
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg1+ incorporation via a Zr4+-anchored metalloligand: fine-tuning catalytic Ag sites in Zr/Ag bimetallic clusters for enhanced eCO2RR-to-CO activity†
Liang-Jun
Li
a,
Wen-Lei
Mu
a,
Yi-Qi
Tian
a,
Wei-Dong
Yu
 *b,
Lan-Yan
Li
*b,
Jun
Yan
a and
Chao
Liu
*a
*b,
Lan-Yan
Li
*b,
Jun
Yan
a and
Chao
Liu
*a
aHunan Provincial Key Laboratory of Chemical Power Sources, College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, Hunan, P. R. China. E-mail: chaoliu@csu.edu.cn
bChina College of Science, Hunan University of Technology and Business, Changsh 410000, P. R. China
First published on 25th March 2024
Abstract
Attaining meticulous dominion over the binding milieu of catalytic metal sites remains an indispensable pursuit to tailor product selectivity and elevate catalytic activity. By harnessing the distinctive attributes of a Zr4+-anchored thiacalix[4]arene (TC4A) metalloligand, we have pioneered a methodology for incorporating catalytic Ag1+ sites, resulting in the first Zr–Ag bimetallic cluster, Zr2Ag7, which unveils a dualistic configuration embodying twin {ZrAg3(TC4A)2} substructures linked by an {AgSal} moiety. This cluster unveils a trinity of discrete Ag sites: a pair ensconced within {ZrAg3(TC4A)2} subunits and one located between two units. Expanding the purview, we have also crafted ZrAg3 and Zr2Ag2 clusters, meticulously mimicking the two Ag site environment inherent in the {ZrAg3(TC4A)2} monomer. The distinct structural profiles of Zr2Ag7, ZrAg3, and Zr2Ag provide an exquisite foundation for a precise comparative appraisal of catalytic prowess across three Ag sites intrinsic to Zr2Ag7. Remarkably, Zr2Ag7 eclipses its counterparts in the electroreduction of CO2, culminating in a CO faradaic efficiency (FECO) of 90.23% at −0.9 V. This achievement markedly surpasses the performance metrics of ZrAg3 (FECO: 55.45% at −1.0 V) and Zr2Ag2 (FECO: 13.09% at −1.0 V). Utilizing in situ ATR-FTIR, we can observe reaction intermediates on the Ag sites. To unveil underlying mechanisms, we employ density functional theory (DFT) calculations to determine changes in free energy accompanying each elementary step throughout the conversion of CO2 to CO. Our findings reveal the exceptional proficiency of the bridged-Ag site that interconnects paired {ZrAg3(TC4A)2} units, skillfully stabilizing *COOH intermediates, surpassing the stabilization efficacy of the other Ag sites located elsewhere. The invaluable insights gleaned from this pioneering endeavor lay a novel course for the design of exceptionally efficient catalysts tailored for CO2 reduction reactions, emphatically underscoring novel vistas this research unshrouds.
Introduction
The electrochemical reduction of CO2 (eCO2RR) stands as a highly promising pathway for the conversion of CO2 into valuable chemical fuels.1,2 Among the diverse range of potential electrocatalysts, silver (Ag)-based nanomaterials have garnered significant attention due to their remarkable selectivity in producing CO.3–6 Despite notable advancements in the synthesis of monodisperse Ag nanoparticles, accurately characterizing their structural attributes and identifying catalytic Ag sites remains a formidable challenge.7,8 This limitation hampers the attainment of a comprehensive understanding of the intricate relationship between structure and activity, consequently impeding the overall progress in this research field. Hence, a pivotal stride toward advancement involves achieving a controlled synthesis of Ag sites' coordination environments. This endeavor would pave way for the creation of meticulously defined catalytic centers. Embracing such a controlled approach holds the potential to unravel the multifaceted interplay between the structure and activity of Ag nanocatalysts, thereby ushering in new avenues for efficient catalysis.9–13Metal oxides have gained prominence as substrates for stabilizing Ag nanoparticles while exposing catalytic sites, thereby contributing significantly to the realm of Ag catalysis.14–16 Zirconia (ZrO2) has demonstrated its potential for effectively stabilizing Ag nanoparticles, offering a diverse range of catalytic applications.17,18 Nevertheless, the intricate interface and surface composition of Ag–ZrO2 nanomaterials remain enigmatic, posing challenges in understanding their complex nature. Extensive research endeavors have been directed towards unraveling the structural and molecular intricacies of Ag–ZrO2 materials, a pursuit that promises valuable insights into binding patterns and atomic-scale electronic configurations. Integrating active catalytic Ag sites into zirconium-oxygen clusters presents a reliable avenue for crafting an Ag–ZrO2 molecular model, offering the prospect of precisely modulating the coordination environment of Ag. Remarkably, such bimetallic clusters, however, have yet to be synthesized. The synthesis of Zr/Ag bimetallic clusters hinges on a strategically designed ligand system that facilitates the orchestrated assembly of Zr and Ag. This design imperative is pivotal in enabling their cooperative engagement to shape bimetallic clusters. Thiacalix[4]arene (TC4A), a subclass of calixarenes, has exhibited exceptional coordination traits with metals due to its distinctive –OH and –S– functional groups.19–22 As such, TC4A emerges as a promising scaffold for constructing nanosized clusters, rendering it a viable candidate for cluster assembly.23–30 By leveraging the principles of soft and hard acid-base theory, Zr4+ is classified as a hard Lewis acid with a propensity for phenolic oxygen, while Ag+ is identified as a soft Lewis acid with an affinity for soft bases such as sulfur atoms.31–34 By capitalizing on these principles, the interaction of Zr4+ and Ag+ with TC4A holds the tantalizing potential to yield uncharted bimetallic clusters characterized by distinctive geometric and electronic configurations.35
This article pioneers the sequential assembly of Zr/Ag bimetallic nanoclusters with clear Ag catalytic sites, utilizing Zr4+-anchored TC4A as a metalloligand and further explores the applications of these clusters in eCO2RR. The groundbreaking synthesis of a dimeric Zr2Ag7, {HNEt3}2{H2Zr2Ag7(TC4A)4(HSal)3}, is reported. This cluster unveils an innovative triad of distinct catalytic Ag+ sites, including a pair nested within the {ZrAg3(TC4A)2} subunits and one situated between the two subunits, each characterized by unique environments, potentially leading to a diverse range of catalytic activities. We employed mass spectrometry to trace the assembly mechanism of Zr2Ag7, successfully isolating two intermediate structures—ZrAg3 and Zr2Ag2 clusters. These structures contain two or one Ag site(s) from the {ZrAg3(TC4A)2} subunits, respectively. By comparing the eCO2RR performance of three clusters, we accurately compared the activity of those three Ag sites. Remarkably, Zr2Ag7 displays remarkable catalytic prowess in eCO2RR, with a CO faradaic efficiency (FECO) of 90.23% at −0.9 V, surpassing ZrAg3 (FECO: 55.45% at −1.0 V) and ZrAg3 (FECO: 13.09% at −1.0 V). To delve deeper, the DFT method was employed to calculate the free energy change during CO2 to CO conversion in different Ag sites. It was demonstrated that the Ag site located on the {Ag(HSal)} moiety could stabilize *COOH in eCO2RR better than those located on the {ZrAg3(TC4A)2} unit.
Results and discussion
Synthesis and characterization
In the initial phase of our investigation, crystals of Zr2Ag7 were synthesized through a solvothermal reaction involving salicylic acid (H2Sal), TC4A, Zr(OAc)4, Ag(CF3CO2) and triethylamine. This reaction took place in MeCN at 80 °C for a duration of 3 days. The ensuing single-crystal analysis unveiled that Zr2Ag7 crystallizes in the monoclinic crystal system with a P21/n space group. The structure comprises a cluster core bearing a total of two negative charges, with counter cations provided by two protonated triethylamines. The molar ratio of Zr to Ag in Zr2Ag7, determined through energy-dispersive X-ray spectroscopy (EDS), is approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.5, which is consistent with the crystal structure. The high-resolution Ag 3d spectrum exhibits two peaks at 374.08 and 368.08 eV, corresponding to the binding energies of Ag 3d5/2 and Ag 3d3/2, respectively, indicating an oxidation state of +1 for Ag (Fig. S21†). The Zr2Ag7 cluster can be dissected into two distinct {ZrAg3(TC4A)2} substructures and an {Ag(HSal)} fragment. Within the {ZrAg3(TC4A)2} unit, the configuration of Zr4+ and three Ag+ takes on a parallelogram arrangement, capped by two deprotonated TC4A ligands and sheltered by a single HSal− ligand, as depicted in Fig. 1A. The Zr4+ site adopts an octahedral coordination geometry, facilitated by coordinating with four phenoxide groups and two S atoms originating from two TC4A ligands. Additionally, coordination involves a carboxylic O and a hydroxyl O from a Sal− ligand. The three Ag+ sites within the {ZrAg3(TC4A)2} unit are categorized into two groups. The two Ag(I) sites in close proximity to Zr4+ exhibit a hexacoordinate environment, while the Ag(II) site, situated farther away from Zr4+, adopts a tetracoordinate configuration. Each site is coordinated by the phenoxide groups and S atoms. The Ag(III) site within the {Ag(HSal)} fragment also exhibits a tetrahedral coordination structure, stabilized by two S atoms from the two {ZrAg3(TC4A)2} units and two O atoms from one HSal− ligand. However, these two O atoms originate from the carboxyl group and, compared to the chelation mode in {Zr(HSal)}, this coordination mode is relatively unstable. The distances involving Ag–O range from 2.297(2) to 2.518(5) Å, while Ag–S distances span from 2.519(4) to 2.493(7) Å. Additionally, the distance between the benzene ring of Sal− and Ag(III) site is 3.420 Å, indicating relatively weak π–d interactions, which provide additional stability to the Ag(III) site (Fig. 1B). From this perspective, this particular Ag site can be regarded as a cluster-stabilized catalytic site, potentially leading to unexpected catalytic effects (Fig. 1C).
:3.5, which is consistent with the crystal structure. The high-resolution Ag 3d spectrum exhibits two peaks at 374.08 and 368.08 eV, corresponding to the binding energies of Ag 3d5/2 and Ag 3d3/2, respectively, indicating an oxidation state of +1 for Ag (Fig. S21†). The Zr2Ag7 cluster can be dissected into two distinct {ZrAg3(TC4A)2} substructures and an {Ag(HSal)} fragment. Within the {ZrAg3(TC4A)2} unit, the configuration of Zr4+ and three Ag+ takes on a parallelogram arrangement, capped by two deprotonated TC4A ligands and sheltered by a single HSal− ligand, as depicted in Fig. 1A. The Zr4+ site adopts an octahedral coordination geometry, facilitated by coordinating with four phenoxide groups and two S atoms originating from two TC4A ligands. Additionally, coordination involves a carboxylic O and a hydroxyl O from a Sal− ligand. The three Ag+ sites within the {ZrAg3(TC4A)2} unit are categorized into two groups. The two Ag(I) sites in close proximity to Zr4+ exhibit a hexacoordinate environment, while the Ag(II) site, situated farther away from Zr4+, adopts a tetracoordinate configuration. Each site is coordinated by the phenoxide groups and S atoms. The Ag(III) site within the {Ag(HSal)} fragment also exhibits a tetrahedral coordination structure, stabilized by two S atoms from the two {ZrAg3(TC4A)2} units and two O atoms from one HSal− ligand. However, these two O atoms originate from the carboxyl group and, compared to the chelation mode in {Zr(HSal)}, this coordination mode is relatively unstable. The distances involving Ag–O range from 2.297(2) to 2.518(5) Å, while Ag–S distances span from 2.519(4) to 2.493(7) Å. Additionally, the distance between the benzene ring of Sal− and Ag(III) site is 3.420 Å, indicating relatively weak π–d interactions, which provide additional stability to the Ag(III) site (Fig. 1B). From this perspective, this particular Ag site can be regarded as a cluster-stabilized catalytic site, potentially leading to unexpected catalytic effects (Fig. 1C).
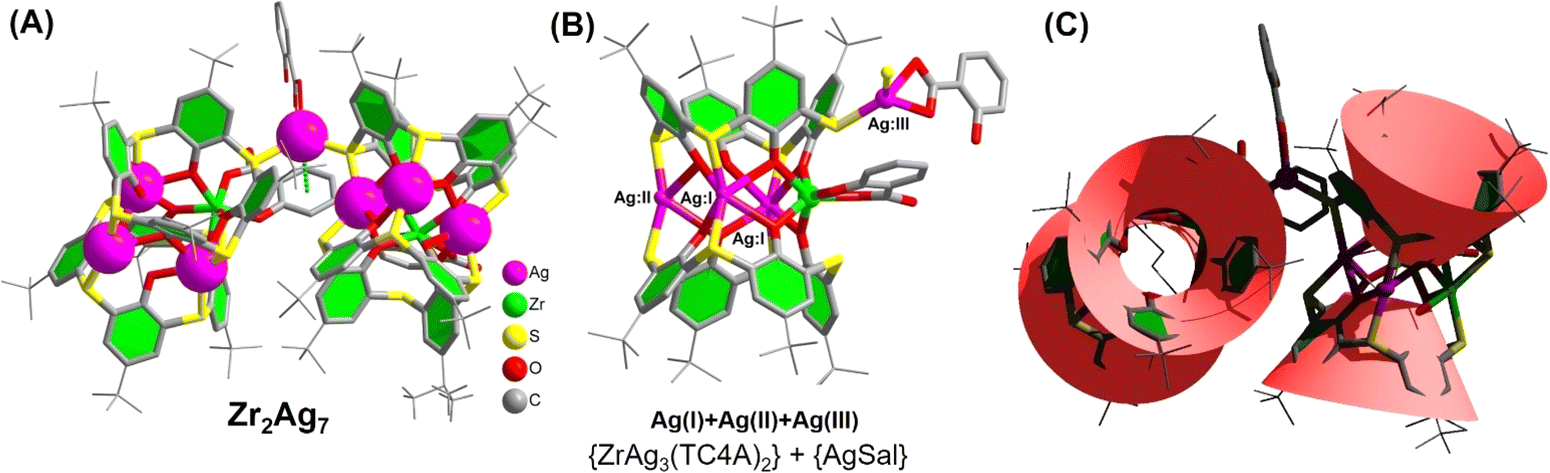 | ||
| Fig. 1 (A) Molecular structure of the Zr2Ag7 cluster; (B) {ZrAg3(TC4A)2} and {AgSal} units; (C) topology structure of the Zr2Ag7 cluster. | ||
The PXRD pattern of Zr2Ag7 crystals exhibits a favorable correspondence with the simulated pattern, providing confirmation of its phase purity (Fig. S11†). Due to intermolecular interactions, crystalline Zr2Ag7 samples are insoluble in most solvents but can dissolve in dichloromethane (CH2Cl2). To investigate its solution stability, Zr2Ag7 crystals were dissolved in CH2Cl2, and the composition was analyzed using ESI-MS in the positive mode. In the ESI-MS spectrum of Zr2Ag7 (Fig. 2), the signal corresponding to the complete cluster was not observed; instead, rich fragmentation information was detected. The signal that appeared at m/z = 4081.07 (1a) represents [H4Zr2Ag7(TC4A)4(HSal)2]+, resulting from the loss of a Sal− ligand from the cluster. Zr2Ag7 contains three Sal− ligands, two of which coordinate with Zr4+, while the other coordinates with Ag1+. The binding strength of this O-containing ligand on the metals follows the order of Zr4+ > Ag+, in accordance with the conventional Hard–Soft Acid–Base (HSAB) theory. This suggests that the Sal2− coordinated with Ag+ in the Ag(HSal) fragment may detach, exposing the Ag(III) active center. Moreover, in the lower m/z range, signals corresponding to the units of {ZrAg3 ± x(TC4A)2} (where x = 0–2) are observed with a high abundance, indicating an equilibrium between parent clusters and fragments in the solution. The most prominent envelope, centered at m/z = 1989.57, can be attributed to the species [H3ZrAg3(TC4A)2(Sal)]+ (calculated m/z = 1989.50), corresponding to half of Zr2Ag7. Additionally, signal peaks corresponding to the species {H2ZrAg4(TC4A)2(Sal)}+ (1b), [H4ZrAg2(TC4A)2(Sal)]+ (1d), and [H4ZrAg(TC4A)2(Sal)]+ (1f) are observed, which are derived from ZrAg3 by either losing a Ag1+ or gaining an additional Ag1+ ion. The gain and loss of Ag1+ from the cluster units indicate that the Zr4+-anchored metalloligand {Zr(TC4A)2} can act as a carrier for Ag1+, with its surface-rich S/O sites providing binding sites.
 | ||
| Fig. 2 Positive ion mode ESI-MS of the Zr2Ag7 clusters in CH2Cl2. | ||
The Zr2Ag7 cluster unveils an innovative triad of distinct Ag+ sites. To conduct precise comparative assessments of the catalytic activity of those sites, the isolation of structural intermediates assumes paramount importance. Through meticulous ESI-MS analysis, a breakthrough surfaced: the identification of the monomeric {ZrAg3(TC4A)2} entity and a discovery that was subsequently subjected to successful crystallization and structural resolution. Synthesized via the interaction between TC4A, Ag(OAc), and Zr(acac)4 in CH3CN, ZrAg3 (Fig. 3B) mirrored the {ZrAg3(TC4A)2} unit within Zr2Ag7. Notably, this structural analog bore two distinct Ag sites, Ag(I) and Ag(II), a remarkable alignment. Intriguingly, in the absence of SalH2 during the reaction, another monomeric configuration emerged: Zr2Ag2 with the composition of {Zr2Ag2(TC4A)2(acac)2} (Fig. 3C). This isomorphic variant, synthesized under solvothermal conditions using Zr(acac)4, TC4A, and Ag(OAc) in MeCN/DMF, exhibited a geometric resemblance to {ZrAg3(TC4A)2}. However, it diverged by substituting one Ag+ and one Zr(Sal) with two Zr(acac), exclusively showcasing the [AgO4S2] coordination geometry, solely encapsulating the Ag(II) sites. Moreover, our investigation extended to an intriguing isomorphic entity, Zr2Na2 (Fig. 3D), wherein the precision replacement of two Ag+ with Na+ showcased an exceptional flexibility within the {Zr2(TC4A)2} core. Notably, Na+ and Ag+ valence similarities rendered this replacement feasible.
 | ||
| Fig. 3 Molecular structures of the {Zr2(TC4A)2} metalloligand (A), as well as ZrAg3 (B), Zr2Ag2 (C) and Zr2Na2 (D) clusters. | ||
From a structural standpoint, the {AgSal} site functions as a bridge, connecting two {ZrAg3(TC4A)2} units and ultimately leading to the formation of the dimeric Zr2Ag7 cluster. The pivotal question at this juncture was whether ZrAg3 could undergo further transformation into Zr2Ag7. To validate this hypothesis, we dispersed ZrAg3 crystals in CH3CN/DCM, introduced Ag(OAc) and H2Sal to the solution, and conducted the reaction at 80 °C, monitoring the process using ESI-MS. Fig. 4 illustrates the time-dependent ESI-MS analysis of the mother liquor at various intervals during the reaction. In the initial stage, only a peak corresponding to {H2ZrAg3(TC4A)2(H2O)}+ (found: m/z = 1851.05) was observed in the ESI-MS, indicating the retention of the integrity of the ZrAg3 cluster in solution. However, as the reaction progressed, new signals emerged. Specifically, peaks corresponding to the units of {H4ZrAg3(TC4A)2(Sal)}+ (m/z = 2011.77) and {H3ZrAg4(TC4A)2(Sal)}+ (m/z = 2095.94) were detected at 30 min and 60 min, respectively. This signal was formed by the binding of Ag1+ to the {ZrAg3(TC4A)2(Sal)} core, suggesting that {ZrAg3(TC4A)2(Sal)} could serve as a seed for further growth. At the 120 min mark, the peak corresponding to {H3Zr2Ag7(TC4A)2(Sal)}+ (m/z = 4083.05) was observed. These findings clearly demonstrate that Zr2Ag7 could be derived from ZrAg3, following a small-to-large assembly pathway. It is worth noting that after two days of reaction, crystals of Zr2Ag7 were obtained, as confirmed by single crystal X-ray diffraction.
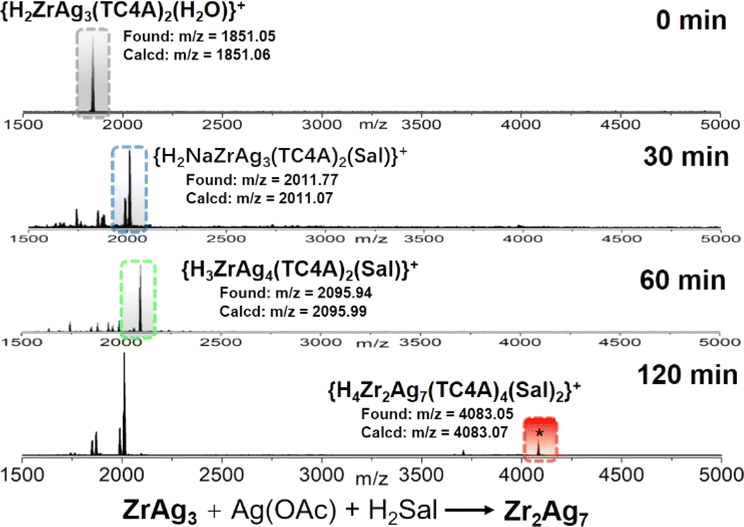 | ||
| Fig. 4 Time-dependent ESI-MS in the range of m/z 1500–5000 for the reaction of ZrAg3, AgOAc and SalH2 in MeCN/DCM at 100 °C at 0 min, 30 min, 60 min and 120 min. | ||
Electrochemical CO2 reduction
The role of Ag sites within Zr2Ag7 unveils a triadic categorization: one nestled amidst a {Ag(HSal)} moiety, and the remaining pair ensconced within two {ZrAg3(TC4A)2} moieties. Clearly, the catalytic potency of Ag sites is intimately entwined with their coordinating milieu. Undertaking a meticulous comparative analysis of different sites at the molecular level assumes paramount importance to fine-tune the catalyst performance.36–47 The structural disparities amongst Zr2Ag2, ZrAg3, and Zr2Ag7 offer an opportune platform for discerning CO2 reduction activity variances across these Ag sites. To ascertain the catalytic prowess of Ag, we also introduced Zr2Na2, an isomorph of Zr2Ag2, into our catalytic reactions, aiming to accentuate the superiority of Ag doping. Furthermore, ZrO2/Ag2O nanoparticles were synthesized and utilized as electrodes for a comparative analysis.48CO2 reduction experiments unfolded within a three-electrode H-type electrochemical cell, employing 0.5 M KHCO3 as the electrolyte. Analyzing products entailed a judicious fusion of gas chromatography (GC) and 1H-NMR techniques. Linear sweep voltammetry (LSV) delved into the heart of the matter, unearthing intriguing revelations. Zr2Ag7 took the center stage, showcasing a remarkable current density amplification and a more optimistic onset potential within CO2-saturated electrolytes, outshining its Ar-purged counterpart—an unmistakable manifestation of its electrocatalytic proficiency (Fig. 5A). ZrAg3 was a close contender, mirroring analogous trends. In stark contrast, the disparity widened significantly upon the entry of Zr2Ag2 and Zr2Na2 into the competition. In addition, the blank electrode and Ag2O/ZrO2 nanoparticles showed no CO2RR activity (Fig. S29 and S30†). The entire potential spectrum bore witness to the dominance of Zr2Ag7, underscoring its unassailable prominence. It is worth nothing that the propensity of Zr2Ag7 for more positive onset potentials (−0.49 V vs. RHE) vis-a-vis ZrAg3 (−0.70 V vs. RHE) and Zr2Ag2 (−0.79 V vs. RHE) in CO2-saturated electrolytes. While Zr2Na2 and Zr2Ag2 remained elusive in terms of catalytic activity inference from LSV curves, their participation in electrolysis experiments presented a differential narrative. Analyzing gaseous products solely revealed CO and H2 for all aforementioned structures, as 1H-NMR failed to detect any liquid byproducts. Remarkably, Zr2Ag7 exhibited an unwavering faradaic efficiency (FE) for CO, culminating in an impressive FECO of 90.23% at −0.9 V vs. RHE, surpassing the performance of most similar Ag-based CO2RR catalysts (Table S2†). In stark contrast, ZrAg3 and Zr2Ag2 lagged, attaining their peak FECO of 55.45% and 13.09%, respectively, at −1.0 V vs. RHE, whereas FECO of Zr2Na2remained insubstantial, almost vanishing across the potential spectrum (Fig. 5B)—an eloquent testament to the preeminence of Ag doping. Fig. 5C paints a revealing picture, juxtaposing the computed CO partial current density (jco) for Zr2Ag7, ZrAg3, and Zr2Ag2. Zr2Ag7 emerged triumphant, boasting a jco of 20.01 mA cm−2 at 1.2 V vs. RHE, dwarfing ZrAg3 (7.19 mA cm−2) and overshadowing Zr2Ag2 by a staggering 36-fold margin (0.55 mA cm−2). The CO2RR turnover frequency calculations further underscored this supremacy, with Zr2Ag7 consistently outperforming ZrAg3 and Zr2Ag2 across all potential domains (Fig. 5D). The unwavering consistency across these findings cements the unequivocal excellence of Zr2Ag7 in selectively electroreducing CO2 to CO, eclipsing its catalyst counterparts.
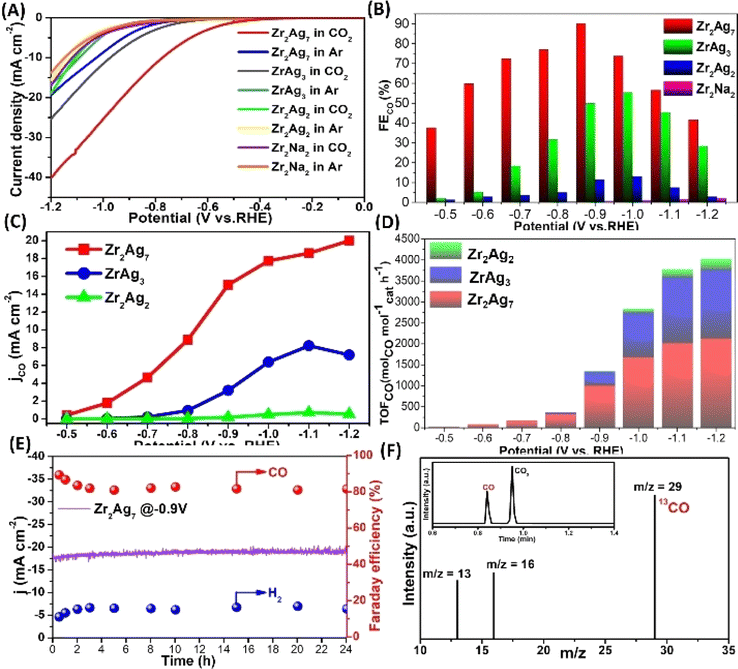 | ||
| Fig. 5 (A) LSV of samples in Ar or CO2 saturated 0.5 M KHCO3 solution; (B) FECO values of Zr2Ag7, ZrAg3, Zr2Ag2 and Zr2Na2 at different voltages; (C) CO partial current density (jCO); (D) TOF values of Zr2Ag7, ZrAg3 and Zr2Ag2 at potentials of −0.5 to −1.2 V vs. RHE; (E) I–t test and FECO values of Zr2Ag7 at −0.9 V in different time periods; (F) GC-MS spectra of 13CO recorded under a13CO2 atmosphere. | ||
To elucidate factors contributing to the difference in catalytic activity between the catalysts, a Tafel analysis was employed to characterize the reaction kinetics in the electrolyte (Fig. S38†). From Zr2Ag7 to ZrAg3 and then to Zr2Ag2, the Tafel slopes of the three species increase sequentially, indicating that the electron transfer energy consumption of the catalyst gradually increases. These findings suggest potential kinetic advantages for Zr2Ag7 in eCO2RR. The electron transfer ability of the catalysts in the electrolyte was further investigated using electrochemical impedance spectroscopy (Fig. S39†). The Nyquist curve clearly indicates that Zr2Ag7 has a lower charge transfer resistance compared to the other two, indicating good electron transfer ability for Zr2Ag7, which is beneficial for the CO2RR process. Moreover, electrochemical measurement of the active surface area (ECSA) revealed that Zr2Ag7 exhibits a higher density of accessible active sites (Fig. S40†). To verify whether Zr2Ag7 exhibits a sustained robustness in electrochemical reactions, in a 0.5 hours electrolysis process across the voltage range of −0.6 V to −1.2 V Zr2Ag7 demonstrated a stable total current density (Fig. S35†). In addition, a consistent current density of about −20 mA and FECO > 80% were kept for 24 hours when operating in the long-term mode at −0.9 V (Fig. 5E). This excellent performance emphasizes the ultrastable character of Zr2Ag7 once again. After the reaction, we recovered the catalyst and conducted ESI-MS measurements. The ESI-MS pattern exhibited a signal corresponding to [H4Zr2Ag7(TC4A)4(HSal)2]+, indicating the continued stability of the Zr2Ag7 structure (Fig. S41†). PXRD and FT-IR revealed that the characteristic signal of the catalyst was preserved after electrolysis (Fig. S42 and S43†). Moreover, EDS analysis of the catalyst post-reaction revealed that Zr and Ag elements maintained a consistent 1:3.5 ratio (Fig. S47†). XPS analysis showed no significant change in the binding energy of Ag in the catalyst after the reaction, indicating the preservation of its coordination environment (Fig. S48†). TEM analysis revealed the presence of clusters in the solution as discrete particles, further confirming the stability of the catalyst (Fig. S49†). To accurately determine the C source of CO, isotope experiments were conducted using 13CO2 as the C source under similar catalytic conditions. The detection of 13CO (m/z = 29) via GC-MS analysis unequivocally confirmed that the generated CO originated from CO2 (Fig. 5F).
The electrocatalytic reduction of CO2 using Ag-based materials typically involves three main steps (Fig. 6A).49 Initially, CO2 is adsorbed on the catalyst surface in the form of *COOH, a process commonly referred to as Proton-Coupled Electron Transfer (PCET) (Step 1). Subsequently, the *COOH species acquires H+ and e−, converting to *CO after releasing H2O (Step 2). Finally, *CO desorbs from the catalyst surface, yielding CO (Step 3). To validate the basic principle of this mechanism, we employed in situ electrochemical attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR).50–54 This technology monitors the real-time absorption of evanescent waves by catalyst surface substances generated by infrared total reflection. We set the potential range between −0.5 V and −1.5 V (vs. RHE) and compared changes in relevant absorption peaks using ZrAg3 and Zr2Ag7 as electrocatalysts (Fig. 6B and C). Both spectra exhibit similarities, and a distinctive peak near 1698 cm−1 is attributed to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in *COOH, becoming more pronounced with increasing voltage. Additionally, the broad peak in the range of 1346–1394 cm−1 represents the stretching of C–OH in *COOH, which increases from −0.5 V to −1.5 V. This trend, akin to the intensity change at 1698 cm−1, indicates a significant increase in surface coverage of *COOH species with rising voltage. At approximately 2104 cm−1, a weak signal is observed, attributed to the Ag–*CO vibration mode. At lower potentials, the *CO band intensity slightly increases, indicating the adsorption of *CO on the catalyst surface. However, at higher potentials (above −0.9 V), CO desorption accelerates, leading to a gradual decrease in the 2104 cm−1 peak until it becomes undetectable. Notably, the intermediate characteristic peak of ZrAg3 becomes distinctly observable only from an electrode potential of approximately −0.8 V, while the corresponding characteristic peaks of Zr2Ag7 are already evident at −0.5 V. This observation indicates that Zr2Ag7 demonstrates a higher reaction activity and stronger catalytic proficiency for eCO2RR, aligning with experimental findings. It should be noted that the position shift of the 1698 cm−1 peak in Zr2Ag7 may be attributed to changes in the coverage of binding species on the catalyst surface caused by different potentials.
O in *COOH, becoming more pronounced with increasing voltage. Additionally, the broad peak in the range of 1346–1394 cm−1 represents the stretching of C–OH in *COOH, which increases from −0.5 V to −1.5 V. This trend, akin to the intensity change at 1698 cm−1, indicates a significant increase in surface coverage of *COOH species with rising voltage. At approximately 2104 cm−1, a weak signal is observed, attributed to the Ag–*CO vibration mode. At lower potentials, the *CO band intensity slightly increases, indicating the adsorption of *CO on the catalyst surface. However, at higher potentials (above −0.9 V), CO desorption accelerates, leading to a gradual decrease in the 2104 cm−1 peak until it becomes undetectable. Notably, the intermediate characteristic peak of ZrAg3 becomes distinctly observable only from an electrode potential of approximately −0.8 V, while the corresponding characteristic peaks of Zr2Ag7 are already evident at −0.5 V. This observation indicates that Zr2Ag7 demonstrates a higher reaction activity and stronger catalytic proficiency for eCO2RR, aligning with experimental findings. It should be noted that the position shift of the 1698 cm−1 peak in Zr2Ag7 may be attributed to changes in the coverage of binding species on the catalyst surface caused by different potentials.
 | ||
| Fig. 6 (A) Schematic depiction of the proposed reaction mechanism of CO2 reduction to CO on catalysts. (B) and (C) The ATR-FTIR results from 2600 to 1300 cm−1 on ZrAg3 and Zr2Ag7. | ||
To further comprehend variations in the catalytic performance among the three Ag sites, specific DFT calculations were conducted to investigate the reaction pathways of different Ag sites on the three clusters. Catalyst models were optimized based on the crystal structures of Zr2Ag7, ZrAg3, and Zr2Ag2, simplifying tBu groups with H atoms to expedite calculation convergence. Additionally, the HSal− ligand in Zr2Ag7 was removed to create the active Ag(III) site. Fig. 7A distinctly illustrates Gibbs free energy changes (ΔG) for each reaction step. For the Ag(I) and Ag(II) sites in Zr2Ag7_m, 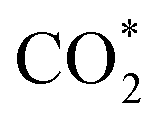 captures a proton–electron pair from the electrolyte to form a *COOH intermediate with ΔG values of 1.68 and 1.92 eV, respectively. These values are significantly larger than the ΔG of the subsequent step to form *CO, implying that the formation of *COOH is the rate-determining step of the reaction. In contrast, the calculated ΔG value for *COOH formation via
captures a proton–electron pair from the electrolyte to form a *COOH intermediate with ΔG values of 1.68 and 1.92 eV, respectively. These values are significantly larger than the ΔG of the subsequent step to form *CO, implying that the formation of *COOH is the rate-determining step of the reaction. In contrast, the calculated ΔG value for *COOH formation via hydrogenation on the Ag(III) site is 0.22 eV, while the ΔG value for the subsequent *CO generation step is 0.40 eV. Both values are much lower than those observed on the Ag(I) and Ag(II) sites. Similar conclusions were drawn when calculating the free energy changes for Ag(II) on ZrAg3_m and Ag(I) on Zr2Ag2_m, which give larger ΔG values of 1.79 and 2.06 eV for *COOH formation, respectively. The calculated free energy of CO2RR suggests that the Ag(III) sites are more energetically favorable for stabilizing the *COOH intermediate compared to the Ag(I) and Ag(II) sites. At the most active Ag(III) site, in addition to the coordination from calixarenes, there is π–d interaction between the benzene ring and Ag site. This unique coordination environment can better disperse the d electron charge of the Ag center, enhancing the ability of the Ag site to stabilize the *COOH intermediate and promote the generation of CO.
hydrogenation on the Ag(III) site is 0.22 eV, while the ΔG value for the subsequent *CO generation step is 0.40 eV. Both values are much lower than those observed on the Ag(I) and Ag(II) sites. Similar conclusions were drawn when calculating the free energy changes for Ag(II) on ZrAg3_m and Ag(I) on Zr2Ag2_m, which give larger ΔG values of 1.79 and 2.06 eV for *COOH formation, respectively. The calculated free energy of CO2RR suggests that the Ag(III) sites are more energetically favorable for stabilizing the *COOH intermediate compared to the Ag(I) and Ag(II) sites. At the most active Ag(III) site, in addition to the coordination from calixarenes, there is π–d interaction between the benzene ring and Ag site. This unique coordination environment can better disperse the d electron charge of the Ag center, enhancing the ability of the Ag site to stabilize the *COOH intermediate and promote the generation of CO.
 | ||
| Fig. 7 (A) Free energy diagrams for the CO2RR pathway of Zr2Ag2_m, ZrAg3_m and Zr2Ag7_m. (B), (C) and (D) optimized structural intermediates of *COOH and *CO for Zr2Ag7_m. | ||
Conclusions
In summary, our study marks the first systematic comparison of the reactivity of three distinct Ag sites in electrochemical CO2 reduction, combining experimental and theoretical perspectives through the construction of cluster models. We synthesized an atomically precise bimetallic Zr2Ag7 cluster, utilizing a calixarene-protected Zr–O/S core as a substrate for loading Ag1+ ions. The Zr2Ag7 cluster features three discrete Ag sites: Ag(I) and Ag(II) within the {ZrAg3(TC4A)2} subunit and Ag(III) between the two subunits. We meticulously traced the assembly pathway of Zr2Ag7 and successfully isolated Ag(II) and Ag(I) sites in ZrAg3, as well as Ag(I) sites in Zr2Ag2, providing accurate structural models for a precise comparison of the activity of the three Ag sites. eCO2RR tests demonstrated that the performance of Zr2Ag7 significantly surpassed those of ZrAg3 and Zr2Ag2. We elucidated the reaction path using in situ ATR-FTIR technology and comprehensively calculated the free energy changes for each elementary step of CO2 conversion to CO at each Ag site using DFT, revealing notable differences in activity among these Ag sites. This study underscores that subtle changes in the coordination geometry of catalytic sites can profoundly influence the catalytic performance. Thus, acquiring atomic structures of nanoclusters is crucial, offering valuable insights for the rational design of cluster structures to achieve efficient catalysis.Data availability
The data that support the findings of this study are available in the main text and the ESI.†Author contributions
C. L. and W. D. Yu supervised the project and conceived the idea. L. J. Li carried out synthesis, characterization and catalytic experiment of clusters. L. Y. L. undertook the calculations for this article. C. L. wrote the manuscript. All the authors discussed the experimental results.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Hunan Province (2023JJ30650), Central South University Innovation-Driven Research Programme (2023CXQD061) and Project Foundation of Hunan Provincial Education Department (22B0641).Notes and references
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, 350–359 CrossRef PubMed
.
- Y. Z. Xu, C. L. Li, Y. Q. Xiao, C. H. Wu, Y. M. Li, Y. B. Li, J. G. Han, Q. H. Liu and J. F. He, ACS Appl. Mater. Interfaces, 2022, 14, 11567–11574 CrossRef CAS PubMed
- Z. Y. Yang, M. Y. Wan, Z. Y. Gu and F. L. Che, J. Phys. Chem. C, 2023, 127, 17685–17693 CrossRef CAS
- M. Abdinejad, E. Irtem, A. Farzi, M. Sassenburg, S. Subramanian, H. P. I. V. Montfort, D. Ripepi, M. R. Li, J. Middelkoop, A. Seifitokaldani and T. Burdyny, ACS Catal., 2022, 12, 7862–7876 CrossRef CAS PubMed
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS
- K. Qi, Y. Zhang, J. Li, C. Charmette, M. Ramonda, X. Q. Cui, Y. Wang, Y. P. Zhang, H. l. Wu, W. S. Wang, X. L. Zhang and D. Voiry, ACS Nano, 2021, 15, 7682–7693 CrossRef CAS PubMed
- S. Q. Liu, S. W. Wu, M. R. Gao, M. S. Li, X. Z. Fu and J. L. Luo, ACS Sustainable Chem. Eng., 2019, 7, 14443–14450 CrossRef CAS
- S. B. Liu, H. B. Tao, L. Zeng, Q. Liu, Z. H. Xu, Q. X. Liu and J. L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed
- L. Qin, F. Sun, X. Ma, G. Ma, Y. Tang, L. Wang, Q. Tang, R. Jin and Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 26136–26141 CrossRef CAS PubMed
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed
- C. M. Zhao, X. Y. Dai, T. Yao, W. X. Chen, X. Q. Wang, J. Wang, J. Yang, S. Q. Wei, Y. Wu and Y. D. Li, J. Am. Chem. Soc., 2017, 139(24), 8078–8081 CrossRef CAS PubMed
- J.-D. Yi, X. P Gao, H. Zhou, W. Chen and Y. Wu, Angew. Chem., Int. Ed., 2022, e202212329 CAS
- X. Q. Wang, Z. Chen, X. Y. Zhao, T. Yao, W. X Chen, R. You, C. M. Zhao, G. Wu, J. Wang, W. X. Huang, J. L. Yang, X. Hong, S. Q. Wei, Y. Wu and Y. D. Li, Angew. Chem., Int. Ed., 2018, 130, 1962–1966 CrossRef
- R. Qi, B. E. Zhu, Z. K. Han and Y. Gao, ACS Catal., 2022, 12, 8269–8278 CrossRef CAS
- X. L. Yuan, Y. S. Wu, B. Jiang, Z. S. Wu, Z. X. Tao, X. Lu, J. Liu, T. Qian, H. P. Lin and Q. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 1664–56649 Search PubMed
- Z. Li, A. A. Haidry, Y. S. Liu, L. C. Sun, L. J. Xie, Q. Fatima and Z. J. Yao, J. Mater. Sci.: Mater. Electron., 2018, 29, 19219–19227 CrossRef CAS
- N. Shimoda, S. Umehara, M. Kasahara, T. Hogo, A. Yamazaki and S. Satokawa, Appl. Catal., A, 2015, 507, 56–64 CrossRef CAS
- X. Wang, Z. Zhao, D. R. Ou, B. F. Tu, D. A. Cui, X. M. Wei and M. J. Cheng, RSC Adv., 2016, 6, 38153–38158 RSC
- C. Shi, M. Zhang, X. X. Hang, Y. F. Bi, L. L. Huang, K. Zhou, Z. H. Xu and Z. P. Zheng, Nanoscale, 2018, 10, 14448–14454 RSC
- H. T. Han, L. Kan, P. Li, G. S. Zhang, Y. Li, W. P. Liao, Y. L. Liu, W. Chen and C. H. T. Hu, Sci. China: Chem., 2021, 64, 426–431 CrossRef CAS
- S. T. Wang, X. H. Gao, X. X. Hang, X. F. Zhu, H. T. Han, W. P. Liao and W. Chen, J. Am. Chem. Soc., 2016, 138, 16236–16239 CrossRef CAS PubMed
- M. M. Deegan, T. S. Ahmed, G. P. A. Yap and E. D. Bloch, Chem. Sci., 2020, 11, 5273–5279 RSC
- Z. Wang, L. Li, L. Feng, Z. Y. Gao, C. H. Tung, L. S. Zheng and D. Sun, Angew. Chem., Int. Ed., 2022, 61, e202200823 CrossRef CAS PubMed
- Z. Wang, H. F. Su, Y. W. Gong, Q. P. Qu, Y. F. Bi, C. H. Tung, D. Sun and L. S. Zheng, Nat. Commun., 2020, 11, 308 CrossRef CAS PubMed
- Z. Wang, F. Alkan, C. M. Aikens, M. Kurmoo, Z. Y. Zhang, K. P. Song, C. H. Tung and D. Sun, Angew. Chem., Int. Ed., 2022, 61, e202206742 CrossRef CAS PubMed
- Z. J. Guan, J. L. Zeng, Z. A. Nan, X. K. Wan, Y. M. Lin and Q. M. Wang, Sci. Adv., 2016, 2, e1600323 CrossRef PubMed
- Z. J. Guan, F. Hu, S. F. Yuan, Z. A. Nan, Y. M. Lin and Q. M. Wang, Chem. Sci., 2019, 10, 3360–3365 RSC
- C. K. Zhang, Z. Wang, W. D. Si, H. X. Chu, L. Zhou, T. Li, X. Q. Huang, Z. Y. Gao, M. Azam, C. H. Tung, P. Cui and D. Sun, Nat. Commun., 2023, 14, 6413 CrossRef CAS PubMed
- S. Q. Li, L. F. Dai, Y. Q. Tian, Y. X. Yi, J. Yan and C. Liu, Chem. Commun., 2023, 59, 575–578 RSC
- L. J. Li, Y. T. Luo, Y. Q. Tian, P. Wang, X. Y. Yi, J. Yan, Y. Pei and C. Liu, Inorg. Chem., 2023, 62, 14377–14384 CrossRef CAS PubMed
- S. Chen, Z. N. Chen, W. H. Fang, W. Zhuang, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 10932–10935 CrossRef CAS PubMed
- X. Fan, F. Yuan, D. Li, S. Chen, Z. Cheng, Z. Zhang, S. Xiang, S.-Q. Zang, J. Zhang and L. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12949–12954 CrossRef CAS PubMed
- M. Y. Gao, K. Wang, Y. Y. Sun, D. J. Li, B. Q. Song, Y. H. Andaloussi, M. J. Zaworotko, J. Zhang and L. Zhang, J. Am. Chem. Soc., 2020, 142, 12784–12790 CrossRef CAS PubMed
- S. Chen, W.-H. Fang, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2018, 57, 11252–11256 CrossRef CAS PubMed
- Y. Q. Tian, W. L. Mu, L. L. Wu, X. Y. Yi, J. Yan and C. Liu, Chem. Sci., 2023, 14, 10212–10218 RSC
- G. Deng, J. Kim, M. S. Bootharaju, F. Sun, K. Lee, Q. Tang, Y. J. Hwang and T. Hyeon, J. Am. Chem. Soc., 2023, 145, 3401–3407 CrossRef CAS PubMed
- S. L. Zhuang, D. Chen, L. W. Liao, Y. Zhao, N. Xia, W. Zhang, C. Wang, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 3073–3077 CrossRef CAS PubMed
- J. Y. Xu, L. Xiong, X. Cai, S. S. Tang, A. C. Tang, X. Liu, Y. Pei and Y. Zhu, Chem. Sci., 2022, 13, 2778–2782 RSC
- X. S. Ma, F. Sun, L. B. Qin, Y. G. Liu, X. W. Kang, L. K. Wang, D. E. Jiang, Q. Tang and Z. H. Tang, Chem. Sci., 2022, 13, 10149–10158 RSC
- J. Wang, F. Xu, Z. Y. Wang, S. Q. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2022, 61, e202207492 CrossRef CAS PubMed
- J. Wang, Z. Y. Wang, S. J. Li, S. Q. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2021, 60, 5959–5964 CrossRef CAS PubMed
- X. Liu, E. D. Wang, M. Zhou, Y. Wan, Y. K. Zhang, H. Q. Liu, Y. Zhao, J. Li, Y. Gao and Y. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202207685 CrossRef CAS PubMed
- X. Liu, X. Cai and Y. Zhu, Acc. Chem. Res., 2023, 56, 1528–1538 CrossRef CAS PubMed
- X. Cai, G. J. Li, W. G. Hu and Y. Zhu, ACS Catal., 2022, 12, 10638–10653 CrossRef CAS
- Y. Zhang, L. Z. Dong, S. Li, X. Huang, J. N. Chang, J. H. Wang, J. Zhou, S. L. Li and Y. Q. Lan, Nat. Commun., 2021, 12, 6390 CrossRef CAS PubMed
- R. Wang, J. Liu, L. Z. Dong, J. Zhou, Q. Huang, Y. R. Wang, J. W. Shi and Y. Q. Lan, CCS Chem., 2023, 5, 2237–2250 CrossRef CAS
- L. Zhang, X. X. Li, Z. L. Lang, Y. Liu, L. Yuan, W. Y. Lu, Y. S. Xia, L. Z. Dong, D. Q. Yuan and Y. Q. Lan, J. Am. Chem. Soc., 2021, 143, 3808–3816 CrossRef CAS PubMed
- J. F. Huang and Y. C. Wu, ACS Sustainable Chem. Eng., 2019, 7, 6352–6359 CrossRef CAS
- X. Zi, Y. J. Zhou, L. Zhu, Q. Chen, Y. Tan, X. Q. Wang, M. Sayed, E. Pensa, R. A. Geioushy, K. Liu, J. W. Fu, E. Cortes and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309351 CrossRef CAS PubMed
- M. J. Zhuansun, Y. Liu, R. H. Lu, F. Zeng, Z. Y. Xu, Y. Wang, Y. Y. Yang, Z. Y. Wang, G. F. Zheng and Y. H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309875 CrossRef CAS PubMed
- A. P. Ayanwale, A. d. J. Ruíz-Baltazar, L. Espinoza-Cristóbal and S. Y. Reyes-López, Dose-Response, 2020, 18, 1–13 CrossRef PubMed
- Z. Z. Liu, X. M. Lv, S. Y. Kong, M. T. Liu, K. H. Liu, J. B. Zhang, B. W. Wu, Q. Zhang, Y. Tang, L. P. Qian, L. J. Zhang and G. F. Zheng, Angew. Chem., Int. Ed., 2023, 62, e202309319 CrossRef CAS PubMed
- Y. Lin, T. Wang, L. L. Zhang, G. Zhang, L. L. Li, Q. F. Chang, Z. F. Pang, H. Gao, K. Huang, P. Zhang, Z. J. Zhao, C. L. Pei and J. L. Gong, Nat. Commun., 2023, 14, 3575 CrossRef CAS PubMed
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. G. Chen, Y. S. Yan, F. Jiao and B. X. Han, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic file in CIF format, full experimental and computational details. CCDC 2290841–2290844. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc07005k |
| This journal is © The Royal Society of Chemistry 2024 |