
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA decade of lessons in the activation of ArIL2 species
Tania
 ,
Marcus
Sceney
and
Jason L.
Dutton
*
,
Marcus
Sceney
and
Jason L.
Dutton
*
Department of Biochemistry and Chemistry, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Victoria, Australia. E-mail: j.dutton@latrobe.edu.au
First published on 9th February 2024
Abstract
Hypervalent iodine(III) compounds of the general structure ArIL2 are widely used as oxidizing agents for a variety of applications across both organic and inorganic chemistry. Considerable work has been done on the activation of these compounds by tuning the ligands at the iodine centre. This perspective summarises the work of our and other groups on rectification of historically misidentified iodine(III) reagents of this class, and the syntheses of activated species. Recent advances focusing on increasing the oxidative capacity of I(III) moieties using Lewis and Brønsted acids and Lewis bases as well as the activation of halogens with I(III) are discussed.
 Tania | Tania is a PhD candidate under the supervision of Prof. Jason Dutton at La Trobe University, Australia. She completed her BSc (honours chemistry) from Guru Nanak Dev University, India and moved to Australia to pursue a Master of Chemical Sciences at La Trobe University. She joined the Dutton lab for her master's research year. Her current research is focused on development and application of phenyl Iodine(III) and Iodine(V) compounds. |
 Marcus Sceney | Marcus Sceney completed his BSc with honours in chemistry at La Trobe University in 2023 and has since been a PhD candidate working under Prof. Jason Dutton at the same. His current research focus is on the application of phenyliodine(III) oxidants in the activation of halogens for electrophilic halogenation of recalcitrant aromatics. |
 Jason L. Dutton | Jason Dutton completed his PhD in 2010 under Prof. Paul Ragogna at The University of Western Ontario, which sparked his interest in the richly funded area of unusual main group compounds. This was followed by postdoctoral studies under Prof. Warren Piers at The University of Calgary. He then made the big move to La Trobe University in Melbourne in 2011 to begin his independent career as a Lecturer where he has remained since and is now Professor of Molecular Curiosities. |
Introduction
Hypervalent iodine compounds are widely used as oxidizing agents for a variety of applications across both organic and inorganic chemistry.1–3 Hypervalent iodine compounds fall into several classes, the two most common motifs being ArIL2 and [Ar–I–Ar]+, where the iodine is in the formal +3 oxidation state. Higher oxidation state compounds, with I in the +5 or even +7 oxidation state are rarer. The focus of this perspective is on the ArIL2 class (Fig. 1). Common compounds here include commercially available PhI(OAc)2, also known as PIDA (phenyliodine bis(acetate) or (diacetoxyiodo)benzene) and PhI(OTFA)2, known as PIFA (phenyliodine bis(trifluoroacetate) or (bis(trifluoro acetoxy)iodo)benzene). Halogen ligated compounds are also widely used, including ArIF2 and ArICl2. These are not commercially available but are easily synthesized. Benziodoxole based compounds, in which the iodine atom is part of a 5-membered ring such as Togni's reagent are also widely used I(III) compounds,4 but out of scope for this perspective. In this perspective, we will focus on strategies to increase the reactivity of ArIL2 compounds by our group and others in the past ∼15 years with a focus on the behaviour at the iodine atom.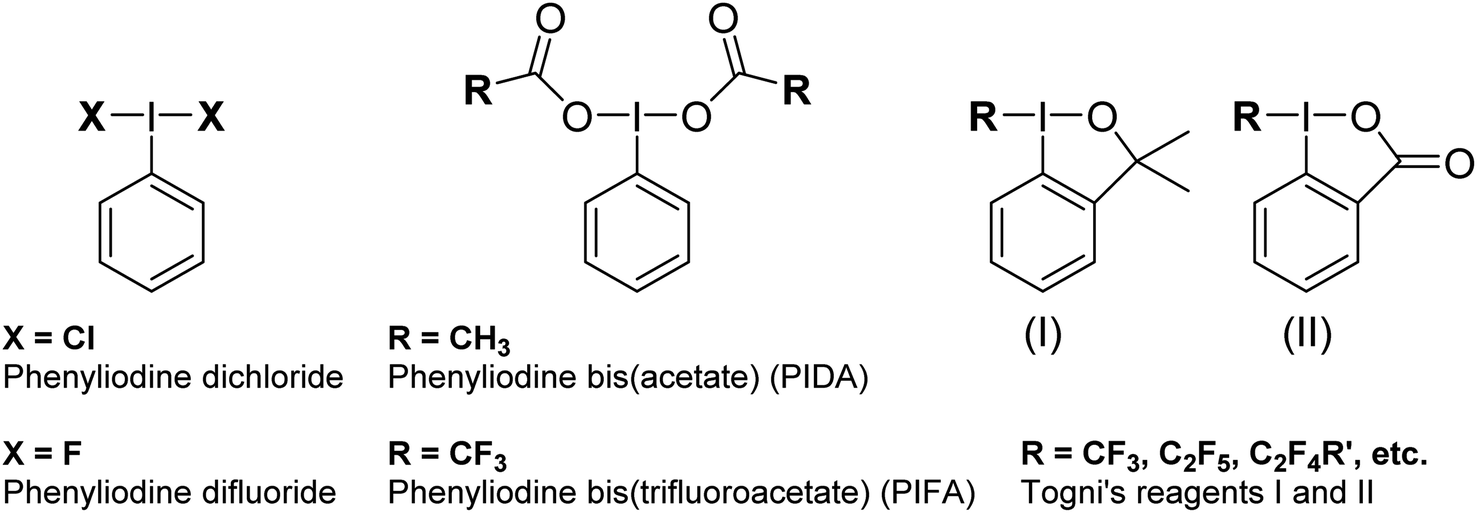 | ||
| Fig. 1 Hypervalent iodine(III) compounds. | ||
As the action of these compounds is oxidative or electrophilic, the important molecular orbitals to consider are the low-lying unoccupied orbitals. For most compounds, the LUMO and LUMO+1, with respect to the iodine, are sigma symmetric antibonding orbitals lying along the L–I–L and Ar–I bond axes. For the orbital with an L–I–L contribution there is typically additional pi-antibonding character with respect to the aryl ring (Fig. 2). The population of this orbital results in rupture of the L–I bonds and formation of Ar–I as I(I) and the lower in energy this orbital is, the more oxidizing the compound will in general be. The antibonding orbital associated with the C–I bond is typically used in halogen bonding interactions as an acceptor.5
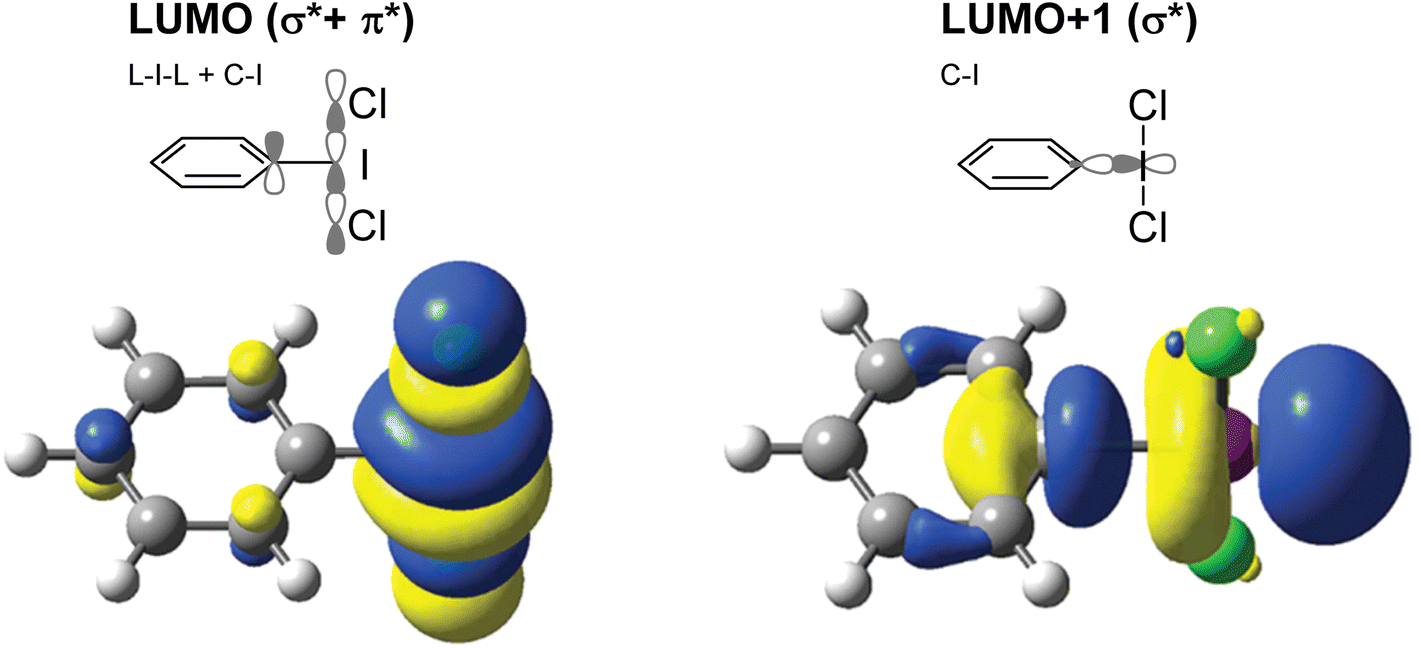 | ||
| Fig. 2 Depictions of the LUMO and LUMO+1 in phenyliodine dichloride and respective computational models generated using B3LYP-D3(BJ)/def2-TZVPPD method/basis set with acetonitrile solvation. In this case, the LUMOs are primarily σ* orbitals with respect to the L–I–L bond (LUMO) and the C–I bond (LUMO+1). | ||
Increasing oxidative capacity
Increasing the oxidative capacity of ArIL2 compounds can be accomplished by modifying two features, the ligand (L) or the substituents on the aryl (Ar) group. This was quantified by Radzhabov and co-workers in a theoretical study where triflate and a variety of acetoxy ligands were considered.6 Commercially available PhI(OAc)2 (0.91 V) was found to be the least oxidizing out of the derivatives considered and PhI(OTf)2 by far the most oxidizing at 2.24 V. PhI(OTFA)2 was intermediate to these at 1.49 V. In general, a less nucleophilic ligand leads to a greater oxidative capacity, as would be expected for a more electron poor iodine centre. Making the aryl group more electron poor or electron rich by incorporating withdrawing or donating group has the expected effect of making the complex more or less oxidizing, respectively. However the effect is more subtle than modifying the ligand, where for ArI(OAc)2 a para-nitro substituent gives 0.97 V and para-methoxy gives 0.88 V. However, as discussed later, substituents on the ligand can have a substantial effect in suppressing decomposition processes.The myth and redemption of ArI(OTf)2
Our entry into this space was in 2011, where our strategy to generate more oxidizing I(III) reagents was the synthesis of dicationic coordination complexes of the type [ArIL2]2+ where L is a neutral rather than the typical anionic ligand. This class of compound has been reported in 1994 by Weiss,7 but sparsely used until the 2010s with only a couple of reports appearing in the next 15 years.8,9 These compounds had also not been structurally confirmed, which as we shall discuss is critically important in I(III) chemistry as misidentifications have been made for a variety of species. The synthetic route to Weiss' reagent is simple, commercially available PhI(OAc)2 is reacted with 2 equivalents of TMS-OTf in CH2Cl2, which was assumed to generate PhI(OTf)2 (much more on this later), followed by the addition of 2 equivalents of a pyridine or quinoline ligand (Scheme 1).10,11 The dicationic coordination complex precipitates immediately from solution and can be easily isolated. We obtained a crystal structure of the 4-DMAP and pyridine analogues and later the 4-cyanopyridine analogue.12 The crystal structure confirmed the structure as proposed by Weiss, with the expected T-shaped geometry about the iodine centre. From (purported) PhI(OTf)2, it has been found that the neutral ligands able to displace the triflates are limited to N-centred ligands resistant to oxidation. Attempted use of N-heterocyclic carbenes led to oxidative coupling of the carbene ligands and attempted use of phosphines led to oxidation of the phosphine rather than formation of a coordination complex for example.12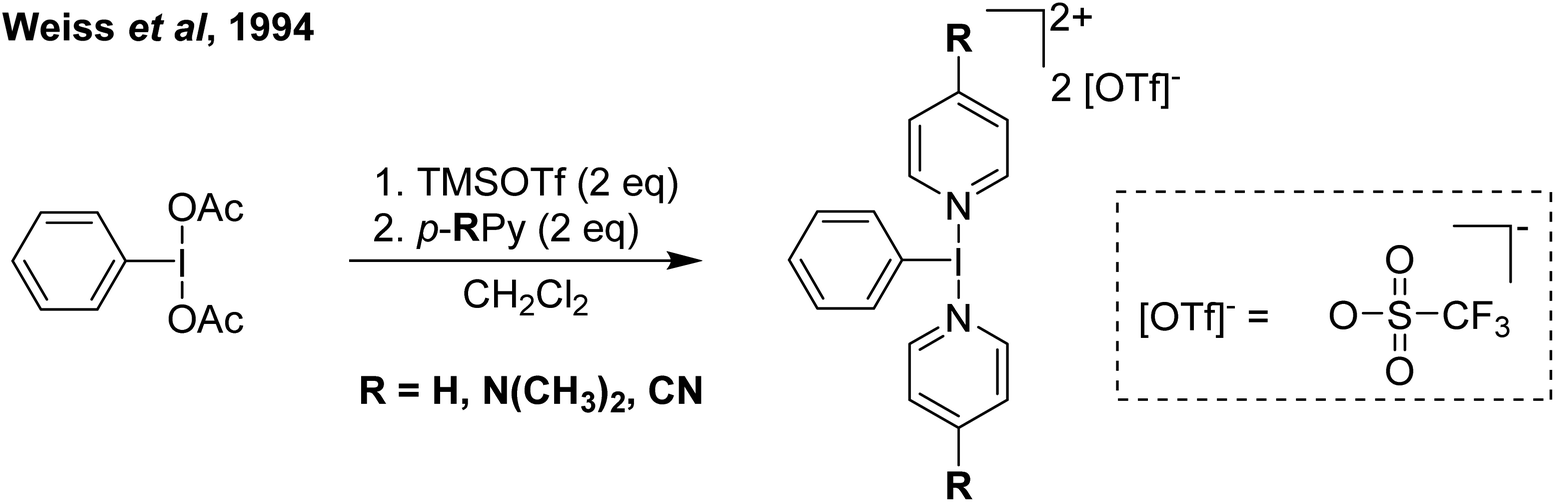 | ||
| Scheme 1 General synthesis of Weiss' reagents. | ||
Our planned use for Weiss' reagent was to perform 2-electron oxidations of transition metals, from which the pyridine ligands could then be displaced by other ligands giving access to a variety of high oxidation state metal complexes. At the same time we commenced our initial studies Ritter and co-workers also rediscovered Weiss' reagent and showed in a report in Science that this oxidation/ligand delivery to metals works very well,13 an early lesson for a young Lecturer/Assistant Professor being that if something is a good idea, someone else is probably working on it! They were able to form an organo Pd(IV) species from Pd(II) from which the delivered pyridine could be displaced by fluoride, which was demonstrated to be effective in the late-stage generation of organofluorinated species using 18F as the fluoride source (Scheme 2). We were subsequently able to show the applicability of Weiss' reagent in the generation of a variety of high oxidation state transition metal complexes, including the first examples of homoleptic trications of Au(III) (Scheme 3).14,15 The Wengryniuk group has found that Weiss' reagents are effective for a variety of transformations in the organic chemistry space, including oxidative rearrangement of tertiary benzyl alcohols and selective oxidation of alcohols, while Huber reported their use for azo couplings.16–18
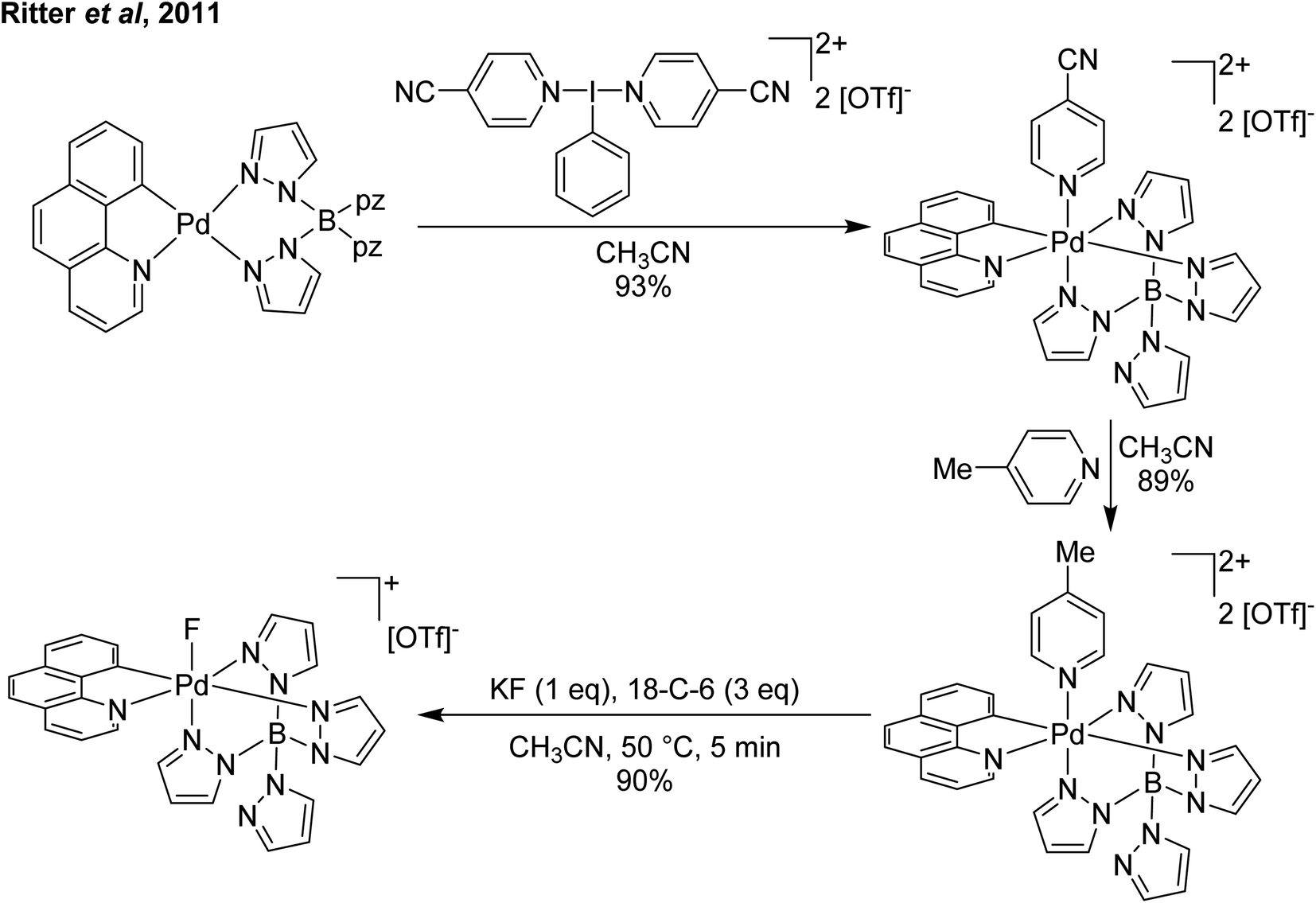 | ||
| Scheme 2 Ritter and coworker's13 oxidation of Pd(II) using a Weiss' reagent to facilitate formation of a Pd(IV) fluoride. | ||
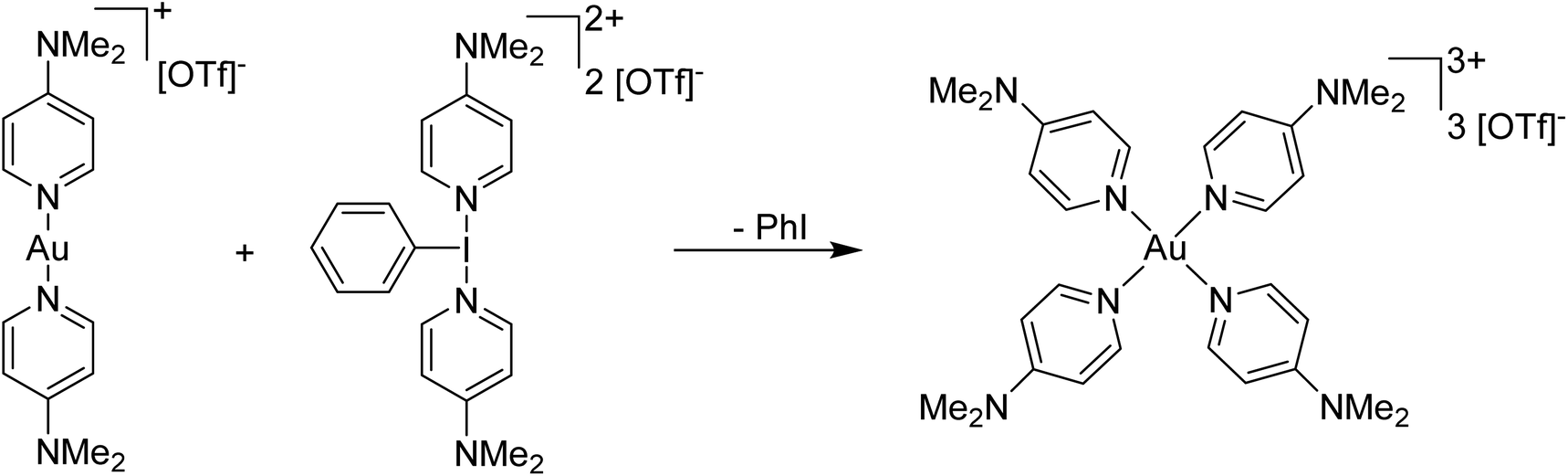 | ||
| Scheme 3 Generation of a homoleptic Au(III) trication using a Weiss' reagent, [PhI(4-DMAP)2][OTf]2. | ||
In main group chemistry, Weiss' reagents are also effective at oxidation and delivery of pyridine ligands, where a 2-electron redox couple is available. The salient example for this story is oxidation of a tellurophene derivative. Unlike lighter thiophene and selenophene, whose chemistry with electron poor reagents is dominated by electrophilic aromatic substitution processes, tellurophene is less aromatic and the Te(II)/Te(IV) redox couple is also accessible. This can be taken advantage of in the formation of switchable functional molecules based on tellurophene, an area of study that has been led by the Seferos group.19–21 We were interested in whether oxidation of 2,5-diphenyltellurophene by delivery of the pyridine ligands to Te and then displacement of the pyridine for other nucleophiles could lead to 2,5-diphenyltellurophene derivatives having interesting properties.22 When the reaction between Weiss' reagent and 2,5-diphenyltellurophene was carried out, a surprising product was obtained in which the 4-DMAP had replaced a C–H at the 3-position, along with generation of protonated 4-DMAP and PhI, an EAS (Electrophilic Aromatic Substitution) like process, where 4-DMAP is acting as the electrophile (Scheme 4). It was hypothesized that this reaction was occurring via Te(IV). Supporting experiments included the reaction of 2,5-diphenyltellurophene with PhI(OAc)2 which resulted in the isolation and X-ray structural characterization of a tellurophene derivative bearing Te(OAc)2 with Te in the +4 oxidation state. Treatment of this species with pyridine effected no change (Scheme 4). Furthermore, reaction of 2,5-diphenyltellurophene with PhI(OTf)2 also gave an isolable Te(IV) derivative and reaction of this species with 2 equivalents of 4-DMAP resulted in the immediate formation of the EAS product. This suggested that with a better leaving group, 4-DMAP could bind to the Te(IV) supporting a high oxidation state coordination complex as the intermediate. The Te(IV) species arising from reaction with PhI(OTf)2 was also characterized by X-ray crystallography, however the compound did not have two triflate anions bound to Te but rather one triflate and one acetate (Scheme 4). This suggested that the oxidizing agent was, in fact, PhI(OTf)(OAc).
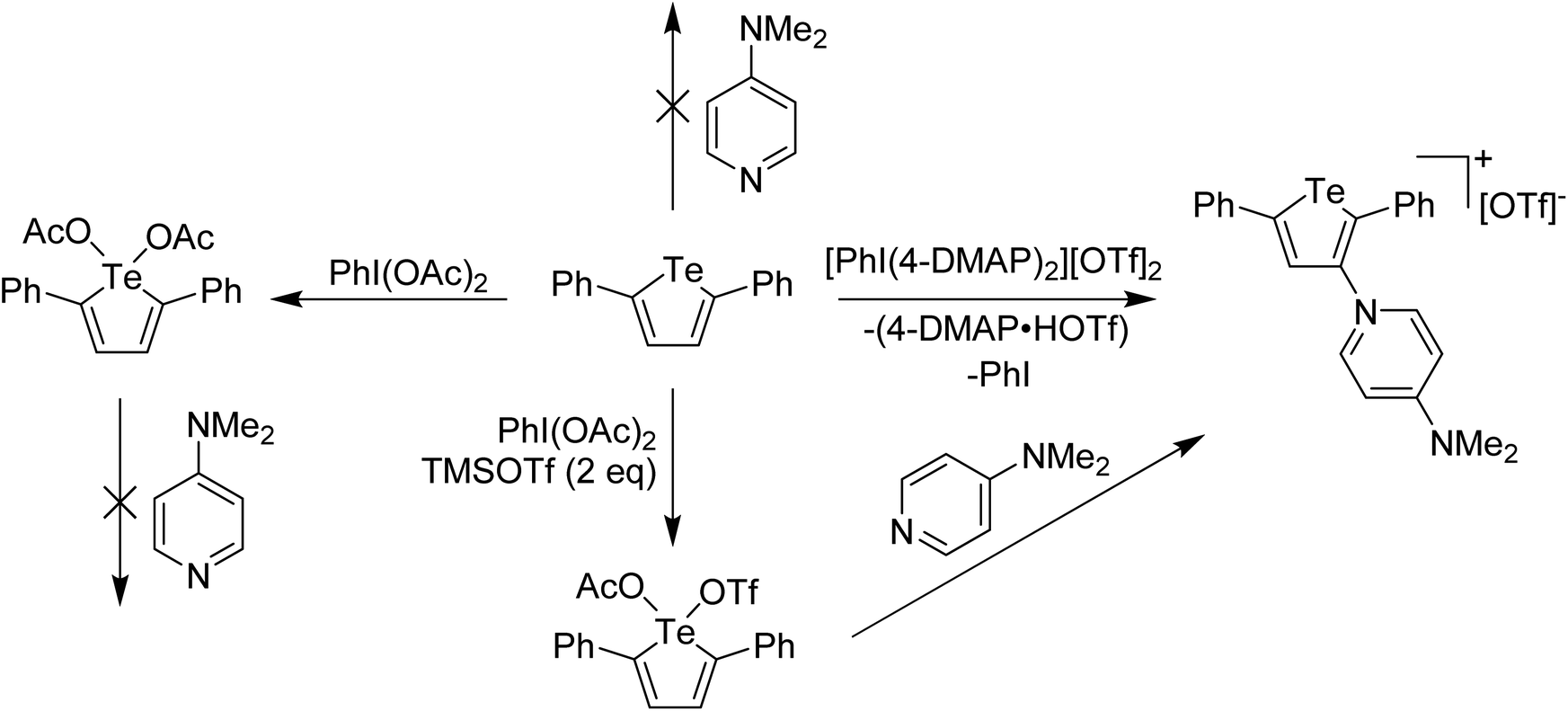 | ||
| Scheme 4 Reaction of 2,5-diphenyltellurophene with various I(III) oxidants and subsequent reaction of Te(IV) products with 4-DMAP. | ||
Examination of the in situ1H NMR for the reaction between PhI(OAc)2 and 2 equivalents of TMS-OTf in CDCl3 showed one phenyl containing species, two acetate containing species (TMS-OAc and PhI(OTf)(OAc)) and one equivalent of TMS-OTf left unconsumed. This was an NMR our group had done many times over several years to confirm clean generation of PhI(OTf)2. Identical spectra also present in the supporting information of other reports purporting to use PhI(OTf)2 clearly showed only 50% consumption of TMS-OTf, a feature that until this point had been overlooked by preceding authors and us. In the initial report of PhI(OTf)2, it was indicated to be an isolable yellow oil and was synthesized via reaction of PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O with TMS-OTf (Scheme 7).23 The PhI(OAc)2 route is far more common (Scheme 7),24 but the route using PhIO has been used by others relatively recently as well in the oxidative coupling of hydrazones and alcohols.12,25,26 As part of our report, in 2015, we included this reassignment of the product of the reaction between PhI(OAc)2 and 2 equivalents of TMS-OTf (Scheme 5).22
O with TMS-OTf (Scheme 7).23 The PhI(OAc)2 route is far more common (Scheme 7),24 but the route using PhIO has been used by others relatively recently as well in the oxidative coupling of hydrazones and alcohols.12,25,26 As part of our report, in 2015, we included this reassignment of the product of the reaction between PhI(OAc)2 and 2 equivalents of TMS-OTf (Scheme 5).22
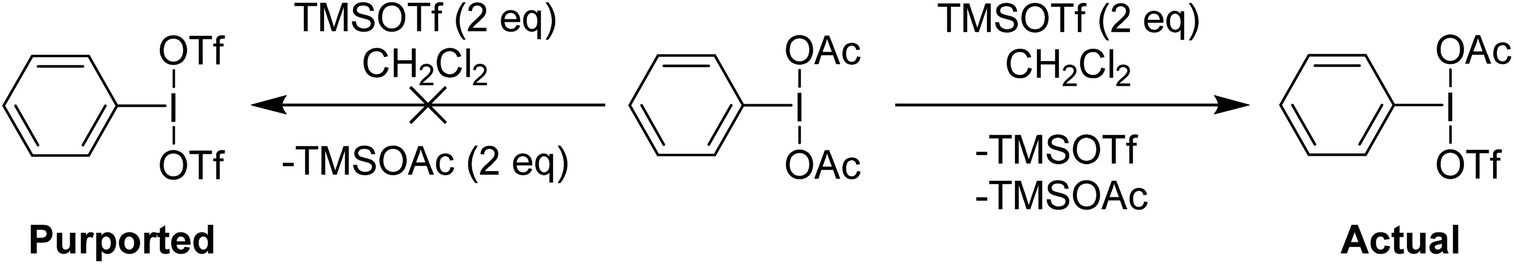 | ||
| Scheme 5 Reaction of PhI(OAc)2 with two equivalents of TMSOTf showing formation of PhI(OAc)(OTf) instead of PhI(OTf)2. | ||
In 2016 the Shafir group was able to isolate a crystalline solid and confirmed the structure via X-ray crystallography to be PhI(OTf)(OAc).27 This was as part of a study examining the effect of the addition of Lewis acids to PhI(OAc)2 and related species. They found that a competing Lewis acid, BF3 in this case, binds to the second oxygen of an acetate and weakens the corresponding I–O bond. Increased reactivity was observed in complexes containing a weaker I–O bond induced by competing coordination to the BF3. Weiss' agent can also be activated by BF3 to increase its reactivity, in this case a p-RPy-BF3 adduct is formed with a more reactive [PhI(p-RPy)]2+ dication left.28 This will react with aryl species to form [Ph–I–Ar]+ iodonium cations, behaviour which Weiss' reagent itself does not display (Scheme 6).
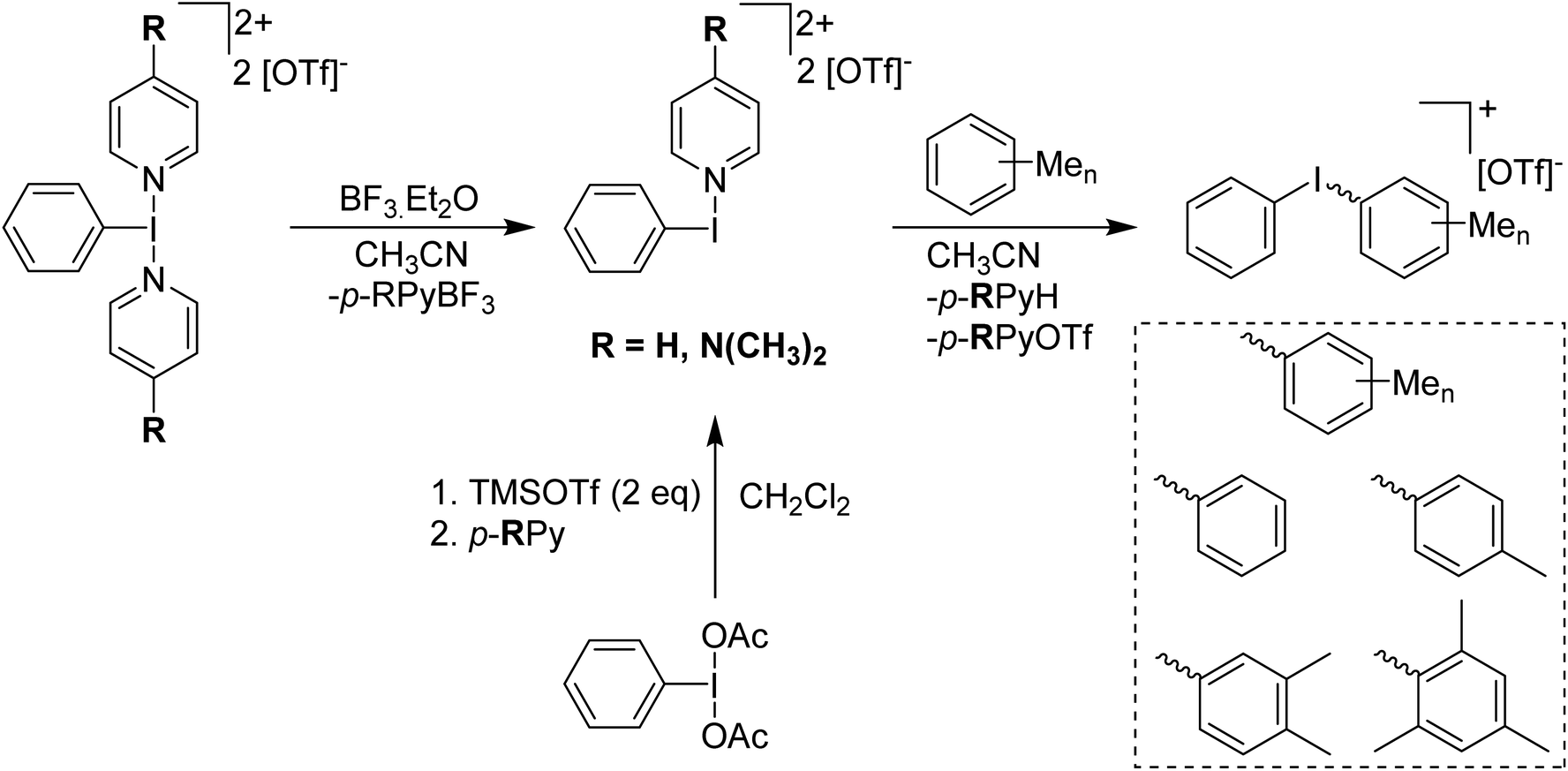 | ||
| Scheme 6 Syntheses of [PhI(p-RPy)][OTf]2 and subsequent reaction with methylbenzenes to form respective diaryliodonium triflate salts. | ||
Subsequent to the reports from Shafir and our group, the invocation of PhI(OTf)2 from PhI(OAc)2 and 2 equivalents of TMS-OTf continued to be used in the literature for a variety of transformations such as oxidative cyclisation reactions, C–H functionalisation reactions at acetyl, alkyl and aryl moieties, addition reactions at alkyne substrates to yield aryl(trifloxyalkenyl)iodonium triflate salts, selective difluoro alkylation of aryliodanes and asymmetric Sigmatropic rearrangement.26,29–32 The identity of the organic products in these reports is not under question but the oxidative agent is PhI(OTf)(OAc), not PhI(OTf)2. This is not innately salient to the reported reactions, evidently PhI(OTf)(OAc) has sufficient oxidative capacity to effect the targeted/observed transformations. However, the reports are often accompanied by detailed theoretical studies to determine the reaction pathway involving relatively large molecules and several transition states using PhI(OTf)2 as the starting point, a molecule which never existed. Many of the reactions include elimination of a proton via loss of HOTf, whereas the presence of acetate would likely result in HOAc being eliminated instead. Misidentifying key I(III) species is not isolated to PhI(OTf)2, another example is a reported thiobenziodoxole used as a trifluoromethylation reagent that was found to not contain I(III) at all upon structural characterization.33,34 Rather an I(I) and a thioperoxy unit in an open configuration is the actual molecule and the thioperoxy acts as the oxidizing agent.
Given the continued invocation of PhI(OTf)2 from PhI(OAc)2 and 2 equivalents of TMS-OTf we sought to clear the issue with a comprehensive investigation.35 The aforementioned studies from our group and Shafir are clear that the experimental outcome of this reaction is a single metathesis to form PhI(OTf)(OAc). Theoretical calculations support this notion, with a calculated ΔG value of PhI(OAc)2 + TMS-OTf → PhI(OTf)(OAc) + TMS-OAc returning −67 kJ mol−1 and the second step of PhI(OTf)(OAc) + TMS-OTf → PhI(OTf)2 + TMS-OAc returning a positive value of +30 kJ mol−1. In the course of this work, it was found that most computational methods do a poor job of optimizing the geometry of bound triflate, both to iodine and in TMS-OTf. ωPBE/def2TZVP did the best job of reproducing the geometry of known bound triflate moieties, and this method was used for the calculations. Calculation of the route from PhIO + 2 TMS-OTf → PhI(OTf)2 + TMS-O-TMS however gave a ΔG of −41 kJ mol−1, indicating that this route might be thermodynamically feasible. Investigating the reaction experimentally via reactions in CD2Cl2 however showed that TMS-OTf is incompletely consumed, and that the dominant iodine containing species to be PhI and iodonium cation [Ph–I–Ph–I]+ and no species consistent with what would be expected for PhI(OTf)2. This led us to the conclusion that PhI(OTf)2 does not exist.35
PhI(OTf)2 is apparently an attractive compound that chemists would like to use, as they have been attempting to use it, and many of these reports are in high profile journals. PhI(OTf)2 is also the most oxidizing of the ArIL2 species, by far, considered by Radzhabov in their study.6 The most obvious limiting factor in the synthesis is the positive ΔG value for the second metathesis reaction from PhI(OTf)(OAc). The Si–F bond is the strongest single bond and an excellent driving force for metathesis reactions, therefore ArIF2 should be a good starting point for the generation of ArI(OTf)2 using TMS-OTf. Calculations for PhIF2 + TMS-OTf → PhI(OTf)F + TMS-F and PhI(OTf)F + TMS-OTf → PhI(OTf)2 + TMS-F returned ΔG values of −34 and −16 kJ mol−1 respectively, indicating the route is thermodynamically feasible.36
Stang and Zefirov reported that reaction of PhIF2 with one equivalent of TMS-OTf at −78 °C gives an unobservable PhI(OTf)F intermediate that can be intercepted for the generation of aryl iodonium cations.37 We found that using 2 equivalents of TMS-OTf resulted in incomplete consumption of TMS-OTf and the formation of PhI as well as [Ph–I–Ph–I]+ as the main iodine containing product evidently arising from EAS of some unobserved electrophilic species onto PhI. Attempting to use methyl groups to block EAS processes failed, with even the pentamethyl variant resulting in formation of iodonium salts as the primary result via cleavage of a C–I bond and generation of I2via some unknown mechanism (Scheme 8).36
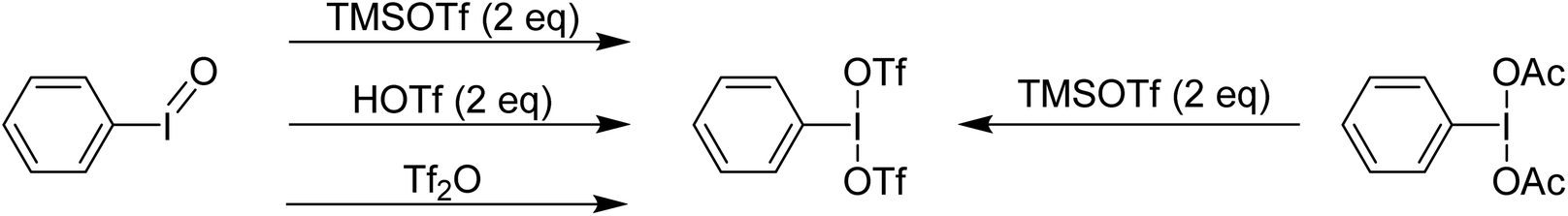 | ||
| Scheme 7 Various synthetic routes purported to yield PhI(OTf)2. | ||
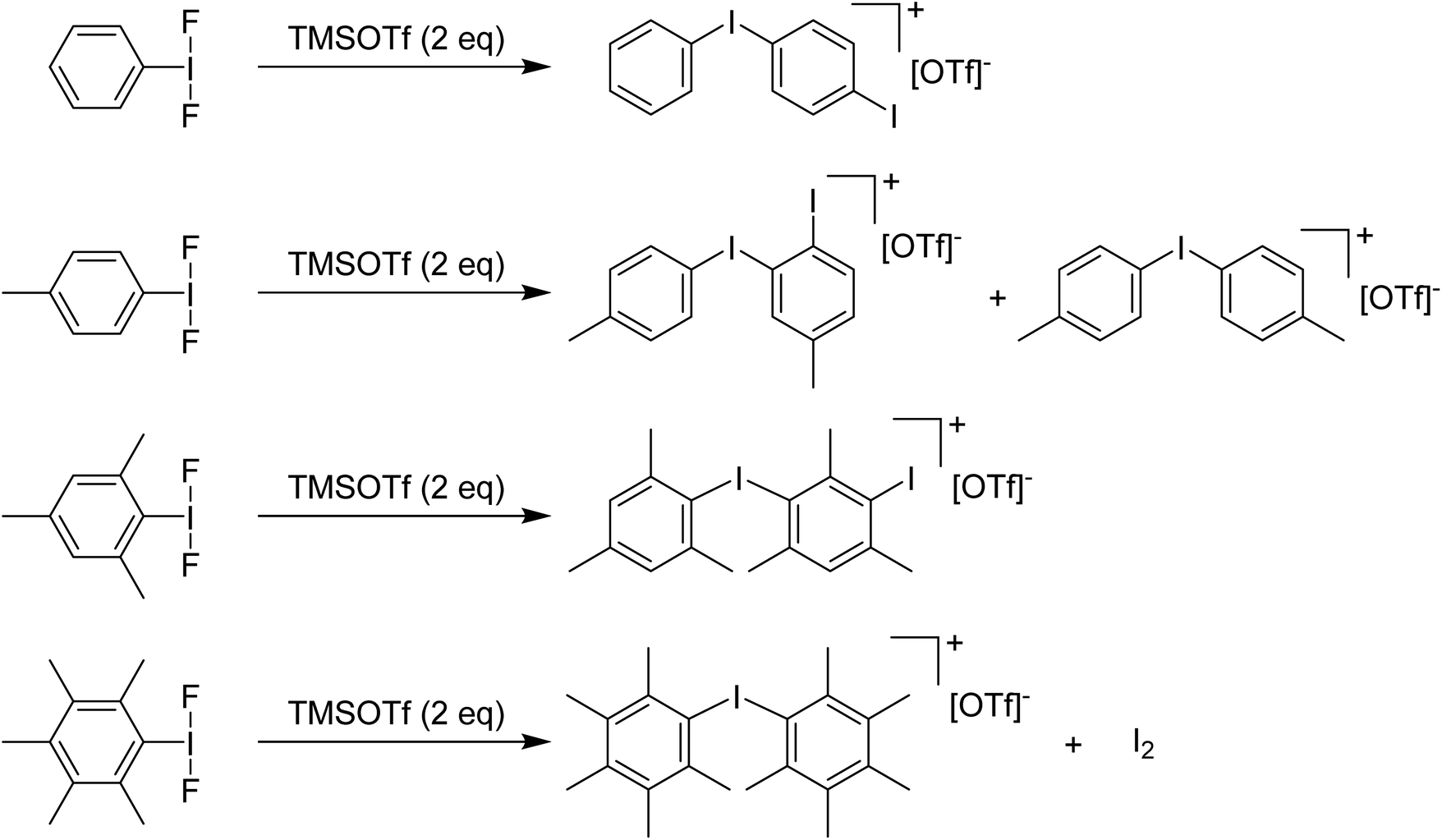 | ||
| Scheme 8 Reactions of various ArI(F)2 analogues with TMSOTf (2 eq.) resulting in corresponding aryl iodonium triflate salts. | ||
Given that electrophilic aromatic substitution appeared to be the key Achilles' Heel, it was reasoned that deactivating the aryl group with an electron withdrawing group might shut this pathway down. Introduction of such a group would make the entire molecule more electron poor and more oxidizing, but the effect is relatively subtle as shown by Radzhabov.6 Nitro was chosen as the deactivating group, with p-NO2-C6H4-IF2 being the new target. This could be accessed by the addition of Olah's reagent to p-NO2-C6H4-I(OTFA)2, a known compound. Alternatively p-NO2-C6H4-IF2 could be synthesized by reaction of p-NO2-C6H4-I with XeF2. Addition of 2 equivalents of TMS-OTf to p-NO2-C6H4-IF2 furnished p-NO2-C6H4-I(OTf)2 in good yield as an isolable solid and the structure was also (finally) confirmed via X-ray crystallography (Scheme 9 and Fig. 3).36
 | ||
| Scheme 9 Complete synthesis of p-NO2-C6H4-I(OTf)2. | ||
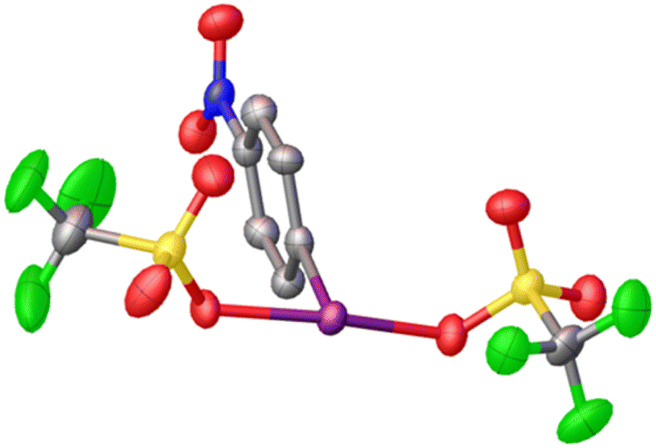 | ||
| Fig. 3 Crystal structure of p-NO2-C6H4-I(OTf)2. | ||
Initial reactivity studies found that p-NO2-C6H4-I(OTf)2 reacts with deactivated aryls to form iodonium cations, but more interestingly reacts with hydride sources at ambient conditions, including HSiEt3 and, also, more significantly hydridic C–H bonds (Scheme 10), giving C-OTf as products, with reduction to I(I) and elimination of HOTf.38 Such activation has only very recently been reported by Periana and co-workers by C6F5-I(NTf2)2 on methane and ethane at elevated temperatures and pressures (see next section).39 We surmised that the intermediate in these reactions was potentially p-NO2-C6H4-I(OTf)(H). To lend support to this hypothesis we reacted p-NO2-C6H4-I(OTf)2 with adamantane and 1-fluoroadamantane. Both reactions generated adamantyl triflate but adamantane reaction gave p-NO2-C6H4-I as side product whereas 1-fluoroadamantane reaction yielded p-NO2-C6H4-I(OTf)(F), which can be isolated and crystalographically characterized.
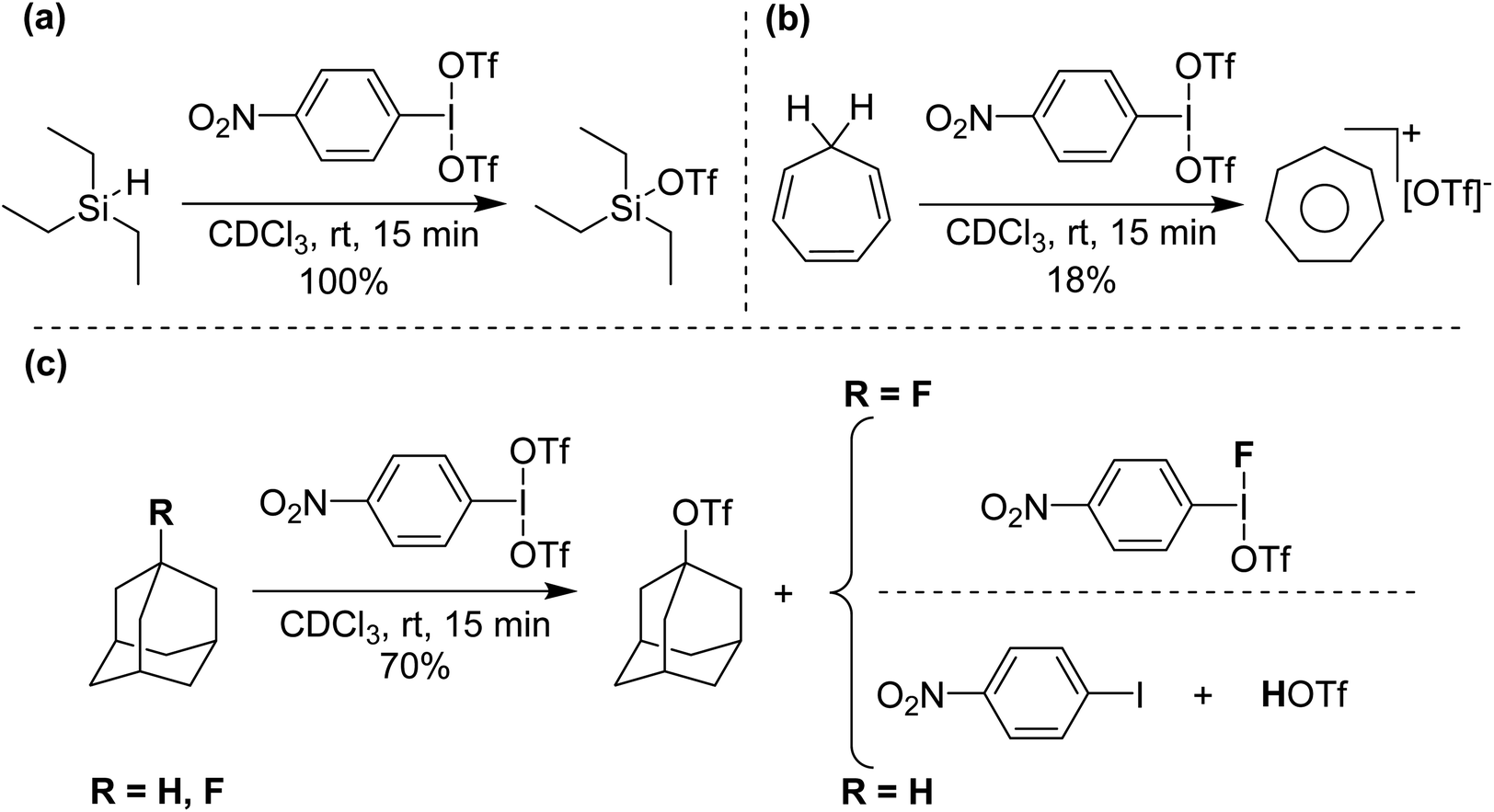 | ||
| Scheme 10 (a) Si–H abstraction from Et3SiH, (b) C–H abstraction from cycloheptatriene and (c) C–H and C–F abstraction from adamantane and 1-fluoroadamantane, respectively, using p-NO2-C6H4-I(OTf)2. | ||
p-NO2-C6H4-I(OTf)2 was also found to perform 4-electron oxidations of cyclohexene derivatives (using 2 equivalents) to aromatic rings, rapidly at −94 °C (Scheme 11). Other methods of doing such oxidations use Pd catalysts, with O2 as the oxidant and require elevated temperatures and extended times.40–43
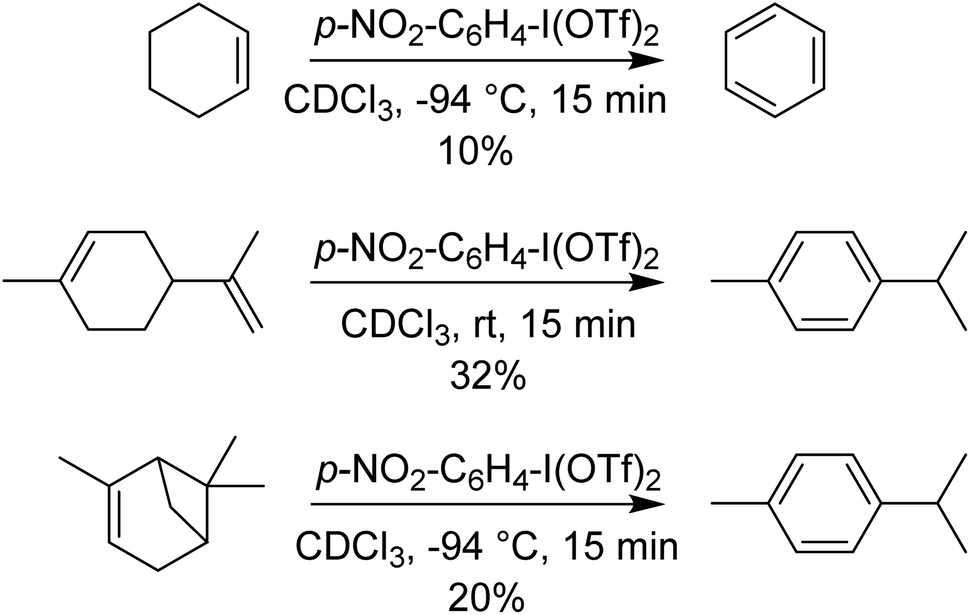 | ||
| Scheme 11 Aromatization of cyclohexene derivatives using p-NO2-C6H4-I(OTf)2. | ||
Periana and co-workers recently reported the isolation of C6F5-I(NTf2)2, albeit without obtaining a crystal structure.39 They found that at elevated temperatures and pressures this compound could effect the activation of simple sp3 C–H bonds in ethane and methane generating the corresponding simple alkyl-NTf2 compounds. Given the above discussion regarding misidentification of reactive I(III) species, we isolated p-NO2-C6H4-I(NTf2)2via reaction of p-NO2-C6H4-IF2 with 2 equivalents of TMS-NTf2 (Scheme 12). We were able to obtain X-ray quality crystals allowing for structure confirmation (Fig. 4).44
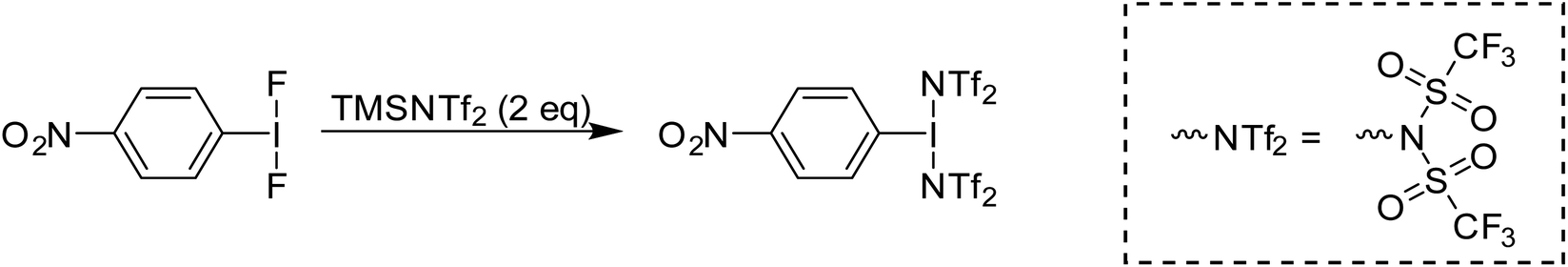 | ||
| Scheme 12 Synthesis of p-NO2-C6H4-I(NTf2)2. | ||
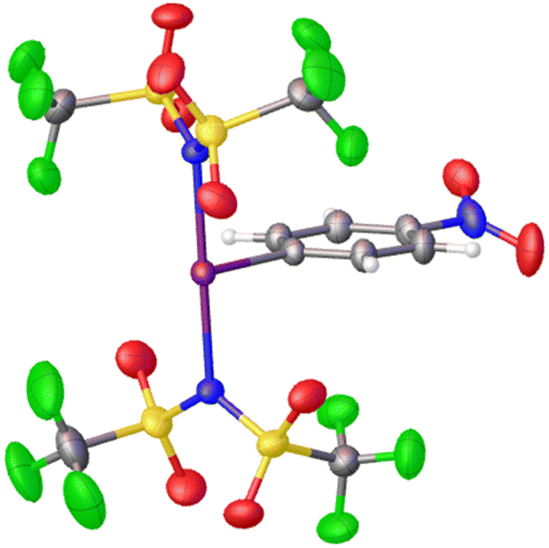 | ||
| Fig. 4 Crystal structure of p-NO2-C6H4-I(NTf2)2. | ||
NTf2− is less coordinating than OTf− and in line with Radzhabov's work this suggests p-NO2-C6H4-I(NTf2)2 should be more oxidizing than p-NO2-C6H4-I(OTf)2, which was confirmed by experimental and theoretical studies. Consideration of even more weakly coordinating anions, using the work of Reed where experimental coordination capacity was determined via R3NH+⋯A− hydrogen bonding interactions, less coordinating anions such as BF4−, PF6−, B(C6F5)4− and carborane anions was carried out via theoretical calculations.45 These studies indicated that the [ArI]2+ fragment abstracts fluorine from the previous 3 anions and for the more robust perfluorinated carborane anion, the theoretically predicted oxidative capacity is “off the charts”. This led to the conclusion that ArI(NTf2)2 is likely, although not certainly, the boundary for oxidative capacity of ArIL2.
This begs the question of where the next frontier for increasing the oxidative capacity of organoiodine compounds might be. The answer could be increasing the oxidation state to I(V). Some recent entries in this area come from Zhdankin and Wengryniuk (Fig. 5). Zhdankin reported the synthesis and structural characterization of I(V) complex 2-iodooxybenzoic acid, which was stated to be the most powerful I(V) oxidant.46 Zhdankin reported the I(V) derivatives of Weiss' reagent as IO using bidentate N-ligands in 2002 and reactivity of these compounds has more recently been explored by Wengryniuk.47–49 Both of these compounds were found to be effective for the oxidation of deactivated or unfunctionalized organic species. Interestingly, both feature phenyl rings without protection from EAS processes. This suggests there is potential scope for increasing the oxidative capacity of these species until decomposition via EAS ensues, and then protection of the ring by incorporating a deactivating group might be possible. These I(V) derivatives of Weiss' reagent have not yet been structurally characterized by X-ray diffraction.
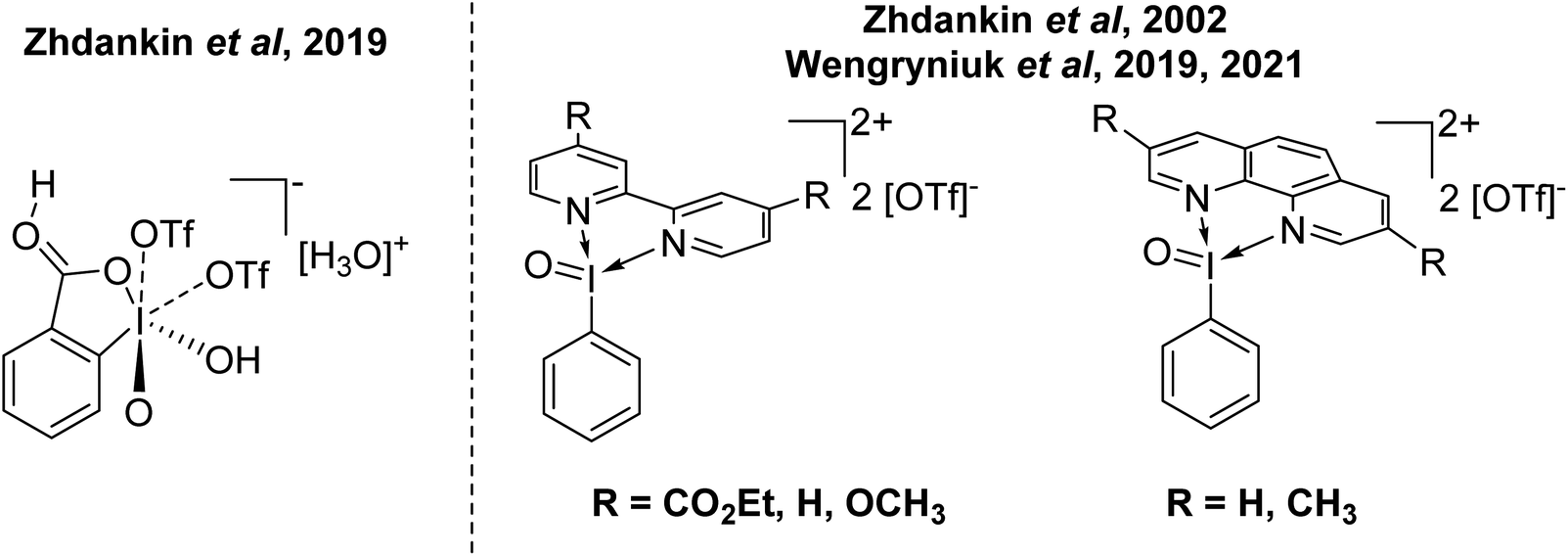 | ||
| Fig. 5 Recently reported hypervalent I(V) compounds possessing high oxidative capacity. | ||
Activation of PhICl2
Phenyliodine dichloride (PhICl2) is one of the most commonly used hypervalent iodine reagents. It is a convenient chlorinating agent, acting as a replacement for Cl2 gas. A major advantage of PhICl2 is that it is an easily weighable solid, and thus can be delivered stoichiometrically for small scale applications which is difficult in case of gaseous Cl2. For laboratory scale purposes PhICl2 also offers an improved safety profile over Cl2 gas.1,3,50 Disadvantages include poor atom economy (PhI is the byproduct and usually discarded), and that PhICl2 is not commercially available as it is unstable at room temperature with respect to decomposition into PhI and Cl2. It is also considered less reactive than Cl2.Synthesis is straightforward and can be done by reaction between PhI and Cl2, although this necessitates handling of Cl2. We find that the most straightforward method is the reaction of PhI, 30% H2O2 and 10 M HCl, where PhICl2 rapidly precipitates and can be easily isolated (Scheme 13). It can be stored at 4 °C as a solid for at least a year.51
 | ||
| Scheme 13 Synthesis of PhICl2via in situ generation of Cl2. | ||
Activation of PhICl2 has been pursued via several methods, including both Lewis bases and Lewis acids.52–55 We became interested in Lewis acid activation accidently, in the pursuit of a [PhI]2+, prior to our realization such a cation was likely unattainable due to deleterious EAS processes as discussed in the previous section. We attempted this by Cl− abstraction from PhICl2 using [Ag]2[B12Cl12]. The [B12Cl12]2− anion is a weakly coordinating anion related to the monoanionic carborane anions with similar robustness and coordinating properties, but is much more synthetically accessible.56
Reaction of PhICl2 with [Ag]2[B12Cl12] in an attempt to generate [PhI]2+ resulted instead in the generation of chlorinated iodobenzene (Scheme 14).57 It was subsequently found that this transformation also occurs using catalytic amounts of [Ag]2[B12Cl12]. Iodobenzene is not chlorinated by Cl2 in the absence of a strong Lewis acid catalyst, therefore it was rationalized that a more activated electrophilic chlorine source was in play and proposed to be transient [PhICl]+. A variety of arenes and other unsaturated molecules that do not react with PhICl2 could be chlorinated using catalytic loadings of [Ag]2[B12Cl12].57 A major limitation is that nothing less activated than PhI can be used as a substrate, as in these cases chlorination of PhI is the dominant product. When pyridine was used as a substrate a complex mixture was obtained. Based on the proposed intermediate of [PhICl]+, which contains a vacant coordination state, we rationalized that nucleophilic pyridine was attacking this site, forming [PhI(Pyr)Cl]+ and undergoing rapid decomposition from here. We had previously attempted to intentionally form this compound via reaction of PhICl2 with TMS-OTf and pyridine as part of our efforts in the examination of coordination chemistry of such species, however this reaction also lead to complex mixtures of products. Interestingly, an examination of the literature revealed that this cation has been proposed as an intermediate in chlorinations, going back decades.58 A series of more recent reports from the group of Murphy reported the activation of PhICl2 with pyridines towards the chlorination of several diazo compounds, with [PhI(Pyr)Cl]+ as the key activated intermediate (Scheme 15).54,59–62
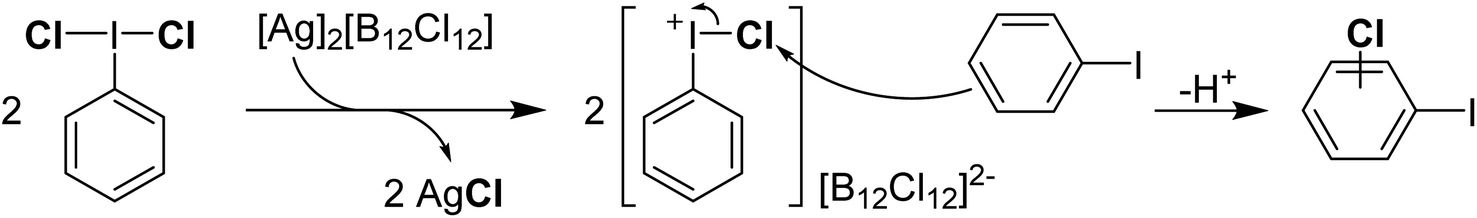 | ||
| Scheme 14 Mechanism of activation of PhICl2 using [Ag]2[B12Cl12] toward chlorination of iodobenzene via a proposed Ar–I+–Cl intermediate. | ||
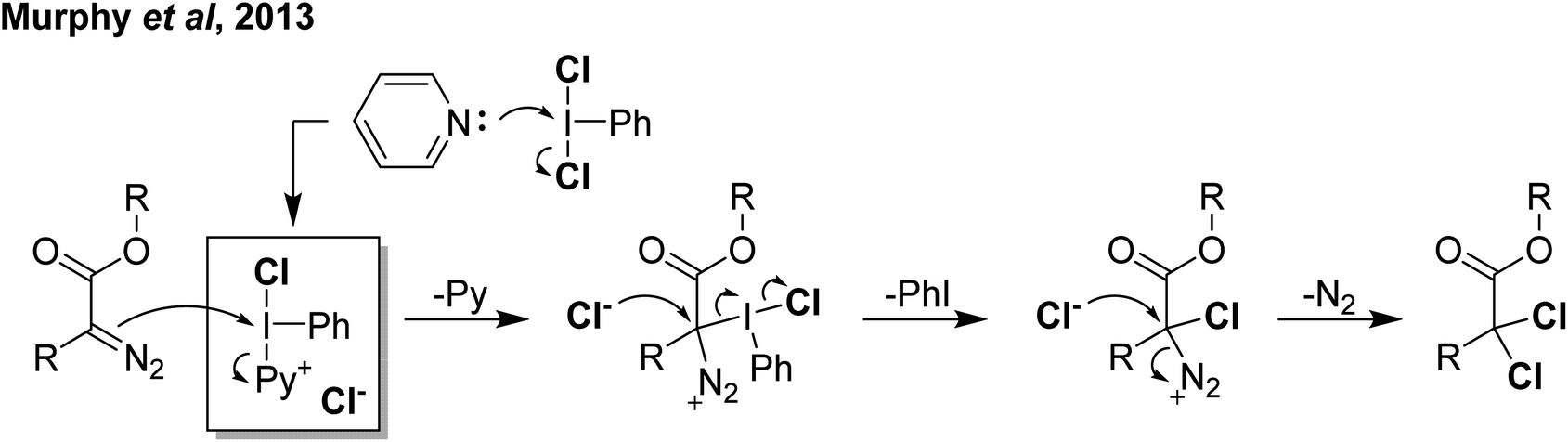 | ||
| Scheme 15 Mechanism proposed for activation of PhICl2 by pyridine via the highlighted intermediate to effect α,α-dichlorination of diazoacetates. | ||
We reacted PhICl2 with pyridine in D-solvents and observed only a very slight shift for the 1H resonances of the pyridine.51 Dissolving PhICl2 in neat pyridine resulted in decomposition at room temperature, however if the solution was held at −30 °C overnight crystal formation was observed (Fig. 6). X-ray diffraction studies of the crystals revealed them to be a weakly halogen bonded PhICl2⋯Pyr complex, with the pyridine trans to the I–C bond, completing a square planar geometry about iodine. The I–N contact was 275 pm, which can be compared with 222 pm for a strong bond in Weiss' reagent and 287 pm for a weaker bond in the diphenyliodonium(tetrafluoroborato)–pyridine complex.51,63
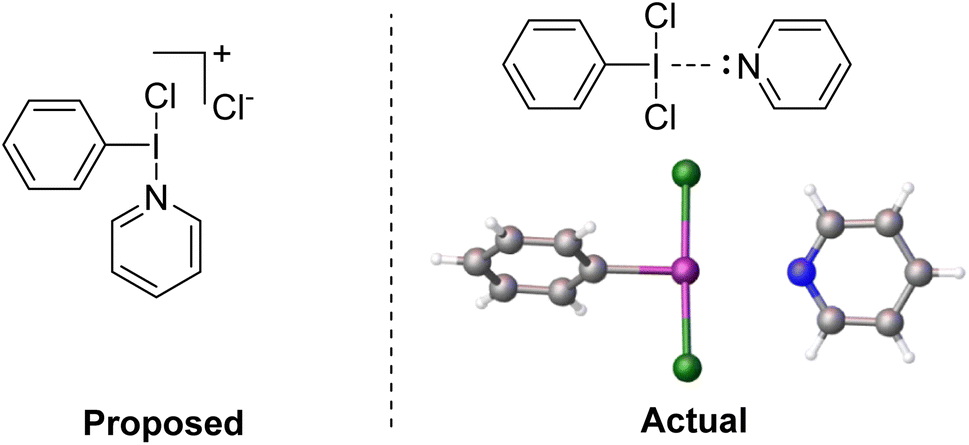 | ||
| Fig. 6 (Left) Proposed structure of the active intermediate for interaction of PhICl2 and pyridine. (Right) Actual structure of the intermediate confirmed by X-ray diffraction. | ||
While pyridine does not activate PhICl2 by the previously suggested method, there is clearly a catalytic activating effect. We chose to examine this via model electrophilic aromatic chlorination reactions, using anisole as an initial model substrate (Scheme 16).64 PhICl2 and anisole react slowly with only 2% conversion after 1 hour and ∼20% conversion after 24 hours to PhI and 4-chloroanisole. Addition of 20 mol% pyridine effected a conversion of 77% after 1 hour. However this reaction generates a molar equivalent of HCl, which readily reacts with pyridine to generate pyridinium chloride, tying up the lone pair. Therefore catalysis should cease, which is not the case. It was then found that chloride is an even more effective catalyst for the reaction, able to effect excellent conversions using [NBu4][Cl] with loadings under 1%. Chloride was found to also interact with the halogen bond accepting site trans to the C–I bond (Fig. 7).
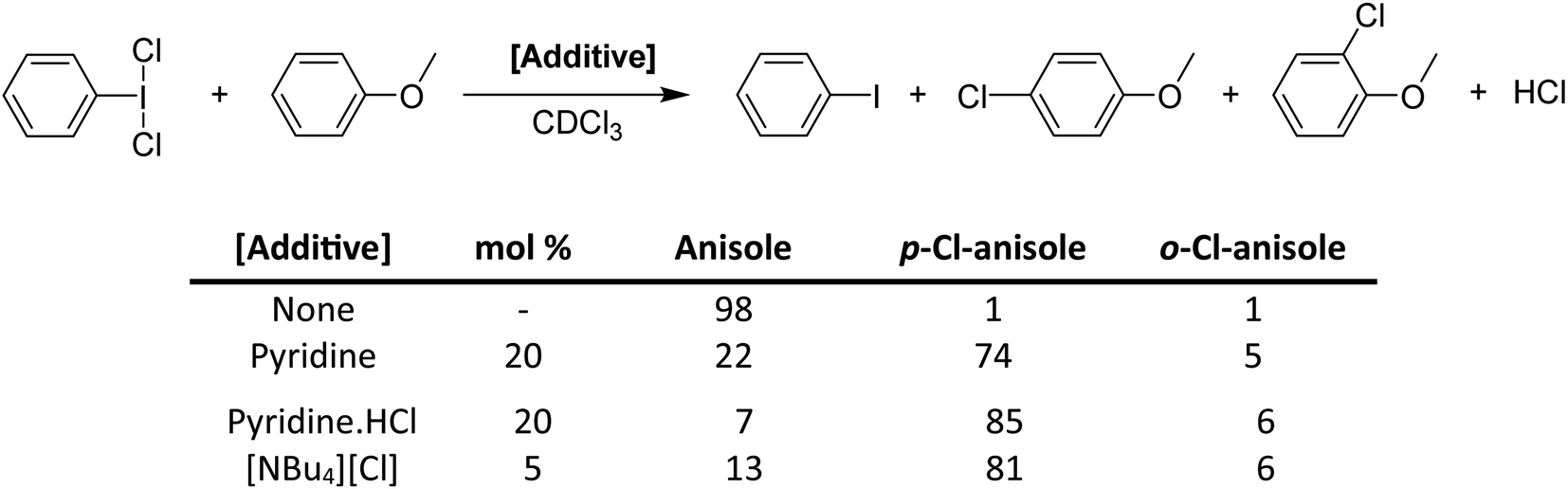 | ||
| Scheme 16 Activation of PhICl2 toward chlorination of anisole using various Lewis base additives, and the resulting conversion ratios at reaction time, t = 1 hour. | ||
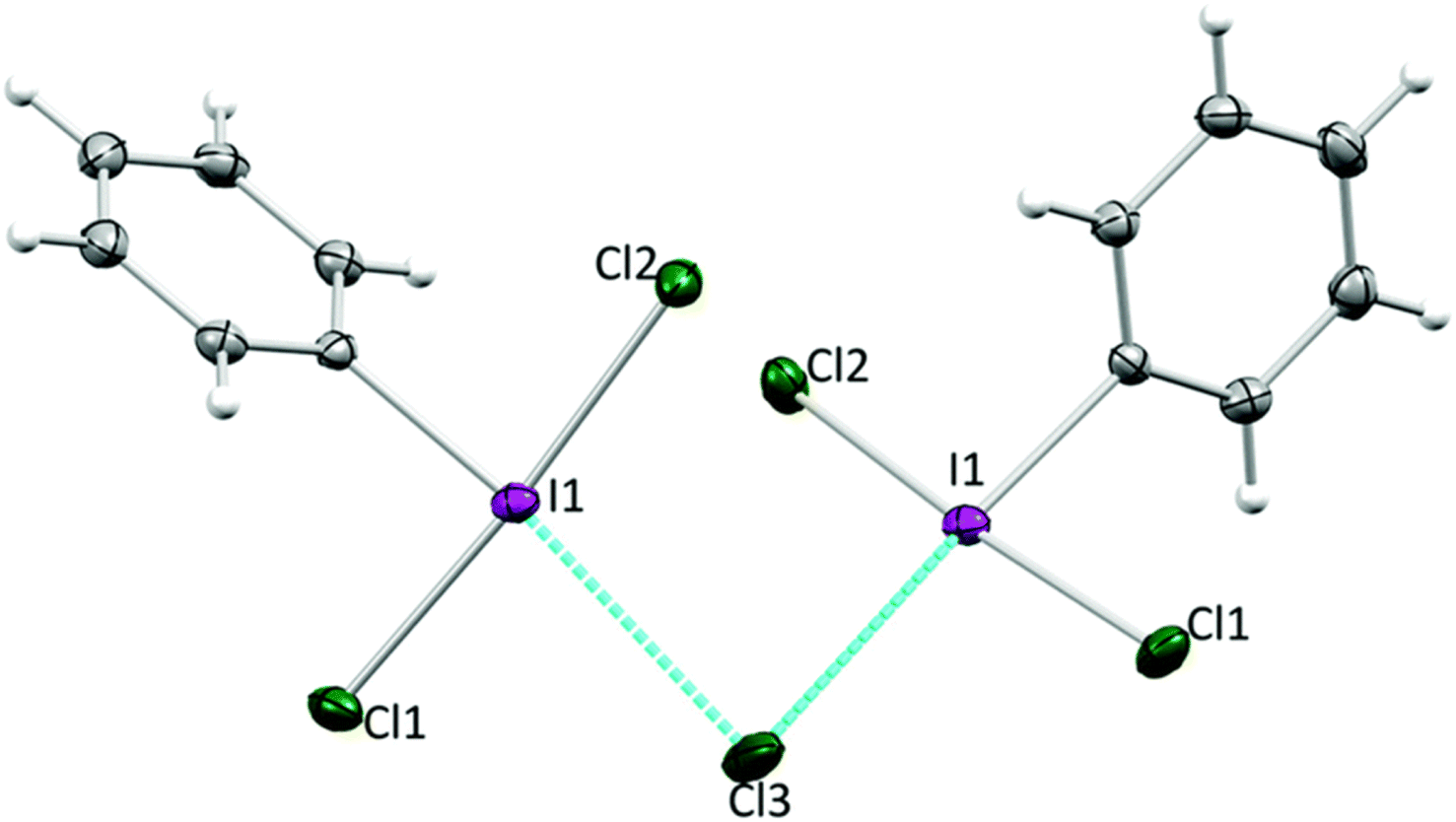 | ||
| Fig. 7 Interaction of PhICl2 and Cl− in the solid state confirmed by X-ray diffraction. | ||
Kinetic analysis using mesitylene as a substrate found the reaction to be first order in PhICl2 and [NBu4][Cl], but zero order in substrate.65 It was also determined that substrates that do not react with Cl2 itself also don't react with PhICl2 under chloride catalysis. Finally, Cl2 could be observed as a product in the absence of substrate. Therefore, it appears that release of Cl2 by the halogen bonding substrate is the reaction mechanism. For chloride we were unable to find a pathway using theoretical methods with energies consistent with experimental conditions. Ariafard and co-workers performed a theoretical study examining the pyridine catalysed reaction reported by Murphy on the chlorination of diazo compounds and also found that Cl2 release and then reaction with substrate is the likely pathway.66 These studies debunk one common notion about PhICl2, that it is less reactive than Cl2. While for these classes of reactions Cl2 is kinetically faster than PhICl2, thermodynamically any aryl chlorination that Cl2 can perform, PhICl2 can also achieve. We could not find a theoretical study on the mechanism of the reaction of PhI with Cl2 to give PhICl2, or the reverse reaction, the well-known tendency for PhICl2 to decompose into PhI and Cl2 in the literature. We have been attempting to find an energetically sensible transition state in silico for reactions in both directions for years without success and are aware of other computational groups also having worked on the problem with equal frustration. The nature of the transition state for this seemingly simple reaction remains an open question.
Having established that nucleophilic activation of PhICl2 is effective but a kinetic effect, we sought to revisit a thermodynamic activation of PhICl2. We had achieved this with Ag+ abstraction of chloride from PhICl2 but found a limiting factor for aryl substrates was self-electrophilic chlorination and no substrate less activated than PhI could be used. However as discussed previously in the pursuit of ArI(OTf)2 it was found incorporation of a nitro group shuts down self-electrophilic substitution reactions, which inspired us to explore the activation of p-NO2-C6H4-ICl2.67 This compound, as well as –CF3 substituted derivatives have been reported to be effective chlorinating agents, without formal activation explored.68–70
We envisioned that p-NO2-C6H4-I(OTf)(Cl) (Fig. 8) would be a stronger chlorinating agent due to increased electrophilicity. In 2019, Nagib reported that the mixed species PhI(OAc)(Cl) is an effective selective chlorinating agent for moderately activated arenes.71 Evans was able to observe this species in 1997 by NMR spectroscopy.72 In 2009 Lupton used the compound as a chlorinating agent for alkenes.73 This species can be synthesized via a number of methods from commercially available PhI(OAc)2, including reaction with HCl, TMS-Cl disproportionation with PhICl2 and chloride salts.
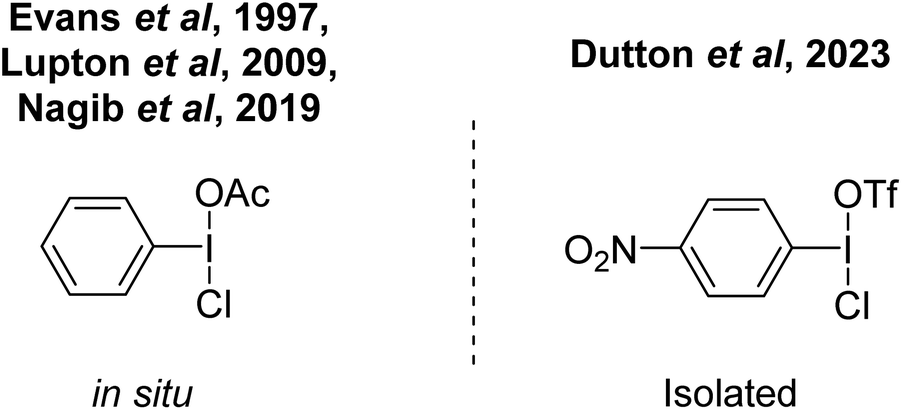 | ||
| Fig. 8 Asymmetric hypervalent I(III) compounds. | ||
We generated the mixed species p-NO2-C6H4-I(OTf)(Cl) by reaction of p-NO2-C6H4-ICl2 with NO2-C6H4-I(OTf)2, which could be isolated and crystalographically characterized and was found to contain the shortest I–Cl bond reported to date at 236 pm, which can be compared to 253 pm in PhICl2 (Fig. 9(a)).74 Calculations showed the chlorine atom in p-NO2-C6H4-I(OTf)(Cl) to have an NPA charge of −0.38 compared to −0.49 in p-NO2-C6H4-ICl2, suggestive of a more electrophilic chlorine.
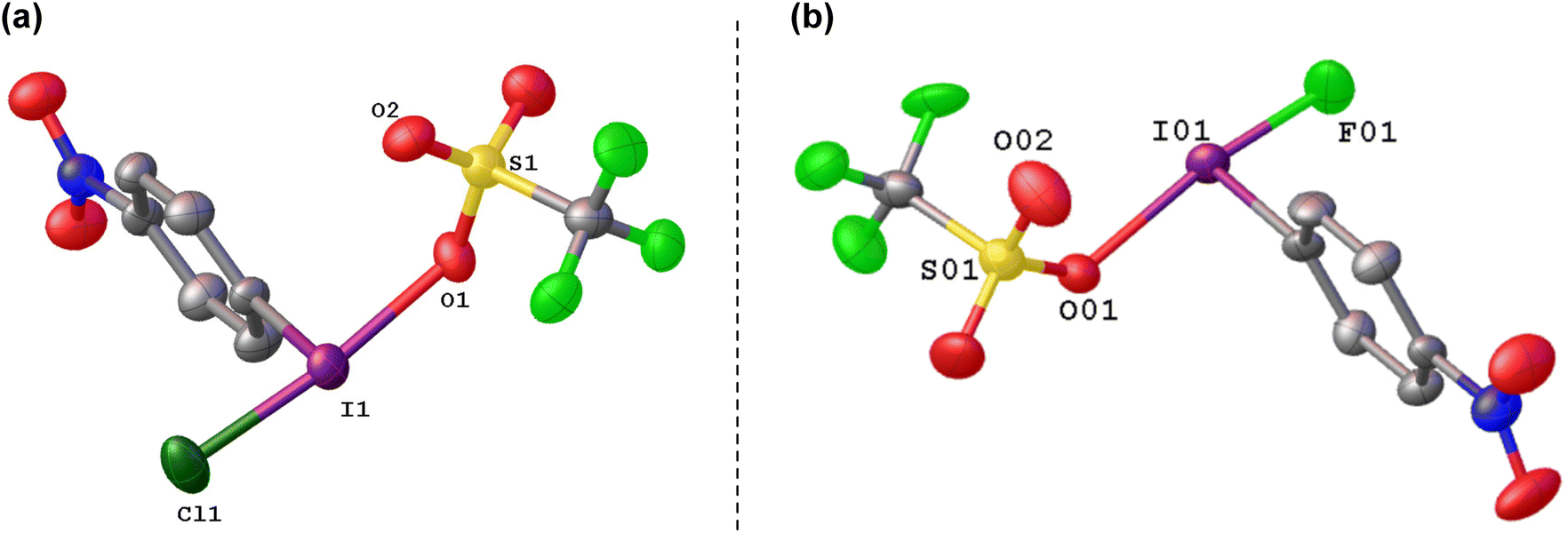 | ||
| Fig. 9 Crystal structures of (a) p-NO2-C6H4-I(OTf)(Cl), and (b) p-NO2-C6H4-I(OTf)(F). | ||
It was found that p-NO2-C6H4-I(OTf)(Cl) could be used for the chlorination of a variety of deactivated aryl substrates that are inert towards PhICl2 and Cl2 (Scheme 17). These reactions also produce HOTf as a product and since p-NO2-C6H4-I(OTf)(Cl) can be generated from HOTf and p-NO2-C6H4-ICl2 can be generated from p-NO2-C6H4-I using Cl2, it was rationalized that the system could be used with catalytic loadings of p-NO2-C6H4-I and Cl2 as the chlorine source. This was found to be effective with catalytic loadings lower than 1% of p-NO2-C6H4-I and HOTf. This is potentially a significant advance as the cost of organoiodine is an issue for larger scale use.
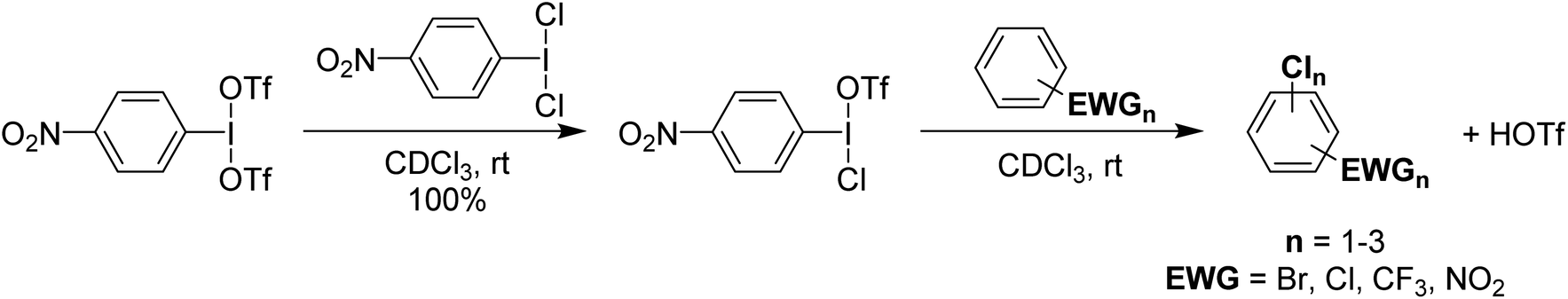 | ||
| Scheme 17 Synthesis of p-NO2-C6H4-I(OTf)(Cl) and its subsequent use in electrophilic chlorination of deactivated aromatics, yielding up to two chlorinations in some species. | ||
Activation of ArIF2
Attempting to activate ArIF2 for fluorinations in the same manner gives a vastly different result. Stang and co-workers invoked PhI(OTf)(F) as a low temperature intermediate in the reaction of PhI with Xe(OTf)(F) and found it to be an efficient source of [PhI]+.37 The proposed PhI(OTf)(F) could not be isolated nor observed. In our efforts to generate PhI(OTf)2 from PhIF2 with TMS-OTf, we also found iodonium salt formation and incomplete consumption of TMS-OTf as well as generation of TMS-F, suggesting that PhI(OTf)(F) reacts with itself.36 However, reaction of p-NO2-C6H4-IF2 with one equivalent of TMS-OTf was a clean reaction and gave p-NO2-C6H4-I(OTf)(F) which could be observed in solution, isolated and crystallographically characterized (Fig. 9), highlighting the effectiveness of –p-NO2 substitution in stabilizing electron deficient aryl organoiodine compounds (Scheme 18).74 Reaction with anisole and toluene, with a target of C–H to C–F aryl fluorination did not result in fluorination, but rather formation of [Ar–I–Ar′]+ iodonium cations. The differing reactivity from p-NO2-C6H4-I(OTf)(Cl) was explored theoretically and it was found that the partial NPA positive charge on iodine is much higher in p-NO2-C6H4-I(OTf)(F) than p-NO2-C6H4-I(OTf)(Cl), with a corresponding more negative charge on the fluoride as compared to the chloride. Ergo p-NO2-C6H4-I(OTf)(Cl) is a source of electrophilic Cl, while p-NO2-C6H4-I(OTf)(F) acts as a source of electrophilic [p-NO2-C6H4-I]+.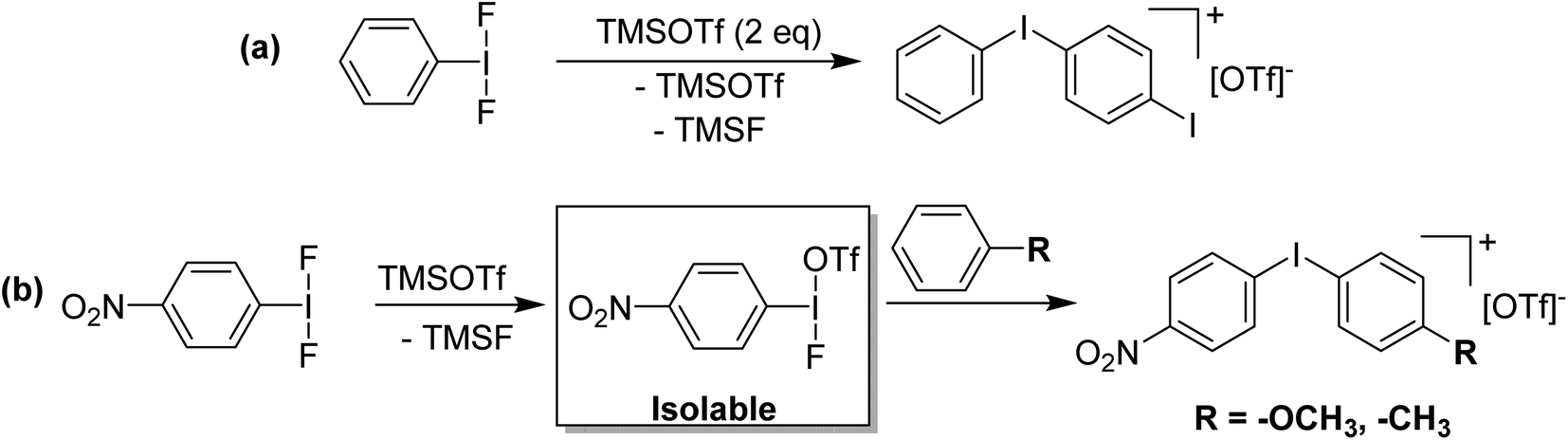 | ||
| Scheme 18 (a) Reaction of PhIF2 with TMSOTf (2 eq.) generating the corresponding diaryliodonium triflate salt. (b) Synthesis of p-NO2-C6H4-I(OTf)(F) via reaction of p-NO2-C6H4-IF2 with TMSOTf and subsequent reaction with activated arenes to give corresponding diaryliodonium triflate salts. | ||
The current effective methods of activating ArIF2 for fluorination reactions is via either BF3 or Brønsted acid activation. These methods have been reported for a variety of transformations including α,α-difluorination of diazoacetate derivatives, cyclisation of allenes, selective fluorination of β-keto esters, geminal difluorination of diazo arenes and selective dearomatization of phenols.75–84 In both cases the proposed method of activation is interaction of BF3 or the A–H with a fluorine atom along the F–I–F bond axis, without formal extraction, which allows for the other I–F to become activated, without rendering the iodine sufficiently electrophilic to result in iodonium cation formation as a competing pathway.
Does electrophilic bromine as I–Br exist?
In contrast to PhIF2 and PhICl2, the literature on PhIBr2 as a brominating agent is limited, likely attributed to the relatively higher stability of bromine (Br2) and weak I–Br bond strength, which favours PhI and Br2 over PhIBr2. Nonetheless there have been several reports dating back over 100 years and with new reports still coming out describing PhIBr2 and its use as a brominating agent.The earliest documentation of synthesis of PhIBr2 dates back a century ago, where its generation was purported through the reaction of PhI with Br2 in petroleum ethers.85 This reaction produced a solid compound under low-temperature conditions. Another report from 1905 involves the synthesis of PhIBr2 by reacting PhI with Br2 in hexanes at a temperature of −5 °C (Scheme 19).86 This methodology has persisted as a reference point for various bromination reactions targeting specific substrates.87,88
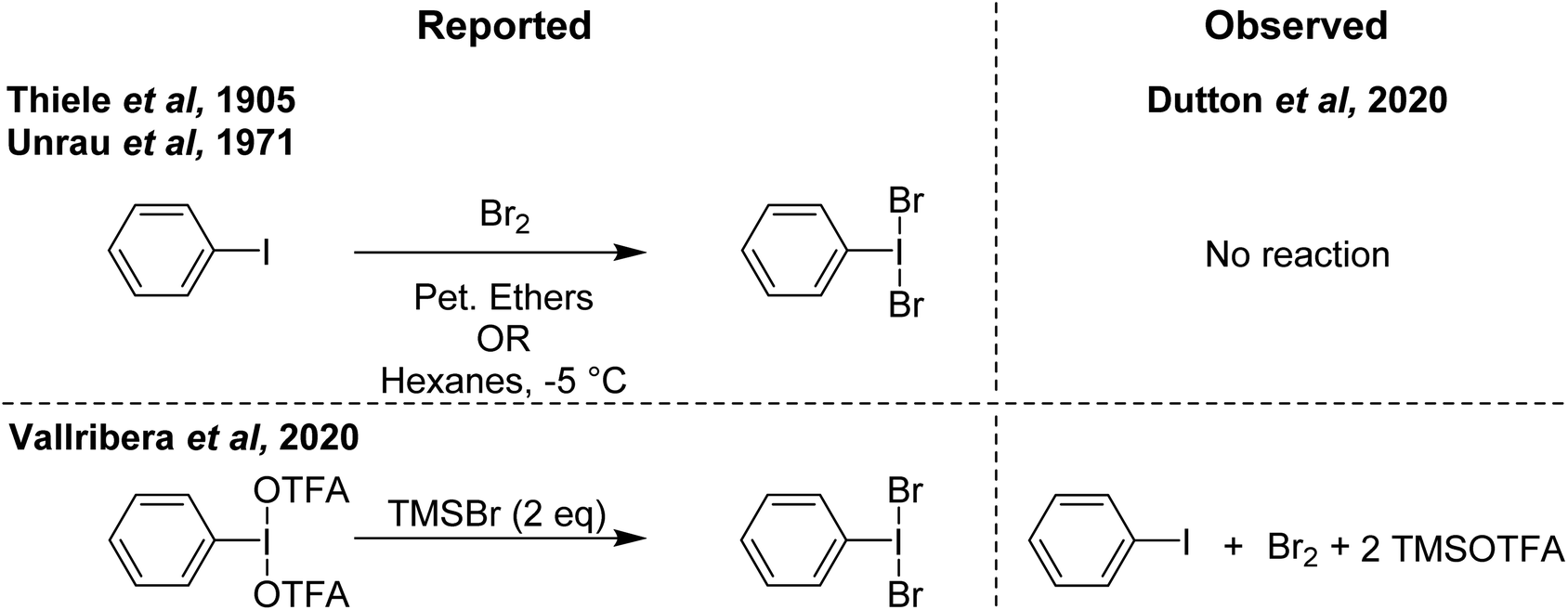 | ||
| Scheme 19 Reported syntheses of PhIBr2 (left) and observed results on attempted replication (right). | ||
More recently in 2020, Cossío and Vallribera proposed an in situ generation of PhIBr2 through the reaction of PhI(OTFA)2 and TMSBr (Scheme 19).89 This freshly formed PhIBr2 was subsequently employed in the bromination of various aryl substrates, with reactions occurring for activated substrates over the course of hours. We had long been interested in using PhIBr2 and this appeared to be a good route to the molecule, however we found that the reaction of PhI(OTFA)2 and 2 equivalents of TMS-Br resulted in immediate generation of Br2 and PhI.90 No spectroscopic evidence was found for detectable amounts of a species consistent with PhIBr2. Evans had previously reported that while PhI(OAc)(Cl) can be observed, reaction of PhI(OAc)2 and TMS-Br resulted in immediate formation of Br2.72,91 Further, it was found that Br2 is capable of performing the reported aryl brominations attributed to PhIBr2 on the timeline of minutes rather than hours (Scheme 20).
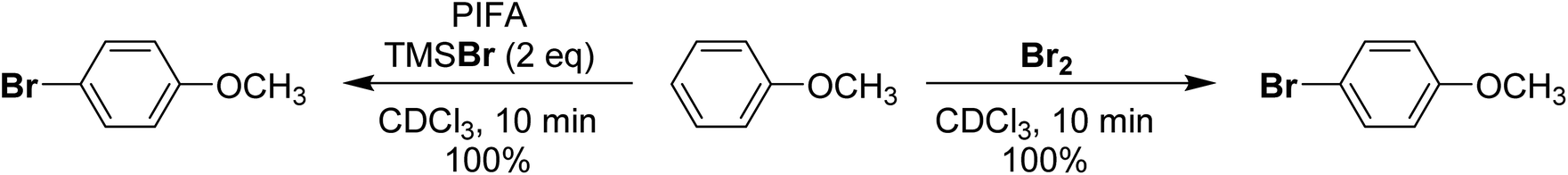 | ||
| Scheme 20 Comparison of bromination of anisole using PhI(OTFA)2 + TMSBr (2 eq.) or Br2 as brominating agents. | ||
Computational studies showed that Br2 and PhI lie far below PhIBr2 in energy, and no feasible pathway between PhIBr2 and PhI/Br2 could be modelled. Another 2020 paper described the detection of PhIBr2, synthesized from PhI(OAc)2 and [NBu4][Br] as an [ONa]− adduct by mass spectrometry.92 The origin of the [ONa]− fragment notwithstanding, examination of their data and repeating the work showed the proposed signal to be one mass unit too light for [PhIBr2-ONa]−, with the lightest isotope at 400 rather than 401 mass units. The isotope pattern also did not exactly match what would be expected. The mass and isotope pattern however did exactly match [NBu4Br2]− and repeating the work with tetra n-hexyl ammonium [N(C6H13)4][Br]confirmed such a cluster was indeed what was being observed. Based on our findings, we do not believe PhIBr2 can be observed, although we cannot rule out its existence as a short lived intermediate. However, we suggest that a mixture of PhI(OTFA)2 or PhI(OAc)2 and 2 equivalents of TMS-Br could serve as a practical source of Br2 in scenarios where handling liquid Br2 is undesirable, or for the generation of very small amounts of Br2.
This invites the question, does I–Br as an electrophilic brominating agent exist? IBr is a well-known interhalogen compound, but acts as a source of electrophilic [I]+.93 A class of organo I(III) compound that contains an I–Br bond is well established in the bromoiodinanes shown in Fig. 10.94 These were first reported in the 1979 and structurally characterized by X-ray crystallography in 2006 by Braddock and co-workers.94,95 The I–Br bonds range from 259 to 269 pm in the compounds. They were found to be competent brominating agents, albeit sluggish, with highly activated anisole requiring 15 hours at room temperature for full conversion to 4-bromoanisole. At the current time, we believe that this is the closest example for a bonified, although weak, system containing an I–Br bond for electrophilic bromination.
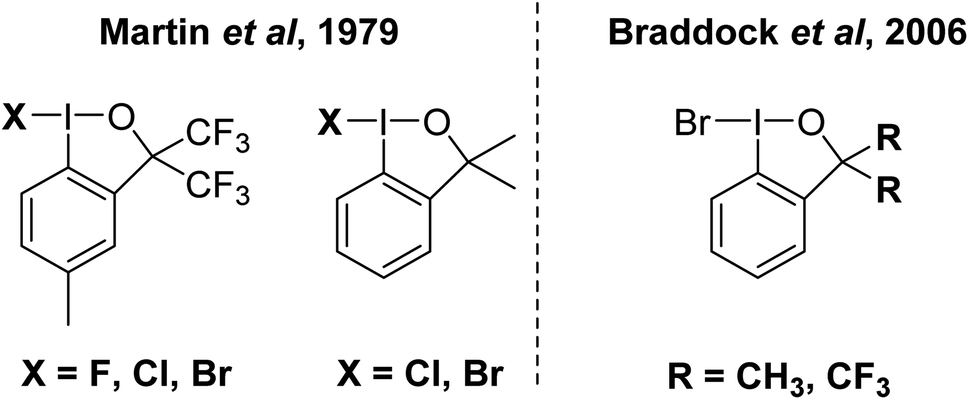 | ||
| Fig. 10 Reported examples of organoiodanes containing an I–Br bond. | ||
For a method using I(III) in the rapid bromination of deactivated arenes we recently found that p-NO2-C6H4-IL2 systems, where L is triflate or trifluoroacetate in concert with Br2 enables the bromination of unreactive species including –CF3 and –p-NO2 substituted arenes (Scheme 21).96 The active intermediate is unknown at present but appears to arise from Lewis acidic activation of Br2 by I(III) and then reoxidation of the HBr by-product by I(III), which is supported by the bromination of 2 equivalents of substrate per Br2 and I(III) equivalent, achieving full consumption of the bromine. The advantage is brominations of deactivated arenes can be carried out rapidly at room temperature in organic solvent, whereas other methods for deactivated species require harsh media such as H2SO4 solvent.97,98 The disadvantage of the method is relatively poor selectivity, with most substrates being brominated at multiple sites.
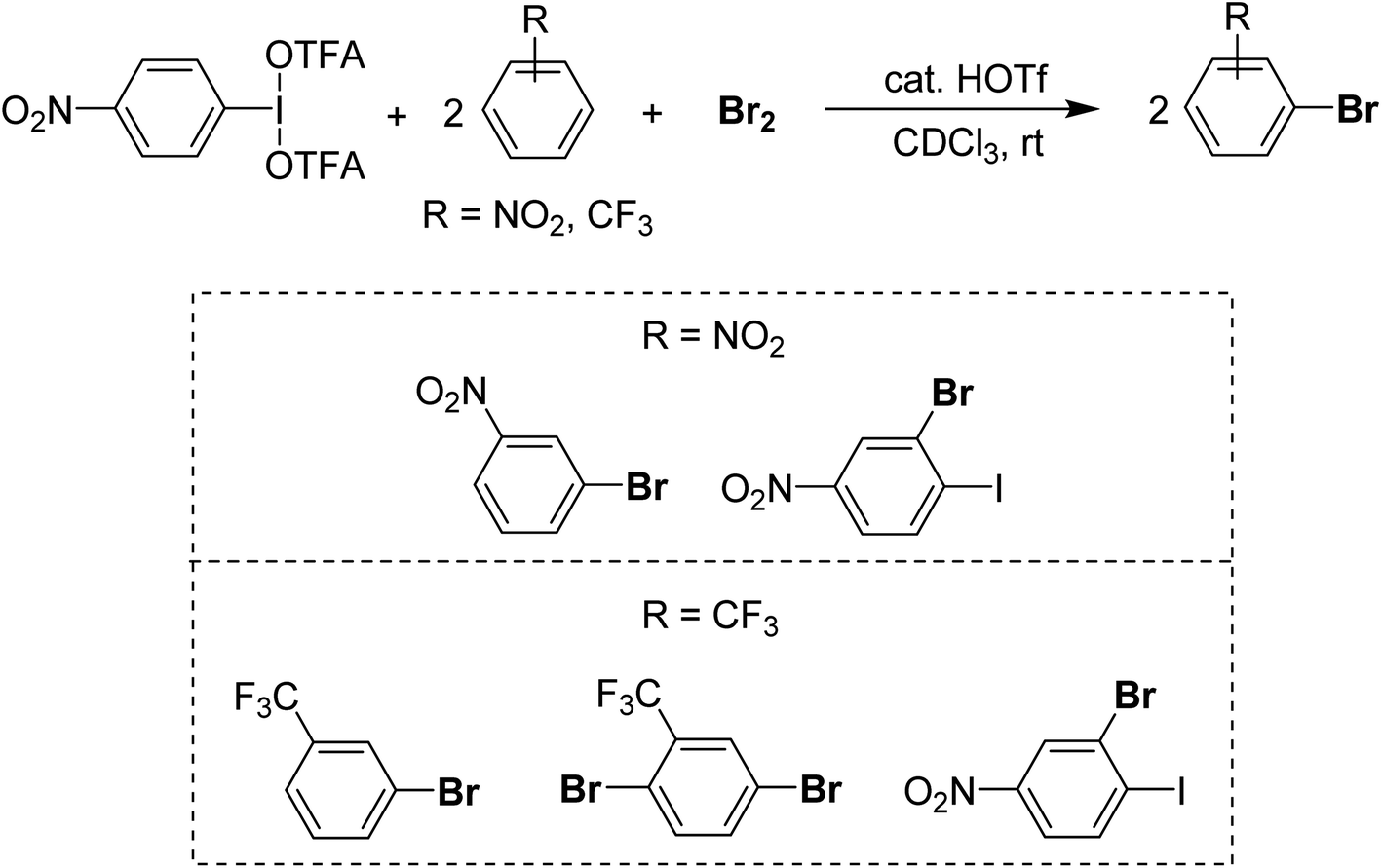 | ||
| Scheme 21 Activation of Br2 and Br−via I(III) resulting in delivery of two equivalents of Br to an electron-poor substrate. | ||
Conclusions
A decade of work on ArIL2, from the perspective of inorganic coordination chemistry has taught us two main lessons, that we hope are also useful for the wider community. The foremost is that it is easy to misidentify hypervalent iodine compounds. Such misidentification does not always affect the organic transformations the iodine is being used for, but not knowing what species one is using is not ideal. Unfortunately, there is no good spectroscopic handle for what is happening at iodine. This often leads to confusion, as observed NMR spectra can be consistent with both actual and purported (but incorrect) compounds. In the absence of an X-ray structure very careful examination of NMR data is needed to avoid misidentification. In our experience, we have found that ArIL2 species, even cationic ones, are not amenable to interrogation by mass spectrometry, with the intact molecule very rarely surviving the mass spectrometry experiment. Additionally, a useful feature of mass spectrometry, characteristic isotope patterns, is not available as there is only one naturally occurring isotope of iodine (127I).The second lesson is that the Achilles Heel for isolation of increasingly electron poor ArIL2 compounds is decomposition via EAS to give [Ar–I–Ar]+ iodonium cations. This can however be suppressed very effectively by the incorporation of deactivating electron withdrawing groups onto the aryl ring.
Author contributions
All authors were involved in the conceptual design and writing of the review. Tania and M. Sceney generated all of the figures.Conflicts of interest
There are no conflicts to declare.References
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS.
- V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds, John Wiley & Sons, 2013 Search PubMed.
- F. C. Sousa e Silva, A. F. Tierno and S. E. Wengryniuk, Molecules, 2017, 22, 780 CrossRef.
- J. Charpentier, N. Fruh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS.
- C. A. Montgomery and G. K. Murphy, Beilstein J. Org. Chem., 2023, 19, 1171–1190 CrossRef CAS PubMed.
- M. R. Radzhabov, A. B. Sheremetev and T. S. Pivina, New J. Chem., 2020, 44, 7051–7057 RSC.
- R. Weiss and J. Seubert, Angew. Chem., Int. Ed., 1994, 33, 891–893 CrossRef.
- V. V. Zhdankin, O. Maydanovych, J. Herschbach, J. Bruno, E. D. Matveeva and N. S. Zefirov, Tetrahedron Lett., 2002, 43, 2359–2361 CrossRef CAS.
- V. V. Zhdankin, O. Maydanovych, J. Herschbach, J. Bruno, E. D. Matveeva and N. S. Zefirov, J. Org. Chem., 2003, 68, 1018–1023 CrossRef CAS PubMed.
- B. Hoblos and S. E. Wengryniuk, Org. Synth., 2021, 98, 391–406 CrossRef PubMed.
- A. Vazquez-Lopez, J. E. Allen and S. E. Wengryniuk, Adv. Synth. Catal., 2023, 365, 2697–2702 CrossRef CAS.
- T. P. Pell, S. A. Couchman, S. Ibrahim, D. J. D. Wilson, B. J. Smith, P. J. Barnard and J. L. Dutton, Inorg. Chem., 2012, 51, 13034–13040 CrossRef CAS PubMed.
- E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639–642 CrossRef CAS PubMed.
- R. Corbo, D. C. Georgiou, D. J. D. Wilson and J. L. Dutton, Inorg. Chem., 2014, 53, 1690–1698 CrossRef CAS.
- R. Corbo, T. P. Pell, B. D. Stringer, C. F. Hogan, D. J. D. Wilson, P. J. Barnard and J. L. Dutton, J. Am. Chem. Soc., 2014, 136, 12415–12421 CrossRef CAS.
- F. Kniep, S. M. Walter, E. Herdtweck and S. M. Huber, Chem.–Eur. J., 2012, 18, 1306–1310 CrossRef CAS.
- B. T. Kelley, J. C. Walters and S. E. Wengryniuk, Org. Lett., 2016, 18, 1896–1899 CrossRef CAS PubMed.
- M. Mikhael, S. A. Adler and S. E. Wengryniuk, Org. Lett., 2019, 21, 5889–5893 CrossRef CAS PubMed.
- E. I. Carrera, T. M. McCormick, M. J. Kapp, A. J. Lough and D. S. Seferos, Inorg. Chem., 2013, 52, 13779–13790 CrossRef CAS PubMed.
- E. I. Carrera and D. S. Seferos, Dalton Trans., 2015, 44, 2092–2096 RSC.
- S. Ye, V. Lotocki, H. Xu and D. S. Seferos, Chem. Soc. Rev., 2022, 51, 6442–6474 RSC.
- A. Aprile, K. J. Iversen, D. J. D. Wilson and J. L. Dutton, Inorg. Chem., 2015, 54, 4934–4939 CrossRef CAS.
- N. S. Zefirov, S. O. Safronov, A. A. Kaznacheev and V. V. Zhdankin, Org. Chem., 1989, 25, 1807–1808 CAS.
- T. Wirth and U. Farid, Angew. Chem., Int. Ed., 2012, 51, 3462–3465 CrossRef.
- K. E. Lutz and R. J. Thomson, Angew. Chem., Int. Ed., 2011, 50, 4437–4440 CrossRef CAS PubMed.
- J. Tian, F. Luo, Q. Zhang, Y. Liang, D. Li, Y. Zhan, L. Kong, Z. Wang and B. Peng, J. Am. Chem. Soc., 2020, 142, 6884–6890 CrossRef CAS.
- S. Izquierdo, S. Essafi, I. Del Rosal, P. Vidossich, R. Pleixats, A. Vallribera, G. Ujaque, A. Lledos and A. Shafir, J. Am. Chem. Soc., 2016, 138, 12747–12750 CrossRef CAS.
- A. Bakro, L. Sharp-Bucknall, T. B. Poynder, J. K. Clegg, D. J. D. Wilson and J. L. Dutton, Chem. Commun., 2021, 57, 12163–12166 RSC.
- K. Kiyokawa, K. Takemoto, S. Yahata, T. Kojima and S. Minakata, Synthesis, 2017, 49, 2907–2912 CrossRef CAS.
- J. Tian, F. Luo, C. Zhang, X. Huang, Y. Zhang, L. Zhang, L. Kong, X. Hu, Z. Wang and B. Peng, Angew. Chem., Int. Ed., 2018, 57, 9078–9082 CrossRef CAS.
- B. L. Tóth, F. Béke, O. Egyed, A. Bényei, A. Stirling and Z. Novák, ACS Omega, 2019, 4, 9188–9197 CrossRef.
- X. Huang, Y. Zhang, C. Zhang, L. Zhang, Y. Xu, L. Kong, Z. Wang and B. Peng, Angew. Chem., Int. Ed., 2019, 58, 5956–5961 CrossRef CAS.
- X. Shao, X. Wang, T. Yang, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 3457–3460 CrossRef CAS PubMed.
- E. V. Vinogradova, P. Müller and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 3125–3128 CrossRef CAS PubMed.
- Tania, S. D. Houston, L. Sharp-Bucknall, T. B. Poynder, M. Albayer and J. L. Dutton, Chem.–Eur. J., 2020, 26, 15863–15866 CrossRef CAS.
- L. Sharp-Bucknall, Tania and J. L. Dutton, Angew. Chem., Int. Ed., 2022, 61, e202212380 CrossRef CAS.
- T. M. Kasumov, N. S. Pirguliyev, V. K. Brel, Y. K. Grishin, N. S. Zefirov and P. J. Stang, Tetrahedron, 1997, 53, 13139–13148 CrossRef CAS.
- Tania, M. Sceney, L. Barwise, J. Bennetts, K. F. White and J. L. Dutton, Dalton Trans., 2023, 52, 15866–15870 RSC.
- N. Gunsalus, A. Koppaka, S. S. Chen, S. H. Park, B. G. Hashiguchi, D. H. Ess and R. A. Periana, Organometallics, 2023, 42, 1505–1512 CrossRef CAS.
- B. M. Trost and P. J. Metzner, J. Am. Chem. Soc., 1980, 102, 3572–3577 CrossRef CAS.
- T. J. Williams, A. J. Caffyn, N. Hazari, P. F. Oblad, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2008, 130, 2418–2419 CrossRef CAS.
- S. R. Kandukuri and M. Oestreich, J. Org. Chem., 2012, 77, 8750–8755 CrossRef CAS PubMed.
- A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 3454–3457 CrossRef CAS PubMed.
- L. Barwise, J. Bennetts, K. F. White and J. L. Dutton, Chem. Commun., 2023, 59, 13340–13343 RSC.
- C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS PubMed.
- M. S. Yusubov, N. S. Soldatova, P. S. Postnikov, R. R. Valiev, A. Yoshimura, T. Wirth, V. N. Nemykin and V. V. Zhdankin, Chem. Commun., 2019, 55, 7760–7763 RSC.
- V. V. Zhdankin, A. Y. Koposov and N. V. Yashin, Tetrahedron Lett., 2002, 43, 5735–5737 CrossRef CAS.
- X. Xiao, N. S. Greenwood and S. E. Wengryniuk, Angew. Chem., Int. Ed., 2019, 58, 16181–16187 CrossRef CAS PubMed.
- X. Xiao, J. M. Roth, N. S. Greenwood, M. K. Velopolcek, J. Aguirre, M. Jalali, A. Ariafard and S. E. Wengryniuk, J. Org. Chem., 2021, 86, 6566–6576 CrossRef CAS PubMed.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS.
- T. B. Poynder, A. I. C. Orué, Tania, L. Sharp-Bucknall, M. T. Flynn, D. J. D. Wilson, K. S. A. Arachchige, J. K. Clegg and J. L. Dutton, Chem. Commun., 2021, 57, 4970–4973 RSC.
- J. Wicha, A. Zarecki and M. Kocór, Tetrahedron Lett., 1973, 14, 3635–3638 CrossRef.
- K. B. Wiberg, W. E. Pratt and M. G. Matturro, J. Org. Chem., 1982, 47, 2720–2722 CrossRef CAS.
- J. Tao, R. Tran and G. K. Murphy, J. Am. Chem. Soc., 2013, 135, 16312–16315 CrossRef CAS PubMed.
- M. S. Carle, G. K. Shimokura and G. K. Murphy, Eur. J. Org Chem., 2016, 2016, 3930–3933 CrossRef CAS.
- V. Geis, K. Guttsche, C. Knapp, H. Scherer and R. Uzun, Dalton Trans., 2009, 2687–2694 RSC.
- T. B. Poynder, S. D. Houston and J. L. Dutton, Chem.–Eur. J., 2021, 2021, 2788–2791 CAS.
- A. Varvoglis, The Organic Chemistry of Polycoordinated Iodine, Wiley VCH, 1992 Search PubMed.
- G. K. Murphy, F. Z. Abbas and A. V. Poulton, Adv. Synth. Catal., 2014, 356, 2919–2923 CrossRef CAS.
- K. E. Coffey, R. Moreira, F. Z. Abbas and G. K. Murphy, Org. Biomol. Chem., 2015, 13, 682–685 RSC.
- K. E. Coffey and G. K. Murphy, Synlett, 2015, 26, 1003–1007 CrossRef CAS.
- Z. Zhao, K. G. Kulkarni and G. K. Murphy, Adv. Synth. Catal., 2017, 359, 2222–2228 CrossRef CAS.
- M. Ochiai, T. Suefuji, M. Shiro and K. Yamaguchi, Heterocycles, 2006, 67, 391 CrossRef.
- Tania, T. B. Poynder, A. Kaur, L. Barwise, S. D. Houston, A. J. Nair, J. K. Clegg, D. J. D. Wilson and J. L. Dutton, Dalton Trans., 2021, 50, 11986–11991 RSC.
- B. A. Davis, Tania and J. L. Dutton, Dalton Trans., 2022, 51, 12384–12388 RSC.
- K. Farshadfar and A. Ariafard, Chem. Commun., 2021, 57, 9108–9111 RSC.
- X.-F. Zhao and C. Zhang, Synthesis, 2007, 551–557 CAS.
- J. Tao and G. K. Murphy, Synthesis, 2019, 51, 3055–3059 CrossRef CAS.
- J. C. Sarie, J. Neufeld, C. G. Daniliuc and R. Gilmour, Synthesis, 2019, 51, 4408–4416 CrossRef CAS.
- P. F. Dai and H. Xu, Eur. J. Org Chem., 2022, e202200779 CrossRef CAS.
- S. C. Fosu, C. M. Hambira, A. D. Chen, J. R. Fuchs and D. A. Nagib, Chem, 2019, 5, 417–428 CAS.
- P. A. Evans and T. A. Brandt, J. Org. Chem., 1997, 62, 5321–5326 CrossRef CAS.
- M. Ngatimin, C. J. Gartshore, J. P. Kindler, S. Naidu and D. W. Lupton, Tetrahedron Lett., 2009, 50, 6008–6011 CrossRef CAS.
- L. Sharp-Bucknall, M. Sceney, K. F. White and J. L. Dutton, Dalton Trans., 2023, 52, 3358–3370 RSC.
- N. Zefirov, A. Koz'min, T. Kasumov, K. Potekhin, V. Sorokin, V. Brel, E. Abramkin, Y. T. Struchkov, V. Zhdankin and P. Stang, J. Org. Chem., 1992, 57, 2433–2437 CrossRef CAS.
- N. Yoneda, J. Fluorine Chem., 2004, 125, 7–17 CrossRef CAS.
- M. Ochiai, M. Hirobe, A. Yoshimura, Y. Nishi, K. Miyamoto and M. Shiro, Org. Lett., 2007, 9, 3335–3338 CrossRef CAS PubMed.
- E. Emer, J. Twilton, M. Tredwell, S. Calderwood, T. L. Collier, B. Liegault, M. Taillefer and V. Gouverneur, Org. Lett., 2014, 16, 6004–6007 CrossRef CAS PubMed.
- G. S. Sinclair, R. Tran, J. Tao, W. S. Hopkins and G. K. Murphy, Eur. J. Org Chem., 2016, 2016, 4603–4606 CrossRef CAS.
- I. n. G. Molnár, C. Thiehoff, M. C. Holland and R. Gilmour, ACS Catal., 2016, 6, 7167–7173 CrossRef.
- F. Scheidt, M. Schäfer, J. Sarie, C. Daniliuc, J. Molloy and R. Gilmour, Angew. Chem., Int. Ed., 2018, 57, 16431–16435 CrossRef CAS PubMed.
- J. Häfliger, O. O. Sokolova, M. Lenz, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2022, 61, e202205277 CrossRef PubMed.
- T. Stünkel, K. Siebold, D. Okumatsu, K. Murata, L. Ruyet, C. G. Daniliuc and R. Gilmour, Chem. Sci., 2023, 14, 13574–13580 RSC.
- Y. J. Yu, M. Schäfer, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2023, 62, e202214906 CrossRef CAS PubMed.
- J. Thiele and W. Peter, Chem. Ber., 1905, 38, 2842–2846 CrossRef CAS.
- M. Fryberg, A. Oehlschlager and A. Unrau, Tetrahedron, 1971, 27, 1261–1274 CrossRef CAS.
- J. G. Cui, L. M. Zeng, J. Y. Su and W. G. Lu, Steroids, 2001, 66, 33–38 CrossRef CAS PubMed.
- J. G. Cui, C. W. Lin, L. M. Zeng and J. Y. Su, Steroids, 2002, 67, 1015–1019 CrossRef CAS PubMed.
- A. Granados, A. Shafir, A. Arrieta, F. P. Cossío and A. Vallribera, J. Org. Chem., 2020, 85, 2142–2150 CrossRef CAS.
- Tania, A. Molino, L. Sharp-Bucknall, D. J. D. Wilson and J. L. Dutton, Org. Biomol. Chem., 2022, 20, 8454–8460 RSC.
- P. A. Evans and T. A. Brandt, Tetrahedron Lett., 1996, 37, 6443–6446 CrossRef CAS.
- A. Watanabe, K. Koyamada, K. Miyamoto, J. Kanazawa and M. Uchiyama, Org. Process Res. Dev., 2020, 24, 1328–1334 CrossRef CAS.
- S. Yannacone, V. Oliveira, N. Verma and E. Kraka, Inorganics, 2019, 7, 47 CrossRef CAS.
- D. C. Braddock, G. Cansell, S. A. Hermitage and A. J. White, Chem. Commun., 2006, 1442–1444 RSC.
- R. L. Amey and J. C. Martin, J. Org. Chem., 1979, 44, 1779–1784 CrossRef CAS.
- L. Sharp-Bucknall, Tania, M. Sceney, L. Barwise and J. L. Dutton, Dalton Trans., 2023, 52, 16472–16479 RSC.
- K. Rajesh, M. Somasundaram, R. Saiganesh and K. Balasubramanian, J. Org. Chem., 2007, 72, 5867–5869 CrossRef CAS PubMed.
- W. Wang, X. Yang, R. Dai, Z. Yan, J. Wei, X. Dou, X. Qiu, H. Zhang, C. Wang and Y. Liu, J. Am. Chem. Soc., 2022, 144, 13415–13425 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |