
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation-induced ambiphilicity triggers N–N bond formation and dinitrogen release in octahedral terminal molybdenum(V) nitrido complexes†
C. Christopher
Almquist
 a,
Thayalan
Rajeshkumar
b,
H. D. A. Chathumal
Jayaweera
a,
Nicole
Removski
a,
Wen
Zhou
a,
Benjamin S.
Gelfand
a,
Laurent
Maron
*b and
Warren E.
Piers
*a
a,
Thayalan
Rajeshkumar
b,
H. D. A. Chathumal
Jayaweera
a,
Nicole
Removski
a,
Wen
Zhou
a,
Benjamin S.
Gelfand
a,
Laurent
Maron
*b and
Warren E.
Piers
*a
aDepartment of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: wpiers@ucalgary.ca
bLPCNO, Université de Toulouse, INSA, UPS, Toulouse, France
First published on 16th February 2024
Abstract
Coupling of octahedral, terminal d1 molybdenum(V) nitrido complexes supported by a dianionic pentadentate ligand via N–N bond formation to give μ-dinitrogen complexes was found to be thermodynamically feasible but faces significant kinetic barriers. However, upon oxidation, a kinetically favored nucleophilic/electrophilic N–N bond forming mechanism was enabled to give monocationic μ-dinitrogen dimers. Computational and experimental evidence for this “oxidation-induced ambiphilic nitrido coupling” mechanism is presented. The factors influencing release of dinitrogen from the resulting μ-dinitrogen dimers were also probed and it was found that further oxidation to a dicationic species is required to induce (very rapid) loss of dinitrogen. The mechanistic path discovered for N–N bond formation and dinitrogen release follows an ECECC sequence (E = “electrochemical step”; C = “chemical step”). Experimental evidence for the intermediacy of a highly electrophilic, cationic d0 molybdenum(VI) nitrido in the N–N bond forming mechanism via trapping with an isonitrile reagent is also discussed. Together these results are relevant to the development of molecular catalysts capable of mediating ammonia oxidation to dihydrogen and dinitrogen.
Introduction
Oxidation of ammonia (NH3) to dihydrogen (H2) and dinitrogen (N2) offers a vector for production of green hydrogen if the energy required to drive the reaction (and the manufacture of NH3) can be obtained from renewable energy sources.1 Catalysts are required for selective reactions and molecular transition metal complexes for the oxidation of NH3 to N2 and H2 (ref. 2) have seen renewed interest in the last few years.1,3–7 This was spurred in part upon recognition that many of the compounds that can effectively mediate water oxidation might be applied to ammonia oxidation8—without the itinerant hazards of generating O2/H2 mixtures. Although not as available as water, ammonia is an abundant feedstock manufactured on a large scale using the Haber–Bosch process via the catalytic reduction of N2 with H2.9 Furthermore, ammonia is easier to oxidize than water and is similarly “carbon free” in terms of the end products of oxidation.Essential steps in the conversion of NH3 to H2 and N2 are the cleavage of three strong N–H bonds, and the formation of an N–N bond.8 Considered superficially, this seems relatively simple, but there are a variety of mechanisms available for each step when using molecular catalysts. For N–H bond cleavage, coordination-induced bond weakening10 of the N–H bond(s) of NH3 upon ligation to a metal can be substantial and the factors that influence the extent to which bond weakening occurs are a subject of current study.5,11–15 When weakened below a threshold of about 52 kcal mol−1, elimination of H2 (BDFE = 104 kcal mol−1) becomes thermodynamically feasible and kinetic pathways involving metal hydrides11 or bimolecular H2 elimination in coordinatively saturated systems15 can lead to “spontaneous” loss of H2 from any of the M–NHn (n = 3, 2, or 1) intermediates formed via loss of hydrogen atoms. Alternatively, use of hydrogen atom abstracting reagents like the 2,4,6-tri-tert-butylphenoxy radical (ArO˙) can be employed if bond weakening of N–H to values below the strength of the ArO–H bond (74.8 kcal mol−1 (ref. 16 and 17)) are achieved.
The other key step, N–N bond formation, can occur from any of these intermediates through nucleophilic attack by external NH3 (ref. 3, 6, 7, 18 and 19) on (usually cationic) M–NHn (n = 2, 1, 0) intermediates or via bimolecular coupling of metal amido20,21 (to bridging hydrazine), imido22 (to μ-diazene) or nitrido (to μ-dinitrogen) complexes.8,14 Nitrogen–nitrogen bond formation via coupling of two terminal nitrido complexes is generally thought to take place via two limiting paths that depend on the electron distribution in the metal nitrido bond, as shown in Scheme 1.8 When the nitrido has significant spin density associated with the terminal nitrogen, such that it behaves as a nitridyl radical, bimolecular homocoupling between these two radicals is possible.4,23–25 Alternatively, in ambiphilic nitrido complexes with energetically matched HOMO nitrido lone pairs and LUMO MN π* orbitals, a nucleophilic/electrophilic N–N coupling mechanism can occur.26 More commonly, this latter mechanism is observed in nitrido pairs where the metal ligand combinations are different, rendering one LnMN nucleophilic and the other electrophilic.27 Between these two limiting extremes there lies a continuum and there is some indication that N–N bond formation between nitridos that utilize both radical character and ambiphilicity make for facile N–N bond formation.28 It is clear that understanding the factors which control the barrier of N–N bond formation is crucial for developing more effective catalysts, since upon coupling the driving force for N2 formation and release can be substantial.
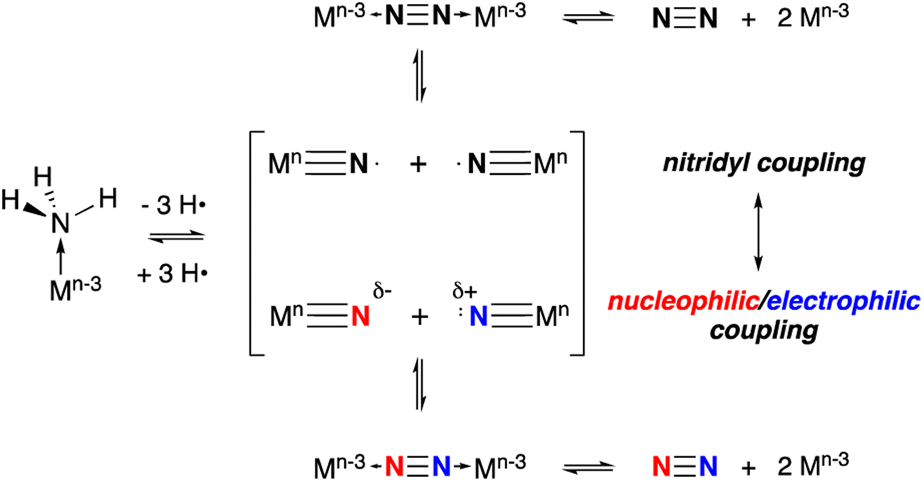 | ||
| Scheme 1 Limiting mechanisms of N–N bond formation via coupling of two terminal metal nitrido complexes. | ||
We recently reported15 the synthesis of a neutral Mo(V) nitrido complex supported by a pentadentate diborate ligand (B2Pz4Py) related to the neutral pentapyridyl “PY5” framework first prepared by the Feringa29 and Stack30,31 groups in the mid 1990's and popularized by Chang and Long as a platform for electrocatalytic applications.32–35 By virtue of its dianionic L3X2 nature, the B2Pz4Py ligand36 lowers the overall charge of its complexes and can stabilize higher oxidation state complexes in metals from across the d block.37–43 The complex (B2Pz4Py)Mo(V)N, 1-Tol, was prepared as the end product of removal of three hydrogen atoms from the Mo(II) neutra ammine complex in the context of probing the coordination-induced bond weakening in these complexes.15 Here we probe the reactivity and properties of the Mo(V) nitrido complexes of two variants of this dianionic pentadentate system in the context of N–N bond formation and provide evidence for a distinct oxidation-induced nucleophilic/electrophilic coupling pathway for N–N bond formation.
Results and discussion
In our previous report,15 we showed that the neutral, d1 molybdenum(V) terminal nitrido complex 1-Tol could be prepared via deprotonation of the cationic imido complex [(B2Pz4Py)Mo(V)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH]+[NTf2]−. However, a one-pot route involving sequential deprotonation of the cationic Mo(III) ammine complex [(B2Pz4Py)Mo(III)–NH3]+[NTf2]− followed by two hydrogen atom transfers using the ArO˙ reagent mentioned above proved a more convenient protocol, particularly for preparing the 15N-labeled nitrido isotopologues (Scheme 2). The nitrido 1-Tol was fully characterized, including via X-ray crystallography.15 The Mo–Nnitrido bond length of 1.729(3) Å was noted to be longer than that observed for the analogous bond length of 1.695(3) Å in the cationic imido precursor and furthermore, the DFT (B3PW91) computed Mo–Nnitrido distance in 1-Tol is 1.660 Å, more in line with expectations. The bonding analysis carried out at the Natural Bonding Orbital (NBO) level indicates as expected the presence of a Mo–N triple bond where the three bonds are highly covalent (see ESI†). In initial studies of the reactivity of 1-Tol, we noted its very poor solubility in most common solvents and found it to be sparingly soluble only in dichloromethane, chloroform or N,N-dimethylformamide (DMF). This hampered reactivity studies, particularly for experiments requiring low temperatures (see below). Examination of the crystal packing diagram of 1-Tol showed an extended ribbon of molecules supported by close contacts (2.576 Å) between the nitrido nitrogen and the meta-aryl protons of the tolyl groups incorporated on the ligand borate moieties (Fig. 1a). We hypothesized that these interactions are likely responsible for the poor solubility of nitrido derivative 1-Tol.
NH]+[NTf2]−. However, a one-pot route involving sequential deprotonation of the cationic Mo(III) ammine complex [(B2Pz4Py)Mo(III)–NH3]+[NTf2]− followed by two hydrogen atom transfers using the ArO˙ reagent mentioned above proved a more convenient protocol, particularly for preparing the 15N-labeled nitrido isotopologues (Scheme 2). The nitrido 1-Tol was fully characterized, including via X-ray crystallography.15 The Mo–Nnitrido bond length of 1.729(3) Å was noted to be longer than that observed for the analogous bond length of 1.695(3) Å in the cationic imido precursor and furthermore, the DFT (B3PW91) computed Mo–Nnitrido distance in 1-Tol is 1.660 Å, more in line with expectations. The bonding analysis carried out at the Natural Bonding Orbital (NBO) level indicates as expected the presence of a Mo–N triple bond where the three bonds are highly covalent (see ESI†). In initial studies of the reactivity of 1-Tol, we noted its very poor solubility in most common solvents and found it to be sparingly soluble only in dichloromethane, chloroform or N,N-dimethylformamide (DMF). This hampered reactivity studies, particularly for experiments requiring low temperatures (see below). Examination of the crystal packing diagram of 1-Tol showed an extended ribbon of molecules supported by close contacts (2.576 Å) between the nitrido nitrogen and the meta-aryl protons of the tolyl groups incorporated on the ligand borate moieties (Fig. 1a). We hypothesized that these interactions are likely responsible for the poor solubility of nitrido derivative 1-Tol.
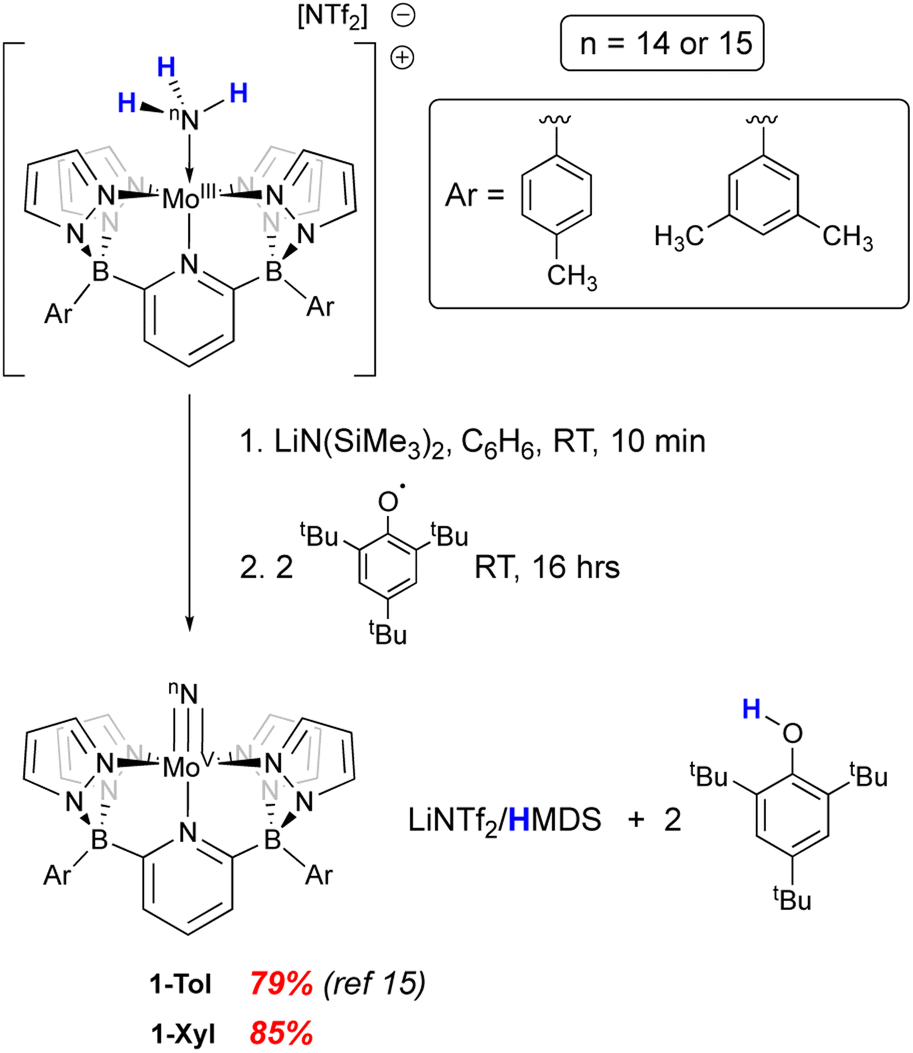 | ||
| Scheme 2 Synthesis of neutral Mo(V) nitrido derivatives 1-Ar (Ar = para-tolyl, meta-xylyl). | ||
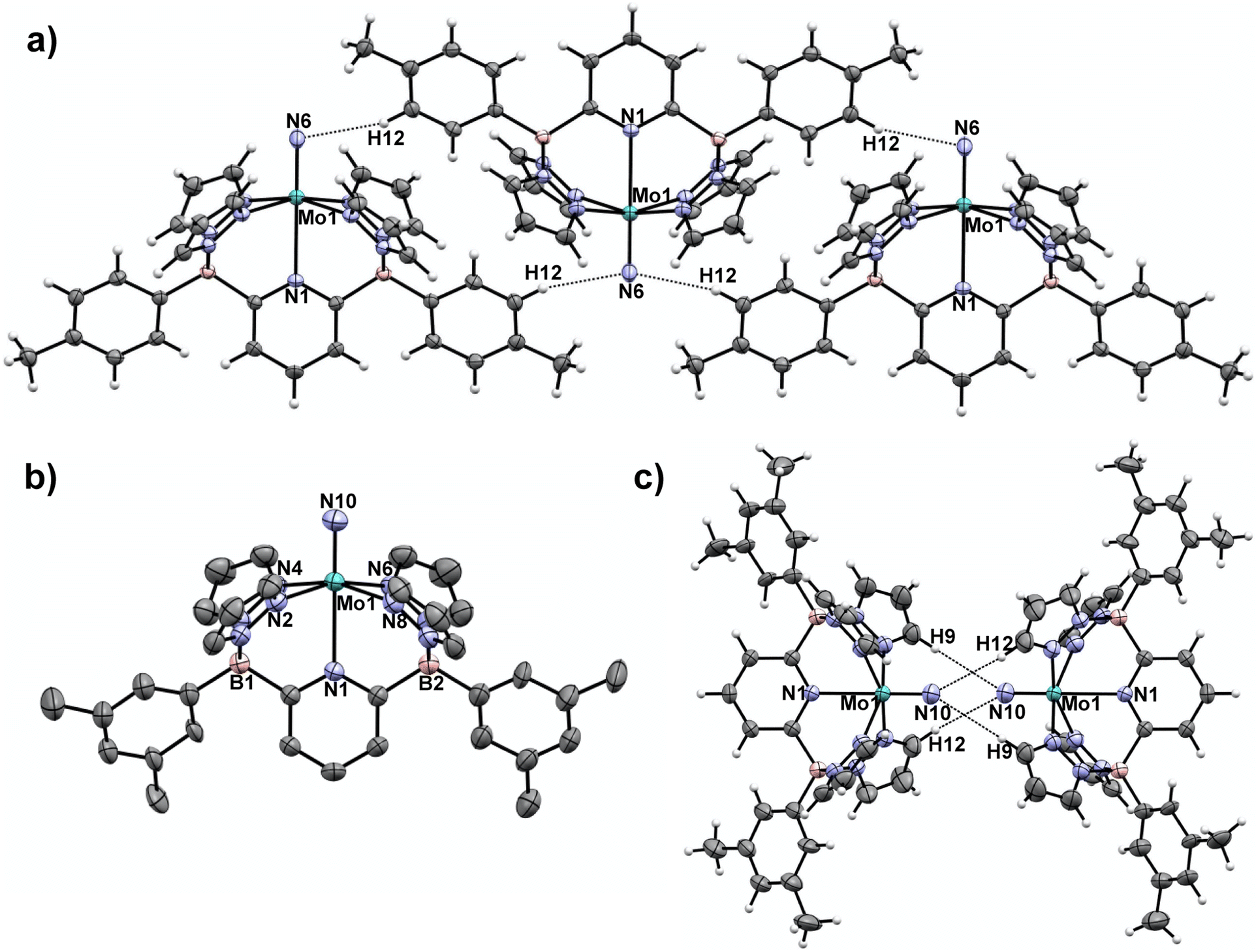 | ||
| Fig. 1 a) Crystal packing motif for nitrido 1-Tol15 with closest intermolecular contacts. Selected intermolecular contact distance (Å): N(6)–H(12), 2.576. (b) Molecular structure of 1-Xyl Selected bond distances (Å): Mo(1)–N(1), 2.4564(14); Mo(1)–N(10), 1.6740(18). (c) Crystal packing motif for nitrido 1-Xyl with closest intermolecular contacts. Selected intermolecular contact distance (Å): N(10)–H(9), 2.930; N(10)–H(12), 3.111. Thermal ellipsoids are shown at the 50% probability level and all H atoms except those involved in intermolecular contacts are omitted for clarity. | ||
The synthetic scheme for preparing the dianionic pentadentate B2Pz4Py ligand system36 is such that modification of the aryl groups on the borates is relatively easy. We postulated that switching from tolyl groups to 3,5-dimethylphenyl substituents would disrupt the intermolecular packing interactions of Fig. 1a and result in a more soluble Mo(V) nitrido derivative. Accordingly, the xylyl-substituted ligand was prepared and used to synthesize the cationic Mo(III) ammine precursor to nitrido 1-Xyl using previously established protocols15 (see Scheme S2 and the ESI† for full synthetic and characterization details of complexes incorporating the xylyl-substituted ligand). Compound 1-Xyl was then prepared in 85% yield as depicted in Scheme 2 and was found to be significantly more soluble in a variety of solvents in comparison to 1-Tol. Like 1-Tol, nitrido 1-Xyl is a d1 (μeff = 1.71, by the Evans method44) species that is largely NMR silent, although the xylyl methyl groups are assignable in the 1H NMR spectra. X-ray quality crystals were obtained by slow evaporation of solvent from a saturated toluene solution, and the molecular and packing structures are shown in Fig. 1b and c, respectively. In contrast to 1-Tol, the shortest intermolecular interactions in 1-Xyl involve the nitrido nitrogen and C–H bonds on an adjacent molecule's pyrazolyl ring and are substantially longer (2.930 and 3.111 Å) than the interactions in 1-Tol. A shorter Mo–Nnitrido distance of 1.674(2) is observed in 1-Xyl, which is more consistent with the computed value for this bond in the gas phase. Both compounds exhibit Mo–Nnitrido stretching frequencies of 1001 cm−1 in the solid state IR spectrum (which shifts to 982 cm−1 in the Mo15Nnitrido isotopologues, see ESI†), suggesting essentially identical bond orders and strengths in these two derivatives.
The d1 Mo(V) nitridos 1-Ar exhibit EPR spectra that indicate the unpaired spin is largely metal-based (see Fig. 2 and S1†), with essentially identical g values of 1.966 and a six-line hyperfine coupling pattern (A = 53 G) to the ≈25% abundant spin 5/2 95/97Mo nuclei. These parameters are typical for d1 Mo(V) compounds.45–48 DFT analysis also indicates there is very little spin density associated with the nitrido nitrogen (16%) with most of it residing in a metal-based d-orbital. The SOMO-1 orbital is Mo–Nnitrido σ-bonding in character and is essentially the nucleophilic nitrido lone pair, accounting for the moderate nucleophilic character of these neutral Mo(V) nitrido complexes.
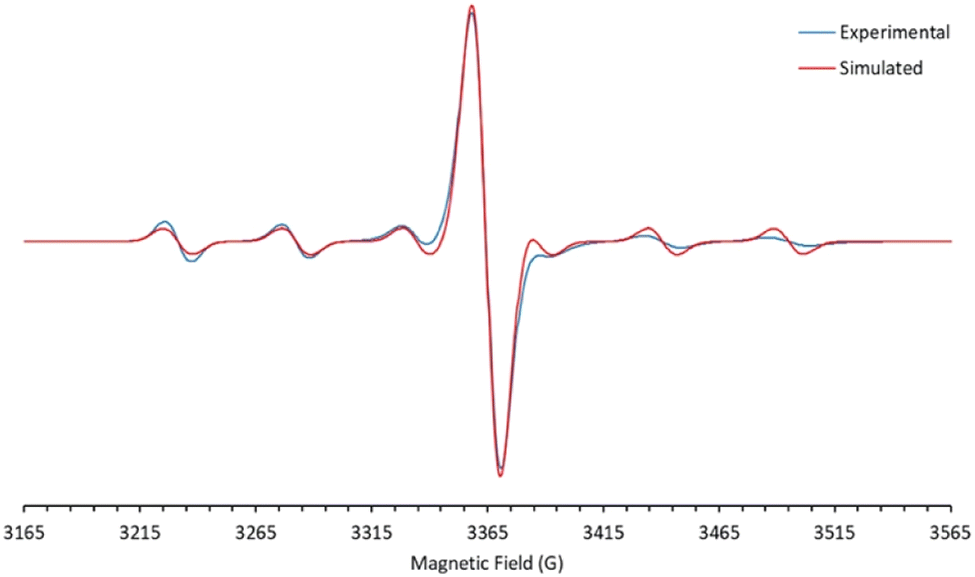 | ||
| Fig. 2 X-band EPR spectrum of 1-Xyl in dry toluene (970 μM), taken at 293 K. The experiment was run with a centre field of 3360.0 G and a sweep width of 500.0 G. The time constant and conversion time were set to 2.56 ms and 8.00 ms, respectively. 16 scans were collected. Frequency: 9.258916 GHz. The simulated spectrum was generated in EasySpin, indicating a Mo-based radical with a central line (IMo = 0) g = 1.966 with a six-line spectrum (IMo = 5/2) AMo = 52.82 G. | ||
Given these characteristics, it is perhaps unsurprising that these terminal nitridos exhibit substantial thermal stability towards N–N bond formation via bimolecular coupling. For 1-Tol, solutions heated at 50 °C in CDCl3 gradually (over the course of 12 weeks) undergo conversion to the previously reported chlorido complex15 [(B2Pz4Py)Mo(III)–Cl]. No intermediates were observed and although N2 is presumed to be eliminated, it was not directly detected in this experiment, which was conducted on a rather small scale. Solutions of 1-Xyl in toluene at 100 °C undergo similarly slow conversion to a new product identified separately (see below) as the neutral μ-dinitrogen dimer 3-Xyl along with traces of the μ-nitrido complex 2-Xyl, which is the xylyl analog of the previously reported dinuclear μ-nitrido complex 2-Tol;15 the spectral characteristics of 2-Xyl were confirmed by its separate synthesis and characterization (see ESI† for details). As shown in Scheme 3, we hypothesize that slow, rate-determining bimolecular coupling occurs to give the bridging dinitrogen dimers 3-Ar, which we assign as having Mo(III)–NN–Mo(III) rather than Mo(II)–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–Mo(II) character based on the observed 11B NMR chemical shift of −153.5 ppm‡ for 3-Xyl and computational interrogation of this species (see below). In addition to this evidence for 3-Xyl, the 15N2 isotopologue was confirmed to be produced by mass spectrometry (Fig. S2†). In the case of coupling from 1-Tol in CDCl3 at 50 °C, 3-Tol is not observed but is oxidized to the [(TolB2Pz4Py)Mo(III)–Cl] product by the solvent. We presume that in the coupling of 1-Xyl, the harsh conditions of the experiment can lead to reaction of 3-Xyl with 1-Xyl to give the traces of μ-nitrido complex we observe spectroscopically, but this is a very slow process even relative to the slow N–N bond formation observed for coupling of 1-Xyl. The bottom line is that, in both complexes, conversion of compounds 1-Ar to dimers 3-Arvia nitrido–nitrido coupling is extremely slow under the conditions employed.
N–Mo(II) character based on the observed 11B NMR chemical shift of −153.5 ppm‡ for 3-Xyl and computational interrogation of this species (see below). In addition to this evidence for 3-Xyl, the 15N2 isotopologue was confirmed to be produced by mass spectrometry (Fig. S2†). In the case of coupling from 1-Tol in CDCl3 at 50 °C, 3-Tol is not observed but is oxidized to the [(TolB2Pz4Py)Mo(III)–Cl] product by the solvent. We presume that in the coupling of 1-Xyl, the harsh conditions of the experiment can lead to reaction of 3-Xyl with 1-Xyl to give the traces of μ-nitrido complex we observe spectroscopically, but this is a very slow process even relative to the slow N–N bond formation observed for coupling of 1-Xyl. The bottom line is that, in both complexes, conversion of compounds 1-Ar to dimers 3-Arvia nitrido–nitrido coupling is extremely slow under the conditions employed.
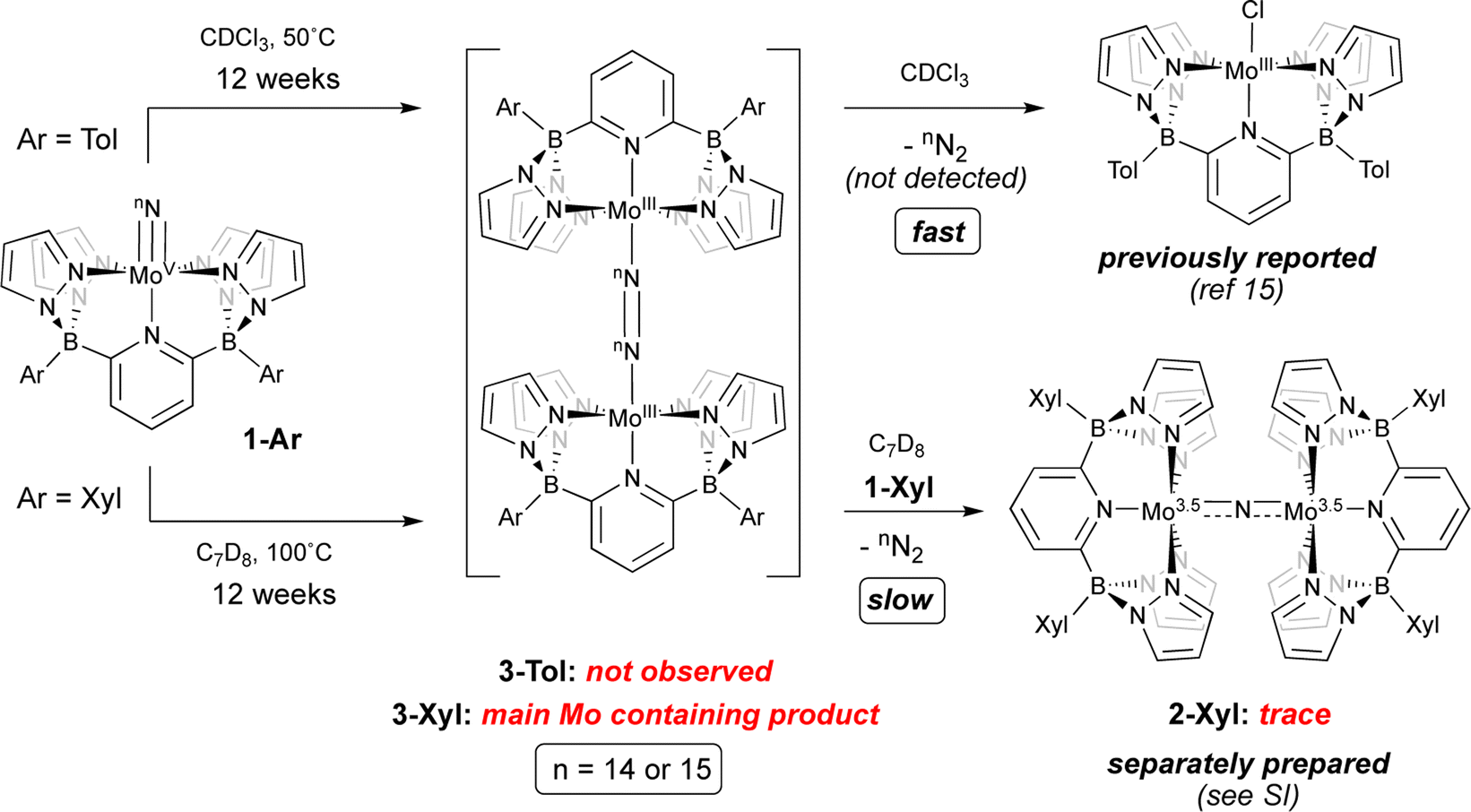 | ||
| Scheme 3 Thermally induced coupling of terminal neutral Mo(V) nitrido complexes 1-Ar. | ||
Since N–N bond formation from these d1 Mo(V) nitridos is so inefficient, we probed their redox properties by cyclic voltammetry. Both complexes 1-Ar are oxidized irreversibly at moderate potentials (Eox = 0.62 V vs. Fc/Fc+ for 1-Xyl and 0.57 V for 1-Tol) and the observed irreversibility is maintained at all scan rates employed (25–1600 mV s−1, see Fig. S3†). This is suggestive of a facile chemical reaction triggered by oxidation and to probe the nature of this reactivity, chemical oxidation using a family of brominated triarylammoniumyl radical cations [N(Ar)3]˙+, partnered with Krossing's perfluorinated tetra-tert-butoxy aluminate weakly coordinating anion49 was undertaken. Here, we have utilized “Magic Blue” (MB˙+, Ar = 4-BrC6H4; E° = + 0.70 V vs. Fc/Fc+ in DCM50), and Magic “Blue's Cousin”51 (BC˙+, Ar = 2-Br-4-tert-butyl-C6H3, E° = + 0.78 V vs. Fc/Fc+ in DCM), the latter being a more soluble derivative for use at lower temperatures. The Krossing anion was used because other WCAs52 were either chemically non-innocent (e.g. [SbX6]−, X = F or Cl) or not well-behaved from a solubility or physical perspective (e.g., [NTf2]− or [B(C6F5)4]−). Krossing's aluminate anion proved both chemically inert and useful in promoting crystallizable products.
Initial experiments involving oxidation of 1-Tol with one equivalent of MB˙+ at room temperature produced a mixture of products but the reactions proceeded much more cleanly when only 0.5 equivalents of oxidant were employed. As shown in Fig. 3, treatment of either nitrido 1-Ar with 0.5 equivalents of MB˙+ result in a rapid elimination of N2 and production of the cationic μ-nitrido dinuclear compounds [2-Ar]+ as the major products (≈95% NMR yields). Although not quantified, the eliminated N2 was confirmed as originating from the nitrido nitrogens by detecting 15N2via NMR spectroscopy (≈−69 ppm (ref. 14 and 53)) and GCMS (Fig. S4†). The cationic dinuclear products were fully characterized by preparing them separately via oxidation of the neutral 2-Ar μ-nitridos, NMR spectroscopy, and X-ray crystallography (Fig. 3). The Mo(1)–N(10)–Mo(2) angle is essentially linear (176.52(19)°) and Evans method measurement of the μeff indicates an S = 2 ground state and therefore a formally Mo(IV)NMo(IV) configuration where electronic coupling across the nitrido bridge is minimal. In comparison, the Mo–N(10) bonds in [2-Ar]+ relative to the analogous bonds in the neutral precursors undergo minimal change. Oxidatively-induced N2 elimination54–56 from terminal nitrido complexes has been observed in the well-studied d2 Mn(V)N complexes supported by tetradentate Schiff base type ligands.57 In these systems, oxidation to a d1 cationic [Mn(VI)N]+ imbues significant nitridyl character to these species and Storr et al. in particular convincingly demonstrated that bimolecular coupling of these nitridyls (Scheme 1) was the likely mechanism of N–N bond formation for that family of compounds.54 In the present molybdenum system, oxidation of d1 nitridos 1-Ar presumably leads to a d0 [Mo(VI)N]+; bimolecular coupling of this species is unlikely on both electrostatic and orbital grounds. Indeed, DFT analysis of the barriers to N–N bond formation from homocoupling of either 1-Tol or [1-Tol]+, show that these reactions, while exergonic, have prohibitively high barriers of 33 and 54 kcal mol−1 at room temperature, respectively (Fig. 4). The former value is certainly consistent with the experimentally observed slow loss of nN2 from compounds 1-Ar, Scheme 3, but the significantly higher barrier to N–N bond formation from [1-Ar]+ initially seemed at odds with the very rapid loss of N2 upon generation of these cations. However, as revealed in Fig. 4, the barrier to heterocoupling of neutral (nucleophilic) nitride 1-Tol with cationic (electrophilic) nitride [1-Tol]+ is much lower at 16 kcal mol−1, strongly suggesting a nucleophilic/electrophilic mechanism for this oxidation induced N–N bond forming reaction as shown in Scheme 4. As shown in the upper right of Fig. 4, the TS adopts a typical “zig-zag” geometry,27,58 and the long N–N separation of 2.277 Å is consistent with an early transition state as befits a low barrier/exothermic reaction profile. Oxidation of 1-Ar removes a mainly d-orbital-based electron and increases the nitrido nitrogen electrophilicity,59–61 such that nucleophilic attack by remaining 1-Ar is now kinetically facile. In support of this, Natural Population Analysis62 (NPA, Tables S19 and S20†) of the natural charges on the Mo and Nnitrido atoms of 1-Tol (Mo = 1.09, N = −0.37) and [1-Tol]+ (1.24, −0.24) show significantly more positive values for the oxidized species indicating a much more electrophilic species. We believe this “induced ambiphilicity” is a distinct mechanistic pathway for N–N bond formation from a single metal nitrido species and contrasts with a path where the nitrido is fully oxidized and undergoes coupling of two nitridyls as in the MnSalen system mentioned above.54
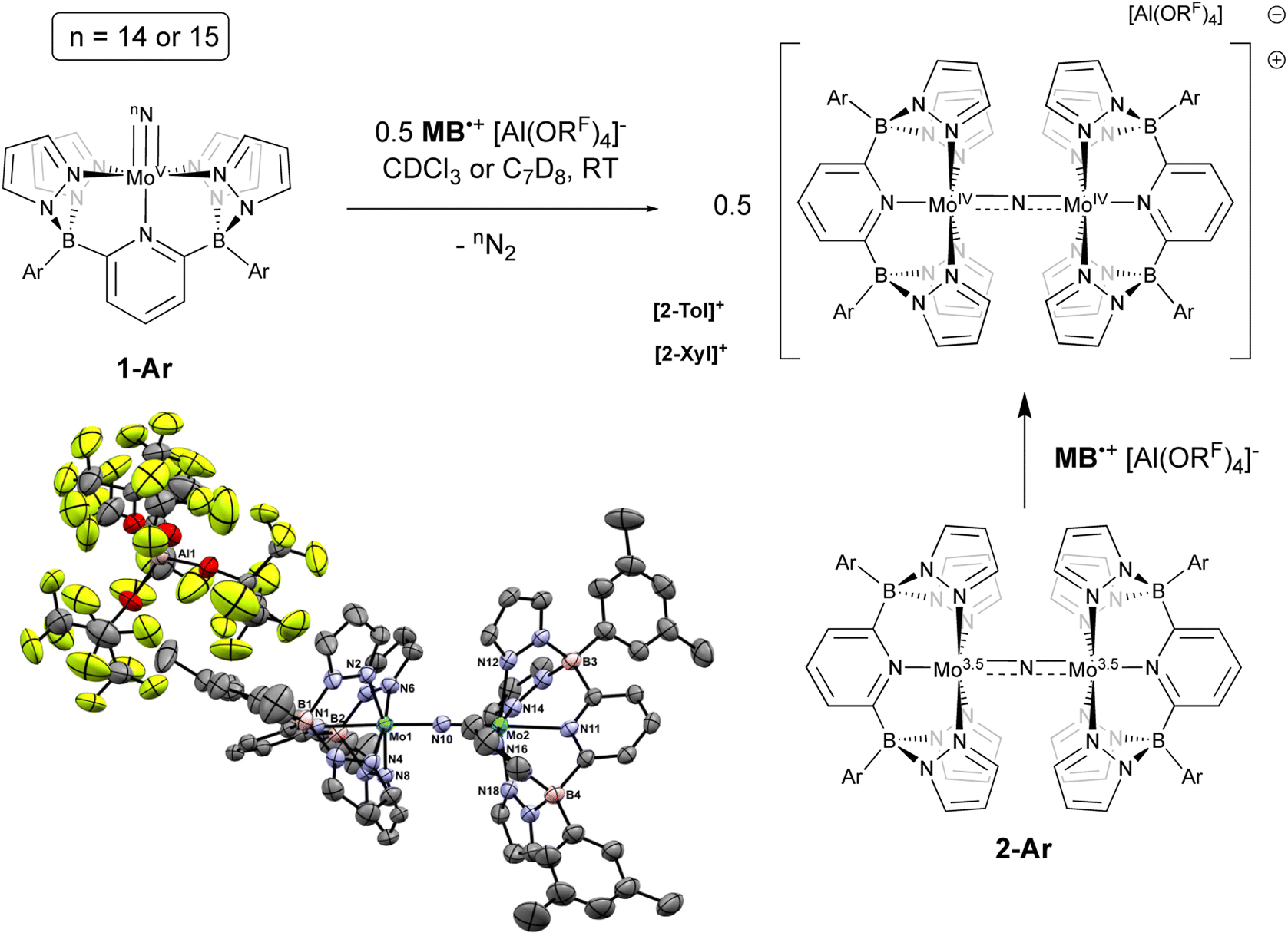 | ||
| Fig. 3 Oxidation of neutral nitridos 1-Ar with 0.5 equivalents of [MB˙]+[Al(ORF)4]. Lower left: molecular structure of [2-Xyl]+[Al(ORF)4]. Thermal ellipsoids are shown at the 50% probability level and all H atoms are omitted for clarity. Selected bond distances (Å): Mo(1)–N(10), 1.856(3); Mo(2)–N(10), 1.859(3); Mo(1)–N(1), 2.260(3); Mo(2)–N(11), 2.265(3). Selected bond angles (°): Mo(1)–N(10)–Mo(2), 176.52(19). | ||
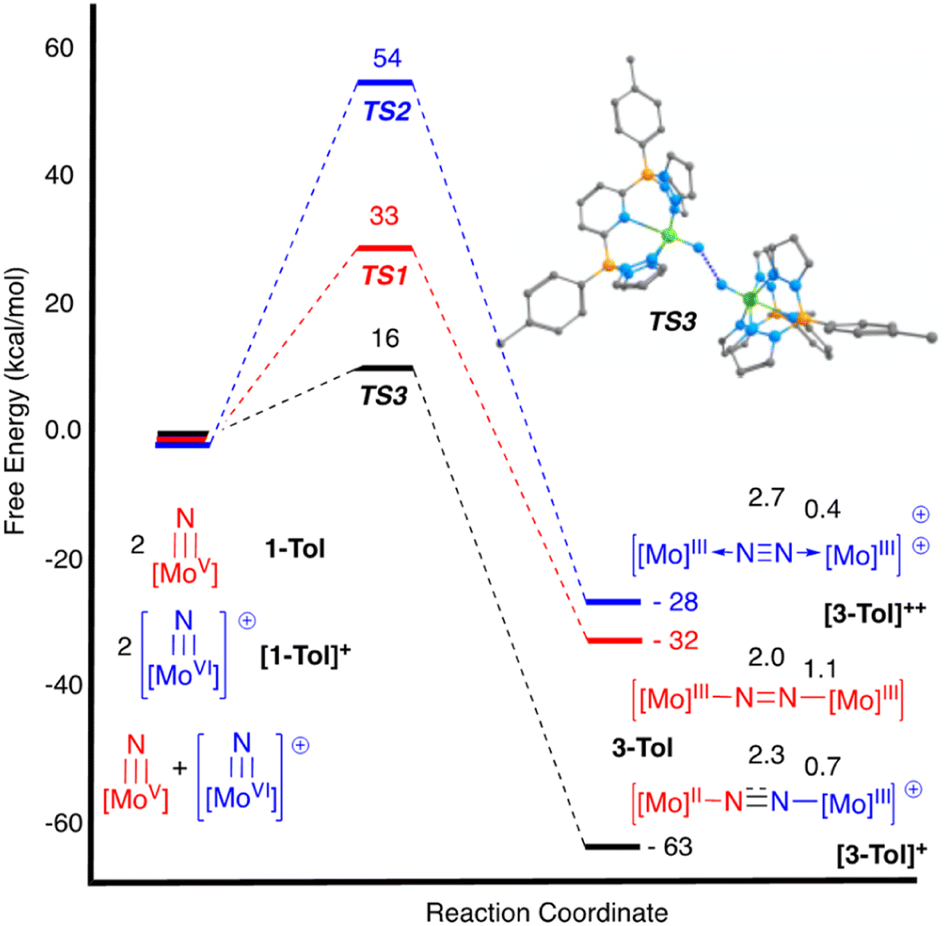 | ||
| Fig. 4 DFT (B3PW91) computed enthalpy profiles for N–N bond formation via bimolecular coupling of 1-Tol (all red), [1-Tol]+ (all blue) and 1-Tol/[1-Tol]+ (mixed red/blue) at room temperature. The energies are given in kcal mol−1 and computed Wiberg Bond Index values for the N–N and Mo–N bonds in the μ-dinitrogen products of coupling are also shown. Upper right: ball and stick depiction of the transition state for ambiphilic coupling of 1-Tol and [1-Tol]+. | ||
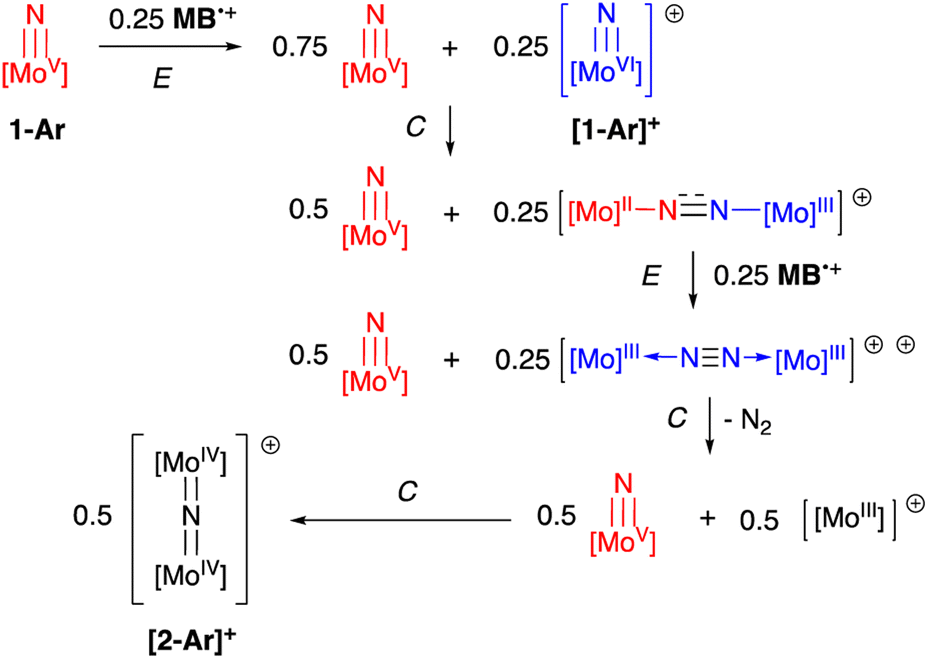 | ||
| Scheme 4 Oxidation-induced nucleophilic/electrophilic N–N bond formation in Mo(V) nitrides: ECECC mechanism. | ||
The fact that only 0.5 equivalents of oxidant is needed for this reaction is not in itself conclusive evidence for the mechanistic proposal in Scheme 4, since the same products and product distribution would result from homocoupling of the cationic nitridos [1-Ar]+ (see Scheme S1†). The DFT results of Fig. 4 also do not directly address the question of which μ-dinitrogen complex most efficiently releases N2, since the energies depicted do not reflect the relative thermodynamic stabilities of the μ-dinitrogen products, nor the barriers to N2 release. Having said that, the WBI values (Table S6†) for the N–N and Mo–N bonds calculated certainly suggest release of N2 from dication [3-Ar]++ should be most favored as this species has a nearly fully formed nitrogen–nitrogen triple bond.63,64 Thus the DFT results are highly suggestive that the ambiphilic coupling is kinetically favored, but that N2 release from the resulting monocationic μ-dinitrogen complex [3-Ar]+ is not necessarily thermodynamically feasible. Therefore, we sought experimental evidence for the oxidation-induced nucleophilic/electrophile N–N coupling mechanism of Scheme 4 by conducting the oxidations of 1-Ar at low temperature and monitoring spectroscopically. At this juncture, the purpose for preparing the more soluble 1-Xyl derivative becomes apparent; 1-Tol could not be employed in these experiments due to extremely low solubility in all solvents below 0 °C. However, 1-Xyl readily dissolves in toluene at −78 °C, and use of the more soluble BC˙+ oxidant allowed us to monitor this process effectively by 1H NMR spectroscopy.
Mixing 1-Xyl with 0.5 equivalents of [BC˙]+[Al(ORF)4]− in toluene at −78 °C leads to a homogeneous solution and no apparent reaction between the reagents (Fig. S5†). The sample was warmed in ≈5 °C increments and as it approached −60 °C, evidence of electron transfer was observed as signals for the triarylamine produced upon reduction of BC˙+ emerged in the diamagnetic region of the spectrum. As further warming took place, at about −40 °C signals for two paramagnetic molybdenum products began to grow in until at room temperature these were present in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The set of broad signals for one of these products matched those found for [2-Xyl]+, while the second, much sharper, set of signals belong to a new, previously unobserved product.
:1 ratio. The set of broad signals for one of these products matched those found for [2-Xyl]+, while the second, much sharper, set of signals belong to a new, previously unobserved product.
Crystals deposited from these experiments proved to be of X-ray quality and their analysis showed them to be mixtures of μ-nitrido cation [2-Xyl]+ and the cationic μ-dinitrogen dimer [3-Xyl]+ (Scheme 5 and Fig. 5), which is the new product that arises from the ambiphilic coupling of 1-Xyl and [1-Xyl]+. The structure of this cationic dinitrogen dimer is depicted in Fig. 5 and shows a moderately activated N–N bond elongated to 1.162(7) Å by the lower valent molybdenum centers. That it is isolable and relatively stable towards loss of N2 is supported by the computed kinetic and thermodynamic properties of N2 release from [3-Xyl]+, which is endothermic by 20 kcal mol−1 and has a barrier of 30 kcal mol−1 (Scheme S4†). In stark contrast, loss of N2 from the dicationic [3-Xyl]2+ is computed to be kinetically facile (barrier of 6 kcal mol−1) and exothermic (by −26 kcal mol−1) as depicted in Scheme S5.† Experimentally consistent with this DFT analysis, simply adding further amounts of 1-Xyl to the 1:1 product mixture does not immediately lead to N2 release and conversion to [2-Xyl]+ (Scheme 5, top arrow, path a). Prolonged heating of this mixture does lead to some conversion (Fig. S38–S46†). However, if more oxidant is added along with the extra 1-Xyl, rapid, clean conversion to [2-Xyl]+ is observed with release of N2 (Scheme 5, bottom arrow, path b, Fig. S48–S54†) presumably from [3-Xyl]2+. Note in this experiment, instead of employing MB˙+, we utilize the acetyl-ferrocenium oxidizing agent (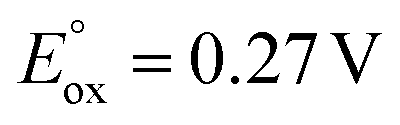 (ref. 50) vs. Fc/Fc+) partnered with Krossing's anion (see ESI† for the full characterization of this reagent), to match with the
(ref. 50) vs. Fc/Fc+) partnered with Krossing's anion (see ESI† for the full characterization of this reagent), to match with the 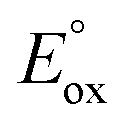 of 0.22 V estimated for the [3-Xyl]+/[3-Xyl]++ couple by cyclic voltammetry (Fig. S37†), while not being strong enough to oxidize either 1-Xyl (0.62 V) or [2-Xyl]+ (0.98 V). Together, these experiments provide strong evidence that N2 release comes via[3-Xyl]++ and confirms the “ECECC” sequence shown in the proposed mechanism of Scheme 4. In more general terms, the observation and isolation of the monocationic μ-dinitrogen intermediate [3-Xyl]+ in the low temperature oxidation of 1-Xyl is convincing experimental evidence that oxidatively-induced N–N bond formation occurs selectively via heterocoupling between the nucleophilic 1-Xyl and electrophilic [1-Xyl]+. As a final note regarding Scheme 4, when the 1:1 mixture of [3-Xyl]+ and [2-Xyl]+ is reduced by
of 0.22 V estimated for the [3-Xyl]+/[3-Xyl]++ couple by cyclic voltammetry (Fig. S37†), while not being strong enough to oxidize either 1-Xyl (0.62 V) or [2-Xyl]+ (0.98 V). Together, these experiments provide strong evidence that N2 release comes via[3-Xyl]++ and confirms the “ECECC” sequence shown in the proposed mechanism of Scheme 4. In more general terms, the observation and isolation of the monocationic μ-dinitrogen intermediate [3-Xyl]+ in the low temperature oxidation of 1-Xyl is convincing experimental evidence that oxidatively-induced N–N bond formation occurs selectively via heterocoupling between the nucleophilic 1-Xyl and electrophilic [1-Xyl]+. As a final note regarding Scheme 4, when the 1:1 mixture of [3-Xyl]+ and [2-Xyl]+ is reduced by 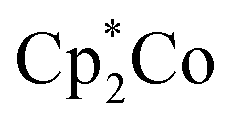 , the products are a 1:1 mixture of the neutral dinuclear species 3-Xyl and 2-Xyl (Fig. S57–S60†). Indeed, it was this experiment that helped to identify the former as the major product in the thermally-induced coupling of 1-Xyl depicted in Scheme 3, since the signals in the 1H NMR spectra for these two experiments indicate the presence of both neutral μ-dinitrogen and μ-nitrido dinuclear compounds. Attempts to separately synthesize pure samples of 3-Xyl have so far failed, but these studies are ongoing.
, the products are a 1:1 mixture of the neutral dinuclear species 3-Xyl and 2-Xyl (Fig. S57–S60†). Indeed, it was this experiment that helped to identify the former as the major product in the thermally-induced coupling of 1-Xyl depicted in Scheme 3, since the signals in the 1H NMR spectra for these two experiments indicate the presence of both neutral μ-dinitrogen and μ-nitrido dinuclear compounds. Attempts to separately synthesize pure samples of 3-Xyl have so far failed, but these studies are ongoing.
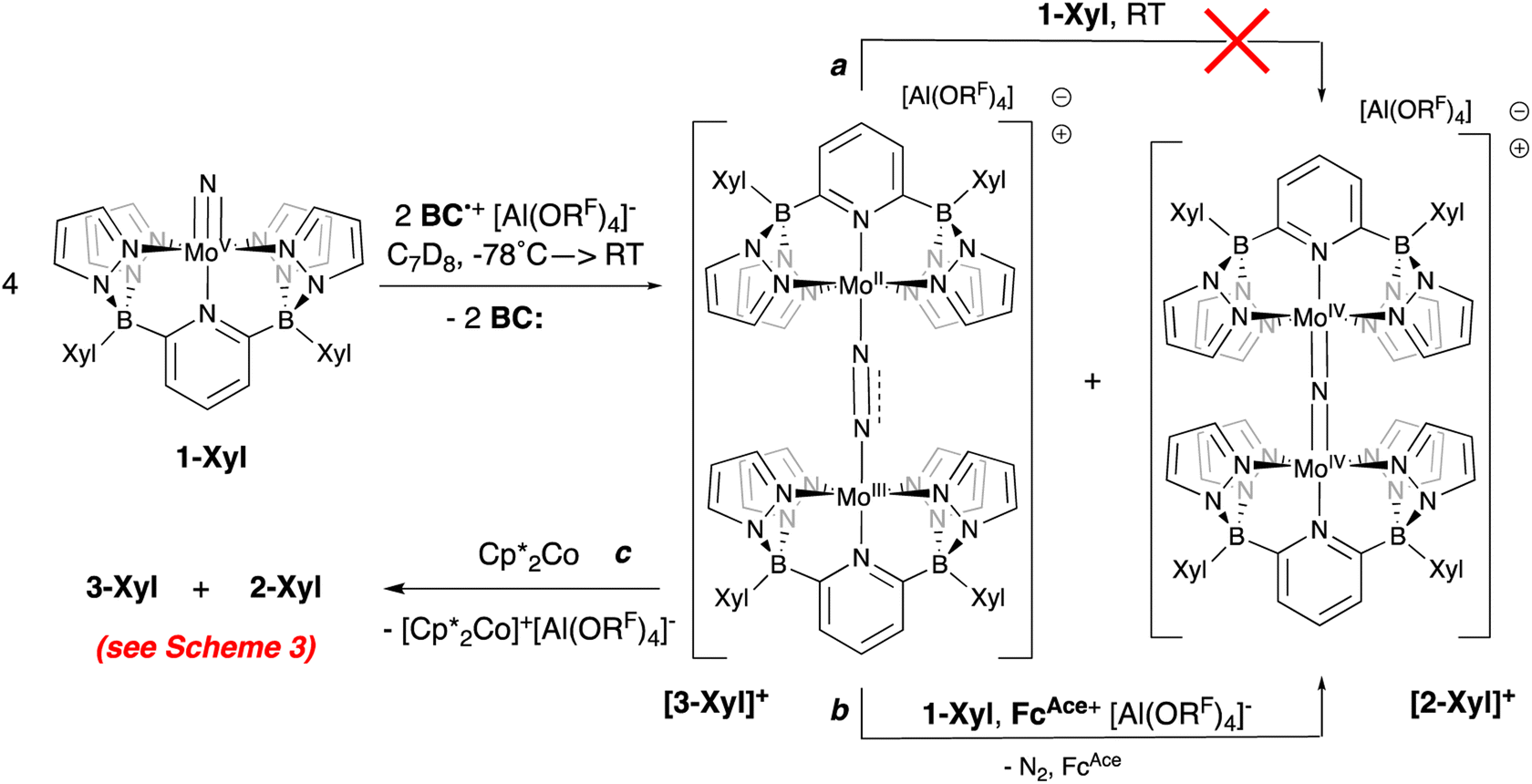 | ||
| Scheme 5 Reaction of 1-Xyl with 0.5 equivalents of oxidant at −78 °C and subsequent transformations of the 1:1 mixture of [3-Xyl]+:[2-Xyl]+. | ||
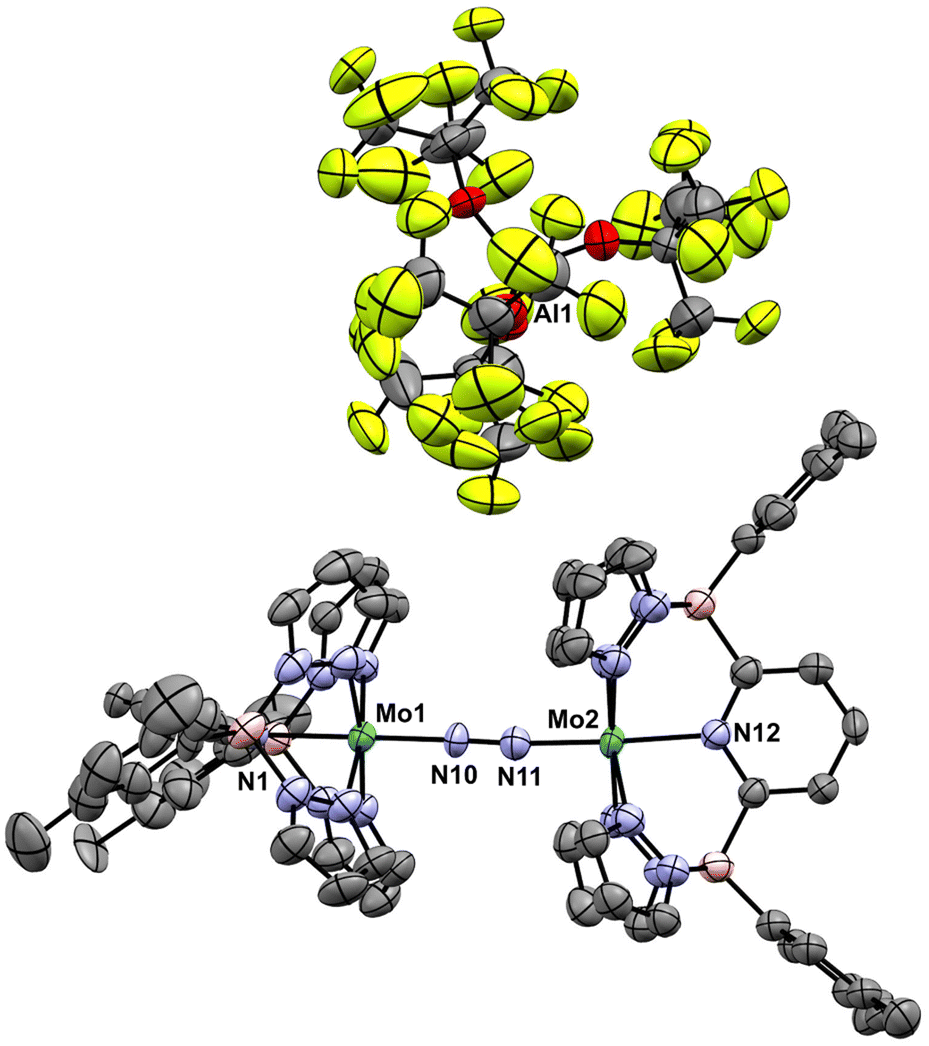 | ||
| Fig. 5 Molecular structure of [3-Xyl]+. Thermal ellipsoids are shown at the 50% probability level and all H atoms are omitted for clarity. Selected bond distances (Å): Mo(1)–N(1), 2.167(5); Mo(1)–N(10), 1.885(5); Mo(2)–N(12), 2.080(5); Mo(2)–N(11), 1.916(6); N(10)–N(11), 1.162(7) cf. N–N distance in free N2 of 1.098. Selected bond angles (°): N(1)–Mo(1)–N(10), 176.6(2); Mo(1)–N(10)–N(11), 173.8(5); N(10)–N(11)–Mo(2), 175.5(4); N(11)–Mo(2)–N(12), 177.2(3). | ||
Although the (evidently) highly reactive d0 nitrido cation [1-Xyl]+ is strongly implicated in the above chemistry, so far direct evidence for it remains elusive. Low temperature oxidation of 1-Ar (−78 °C, CDCl3 or CD2Cl2) with one equivalent of [MB˙]+[Al(ORF)4]− gave mixed results. Although no diamagnetic products were observed, consumption of the oxidizing agent was implied by an observed color change to a deep green solution as the solutions slowly warmed. Since we do not observe signals for reduced triarylamine under these conditions, we speculate that the cationic [1-Ar]+ products interact with this NAr3 by-product in some as yet unidentified way. However, if these reactions were carried out in the presence of an isonitrile trapping agent65 (2,6-dimethylphenylisocyanide), clean conversion to cationic Mo(IV) carbodiimide complexes [4-Ar]+ were observed, along with the emergence of signals for NAr3 (Scheme 6). No reaction was observed between xylyl isocyanide and neutral nitridos 1-Ar, but upon addition of one equivalent of [MB˙]+[Al(ORF)4]− at −78 °C, a color change to deep purple was observed as the sample was gradually warmed and production of carbodiimides [4-Ar]+ occurred. A separate control experiment showed that xylyl isocyanide is not oxidized by the [MB˙]+[Al(ORF)4]− oxidant, so we take this as evidence of formation of cations [1-Ar]+, which are trapped by the isocyanide reagent to give the observed products.
 | ||
| Scheme 6 Reaction of 1-Ar with 1 equivalent of oxidant at −78 °C in the presence of xylyl isocyanide. | ||
These Mo(IV) carbodiimide products give assignable proton NMR spectra (Fig. S6†) that clearly indicate incorporation of the xylyl isocyanide reagent. This is also supported by stretching bands (both at 1940 cm−1) in the FTIR spectra of [4-Tol]+ and [4-Xyl]+, respectively. Dark purple crystals of the latter derivative confirm the structures of these compounds as shown by the molecular structure depicted in Fig. 6. The bond distances and angles (see caption) within the Mo–NCN–Xyl unit (particularly the Mo(1)–N(10) distance of 1.921(3) Å and the near linear Mo(1)–N(10)–C(34) angle of 171.8(3)°) are consistent with some double bond character to the Mo(1)–N(10) linkage and CN double bonds as depicted in Scheme 6.
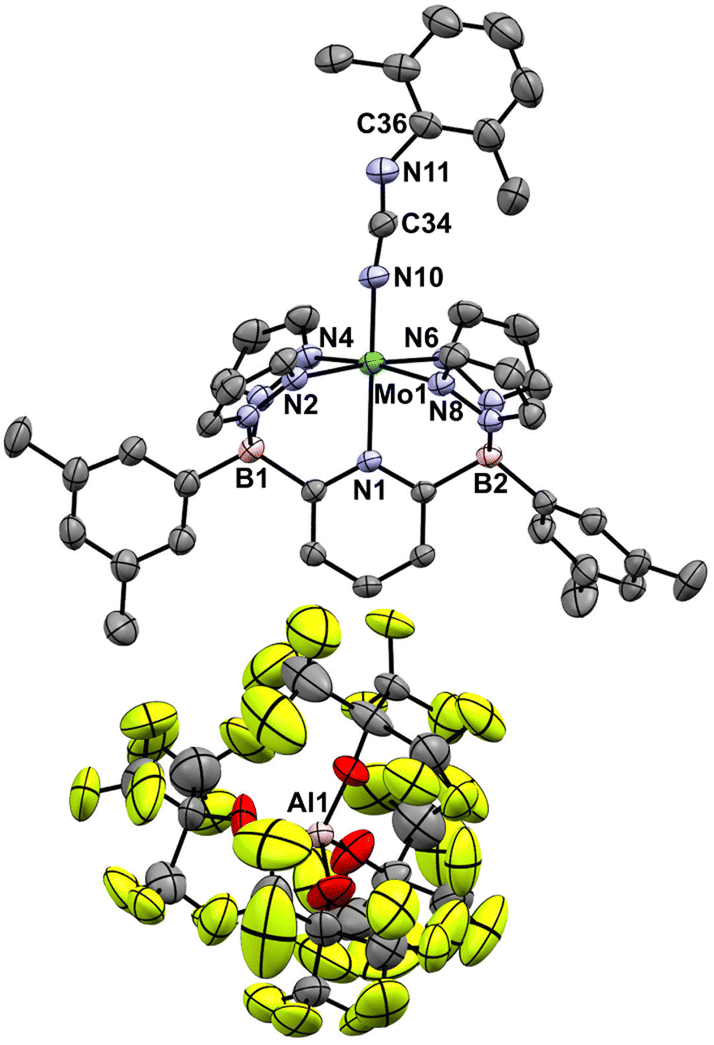 | ||
| Fig. 6 Molecular structure of [4-Xyl]+. Thermal ellipsoids are shown at the 50% probability level and all H atoms are omitted for clarity. Selected bond distances (Å): Mo(1)–N(1), 2.167(3); Mo(1)–N(10), 1.921(3); N(10)–C(34), 1.210(5); C(34)–N(11), 1.207(5); N(11)–C(36), 1.391(6) selected bond angles (°): N(1)–Mo(1)–N(10), 176.82(12); Mo(1)–N(10)–C(34), 171.8(3); N(10)–C(34)–N(11), 170.1(5); C(34)–N(11)–C(36), 137.1(4). | ||
Conclusions
Nitrogen–nitrogen bond formation via coupling of two terminal metal nitrido complexes is one possible pathway for generating N2 in an ammonia oxidation catalytic scheme. The lowest barrier path for nitrido coupling is to a large degree dictated by the electronic configuration of the LnMN species (geometry, d electron count, M–N bond order) and the extent to which spin density is or is not localized on the nitrido nitrogen. In the d1 Mo(V) nitrido complexes 1-Ar described here, there is very little nitridyl character associated with the MN moiety and so the barrier to N–N bond formation is prohibitively high for this homocoupling path. However, we observe facile loss of N2 from these compounds upon oxidation with 0.5 equivalents of a one electron oxidant and convincingly demonstrate that this opens up a much lower barrier nucleophilic/electrophilic pathway for N–N bond formation. Oxidation of 1-Ar to [1-Ar]+ flips the nitrido from being nucleophilic in the neutral Mo(V) species to strongly electrophilic in the Mo(VI) cation and induces ambiphilicity into the system, allowing for rapid N–N bond formation.We believe the “oxidation-induced ambiphilicity” mechanism depicted in Scheme 4 to be a new mechanistic variation for N–N bond formation from metal nitridos. Some years ago, Che and co-workers observed facile photoinduced N2 formation from [(NH3)4Os(VI)N]3+ which was accelerated in the presence of sacrificial electron donors.66 It was proposed that the N–N bond formation took place by bimolecular coupling between the excited state [(NH3)4Os(VI)N]3+* and the ground state [(NH3)4Os(VI)N]3+, which also could be considered a type of “induced ambiphilicity”, triggered in this instance by reduction. However, under oxidation catalysis, the path discussed here would likely be more relevant. It is also worth noting that the ECECC path uncovered here is one of several potential N–N bond forming steps in AO catalytic schemes. The favored path (or paths operating in parallel67) will be to a large extent dependent on the conditions of the ammonia oxidation catalysis experiments.
The coupling of two metal nitridos to form dinitrogen complexes is the reverse of metal-mediated dinitrogen cleavage,68 a well-studied reaction of interest in the context of nitrogen fixation to ammonia or other nitrogen containing compounds.69 While the direction of the reaction is dictated mostly by thermodynamic factors, the kinetics of the preferred process can be significantly influenced by electron configuration and metal geometry. Here, one electron oxidation significantly lowers the barrier of N–N bond formation. In some systems where dinitrogen cleavage is thermodynamically favored, a one electron reduction can lead to significant rate enhancements for metal nitrido formation.70–72
Another conclusion from this work re-emphasizes that formation of μ-nitrido bridged dinuclear complexes is a potential pitfall to be considered in the development of catalysts for ammonia oxidation. They can arise in situations where the dominant N–N bond forming step is coupling of two higher valent LmMnN compounds. If N2 release from the μ-dinitrogen product of coupling is more rapid than the N–N coupling step (which is likely on thermodynamic and kinetic grounds) then lower valent LmMn−3 species are formed in the presence of nucleophilic nitrido starting material, leading to the M–N–M products observed here and elsewhere.4,27,55 Under catalytic conditions, NH3 would likely effectively compete with LmMn−3N for the low valent metal centers but ways to avoid formation of μ-nitrido complexes that involved ligand designs aimed at sterically allowing formation of bridging dinitrogen complexes while disfavoring bridging nitrido side products would be worth pursuing.
Data availability
All data related to this article can be found in the ESI document published with this article.†Author contributions
The manuscript was written through contributions of all authors. CCA and WEP conceived the study and CCA conducted the bulk of the experiments. HDACJ contributed to the Xyl ligand synthesis and Mo starting materials and NR conducted preliminary N2 elimination experiments for 1-Tol. LM directed the computational work, which was performed by TR. WZ and BSG contributed some of the X-ray structure determinations. WEP and CCA wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was provided by NSERC of Canada in the form of a Discovery Grant to W. E. P. who also acknowledges the Canada Research Chair secretariat for a Tier I CRC (2020–2027). C. C. A. thanks Alberta Innovates for scholarship support. The computational work was supported by the HPCs CALcul en Midi-Pyrénées (CALMIP-EOS grant 1415); L. M. is a senior member of the Institut Universitaire de France.Notes and references
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- M. S. Thompson and T. J. Meyer, J. Am. Chem. Soc., 1981, 103, 5577–5579 CrossRef CAS.
- F. Habibzadeh, S. L. Miller, T. W. Hamann and M. R. Smith, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2849–2853 CrossRef CAS PubMed.
- K. Nakajima, H. Toda, K. Sakata and Y. Nishibayashi, Nat. Chem., 2019, 11, 702–709 CrossRef CAS PubMed.
- P. Bhattacharya, Z. M. Heiden, G. M. Chambers, S. I. Johnson, R. M. Bullock and M. T. Mock, Angew. Chem., Int. Ed., 2019, 58, 11618–11624 CrossRef CAS PubMed.
- M. D. Zott, P. Garrido-Barros and J. C. Peters, ACS Catal., 2019, 9, 10101–10108 CrossRef CAS.
- Y. Li, J.-Y. Chen, Q. Miao, X. Yu, L. Feng, R.-Z. Liao, S. Ye, C.-H. Tung and W. Wang, J. Am. Chem. Soc., 2022, 144, 4365–4375 CrossRef CAS PubMed.
- P. L. Dunn, B. J. Cook, S. I. Johnson, A. M. Appel and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 17845–17858 CrossRef CAS PubMed.
- V. Smil, Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, MIT Press, Cambridge, MA, 2001 Search PubMed.
- M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730 CrossRef CAS PubMed.
- M. J. Bezdek, I. Pelczer and P. J. Chirik, Organometallics, 2020, 39, 3050–3059 CrossRef CAS.
- P. Bhattacharya, Z. M. Heiden, E. S. Wiedner, S. Raugei, N. A. Piro, W. S. Kassel, R. M. Bullock and M. T. Mock, J. Am. Chem. Soc., 2017, 139, 2916–2919 CrossRef CAS PubMed.
- B. J. Cook, M. Barona, S. I. Johnson, S. Raugei and R. M. Bullock, Inorg. Chem., 2022, 61, 11165–11172 CrossRef CAS PubMed.
- S. I. Johnson, S. P. Heins, C. M. Klug, E. S. Wiedner, R. M. Bullock and S. Raugei, Chem. Commun., 2019, 55, 5083–5086 RSC.
- C. C. Almquist, N. Removski, T. Rajeshkumar, B. S. Gelfand, L. Maron and W. E. Piers, Angew. Chem., Int. Ed., 2022, 61, e202203576 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Chem. Rev., 2022, 122, 1–49 CrossRef CAS PubMed.
- H. Toda, K. Kuroki, R. Kanega, S. Kuriyama, K. Nakajima, Y. Himeda, K. Sakata and Y. Nishibayashi, ChemPlusChem, 2021, 86, 1511–1516 CrossRef CAS PubMed.
- M. J. Trenerry, C. M. Wallen, T. R. Brown, S. V. Park and J. F. Berry, Nat. Chem., 2021, 13, 1221–1227 CrossRef CAS PubMed.
- N. X. Gu, P. H. Oyala and J. C. Peters, Angew. Chem., Int. Ed., 2021, 60, 4009–4013 CrossRef CAS PubMed.
- M. E. Ahmed, M. Raghibi Boroujeni, P. Ghosh, C. Greene, S. Kundu, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2022, 146, 21136–21145 CrossRef PubMed.
- S.-M. Yiu, W. W. Y. Lam, C.-M. Ho and T.-C. Lau, J. Am. Chem. Soc., 2007, 129, 803–809 CrossRef CAS PubMed.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552–558 CrossRef CAS PubMed.
- M. G. Scheibel, J. Abbenseth, M. Kinauer, F. W. Heinemann, C. Würtele, B. de Bruin and S. Schneider, Inorg. Chem., 2015, 54, 9290–9302 CrossRef CAS PubMed.
- T. Miyazaki, H. Tanaka, Y. Tanabe, M. Yuki, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2014, 53, 11488–11492 CrossRef CAS PubMed.
- D. C. Ware and H. Taube, Inorg. Chem., 1991, 30, 4605–4610 CrossRef CAS.
- S. B. Seymore and S. N. Brown, Inorg. Chem., 2002, 41, 462–469 CrossRef CAS PubMed.
- S. V. Park, A. R. Corcos, A. N. Jambor, T. Yang and J. F. Berry, J. Am. Chem. Soc., 2022, 144, 3259–3268 CrossRef CAS PubMed.
- M. E. de Vries, R. M. La Crois, G. Roelfes, H. Kooijman, A. L. Spek, R. Hage and B. L. Feringa, Chem. Commun., 1997, 1549–1550, 10.1039/A702804K.
- R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 1997, 119, 8566–8567 CrossRef CAS.
- R. J. M. Klein Gebbink, R. T. Jonas, C. R. Goldsmith and T. D. P. Stack, Inorg. Chem., 2002, 41, 4633–4641 CrossRef CAS PubMed.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- D. Z. Zee, T. Chantarojsiri, J. R. Long and C. J. Chang, Acc. Chem. Res., 2015, 48, 2027–2036 CrossRef CAS PubMed.
- D. M. Spasyuk, S. H. Carpenter, C. E. Kefalidis, W. E. Piers, M. L. Neidig and L. Maron, Chem. Sci., 2016, 7, 5939–5944 RSC.
- D. W. Beh, W. E. Piers, I. del Rosal, L. Maron, B. S. Gelfand, C. Gendy and J. B. Lin, Dalton Trans., 2018, 47, 13680–13688 RSC.
- D. W. Beh, W. E. Piers, B. S. Gelfand and J.-B. Lin, Dalton Trans., 2020, 49, 95–101 RSC.
- D. W. Beh, W. E. Piers, L. Maron, Y. Yang, B. S. Gelfand and J.-B. Li, Polyhedron, 2020, 179, 114410 CrossRef CAS.
- L. Nurdin, D. M. Spasyuk, L. Fairburn, W. E. Piers and L. Maron, J. Am. Chem. Soc., 2018, 140, 16094–16105 CrossRef CAS PubMed.
- L. Nurdin, D. M. Spasyuk, W. E. Piers and L. Maron, Inorg. Chem., 2017, 56, 4157–4168 CrossRef CAS PubMed.
- L. Nurdin, Y. Yang, P. G. N. Neate, W. E. Piers, L. Maron, M. L. Neidig, J. B. Lin and B. S. Gelfand, Chem. Sci., 2021, 12, 2231–2241 RSC.
- D. W. Beh, A. J. Cuellar De Lucio, I. del Rosal, L. Maron, D. Spasyuk, B. S. Gelfand, J.-B. Li and W. E. Piers, Organometallics, 2023, 42, 1149–1157 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005, 10.1039/JR9590002003.
- J. C. Kim, W. S. Rees Jr and V. L. Goedken, Inorg. Chem., 1995, 34, 2483–2486 CrossRef CAS.
- P. Basu, J. Chem. Educ., 2001, 78, 666 CrossRef CAS.
- S. Verma and M. Hanack, Z. Anorg. Allg. Chem., 2003, 629, 880–892 CrossRef CAS.
- E. Seikel, B. Oelkers, O. Burghaus and J. Sundermeyer, Inorg. Chem., 2013, 52, 4451–4457 CrossRef CAS PubMed.
- I. Krossing, Chem.–Eur. J., 2001, 7, 490–502 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- M. R. Talipov, M. M. Hossain, A. Boddeda, K. Thakur and R. Rathore, Org. Biomol. Chem., 2016, 14, 2961–2968 RSC.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS PubMed.
- P. L. Dunn, S. I. Johnson, W. Kaminsky and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 3361–3365 CrossRef CAS PubMed.
- R. M. Clarke and T. Storr, J. Am. Chem. Soc., 2016, 138, 15299–15302 CrossRef CAS PubMed.
- M. Keener, M. Peterson, R. Hernández Sánchez, V. F. Oswald, G. Wu and G. Ménard, Chem.–Eur. J., 2017, 23, 11479–11484 CrossRef CAS PubMed.
- T. Chantarojsiri, A. H. Reath and J. Y. Yang, Angew. Chem., Int. Ed., 2018, 57, 14037–14042 CrossRef CAS PubMed.
- J. Du Bois, C. S. Tomooka, J. Hong and E. M. Carreira, Acc. Chem. Res., 1997, 30, 364–372 CrossRef CAS.
- C. E. Laplaza, M. J. A. Johnson, J. C. Peters, A. L. Odom, E. Kim, C. C. Cummins, G. N. George and I. J. Pickering, J. Am. Chem. Soc., 1996, 118, 8623–8638 CrossRef CAS.
- C. Schiller, D. Sieh, N. Lindenmaier, M. Stephan, N. Junker, E. Reijerse, A. A. Granovsky and P. Burger, J. Am. Chem. Soc., 2023, 145, 11392–11401 CrossRef CAS PubMed.
- G. P. Connor, B. Q. Mercado, H. M. C. Lant, J. M. Mayer and P. L. Holland, Inorg. Chem., 2019, 58, 10791–10801 CrossRef CAS PubMed.
- D. Martelino, S. Mahato, W. VandeVen, N. M. Hein, R. M. Clarke, G. A. MacNeil, F. Thomas and T. Storr, J. Am. Chem. Soc., 2022, 144, 11594–11607 CrossRef CAS PubMed.
- E. D. Hedegård, J. Bendix and S. P. A. Sauer, J. Mol. Struct.: THEOCHEM, 2009, 913, 1–7 CrossRef.
- F. Hasanayn, P. L. Holland, A. S. Goldman and A. J. M. Miller, J. Am. Chem. Soc., 2023, 145, 4326–4342 CrossRef CAS PubMed.
- L. S. Yamout, M. Ataya, F. Hasanayn, P. L. Holland, A. J. M. Miller and A. S. Goldman, J. Am. Chem. Soc., 2021, 143, 9744–9757 CrossRef CAS PubMed.
- A. K. Maity, J. Murillo, A. J. Metta-Magaña, B. Pinter and S. Fortier, J. Am. Chem. Soc., 2017, 139, 15691–15700 CrossRef CAS PubMed.
- H.-W. Lam, C.-M. Che and K.-Y. Wong, J. Chem. Soc., Dalton Trans., 1992, 1411–1416, 10.1039/DT9920001411.
- C.-P. Chen, W. Alharbi, T. R. Cundari, T. W. Hamann and M. R. Smith III, J. Am. Chem. Soc., 2023, 145, 26339–26349 CrossRef CAS PubMed.
- S. J. K. Forrest, B. Schluschaß, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed.
- S. Kuriyama and Y. Nishibayashi, Tetrahedron, 2021, 83, 131986 CrossRef CAS.
- J. C. Peters, J.-P. F. Cherry, J. C. Thomas, L. Baraldo, D. J. Mindiola, W. M. Davis and C. C. Cummins, J. Am. Chem. Soc., 1999, 121, 10053–10067 CrossRef CAS.
- D.-D. Zhai, S.-Q. Zhang, S.-J. Xie, R.-K. Wu, F. Liu, Z.-F. Xi, X. Hong and Z.-J. Shi, J. Am. Chem. Soc., 2022, 144, 14071–14078 CrossRef CAS PubMed.
- G. P. Connor, D. Delony, J. E. Weber, B. Q. Mercado, J. B. Curley, S. Schneider, J. M. Mayer and P. L. Holland, Chem. Sci., 2022, 13, 4010–4018 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and characterization details for all new compounds, including spectroscopic data, X-ray crystallographic data and computational details with the cartesian coordinates for calculated structures (PDF). CCDC 2303932–2303944. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00090k |
| ‡ We have found 11B NMR spectral data for the ligand borate moieties to be indicative of the formal oxidation state of the molybdenum centers in these compounds. For example, Mo(III) derivatives fall in the range of −150 to −175 ppm, while the d1 Mo(V) complexes are generally 11B NMR silent. Mo(IV) complexes, on the other hand, generally resonate in the −40 to −50 ppm range. |
| This journal is © The Royal Society of Chemistry 2024 |