Contact angle of electrodes with 0–70% mass ratio of PTFE. (d) Partial current density of CO on electrodes with 0–70% mass ratio of PTFE. CO is detected by DEMS during LSV tests at 5 mV s−1 scan rate. 4 M KCl (pH = 2) is used to reduce the solution resistance. (e) CO/H2 ratio and ionic current of CO at −100 mA cm−2 constant applied current density at the 30%-PTFE/Ag electrode. The electrolyte is 0.5 M Li2SO4 (pH = 2) with a flow rate of 13 sccm. (f) Post-reaction cross-section SEM image of the 30%-PTFE/Ag electrode. | ||
The above results clearly indicate the importance of regulating the local proton availability for the CO2RR. In the flow cell, the electrolyte is introduced into the reaction chamber through a peristaltic pump, where the fluid flow determines proton transport. Reynold's number corresponding to the typical flow rate employed ranges from 14 to 69 (ESI Table 4†), suggesting the reasonable simplification to use laminar flow for a uniform diffusion layer.20 In the commonly used flow rate of 13 standard cubic centimetres per minute (sccm), the diffusion layer of protons can be as thick as 182 μm and the surface concentration of carbonate rapidly increases to above 0.89 M at a current density above −100 mA cm−2 (Fig. 4a). This explains fast decay of CO2-to-CO observed in Li+ electrolyte (solubility of Li2CO3 is 0.18 M, Fig. 3e and f). When the flow rate increases to 63 sccm, the proton diffusion layer decreases to 37 μm, leading to around 90% reduction in the carbonate (Fig. 4b). The CO2 recovery by the bulk electrolyte is more effective at 63 sccm, with the CO2 regeneration occurring at 25 μm, 7 times shorter than that for 13 sccm (Fig. 4c). It replenished the interfacial CO2 to a higher concentration. Based on the above results, we perform the CO2RR in mild acid with 30%-PTFE/Ag electrodes at a flow rate of 13–63 sccm. Under slow CO2 supply rates, the CO2RR becomes diffusion-limited due to inadequate gaseous supply, where the regenerated CO2 at the electrode vicinity plays a significant role in determining the activity. As shown in Fig. 4d, the FEco has a higher supply rate of 1.3 sccm. This can be attributed to the enhanced mass transport of protons for a more efficient CO2 recovery. Simultaneously, the single-pass carbon utilization efficiency (the fraction of electrochemically converted CO2 during a single pass) is improved when the electrolyte flow rate is fast.
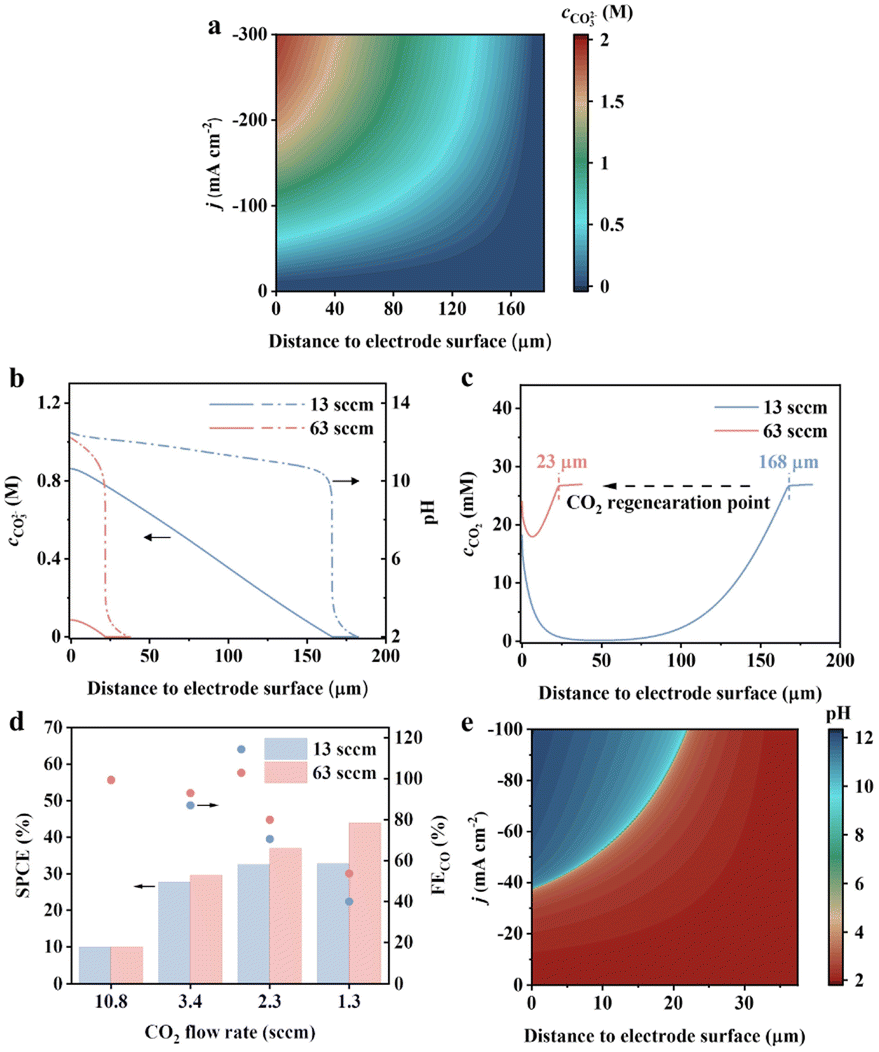 | ||
| Fig. 4 (a) Simulated carbonate concentration profile at different current densities in 0.5 M K2SO4 (pH = 2), at 13 sccm electrolyte flow rate. (b) Simulated carbonate concentration and pH profile at 13 and 63 sccm electrolyte flow rates, at −100 mA cm—2 applied current density. (c) Simulated CO2 concentration profiles at 13 and 63 sccm electrolyte flow rates, at −100 mA cm—2 applied current density. (d) FE and single pass CO2 efficiency towards CO on 30%-PTFE/Ag at −100 mA cm—2 applied current density. The electrolyte is 0.5 M K2SO4 (pH = 2). (e) Simulated pH profile at different current densities at 63 sccm electrolyte flow rate. | ||
With the regulation of mass transport of CO2 and H+, the selectivity and SPCE can be substantially improved. The remaining question is whether the carbonate issue can be avoided under mild acidic conditions by controlling mass transport. Unfortunately, simulation results indicate that even at an elevated electrolyte flow rate of 63 sccm, the surface pH jumps from below 4 at −37 mA—m−2 to above 10 at −38 mA—m−2 (Fig. 4e), making it challenging to shield the H+-HER while keeping the local pH below pKa of carbonic acid. Therefore, the pursuit of a carbonate-free interface has to be performed in metal cation-free acidic media to remove the counterion for salt. Future catalyst design may aim to break the scaling relationship between C, and H intermediates to optimize their relative binding energy, thereby enhancing the intrinsic selectivity towards the CO2RR in a metal cation-free acidic case.25
Conclusions
In this work, we thoroughly investigated the electrode process and local environment of the CO2RR under mild acidic conditions. The fast consumption rate of protons for the HER-H+ and generation rate of OH− from the CO2RR and HER-H2O at −5.5 mA cm−2 elevated the local pH to 11 for unavoidable carbonate formation at the interface. As the proton supply rate is determined by the mass transport, regulating hydrodynamics enhanced the mass transport of protons by 5 times. The single-pass carbon utilization efficiency substantially increases to 44% at −100 mA cm−2, benefiting from closer CO2 regeneration at the electrode vicinity at a high flow rate. Although regulating mass transport effectively alleviates the carbonate issue, it is difficult to solve the carbonate formation completely. Strong acidic conditions or metal-cation-free systems should be explored for this purpose. Thus, designing intrinsically active catalysts in strong acid or metal-cation-free media is necessary in the future.Data availability
All relevant data are presented in the main text and ESI.†Author contributions
Y. W. designed and supervised the project. Z. Z. performed the electrochemical measurements, COMSOL simulations and wrote the paper. L. X. performed SEM characterization. All authors discussed, commented on, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Excellent Young Scientist Fund (Hongkong and Macau) from the National Natural Science Foundation of China (project no. 22222208) and the Research Grants Council of the Hong Kong Special Administrative Region (project no. 14307322). The authors acknowledge the Department of Mechanical and Automation Engineering, The Chinese University of Hong Kong for the support of COMSOL.Notes and references
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Electrochemical CO2-to-ethylene conversion on polyamine-incorporated Cu electrodes, Nat. Catal., 2020, 4, 20–27 CrossRef.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- C. J. Bondue, M. Graf, A. Goyal and M. T. M. Koper, Suppression of Hydrogen Evolution in Acidic Electrolytes by Electrochemical CO2 Reduction, J. Am. Chem. Soc., 2021, 143, 279–285 CrossRef CAS PubMed.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Competition between Hydrogen Evolution and Carbon Dioxide Reduction on Copper Electrodes in Mildly Acidic Media, Langmuir, 2017, 33, 9307–9313 CrossRef CAS PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- B. Mayerhöfer, D. McLaughlin, T. Böhm, M. Hegelheimer, D. Seeberger and S. Thiele, Bipolar Membrane Electrode Assemblies for Water Electrolysis, ACS Appl. Energy Mater., 2020, 3, 9635–9644 CrossRef.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Efficient Electrocatalytic CO2 Reduction to C2+ Alcohols at Defect-Site-Rich Cu Surface, Joule, 2021, 5, 429–440 CrossRef CAS.
- R. L. Pindar, C. Batchelor-McAuley, M. Yang and R. G. Compton, Towards Direct Electroanalysis in Seawater: Understanding the Role of the Buffer Capacity of Seawater in Proton-Coupled Electron Transfer Reactions, J. Phys. Chem. C, 2021, 125, 27949–27958 CrossRef CAS.
- J. A. Rabinowitz and M. W. Kanan, The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- A. Ozden, F. P. García de Arquer, J. E. Huang, J. Wicks, J. Sisler, R. K. Miao, C. P. O'Brien, G. Lee, X. Wang, A. H. Ip, E. H. Sargent and D. Sinton, Carbon-efficient carbon dioxide electrolysers, Nat. Sustain., 2022, 5, 563–573 CrossRef.
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. G. d. Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, CO2 electrolysis to multicarbon products in strong acid, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, High carbon utilization in CO2 reduction to multi-carbon products in acidic media, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- Z. Liu, T. Yan, H. Shi, H. Pan, Y. Cheng and P. Kang, Acidic Electrocatalytic CO2 Reduction Using Space-Confined Nanoreactors, ACS Appl. Mater. Interfaces, 2022, 14, 7900–7908 CrossRef CAS PubMed.
- X. Sheng, W. Ge, H. Jiang and C. Li, Engineering Ni-N-C Catalyst Microenvironment Enabling CO2 Electroreduction with Nearly 100% CO Selectivity in Acid, Adv. Mater., 2022, 34, 2201295 CrossRef CAS PubMed.
- M. R. Singh, E. L. Clark and A. T. Bell, Effects of electrolyte, catalyst, and membrane composition and operating conditions on the performance of solar-driven electrochemical reduction of carbon dioxide, Phys. Chem. Chem. Phys., 2015, 17, 18924–18936 RSC.
- A. B. Moss, S. Garg, M. Mirolo, C. A. Giron Rodriguez, R. Ilvonen, I. Chorkendorff, J. Drnec and B. Seger, In operando investigations of oscillatory water and carbonate effects in MEA-based CO2 electrolysis devices, Joule, 2023, 7, 350–365 CrossRef CAS.
- Z. Xu, Y. Xie and Y. Wang, Pause electrolysis for acidic CO2 reduction on 3-dimensional Cu, Mater. Rep.: Energy, 2023, 3, 100173 CAS.
- H.-G. Qin, F.-Z. Li, Y.-F. Du, L.-F. Yang, H. Wang, Y.-Y. Bai, M. Lin and J. Gu, Quantitative Understanding of Cation Effects on the Electrochemical Reduction of CO2 and H+ in Acidic Solution, ACS Catal., 2022, 13, 916–926 CrossRef.
- Z. Ma, Z. Yang, W. Lai, Q. Wang, Y. Qiao, H. Tao, C. Lian, M. Liu, C. Ma, A. Pan and H. Huang, CO2 electroreduction to multicarbon products in strongly acidic electrolyte via synergistically modulating the local microenvironment, Nat. Commun., 2022, 13, 7596 CrossRef CAS PubMed.
- R. G. Compton and C. E. Banks, Understanding voltammetry, World Scientific, 2018 Search PubMed.
- V. Vedharathinam, Z. Qi, C. Horwood, B. Bourcier, M. Stadermann, J. Biener and M. Biener, Using a 3D Porous Flow-Through Electrode Geometry for High-Rate Electrochemical Reduction of CO2 to CO in Ionic Liquid, ACS Catal., 2019, 9, 10605–10611 CrossRef CAS.
- C. J. Bondue and M. T. M. Koper, A DEMS approach for the direct detection of CO formed during electrochemical CO2 reduction, J. Electroanal. Chem., 2020, 875, 113842 CrossRef CAS.
- S. Ringe, C. G. Morales-Guio, L. D. Chen, M. Fields, T. F. Jaramillo, C. Hahn and K. Chan, Double layer charging driven carbon dioxide adsorption limits the rate of electrochemical carbon dioxide reduction on Gold, Nat. Commun., 2020, 11, 33 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Enhancing Hydrogen Evolution Activity in Water Splitting by Tailoring Li+-Ni(OH)2-Pt Interfaces, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- E. R. Cave, C. Shi, K. P. Kuhl, T. Hatsukade, D. N. Abram, C. Hahn, K. Chan and T. F. Jaramillo, Trends in the Catalytic Activity of Hydrogen Evolution during CO2 Electroreduction on Transition Metals, ACS Catal., 2018, 8, 3035–3040 CrossRef CAS.
- D.-J. Chen, R. W. Penhallurick and Y. J. Tong, A versatile and robust surface-poison-resisting Scanning Amperometric Proton Microscopy, J. Electroanal. Chem., 2020, 875, 113918 CrossRef CAS.
- M. C. O. Monteiro, L. Jacobse and M. T. M. Koper, Understanding the Voltammetry of Bulk CO Electrooxidation in Neutral Media through Combined SECM Measurements, J. Phys. Chem. Lett., 2020, 11, 9708–9713 CrossRef CAS PubMed.
- Y. Luo, K. Zhang, Y. Li and Y. Wang, Valorizing carbon dioxide via electrochemical reduction on gas-diffusion electrodes, InfoMat, 2021, 3, 1313–1332 CrossRef CAS.
- Y. Lin, T. Wang, L. Zhang, G. Zhang, L. Li, Q. Chang, Z. Pang, H. Gao, K. Huang, P. Zhang, Z. J. Zhao, C. Pei and J. Gong, Tunable CO2 electroreduction to ethanol and ethylene with controllable interfacial wettability, Nat. Commun., 2023, 14, 3575 CrossRef CAS PubMed.
- X. Du, P. Zhang, G. Zhang, H. Gao, L. Zhang, M. Zhang, T. Wang and J. Gong, Confinement of ionomer for electrocatalytic CO2 reduction reaction via efficient mass transfer pathways, Natl. Sci. Rev., 2023, 11, nwad149 CrossRef PubMed.
- Z. Xing, X. Hu and X. Feng, Tuning the Microenvironment in Gas-Diffusion Electrodes Enables High-Rate CO2 Electrolysis to Formate, ACS Energy Lett., 2021, 6, 1694–1702 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Modelling details and the experimental setup for scanning electrochemical microscopy. See DOI: https://doi.org/10.1039/d3sc06583a |
| This journal is © The Royal Society of Chemistry 2024 |