
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodoarene-directed photoredox β-C(sp3)–H arylation of 1-(o-iodoaryl)alkan-1-ones with cyanoarenes via halogen atom transfer and hydrogen atom transfer†
Liang
Zeng‡
ab,
Chong-Hui
Xu‡
a,
Xiu-Yuan
Zou
 b,
Qing
Sun
*b,
Ming
Hu
*ab,
Xuan-Hui
Ouyang
b,
De-Liang
He
*a and
Jin-Heng
Li
*acde
b,
Qing
Sun
*b,
Ming
Hu
*ab,
Xuan-Hui
Ouyang
b,
De-Liang
He
*a and
Jin-Heng
Li
*acde
aState Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University, Changsha 410082, China. E-mail: huming0731@163.com; delianghe@hnu.edu.cn; jhli@hnu.edu.cn
bKey Laboratory of Jiangxi Province for Persistent Pollutants Control and Resources Recycle, Nanchang Hangkong University, Nanchang 330063, China. E-mail: sunqing@nchu.edu.cn
cState Key Laboratory Base of Eco-Chemical Engineering, College of Chemical Engineering, Qingdao University of Science and Technology, Qingdao 266042, China
dState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, China
eSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 475004, China
First published on 26th March 2024
Abstract
Site selective functionalization of inert remote C(sp3)–H bonds to increase molecular complexity offers vital potential for chemical synthesis and new drug development, thus it has been attracting ongoing research interest. In particular, typical β-C(sp3)–H arylation methods using chelation-assisted metal catalysis or metal-catalyzed oxidative/photochemical in situ generated allyl C(sp3)–H bond processes have been well developed. However, radical-mediated direct β-C(sp3)–H arylation of carbonyls remains elusive. Herein, we describe an iodoarene-directed photoredox β-C(sp3)–H arylation of 1-(o-iodoaryl)alkan-1-ones with cyanoarenes via halogen atom transfer (XAT) and hydrogen atom transfer (HAT). The method involves diethylaminoethyl radical-mediated generation of an aryl radical intermediate via XAT, then directed 1,5-HAT to form the remote alkyl radical intermediate and radical–radical coupling with cyanoarenes, and is applicable to a broad scope of unactivated remote C(sp3)–H bonds like β-C(sp3)–H bonds of o-iodoaryl-substituted alkanones and α-C(sp3)–H bonds of o-iodoarylamides. Experimental findings are supported by computational studies (DFT calculations), revealing that this method operates via a radical-relay stepwise mechanism involving multiple SET, XAT, 1,5-HAT and radical–radical coupling processes.
Site selective functionalization of inert C(sp3)–H bonds for targeted increase of molecular complexity and upgrading abundant hydrocarbon feedstocks into value-added compounds represents a prominent theme in chemistry.1–3 In the field, direct C(sp3)–H arylation of saturated carbonyl compounds, such as ketones, amides, aldehydes, esters and acids, has been recognized as one of the most powerful methodologies for the site selective incorporation of an aryl functionality into the alkyl chain to construct a wealth of biologically relevant aryl motif-containing molecules and synthetic intermediates.2,3 In contrast to the well-explored arylation of C(sp3)–H bonds at the α-position in carbonyl compounds,1,2 direct arylation of C(sp3)–H bonds at the β-position or at remote positions in the carbonyl compounds is largely underdeveloped and remains a great challenge in site selectivity control due to the competitive reactions, such as the more acidic α-C(sp3)–H bond arylation.3–6 Typically, transition-metal-catalyzed chelation-assisted β-C(sp3)–H arylation reactions as reliable methods for producing β-aryl carbonyl systems have been intensively developed, where the vast majority of which concerned directing group (DG)-possessing amides,3,4a–d and ketones/aldehydes (using amino-acid based transient directing groups).4e–m Alternatively, the β-C(sp3)–H arylation of ketones and esters via α,β-dehydrogenation using transition-metal redox catalysis obviating the need for the directing group installation and removal redundant phases has been developed.5 However, these strategies suffer from the limited examples for heteroarylation, and the need for precious transition metal catalysts and stoichiometric additives (such as lithium amides and silver salts), thus hampering wide synthetic application.
Recent advances have indicated that manipulations of the radical strategy, especially photoredox catalysis, provide a broader scope of substrates, lower cost and milder conditions, and more precise site selectivity than what can be accessed using the conventional transition-metal catalysis.6 For example, a breakthrough has been achieved by the MacMillan group in radical-mediated β-C(sp3)–H arylation of ketones and aldehydes with electron-deficient cyanoarenes as the aryl coupling partners via radical–radical coupling using a cooperative photoredox and enamine catalysis, enabling the construction of β-aryl ketones and aldehydes (Scheme 1a(i)).6a Along this radical strategy, Ooi and coworkers have developed a photoredox β-C(sp3)–H arylation of preformed silyl enol ethers with 4-cyanopyridines through a sequence of single-electron transfer (SET)/deprotonation to generate allyl β-carbon-centered radicals and radical–radical coupling to achieve β-heteroarylation of ketones (Scheme 1a(ii)).6b Chang and co-workers have established a new copper-catalyzed β-C(sp3)–H arylation of carbonyls, including ketones, aldehydes and esters, in which silyl enol ethers were initially prepared from carbonyls followed by copper-catalyzed oxidative dehydrogenative coupling with polyfluoroarenes via a pathway of allyl radical generation and C–Cu single-electron oxidative formation, affording highly valuable β-polyfluoroaryl carbonyl compounds (Scheme 1b(iii)).6c Studer and Liu have reported a formal β-C–H arylation of readily prepared silyl enol ethers with (hetero)aryl bromides (such as aryl bromides, vinyl bromides and alkynyl bromides) using a cooperative nickel and photoredox catalysis though photoredox generation of the silyl enol ether radical cations, deprotonation, single electron oxidation with the Ni0 species to form the C–NiI intermediates and cross-coupling cascades, realizing β-C(sp3)–H arylation of aldehydes and ketones (Scheme 1b(iv)).6d Despite being efficient and applicable for synthetic purposes, these methods necessitate preinstallation of specific feedstocks (such as enamines and silyl enol ethers) for ready oxidative generation of the radical cations and then deprotonation to form the crucial allylic β-sp3-carbon-centered radicals, which may be actually considered as a mode of the activated allyl C(sp3)–H functionalization. Until now, it is still difficult to render direct functionalization of C(sp3)–H bonds at the β-position or at remote positions of saturated carbonyls in radical-mediated reactions. Thus, there is a high demand for innovation of new radical strategies that directly access the crucial carbon-centered radicals at the β-position or at remote positions, preferably with readily accessible carbonyl feedstocks, for their site-selective remote C(sp3)–H arylation.
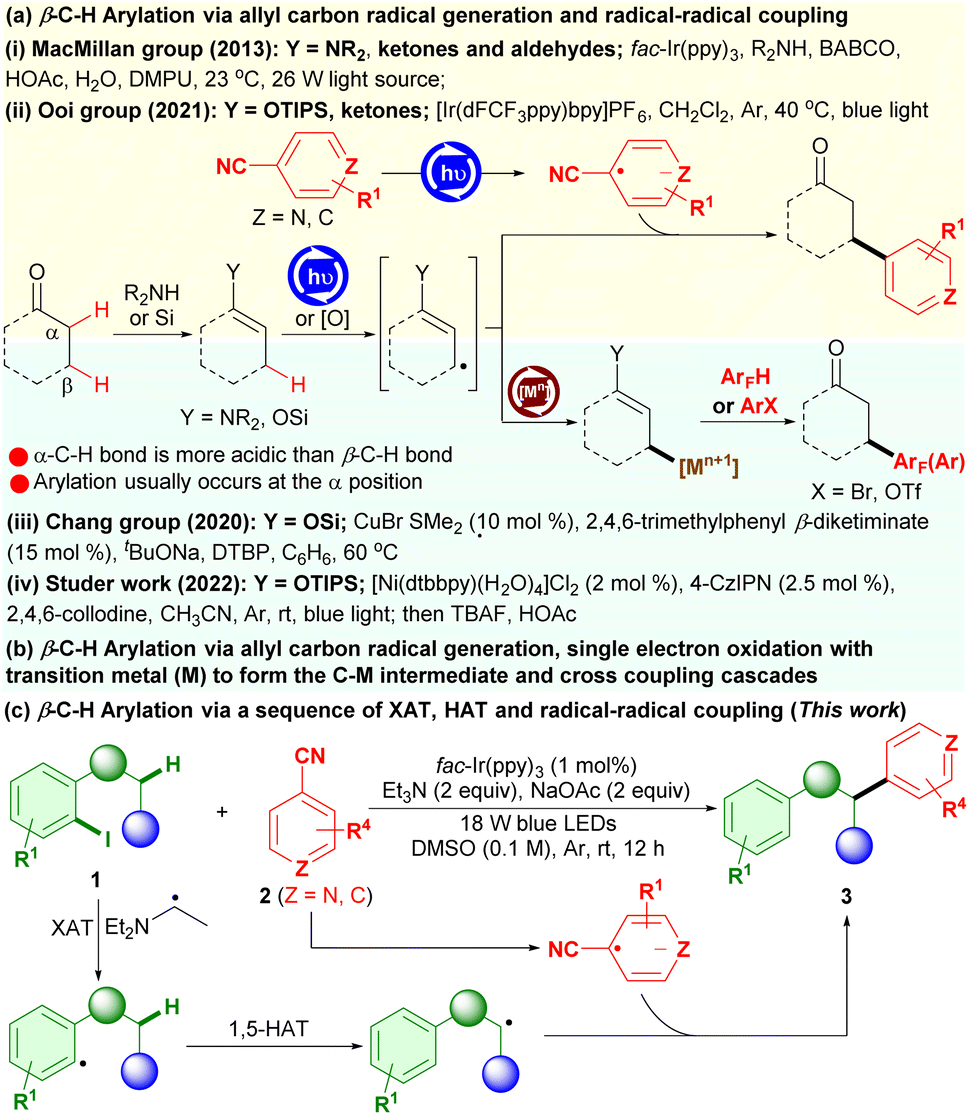 | ||
| Scheme 1 β-C(sp3)–H radical arylation of carbonyls. | ||
Herein, we report the first iodoarene-directed photoredox β-C–H arylation of 1-(o-iodoaryl)alkan-1-ones, which can be conveniently prepared from readily available 2-iodoarylaldehydes, with cyanoarenes via halogen atom transfer (XAT) and 1,5-hydrogen atom transfer (HAT), enabling the preparation of a diverse array of β-aryl arylalkanones with high efficiency (Scheme 1c). Therein, photooxidation/deprotonation of a triethylamine generates the diethylamino-ethyl radical, which serves as the halogen-atom transfer agent for the activation of aryl iodides to undergo XAT7 and generate the aryl radicals, followed by directed 1,5-HAT8 to afford the remote β-sp3-carbon-centered radicals and then radical–radical coupling. This iodoarene-directed photoredox strategy can also be applicable to α-C(sp3)–H arylation of N-(o-iodoaryl)amides for functionalized (hetero)arenes.
We commenced our studies by exploring the photoredox remote C(sp3)–H arylation of 1-(2-iodophenyl)-3-methyl-butan-1-one 1a with isonicotinonitrile 2a (Table 1). After extensive screening of the reaction parameters, a combination of 1 mol% fac-Ir(ppy)3, 2 equiv. Et3N, 2 equiv. NaOAc, DMSO solvent and 18 W blue LEDs light was found to be optimal for the photoredox remote C(sp3)–H arylation of substrate 1a with isonicotinonitrile 2a, affording the desired product 3aa in 71% yield (entry 1). Other photocatalysts, such as Ir(ppy)2(dtbbpy)PF6, Ru(bpy)3Cl2, 4-CZIPN, 3DPA2FBN and eosin Y, were evaluated (entries 2–6): they showed less efficiency than fac-Ir(ppy)3. Control experiments show that both Ir(ppy)3 (entry 7) and Et3N (entry 8) are crucial for success as omitting either led to no detectable product 3aa. A series of amines, including iPr2NEt, 1,4-diazabicyclo[2.2.2]octane (DABCO), 2,2,6,6-tetramethyl-piperidine (HTMP), 1,2,2,6,6-pentamethylpiperidine (PMP) and iPr2NH, were reactive, but all were inferior to Et3N (entries 9–12). Notably, the reaction could occur in the absence of NaOAc, albeit with diminished yield (45% yield; entry 13). In contrast, other inorganic bases like K2CO3 and NaOtBu were found to inhibit the reaction (entries 14 and 15). These results indicate that the organic bases like Et3N mainly serve as the electron transfer reagents for photoreductive cleavage of the C(sp2)–I bonds, and the inorganic bases as the iodine removal reagents. Further screening of the solvent effect demonstrated DMSO as the optimal option (entry 1 versus entries 16 and 17). The reaction cannot occur without blue LEDs light (in the dark) (entry 18). Gratifyingly, the optimal conditions are amenable to a scale up to 1 mmol 1a, giving 3aa in satisfactory yield (entry 19).
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Standard reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), fac-Ir(ppy)3 (1 mol%), Et3N (2 equiv.), NaOAc (2 equiv.), DMSO (2 mL), 18 W blue LEDs, argon, room temperature, and 12 h. Some by-products, including 3-methyl-1-phenylbutan-1-one 4a, 3,3-dimethyl-2,3-dihydro-1H-inden-1-one 5a and N,N-diethyl-1-iodoethan-1-amine 6, were detected by GC-MS analysis and HRMS analysis. b Isolated yield. c 1a (1 mmol) and 24 h. | ||
| 1 | None | 71 |
| 2 | Ir(ppy)2(dtbbpy)PF6 instead of fac-Ir(ppy)3 | 65 |
| 3 | Ru(bpy)3Cl2 instead of fac-Ir(ppy)3 | Trace |
| 4 | 4-CZIPN instead of fac-Ir(ppy)3 | 33 |
| 5 | 3DPA2FBN instead of fac-Ir(ppy)3 | 42 |
| 6 | Eosin Y instead of fac-Ir(ppy)3 | Trace |
| 7 | Without fac-Ir(ppy)3 | 0 |
| 8 | Without Et3N | 0 |
| 9 | iPr2NEt instead of Et3N | 68 |
| 10 | DABCO instead of Et3N | 11 |
| 11 | HTMP or PMP instead of Et3N | <5 |
| 12 | iPr2NH instead of Et3N | 34 |
| 13 | Without NaOAc | 45 |
| 14 | K2CO3 instead of NaOAc | 43 |
| 15 | NaOtBu instead of NaOAc | 10 |
| 16 | DMF instead of DMSO | 53 |
| 17 | CH3CN instead of DMSO | 31 |
| 18 | Without light (in the dark) | 0 |
| 19c | None | 62 |

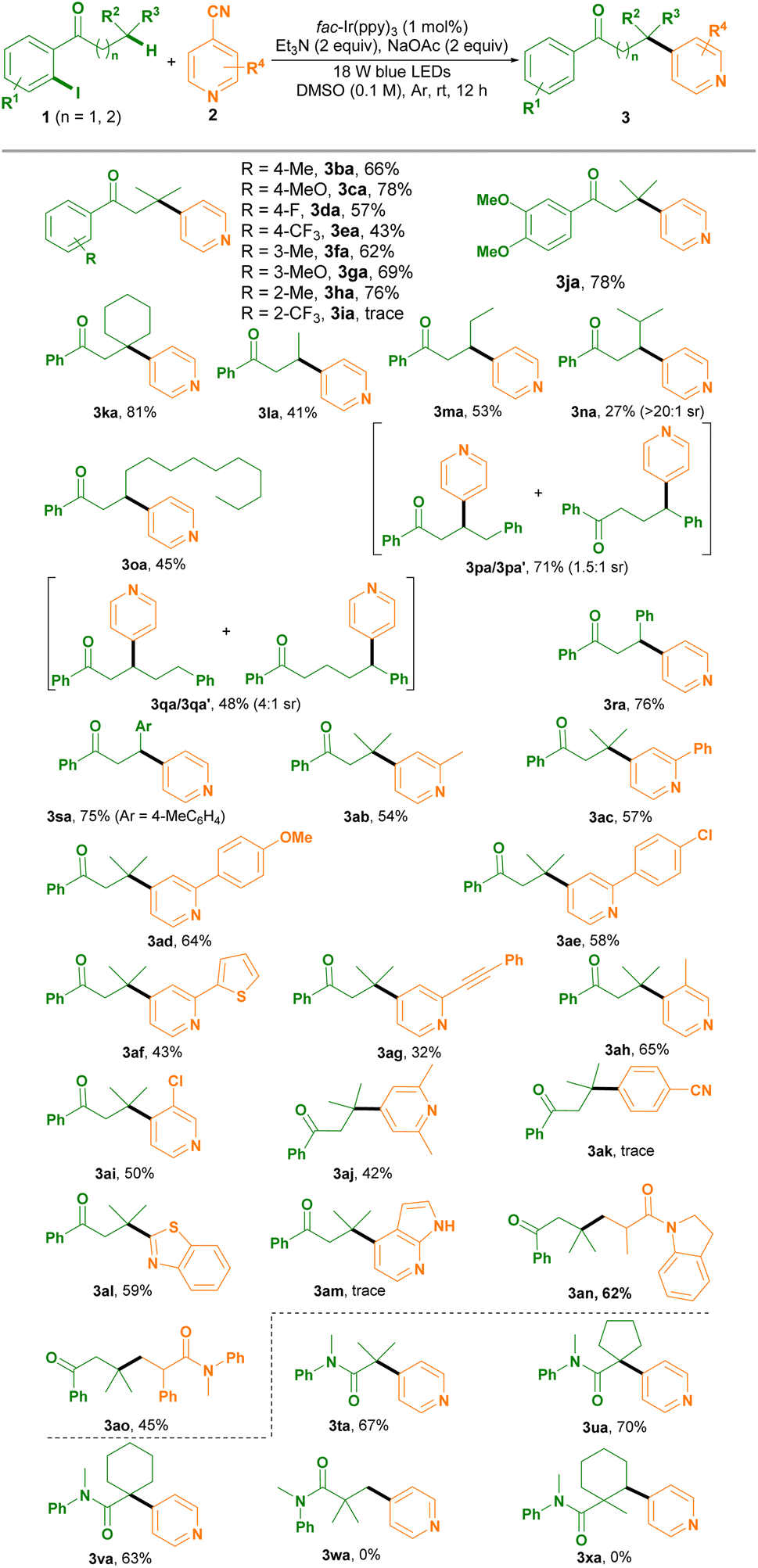
After establishing the optimized conditions, we set out to investigate the scope of this photoredox remote C(sp3)–H arylation protocol (Table 2). As shown in Table 2, a variety of 1-(2-iodoaryl)-1-ketones 1b–h bearing a functionality, such as Me, MeO, F and CF3, on the aryl ring at the para- or meta-position were compatible with the optimized conditions, affording 3ba–ha in moderate to good yields. Moreover, the electron nature and the steric hindrance effect affected the reactivity. While in the presence of isonicotinonitrile 2a, fac-Ir(ppy)3, Et3N and NaOAc the electron-donating 4-MeO-substituted 1-(2-iodoaryl)-1-ketone 1c, for example, was converted to 3ca in 78% yield; using the electron-withdrawing 4-CF3-substituted substrate 1e or the more sterically hindered 3-MeO-substituted one 1g resulted in slightly diminished yields (3ea, 3ga). 1-(2-Iodoaryl)-1-ketone 1h with a Me group at the ortho-position of the aryl ring also exhibited high reactivity, giving 3ha in 76% yield. However, the 1-(2-iodoaryl)-1-ketone possessing the electron-withdrawing CF3 group substituted at the ortho-position of the aryl ring 1i showed no reactivity for the reaction. 3,4-DiMeO-substituted (2-iodoaryl)-1-ketone 1j was suitable for efficiently constructing 3ja. Using 2-cyclohexyl-1-(2-iodophenyl)ethan-1-one 1k delivered 3ka in 81% yield. For chain extended 1-(2-iodophenyl)-1-ketones 1l–m, site selective 3-arylation efficiently occurred via XAT and 1,5-HAT cascades (3la–oa). Interestingly, 1-(2-iodophenyl)-4-methyl-pentan-1-one 1n containing a tertiary carbon at position 4 site selectively underwent 3-arylation, not 4-arylation, albeit with a low yield (3na). Whereas 1-(2-iodoaryl)-1-ketone 1p possessing a phenyl ring at position 4 underwent 3-arylation and 4-arylation in 71% yield and 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 site selectivity (sr) (3pa/3pa′). In the case of 1-(2-iodoaryl)-1-ketone 1q containing a phenyl ring at position 5, both 3-arylation and 5-arylation happened, giving 3qa and 3qa′ in 48% yield and 4:1 sr. 1-(2-Iodoaryl)-1-ketones 1r–s having an aryl ring (such as phenyl or 4-methylphenyl) exhibited high reactivity for furnishing 3ra–sa in good yields.
:1 site selectivity (sr) (3pa/3pa′). In the case of 1-(2-iodoaryl)-1-ketone 1q containing a phenyl ring at position 5, both 3-arylation and 5-arylation happened, giving 3qa and 3qa′ in 48% yield and 4:1 sr. 1-(2-Iodoaryl)-1-ketones 1r–s having an aryl ring (such as phenyl or 4-methylphenyl) exhibited high reactivity for furnishing 3ra–sa in good yields.
| a Reaction conditions: 1 (0.2 mmol), 2 (0.2 mmol), fac-Ir(ppy)3 (1 mol%), Et3N (2 equiv.), NaOAc (2 equiv.), DMSO (0.1 M; 2 mL), 18 W blue LEDs, argon, room temperature, and 12 h. The site-selectivity ratio (sr) is given in parentheses. |
|---|
|
A variety of isonicotinonitriles 2b–j were found to be compatible with the optimized conditions, site selectively delivering 3ab–aj in moderate yields. We found that a wide range of functionalities, namely Me, Ph, 4-MeOC6H4, 4-ClC6H4, thiophen-3-yl and phenylethynyl, on the pyridine ring at position 2 were compatible for site selectively assembling the 3-arylation products 3ab–ag in 32–64% yields. Isonicotinonitriles 2h–i bearing a Me or a Cl group at position 3 executed the photoredox remote C(sp3)–H arylation protocol smoothly (3ah–ai). For 2,6-diMe-substituted isonicotinonitrile 2j the reaction successfully occurred and delivered 3aj in moderate yield. However, terephthalonitrile 1k had no reactivity (3ak). Additionally, heterocyclic cyanoarenes 2l and 2m were investigated. Benzo[d]thiazole-2-carbonitrile (2l) was converted efficiently into 3al in 59% yield, whereas 1H-pyrrolo[2,3-b]pyridine-4-carbonitrile (2m) had no reactivity. Notably, we investigated electron-deficient alkenes 2n and 2o under the standard conditions, affording the alkylated products in 45–62% yields.
This photoredox remote C(sp3)–H arylation protocol was applicable to N-(2-iodoaryl)amides (3ta–va), thus offering a new α-C(sp3)–H arylation strategy. For example, N-(2-iodophenyl)-isobutyramide 1t executed α-C(sp3)–H arylation via a sequence of XAT and 1,5-HAT, affording 3ta in 67% yield. However, N-(2-iodophenyl)-N-methylpivalamide (1w) and N-(2-iodophenyl)-N-1-dimethylcyclohexane-1-carboxamide (1x) showed no reactivity for the construction of β-C(sp3)–H arylation products.
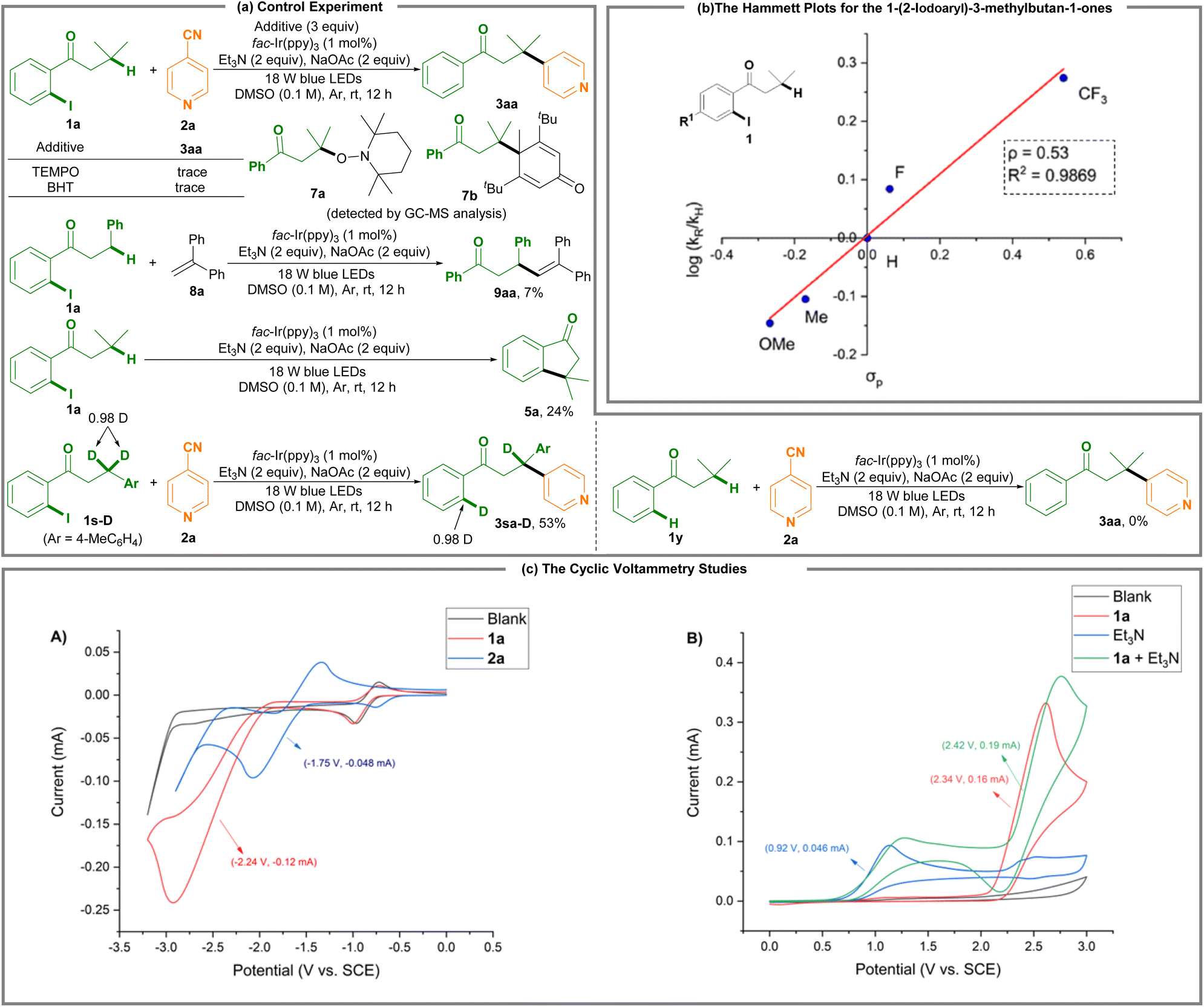
Using a radical scavenger like TEMPO or BHT led to complete suppression of the remote C(sp3)–H photoredox arylation reaction, along with the detectable 7a and 7b by GC-MS analysis (Scheme 2a). Using 1,1-diphenylalkene 8a as a radical scavenger delivered alkyl alkene 9aa in 7% yield. The results indicate that the alkyl radical may be formed, which is supported by the results of the intramolecular cyclization of 1-(2-iodophenyl)-3-methylbutan-1-one 1a where 3,3-dimethyl-2,3-dihydro-1H-inden-1-one 5a is produced in 24% yield. The deuterium-labeled experiments suggest a 1,5-hydride shift process. However, 3-methyl-1-phenylbutan-1-one 1y, a non-o-iodo-substituted substrate, could not undergo the target reaction (3aa).
 | ||
| Scheme 2 Mechanistic investigations. | ||
Hammett studies were performed to determine the electronic effect of the substituents of the 1-(2-iodoaryl)-3-methylbutan-1-ones 1 on the reaction rates (Scheme 2b and Fig. S5; ESI†). The Hammett curve indicated an increase in the k value with the strong electron-withdrawing substrates (k1-(2-iodo-4-(trifluoromethyl)phenyl)-3-methylbutan-1-one ≫ k1-(2-iodo-4-methoxyphenyl)-3-methylbutan-1-one). The relative rates of such substituted 1-(2-iodoaryl)-3-methylbutan-1-ones 1 appear to have a good linear correlation (R2 = 0.9869) between the log(kX/kH) and the σp values of the respective substituents. The Hammett plot gives a slope of ρ = 0.53. The linear relationship in the Hammett plot with a positive slope suggests the generation of a negative charge on the aryl ring in the reaction.
Scheme 2c shows the cyclic voltammetry (CV) studies, revealing a reduction potential peak at Ep1/2red = −2.24 V vs. standard calomel electrode (SCE) for 1-(2-iodophenyl)-3-methylbutan-1-one 1a and a reduction potential peak at Ep1/2red = −1.75 V vs. SCE for isonicotinonitrile 2a (Fig. S6 and S7; ESI†). Upon the catalytic activity of the fac-Ir(ppy)3 catalyst (Ep1/2red = −1.73 V vs. SCE), the reductive conversion of 1a is very difficult because of its lower negative reduction potential, and thus photoreduction of isonicotinonitrile 2a takes precedence over that of 1a. On the other hand, 1-(2-iodophenyl)-3-methylbutan-1-one 1a exhibited an oxidation potential peak at Ep1/2ox = 2.34 V vs. SCE (red line; Scheme 2c(B)) and Et3N has an oxidation potential peak at Ep1/2ox = 0.92 V vs. SCE (blue line; Scheme 2c(B)). Moreover, a mixture of 1a and Et3N reveals an increasing oxidation potential and current (from Ep1/2red = 2.34 V, 0.16 mA to Ep1/2red = 2.42 V, 0.19 mA) (green line; Scheme 2c(B)). These results suggest that Et3N is more reactive than 1a for anodic oxidation, and this protocol involves a Et3N reacting with 1a process.
The Stern–Volmer fluorescence quenching experiments revealed that the Ir(ppy)3 luminescence was obviously decreased by Et3N (Fig. S1; ESI†), and slightly decreased by 1-(2-iodophenyl)-3-methylbutan-1-one 1a (Fig. S2†) or cyanopyridine 2a (Fig. S3†), suggesting that the reaction operates via a canonical photoredox radical cycle consisting of a photoreductive quenching by Et3N. The light on–off experiments show that visible light is crucial as no desired reaction was detected in the absence of the additional blue light (Fig. S4†).
According to the present results and previous reports, state-of-the-art density functional theory (DFT) calculations were performed at the SMD-M06-2X/6-311+G(d,p)/SDD//SMD-M06-2X/6-31G(d,p)/Lanl2DZ theoretical level to further elucidate the reaction mechanism (Fig. 1). Initially, the photocatalyst PC (fac-Ir(ppy)3) is excited to the excited state PC*-T1 under blue LED illumination. Following this, PC undergoes two consecutive single-electron transfer (SET) processes. In the first SET, the excited state PC*-T1 reacts with Et3N to generate the radical anion PC˙−. Simultaneously, Et3N loses a hydrogen proton, which is abstracted by AcO−, leading to the creation of the radical intermediate A.9 In the second SET, 2a reacts with PC˙−, resulting in the formation of the radical 2a′ and the return of PC to its ground state PC. Next, the radical intermediate A engages in iodine atom transfer (XAT) with the substrate 1a, overcoming an energy barrier of 7.9 kcal mol−1, generating intermediates B and C. Radical intermediate B then undergoes 1,5-hydrogen atom transfer (1,5-HAT) via transition state TSD, surpassing a moderate activation energy of 7.9 kcal mol−1, thereby yielding the radical intermediate D. Subsequently, a barrierless radical–radical coupling between intermediate D and 2a′ leads to the formation of intermediate E (Fig. S8†). Finally, intermediate E overcomes a 7.0 kcal mol−1 energy barrier, facilitating the cleavage of the C–C bond and releasing 34.5 kcal mol−1 of free energy. Ultimately, this process yields the final product 3aa after the departure of the cyanide group. The entire process is highly exergonic, with a free energy release of 153.2 kcal mol−1. Therefore, our computational study provides additional support for the experimental findings, demonstrating that the reaction proceeds through a stepwise mechanism involving multiple SET, XAT, 1,5-HAT, and radical–radical coupling processes.
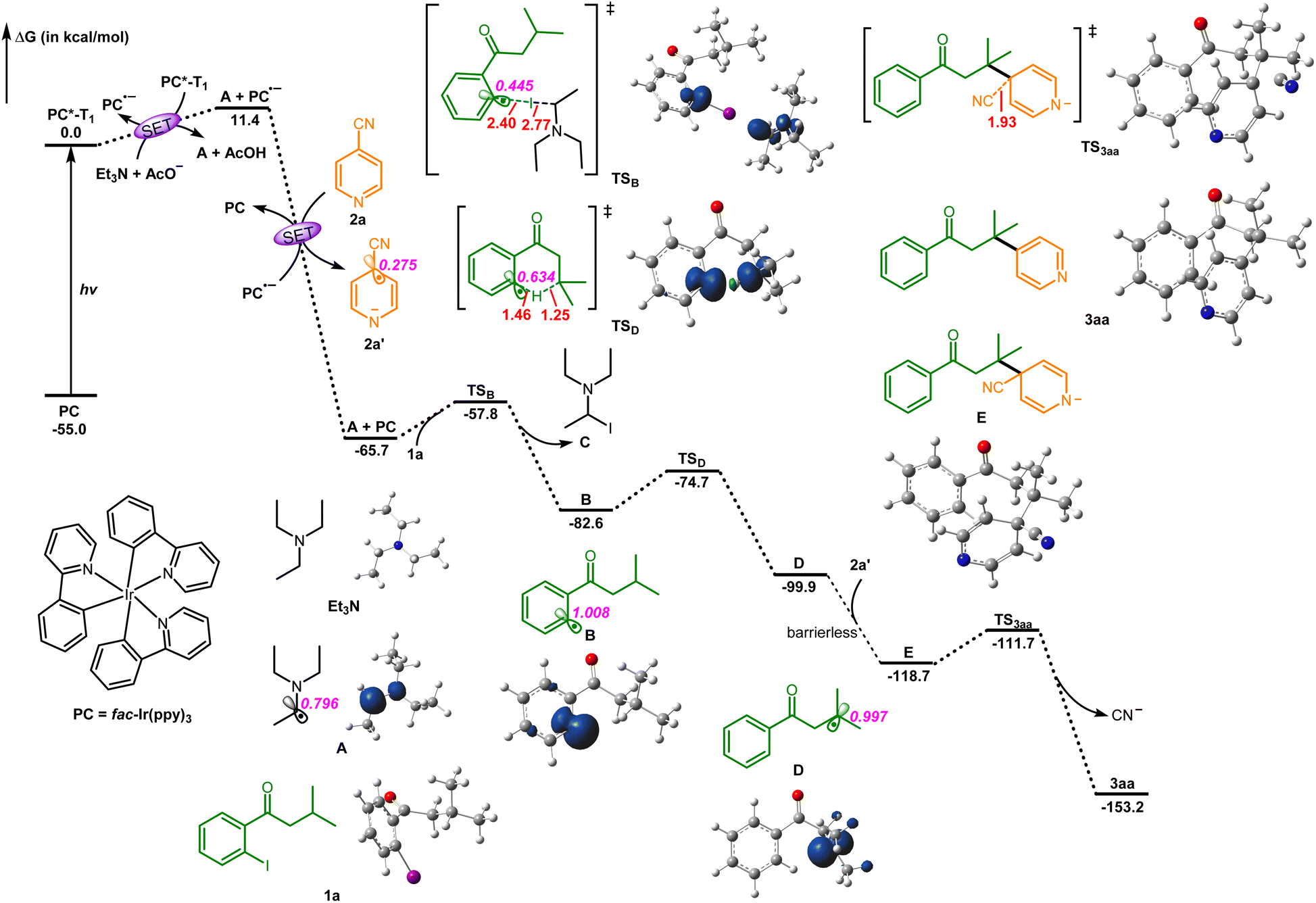 | ||
| Fig. 1 DFT-calculated energy profiles of the mechanistic pathway. Optimized intermediate and transition-state structures (key bond distances in Å) as well as the spin density plots (isovalue: 0.008 au) of key radical species are given. The Gibbs free energies (ΔG) are given in kcal mol−1. The italic numbers in purple are the corresponding Mulliken atomic spin populations of the radical centered atom. | ||
Consequently, a plausible mechanism for this photoredox remote C(sp3)–H arylation protocol as outlined in Scheme 3 was proposed.6–8 Photooxidation of Et3N by the excited state Ir(ppy)3* affords the Ir(ppy)3 radical anion and the Et3N radical cation, followed by deprotonation to generates the diethylaminoethyl radical intermediate A with the aid of bases (such as NaOAc and cyanoarenes).6–8 The XAT between the diethylaminoethyl radical intermediate B and 1-(2-iodophenyl)-3-methylbutan-1-one 1a produces the phenyl radical intermediate B and N,N-diethyl-1-iodoethan-1-amine C (6) (determined by HRMS analysis).7 The intermediate B subsequently undergoes an intramolecular 1,5-HAT to deliver the alkyl radical intermediate D.8 Meanwhile, single electron reduction of isonicotinonitrile 2a by the Ir(ppy)3 radical anion gives rise to the formation of the isonicotinonitrile radical anion 2a′ and regeneration of the Ir(ppy)3 photocatalyst.6 Radical–radical coupling between the alkyl radical intermediate D and the isonicotinonitrile radical anion 2a′ affords the anion intermediate E, followed by decyanation with the aid of bases to access the desired product 3aa.
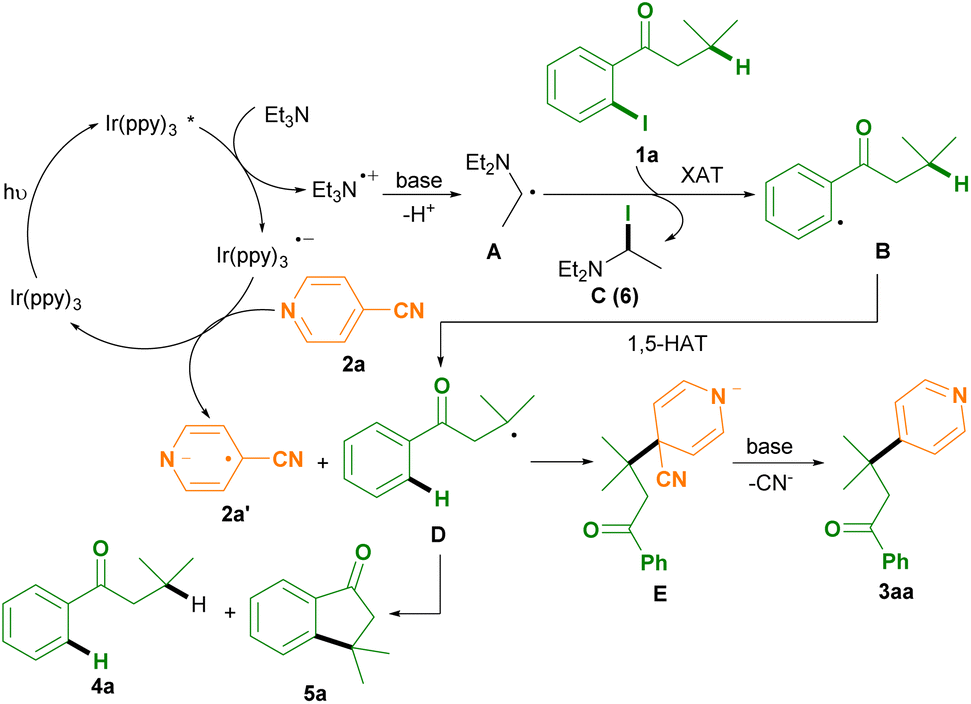 | ||
| Scheme 3 Possible reaction mechanism. | ||
Conclusions
In summary, we have developed a new, site selective iodoarene-directed photoredox β-C(sp3)–H arylation of 1-(o-iodoaryl)alkan-1-ones with cyanoarenes via XAT and 1,5-HAT for producing β-aryl arylalkanones. Using an aryl radical, which is generated from XAT between an iodoarene and the diethylaminoethyl radical, renders site selective 1,5-HAT to form the remote sp3-carbon radical and then radical–radical coupling, thus achieve iodoarene-directed remote C(sp3)–H arylation and prohibiting undesired radical pathways. This mild and selective iodoarene-directed photoredox XAT and HAT method for remote C(sp3)–H arylation delivers a diverse array of functionalized (hetero)arenes with high site-selectivity, a good tolerance of functionalization and a broad scope of remote C(sp3)–H bonds.Data availability
The datasets supporting this article have been uploaded as part of the ESI† material.Author contributions
Q. S., M. H., D.-L. H. and J.-H. L. conceptualized the project. L. Z., C.-H. X., M. H. and X.-H. O. performed the experimental studies. X.-Y. Z. and Q. S. performed the DFT calculations. Q. S., M. H., D.-L. H. and J.-H. L. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 22271245), and the Jiangxi Province Science and Technology Project (No. 20212AEI91002, 20202ACBL213002 and 20224BAB213013), the Jiangxi Educational Committee Foundation of China (No. GJJ210906), and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University (No. 2021ZD01) for their financial support.Notes and references
- For selected reviews, see: (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (c) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (d) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed; (e) X. Wu, Z. Ma, T. Feng and C. Zhu, Chem. Soc. Rev., 2021, 50, 11577–11613 RSC; (f) B. Yue, X. Wu and C. Zhu, Chin. J. Org. Chem., 2022, 42, 458–470 CrossRef CAS; (g) S. Saha, J. Das, S. A. Al-Thabaiti, S. M. Albukhari, Q. A. Alsulami and D. Maiti, Catal. Sci. Technol., 2023, 13, 11–27 RSC.
- For selected reviews on α-arylation of saturated carbonyls, see: (a) Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, Germany, 2000, pp. 227–483 Search PubMed; (b) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS PubMed; (c) P. Noväk and R. Martin, Curr. Org. Chem., 2011, 15, 3233–3262 CrossRef; (d) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676–707 CrossRef CAS PubMed; (e) Y.-J. Hao, X.-S. Hu, Y. Zhou, J. Zhou and J.-S. Yu, ACS Catal., 2020, 10, 955–993 CrossRef CAS.
- For selected reviews on β-C(sp3)–H functionalization of saturated carbonyls involving β-C(sp3)–H arylation, see: (a) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC; (b) G. Yan and A. J. Borah, Org. Chem. Front., 2014, 1, 838–842 RSC; (c) Z. Huang and G. Dong, Tetrahedron Lett., 2014, 55, 5869–5889 CrossRef CAS; (d) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764–7786 RSC; (e) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635–645 CrossRef CAS PubMed; (f) Q. Zhang and B.-F. Shi, Chin. J. Chem., 2019, 37, 647–656 CrossRef CAS; (g) C. Wang and G. Dong, ACS Catal., 2020, 10, 6058–6070 CrossRef CAS; (h) B. Liu, A. M. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957–15074 CrossRef CAS PubMed; (i) S. Dutta, T. Bhattacharya, F. J. Geffers, M. Burger, D. Maiti and D. B. Werz, Chem. Sci., 2022, 13, 2551–2573 RSC.
- For representative papers on transition-metal-catalyzed chelation-assisted β-arylation of carbonyls, see: amides: (a) Y. Feng, Y. Wang, B. Landgraf, S. Liu and G. Chen, Org. Lett., 2010, 12, 3414–3417 CrossRef CAS PubMed; (b) M. Wasa, K. S. L. Chan, X.-G. Zhang, J. He, M. Miura and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 18570–18572 CrossRef CAS PubMed; (c) R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030–6032 CrossRef CAS PubMed; (d) M. Tomanik and J.-Q. Yu, J. Am. Chem. Soc., 2023, 145, 17919–17925 CrossRef CAS PubMed , and references cited therein. Ketones/aldehydes: (e) R.-Y. Zhu, L.-Y. Liu, H. S. Park, K. Hong, Y. Wu, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 16080–16083 CrossRef CAS PubMed; (f) L. Pan, K. Yang, G. Li and H. Ge, Chem. Commun., 2018, 54, 2759–2762 RSC; (g) C. Dong, L. Wu, J. Yao and K. Wei, Org. Lett., 2019, 21, 2085–2089 CrossRef CAS PubMed; (h) L.-J. Xiao, K. Hong, F. Luo, L. Hu, W. R. Ewing, K.-S. Yeung and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 9594–9600 CrossRef CAS PubMed; (i) P. A. Provencher, J. F. Hoskin, J. J. Wong, X. Chen, J.-Q. Yu, K. N. Houk and E. J. Sorensen, J. Am. Chem. Soc., 2021, 143, 20035–20041 CrossRef CAS PubMed; (j) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS PubMed; (k) K. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775–12778 CrossRef CAS PubMed; (l) X.-L. Zhang, G.-F. Pan, X.-Q. Zhu, R.-L. Guo, Y.-R. Gao and Y.-Q. Wang, Org. Lett., 2019, 21, 2731–2735 CrossRef CAS PubMed; (m) K. Yang, Z. Li, C. Liu, Y. Li, Q. Hu, M. Elsaid, B. Li, J. Das, Y. Dang, D. Maiti and H. Ge, Chem. Sci., 2022, 13, 5938–5943 RSC. Acids: (n) R. Giri, N. Maugel, J. J. Li, D. H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS PubMed; (o) L. Hu, G. Meng and J.-Q. Yu, J. Am. Chem. Soc., 2022, 144, 20550–20553 CrossRef CAS PubMed.
- For representative papers on β-arylation of simple carbonyls, such as esters, amides and silyl ketene acetals, using a transition-metal redox catalysis, see: (a) A. Renaudat, L. Jean-Gérard, R. Jazzar, C. E. Kefalidis, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2010, 49, 7261–7265 CrossRef CAS PubMed; (b) S. Aspin, A.-S. Goutierre, P. Larini, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 51, 10808–10811 CrossRef CAS PubMed; (c) M. V. Leskinen, K.-T. Yip, A. Valkonen and P. M. Pihko, J. Am. Chem. Soc., 2012, 134, 5750–5753 CrossRef CAS PubMed; (d) M. V. Leskinen, K.-T. Yip, A. Valkonen and P. M. Pihko, J. Am. Chem. Soc., 2012, 134, 5750–5753 CrossRef CAS PubMed; (e) Z. Huang and G. Dong, J. Am. Chem. Soc., 2013, 135, 17747–17750 CrossRef CAS PubMed; (f) Z. Huang, Q. P. Sam and G. Dong, Chem. Sci., 2015, 6, 5491–5498 RSC; (g) Z. Huang and G. Dong, Tetrahedron, 2018, 74, 3253–3265 CrossRef CAS; (h) M. Chen, F. Liu and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 3815–3819 CrossRef CAS PubMed; (i) P. Gandeepan, P. Rajamalli and C.-H. Cheng, ACS Catal., 2014, 4, 4485–4489 CrossRef CAS; (j) S. Aspin, L. López-Suárez, P. Larini, A. S. Goutierre, R. Jazzar and O. Baudoin, Org. Lett., 2013, 15, 5056–5069 CrossRef CAS PubMed; (k) C.-H. Xu, L. Zeng, G.-F. Lv, J.-H. Qin, X.-H. Xu and J.-H. Li, Org. Lett., 2023, 25, 7645–7649 CrossRef CAS PubMed; (l) X. Jie, Y. Shang, X. Zhang and W. Su, J. Am. Chem. Soc., 2016, 138, 5623–5633 CrossRef CAS PubMed; (m) Z. Chen, H. Li, Y. Liao, M. Wang and W. Su, Chem. Commun., 2023, 59, 6686–6689 RSC.
- (a) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed; (b) T. Nakashima, H. Fujimori, K. Ohmatsu and T. Ooi, Chem.–Eur. J., 2021, 27, 9253–9256 CrossRef CAS PubMed; (c) W. Xie, D. Kim and S. Chang, J. Am. Chem. Soc., 2020, 142, 20588–20593 CrossRef CAS PubMed; (d) K. Liu and A. Studer, Angew. Chem., Int. Ed., 2022, 61, e202206533 CrossRef CAS PubMed.
- For reviews and representative papers, see: (a) W. P. Neumann, Synthesis, 1987, 665–683 CrossRef CAS; (b) C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed; (c) X.-S. Zhou, D.-M. Yan and J.-R. Chen, Chem, 2020, 6, 808–831 CrossRef; (d) C.-Y. Huang, J. Li and C.-J. Li, Chem. Sci., 2022, 13, 5465–5504 RSC; (e) F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed; (f) H. F. Piedra, C. Valdés and M. Plaza, Chem. Sci., 2023, 14, 5545–5568 RSC; (g) K. Sachidanandan, B. Niu and S. Laulhé, ChemCatChem, 2023, 15, e20230086 CrossRef; (h) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed; (i) H. Zhao, A. J. McMillan, T. Constantin, R. C. Mykura, F. Juliá and D. Leonori, J. Am. Chem. Soc., 2021, 143, 14806–14813 CrossRef CAS PubMed; (j) B. Górski, A.-L. Barthelemy, J. J. Douglas, F. Juliá and D. Leonori, Nat. Catal., 2021, 4, 623–630 CrossRef; (k) Z. Zhang, B. Górski and D. Leonori, J. Am. Chem. Soc., 2022, 144, 1986–1992 CrossRef CAS PubMed; (l) X. Tian, J. Kaur, S. Yakubov and J. P. Barham, ChemSusChem, 2022, 15, e202200906 CrossRef CAS PubMed; (m) N. Sanosa, B. Peñín, D. Sampedro and I. Funes-Ardoiz, Eur. J. Org Chem., 2022, 2022, e202200420 CrossRef CAS; (n) B. Mao, X.-Y. Zhang, Y. Wei and M. Shi, Chem. Commun., 2022, 58, 3653–3656 RSC; (o) V. S. Kostromitin, A. O. Sorokin, V. V. Levin and A. D. Dilman, Chem. Sci., 2023, 14, 3229–3234 RSC.
- For selected reviews and papers, see: (a) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569–1586 CrossRef CAS PubMed; (b) L. Xiao, J. Li and T. Wang, Acta Chim. Sin., 2019, 77, 841–849 CrossRef CAS; (c) H. Chen and S. Yu, Org. Biomol. Chem., 2020, 18, 4519–4532 RSC; (d) G. Kumar, S. Pradhan and I. Chatterjee, Chem.–Asian J., 2020, 15, 651–672 CrossRef CAS PubMed; (e) W. Guo, Q. Wang and J. Zhu, Chem. Soc. Rev., 2021, 50, 7359–7377 RSC; (f) X. Wu, Z. Ma, T. Feng and C. Zhu, Chem. Soc. Rev., 2021, 50, 11577–11613 RSC; (g) J.-M. Xi and W.-W. Liao, Org. Chem. Front., 2022, 9, 4490–4506 RSC; (h) X. F. Xia, Q. Huang, T. Y. Sun, Y. Jiang and G. Ran, ACS Catal., 2022, 12, 8868–8876 CrossRef; (i) S. Sarkar, W. Sidhant, X. Jia and V. Gevorgyan, Chem, 2022, 8, 3096–3108 CrossRef CAS PubMed; (j) R. Guo, H. Xiao, S. Li, Y. Luo, J. Bai, M. Zhang, Y. Guo, X. Qi and G. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208232 CrossRef CAS PubMed; (k) M. G. Pizzio, E. G. Mata, P. Dauban and T. Saget, Eur. J. Org. Chem., 2023, 26, e202300616 CrossRef CAS; (l) W.-X. Tang, K.-Q. Chen, D.-Q. Sun and X.-Y. Chen, Org. Biomol. Chem., 2023, 21, 715–718 RSC; (m) J. Wang, Q. Xie, G. Gao, H. Li, W. Lu, X. Cai, X. Chen and B. Huang, Org. Chem. Front., 2023, 10, 4394–4399 RSC; (n) X.-Y. Wang, Y.-Q. He, Y. Zhou, L. Lu, X.-R. Song, Z.-Z. Zhou, W.-F. Tian and Q. Xiao, Org. Lett., 2023, 25, 3847–3852 CrossRef CAS PubMed.
- Y. Li and D. Wang, Phys. Chem. Chem. Phys., 2018, 20, 12106–12111 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06637a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |