DOI:
10.1039/D3SC06619C
(Edge Article)
Chem. Sci., 2024,
15, 3711-3720
Efficient O- and S-glycosylation with ortho-2,2-dimethoxycarbonylcyclopropylbenzyl thioglycoside donors by catalytic strain-release†
Received
9th December 2023
, Accepted 18th January 2024
First published on 7th February 2024
Abstract
We herein present a strain-release glycosylation method employing a rationally designed ortho-2,2-dimethoxycarbonylcyclopropylbenzyl (CCPB) thioglycoside donor. The donor is activated through the nucleophilic ring-opening of a remotely activable donor–acceptor cyclopropane (DAC) catalyzed by mild Sc(OTf)3. Our new glycosylation method efficiently synthesizes O-, N-, and S-glycosides, providing facile chemical access to the challenging S-glycosides. Because the activation conditions of conventional glycosyl donors and our CCPB thioglycoside are orthogonal, our novel donor is amenable to controlled one-pot glycosylation reactions with conventional donors for expeditious access to complex glycans. The strain-release glycosylation is applied to the assembly of a tetrasaccharide of O-polysaccharide of Escherichia coli O-33 in one pot and the synthesis of a 1,1′-S-linked glycoside oral galectin-3 (Gal-3) inhibitor, TD139, to demonstrate the versatility and effectiveness of the novel method for constructing both O- and S-glycosides.
Introduction
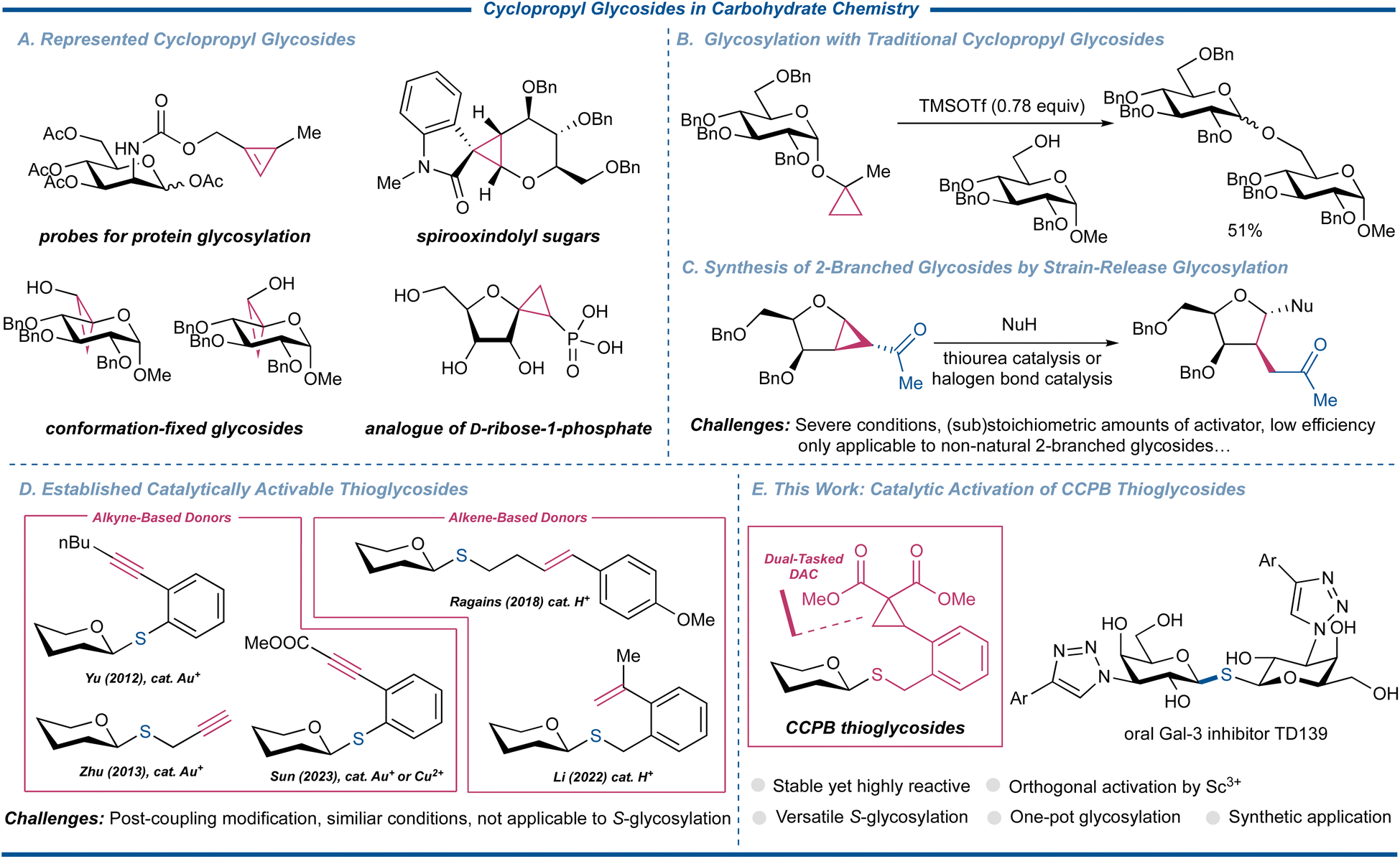
The exploration of cyclopropyl glycostructures significantly expands the horizons of glycoscience, showcasing their multifaceted utilities in diverse domains of imaging and profiling, conformational studies, and medicinal chemistry (Fig. 1A).1,2 Within the realm of synthetic carbohydrate chemistry, these versatile substrates are important synthons for crafting unnatural sugar mimetics with the merits of the high reactivity of cyclopropanes, and the rich functionality and unambiguous stereochemistry of carbohydrates.3 Of note, although cyclopropyl glycosides have been conceptualized for chemical glycosylation, this reaction has so far been limited to a single armed glycosyl donor, characterized by a restricted acceptor scope and unsatisfactory yields (Fig. 1B).4
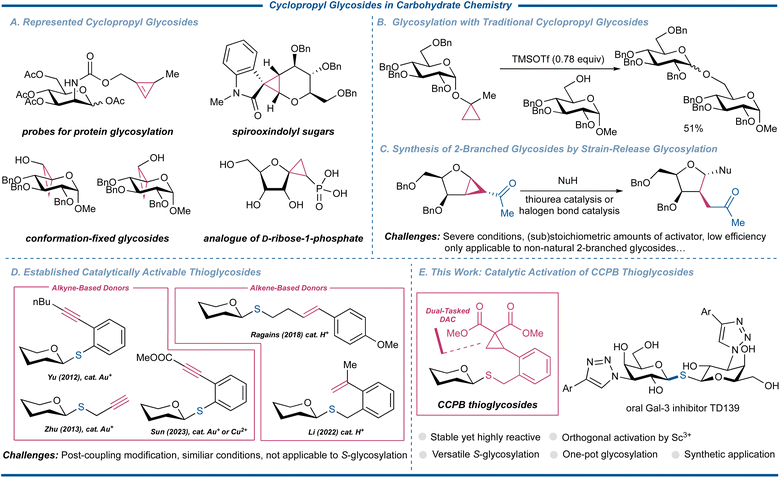 |
| | Fig. 1 Cyclopropyl glycosides in carbohydrate chemistry. | |
Donor–acceptor cyclopropanes (DACs) have been recognized as a special and valuable cyclopropane building block that can undergo facile nucleophilic ring-opening reactions of various C-, N-, O-, S-, and Se-nucleophiles facilitated by the liberation of the ring strain, rendering their chemistry one of the most important pillars in the modern synthesis of carbo- and heterocyclic compounds.5 Chemical glycosylation assumes paramount importance in synthetic carbohydrate chemistry to acquire sufficient amounts of structurally well-defined and compositionally homogeneous materials for unveiling their key functions in numerous biological processes as well as their potential as the new generation of therapeutics.6–8 Thus, the introduction of DAC-fused glycosyl donors has surfaced as a significant advancement in the synthesis of glycomimetics. These specialized donors manifest the capacity to be activated by either Lewis acids or halonium cations, imparting impeccable stereocontrol in the formation of 1,2-trans C-2 branched sugar amino acid derivatives or C-2 branched glycosides.9 In this field, Loh's group achieved the activation of such donors using a thiourea catalyst or a halogen bond catalyst capitalizing on non-covalent interactions (Fig. 1C).10–12 While these methodologies have predominantly led to the generation of non-natural oligosaccharides or glycoconjugates, the realm of chemical glycosylation encompassing strain-driven reactions to access naturally occurring glycoarchitectures remains underexplored, with sporadic protocols having been reported.
Among myriad glycosyl donors, thioglycosides, despite their infrequent occurrence in nature, are an important category of glycosyl donors for the synthesis of oligosaccharides and glycoconjugates as they hold excellent thermostability and tunable reactivities.13,14 They can withstand a wide array of protecting group manipulations, endowing their anomeric thioether as not only a good leaving group but a qualified temporary anomeric protecting group. Although there have been reports on Bi(V)-15 and Au(I)-catalyzed16 activation of conventional thioglycosides, substantial efforts have been invested in the modification of thioglycosides to enable catalytic glycosylation more efficiently. However, it was only in the past decade that a handful of successful examples were reported, predominantly centered around the well-established alkyne or alkene chemistry hinging on Au(I) or superacid catalysis, pioneered by the groups of Yu17 and Ragains,18,19 respectively. These methods were subsequently carried forward and glorified by the groups of Zhu,20 Sun,21 Li,22 and Chai,23 whose elegant protocols significantly expanded the donor and acceptor scopes of catalytic glycosylation of thioglycosides (Fig. 1D). Many of these advancements also allowed the latent-active protocol to streamline the assembly of oligosaccharides.24 Furthermore, these glycosylation reactions operate through similar reaction mechanisms of Au(I)-initiated or protonation-triggered nucleophilic cyclization.
S-Glycosylation is an important branch of chemical glycosylation reactions as the produced S-linked glycosides can express improved biological properties compared to natural O-glycan due to their flexibility and affinity while exhibiting significant stability toward enzymatic hydrolysis, making them vital biologically active molecules.25 Among these attractive substrates, 1,1′-S-linked glycosides, a family of bioisosteric counterparts of 1,1′-disaccharides, have been widely used for biological and medicinal studies, thus attracting much synthetic interest toward such structures.26–30 Presently, only a limited assortment of glycosylation protocols possess the capacity for both O- and S-glycosylation. This scarcity is primarily rooted in the inherent incompatibility of thiol acceptors with commonly employed promoters, such as halonium sources or Au(I) catalysts.31 Moreover, to the best of our knowledge, the repertoire of effective methodologies for the synthesis of S-glycosides from thioglycosides remains notably restricted due to the comparable reactivity profiles of anomeric sulfides and acceptor thiols, with only one example utilizing a modified Yu's N-phenyl trifluoroacetamide and 1.0 equiv. of trimethylsilyl trifluoromethanesulfonate (TMSOTf) as the promoter, further underscoring the inherent challenge and complexity in the endeavor to synthesize S-glycosides from thioglycosides.32 In continuation of our interest in strain-release glycosylation,33,34 we herein successfully merged the diverse reactivities of thioglycosides and DAC to engender a new genre of thioglycoside, termed ortho-2,2-dimethoxycarbonylcyclopropylbenzyl (CCPB) thioglycoside, for versatile O-, S-, and N-glycosylation (Fig. 1E). The unique reaction mode of Lewis acid-catalyzed ring-strain release glycosylation makes it compatible with published catalytically activable thioglycosides, which was harnessed to yield orthogonal one-pot glycosylation and kinetic one-pot strain-release glycosylation. The synthetic utility of this method was demonstrated by the concise and expedient assembly of the tetrasaccharide repeating unit of O-polysaccharide of Escherichia coli O-33 in one pot for the first time and the synthesis of the 1,1′-S-linked glycoside oral Gal-3 inhibitor TD139 with the potential ability to treat COVID-19, respectively.
Results and discussion
Working hypothesis
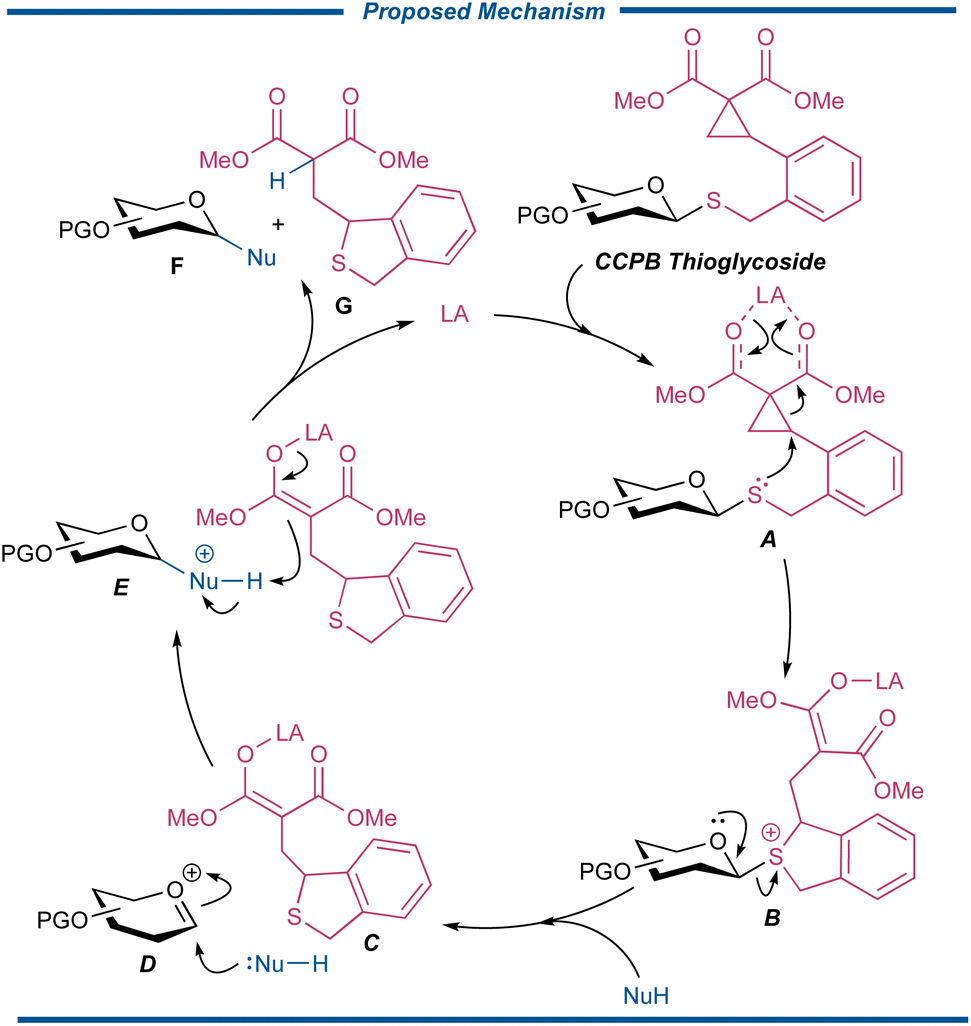
The working hypothesis for the activation of CCPB thioglycosides is illustrated as follows in Fig. 2. A metal-centered Lewis acid can first recognize and chelate with the two carbonyl groups of the intramolecularly incorporated DAC, which is then attacked by the sulfide to form the sulfonium ion B with a metal enolate motif. The intermediate B subsequently disassociates to generate cyclic thioether enolate C as the in situ formed acid scavenger and oxocarbenium D. The nucleophile is then engaged in the glycosylation event to form the protonated glycoside E, which is further deprotonated by enolate C to finally release glycosylated product F and sulfide G as well as regenerating the Lewis acid catalyst for the next cycle of glycosylation reaction.
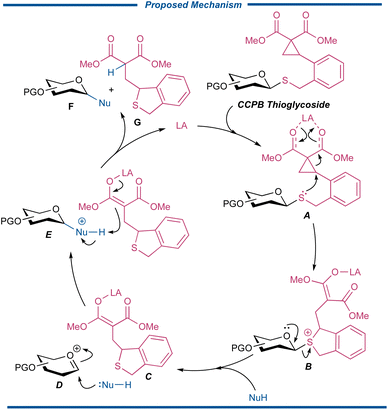 |
| | Fig. 2 The proposed working mechanism of CCPB thioglycosides. | |
Synthesis of CCPB thioglycosides
Building upon our conceptual design, we commenced testing our idea by exploring the synthesis of CCPB thioglycosides (Fig. 3). Thus various glycosyl thiols 1a–h derived from D-glucose, D-galactose, D-mannose, D-xylose, L-rhamnose, D-glucosamine, and lactose, which could be easily prepared by the coupling of the corresponding glycosyl bromide and potassium thioacetate or thiourea, and the following transformation (for more details, see the ESI†), were coupled with known CCPB bromide 235 in the presence of triethylamine (TEA) in anhydrous MeCN, and all resulting CCPB thioglycosides 3a–h were separated as white foams in excellent yields regardless of the protecting fashion and sugar types. The donors showed excellent stability under ambient conditions. It should be noted that the coupling reaction was configuration-retained at the anomeric position. The straightforward installation of the CCPB group directly avoided the extra labor work of modification of aglycon within a densely functionalized sugar molecule. These results set a solid foundation for subsequent studies on strain-release glycosylation.
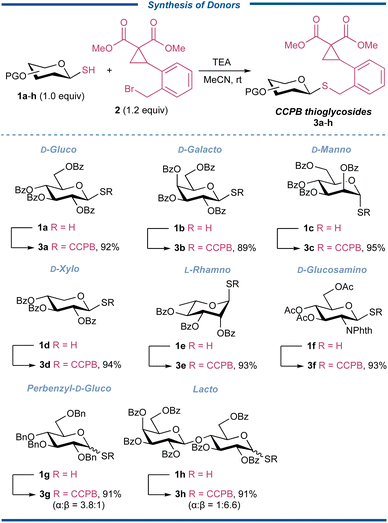 |
| | Fig. 3 Synthesis of CCPB thioglycosides. | |
Reaction optimization
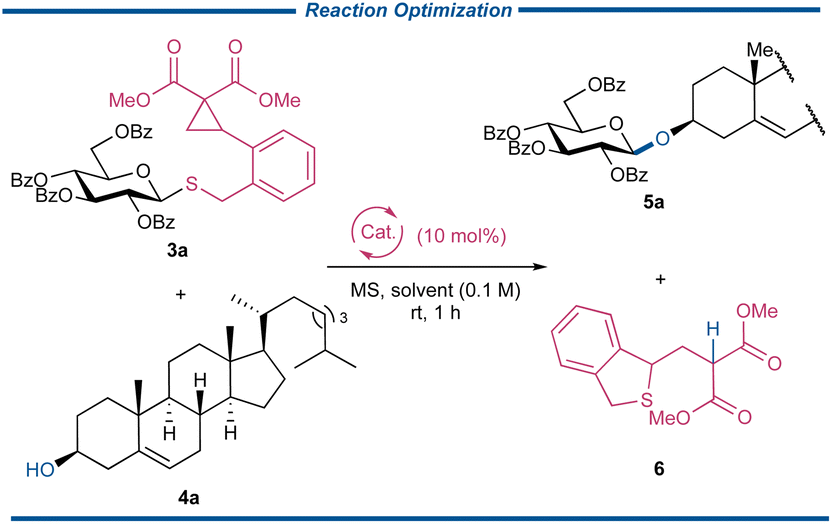
With a series of CCPB thioglycosides in hand, the key strain-release glycosylation reaction was optimized with the glycosylation reaction between donor 3a and acceptor cholesterol 4a (Table 1). To our delight, the glycosylation reaction performed well in the presence of Sc(OTf)3 and a 5 Å molecular sieve (MS) in 1,2-dichloroethane (DCE), and product 5a was obtained in the yield of 86% along with the separation of departing product 6 in 90%, attesting to our proposal (entry 1). A panel of Lewis acids was then subjected to the glycosylation reactions (entries 2–10), and varied chelatable Lewis acids, including Fe(OTf)3, FeCl3, Cu(OTf)2, and Sn(OTf)2 could catalyze the glycosylation reactions to give 5a in synthetically acceptable yields. These results distinctly demonstrated the superior reactivity of CCPB thioglycoside over established strain-release glycosylation. A sharp contrast was observed when Ni(OTf)2 and BiPh3(OTf)2 were used as Lewis acids and no reactions occurred with the complete recovery of the unreacted donors under these conditions. The ineffectiveness of Ph3Bi(OTf)2 in our reaction might be attributed to its bulky nature, making it difficult to approach either the dicarbonyl group of the DAC site or the sulfide surrounded by the sugar ring and DAC. The glycosylation reactions with non-chelatable Lewis acids or protonic acid were proved to be futile (entries 8–10). The catalyst loading had a negligible effect on the reaction efficiency (entry 11). Different types of organic solvents were also screened, and CH2Cl2 stood out as the optimal solvent to afford the product with the best yield of 93%. Note that the unsatisfactory result in MeCN was from the poor solubility of 4a instead of the reaction itself. Another indication of the high reactivity of CCPB thioglycoside was that the types of molecular sieves had a marginal effect on the yields. Finally, the low-temperature glycosylation reaction was tried, and no significant loss of yield was observed until the temperature was below −40 °C although with the sacrifice of reaction time. As above, the conditions listed in entry 12, which include 1.2 equiv. of CCPB thioglycoside 3a, acceptor 4a, 0.1 equiv. of Sc(OTf)3 and 5 Å MS in anhydrous CH2Cl2, were selected to study the strain-release glycosylation of CCPB thioglycosides.
Table 1 Reaction optimization of strain-release glycosylationa
|
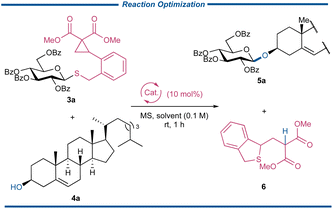
|
| Entry |
Lewis acids |
Solvents |
MS |
Yieldsb (%) |
|
Unless otherwise noted, all reactions were carried out with 3a (0.06 mmol, 1.2 equiv.), 4a (1.0 equiv.), a Lewis acid catalyst (0.1 equiv.) and molecular sieve (MS, 50 mg) in a corresponding solvent (0.5 mL) under an argon atmosphere for 1 h.
Isolated yields.
0.2 equiv. of catalyst.
The reaction was performed in an ice bath.
|
| 1 |
Sc(OTf)3 |
DCE |
5 Å |
86 |
| 2 |
Fe(OTf)3 |
DCE |
5 Å |
78 |
| 3 |
FeCl3 |
DCE |
5 Å |
67 |
| 4 |
Cu(OTf)2 |
DCE |
5 Å |
79 |
| 5 |
Sn(OTf)2 |
DCE |
5 Å |
81 |
| 6 |
Ni(OTf)2 |
DCE |
5 Å |
NR |
| 7 |
BiPh3(OTf)2 |
DCE |
5 Å |
NR |
| 8 |
B(C6F5)3 |
DCE |
5 Å |
NR |
| 9 |
TMSOTf |
DCE |
5 Å |
Trace |
| 10 |
TfOH |
DCE |
5 Å |
Trace |
| 11c |
Sc(OTf)3 |
DCE |
5 Å |
86 |
| 12 |
Sc(OTf)3 |
CH2Cl2 |
5 Å |
93 |
| 13 |
Sc(OTf)3 |
Toluene |
5 Å |
88 |
| 14 |
Sc(OTf)3 |
Et2O |
5 Å |
90 |
| 15 |
Sc(OTf)3 |
MeCN |
5 Å |
Trace |
| 16 |
Sc(OTf)3 |
CH2Cl2 |
4 Å |
88 |
| 17 |
Sc(OTf)3 |
CH2Cl2 |
3 Å |
91 |
| 18d |
Sc(OTf)3 |
CH2Cl2 |
5 Å |
91 |
Substrate scope studies
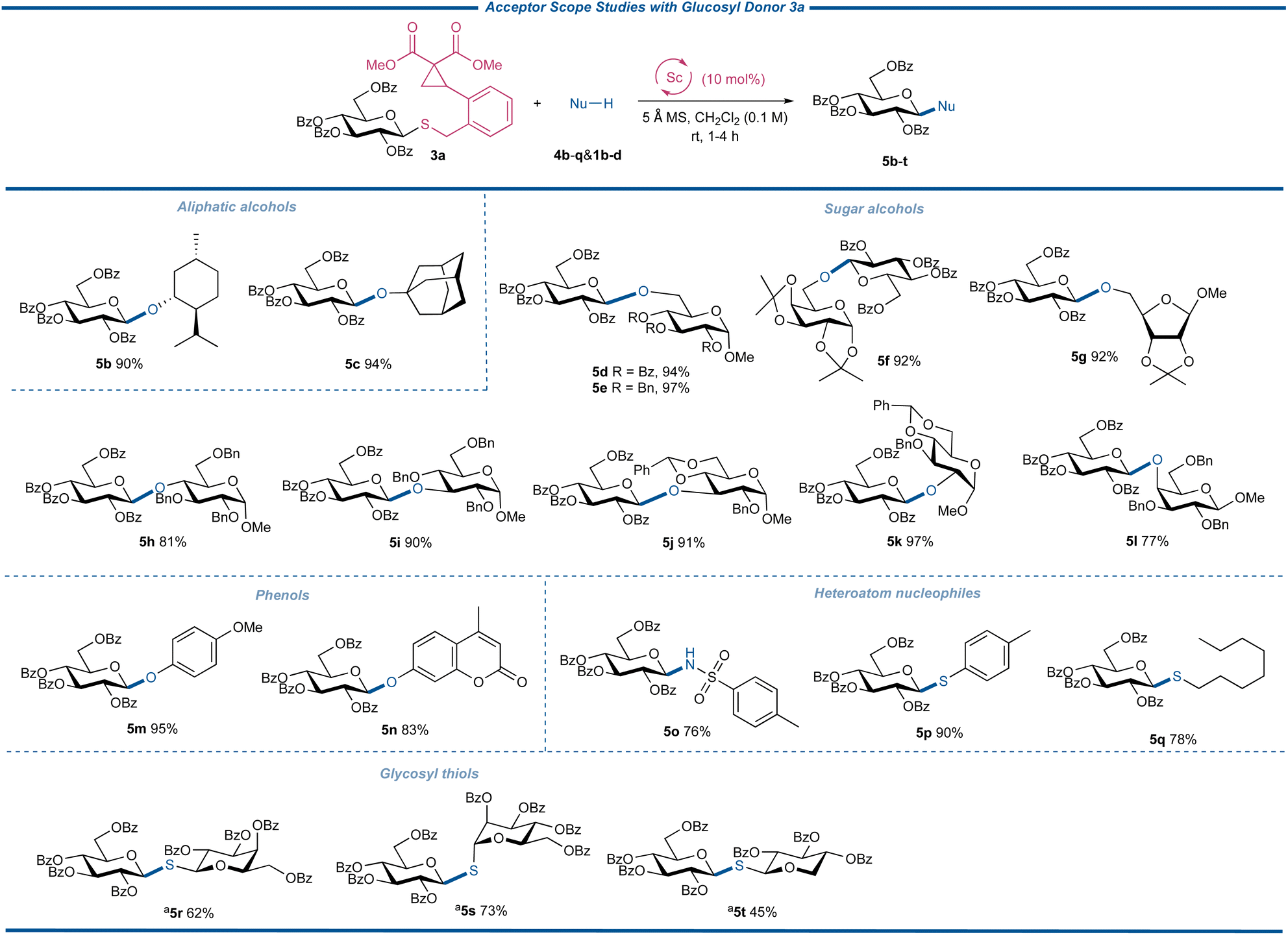
With the optimal conditions for the glycosylation reactions with CCPB thioglycosides as glycosylating agents established, the strain-release glycosylation was investigated by testing the acceptor scope with donor 3a under the catalyzation of the Sc(OTf)3 catalyst (Fig. 4). Thus nucleophiles 4b–q were coupled with 3a to afford disaccharides or glycosides 5b–q in 76 to 97% yields, demonstrating the exceptional reactivity of CCPB thioglycosides. Both aliphatic alcohols and sugar alcohols with primary, secondary, and tertiary hydroxyl groups are competent coupling partners. Various protecting groups are stable under these mild reaction conditions. To our delight, the acid-labile benzylidene group and acetonide survived. The secondary sugar alcohols of C-2-OH, C-3-OH, and C-4-OH were well glycosylated with donor 3a. Acceptors 4i and 4j are both C-3-OH nucleophiles with different protecting fashions, while 4j is a disarmed acceptor resulting from the electron-withdrawing benzylidene group. The comparably high yields of 5i and 5j showed the generality of our glycosylation reaction. The axial-oriented hydroxyl group is a poor nucleophile and the glycosylation between galactosyl acceptor 4l and 3a gave the disaccharide 5l in satisfactory yield with the new type of remotely activable thioglycoside donor. The glycosylation of phenol acceptors is of great importance due to the varied bioactivities of phenol glycosides as both potential drug molecules and natural products. To test the ability of the glycosylation with CCPB thioglycoside in forging the phenolic glycosidic linkages, electron-rich acceptor 4m and electron-deficient 4-methylumbelliferone 4n were engaged in the glycosylation reaction, respectively, and both coupling products were obtained in excellent yields. Note that 4-methylumbelliferyl β-D-glucopyranoside is a useful probe for the measurement of spore-associated enzyme activity36–39 and the derivatives of aglycon 4n exhibit antimicrobial activity.40 The previous coupling methods to synthesize such compounds usually entailed the unstable glycosyl halides as donors and a stoichiometric amount of silver salt as the promoter. The current synthesis supplied a catalytically feasible alternative to 4-methylumbelliferyl glycosides of biological interest utilizing stable glycosyl donors. Next, we attempted to test our glycosylation reaction using heteroatom nucleophiles 4o–q. The successful transformation of sulfonamide into glycoside 5o underlined the capacity of N-glycosylation. Importantly, both aromatic and aliphatic thiols 4p and 4q could be transformed into corresponding thioglycosides in high yields, indicating that the thiol acceptors with strong nucleophilicities did not intervene in the activation of the CCPB thioglycosides. These examples also offered a new entry to various thioglycosides from a catalytically activable thioglycoside donor. With these encouraging results, we were delighted to observe that the challenging glycosyl thiols 1b–d, with both axial- and equatorial-oriented thiol groups, were competent coupling partners to synthesize valuable 1,1′-thiodisaccharides 5t–v with ease, granting efficient and convenient access to these biologically and medicinally critical glycosyl mimics. These satisfactory results demonstrated that our new strain-release glycosylation can be used for versatile S-glycosylation with different S-nucleophiles, representing its extraordinary reactivity.
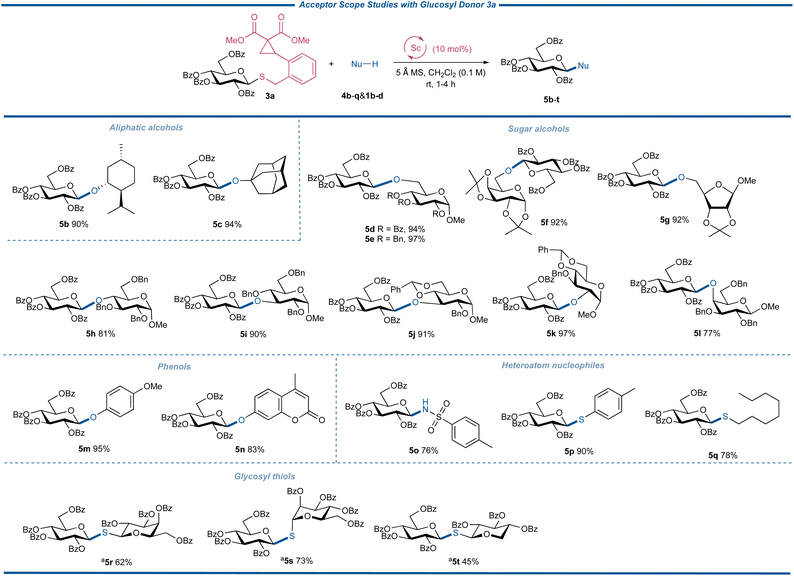 |
| | Fig. 4 Acceptor scope studies. a2.0 equiv. of acceptor. | |
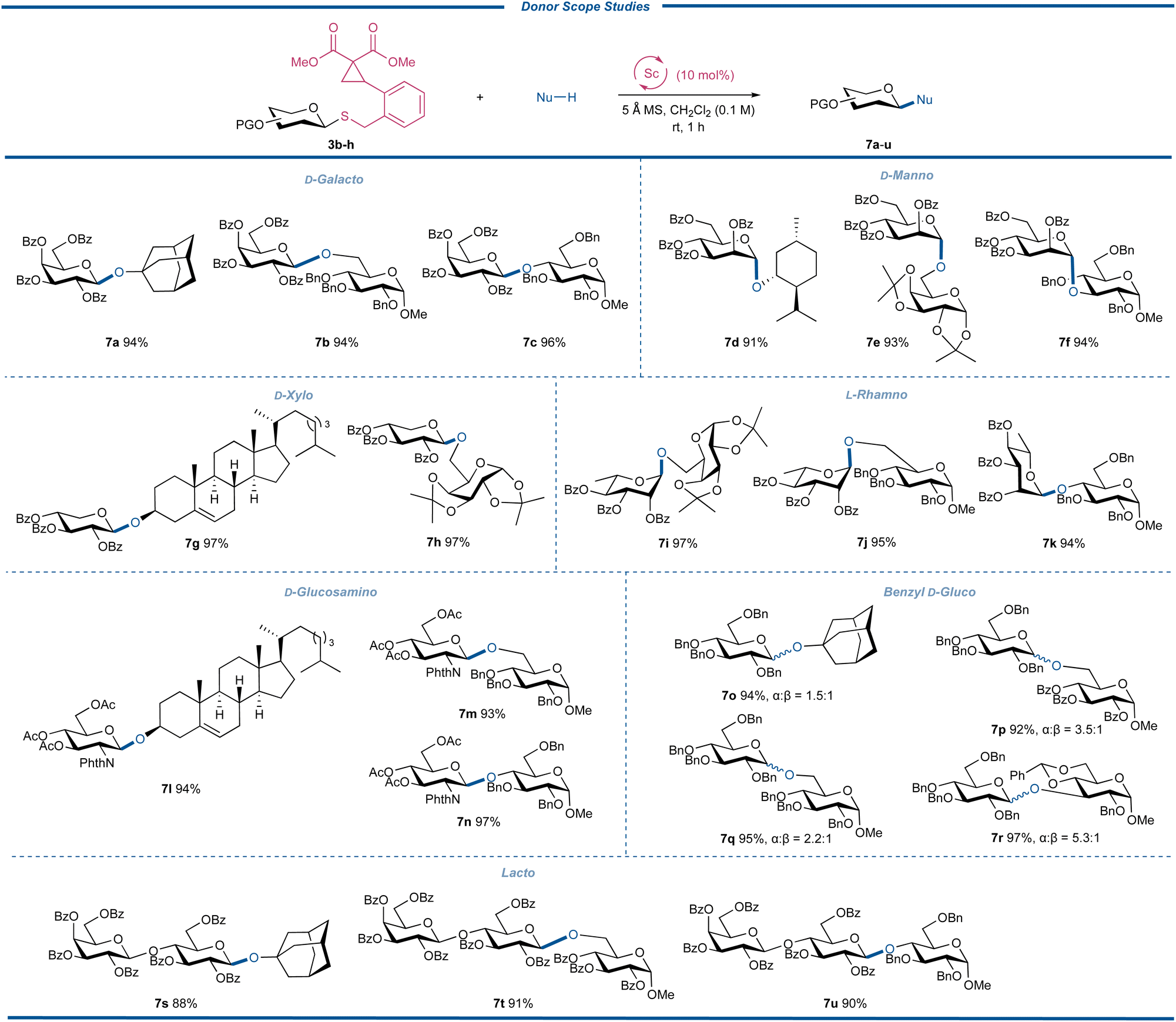
Subsequently, by randomly pairing the donors 3b–h with different acceptors, the generality of different CCPB thioglycosides with varied sugars and protecting fashions was further evaluated and the results are depicted in Fig. 5. All glycosylation reactions with D-galactosyl, D-mannosyl, D-xylosyl, L-rhamnosyl, D-glucosaminyl, D-glucosyl, and lactosyl donors (3b–h) gave the oligosaccharides or glycosides (7a–u) in excellent yields ranging from 88 to 97%. These donors represented most of the available sugar types like deoxy sugar, L-sugar, and amino sugar. The glycosyl donors with the C-2 participatory group unexceptionally delivered the desired products with 1,2-trans glycosidic bonds without any formation of orthoester or 1,2-cis byproducts. Amino sugars typically show moderate reactivity due to the electron-withdrawing induction effect of C-2 N-functionalities.41 The successful preparation of 7l–n not only suggested that the acetyl group is a qualified protecting group but the disarmed glucosaminyl donor could be glycosylated efficiently. We also examined the reactivity and stereoselectivity of the glycosylation with benzyl-protected CCPB thioglucoside 3g. By employing different acceptors, we found that all reactions had excellent yields with the α-isomers as the major products regardless of the structure and nucleophilicity of the acceptors with the ratios of anomers ranging from α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β = 1.5:1 to 5.3:1. Considering the difficulty of α-glycosylation of D-glucose, these interesting results might provide new solutions to the challenging 1,2-cis-glycosylation reactions.42–45 Additionally, the excellent yields of the glycosylation with lactose-derived CCPB thioglycoside displayed the practicability of our method in the assembly of complex oligosaccharides. An important feature of the current glycosylation reactions was that there was no insertion of the acceptor into DAC sites, indicating that the intramolecular ring-opening of DAC by the anomeric sulfide was favorable over the intermolecular reaction with acceptors of O-, S- and N-nucleophiles.
:β = 1.5:1 to 5.3:1. Considering the difficulty of α-glycosylation of D-glucose, these interesting results might provide new solutions to the challenging 1,2-cis-glycosylation reactions.42–45 Additionally, the excellent yields of the glycosylation with lactose-derived CCPB thioglycoside displayed the practicability of our method in the assembly of complex oligosaccharides. An important feature of the current glycosylation reactions was that there was no insertion of the acceptor into DAC sites, indicating that the intramolecular ring-opening of DAC by the anomeric sulfide was favorable over the intermolecular reaction with acceptors of O-, S- and N-nucleophiles.
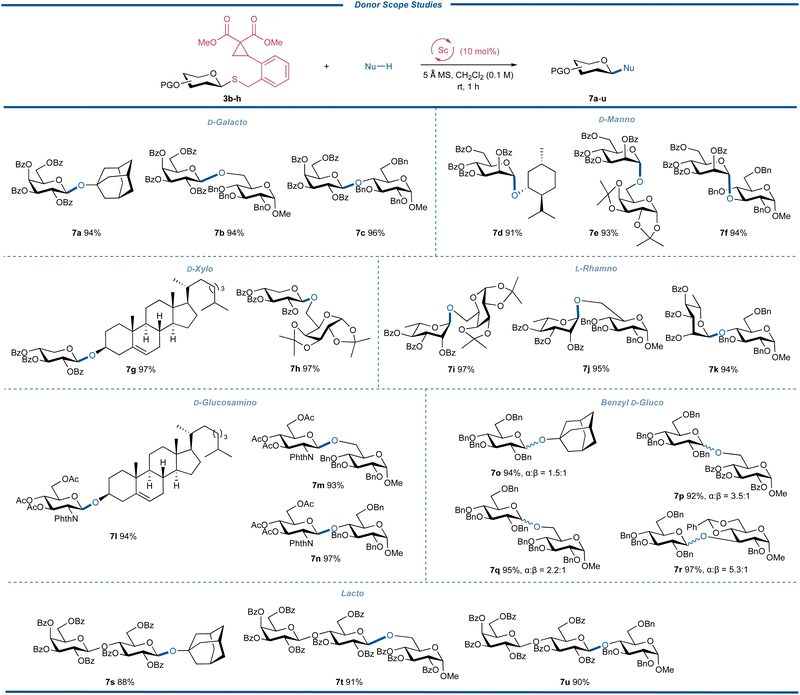 |
| | Fig. 5 Donor scope studies. | |
One-pot glycosylation studies
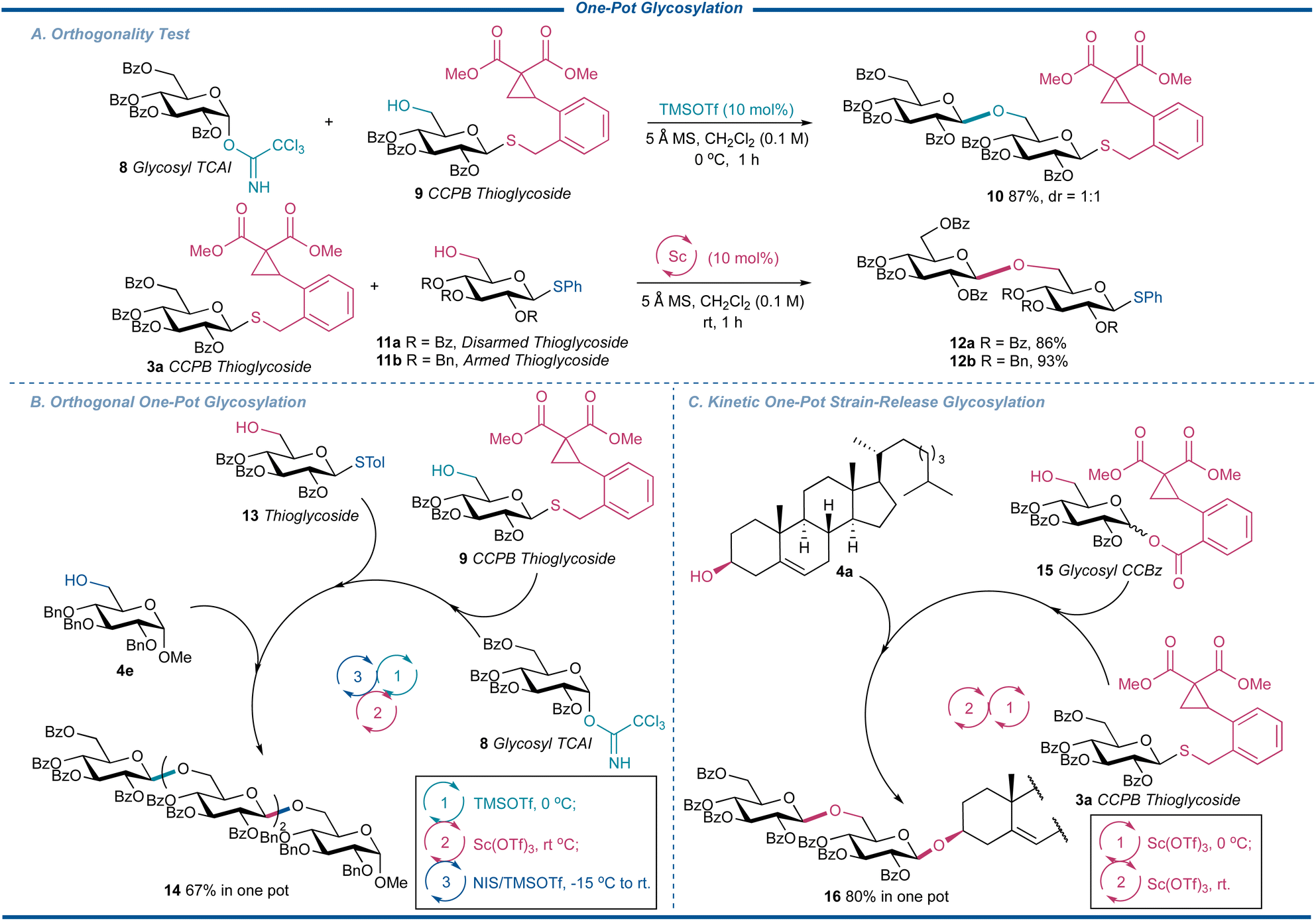
During our optimization of the reaction, we noticed the current glycosylation showed excellent compatibility with the existing glycosylation protocols. In addition, the glycosylation of CCPB thioglycoside showed superior reactivity over our published glycosylation reaction with glycosyl CCBz as the donors. These interesting natures of CCPB thioglycoside thus spurred us to consider the possibility of absorbing CCPB thioglycoside into the one-pot glycosylation, which plays an irreplaceable role in the synthesis of complex oligosaccharides and glycoconjugates.46,47 On one hand, it simplifies the operation and purification process during the oligosaccharide preparation. More importantly, compared to normal iterative glycosylation, the higher overall efficiency of synthesis in a single pot is secured by minimizing the tedious glycosylation-deprotection-glycosylation program. To explore the potency of CCPB thioglycoside, we first conducted the orthogonality test to figure out the compatibility of CCPB thioglycoside with commonly used donors (Fig. 6A). Schmidt's donor 8 was first used as the donor, which could be activated over the CCPB thioglycoside acceptor 9 to uneventfully generate the disaccharide CCPB thioglycoside 10 in an excellent yield of 87% with 1:1 d.r. in the presence of TMSOTf. To our delight, both configurated CCPB thioglycosides could participate in the coupling reaction. Then both armed and disarmed thioglycosides 11a and 11b were exposed to strain-release glycosylation as the acceptors to examine the ability of the CCPB thioglycosides as donors and the desired disaccharide thioglycosides 12a and 12b were separated in excellent yields. Of note, the aglycon transfer side reaction was not detected for both cases. It is well known that this side reaction often takes place during the glycosylation of a disarmed glycosyl donor with an armed thioglycoside acceptor 11b while disaccharide 12b was obtained in an excellent yield of 93%, showcasing the unique reactivity of the current strain-release glycosylation.
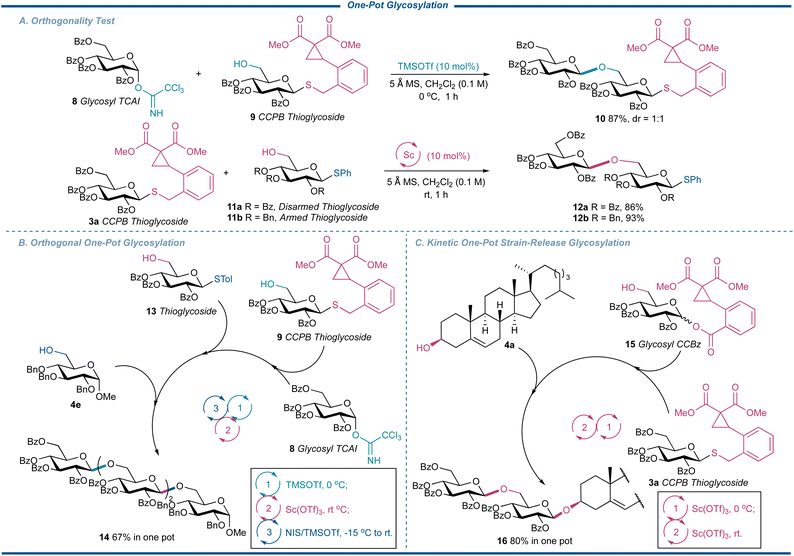 |
| | Fig. 6 One-pot glycosylation studies. | |
Encouraged by these results, we next performed the orthogonal one-pot glycosylation reaction utilizing the ring-strain release glycosylation (Fig. 6B). Thus 8 was catalytically activated in the presence of TMSOTf over the CCPB thioglycoside 9. When the full consumption of 9 was detected, the mixture was then treated with thioglycoside 13 as an acceptor and Sc(OTf)3 to afford the trisaccharide without any aglycon transfer. Finally, by adding the terminal acceptor 4e and N-iodosuccinimide (NIS)/TMSOTf to activate the trisaccharide thioglycoside at −15 °C, the tetrasaccharide 14 corresponding to β-1,6-glucan with immunomodulatory function, antitumor, and antioxidant activities,48 was obtained in the yield of 67% in one pot, demonstrating the versatility of CCPB thioglycoside. We further considered that the reactivities of CCPB thioglycoside and glycosyl CCBz can be kinetically discriminated by the reaction temperature even if both the donors are disarmed (Fig. 6C). Thus the CCPB thioglycoside 3a was activated over glucosyl CCBz 15 at 0 °C with Sc(OTf)3, which was then treated with cholesterol 4a by raising the temperature to room temperature. The glycoconjugate 16 was separated in the yield of 80%, showing the higher reactivity of CCPB thioglycoside over glycosyl CCBz as well as the concept of kinetic one-pot strain-release glycosylation. On average, each step of strain-release glycosylation was up to 90%. These results indicated that our current protocol is compatible with both orthogonal one-pot glycosylation and kinetic one-pot glycosylation for the facile assembly of complex oligosaccharides.
Total synthesis of TD139
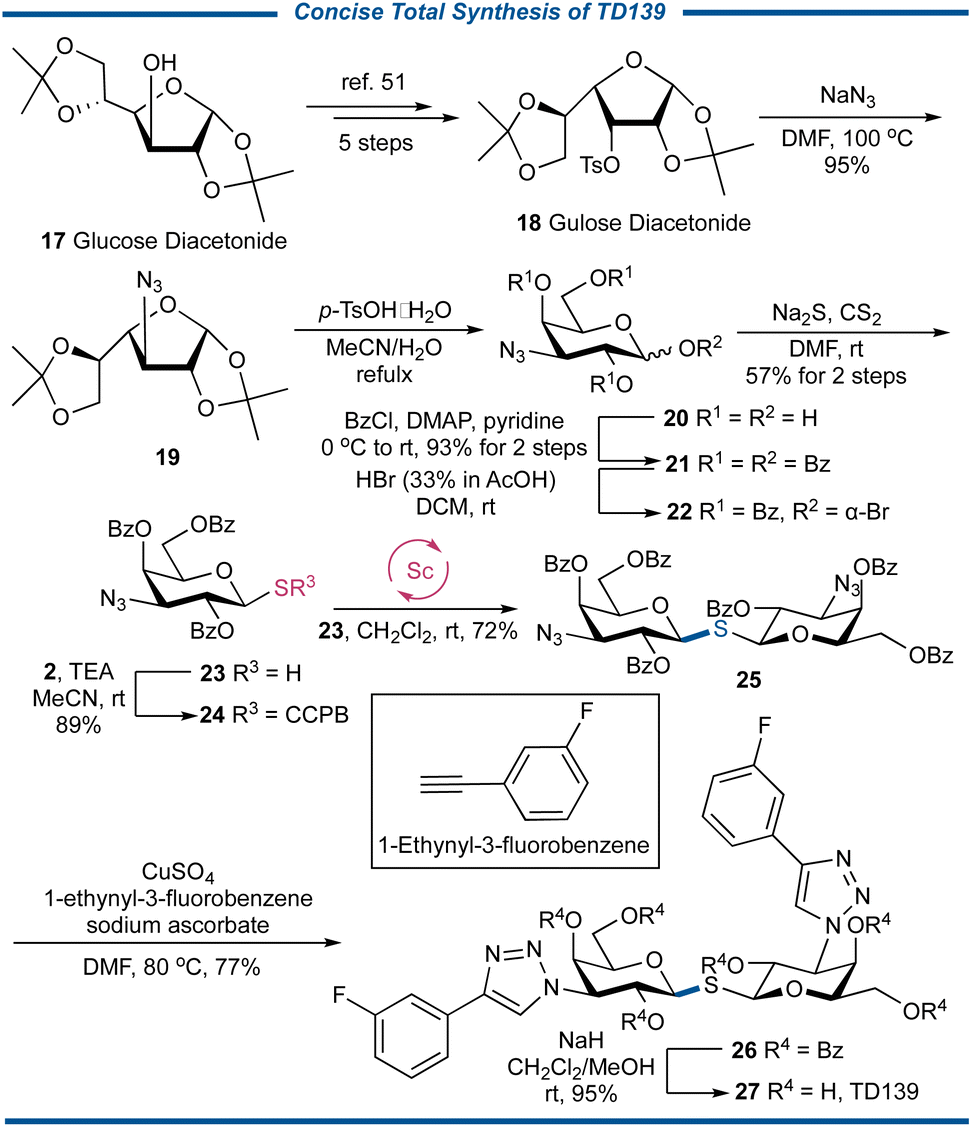
Galectin (Gal)-3 is a profibrotic β-galactoside-binding activating monocyte and macrophage which can be implicated in COVID-19 immunopathology and the cytokine storm. TD139 (also known as olitigaltin or GB0139) is an oral 1,1′-thiodisaccharide-type small molecule drug that can safely suppress Gal-3 expression on bronchoalveolar lavage macrophages and, in a concerted fashion, decrease plasma biomarkers related to IPF progression.49 Recent clinical studies have demonstrated that treating COVID-19 patients with TD139 resulted in a reduction of inflammation, coagulation, and fibrosis markers, compared to patients who received the standard of care, showing the therapeutic potential for TD139 in hospitalized patients with COVID-19.50 To showcase the unique usefulness of the CCPB thioglycoside in the effective S-glycosylation of glycosyl thiols, we decided to conduct the synthesis of the TD139 (Fig. 7). Thus, commercially available glucose diacetonide 17 was transformed into the known gulose diacetonide 18 with O-3 tosylated in five steps.51 The SN2 substitution with NaN3 gave the 3-deoxy-3-azidogalactose 19 in an almost quantitative yield, which was then subjected to acid-mediated hydrolysis to obtain the pyranose-conformed product 20. The benzoylation followed by the bromination gave the bromide 22, which was treated with Na2S in the presence of CS2 in anhydrous DMF52 to afford thiol 23 in the yield of 57% in two steps. After the coupling reaction between 23 and 2 under basic conditions to generate CCPB thioglycoside 24, its key strain-release glycosylation with 23 led to the formation of 1,1′-thiodisaccharide 25 in 72% yield. It should be noted we also conducted the traditional synthesis utilizing the coupling reaction between bromide 22 and thiol 23, and 25 was obtained in an inferior yield of 37%, further demonstrating the important role of stable CCPB thioglycoside donor 24. Finally, after the “click” reaction of 25 with 1-ethynyl-3-fluorobenzene to engender triazole 26 in a satisfactory yield of 77%, the synthesis of TD139 27 was completed by transesterification with in situ generated NaOMe in MeOH. The concise synthesis of TD139 vividly showcases the ability of CCPB thioglycoside in the preparation of complex S-linked sugar molecules of biological and medicinal interest.
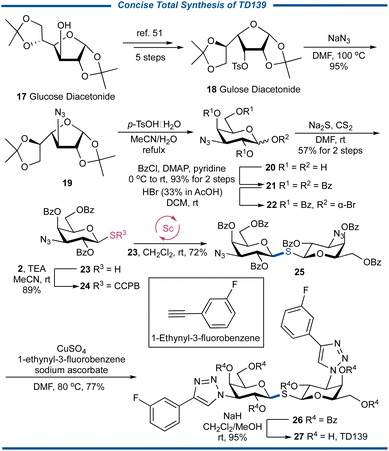 |
| | Fig. 7 Total synthesis of TD139. | |
Assembly of the tetrasaccharide of Escherichia coli O33
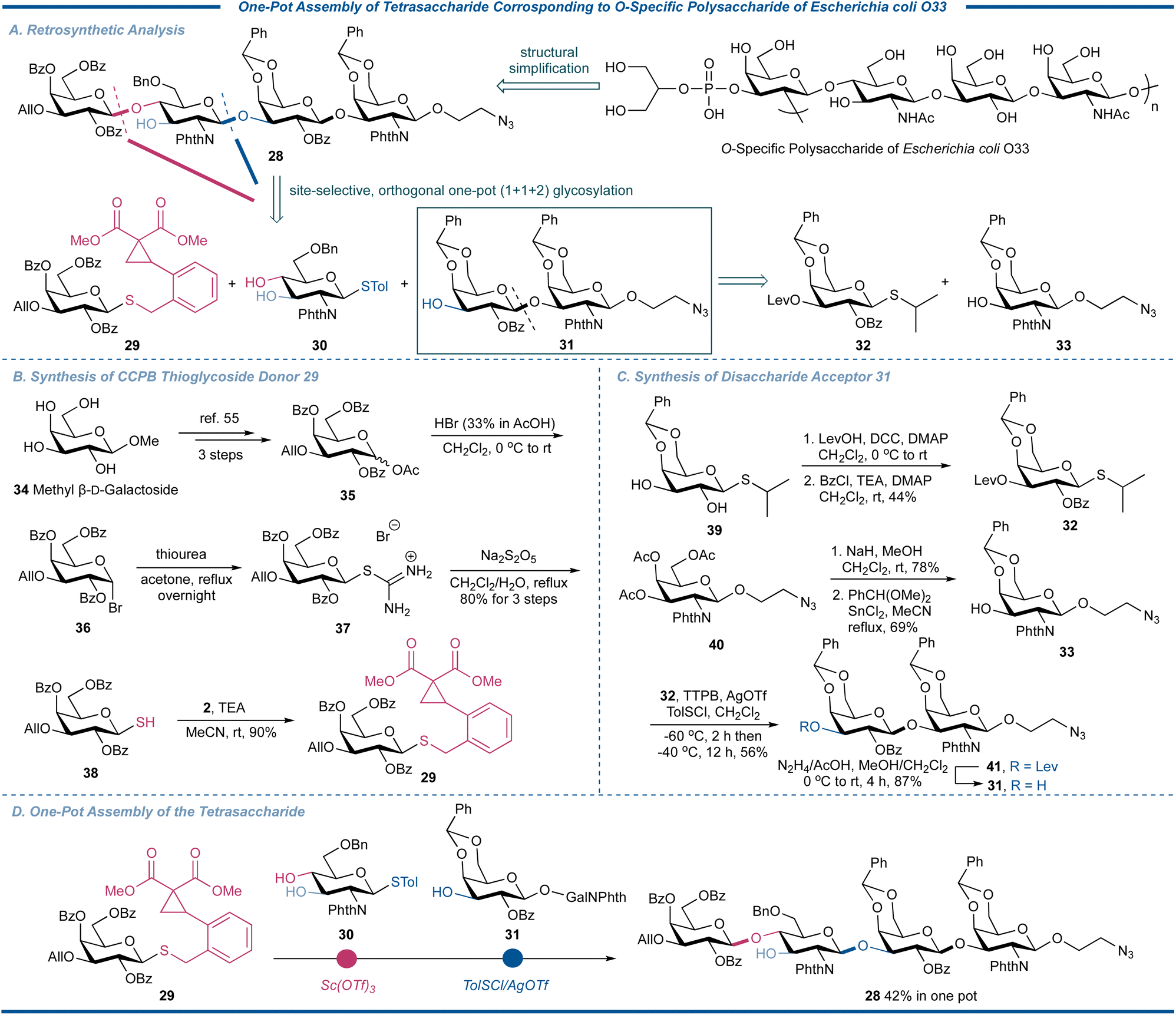
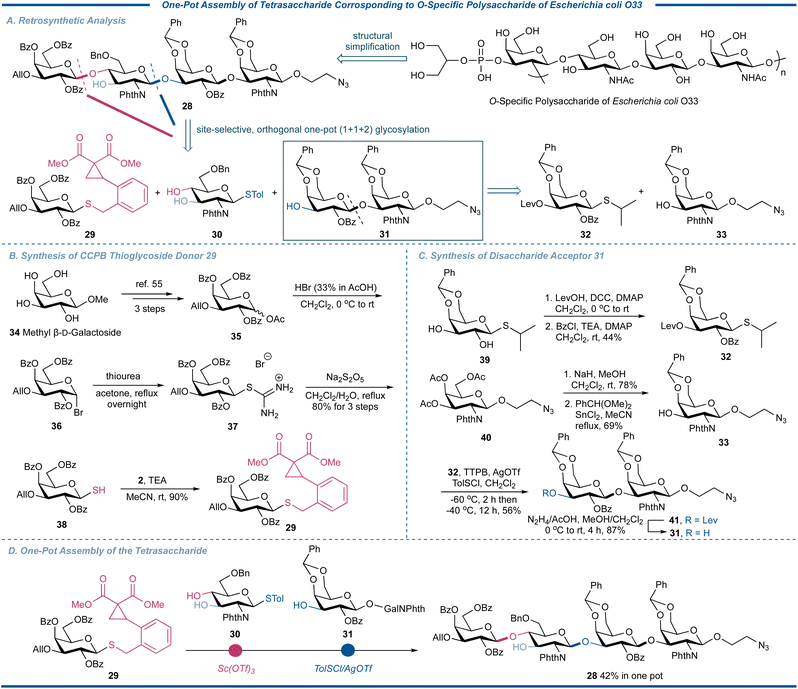
To further demonstrate the synthetic utility of the strain-release glycosylation with CCPB thioglycosides in preparation of complex natural oligosaccharides, we decided to apply our one-pot strategy to the assembly of a tetrasaccharide repeating unit corresponding to the polysaccharide of Escherichia coli O33,53 the predominant facultative anaerobe of the colonic flora of many mammals, including humans (Fig. 8). This tetrasaccharide has a backbone of →2)-3-P-2-Gro-β-D-Galp-(1→4)-β-D-GlcpNAc-(1→3)-β-D-Galp-(1→3)-β-D-GalpNAc-(1→, which could be reduced to our designed target molecule 28 with a readily conjugable 2-aminoethoxy spacer at the reducing end and modifiable site in the Gal unit. Retrosynthetically, this tetrasaccharide could be assembled in one pot using advanced building blocks 29, 3054 and 31 through a [1 + 1 + 2] strategy. The terminal disaccharide acceptor 31 could be synthesized by the glycosylation of easily obtained thioglycoside 32 and galactosaminyl acceptor 33 by the traditional thioglycoside activation protocol. As shown in Fig. 8B, starting from commercially available methyl galactoside 34, acetate 35 was prepared through 3 steps according to the literature.55 After bromination, coupling with thiourea, and reductive cleavage to generate glycosyl thiol 38, its coupling reaction with CCPB bromide 2 under basic conditions efficiently gave the desired CCPB thioglycoside 29 in overall 72% yield in 4 steps, further demonstrating the convenience of preparing this new type of catalytically activable thioglycoside donor. The building block 32 was synthesized by the sequentially regioselective protection of known thioglycoside 39 by levulinoyl (Lev) and Bz, respectively, while 33 was obtained by the transesterification and installation of the benzylidene group of 2-azidoethyl galactosaminoside 33. The disaccharide acceptor 33 was then synthesized by the p-methylbenzenesulfenyl chloride (TolSCl)/AgOTf-promoted coupling reaction of donor 32 and acceptor 33 in 87% yield. With all three synthons in hand, we commenced to proceed with the synthesis of tetrasaccharide. Thus, the CCPB thioglycoside 29 was activated over the thioglycoside acceptor 30 in the presence of two free hydroxyl groups to regioselectively afford the desired disaccharide; the regioselectivity could be attributed to the low nucleophilicity of O-3 resulting from the induction effect and shielding effect of the C-2 bulky phthalimido group. After the indication of the full consumption of the CCPB thioglycoside donor, the terminal disaccharide acceptor 31 and promoter were added to trigger the activation of thioglycoside. By using a combination of TolSCl/AgOTf at −60 °C, the tetrasaccharide 28 was assembled for the first time in an overall yield of 42% in one pot, demonstrating the potency of CCPB thioglycoside as an efficient glycosylating agent in the synthesis of complex natural oligosaccharides.
 |
| | Fig. 8 One-pot assembly of the tetrasaccharide of E. coli O33. | |
Conclusions
In summary, we successfully devised CCPB thioglycosides as stable yet highly reactive glycosylating agents for the next generation of catalytic strain-release glycosylation reactions, applicable to wide scopes of donors and acceptors. This novel donor combines the versatile reaction modes of thioglycosides and DACs, as evidenced by the facile assembly of a library of diverse oligosaccharides and glycoconjugates in high yields. A salient feature of the CCPB donors is that they couple efficiently with thiols to afford structurally diverse S-linked glycosides, remarkably extending the chemical space of S-glycosylation using thioglycoside as the donor. The orthogonality test demonstrated the compatibility of CCPB thioglycosides with commonly used glycosyl donors, which was further supported by the orthogonal one-pot glycosylation and temperature-controlled kinetic one-pot strain-release glycosylation reaction to fast generate molecular complexity. The synthetic merits of the current glycosylation reaction in effective S-glycosylation and one-pot glycosylation were showcased by the expedient synthesis of an oral Gal-3 inhibitor, TD139, and the convenient one-pot assembly of a tetrasaccharide corresponding to the polysaccharide of Escherichia coli O33, respectively. We believe the desirable properties of this donor will render it a powerful alternative to other glycosylations and enable it to be extensively employed in the synthesis of oligosaccharides and glycoconjugates, contributing to the study of glycobiology in the future.
Data availability
The synthetic procedures and the characterization of all products, ESI figures and NMR spectra of the obtained molecules can be found in the ESI.†
Author contributions
Conceptualization: X. W. L. and H. D.; methodology: H. D. and J. L.; investigation: H. D., J. L., X. L. Z., Y. X. and Y. H. Z.; resources: H. D., J. L., X. L. Z., Y. X. and Y. H. Z.; original draft: X. W. L. and H. D.; review and editing: all authors; funding acquisition: X. W. L.; project administration: X. W. L.; supervision: X. W. L.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to the National Research Foundation (NRF-CRP22-2019-0002), Ministry of Education (MOE-T2EP30120-0007), and A*A*STAR (A20E5c0087) for financial support to this study.
References
- D. M. Patterson, K. A. Jones and J. A. Prescher, Mol. BioSyst., 2014, 10, 1693–1697 RSC.
- J. Cao and S. P. Vincent, Tetrahedron, 2023, 140, 133465 CrossRef CAS.
- J. E. Harvey, R. J. Hewitt, P. W. Moore and K. K. Somarathne, Pure Appl. Chem., 2014, 86, 1377–1399 CAS.
- C. Scholl, T. Licisyn, C. Cummings, K. Hughes, D. Johnson, W. Boyko and R. Giuliano, Carbohydr. Res., 2012, 356, 288–294 CrossRef CAS.
- T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS PubMed.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS PubMed.
- T. J. Boltje, T. Buskas and G.-J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed.
- V. Ganesh, P. R. Sridhar and S. Chandrasekaran, Isr. J. Chem., 2016, 56, 417–430 CrossRef CAS.
- C. Xu and C. C. J. Loh, Nat. Commun., 2018, 9, 4057 CrossRef PubMed.
- C. Xu and C. C. J. Loh, J. Am. Chem. Soc., 2019, 141, 5381–5391 CrossRef CAS PubMed.
- W. Ma, J.-L. Kirchhoff, C. Strohmann, B. Grabe and C. C. J. Loh, J. Am. Chem. Soc., 2023, 145, 26611–26622 CrossRef CAS PubMed.
- J. D. C. Codée, R. E. J. N. Litjens, L. J. van den Bos, H. S. Overkleeft and G. A. van der Marel, Chem. Soc. Rev., 2005, 34, 769–782 RSC.
- G. Lian, X. Zhang and B. Yu, Carbohydr. Res., 2015, 403, 13–22 CrossRef CAS PubMed.
- M. Goswami, A. Ellern and N. L. B. Pohl, Angew. Chem., Int. Ed., 2013, 52, 8441–8445 CrossRef CAS PubMed.
- A. M. Vibhute, A. Dhaka, V. Athiyarath and K. M. Sureshan, Chem. Sci., 2016, 7, 4259–4263 RSC.
- F. Yang, Q. Wang and B. Yu, Tetrahedron Lett., 2012, 53, 5231–5234 CrossRef CAS.
- K. D. Lacey, R. D. Quarels, S. Du, A. Fulton, N. J. Reid, A. Firesheets and J. R. Ragains, Org. Lett., 2018, 20, 5181–5185 CrossRef CAS PubMed.
- S. Du and J. R. Ragains, Org. Lett., 2019, 21, 980–983 CrossRef CAS PubMed.
- S. Adhikari, K. N. Baryal, D. Zhu, X. Li and J. Zhu, ACS Catal., 2013, 3, 57–60 CrossRef CAS.
- H. Liu, Z.-F. Liang, H.-J. Liu, J.-X. Liao, L.-J. Zhong, Y.-H. Tu, Q.-J. Zhang, B. Xiong and J.-S. Sun, J. Am. Chem. Soc., 2023, 145, 3682–3695 CrossRef CAS PubMed.
- Z. Qiao, P. Wang, J. Ni, D. Li, Y. Sun, T. Li and M. Li, Eur. J. Org Chem., 2022, e202101367 CrossRef CAS.
- J. Wang, X. Lan, S. Zhang, C. Cai, Q. Zhang, Y. Feng and Y. Chai, Org. Lett., 2023, 25, 6116–6121 CrossRef CAS.
- X. Xiao, Y. Zhao, P. Shu, X. Zhao, Y. Liu, J. Sun, Q. Zhang, J. Zeng and Q. Wan, J. Am. Chem. Soc., 2016, 138, 13402–13407 CrossRef CAS.
- M. Qiao, L. Zhang, R. Jiao, S. Zhang, B. Li and X. Zhang, Tetrahedron, 2021, 81, 131920 CrossRef CAS.
- T. Vašíček, V. Spiwok, J. Červený, L. Petrásková, L. Bumba, D. Vrbata, H. Pelantová, V. Křen and P. Bojarová, Chem.–Eur. J., 2020, 26, 9620–9631 CrossRef.
- G. R. Morais, A. J. Humphrey and R. A. Falconer, Carbohydr. Res., 2009, 344, 1039–1045 CrossRef CAS.
- B. V. Rao, S. Manmode and S. Hotha, Carbohydr. Res., 2015, 417, 103–108 CrossRef.
- X. Zeng, R. Smith and X. Zhu, J. Org. Chem., 2013, 78, 4165–4170 CrossRef CAS PubMed.
- X. Zhao, B. Wu, P. Shu, L. Meng, J. Zeng and Q. Wan, Carbohydr. Res., 2021, 508, 108415 CrossRef CAS PubMed.
- S. Halder, R. B. Addanki, S. Moktan and P. K. Kancharla, J. Org. Chem., 2022, 87, 7033–7055 CrossRef CAS PubMed.
- G. Xin and X. Zhu, Tetrahedron Lett., 2012, 53, 4309–4312 CrossRef CAS.
- X. Xiao, H. Ding, L.-C. Peng, X.-Y. Fang, Y.-Y. Qin, Q.-Q. Mu and X.-W. Liu, CCS Chem., 2023, 5, 2910–2921 CrossRef CAS.
- H. Ding, J. Lyu, X.-L. Zhang, X. Xiao and X.-W. Liu, Nat. Commun., 2023, 14, 4010 CrossRef CAS PubMed.
- D. A. Dias and M. A. Kerr, Org. Lett., 2009, 11, 3694–3697 CrossRef CAS PubMed.
- X. Wei, Y. Ma, Q. Wu, J. Zhang, Z. Cai and M. Lu, Molecules, 2015, 20, 21681–21699 CrossRef CAS PubMed.
- D. Gomes, M. Gama and L. Domingues, Biotechnol. Biofuels, 2018, 11, 111 CrossRef PubMed.
- A. Hernández-Guzmán, A. Flores-Martínez, P. Ponce-Noyola and J. C. Villagómez-Castro, FEBS Open Bio, 2016, 6, 1067–1077 CrossRef PubMed.
- B. Setlow, R.-M. Cabrera-Martinez and P. Setlow, J. Appl. Microbiol., 2004, 96, 1245–1255 CrossRef CAS PubMed.
- Y. Shi and C.-H. Zhou, Bioorg. Med. Chem. Lett., 2011, 21, 956–960 CrossRef CAS PubMed.
- Y. Yang and B. Yu, Tetrahedron, 2014, 70, 1023–1046 CrossRef CAS.
- Y. Zhang, H. He, Z. Chen, Y. Huang, G. Xiang, P. Li, X. Yang, G. Lu and G. Xiao, Angew. Chem., Int. Ed., 2021, 60, 12597–12606 CrossRef CAS PubMed.
- X. Liu, Y. Song, A. Liu, Y. Zhou, Q. Zhu, Y. Lin, H. Sun, K. Zhu, W. Liu, N. Ding, W. Xie, H. Sun, B. Yu, P. Xu and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202201510 CrossRef CAS PubMed.
- C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q.-D. Dang, K. N. Houk and D. Niu, Nat. Chem., 2022, 14, 686–694 CrossRef CAS.
- H. Zhang, A. Guo, Y. Meng, Y. Wang, J. Gao and X.-W. Liu, Chin. J. Chem., 2023, 41, 651–656 CrossRef CAS.
- S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS PubMed.
- M. M. L. Zulueta, D. Janreddy and S.-C. Hung, Isr. J. Chem., 2015, 55, 347–359 CrossRef CAS.
- X. Wang, Y. Qu, Y. Wang, X. Wang, J. Xu, H. Zhao, D. Zheng, L. Sun, G. Tai, Y. Zhou and H. Cheng, Front. Immunol., 2022, 13, 859923 CrossRef CAS PubMed.
- N. Hirani, A. C. MacKinnon, L. Nicol, P. Ford, H. Schambye, A. Pedersen, U. J. Nilsson, H. Leffler, T. Sethi, S. Tantawi, L. Gravelle, R. J. Slack, R. Mills, U. Karmakar, D. Humphries, F. Zetterberg, L. Keeling, L. Paul, P. L. Molyneaux, F. Li, W. Funston, I. A. Forrest, A. J. Simpson, M. A. Gibbons and T. M. Maher, Eur. Respir. J., 2021, 57, 2002559 CrossRef CAS PubMed.
- E. E. Gaughan, T. M. Quinn, A. Mills, A. M. Bruce, J. Antonelli, A. C. MacKinnon, V. Aslanis, F. Li, R. O’Connor, C. Boz, R. Mills, P. Emanuel, M. Burgess, G. Rinaldi, A. Valanciute, B. Mills, E. Scholefield, G. Hardisty, E. G. Findlay, R. A. Parker, J. Norrie, J. W. Dear, A. R. Akram, O. Koch, K. Templeton, D. H. Dockrell, T. S. Walsh, S. Partridge, D. Humphries, J. Wang-Jairaj, R. J. Slack, H. Schambye, D. Phung, L. Gravelle, B. Lindmark, M. Shankar-Hari, N. Hirani, T. Sethi and K. Dhaliwal, Am. J. Respir. Crit. Care Med., 2023, 207, 138–149 CrossRef CAS PubMed.
- G. Legler and S. Pohl, Carbohydr. Res., 1986, 155, 119–129 CrossRef CAS PubMed.
- Y. Ishido, N. Kanbayashi, N. Fujii, T.-a. Okamura, T. Haino and K. Onitsuka, Chem. Commun., 2020, 56, 2767–2770 RSC.
- S. N. Senchenkova, W. Hou, O. I. Naumenko, P. Geng, A. S. Shashkov, A. V. Perepelov, B. Yang and Y. A. Knirel, Carbohydr. Res., 2018, 460, 47–50 CrossRef CAS PubMed.
- A. Miermont, Y. Zeng, Y. Jing, X.-S. Ye and X.-F. Huang, J. Org. Chem., 2007, 72, 8958–8961 CrossRef CAS PubMed.
- G. Ekborg, T. Curenton, N. R. Krishna and L. Rodén, J. Carbohydr. Chem., 1990, 9, 15–37 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental details. See DOI: https://doi.org/10.1039/d3sc06619c |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jian
Lv‡
a,
Xiao-Lin
Zhang
a,
Jian
Lv‡
a,
Xiao-Lin
Zhang