
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOpportunities and challenges for plastic depolymerization by biomimetic catalysis
Yanfen
Wu†
,
Qikun
Hu†
,
Yizhen
Che†
and
Zhiqiang
Niu
 *
*
State Key Laboratory of Chemical Engineering, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: niuzq@tsinghua.edu.cn
First published on 20th March 2024
Abstract
Plastic waste has imposed significant burdens on the environment. Chemical recycling allows for repeated regeneration of plastics without deterioration in quality, but often requires harsh reaction conditions, thus being environmentally unfriendly. Enzymatic catalysis offers a promising solution for recycling under mild conditions, but it faces inherent limitations such as poor stability, high cost, and narrow substrate applicability. Biomimetic catalysis may provide a new avenue by combining high enzyme-like activity with the stability of inorganic materials. Biomimetic catalysis has demonstrated great potential in biomass conversion and has recently shown promising progress in plastic degradation. This perspective discusses biomimetic catalysis for plastic degradation from two perspectives: the imitation of the active centers and the imitation of the substrate-binding clefts. Given the chemical similarity between biomass and plastics, relevant work is also included in the discussion to draw inspiration. We conclude this perspective by highlighting the challenges and opportunities in achieving sustainable plastic recycling via a biomimetic approach.
1. Introduction
The exponential growth of plastic production and mismanagement of plastic waste over the past few decades have caused escalating concerns across the globe.1–3 To mitigate the environmental impact of plastic waste, it is imperative to develop plastic recycling approaches beyond landfill and incineration. While mechanical recycling has been used for recovery of thermoplastics, the regenerated raw materials often suffer from deterioration.4,5 Chemical recycling has attracted increasing research interest in recent years.4,6–9 Breaking the C–C, C–O, or C–N bonds in polymer backbones enables the transformation of post-consumer plastics into building blocks for new materials. For example, hydrogenolysis of polyolefins produces valuable products such as liquid fuels, waxes, and lubricants;10–12 solvolysis of polyesters yields corresponding monomers or value-added derivatives;4,13–16 and polyamides can be catalytically converted to either ε-caprolactam by a chain-end back-biting process or alcohols and amines by hydrogenolysis.17,18 Despite the progress, the depolymerization of waste plastics often requires harsh conditions to achieve bond cleavage, undermining the economic viability of these processes. Therefore, developing cost-effective and energy-efficient chemical recycling processes plays a significant role in achieving a more sustainable future.While chemical recycling is energy intensive, enzymatic depolymerization is performed under environmentally benign conditions. Several enzymes, such as hydrolase, peroxidase, and oxidase, have been demonstrated to have the ability to degrade polymers in the ambient environment. Enzymatic depolymerization of polyesters has been intensively investigated. Various types of hydrolases have been identified to degrade polyethylene terephthalate (PET), such as lipases,19,20 cutinases,21–23 and PETases.24 Meanwhile, the protein engineering strategy has been employed to improve catalytic activity and thermostability of these PET-degrading enzymes. For instance, a disulfide bridge was introduced into leaf-branch compost cutinase (LCC) to improve its thermostability, and site-specific saturation mutagenesis was conducted to improve its specific activity.25 Very recently, the highly labile PETase was engineered through a machine learning algorithm, resulting in a robust and active variant termed FAST-PETase.26 FAST-PETase completely depolymerized many kinds of commercial post-consumer PET at 50 °C within one week. Polyolefins and polyamides are more challenging to depolymerize by enzymes compared with polyesters. Manganese peroxidase (MnP), lignin peroxidase, and laccase have been shown to cleave sp3 C–C bonds in polyethylene (PE) at 37 °C,27–29 although oxidation pretreatment or redox mediators are required in these processes.30–32 In the case of low molecular weight PE, alkane hydroxylases from Pseudomonas putida can directly catalyze the depolymerization through the terminal oxidation mechanism.33,34 Furthermore, a purified hydroquinone peroxidase has been demonstrated to accelerate the cleavage of C–C bonds in polystyrene (PS) in a dichloromethane–water system.35 For polyamides, although various hydrolases (proteases, cutinases, and amidases) can facilitate their surface modification,36–39 obvious mass loss and molecular weight decrease of polyamide can only be observed using MnP or a laccase-mediated system at 30 °C.40 While these laboratory results suggest the feasibility of enzymatic plastic recycling, challenges related to the enzyme's activity, stability, and cost need to be further addressed. The main problem using enzymes is their poor stability upon temperature changes, pH fluctuations, and the presence of contaminants and additives in the plastic wastes. Furthermore, most enzymes can only catalyze the degradation of amorphous polymers.
The high efficiency of enzymatic plastic degradation inspires the development of biomimetic catalysts to reduce the energy consumption of plastic recycling. The field of synthetic enzymes is rapidly growing. Cyclodextrins,29,41 crown ethers,42 metalloporphyrins,43 molecularly imprinted polymers44,45 and other materials46–48 have shown enzyme-like activities. These catalysts are generally more stable than natural enzymes and capable of functioning under a wide range of conditions. Designing enzyme-mimicking catalysts for plastic depolymerization has the potential to circumvent the need for harsh reaction conditions. So far, there are limited research studies on the mimicry of plastic-degrading enzymes. However, biomimetic catalysis applied to cellulose upgrading and deoxyribonucleic acid (DNA) hydrolysis is expected to promote the advancement of biomimetic plastic recycling due to the structural similarities.
In this perspective, we examine the current landscape of biomimetic catalysis for bond cleavage in plastics and related macromolecular substrates with specific focus on simulating both the active sites and the substrate-binding clefts of relevant enzymes. For the active sites, we discuss the mimicry of hydrolases for polyester hydrolysis, glycosidase for cellulose upgrading, peroxidase for plastic depolymerization and purple acid phosphatase mimetics for DNA hydrolysis. The role of the second coordination sphere is also discussed. In terms of substrate-binding clefts, emphasis is placed on molecular imprinting and diffusion control in porous materials to enhance the efficiency and selectivity toward polymer degradation. Through this discussion, we hope to elucidate the underlying principles and strategies for developing more efficient plastic-degrading catalysts.
2. Mimicking the active sites
The active site of an enzyme is a small part of the protein that directly binds to a substrate and promotes bond cleavage and formation. It is composed of several amino acid residues located in a three-dimensional cleft of the protein, sometimes including metal cations as co-factors.49 The active site interacts with a substrate through non-covalent forces such as hydrogen bonding, π–π stacking, hydrophobic effects, electrostatic interactions, and van der Waals forces. These interactions are crucial for biocatalytic processes in terms of substrate confinement, organization and activation as well as transition state stabilization.50,51Recent advances in protein isolation, purification, and characterization have uncovered the structure of the active sites in enzymes for macromolecule depolymerization.52–55 This provides helpful information for chemists to mimic the structure or the function of these enzymes.56 Organometallic complexes are frequently employed to mimic the functions of metalloenzymes.57–61 In comparison, imitating the active sites composed solely of amino acids requires more efforts to accurately position the related functional groups in three dimensions. Previous studies have highlighted the catalyst design principles based on model substrates.62,63 In this section, we will focus on the related work for plastic and cellulose depolymerization.
2.1. Hydrolase mimics for polyester depolymerization
Chemical depolymerization of PET needs high temperature (150–200 °C), increased pressure (20–40 atm), or concentrated acids and bases. Recent research has revealed that some microorganisms can break down PET under ambient conditions by producing hydrolytic enzymes such as cutinases,21,22,25 lipases,19,20 and PETases.24 PETase has similar characteristics to lipases and cutinases such as having an α/β-hydrolase fold, but its active-site cleft is more open than those of cutinases. Fig. 1a shows the proposed mechanism for PET binding in this shallow cleft: when PET enters the groove, the ester carbonyl group is polarized by an anion hole and ready to be attacked by a nucleophilic hydroxyl group carried by a catalytic triad (serine, histidine, and aspartic acid).55 The binding cleft confines the PET and nucleophile in close proximity to accelerate the hydrolysis rate.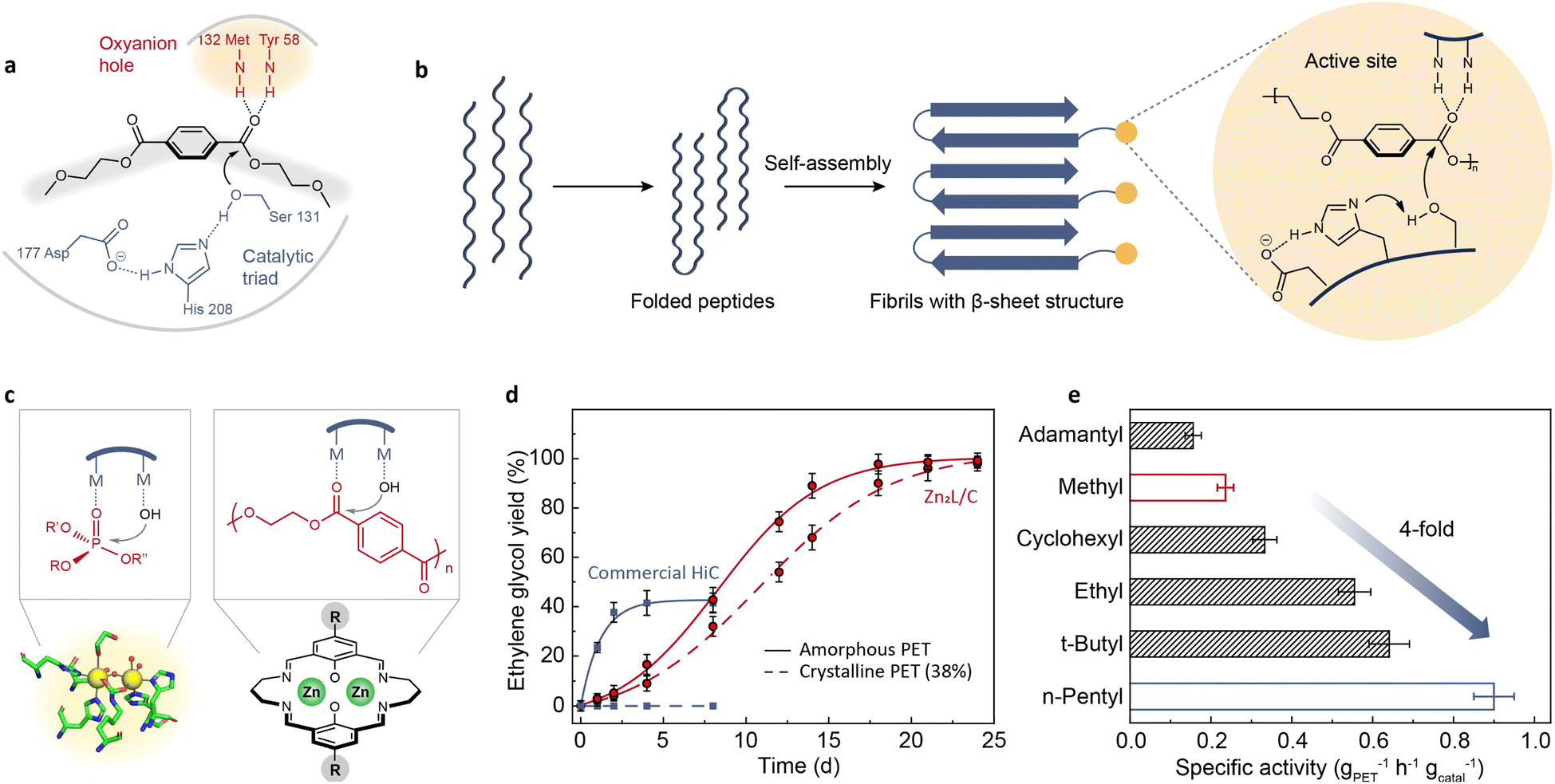 | ||
Fig. 1 (a) Schematic representation of the hydrolysis mechanism proposed for PETase.55 Adapted with permission from ref. 55. Copyright 2017 Springer Nature. (b) PET hydrolase mimic obtained by self-assembly of polypeptide MAX.64 Adapted with permission from ref. 64. Copyright 2023 Elsevier Inc. (c) Schematic diagram of the hydrolysis mechanism of OpdA and a hypothesized binuclear catalyst.65 (d) Reaction kinetics of PET hydrolysis catalyzed by Zn2L and commercial Humicola insolens cutinase (HiC) at pH 8 and 60 °C.65 Adapted with permission from ref. 65. Copyright 2023 Springer Nature. (e) The specific activities of Zn2L with different alkyl substituents for PET hydrolysis in a mixed solution of NaOH aqueous solution (0.1 M) and methanol (v/v = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 60 °C.66 Adapted with permission from ref. 66. Copyright 2023 The Royal Society of Chemistry. :1) at 60 °C.66 Adapted with permission from ref. 66. Copyright 2023 The Royal Society of Chemistry. | ||
Attempts have been made to directly replicate or modify the active site of PETase. Li et al.64 combined PETase's catalytic triad with a self-assembling polypeptide (Fig. 1b). This enzyme mimic achieved a 16% weight reduction of a commercial PET film after a two-week incubation at pH 9 and 65 °C. However, the observed activity is not comparable to the performance of PETase. This implies that the amino acid residues can only exert optimal hydrolytic activity in a specific spatial conformation. Moreover, the application of this enzyme mimic is still constrained by limited thermostability and a narrow pH range. Hence, developing stable and highly efficient enzyme mimics by imitating the active site of PET hydrolase remains a challenge.
Instead of replicating the active site, imitating the mechanism of enzymatic depolymerization is also explored. As mentioned, an important reason why PET-degrading enzymes can achieve ester bond cleavage under ambient conditions is that the binding cleft can bring PET and nucleophile into proximity, thus increasing the effective concentration of the reactants. It is recognized that the proximity effect is also involved in other hydrolysis processes catalyzed by metallohydrolases, such as the organophosphate-degrading enzyme from Agrobacterium radiobacter (OpdA).67 The active site of OpdA comprises two metal ions, one of which binds to the hydroxyl group while the other binds to the phosphate group (Fig. 1c), enabling facile nucleophilic attack. Synthetic analogs of OpdA are much easier to develop than those of PETase. Based on these considerations, a binuclear zinc complex (Zn2L) was synthesized and exhibited catalytic activity for PET hydrolysis under a wide range of reaction conditions.65 One advantage of Zn2L was that its catalytic activity was not compromised too much for high-crystallinity PET, whereas the enzyme was completely inactive for the same substrate (Fig. 1d). Furthermore, Zn2L was effective toward various post-consumer PET wastes and other types of polyesters. The catalytic activity of Zn2L was further optimized by tuning the substituents in the para position of the macrocyclic ligand.66 The electronic effects of the para-substituents can influence the binding strength of metal centers to the substrates. Hammett studies revealed a volcano-shaped correlation between the reaction rate and the Hammett constant. Guided by this structure–activity relationship, a more efficient catalyst (n-pentyl-substituted binuclear zinc complex) was identified, with a specific activity four times higher than that of the original methyl substitution (Fig. 1e). In addition to substituent modulation, changing the metal species in the metal complexes provides another way for enhancing activity, which has been verified in the literature.65
2.2. Glycosidase mimics for cellulose upcycling
Cellulose is a linear polysaccharide with glucose monosaccharide units connected by C–O–C linkages. Since C–O–C linkages are ubiquitous in many plastics (Fig. 2a), approaches for cellulose conversion could be used for plastic recycling. The upgrading of cellulose requires first depolymerizing it into soluble monomer sugars and oligosaccharides by breaking the stubborn glucoside bond, and then further transforming them into biofuels.68 The enzymatic cleavage of glycosidic bonds in cellulose is typically facilitated by two carboxylate residues within the active sites of glycosidases.69 The majority of glycosidases hydrolyze cellulose via either a double or single displacement mechanism, resulting in retention or inversion of the anomeric configuration, respectively.69–71 The catalytic process via the double displacement mechanism involves one carboxylate amino acid residue serving as an acid/base and the other serving as a nucleophile (Fig. 2b). The single displacement mechanism entails typical acid–base catalysis, in which one amino acid residue activates water and the other protonates the glycosidic oxygen (Fig. 2c).69–71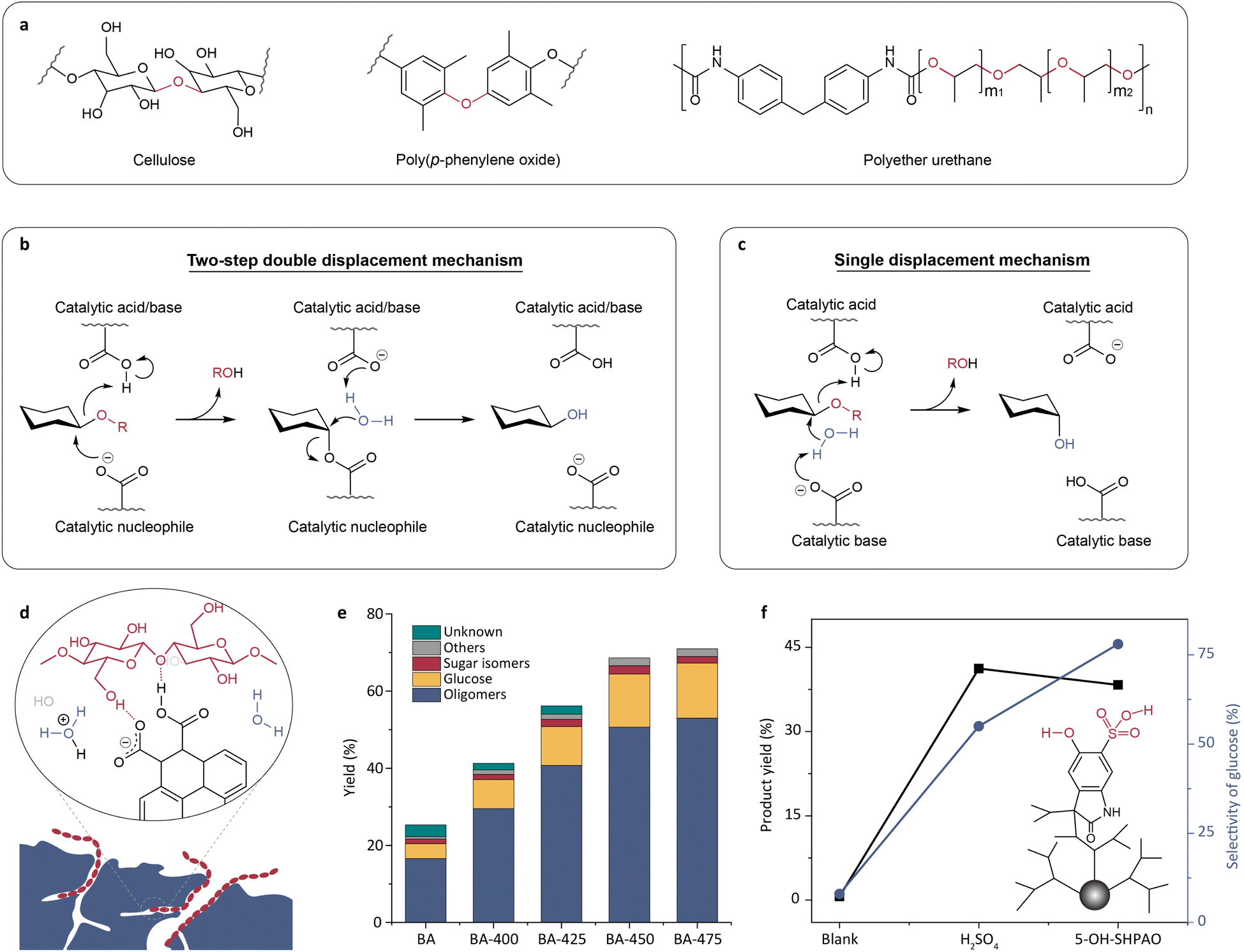 | ||
| Fig. 2 (a) The structures of cellulose and plastics containing C–O–C linkages. (b and c) Schematic illustrations of the double displacement mechanism (b) and the single displacement mechanism (c) proposed for cellulose hydrolysis catalyzed by glycosidases.71 Adapted with permission from ref. 71. Copyright 2022 Springer Nature. (d) A glycosidase mimic made by introducing carboxyl groups through oxidation of activated carbon.73 (e) The effect of oxidation temperature on cellulose hydrolysis activity.73 Adapted with permission from ref. 73. Copyright 2016 Wiley-VCH. (f) The comparison of cellulose hydrolysis with 5-OH-SHAPO, H2SO4, and without a catalyst.78 Adapted with permission from ref. 78. Copyright 2016 The Royal Society of Chemistry. | ||
As a pair of carboxylate residues is highly conserved in the active sites of most glycosidases,69,72 the emulation of the active site of glycosidases was attempted by simply introducing carboxyl groups. For example, Shrotri et al. introduced carboxyl functional groups into activated carbon by oxidation (Fig. 2d).73 They observed that increasing the oxidation temperature led to an increased density of carboxyl groups and thus the yield of hydrolysate (Fig. 2e). Similarly, acid–base pairs (e.g. COOH–COO−, COOH–NH2, and COOH-imidazole) have also been introduced into carbon materials or magnetic nanomaterials to mimic the active sites of glycosidase, aiming to obtain recyclable and stable biomimetic catalysts.74–76 Although these catalysts can catalyze the cleavage of glycosidic bonds, their catalytic activity is far below that of the natural enzymes, because the acid–base pairs are randomly distributed on the material surface. The importance of spatial positioning of the acid–base pairs has been validated in cellobiose hydrolysis catalyzed by organic acids.77 The comparison of turnover frequency (TOF) values between ortho-, meta-, and para-hydroxybenzoic acids suggests that a vicinal acid–base pair significantly contributes to catalytic activity. High catalytic performance of adjacent acid–base pairs was also observed in the enzyme mimic designed by Yu et al., using a sulfonated, hyperbranched poly(arylene oxindole).78 The hydroxyl group and the neighboring sulfonic acid in the glycosidase mimic, 5-OH-SHAPO, synergistically catalyzed the depolymerization of cellulose. This enzyme mimic exhibited high catalytic activity and glucose selectivity comparable to those of inorganic acids (Fig. 2f). The above results suggest that developing enzyme mimics capable of reproducing the accurate spatial conformation of the active site will improve catalytic activity. Further discussion on this strategy will be presented in Section 3.
In addition to the precise positioning of acid–base pairs, researchers have developed another strategy to enhance the catalytic activity by introducing cellulose-binding domains in the catalyst design.79 For example, a series of solid acid catalysts containing –Cl groups have shown enhanced cellulose hydrolysis rates compared to the corresponding catalysts without –Cl.80–82 Similarly, the rate enhancement can also be achieved by introducing boronic acid groups (–B(OH)2) into solid acid catalysts.83 The functional groups (–Cl or –B(OH)2) designed in these glycosidase mimics can bind cellulose by forming hydrogen bonds80–82 or reversible covalent bonds83 with the hydroxyl groups of cellulose, and thus improve mass transfer between the catalysts and cellulose and catalytic performance.
2.3. Peroxidase mimics for plastic depolymerization
As early as 1997, Deguchi and coworkers discovered that lignin-degrading fungi had the capability to degrade nylon.84 Subsequently, MnP was identified from the fungi to be the enzyme that catalyzes nylon degradation.85 After two days of incubation with MnP at 30 °C, a nylon film showed a 57% reduction in average molecular weight, and oxidative degradation introduced new functional groups (–CH3, –CHO, and –NHCHO). Peroxidase has also been reported to degrade acrylate polymers.86–89 The hydroquinone peroxidase of the bacterium Azotobacter beijerinckii HM121 can rapidly degrade polyacrylamide and polyacrylic acid into small molecules in the presence of H2O2 and tetramethylhydroquinone at 30 °C.87 Horseradish peroxidase was demonstrated to catalyze the radical degradation of partially hydrolyzed polyacrylamide.88 Additionally, the crude peroxidase from Trametes versicolor CBR-04 has been reported to break down poly(potassium acrylate) at room temperature.89Although research on mimicking peroxidase for plastic degradation remains limited to date, progress has been made for lignin depolymerization. Discussion in this field may provide valuable guidance for designing peroxidase mimics for plastic depolymerization or functionalization.
Peroxidases catalyze the oxidation of a substrate by peroxides such as H2O2. Most peroxidases contain the heme protein, with a central iron ion coordinated to a porphyrin ring.90 Therefore, synthetic iron porphyrins are often used as models for heme peroxidases. The structure of synthetic iron porphyrins can be fine-tuned for enhanced activity and stability by adjusting axial ligands and substituents on the porphyrin ring, or even replacing an iron center with other metals. For instance, Zhu et al. synthesized a series of metallo-deuteroporphyrins as biomimetic catalysts for the oxidative conversion of lignin to aromatics (Fig. 3a–c).91 By studying the catalytic activity on model substrates (2-(2-methoxyphenoxy)-1-phenylethanol), Co(DPDME) was identified as the best one in the M(DPDME) series (Fig. 3a). Furthermore, six Co-deuteroporphyrins with substituents of varying electron-donating capacities on the propionate side chains were designed to investigate the impact of these substituents on reaction activity and catalyst stability (Fig. 3b,c). Co(DPCys) was ultimately chosen as the catalyst for real lignin depolymerization owing to its capacity to catalyze the cleavage of C–O and C–C bonds in diverse lignin model compounds at room temperature (Fig. 3c). The liquid product yield from the real lignin depolymerization catalyzed by Co(DPCys) reached 31.2% after 10 hours at 150 °C, showcasing a 5.5-fold increase compared to the blank control.
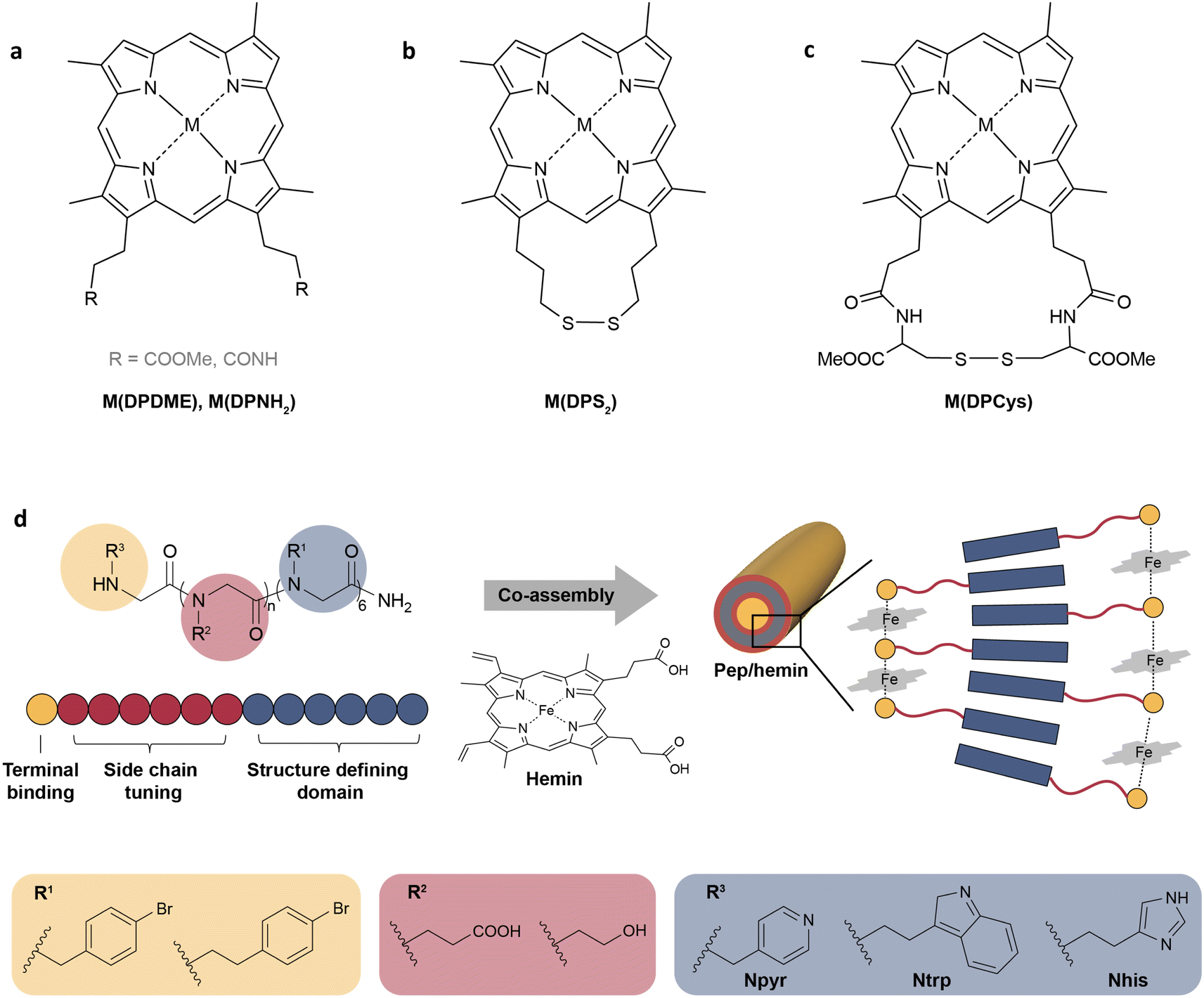 | ||
| Fig. 3 (a–c) The structures of metallo-deuteroporphyrins designed for the catalytic depolymerization of lignin.91 Adapted with permission from ref. 91. Copyright 2015 Wiley-VCH. (d) The schematic representation of self-assembled peptoid/hemin nanotubes.92 Adapted with permission from ref. 92. Copyright 2022 Springer Nature. | ||
In addition to regulating the central metal and the substituents of metalloporphyrin complexes, optimizing the microenvironment around the active site is another strategy to improve the catalytic performance. In natural peroxidases, the amino acid residues around the ferric center can help locate H2O2, facilitate the cleavage of the O–O bond, and stabilize the high oxidation state of iron.90 To imitate the function of the surrounding residues, a self-assembled peptide/hemin (Pep/hemin) nanomaterial was developed.92 As shown in Fig. 3d, peptoid sequences with variations in the terminal binding site, polar side-chain domain length, and side chain group were synthesized, and then the peptoids were mixed and incubated with equimolar hemin to prepare Pep/hemin nanotubes (Fig. 3d). Among all the as-prepared nanotubes, Pep-1/hemin with the active site located at an optimal distance from the hydrophobic domain (n = 6), a carboxyl group on the side chain, and Npyr as terminal binding sites, showed the highest catalytic activity toward the oxidation of model compounds and surpassed all hemin-containing peroxidase mimics at the time. The self-assembled Pep/hemin not only enhanced catalytic activity but also demonstrated robustness at elevated temperatures in a wide pH range. Pep-1/hemin caused a 65.7% mass loss of lignin with a 61.7% yield of phenolic compounds after incubation at 60 °C for 2 h in the presence of H2O2. In addition to self-assembled peptides, the microenvironment has also been regulated by combining hemin-containing peroxidase mimics with metal–organic frameworks (MOFs),93,94 graphene,95 and supramolecular polymers.96
Various Fe-based, V-based and MOF-based materials have been reported to exhibit peroxidase-like activity,93,97–100 providing additional references for developing peroxidase mimics for plastic depolymerization. Iron oxide nanoparticles are widely used in the biomedical field and environmental remediation due to their outstanding peroxidase-like activity.99–101 This activity is attributed to the Fe2+-induced Fenton and/or Haber–Weiss reactions.97 In 2022, Liu and coworkers applied Fe3O4 nanoparticles for the removal and degradation of microplastics.102 Fe3O4 nanoparticles can adsorb polypropylene (PP), PE, polyvinyl chloride, PS and PET microplastics from water through hydrogen bonding, and then they can be removed by magnetic separation. When heated at the melting temperatures of the respective plastics, almost 100% degradation of PP, PE, and PS was achieved by Fe3O4 in the presence of H2O2. The Fe3O4 nanoparticles can be easily recovered from the reaction solution and retained their original performance. Its stability and recyclability make it suitable for large-scale applications.
2.4. The second coordination sphere: lessons from phosphatase mimics
Metalloenzymes play crucial roles in essential life processes such as oxygen reduction and DNA hydrolysis. Although the metal ions and coordinating amino acids that make up metallohydrolases are limited, the functions of metallohydrolases vary greatly even with the same metal ions and ligands.103,104 A typical example is that heme B serves as the active centers in various enzymes such as globulins, catalases, oxidases, oxygenases and so on.90 What makes the difference is the second coordination sphere, which involves noncovalent interactions between ligands and substrates or ligands and ligands. Many reviews have detailed the roles played by the second coordination sphere in enzymatic reactions.60,105 In general, the interactions within the second coordination sphere affect biocatalytic processes by orienting substrates, stabilizing transition states, changing the redox properties, and aiding product release.60,106 Hence, many research groups are interested in designing synthetic complexes with a modified second coordination sphere to investigate their functions in activity and selectivity.Here we use purple acid phosphatases (PAPs, E.C. 3.1.3.2) as an example to illustrate the importance of the second coordination sphere. PAPs are known for their ability to enzymatically hydrolyze a diverse range of phosphorylated compounds, a process heavily involving the participation of the second coordination sphere.103 Mimicking PAPs has a theoretical basis and can promote the understanding of the function of the second shell in turn. PAPs are a kind of binuclear hydrolase characterized by heterovalent dual metal sites containing Fe(III) and M(II), where M can be Fe, Mn, or Zn. Taking kidney bean PAP for example, its active centers are composed of Fe(III) and Zn(II) sites which are bonded with oxygen and nitrogen atoms from tyrosine, aspartate, histidine residues and asparagine (Fig. 4a).107 However, the majority of components within the first coordination sphere do not actively participate in the catalytic cycle as shown in Fig. 4. In contrast, the second coordination sphere including two protonated histidine residues assumes a pivotal role in accelerating the hydrolysis process. During the reaction (Fig. 4c), His202 and His295 bind to two oxygen atoms from phosphate ester by hydrogen bonding to stabilize the transition state, while His296 functions as a proton donor, facilitating the departure of the leaving group. Mutagenesis studies have validated the importance of histidine residues in the hydrolysis process.108 In the following part, we will discuss the synthetic complexes as the mimics of PAPs and their role in accelerating the hydrolysis of DNA. Although the substrates are different, related work can provide a reference for the design of plastic hydrolysis catalysts.
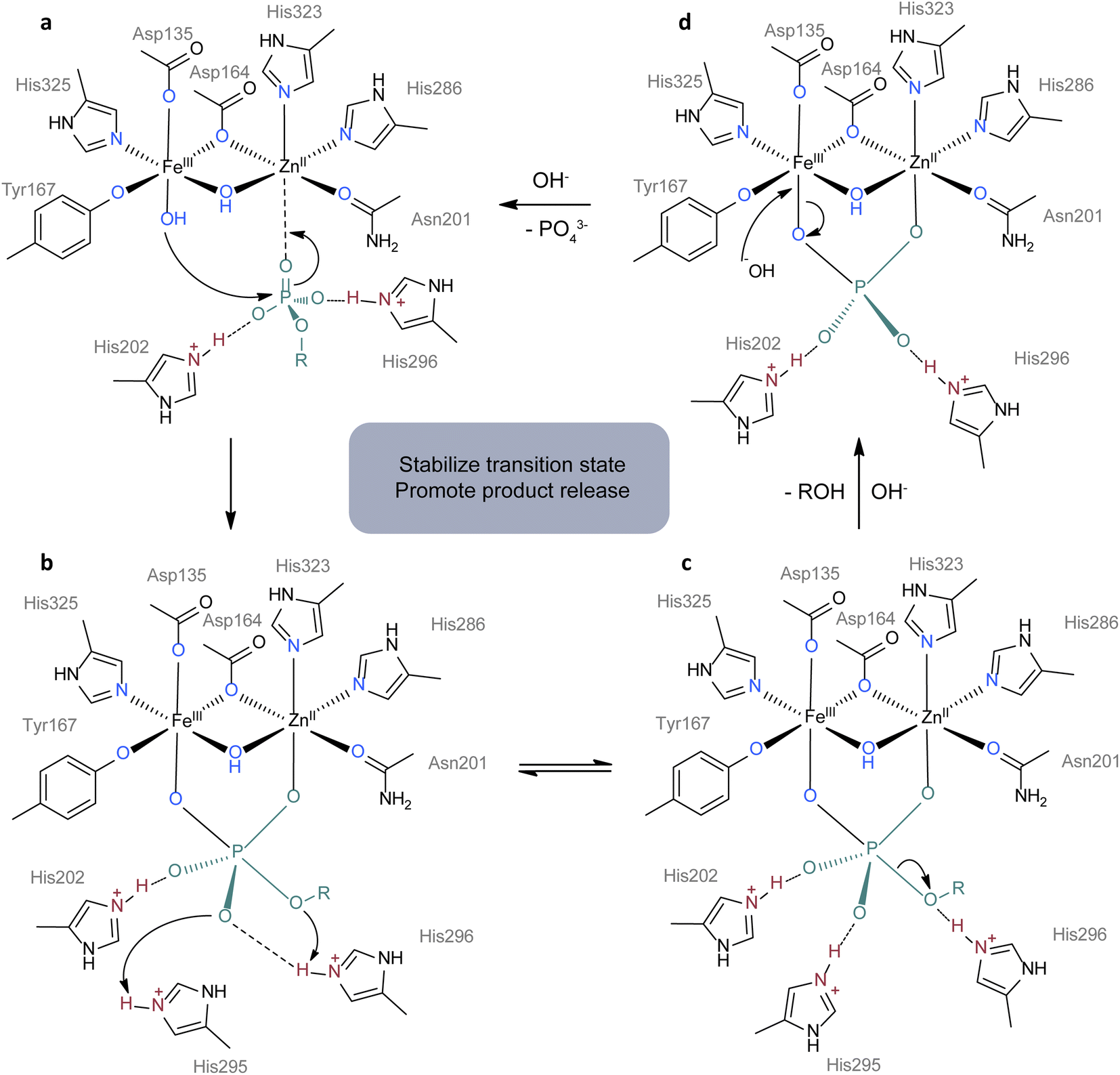 | ||
| Fig. 4 Proposed mechanism for the hydrolysis of phosphomonoesters by kidney bean purple acid phosphatase.107 (a) The substance is oriented for attackment of FeIII–OH by interactions with ZnII, His202 and His296. (b) RO group in subtance attacks protonated His296 and O atom attacks protonated His295. (c) P–O bond breaks with the release of ROH. (d) A hydroxide ion attacks FeIII with the release of the phosphate group to start another cycle of catalysis. The first coordination sphere, second coordination sphere and substrate are colored in blue, red and green. Adapted with permission from ref. 107. Copyright 1996 Academic Press. | ||
Over the past few decades, considerable efforts have been dedicated to synthesizing analogs of PAPs. One research focus is to increase substrate affinity and direct the nucleophile to the phosphodiester linkages by introducing functional groups in the second coordination sphere. An et al. designed [Cu(L1)2(Br)](ClO4)5·6H2O with quaternary ammonium-modified bipyridines as ligands to promote DNA hydrolysis.109 The Br− is easily replaced by OH− in aqueous solution to form the active Cu–OH species (Fig. 5a). The distance between the hydroxyl and the quaternary ammonium ion is around 5.5–5.7 Å, similar to that of adjacent phosphodiesters in DNA (ca. 6 Å). Through electrostatic interactions, the quaternary ammonium binds to the neighboring phosphoryl oxygen atoms, allowing the DNA to be cleaved by the spatially adjacent nucleophile (OH or H2O). Apart from a quaternary ammonium ion, easily protonated guanidyl groups can also electrostatically interact with phosphodiester groups. Tjioe et al. introduced protonated guanidyl groups to copper(II) complexes with 1,4,7-triazacyclononane ligands.110 Compared with non-guanidinylated analogs, a 22-fold enhancement in the hydrolysis rate has been achieved. This is because protonated guanidyl attracts the phosphodiester linkage and orients the nucleophilic Cu–OH to the phosphodiester group.
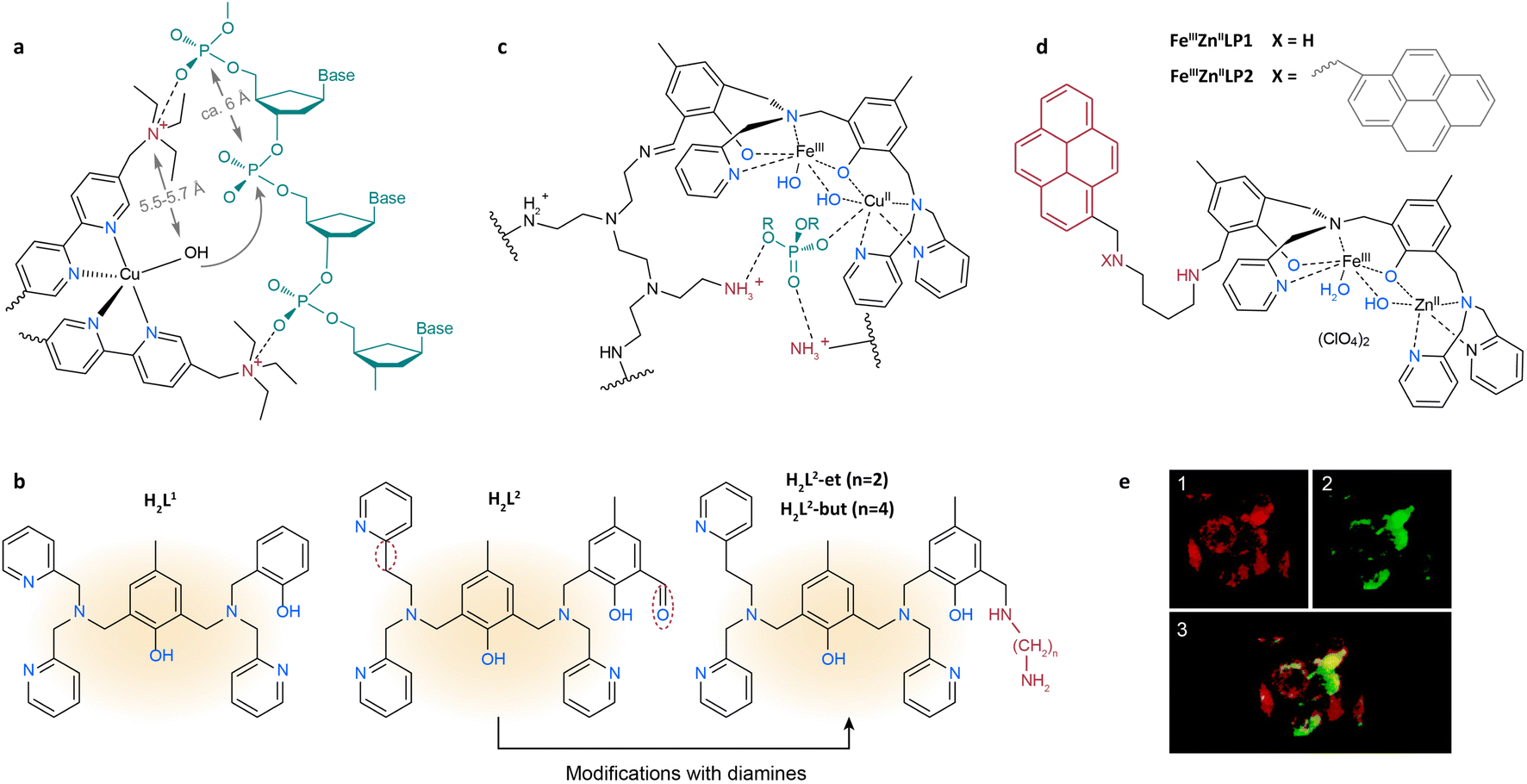 | ||
| Fig. 5 (a) The proposed intermediate of DNA cleavage by [Cu(L1)2(Br)](ClO4)5·6H2O, where L1 is 5,5′-di(1-(triethylammonio)methyl)-2,2′-dipyridyl.109 Adapted with permission from ref. 109. Copyright 2006 The Royal Society of Chemistry. (b) The structures of H2L1, H2L2, H2L2-et and H2L2-but.112 Adapted with permission from ref. 112. Copyright 2017 The Royal Society of Chemistry. (c) The proposed structure for the FeCuL1-PEI system with a bound phosphate diester.111 Adapted with permission from ref. 111. Copyright 2013 American Chemical Society. (d) The structures of FeIIIZnIILP1 and FeIIIZnIILP2.113 (e) Accumulation of FeIIIZnIILP1 in lysosomes, where FeIIIZnIILP1 is pseudocolored in green (1) and the lysotracker in red (2).113 Adapted with permission from ref. 113. Copyright 2017 American Chemical Society. The substrate and first and second coordination spheres are colored in green, blue and red, respectively, unless specified. | ||
Another research endeavor is focused on stabilizing the transition state by using the second coordination sphere. Since DNA is negatively charged in aqueous solution, positively charged groups in the catalyst can electrostatically attract substrates and accelerate the release of products. Aliphatic amines are of particular interest due to their ability as hydrogen donors and their electrostatic interactions with the phosphodiester backbone of DNA. As shown in Fig. 5b, the first coordination sphere of PAPs is extensively mimicked by an asymmetric chelating ligand H2L1, whereas the second coordination sphere can be reproduced by chemical modification with aliphatic amines. Based on this ligand, de Souza et al. synthesized a PAP analogue and covalently grafted it onto polyethyleneimine (PEI) to mimic the histidine residues in the polypeptide chain (Fig. 5c).111 The as-obtained catalyst was termed FeCuL1-PEI. During DNA hydrolysis, the terminal amino groups of FeCuL1-PEI were protonated, facilitating the electrostatic interactions between the catalyst and DNA substrate. Moreover, the activation energy for DNA hydrolysis was lower on FeCuL1-PEI than on FeCuL1, which can be attributed to the stabilization of the negatively charged transition state and the formation of hydrogen bonds. A kcat/KM value of 2.7 × 104 h−1 M−1 for FeCuL1-PEI was record high among PAP molecular models at the time. In another example, Silva et al. introduced ethylene diamine and butene diamine to a H2L2 ligand and synthesized H2L2-et and H2L2-but (Fig. 5b), respectively.112 They employed these two modified ligands to construct FeIIIZnII-based PAP analogues and studied the influence of the length of diamine on their hydrolysis activity. Compared with FeIIIZnIIL2 (without diamines), the diamine group enhanced the kcat/KM by 134% and 114% for FeIIIZnIIL2-et and FeIIIZnIIL2-but, respectively. According to the calculated energy minima, the terminal amino group of FeIIIZnIIL2-et was spatially closer to DNA than FeIIIZnIIL2-but's, leading to more stable interaction between DNA and the complex.112
Studies on PAP mimics can help develop antibiotics and antitumor drugs for the cleavage of target DNA. Camargo et al. synthesized FeIIIZnIILP1 and FeIIIZnIILP2 by introducing a pyrene group to the H2L1 ligand (Fig. 5d) to enhance the affinity between DNA and the catalysts.113 Due to the π–π stacking interaction in the second coordination sphere, all of the supercoiled plasmid DNA was converted to the open circular and linear forms at pH 7.0 in the presence of FeIIIZnIILP2 after 16 hours. The apoptosis ratio of K562 cells (from a human chronic myelogenous leukemia cell line) treated with FeIIIZnIILP1 was six times higher than the apoptosis ratio of those treated with FeIIIZnIIL1. The colocalization of FeIIIZnIILP1 with lysosomes further indicated its potential as a novel antitumor drug (Fig. 5e).114
Asides from PAP mimics, tuning the second coordination sphere of natural enzymes has also been demonstrated to accelerate various reactions, such as CO2 reduction, hydrogen evolution, hydrolysis of glycosidic bonds and so on.115–117 Although the second coordination sphere effects have hardly been studied in the field of plastic depolymerization, many kinds of groups in polymer substrates can be involved in non-covalent interactions. For example, the aromatic groups in PS and PET can potentially participate in π–π stacking. The ester groups in polyesters and the amide groups in polyamides such as nylon-6 can be regarded as hydrogen bond acceptors and donors. We envision that mimicking the second coordination sphere through ligand design may be a new opportunity to achieve catalytic depolymerization of plastics under mild conditions.
3. Mimicking the substrate-binding cleft
Substrate-binding clefts significantly influence substrate specificity and reaction selectivity in enzyme catalysis. For instance, adenosine triphosphate (ATP)-dependent proteases feature protease domains with a unique six-fold symmetry (Fig. 6a).118 This symmetry is altered by ATP to switch between “open” and “closed” interdomain contacts. These ATP-regulated orientations effectively restrict the access of a substrate to catalytic sites in a specialized tunnel. Cellobiohydrolase follows a similar mechanism for cellulose degradation.119,120 It possesses a long tunnel where cellobioside binding occurs. As the cellulose chain threads further into this tunnel, it undergoes catalytic transformation. Interactions between the substrate and enzymes' cleft have also been identified in the enzymatic degradation of plastics. Mechanistic studies of PETase demonstrated that PET chains are guided to the vicinity of the catalytic triad through a π–π stacking between the PET benzene ring and the tryptophan residues in the shallow cleft (Fig. 6b).121 This action allows for the selective, processive depolymerization of the polymer. These insights into substrate-binding clefts offer catalyst design principles for the degradation of targeted substrates ranging from biological macromolecules to plastic wastes.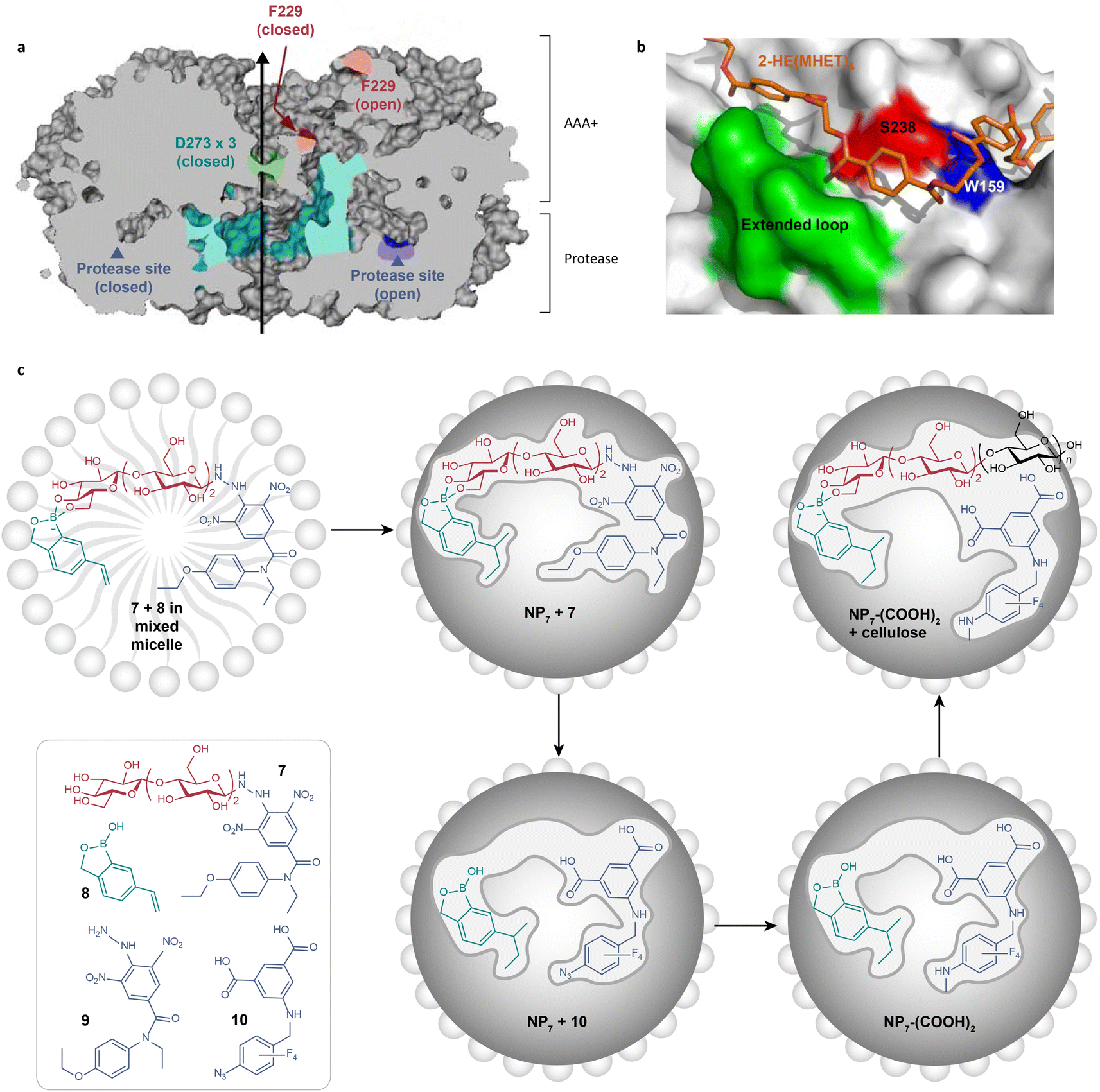 | ||
| Fig. 6 (a) A diagram showing a cross-section of triple-symmetric FtsH, including the three-fold axis (depicted by a black line) and two protease active sites (indicated by Aegean blue arrowheads). AAA+ means ATPases associated with various cellular activities.118 Adapted with permission from ref. 118. Copyright 2006 Elsevier Inc. (b) The structure of subsite II of I. sakaiensis PETase (IsPETase).121 The structure of IsPETase is presented as surface models in gray color. The 2-hydroxyethyl-(monohydroxyethyl terephthalate)4 docking model is shown in orange color. The residues S328 and W159, as well as the extended loop of IsPETase, are specifically labeled, which are crucial for forming the shallow and continuous clefts in subsite II and high hydrolytic activity of IsPETase. Adapted with permission from ref. 121. Copyright 2018 Springer Nature. (c) The preparation route of exocellulase mimics (NP7–(COOH)2) through the molecular imprinting approach.72 Adapted with permission from ref. 72. Copyright 2022 American Chemical Society. | ||
3.1. Creating clefts via molecular imprinting
Molecularly imprinted polymers (MIPs) have been developed to emulate substrate-binding clefts. The imprinting strategy allows for a desired cavity that has multiple interactions with a specific template molecule in the polymer matrix.122 Zhao and coworkers synthesized a variety of glycosidase mimics with cellulose hydrolysis activity using molecularly imprinted polymers.72,123 As shown in Fig. 6c, a mixed micelle containing template molecules, free radical cross-linkers, and photo initiators was initially formed. Subsequently, the template molecules were removed, resulting in a cleft, which was then post-modified to attain the desired geometrically and chemically fitting structure. Designing enzyme mimics via this strategy not only introduces the active site, but also tailors the size and depth of the cleft in the matrix by changing the template molecules. Nevertheless, molecularly imprinted enzyme mimics were less active than natural enzymes.123 This gap could be attributed to several limitations of imprinted polymers, such as structural rigidity, inhomogeneity in binding sites, and limited mass transfer within the system.44,122 These limitations particularly affect the catalytic activity when the substrates are macromolecules like plastics. Promising strategies have emerged to address these issues, such as downsizing MIPs using microgels,122 integrating porous materials to produce highly cross-linked MIPs,124,125 incorporating highly active nanozymes126 and so on. These advances hold great promise for the application of the molecular imprinting approach in plastic degradation.3.2. Mimicking clefts using porous materials
In the context of polyolefin depolymerization, the emulation of a substrate-binding cleft has been focused on tuning the interactions between polymer chains and porous materials. The interfacial interactions are initially investigated between porous filters and polymers in early research.127,128 Duan et al. offered insights into this behavior by comparing the infiltrations of poly(vinylidene fluoride) (PVDF) and poly(ethylene oxide) (PEO) into porous UiO-66.129 As shown in Fig. 7a, PEO fully infiltrated into the pores of UiO-66 due to stronger site-to-site interaction with O-(PEO) and the terminal OH groups on the Zr nodes of UiO-66. In contrast, PVDF exhibited limited infiltration into UiO-66 because of the weaker interaction between F-(PVDF) and the terminal OH groups.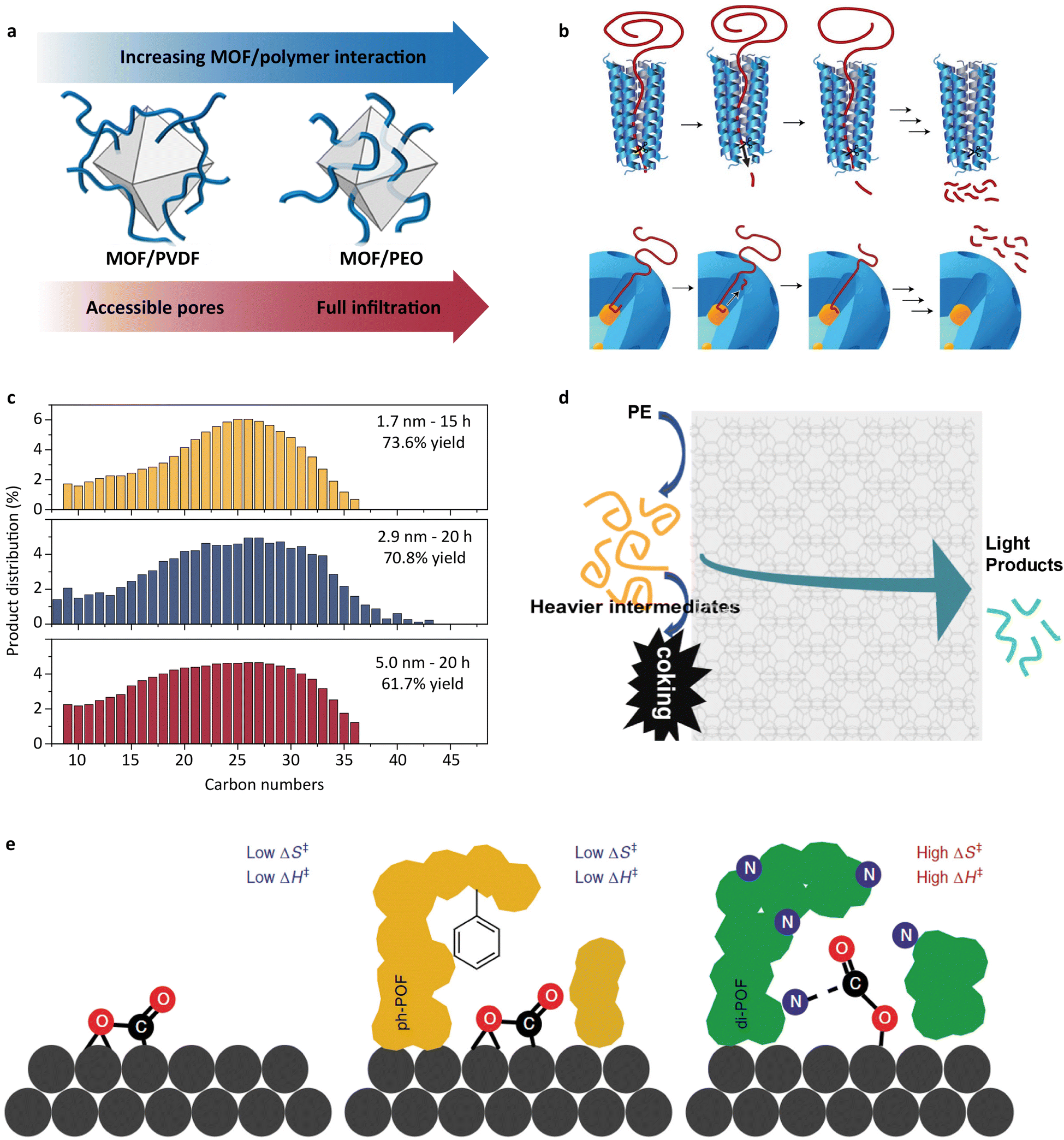 | ||
| Fig. 7 (a) Schematic illustration of the infiltration of PVDF and PEO into UiO-66.129 Adapted with permission from ref. 129. Copyright 2019 American Chemical Society. (b) The processive mechanism through which many enzymes deconstruct large macromolecules and an analogous mechanism proposed for the mSiO2/Pt/SiO2 catalyst.11 Adapted with permission from ref. 11. Copyright 2020 Springer Nature. (c) Carbon number distributions of wax products from polyethylene hydrogenolysis using mSiO2/Pt-x/SiO2 (x: the mean Pt nanoparticle diameter). Top: mSiO2/Pt-1.7/SiO2; center: mSiO2/Pt-2.9/SiO2; bottom: mSiO2/Pt-5.0/SiO2.130 Adapted with permission from ref. 130. Copyright 2022 American Chemical Society. (d) Schematic illustration of the cascade cracking steps on the external zeolite surface and within zeolite micropores on n-ZSM-5.131 Adapted with permission from ref. 131. Copyright 2022 American Chemical Society. (e) The proposed transition state models for exposed Pd surfaces (such as in Pd/Al2O3 and Pd/POF), non-interacting POF-covered Pd surfaces (such as in ph-POF/Pd/POF) and strongly interacting POF layers (such as in POF/Pd/POF and di-POF/Pd/POF), respectively.132 Adapted with permission from ref. 132. Copyright 2019 Springer Nature. | ||
The studies of the interfacial interactions between the inorganic porous phase and continuous polymeric phase have inspired researchers to introduce the concept of a substrate-binding cleft into the catalyst design for plastic depolymerization. Perras and coworkers designed a core–shell material for the degradation of waste polyolefins.11 This core–shell catalyst consists of Pt nanoparticles supported on SiO2 as the core and porous SiO2 as the shell. Through solid-state nuclear magnetic resonance analysis, they confirmed that polyethylene chains diffused into the pores of the SiO2 shell and interacted with the active sites in the core. Importantly, the diffusion rate of the polymer chains could be controlled by tuning the pore size, thereby influencing the product distribution. They likened this to an enzyme-like progressive depolymerization mechanism and correlated the selectivity with a “headfirst” adsorption of the polymer chains (Fig. 7b). In specific, the polymer chains adopt a straightened conformation navigating the narrow pores of the shell to reach the active sites situated at the core. The cleavage of the C–C bonds occurs at regular intervals, a process regulated by the diffusion rate of the polymer chains. Further research on the core–shell structure was conducted by Wu et al.,130 focusing on how changes in the activity affect this progressive mechanism. It was found that the rate of C–C bond cleavage can be modulated by changing the size of Pt nanoparticles, while the product distribution remained nearly constant (Fig. 7c). This phenomenon suggests that the progressive mechanism is independent of the activity of the core–shell catalysts. Another research study on an “open–open” pore structure was recently reported by Tennakoon et al.133 This structure facilitated a faster diffusion rate of the products from the active sites compared with the traditional “open-end” core–shell structure. This diffusion behavior can avoid over-hydrogenolysis and thus achieves a very low yield of volatile products. These results highlight that the structure designed substrate-binding clefts tuned the selectivity via controlling the polymer chains' diffusion rate in its designed structure.
Designing porous materials to mimic substrate-binding clefts can also alter selectivity by influencing reaction mechanisms. Zhao et al. found that the product distribution of PP pyrolysis narrowed from the C3–C15 range in the blank group to C4–C9 in the presence of HY zeolites.134 Meanwhile, cyclic hydrocarbons were detected in the presence of HY, whereas they were absent in the blank group. This suggests that PP pyrolysis follows not only the free radical mechanism but also the ionic mechanism. The effect of pore size on the product distribution was also observed in the catalytic pyrolysis of mixed PP and PE.135 ZSM-5 showed higher selectivity for olefins than USY, while USY generated more paraffins and coke than ZSM-5. This difference may result from hindrance of bimolecular reactions in the 10-membered-ring channels of ZSM-5, whereas the 12-membered-ring pores of USY allowed for these reactions to occur. This implies that the diffusion limitation of intermediate products in zeolites can change the product distribution by influencing reaction types. Catalytic pyrolysis of model compounds clearly demonstrated this effect.136 Small pore size (<6.1 Å) zeolites mainly promoted the cleavage of long-chain hydrocarbons with little benzene, toluene, and xylene (BTX) produced, because the pore size is insufficient for these aromatic products to diffuse. Large pore size (≥6.1 Å) zeolites can further catalyze aromatization and hydrogen transfer reactions to produce BTX and paraffins. By mimicking the substrate-binding cleft using porous materials, the diffusion and reaction of polymers and intermediates can be tuned, thereby altering the product distribution.
3.3. The challenge in selectivity control
The role of substrate-binding clefts in affecting reaction activity and selectivity is crucial. However, the efficiency of this strategy lies in the precise positioning of the active sites. Often times, a polymer chain can navigate and react within a pore, but the desired selectivity cannot be achieved in the absence of accurate positioning. Polyethylene hydrocracking catalyzed by zeolites evidences this point (Fig. 7d). The Brønsted acid sites in zeolites act as the primary active centers for cracking. When Brønsted acid sites are uniformly distributed across the zeolite structure, the product distribution for polyethylene cracking appears similar among different zeolites, predominantly falling within the C2 to C9 range.131,137,138 Future studies need to give more considerations to the appropriate positions of the active sites in the porous matrix to regulate the product distribution.While it has been shown that the diffusion of polymers in porous materials is pore-size-dependent, the chemical feature of the functional groups on the inner surface of the pores can be another important factor.129,139 Riscoe et al. offered an approach into this dimension (Fig. 7e). They crafted a polymer–nanocrystal hybrid to tailor CO2 diffusion by modulating the functional groups inside the pores including the phenyl and the amino groups.132 It turned out that the amino group has lower adsorption enthalpy toward the CO2 product, because N atoms interact with the CO2 product and reduce its diffusion rate in the pores. The strategy of surface functionalization could be more widely used in catalyst design in the future. For instance, adding hydrophilic functional groups into the SiO2 pores might reduce the PE diffusion rate thus altering the selectivity. In addition, tuning the density and chemical feature of the functional groups is more precise than controlling the pore size. It is expected that more efforts will be dedicated to this area.
4. Conclusions and perspectives
The urgency of plastic pollution has stimulated extensive research into chemical recycling processes and plastic-degrading enzymes. Biomimetic catalysis has been demonstrated to be effective in degrading macromolecules under mild conditions (Table 1). This perspective has underscored two main approaches to mimic the plastic-degrading enzymes: imitating the active sites and the substrate-binding cleft. The active sites play a key role in activating and transforming substrates. Directly replicating the active site of plastic-degrading enzymes is highly challenging because their active sites are composed of a network of amino acid residues with intricate positioning. Alternatively, the structural emulation of binuclear esterases has been well-developed by following similar catalytic mechanisms. These esterases contain two metal centers and utilize the proximity effect to enhance the effective concentration of substrates locally during hydrolysis. The mimicry of the active centers of binuclear esterase has shown excellent polyester hydrolysis activity even under weakly alkaline or near-neutral conditions.140 In addition to metallic centers, the second coordination sphere often plays subtle yet crucial roles in stabilizing key intermediates through non-covalent interactions. Therefore, it is anticipated that the regulation of the functional groups in the second coordination sphere, for instance by introducing auxiliary ligands in metal complexes, would further improve the catalytic activity of enzyme mimics thus facilitating plastic depolymerization under mild conditions.| Enzyme mimics | Conversion (%) | Conditions | Ref. | |
|---|---|---|---|---|
| a Weight loss. b Yield of levulinic acid. c Yield of glucose. d Yield of reducing sugar. e Yield of phenolic compounds. f Product: open circular DNA. g High density PE as the reactant. | ||||
| PET hydrolysis | Self-assembled peptide | 16a | pH 9, 65 °C, 2 weeks | 64 |
| Binuclear zinc complex | 100 | pH 8, 40 °C, 10 weeks | 65 | |
| n-Pentyl-substituted binuclear zinc complex | 100 | 80% 0.1 M NaOH solution + 20% CH3OH, 60 °C, 10 h | 66 | |
| Cellulose hydrolysis | COOH-functionalized carbon | 96 | 180 °C, 20 min | 73 |
| 5-OH-SHAPO | 62 | 150 °C, 5 h | 78 | |
| Solid acid containing –Cl and –SO3H | 46b | 180 °C, 12 h | 80 | |
| Porous polymers bearing boronic and sulfonic acids | 94.6c | 120 °C, 48 h | 83 | |
| Molecularly imprinted synthetic glucosidase | 15.8d | pH 5, 90 °C, 12 h | 123 | |
| A blend of cellulase-mimicking polymeric nanoparticles | 40.4d | pH 6.5, 90 °C, 12 h | 72 | |
| Lignin oxidation | Co(DPCys) | 31.2 | 150 °C, 10 h, oxone | 91 |
| Pep-1/hemin | 61.7e | 60 °C, 3 h, H2O2 | 92 | |
| Microplastic degradation | Fe3O4 nanoaggregates | 100 for PE and PS, 95 for PP, 64 for PETa | 12 h, H2O2, 150 °C for PE, 200 °C for PS, 140 °C for PP, 240 °C for PET | 102 |
| DNA hydrolysis | [Cu(L1)2(Br)](ClO4)5 | 100 | pH 7.2, 37 °C, 1 h | 109 |
| FeCuL1-PEI | 45f | pH 7.0, 37 °C, 24 h | 111 | |
| FeZnL2-et | 63f | pH 7.0, 50 °C, 4 h | 112 | |
| FeZnLP1 | 95 | pH 7.0, 37 °C, 16 h | 113 | |
| FeZnLP2 | 100 | |||
| PE hydrogenolysis | mSiO2/Pt/SiO2 | 6.7g | 1.38 MPa H2, 250 °C, 6 h | 11 |
| mSiO2/Pt/MCM-48 | 35.4g | 2.06 MPa H2, 300 °C, 6 h | 133 | |
On the other hand, enzymes typically feature a substrate-binding cleft ensuring the specificity for the substrate and the selectivity of the product. This structure facilitates enzymes to degrade polymers in a processive manner in some cases. The molecular imprinting approach has been employed to emulate the enzyme's cleft, proving effective in cellulose hydrolysis. Nevertheless, this technique encounters limitations when applied to plastics. Porous materials have been introduced to mimic the interaction between the substrate and clefts in the enzyme, as demonstrated in core–shell materials, molecular sieves, and metal organic frameworks. These porous materials exhibit good stability, ease of synthesis, and mouldability of sites compared to molecule enzyme mimics. It has been verified that the product distribution of PE depolymerization is significantly affected by the pore size of the catalyst. It should be noted that the substrate-binding cleft in these biomimetic catalysts lacks the flexibility observed in natural enzymes, which is crucial for adjusting the active site and accommodating substrates.
From the above discussion, we can distill all these studies down to the following design principles:
(1) Active site: The essence of mimicking active sites is not just in replicating their exact structural nuances but also in emulating their reaction pathways. A profound understanding of the enzymatic reaction mechanism is paramount and should be the cornerstone of the catalyst design.
(2) Cleft: Porous materials are often employed to mimic the function of enzyme clefts. Adjusting pore sizes has proven effective in modulating polymer chain diffusion rates and enhancing selectivity to some extent. Altering the functional groups within the pores could be promising to achieve improved diffusion control of polymeric substrates.
(3) Synergy: The collaboration between the active site and the substrate-binding cleft is more important than imitating the function of the two alone. However, this synergy relies on the precise positioning of both the active site and the cleft. Their orchestrated alignment might unlock unprecedented catalytic efficiencies. Although the molecular imprinting strategy has shown great potential in simulating synergistic interactions during catalytic conversion of cellulose and small molecules, polymer depolymerization may require the integration of other approaches to address constrained mass transfer and poor stability issues. Developing biomimetic catalysts with a flexible cleft near active sites may potentially contribute to addressing constrained mass transfer issues. This may be achieved by incorporating materials such as peptides and peptoids into biomimetic catalysts.
Applying the aforementioned design principles, scientists have demonstrated the significance of biomimetic catalysis in recycling polyester and polyolefin plastics. A persistent challenge still lies in tailoring enzyme mimics to achieve substrate specificity, product selectivity, and high activity for plastic depolymerization, which are crucial for addressing complex real-world solid wastes. The application of biomimetic catalysis in plastic recycling is steadily gaining momentum, and we hope that this perspective will help bridge the gap between chemical recycling of plastics and enzyme-catalyzed macromolecular depolymerization.
Author contributions
Z. N. conceived the perspective and edited the manuscript. Y. W., Q. H., and Y. C. conducted literature review and prepared the first draft of the manuscript. The manuscript was written through contributions of all authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22375113 and 22075162), the National Key R&D Program of China (2019YFA0709200), the Tsinghua University Initiative Scientific Research Program (20221080067), and Ordos Laboratory (Ordoslab-2024014).References
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Plastic waste inputs from land into the ocean, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, The global threat from plastic pollution, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, Degradation Rates of Plastics in the Environment, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. Chen, F. A. Leibfarth and H. Sardon, Critical advances and future opportunities in upcycling commodity polymers, Nature, 2022, 603, 803–814 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Beyond Mechanical Recycling: Giving New Life to Plastic Waste, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Catalytic processing of plastic waste on the rise, Chem, 2021, 7, 1487–1533 Search PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Chemical recycling to monomer for an ideal, circular polymer economy, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- F. Zhang, F. Wang, X. Wei, Y. Yang, S. Xu, D. Deng and Y.-Z. Wang, From trash to treasure: Chemical recycling and upcycling of commodity plastic waste to fuels, high-valued chemicals and advanced materials, J. Energy Chem., 2022, 69, 369–388 CrossRef CAS.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. Laiq Ur Rehman, Q. Li and M. Ju, Upcycling and catalytic degradation of plastic wastes, Cell Rep. Phys. Sci., 2021, 2, 100514 CrossRef CAS.
- W. T. Lee, A. van Muyden, F. D. Bobbink, M. D. Mensi, J. R. Carullo and P. J. Dyson, Mechanistic classification and benchmarking of polyolefin depolymerization over silica-alumina-based catalysts, Nat. Commun., 2022, 13, 4850 CrossRef CAS PubMed.
- A. Tennakoon, X. Wu, A. L. Paterson, S. Patnaik, Y. Pei, A. M. LaPointe, S. C. Ammal, R. A. Hackler, A. Heyden, I. I. Slowing, G. W. Coates, M. Delferro, B. Peters, W. Huang, A. D. Sadow and F. A. Perras, Catalytic upcycling of high-density polyethylene via a processive mechanism, Nat. Catal., 2020, 3, 893–901 CrossRef CAS.
- I. L. Peczak, R. M. Kennedy, R. A. Hackler, B. Lee, M. Meirow, E. Luijten, K. R. Poeppelmeier and M. Delferro, Treasuring trash: Pt/SrTiO3 catalysts process plastic waste into high-value materials, Matter, 2023, 6, 3296–3321 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Chemical and biological catalysis for plastics recycling and upcycling, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Catalysis as an Enabling Science for Sustainable Polymers, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- R. Esquer and J. J. García, Metal-catalysed poly(ethylene) terephthalate and polyurethane degradations by glycolysis, J. Organomet. Chem., 2019, 902, 120972 CrossRef CAS.
- F. R. Veregue, C. T. Pereira da Silva, M. P. Moisés, J. G. Meneguin, M. R. Guilherme, P. A. Arroyo, S. L. Favaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, Ultrasmall Cobalt Nanoparticles as a Catalyst for PET Glycolysis: A Green Protocol for Pure Hydroxyethyl Terephthalate Precipitation without Water, ACS Sustainable Chem. Eng., 2018, 6, 12017–12024 CrossRef CAS.
- L. Wursthorn, K. Beckett, J. O. Rothbaum, R. M. Cywar, C. Lincoln, Y. Kratish and T. J. Marks, Selective Lanthanide-Organic Catalyzed Depolymerization of Nylon-6 to ε-Caprolactam, Angew. Chem., Int. Ed., 2022, 62, e202212543 CrossRef PubMed.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, Hydrogenative Depolymerization of Nylons, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed.
- M. A. M. E. Vertommen, V. A. Nierstrasz, M. v. d. Veer and M. M. C. G. Warmoeskerken, Enzymatic surface modification of poly(ethylene terephthalate), J. Biotechnol., 2005, 120, 376–386 CrossRef CAS PubMed.
- K. Świderek, S. Velasco-Lozano, M. À. Galmés, I. Olazabal, H. Sardon, F. López-Gallego and V. Moliner, Mechanistic studies of a lipase unveil effect of pH on hydrolysis products of small PET modules, Nat. Commun., 2023, 14, 3556 CrossRef.
- R.-J. Müller, H. Schrader, J. Profe, K. Dresler and W.-D. Deckwer, Enzymatic Degradation of Poly(ethylene terephthalate): Rapid Hydrolyse using a Hydrolase from T. fusca, Macromol. Rapid Commun., 2005, 26, 1400–1405 CrossRef.
- Å. M. Ronkvist, W. Xie, W. Lu and R. A. Gross, Cutinase-Catalyzed Hydrolysis of Poly(ethylene terephthalate), Macromolecules, 2009, 42, 5128–5138 CrossRef.
- A. M. de Castro, A. Carniel, J. Nicomedes Junior, A. da Conceicao Gomes and E. Valoni, Screening of commercial enzymes for poly(ethylene terephthalate) (PET) hydrolysis and synergy studies on different substrate sources, J. Ind. Microbiol. Biotechnol., 2017, 44, 835–844 CrossRef PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, A bacterium that degrades and assimilates poly(ethylene terephthalate), Science, 2016, 351, 1196 CrossRef CAS.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guemard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. Andre, S. Duquesne and A. Marty, An engineered PET depolymerase to break down and recycle plastic bottles, Nature, 2020, 580, 216–219 CrossRef CAS.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Machine learning-aided engineering of hydrolases for PET depolymerization, Nature, 2022, 604, 662–667 CrossRef CAS.
- R. Wei and W. Zimmermann, Microbial enzymes for the recycling of recalcitrant petroleum-based plastics: how far are we?, Microb. Biotechnol., 2017, 10, 1308–1322 CrossRef CAS PubMed.
- M. Santo, R. Weitsman and A. Sivan, The role of the copper-binding enzyme – laccase – in the biodegradation of polyethylene by the actinomycete Rhodococcus ruber, Int. Biodeterior. Biodegrad., 2013, 84, 204–210 CrossRef CAS.
- M. Fujisawa, H. Hirai and T. Nishida, Degradation of Polyethylene and Nylon-66 by the Laccase-Mediator System, J. Polym. Environ., 2001, 9, 103–108 CrossRef CAS.
- S. Ghatge, Y. Yang, J.-H. Ahn and H.-G. Hur, Biodegradation of polyethylene: a brief review, Appl. Biol. Chem., 2020, 63, 27 CrossRef CAS.
- M. Koutny, M. Sancelme, C. Dabin, N. Pichon, A.-M. Delort and J. Lemaire, Acquired biodegradability of polyethylenes containing pro-oxidant additives, Polym. Degrad. Stab., 2006, 91, 1495–1503 CrossRef CAS.
- A. Arkatkar, A. A. Juwarkar, S. Bhaduri, P. V. Uppara and M. Doble, Growth of Pseudomonas and Bacillus biofilms on pretreated polypropylene surface, Int. Biodeterior. Biodegrad., 2010, 64, 530–536 CrossRef CAS.
- H. J. Jeon and M. N. Kim, Functional analysis of alkane hydroxylase system derived from Pseudomonas aeruginosa E7 for low molecular weight polyethylene biodegradation, Int. Biodeterior. Biodegrad., 2015, 103, 141–146 CrossRef CAS.
- M. Yoon and H. Jeon, Biodegradation of Polyethylene by a Soil Bacterium and AlkB Cloned Recombinant Cell, J. Biorem. Biodegrad., 2012, 3, 145 CAS.
- K. Nakamiya, G. Sakasita, T. Ooi and S. Kinoshita, Enzymatic degradation of polystyrene by hydroquinone peroxidase of Azotobacter beijerinckii HM121, J. Ferment. Bioeng., 1997, 84, 480–482 CrossRef CAS.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Enzymes' Power for Plastics Degradation, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- M. Parvinzadeh, R. Assefipour and A. Kiumarsi, Biohydrolysis of nylon 6,6 fiber with different proteolytic enzymes, Polym. Degrad. Stab., 2009, 94, 1197–1205 CrossRef CAS.
- C. Silva, R. Araújo, M. Casal, G. M. Gübitz and A. Cavaco-Paulo, Influence of mechanical agitation on cutinases and protease activity towards polyamide substrates, Enzyme Microb. Technol., 2007, 40, 1678–1685 CrossRef CAS.
- M. Kanelli, S. Vasilakos, S. Ladas, E. Symianakis, P. Christakopoulos and E. Topakas, Surface modification of polyamide 6.6 fibers by enzymatic hydrolysis, Process Biochem., 2017, 59, 97–103 CrossRef CAS.
- M. Fujisawa, H. Hirai and T. Nishida, Degradation of Polyethylene and Nylon-66 by the Laccase-Mediator System, J. Polym. Environ., 2001, 9, 103–108 CrossRef CAS.
- Y. Lee and N. K. Devaraj, Lipase mimetic cyclodextrins, Chem. Sci., 2021, 12, 1090–1094 RSC.
- G. W. Gokel, S. Negin and R. Cantwell, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, Elsevier, Oxford, 2017, pp. 3–48, DOI:10.1016/B978-0-12-409547-2.12519-3.
- M. M. Pereira, L. D. Dias and M. J. F. Calvete, Metalloporphyrins: Bioinspired Oxidation Catalysts, ACS Catal., 2018, 8, 10784–10808 CrossRef CAS.
- R. Tian, Y. Li, J. Xu, C. Hou, Q. Luo and J. Liu, Recent development in the design of artificial enzymes through molecular imprinting technology, J. Mater. Chem. B, 2022, 10, 6590–6606 RSC.
- J. Li, M. Zhu, M. Wang, W. Qi, R. Su and Z. He, Molecularly imprinted peptide-based enzyme mimics with enhanced activity and specificity, Soft Matter, 2020, 16, 7033–7039 RSC.
- Z. Yu, R. Lou, W. Pan, N. Li and B. Tang, Nanoenzymes in disease diagnosis and therapy, Chem. Commun., 2020, 56, 15513–15524 RSC.
- S. Li, Z. Zhou, Z. Tie, B. Wang, M. Ye, L. Du, R. Cui, W. Liu, C. Wan, Q. Liu, S. Zhao, Q. Wang, Y. Zhang, S. Zhang, H. Zhang, Y. Du and H. Wei, Data-informed discovery of hydrolytic nanozymes, Nat. Commun., 2022, 13, 827 CrossRef CAS.
- B. Li, R. Ma, L. Chen, C. Zhou, Y.-X. Zhang, X. Wang, H. Huang, Q. Hu, X. Zheng, J. Yang, M. Shao, P. Hao, Y. Wu, Y. Che, C. Li, T. Qin, L. Gao, Z. Niu and Y. Li, Diatomic iron nanozyme with lipoxidase-like activity for efficient inactivation of enveloped virus, Nat. Commun., 2023, 14, 7312 CrossRef CAS PubMed.
- E. Ghanem and F. M. Raushel, in eLS, 2012, DOI:10.1002/9780470015902.a0000714.pub2.
- C. Selvaraj, O. Rudhra, A. S. Alothaim, M. Alkhanani and S. K. Singh, in Advances in Protein Chemistry and Structural Biology, ed. R. Donev, Academic Press, 2022, vol. 130, pp. 59–83 Search PubMed.
- F. Yabukarski, J. T. Biel, M. M. Pinney, T. Doukov, A. S. Powers, J. S. Fraser and D. Herschlag, Assessment of enzyme active site positioning and tests of catalytic mechanisms through X-ray-derived conformational ensembles, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 33204–33215 CrossRef CAS PubMed.
- B. C. Knott, E. Erickson, M. D. Allen, J. E. Gado, R. Graham, F. L. Kearns, I. Pardo, E. Topuzlu, J. J. Anderson, H. P. Austin, G. Dominick, C. W. Johnson, N. A. Rorrer, C. J. Szostkiewicz, V. Copié, C. M. Payne, H. L. Woodcock, B. S. Donohoe, G. T. Beckham and J. E. McGeehan, Characterization and engineering of a two-enzyme system for plastics depolymerization, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25476–25485 CrossRef CAS PubMed.
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. L. Woodcock, J. E. McGeehan and G. T. Beckham, Characterization and engineering of a plastic-degrading aromatic polyesterase, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4350–E4357 CrossRef CAS PubMed.
- H. Hong, D. Ki, H. Seo, J. Park, J. Jang and K.-J. Kim, Discovery and rational engineering of PET hydrolase with both mesophilic and thermophilic PET hydrolase properties, Nat. Commun., 2023, 14, 4556 CrossRef CAS PubMed.
- X. Han, W. Liu, J. W. Huang, J. Ma, Y. Zheng, T. P. Ko, L. Xu, Y. S. Cheng, C. C. Chen and R. T. Guo, Structural insight into catalytic mechanism of PET hydrolase, Nat. Commun., 2017, 8, 2106 CrossRef PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Supramolecular catalysis. Part 2: artificial enzyme mimics, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- A. Silakov, M. T. Olsen, S. Sproules, E. J. Reijerse, T. B. Rauchfuss and W. Lubitz, EPR/ENDOR, Mossbauer, and quantum-chemical investigations of diiron complexes mimicking the active oxidized state of [FeFe]hydrogenase, Inorg. Chem., 2012, 51, 8617–8628 CrossRef CAS PubMed.
- C. J. Allpress, K. Grubel, E. Szajna-Fuller, A. M. Arif and L. M. Berreau, Regioselective aliphatic carbon-carbon bond cleavage by a model system of relevance to iron-containing acireductone dioxygenase, J. Am. Chem. Soc., 2013, 135, 659–668 CrossRef CAS PubMed.
- S. Zheng, T. C. Berto, E. W. Dahl, M. B. Hoffman, A. L. Speelman and N. Lehnert, The functional model complex [Fe2(BPMP)(OPr)(NO)2](BPh4)2 provides insight into the mechanism of flavodiiron NO reductases, J. Am. Chem. Soc., 2013, 135, 4902–4905 CrossRef CAS PubMed.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Insights into metalloenzyme microenvironments: biomimetic metal complexes with a functional second coordination sphere, Chem. Soc. Rev., 2013, 42, 8360–8375 RSC.
- F. Mancin, P. Scrimin and P. Tecilla, Progress in artificial metallonucleases, Chem. Commun., 2012, 48, 5545–5559 RSC.
- M. D. Nothling, Z. Xiao, A. Bhaskaran, M. T. Blyth, C. W. Bennett, M. L. Coote and L. A. Connal, Synthetic Catalysts Inspired by Hydrolytic Enzymes, ACS Catal., 2018, 9, 168–187 CrossRef.
- J. Bjerre, C. Rousseau, L. Marinescu and M. Bols, Artificial enzymes, “chemzymes”: current state and perspectives, Appl. Microbiol. Biotechnol., 2008, 81, 1–11 CrossRef CAS PubMed.
- X. Li, Y. Zhou, Z. Lu, R. Shan, D. Sun, J. Li and P. Li, Switchable enzyme mimics based on self-assembled peptides for polyethylene terephthalate degradation, J. Colloid Interface Sci., 2023, 646, 198–208 CrossRef CAS PubMed.
- S. Zhang, Q. Hu, Y.-X. Zhang, H. Guo, Y. Wu, M. Sun, X. Zhu, J. Zhang, S. Gong, P. Liu and Z. Niu, Depolymerization of polyesters by a binuclear catalyst for plastic recycling, Nat. Sustain., 2023, 6, 965–973 CrossRef.
- S. Zhang, Y. Xue, Y. Wu, Y.-X. Zhang, T. Tan and Z. Niu, PET recycling under mild conditions via substituent-modulated intramolecular hydrolysis, Chem. Sci., 2023, 14, 6558–6563 RSC.
- G. Schenk, N. Mitic, L. R. Gahan, D. L. Ollis, R. P. McGeary and L. W. Guddat, Binuclear metallohydrolases: complex mechanistic strategies for a simple chemical reaction, Acc. Chem. Res., 2012, 45, 1593–1603 CrossRef CAS.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Targeted chemical upgrading of lignocellulosic biomass to platform molecules, Green Chem., 2014, 16, 4816–4838 RSC.
- D. L. Zechel and S. G. Withers, Glycosidase mechanisms: anatomy of a finely tuned catalyst, Acc. Chem. Res., 2000, 33, 11–18 CrossRef CAS PubMed.
- S. Pengthaisong, B. Piniello, G. J. Davies, C. Rovira and J. R. Ketudat Cairns, Reaction Mechanism of Glycoside Hydrolase Family 116 Utilizes Perpendicular Protonation, ACS Catal., 2023, 13, 5850–5863 CrossRef CAS PubMed.
- J. F. Wardman, R. K. Bains, P. Rahfeld and S. G. Withers, Carbohydrate-active enzymes (CAZymes) in the gut microbiome, Nat. Rev. Microbiol., 2022, 20, 542–556 CrossRef CAS PubMed.
- M. Zangiabadi and Y. Zhao, Synergistic Hydrolysis of Cellulose by a Blend of Cellulase-Mimicking Polymeric Nanoparticle Catalysts, J. Am. Chem. Soc., 2022, 144, 17110–17119 CrossRef CAS PubMed.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Air Oxidation of Activated Carbon to Synthesize a Biomimetic Catalyst for Hydrolysis of Cellulose, ChemSusChem, 2016, 9, 1299–1303 CrossRef CAS PubMed.
- Z. Chen, Q. Li, Y. Xiao, C. Zhang, Z. Fu, Y. Liu, X. Yi, A. Zheng, C. Li and D. Yin, Acid–base synergistic catalysis of biochar sulfonic acid bearing polyamide for microwave-assisted hydrolysis of cellulose in water, Cellulose, 2018, 26, 751–762 CrossRef.
- E. J. Cho, S. J. Lee, K. Lee, D.-S. Lee, Y. J. Lee and H.-J. Bae, A Reusable Biomimetic Magnetic Nanoenzyme for Cellulosic Biomass Degradation, BioEnergy Res., 2014, 8, 788–795 CrossRef.
- E. J. Cho, Y. Song, Y. J. Lee and H.-J. Bae, Preparation and characterization of novel green magnetic nanocatalyst for cellulosic biomass degradation under mild conditions, J. Ind. Eng. Chem., 2016, 40, 185–190 CrossRef CAS.
- H. Kobayashi, M. Yabushita, J.-y. Hasegawa and A. Fukuoka, Synergy of Vicinal Oxygenated Groups of Catalysts for Hydrolysis of Cellulosic Molecules, J. Phys. Chem. C, 2015, 119, 20993–20999 CrossRef CAS.
- F. Yu, M. Smet, W. Dehaen and B. F. Sels, Water-soluble sulfonated hyperbranched poly(arylene oxindole) catalysts as functional biomimics of cellulases, Chem. Commun., 2016, 52, 2756–2759 RSC.
- G. Yang, X. Luo and L. Shuai, Bioinspired Cellulase-Mimetic Solid Acid Catalysts for Cellulose Hydrolysis, Front. Bioeng. Biotechnol., 2021, 9, 770027 CrossRef PubMed.
- F. Shen, R. L. Smith, L. Li, L. Yan and X. Qi, Eco-friendly Method for Efficient Conversion of Cellulose into Levulinic Acid in Pure Water with Cellulase-Mimetic Solid Acid Catalyst, ACS Sustainable Chem. Eng., 2017, 5, 2421–2427 CrossRef CAS.
- H.-X. Li, X. Zhang, Q. Wang, D. Yang, Q. Cao and L. Jin, Study on the hydrolysis of cellulose with the regenerable and recyclable multifunctional solid acid as a catalyst and its catalytic hydrolytic kinetics, Cellulose, 2019, 27, 285–300 CrossRef.
- C. Zhang, Z. Fu, Y. C. Liu, B. Dai, Y. Zou, X. Gong, Y. Wang, X. Deng, H. Wu, Q. Xu, K. R. Steven and D. Yin, Ionic liquid-functionalized biochar sulfonic acid as a biomimetic catalyst for hydrolysis of cellulose and bamboo under microwave irradiation, Green Chem., 2012, 14, 1928–1934 RSC.
- Q. Yang and X. Pan, Bifunctional Porous Polymers Bearing Boronic and Sulfonic Acids for Hydrolysis of Cellulose, ACS Sustainable Chem. Eng., 2016, 4, 4824–4830 CrossRef CAS.
- T. Deguchi, M. Kakezawa and T. Nishida, Nylon biodegradation by lignin-degrading fungi, Appl. Environ. Microbiol., 1997, 63, 329–331 CrossRef CAS PubMed.
- T. Deguchi, Y. Kitaoka, M. Kakezawa and T. Nishida, Purification and characterization of a nylon-degrading enzyme, Appl. Environ. Microbiol., 1998, 64, 1366–1371 CrossRef CAS PubMed.
- A. Nyyssölä and J. Ahlgren, Microbial degradation of polyacrylamide and the deamination product polyacrylate, Int. Biodeterior. Biodegrad., 2019, 139, 24–33 CrossRef.
- K. Nakamiya, T. Ooi and S. Kinoshita, Degradation of synthetic water-soluble polymers by hydroquinone peroxidase, J. Ferment. Bioeng., 1997, 84, 213–218 CrossRef CAS.
- W. J. R. Gilbert, S. J. Johnson, J. S. Tsau, J. T. Liang and A. M. Scurto, Enzymatic degradation of polyacrylamide in aqueous solution with peroxidase and H2O2, J. Appl. Polym. Sci., 2016, 134, 44560 CrossRef.
- W. Bankeeree, C. Samathayanon, S. Prasongsuk, P. Lotrakul and S. Kiatkamjornwong, Rapid Degradation of Superabsorbent Poly(Potassium Acrylate) and its Acrylamide Copolymer Via Thermo-Oxidation by Hydrogen Peroxide, J. Polym. Environ., 2021, 29, 3964–3976 CrossRef CAS.
- T. L. Poulos, Heme Enzyme Structure and Function, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS.
- C. Zhu, W. Ding, T. Shen, C. Tang, C. Sun, S. Xu, Y. Chen, J. Wu and H. Ying, Metallo-deuteroporphyrin as a biomimetic catalyst for the catalytic oxidation of lignin to aromatics, ChemSusChem, 2015, 8, 1768–1778 CrossRef CAS.
- T. Jian, Y. Zhou, P. Wang, W. Yang, P. Mu, X. Zhang, X. Zhang and C. L. Chen, Highly stable and tunable peptoid/hemin enzymatic mimetics with natural peroxidase-like activities, Nat. Commun., 2022, 13, 3025 CrossRef CAS.
- N. Zhu, C. Liu, R. Liu, X. Niu, D. Xiong, K. Wang, D. Yin and Z. Zhang, Biomimic Nanozymes with Tunable Peroxidase-like Activity Based on the Confinement Effect of Metal–Organic Frameworks (MOFs) for Biosensing, Anal. Chem., 2022, 94, 4821–4830 CrossRef CAS PubMed.
- F.-X. Qin, S.-Y. Jia, F.-F. Wang, S.-H. Wu, J. Song and Y. Liu, Hemin@metal–organic framework with peroxidase-like activity and its application to glucose detection, Catal. Sci. Technol., 2013, 3, 2761–2768 RSC.
- Q. Xin, X. Jia, A. Nawaz, W. Xie, L. Li and J. R. Gong, Mimicking peroxidase active site microenvironment by functionalized graphene quantum dots, Nano Res., 2020, 13, 1427–1433 CrossRef CAS.
- M. A. Castriciano, A. Romeo, M. C. Baratto, R. Pogni and L. M. Scolaro, Supramolecular mimetic peroxidase based on hemin and PAMAM dendrimers, Chem. Commun., 2008, 6, 688–690 RSC.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Nanomaterials with enzyme-like characteristics (nanozymes): next-generation artificial enzymes (II), Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- Q. Diao, X. Chen, Z. Tang, S. Li, Q. Tian, Z. Bu, H. Liu, J. Liu and X. Niu, Nanozymes: powerful catalytic materials for environmental pollutant detection and degradation, Environ. Sci.: Nano, 2024, 11, 766–796 RSC.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Intrinsic peroxidase-like activity of ferromagnetic nanoparticles, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- K. Fan, H. Wang, J. Xi, Q. Liu, X. Meng, D. Duan, L. Gao and X. Yan, Optimization of Fe3O4 nanozyme activity via single amino acid modification mimicking an enzyme active site, Chem. Commun., 2017, 53, 424–427 RSC.
- M. Liang, K. Fan, Y. Pan, H. Jiang, F. Wang, D. Yang, D. Lu, J. Feng, J. Zhao, L. Yang and X. Yan, Fe3O4 Magnetic Nanoparticle Peroxidase Mimetic-Based Colorimetric Assay for the Rapid Detection of Organophosphorus Pesticide and Nerve Agent, Anal. Chem., 2013, 85, 308–312 CrossRef CAS PubMed.
- M. Zandieh and J. Liu, Removal and Degradation of Microplastics Using the Magnetic and Nanozyme Activities of Bare Iron Oxide Nanoaggregates, Angew. Chem., Int. Ed., 2022, 61, e202212013 CrossRef CAS.
- N. Mitić, S. J. Smith, A. Neves, L. W. Guddat, L. R. Gahan and G. Schenk, The Catalytic Mechanisms of Binuclear Metallohydrolases, Chem. Rev., 2006, 106, 3338–3363 CrossRef.
- Y.-W. Lin, Rational design of metalloenzymes: From single to multiple active sites, Coord. Chem. Rev., 2017, 336, 1–27 CrossRef CAS.
- J. D. McCarter and G. Stephen Withers, Mechanisms of enzymatic glycoside hydrolysis, Curr. Opin. Struct. Biol., 1994, 4, 885–892 CrossRef CAS PubMed.
- J. N. H. Reek, B. De Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Transition Metal Catalysis Controlled by Hydrogen Bonding in the Second Coordination Sphere, Chem. Rev., 2022, 122, 12308–12369 CrossRef CAS PubMed.
- T. Klabunde, H. Witzel and B. Krebs, Mechanism of Fe(III)–Zn(II) Purple Acid Phosphatase Based on Crystal Structures, J. Mol. Biol., 1996, 259, 737–748 CrossRef CAS.
- H. Kaija, S. L. Alatalo, J. M. Halleen, Y. Lindqvist, G. Schneider, H. Kalervo Väänänen and P. Vihko, Phosphatase and Oxygen Radical-Generating Activities of Mammalian Purple Acid Phosphatase Are Functionally Independent, Biochem. Biophys. Res. Commun., 2002, 292, 128–132 CrossRef CAS.
- Y. An, M.-L. Tong, L.-N. Ji and Z.-W. Mao, Double-strand DNA cleavage by copper complexes of 2,2′-dipyridyl with electropositive pendants, Dalton Trans., 2006, 2066–2071 RSC.
- L. Tjioe, A. Meininger, T. Joshi, L. Spiccia and B. Graham, Efficient Plasmid DNA Cleavage by Copper(II) Complexes of 1,4,7-Triazacyclononane Ligands Featuring Xylyl-Linked Guanidinium Groups, Inorg. Chem., 2011, 50, 4327–4339 CrossRef CAS.
- B. De Souza, G. L. Kreft, T. Bortolotto, H. Terenzi, A. J. Bortoluzzi, E. E. Castellano, R. A. Peralta, J. B. Domingos and A. Neves, Second-Coordination-Sphere Effects Increase the Catalytic Efficiency of an Extended Model for FeIIIMII Purple Acid Phosphatases, Inorg. Chem., 2013, 52, 3594–3596 CrossRef CAS.
- G. A. D. S. Silva, A. L. Amorim, B. D. Souza, P. Gabriel, H. Terenzi, E. Nordlander, A. Neves and R. A. Peralta, Synthesis and characterization of FeIII(μ-OH)ZnII complexes: effects of a second coordination sphere and increase in the chelate ring size on the hydrolysis of a phosphate diester and DNA, Dalton Trans., 2017, 46, 11380–11394 RSC.
- T. P. Camargo, A. Neves, R. A. Peralta, C. Chaves, E. C. P. Maia, E. H. Lizarazo-Jaimes, D. A. Gomes, T. Bortolotto, D. R. Norberto, H. Terenzi, D. L. Tierney and G. Schenk, Second-Sphere Effects in Dinuclear FeIIIZnII Hydrolase Biomimetics: Tuning Binding and Reactivity Properties, Inorg. Chem., 2018, 57, 187–203 CrossRef CAS PubMed.
- M. Borkowska, M. Siek, D. V. Kolygina, Y. I. Sobolev, S. Lach, S. Kumar, Y.-K. Cho, K. Kandere-Grzybowska and B. A. Grzybowski, Targeted crystallization of mixed-charge nanoparticles in lysosomes induces selective death of cancer cells, Nat. Nanotechnol., 2020, 15, 331–341 CrossRef CAS PubMed.
- C. Zhang, P. Gotico, R. Guillot, D. Dragoe, W. Leibl, Z. Halime and A. Aukauloo, Bio-Inspired Bimetallic Cooperativity Through a Hydrogen Bonding Spacer in CO2 Reduction, Angew. Chem., Int. Ed., 2023, 62, e202214665 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s−1 for H2 Production, Science, 2011, 333, 863–866 CrossRef CAS.
- B. Sharma and S. Striegler, Tailored Interactions of the Secondary Coordination Sphere Enhance the Hydrolytic Activity of Cross-Linked Microgels, ACS Catal., 2019, 9, 1686–1691 CrossRef CAS.
- R. Suno, H. Niwa, D. Tsuchiya, X. Zhang, M. Yoshida and K. Morikawa, Structure of the whole cytosolic region of ATP-dependent protease FtsH, Mol. Cell, 2006, 22, 575–585 CrossRef CAS.
- C. Divne, J. Ståhlberg, T. Reinikainen, L. Ruohonen, G. Pettersson, J. K. C. Knowles, T. T. Teeri and T. A. Jones, The Three-Dimensional Crystal Structure of the Catalytic Core of Cellobiohydrolase I from Trichoderma reesei, Science, 1994, 265, 524–528 CrossRef CAS PubMed.
- R. L. Silveira and M. S. Skaf, Concerted motions and large-scale structural fluctuations of Trichoderma reesei Cel7A cellobiohydrolase, Phys. Chem. Chem. Phys., 2018, 20, 7498–7507 RSC.
- S. Joo, I. J. Cho, H. Seo, H. F. Son, H.-Y. Sagong, T. J. Shin, S. Y. Choi, S. Y. Lee and K.-J. Kim, Structural insight into molecular mechanism of poly(ethylene terephthalate) degradation, Nat. Commun., 2018, 9, 382 CrossRef PubMed.
- A. Mujahid and F. L. Dickert, in Molecularly Imprinted Catalysts, 2016, pp. 79–101, DOI:10.1016/b978-0-12-801301-4.00005-0.
- X. Li, M. Zangiabadi and Y. Zhao, Molecularly Imprinted Synthetic Glucosidase for the Hydrolysis of Cellulose in Aqueous and Nonaqueous Solutions, J. Am. Chem. Soc., 2021, 143, 5172–5181 CrossRef CAS PubMed.
- Y. Yuan, Y. Yang, M. Faheem, X. Zou, X. Ma, Z. Wang, Q. Meng, L. Wang, S. Zhao and G. Zhu, Molecularly Imprinted Porous Aromatic Frameworks Serving as Porous Artificial Enzymes, Adv. Mater., 2018, 30, 1800069 CrossRef PubMed.
- L. Wan, H. Liu, C. Huang and X. Shen, Enzyme-like MOFs: synthetic molecular receptors with high binding capacity and their application in selective photocatalysis, J. Mater. Chem. A, 2020, 8, 25931–25940 RSC.
- Z. Zhang, X. Zhang, B. Liu and J. Liu, Molecular Imprinting on Inorganic Nanozymes for Hundred-fold Enzyme Specificity, J. Am. Chem. Soc., 2017, 139, 5412–5419 CrossRef CAS PubMed.
- J. M. Duval, A. J. B. Kemperman, B. Folkers, M. H. V. Mulder, G. Desgrandchamps and C. A. Smolders, Preparation of zeolite filled glassy polymer membranes, J. Appl. Polym. Sci., 1994, 54, 409–418 CrossRef CAS.
- I. F. J. Vankelecom, E. Merckx, M. Luts and J. B. Uytterhoeven, Incorporation of Zeolites in Polyimide Membranes, J. Phys. Chem., 1995, 99, 13187–13192 CrossRef CAS.
- P. Duan, J. C. Moreton, S. R. Tavares, R. Semino, G. Maurin, S. M. Cohen and K. Schmidt-Rohr, Polymer Infiltration into Metal-Organic Frameworks in Mixed-Matrix Membranes Detected in Situ by NMR, J. Am. Chem. Soc., 2019, 141, 7589–7595 CrossRef CAS PubMed.
- X. Wu, A. Tennakoon, R. Yappert, M. Esveld, M. S. Ferrandon, R. A. Hackler, A. M. LaPointe, A. Heyden, M. Delferro, B. Peters, A. D. Sadow and W. Huang, Size-Controlled Nanoparticles Embedded in a Mesoporous Architecture Leading to Efficient and Selective Hydrogenolysis of Polyolefins, J. Am. Chem. Soc., 2022, 144, 5323–5334 CrossRef CAS PubMed.
- J. Duan, W. Chen, C. Wang, L. Wang, Z. Liu, X. Yi, W. Fang, H. Wang, H. Wei, S. Xu, Y. Yang, Q. Yang, Z. Bao, Z. Zhang, Q. Ren, H. Zhou, X. Qin, A. Zheng and F.-S. Xiao, Coking-Resistant Polyethylene Upcycling Modulated by Zeolite Micropore Diffusion, J. Am. Chem. Soc., 2022, 144, 14269–14277 CrossRef CAS PubMed.
- A. R. Riscoe, C. J. Wrasman, A. A. Herzing, A. S. Hoffman, A. Menon, A. Boubnov, M. Vargas, S. R. Bare and M. Cargnello, Transition state and product diffusion control by polymer–nanocrystal hybrid catalysts, Nat. Catal., 2019, 2, 852–863 CrossRef CAS.
- A. Tennakoon, X. Wu, M. Meirow, D. Howell, J. Willmon, J. Yu, J. V. Lamb, M. Delferro, E. Luijten, W. Huang and A. D. Sadow, Two Mesoporous Domains Are Better Than One for Catalytic Deconstruction of Polyolefins, J. Am. Chem. Soc., 2023, 145, 17936–17944 CrossRef CAS PubMed.
- W. Zhao, S. Hasegawa, J. Fujita, F. Yoshii, T. Sasaki, K. Makuuchi, J. Sun and S.-i. Nishimoto, Effects of zeolites on the pyrolysis of polypropylene, Polym. Degrad. Stab., 1996, 53, 129–135 CrossRef CAS.
- T.-T. Wei, K.-J. Wu, S.-L. Lee and Y.-H. Lin, Chemical recycling of post-consumer polymer waste over fluidizing cracking catalysts for producing chemicals and hydrocarbon fuels, Resour., Conserv. Recycl., 2010, 54, 952–961 CrossRef.
- J. Gancedo, H. Li, J. S. Walz, L. Faba, S. Ordoñez and G. W. Huber, Investigation into the shape selectivity of zeolites for conversion of polyolefin plastic pyrolysis oil model compound, Appl. Catal., A, 2024, 669, 119484 CrossRef CAS.
- Y. Nakaji, M. Tamura, S. Miyaoka, S. Kumagai, M. Tanji, Y. Nakagawa, T. Yoshioka and K. Tomishige, Low-temperature catalytic upgrading of waste polyolefinic plastics into liquid fuels and waxes, Appl. Catal., B, 2021, 285, 119805 CrossRef CAS.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Plastic waste to fuels by hydrocracking at mild conditions, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS PubMed.
- R. Lin, B. Villacorta Hernandez, L. Ge and Z. Zhu, Metal organic framework based mixed matrix membranes: an overview on filler/polymer interfaces, J. Mater. Chem. A, 2018, 6, 293–312 RSC.
- Y. Wu, J. Tian, M. Sun, L. Gao, J. Xu and Z. Niu, Embedding an esterase mimic inside polyesters to realize rapid and complete degradation without compromising their utility, Green Chem., 2024, 26(5), 2851–2857 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |