
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePnictogen bonding in imide derivatives for chiral folding and self-assembly†
Zhuoer
Wang
,
Zhaozhen
Cao
,
Aiyou
Hao
 and
Pengyao
Xing
*
and
Pengyao
Xing
*
Key Laboratory of Colloid and Interface Chemistry of Ministry of Education and School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, People's Republic of China. E-mail: xingpengyao@sdu.edu.cn
First published on 5th April 2024
Abstract
Pnictogen bonding (PnB) is an attraction interaction that originates from the anisotropic distribution of electron density of pnictogen elements, which however has been rarely found in nitrogen atoms. In this work, for the first time, we unveil the general presence of N-involved PnB in aromatic or aliphatic imide groups and reveal its implications in chiral self-assembly of folding. This long-neglected interaction was consolidated by Cambridge structural database (CSD) searching as well as subsequent computational studies. Though the presence of PnB has limited effects on spectroscopic properties in the solution phase, conformation locking effects are sufficiently expressed in the chiral folding and self-assembly behavior. PnB anchors the chiral conformation to control the emergence and inversion of chiroptical signals, while intramolecular PnB induces the formation of supramolecular tilt chirality. It also enables the chiral folding of imide-containing amino acid or peptide derivatives, which induces the formation of unique secondary structural sequences such as β-sheets. Finally, the effects of PnB in directing folded helical structures were revealed. Examples of cysteine and cystine derivatives containing multiple N⋯O and N⋯S PnBs constitute an α-helix like secondary structure with characteristic circular dichroism. This work discloses the comprehensive existence of imide-involved PnB, illustrates its important role in folding and self-assembly, and sheds light on the rational fabrication of conformation-locked compounds and polymers with controllable chiroptical activities.
Introduction
Noncovalent forces play vital roles in the folding and subsequent self-assembly of biomolecules such as proteins and nucleic acids.1–4 Besides the famous hydrogen bonds, σ-hole based interactions were also found in many protein structures.5 Due to the polarizability of elements from Group 17 to 14, the covalent appendance of electro-withdrawing groups introduces electrophilic regions, which are assigned as σ-holes (or π-holes). σ-Holes and π-holes could occur individually or on the same molecule.6a Based on the groups, they are classified as the halogen bond (HaB), chalcogen bond (ChB), pnictogen bond (PnB) and tetrel bond (TeB).6b Density functional theory (DFT) based calculations consolidate that the electrostatic interaction contributes mainly to the above forces, similar to the hydrogen bond (HB), while the σ-hole based interactions show enhanced directionality, which leads to intensive applications in crystal engineering, catalysis and coordination chemistry.7–9 The interactions are modulable towards the nature of elements, polarizability, electronegativity, the electro-withdrawing effect of substituents, the directional angle between Nu and electrophilic holes as well as valency. Based on the above determining factors, HaB based on Br and I, ChB based on Se and Te, as well as PnB based on As and Sb were investigated more than other elements in the same groups.10–14 Especially for PnB, P and N-based donors are rarely reported due to their relatively strong electronegativity and low polarizability.15 As a poor PnB donor, N could only exhibit positive holes when covalently attaching strong electron-withdrawing groups or coordinating to the metals.16,17 Cambridge structural database (CSD) searching indicates the presence of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N⋯F and C–N⋯F PnB based on σ-holes and N⋯O and N⋯F based on π-holes of –NO2 and –CN– segments in CuII and AuIII coordination complexes.18 Other types of N involved PnB donors are extremely rare.
N⋯F and C–N⋯F PnB based on σ-holes and N⋯O and N⋯F based on π-holes of –NO2 and –CN– segments in CuII and AuIII coordination complexes.18 Other types of N involved PnB donors are extremely rare.
σ-hole (or π-hole) based interactions have implications on the self-assembly, which have shown great impacts on the properties of self-assembled arrays, phase behaviors and supramolecular chirality.19–22 Inspired by σ-holes in some important biomacromolecules, exploration of weak force driven chiral self-assembly provides a novel methodology for functional supramolecular aggregated or folded chiral molecules.23–25 Developing new types of weak forces and expanding rarely explored elements as donors would initiate improved functionality, which however remains considerably challenging. Nitrogen widely exists in amino acids, peptides and proteins, which are in the forms of amides and amines.26–28 Though in the amide form, the appended carbonyl groups would withdraw electrons from nitrogen, it could hardly induce the emergence of practical σ-holes, and thus no amide-based PnBs are found. Bickelhaupt et al. obtained the interaction energy (ΔEint) values of halogenated nitrogen derivatives with halides, suggesting the ΔEint of X3N⋯X- (X = F, Cl, Br) ranging from −3.5 to −86.1 kcal mol−1.29 This work provides theoretical hints that by appropriately controlling electron-withdrawing groups, intermolecular N-involved PnB might be realized in the solution or solid phase.
On top of the above developments, we considered that imides consisting of two acyl groups bound to nitrogen would exert a great electron-withdrawing effect, which might induce the emergence of positive hole regions. Imides were widely used in biochemical agents, pharmacy, industrial chemical intermediates and some high strength or conductive polymers such as Kapton .30–34 Imide containing compounds such as succinimide, maleimide, glutarimide, phthalimide and naphthalimide greatly facilitate the fabrication of functional compounds such as the famous perylene diimide (PDI)-based fluorophores.35 Arylene imides and diimides are also widely employed as electron-rich and electron-deficient scaffolds.36 Nevertheless, the potential imide N-involved PnB has seldom been referred to, which although may have great impacts on the folding and self-assembly of imide-containing compounds and biomacromolecules (Scheme 1). The imide-based PnB interaction can be found in some proteins (Scheme 1a), which stabilize structural formation and stabilization, as well as their biofunctions.37 Through this inspiration, in this work, we for the first time unveil the general presence of intra- and intermolecular PnB in imide-based compounds where imide N atoms act as donors. CSD searching indicates that there are thousands of X-ray structures containing imide-based PnB, most of which occur intramolecularly. The occurrence of intramolecular PnB is in most cases aided by the short tether (less than 3 atoms) between PnB acceptors (N, O, and S) and imide N. And the N⋯O distances (for instance) are within 2.5∼3.0 Å, shorter than the sum of the van der Waals radii (3.07 Å). The PnB locks the conformation by anchoring the side substitutes, which, in the chiral compounds, enables the fixation of chiral conformation and subsequently allows for chirality transfer with controlled chiroptical activities (Scheme 1b and c). The implications for chiral properties at the molecular and supramolecular level were illustrated in intra- and intermolecular PnB by highlighting several representative examples, especially in the simple folding of amino acid or peptide imide derivatives and the formation of supramolecular tilt chirality initiated by intermolecular PnB (Scheme 1d). This work systematically unveils the occurrence, properties and applications of a new type of PnB in chiral folding and self-assembly and sheds light on the rational design of imide-based functional small organic compounds or polymers.
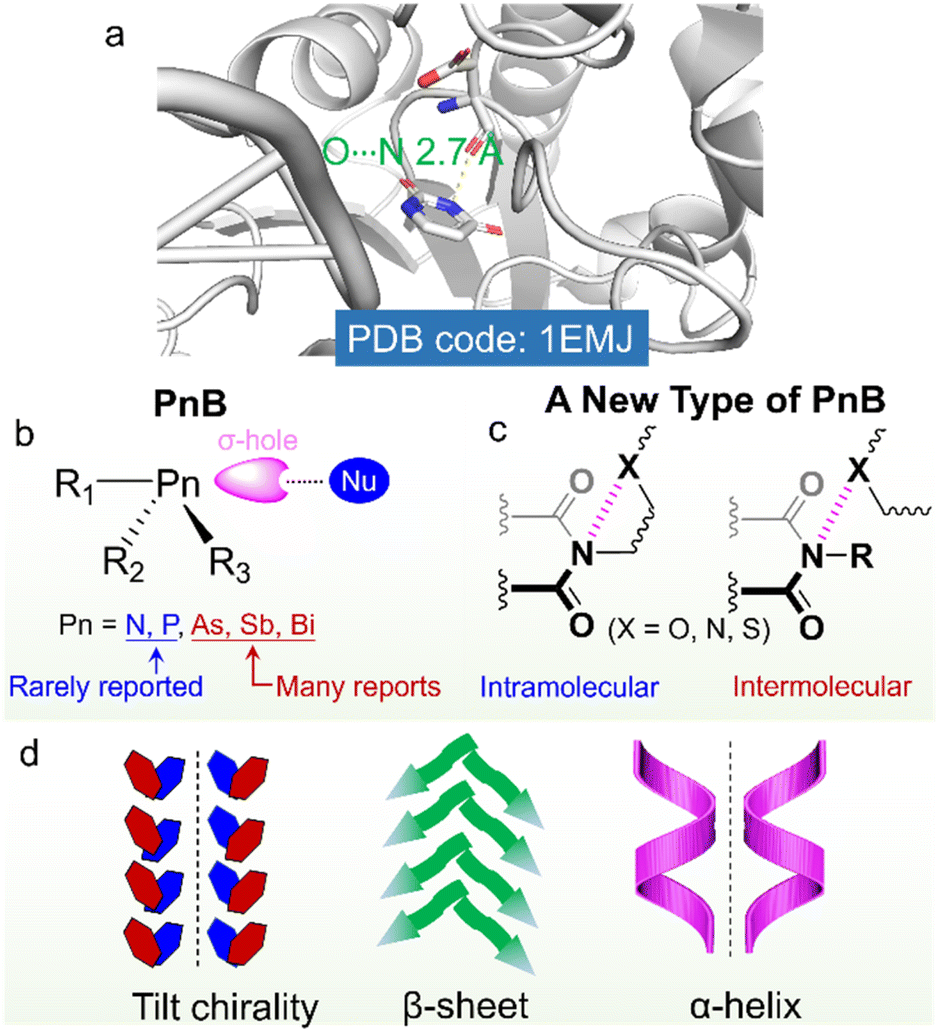 | ||
| Scheme 1 (a) A O⋯N PnB found in a hydrolase where imide and carbonyl oxygen act as the PnB donor and acceptor respectively. (b) General form of PnB based on σ-hole interaction. N and P-based PnB donors are rarely reported. (c) An imide-based PnB that can occur intra- or intermolecularly. (d) The imide-involved PnB drives the formation of supramolecular tilt chirality and secondary structures of amino acids and peptides. | ||
Results and discussion
Our previous studies on imide structures inspired us to validate the general existence of PnB short contacts.38 On top of that, a search for organic crystal structures involving intramolecular and intermolecular short contacts of imides N⋯X (X = O, N, S) was performed on the CSD (Fig. 1a–c). To ensure accurate results, all results obtained were manually checked and further analyzed using statistical methods. CSD searches for intramolecular interactions for N⋯O, N⋯N and N⋯S yielded a total of 1666, 555 and 123 search results, respectively, which occur mainly among compounds of amino acids and amino alcohol derivatives. And in most cases, the spacer between imide N and X (X = O, N, S) contains two atoms and the proportion of the spacer is dramatically decreased when the length is longer than 4 atoms. This suggests the crucial role of spacer length in confining the short contacts.39 The distribution of space groups that crystals adopted is shown in Fig. 1b. It is interestingly found that a considerable proportion falls into the chiral space group of P21, P212121, which contain the 21 spiral axis that could enable the formation of helical structures.40–42 The finding illustrates the potential of N⋯X short contacts in manipulation of chiroptical helical structures. We further summarized the N⋯X distance and C–N–X angle θ in the polar coordinate diagrams (Fig. 1d). As most of the distances are restricted within 3.0 Å, the C–N–O angles are distributed between 45 and 150°, implying that multi-modal interactions occurred in N⋯X intramolecularly folded compounds.43 In contrast, C–N–N angles are distributed mostly at 60°, quite similar to that of C–N–S angles. If not restrained by the tether between N and X, the N⋯X short contacts that occur intermolecularly would adopt a perpendicular direction such that C–N–O and C–N–N angles are mostly distributed at around 90°. This perpendicular direction largely utilizes the potential positive electrophilic regions on imide N atoms. The statistical results based on CSD searching validate the general existence of N⋯X short contacts, and a considerable proportion of structures are chiral, which shall broaden the rational control and design of chiroptical materials.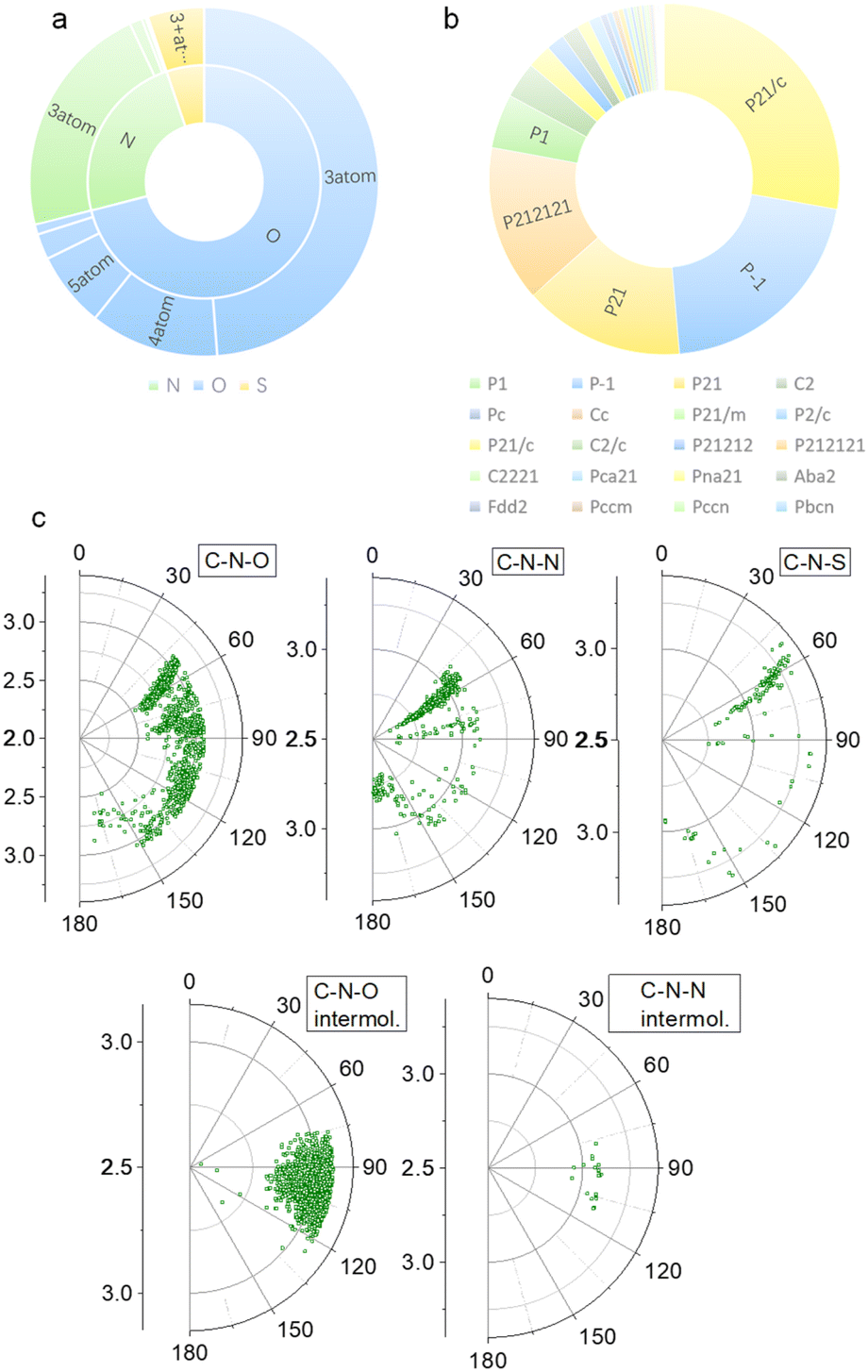 | ||
| Fig. 1 CSD search results. (a) Distribution chart of the search results of N⋯N (green), N⋯O (blue) and N⋯S (yellow) short contacts (distances within the sum of vdWs radii) based on the CSD database. 3, 4 and 5 atoms represent the positions of X atoms, which means that the spacer atom numbers between N and X are 2, 3 and 4 respectively. (b) Distribution chart of space groups. (C) Polar coordinate diagrams of the N⋯X distance and C–N–X angle θ that occur intra- or intermolecularly. | ||
Representatively, we screened some typical compounds with imide groups and potential PnB shown in Chart 1. Except for 3, other compounds possess imides conjugated with aromatic groups. Most of these compounds show short contacts between O and N either in an intramolecular or intermolecular manner (Fig. 2a and S10†). Phthalimide derivatives 1 and 2 whose alcohol moieties are conjugated to imide N via two carbon atom tethers feature N⋯O short contacts. The distances are determined to be 2.871 and 2.793 Å respectively. Structural extension to naphthalimide groups leads to such a short contact as well (compound 6, d = 2.946 Å). It also applies for other types of aromatic imide structures of compounds 5 and 8, giving N⋯O contacts of 2.709 and 2.750 Å respectively. Nonaromatic succinimide derivative 3 contains a similar N⋯O short contact (2.772 Å), indicating that the aromatic imide structure is not a necessity. Electrostatic potential (ESP) maps were calculated based on the X-ray structures of 6 and 7 (Fig. 2b, S11 and Table S5†).44 On both sides of imide N, two most positive (VS, max) regions were found with values ranging from 7.3 to 13.9 kcal mol−1, which might be related to the strong electron-withdrawing effect of the carbonyl group that induces the emergence of π-holes. It is quite similar to the π-holes on nitro groups. The uneven distribution of VS, max on both sides of N is possibly attributed to the neighbouring groups. And in both cases, O points to the regions with the highest VS, max values (12.7 and 13.9 kcal mol−1 respectively for 6 and 7). Noncovalent interaction (NCI) analysis was performed on compounds 4, 7 and 9 respectively. Compound 4 features intermolecular stacking supported by the N⋯O short contact, which corresponds to the van der Waals forces (vdWs, green region). In an intramolecular manner, 7 and 9 comprise attraction force between O and N regions. NCI analysis verifies the crucial role of N⋯O interaction in directing the self-assembly and folding of imide structures. By rotating the dihedral angle θ and freezing other parts, the changes in electronic energy are summarized in Fig. 2d. The lowest energy (θ ≈ 90°) corresponds to the formation of a N⋯O short contact, while the highest energy (θ ≈ 0°) is contributed by the repulsion between amide O and hydroxyl groups. The energy gap is determined to be 35.3 kcal mol−1, which effectively hampers the free rotation of the N–C bond. This energy is contributed by the O/O repulsion and N⋯O attraction, which in a synergistic manner anchor the very conformation. The rotary transition state (TS) of compound 7 was further calculated at the B3LYP-D3/def2SVP level of theory, which afforded a rotary energy barrier of 15.66 kcal mol−1, lower than the extremum energy gap. NBO calculations on the compounds 1, 9 and 10 were performed. However, the values are too small to be observed, which means that the orbital–orbital components in the N–O type PnB are negligible. We also checked the possibility of n to π* type of interactions and the Burgi–Dunitz angle from such interactions, as shown in Table S6.† It shows that most of the studied compounds do not contain n–π* interaction, while only compounds 4, 5 and 11 show n–π* interaction. These interactions synergistically contribute to the stability of PnB.
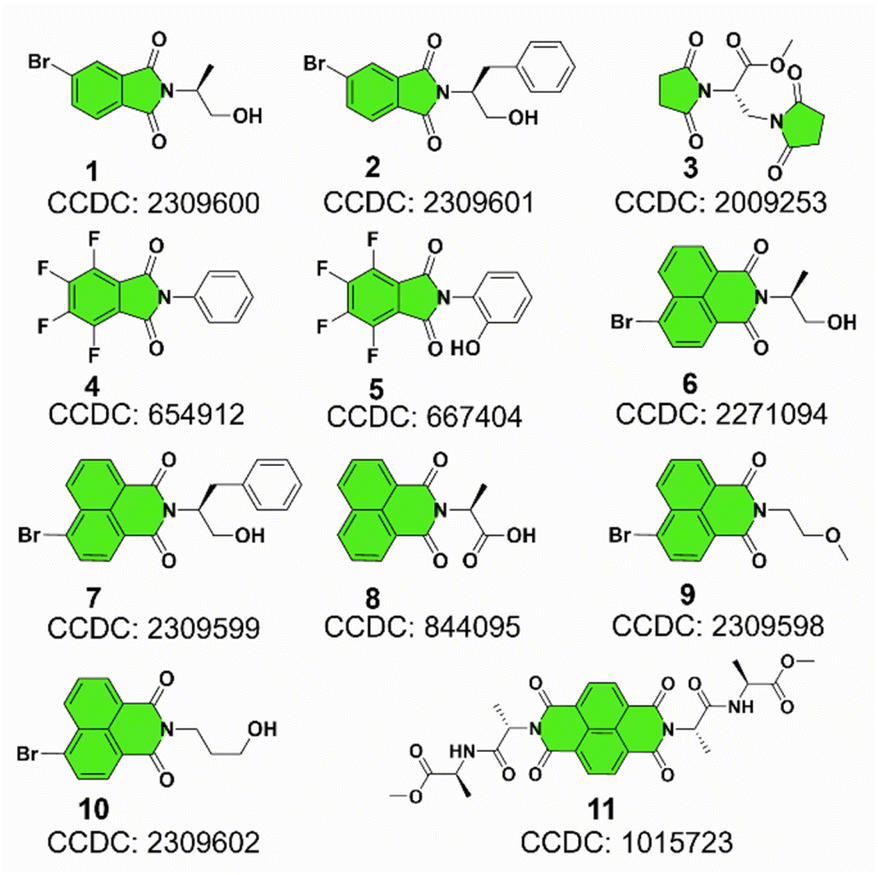 | ||
| Chart 1 Molecular structures of some representative PnB containing imide derivatives. | ||
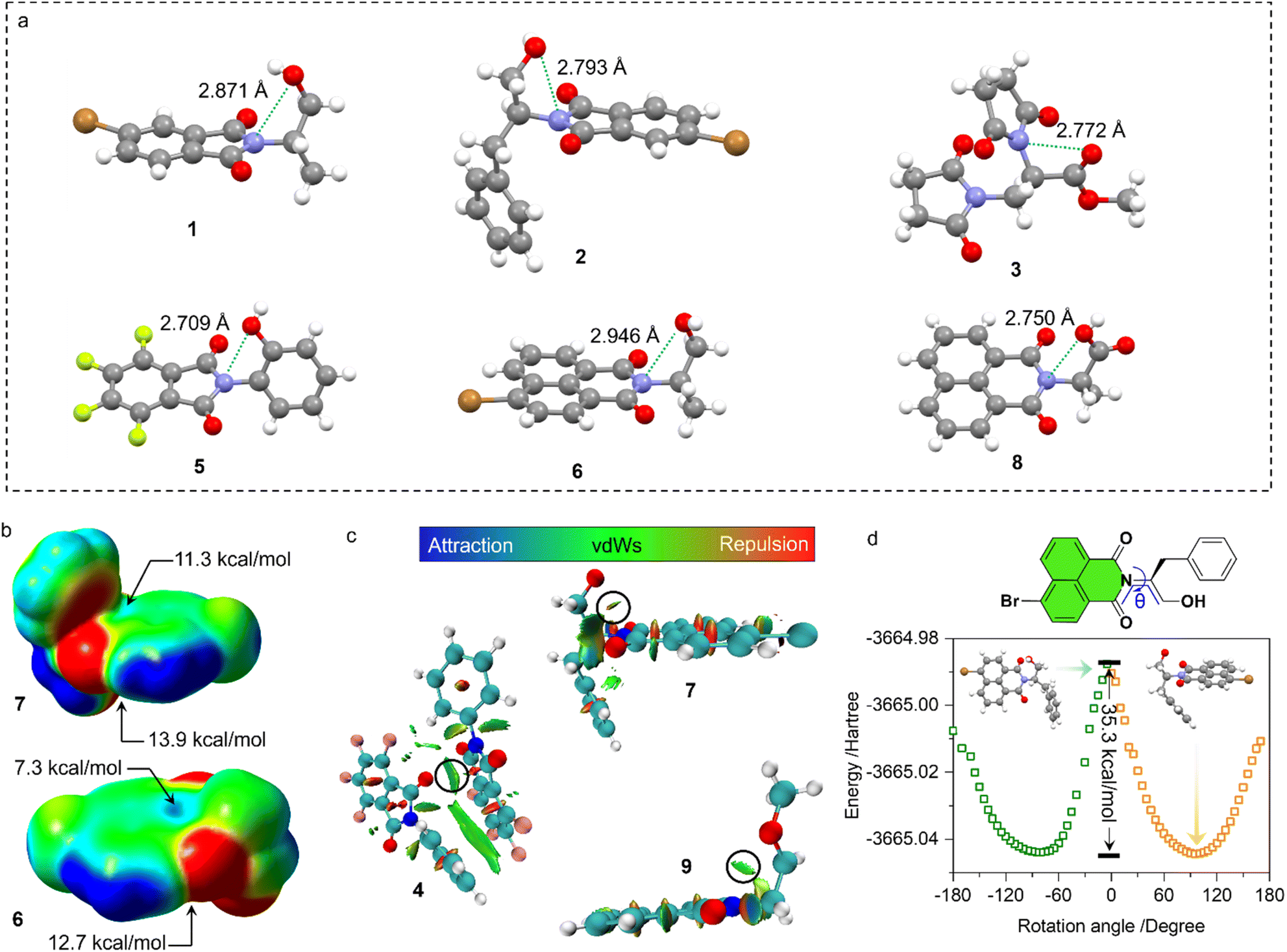 | ||
| Fig. 2 (a) X-ray structures of some PnB-containing imide derivatives (1, 2, 3, 5, 6, and 8). (b) ESP maps of 6 and 7 respectively, as well as their extreme points on both sides of imide N. (c) NCI analysis of 4, 7 and 9 to reveal the interaction between O and N. (d) Electronic energy diagram of compound 7 as a function of rotational dihedral angle θ by freezing other molecular parts. | ||
Will the weak N⋯O short contacts influence the properties in the solution and solid states? To figure out this question, we compared compounds 9 and 10 with respect to their folding and self-assembly arrangements as well as the spectroscopic data in the solution and solid phases (Fig. 3). Compounds 9 and 10 are isomerides whose O atoms are at different positions (Fig. 3a). This results in the formation of N⋯O PnB (2.957 Å) and H⋯O HB (2.046 Å) respectively for 9 and 10. In compound 10, the N⋯O distance of 3.632 Å is out of the sum of vdWs distances, and thus no PnB is expected. The different types of folding influence the self-assembled arrangements. The presence of Br shall introduce HaB that occurs between Br⋯O. Complementary Br⋯O HaB (3.184 Å) aids in the formation of box-like dimers which further stack through π–π stacking (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group). The complementary HaB is apparently facilitated by the presence of PnB that draws the distance between Br and O. In comparison, compound 10 with HB would not favor complementary HaB, yet adopts a parallelly packed arrangement within a P21/c space group. The distinct packing in the solid state reflects the crucial role of PnB in directing self-assembly arrays. In the solution phase (CDCl3), chemical shifts of aromatic protons of 9 and 10 show differences (Fig. 3b) that protons of 9 shift to lower fields. As the tether between O and N contains two or more CH2, the inductive effect of O is negligible. The shifts might be associated with the N⋯O PnB and H⋯O HB. The nature of PnB is that O donates electrons to electrophilic regions of imide N, while for HB, imide O donates electrons to the electrophilic H. The above difference will slightly alter the electronic properties of the naphthalimide core both in solution and solid states. In the Fourier transform infrared spectroscopy (FT-IR) spectra, two characteristic absorption peaks at 1656 and 1342 cm−1 respectively are assigned to the CO and C–N stretching vibrational bands, which shift to 1646 and 1331 cm−1 ascribed to the formation of PnB (Fig. 3c). In the diluted solution, we also observed slight emission and absorption spectroscopic changes (Fig. 3d). Compound 10 shows bathochromic shifts with respect to the absorption and emission spectra ascribed to the distinct properties of PnB and HB. The effect was more pronounced in the solid-state diffusion reflectance spectra (Fig. 3e). Compound 10 exhibits a more profound bathochromic shift with a newly emerged peak at around 450 nm. This means that the design of PnB may realize chromic properties. The heavy atomic effects of Br enable room temperature phosphorescence in the solid state (Fig. 3f). Compounds 9 and 10 show different decay rates that afford a lifetime of 6.898 ms and 5.398 ms respectively. And in the corresponding delayed emission spectra, 9 also gives rise to a slight hypsochromic shift compared to 10 as well. Through the above comparison, the differences are less pronounced for 9 and 10 due to the weak nature of the PnB. Nevertheless, such a difference is potentially boosted in future design such as introducing PnB in polymeric materials, which may significantly influence self-assembly, photophysical properties and performances.
space group). The complementary HaB is apparently facilitated by the presence of PnB that draws the distance between Br and O. In comparison, compound 10 with HB would not favor complementary HaB, yet adopts a parallelly packed arrangement within a P21/c space group. The distinct packing in the solid state reflects the crucial role of PnB in directing self-assembly arrays. In the solution phase (CDCl3), chemical shifts of aromatic protons of 9 and 10 show differences (Fig. 3b) that protons of 9 shift to lower fields. As the tether between O and N contains two or more CH2, the inductive effect of O is negligible. The shifts might be associated with the N⋯O PnB and H⋯O HB. The nature of PnB is that O donates electrons to electrophilic regions of imide N, while for HB, imide O donates electrons to the electrophilic H. The above difference will slightly alter the electronic properties of the naphthalimide core both in solution and solid states. In the Fourier transform infrared spectroscopy (FT-IR) spectra, two characteristic absorption peaks at 1656 and 1342 cm−1 respectively are assigned to the CO and C–N stretching vibrational bands, which shift to 1646 and 1331 cm−1 ascribed to the formation of PnB (Fig. 3c). In the diluted solution, we also observed slight emission and absorption spectroscopic changes (Fig. 3d). Compound 10 shows bathochromic shifts with respect to the absorption and emission spectra ascribed to the distinct properties of PnB and HB. The effect was more pronounced in the solid-state diffusion reflectance spectra (Fig. 3e). Compound 10 exhibits a more profound bathochromic shift with a newly emerged peak at around 450 nm. This means that the design of PnB may realize chromic properties. The heavy atomic effects of Br enable room temperature phosphorescence in the solid state (Fig. 3f). Compounds 9 and 10 show different decay rates that afford a lifetime of 6.898 ms and 5.398 ms respectively. And in the corresponding delayed emission spectra, 9 also gives rise to a slight hypsochromic shift compared to 10 as well. Through the above comparison, the differences are less pronounced for 9 and 10 due to the weak nature of the PnB. Nevertheless, such a difference is potentially boosted in future design such as introducing PnB in polymeric materials, which may significantly influence self-assembly, photophysical properties and performances.
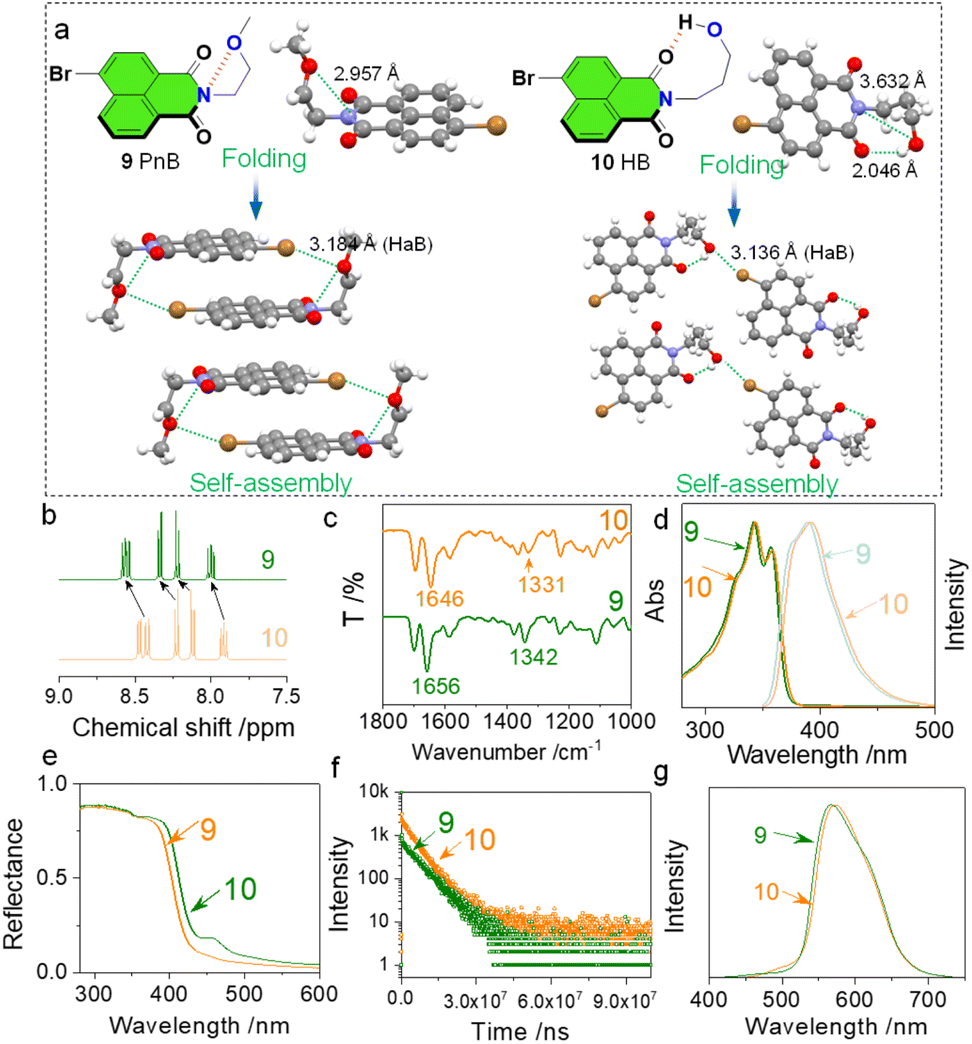 | ||
| Fig. 3 (a) Solid-state structure comparison between 9 and 10, which give a N⋯O short contact and a H⋯O hydrogen bond respectively. (b) 1H NMR spectrum comparison in CDCl3. (c) FT-IR spectrum comparison. (d) Absorption and emission spectrum comparison (c = 0.4 mM, λex = 320 nm). (e) Diffusion reflectance spectrum comparison of 9 and 10 in the solid-state. (f) Emission decay curves in the solid state. (g) Delayed emission (1 ms) spectra in the solid state (λex = 320 nm). | ||
Then the chiroptical properties were probed using circular dichroism (CD) spectra (Fig. 4a, S13–S18†). In the solution state, compounds 1 and 2 with similar chromophores and absolute chirality yet with different substitutes yielded active Cotton effects (Fig. 4a and b). The active Cotton effects range from 230 to 300 nm and fall into the absorption of phthalimides, which are not intrinsically chiral. The conformation-locked structure by N⋯O PnB would facilitate chirality transfer to aromatic entities from amino alcohols. L- and D-enantiomers afforded positive and negative CD signs respectively, and this principle is also observed in an analogue shown in Fig. 4c. We also explored the chiroptical properties of 6, 7 and 8 in the solution and solid state (Fig. 4d–f). All three compounds share a similar naphthalimide aromatic core conjugated to the chiral entities. Using the nanoprecipitation method, after pouring the DMSO stock solutions into poor solvent water, self-assemblies were obtained. As shown in Fig. S19,† most CD spectra show great similarity compared to the solid-state CD spectra, indicating that the molecular arrangements in the solution-based self-assemblies follow a similar modality compared to the solid or crystalline states. Nevertheless, compound 7 exhibits an opposite sign to 6 though 6 and 7 possess identical bromo-naphthalimide chromophores (Fig. 4d and e). Their single crystal structures displayed in Fig. 4g suggest that the Br group has different relative orientation to the chiral parts. And the two relative orientations could be switched by rotating the N–C bond, which however is anchored by PnB and CO/O electrostatic repulsion forces (Fig. 2d). Calculated ECD spectra also verified the opposite mirror CD signal (Fig. S20†). The difference caused by the conformation is covered by the solid-state assembly where supramolecular packing leads to pronounced supramolecular chirality (7 and 8 exhibited a similar sign in the solid state). The above results illustrate the fact that N⋯O PnB could be used to anchor or manipulate the chiral conformation with active chiroptical properties. The high rotational barrier illustrated the integrity of PnB-locked conformation that might be resistant to temperature. Temperature-variable CD spectra of 6 and 7 in solution were collected (Fig. 4h and i). Heating from 0 to 70 °C produces the slightly declined CD signal. Subsequent cooling from 70 °C to 0 °C fully recovers the Cotton effect intensity and the corresponding absorption (Fig. S21†), suggesting that the chiral conformation has good resistance to temperature variations.
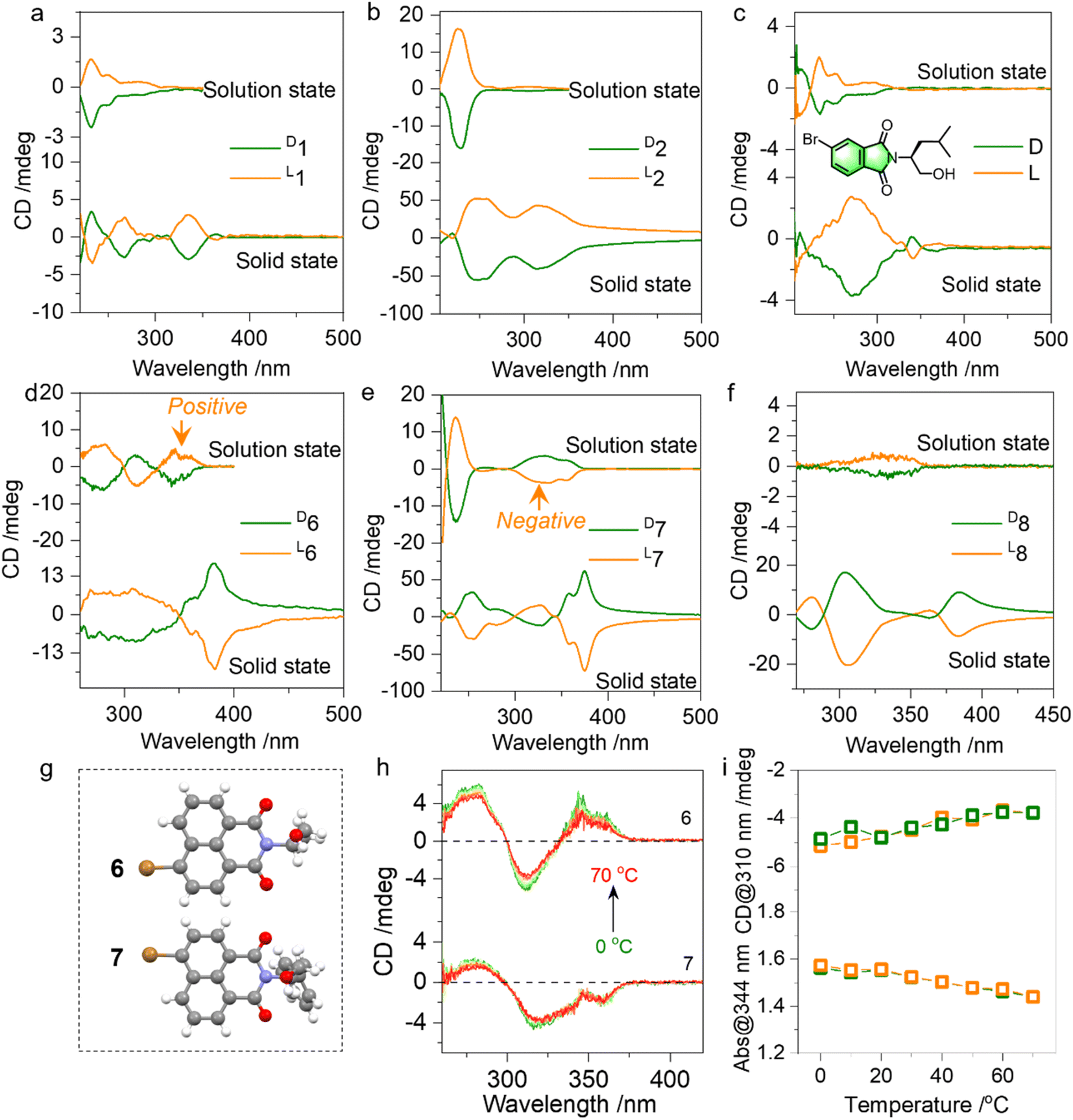 | ||
| Fig. 4 (a–f) CD spectra comparison between solution (dichloroethane, c = 0.4 mM) and solid phases for 1, 2, 3, 6, 7, 8 and an analogue respectively. Solid state CD spectra were collected in KBr tablets to eliminate any artifacts. (g) X-ray structures of 6 and 7. (h) Temperature-variable CD spectra of 6 and 7 (dichloroethane, c = 0.4 mM) from 0 to 70 °C. (i) Variations of CD and absorption spectra of 6 during heating (yellow) and cooling (green) processes. | ||
Then we explored the impact of PnB occurring intermolecularly in self-assemblies. Compounds 4 and 5 feature N⋯O PnB between imide CO and N respectively with distances of 2.979 and 3.040 Å (Fig. 5). The self-assembly is also aided by other supramolecular forces such as arene-perfluoroarene (AP) force between benzene and perfluorinated phthalimide moieties, which finally afforded one-dimensional packing arrays with specific homochirality (Fig. 5a and d). Within the one-dimensional growth, adjacent perfluorinated phthalimide moieties demonstrated a discontinuous tilt chirality with a designated 21 M-handedness. As the compound 4 or 5 has no inherent chirality, the self-resolution during crystallization can be regarded as the conglomeration behaviour (Fig. S22†).45 Conglomerate crystals tend to form chiral assemblies with unbiased M- or P-handedness. Consequently, the random measurement of different crystals or solids produced opposite handedness with mirror CD signs (Fig. 5b and e). Solid B and solid A respectively possessed positive and negative Cotton effects. To correlate the CD signal and handedness, ECD calculation was performed based on the tilt chirality found in the crystals. The fragmented M-helices of 4 and 5 gave exciton-type positive signals centered at around 350 nm (Fig. 5c and f). This strongly evidences the correlation between handedness and CD signs that solid A and solid B from 4 are P- and M-helices respectively. The observations imply the rational usage of N⋯O PnB in designing supramolecular chiral helices with predicted chiroptical properties.
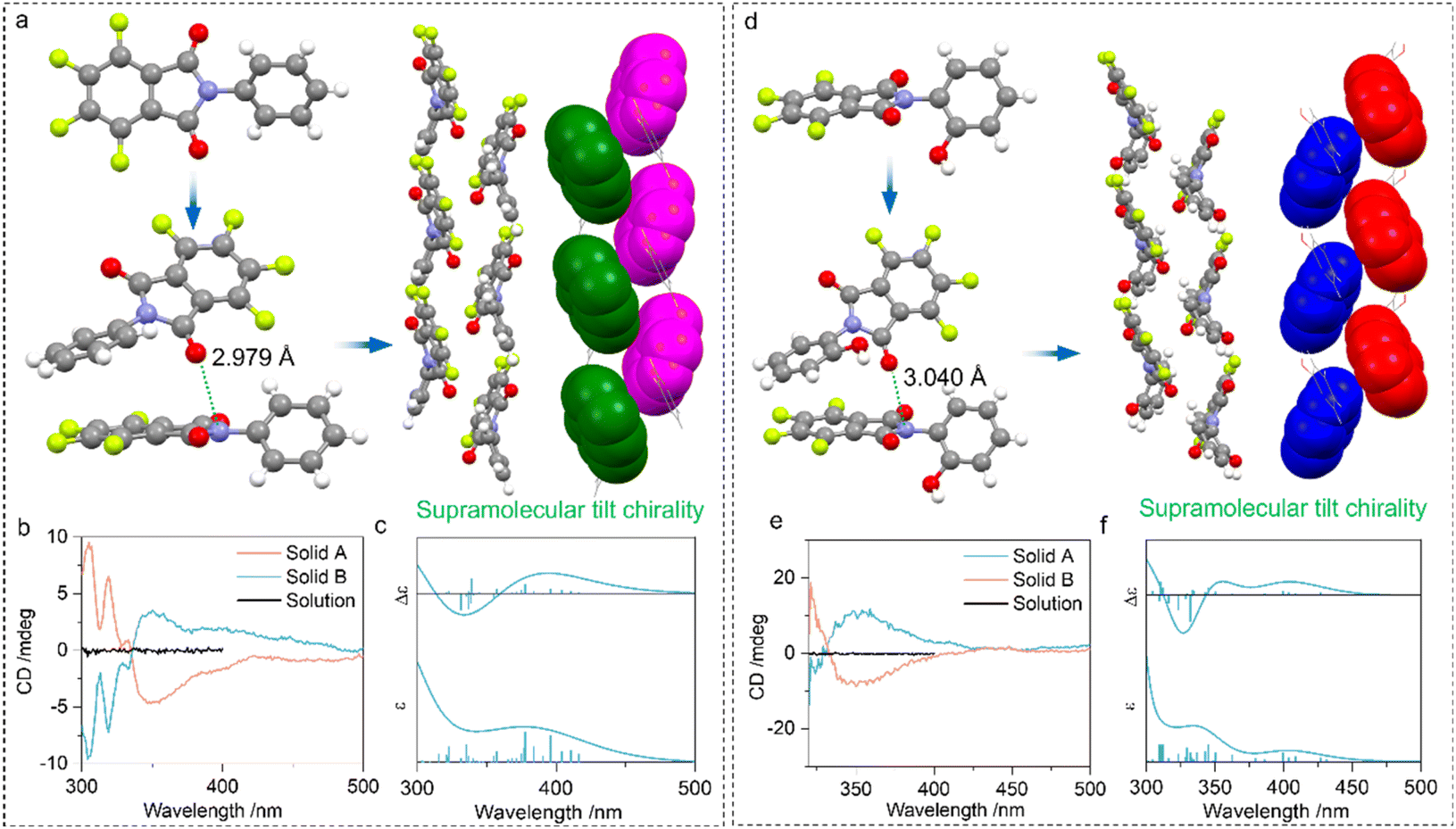 | ||
| Fig. 5 (a) X-ray structure of 4 as well as the illustration of PnB directed hierarchical self-assembly into supramolecular chiral helices. (b) CD spectra of different solid assemblies and solution phase of 4. (c) Calculated electronic CD spectrum based on the structure of (a). (d–f) The corresponding X-ray crystal structure, CD spectra and calculated electronic CD spectrum of 5 respectively (basis set: B3LYP-D3/def2SVP). | ||
The intramolecular or intermolecular N⋯O PnB favors the folding or self-assembly of amino acids or peptides into secondary structures. Some examples are shown in Fig. 6a, which are folded amino acid imide derivatives including Ala, Val, His, Gln, Phe and Leu residues. The N⋯O distances range from 2.599 to 2.774 Å. Due to the presence of carboxylic acid ester, there are two PnB acceptor O atoms, which did not show apparent selectivity. Adaptative to the structures of substituents, two O atoms form PnB in a nonpreferred selective manner. The conformation-locked geometries generally exist in the amino acid-containing imide derivatives, which shall facilitate the fabrication of chiroptical materials.
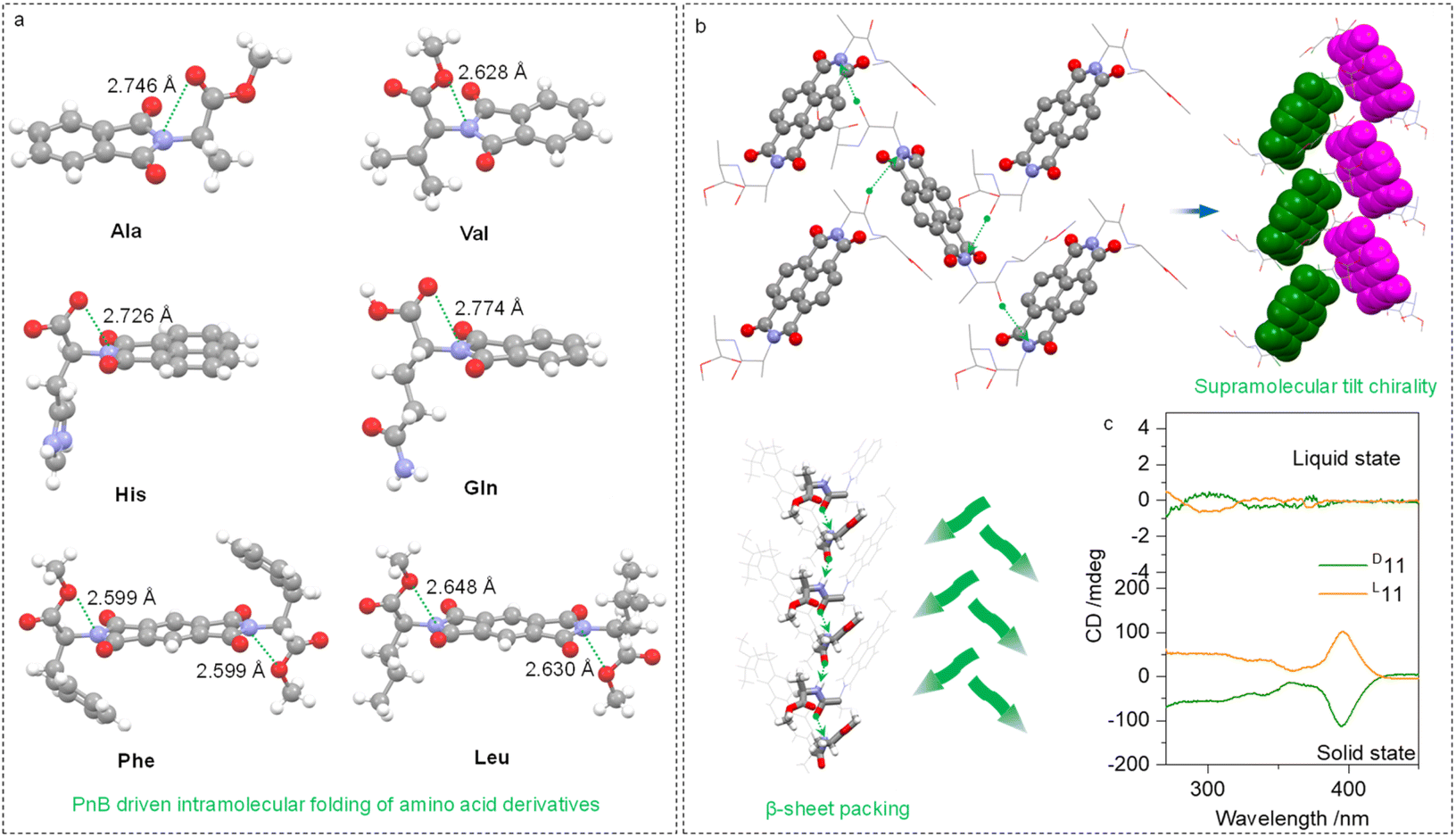 | ||
| Fig. 6 (a) X-ray structures of representative PnB-containing amino acid-imide derivatives. (b) Self-assembly of 11 into supramolecular tilt chiral helices and β-sheet structures. (c) CD spectrum comparison between solution (solvent: dichloroethane, c = 0.4 mM) and solid states. | ||
Intermolecular N⋯O PnBs between Ala–Ala dipeptides (compound 11) conjugated to aromatic imide structures were found to assist in the formation of supramolecular secondary structures. The N⋯O interaction between carbonyl and imide N crosslinks adjacent structures to afford supramolecular helices (Fig. 6b). By highlighting aromatic imide moieties, the 21 tilt helix was observed within a P212121 space group, and importantly, the dipeptide sequences adopt β-sheet arrangements. Interestingly, compound 11 does not have intramolecular N⋯O PnB in the X-ray structure, which correspondingly barely showed active Cotton effects (Fig. 6c) in the solution phase. This phenomenon further establishes the relationship between the active Cotton effects and PnB-locked conformations displayed in Fig. 4. In the solid state, an active CD signal of 11 was observed thanks to the presence of tilt chirality induced by PnB and other noncovalent forces.
Further implications of PnB on the intramolecular conformation locking or folding are given by the cystine and cysteine-based compounds 12 and 13 (Fig. 7a). Single and double functionalities of sulfur atoms perform as PnB acceptors, in addition to the copresence of ester oxygen atoms. Single crystal analysis indicated the presence of four PnB interactions that occurred intramolecularly for 12 and 13 respectively (Fig. 7b and c). The PnBs are continuous and adjacent. Compound 12 gives rise to CO‧‧‧N‧‧‧S‧‧‧N‧‧‧O arrays with distances of 2.816, 3.211, 3.211 and 2.846 Å respectively. Compound 13 gives rise to CO‧‧‧N‧‧‧S and S‧‧‧N‧‧‧OC arrays with distances of 2.764, 3.168, 3.062 and 2.649 Å respectively. Clearly, the S‧‧‧N distances are shorter than the sum of van der Waals radii of 332 pm. The conformation locking of cysteine and cystine derivatives through multiple PnBs is interesting and provides hints for the design of peptides or peptoids with special folding geometries. Highlighting the skeleton of compounds 12 and 13 witnessed the formation of P- and M-helical geometry respectively (Fig. 7b and c). It is not surprising because the absolute configuration of 12 and 13 is R,R and S,S respectively. Folding shall transfer the absolute chirality of cystine to the skeletons. We further synthesized compound 13 and measured the CD spectra (Fig. 7d). In diluted solution, RR13 and SS13 respectively gave negative and positive CD bands at around 250 nm, indicating the transfer of chirality to imide chromophores. This characteristic CD signal is verified using the calculated ECD spectra, showing similar curves at around 250 nm. Indeed, the derivation of either aromatic or aliphatic imide structures shall indiscriminately introduce PnB to enable new functionalities. Normally the formation of helical structures for peptides or peptoids requires at least four amino acid sequences with sufficient skeleton length and hydrogen bonds. The present cases shown by representatives 12 and 13 illustrate the feasible realization of helically folded structures using dimers.
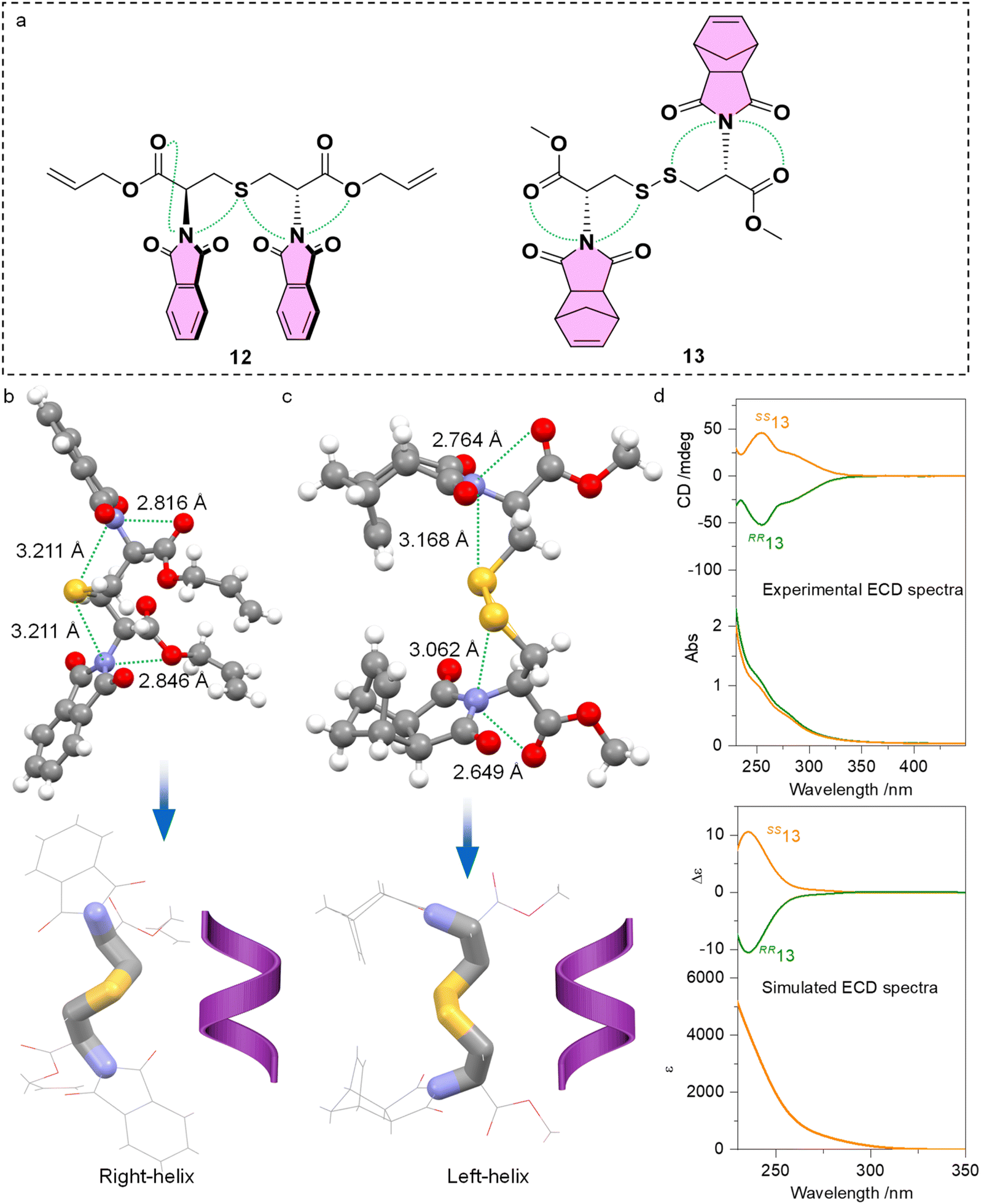 | ||
| Fig. 7 (a) Molecular structures of compounds 12 and 13, as well as their intramolecular PnB (dashed green lines). (b and c) X-ray structures of 12 and 13, and their folded chiral skeletons by N⋯O and N⋯S PnB. (d) Experimental and simulated ECD spectra of 13 in the solution phase (solvent: methanol, c = 0.4 mM). ECD was calculated at the B3LYP-D3/def2SVP level of theory. | ||
The formation of intramolecular hydrogen bonding involving imide would show the shift of CO bands to lower energies, which shall appear at 1656 cm−1.46 As shown in Fig. S28,† the CO stretching bands of compounds 1, 2, 5 and 8 could be found at above 1700 cm−1, indicative of the failed formation of H-bonded pseudocycles. In contrast, other compounds 6, 7, 9, 10 and 11 demonstrate CO stretching bands at 1650, 1653, 1659, 1652 and 1668 cm−1 respectively, which verify the formation of intramolecular hydrogen bonding, accompanied by the potential formation of PnB. We also carried out temperature-variable 1H NMR tests of compounds 1 and 10 for comparison in dichloroethane-d4. As shown in 1H NMR spectra (Fig. S29†), increasing temperature from 298 K to 333 K led to the shifts of hydroxyl protons as well as the aromatic protons, which shift to high fields and lower fields respectively. Compound 1 mainly forms PnB without an intramolecular hydrogen bond, while compound 10 mainly forms a hydrogen bond. Increasing temperature would disassociate both the hydrogen bond and PnB, causing a 0.183 ppm shift of the OH group for compound 1. This value is larger (0.269 ppm) in compound 10 due to the breakdown of hydrogen bonds, and thus the shifts of aromatic protons (0.03 ppm) are more prominent than that of compound 1 (0.013 ppm). The different shift values verify the dominant formation of PnB and hydrogen bonds in 1 and 10 respectively. Based on the single crystal X-ray results, though the PnB and hydrogen bonds might coexist in solutions, they could hardly coexist in solid states ascribed to the highly directional angle of PnB or hydrogen bonds.
Conclusions
In summary, this work via CSD searching and analysis, aided by computational studies, illustrates the general presence of PnB in imide derivatives. Besides occurring intermolecularly, intramolecular imide-involved PnBs are facilitated by short tethers between N and PnB acceptors. The relative high rotary energy barrier indicates a conformation-locked geometry, which provides hints for controlled molecular folding and fixed chirality. Chiroptical properties of representative examples were investigated, which reflected that PnB anchors the chiral geometry and enables chirality transfer from chiral centers to the skeleton. It also demonstrated that the directional PnBs induced the formation of supramolecular tilt chirality for aromatic moieties. Secondary structures such as β-sheets were also found. Specifically, cystine- and cysteine based derivatives were supported by quadruple PnB bonds which robustly anchor the conformation and improve the chirality transfer from cystine to the skeleton. Compounds 12 and 13 gave rise to a P- and M-helix respectively depending on the absolute chirality of cystine. This work, with an emphasis on the general existence of PnB, suggests its vital role in chiral folding and self-assembly with controllable chirality and chiroptical properties. It provides constructive thoughts for the rational design of imide-based chiral functional compounds and polymers.Experimental
Materials, experimental details, and additional CD, 1H NMR, and MS spectra can be found in the ESI.†Data availability
All the experimental data related to this study are available from the corresponding author upon reasonable request.Author contributions
Z. Wang and Z. Cao carried out the main experiments and data analysing. P. Xing and A. Hao proposed the assumption and wrote the paper. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was also supported by the National Natural Science Foundation of China (No. 21901145, 22171165, and 22371170). We also acknowledge the financial support from the Youth cross-scientific innovation group of Shandong University (2020QNQT003) and the project of construction and management research of laboratory of Shandong University (sy20202202). We thank Prof. Di Sun at Shandong University for assistance with the data collection of X-ray crystal structures.Notes and references
- J. Chen, Q. Peng, X. Peng, H. Zhang and H. Zeng, Chem. Rev., 2022, 122, 14594–14678 CrossRef CAS PubMed.
- L. E. Mackenzie and P. Stachelek, Nat. Chem., 2021, 13, 521–522 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed.
- K. E. Riley and P. Hobza, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 3–17 CAS.
- R. W. Newberry and R. T. Raines, ACS Chem. Biol., 2019, 14, 1677–1686 CrossRef CAS PubMed.
- (a) W. Zierkiewicz, M. Michalczyk and S. Scheiner, Molecules, 2021, 26, 1740 CrossRef CAS PubMed; (b) M. G. Walker, C. G. Mendez and P. S. Ho, Chem.–Asian J., 2023, 18, e202300026 CrossRef CAS PubMed.
- Z. Wang, A. Hao and P. Xing, Small, 2023, 19, 2302517 CrossRef CAS PubMed.
- Z. Zhao and Y. Wang, Acc. Chem. Res., 2023, 56, 608–621 CrossRef CAS PubMed.
- P. Weng, X. Yan, J. Cao, Z. Li and Y.-B. Jiang, Chem. Commun., 2022, 58, 6461–6464 RSC.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS PubMed.
- S. Zheng, J. Han, X. Jin, Q. Ye, J. Zhou, P. Duan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 22711–22716 CrossRef CAS PubMed.
- N. M. Kreienborg, F. Otte, C. Strohmann and C. Merten, Phys. Chem. Chem. Phys., 2023, 25, 15110–15114 RSC.
- L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed.
- Y. Xia, A. Hao and P. Xing, ACS Nano, 2023, 17, 21993–22003 CrossRef PubMed.
- K. Lisac, F. Topić, M. Arhangelskis, S. Cepić, P. A. Julien, C. W. Nickels, A. J. Morris, T. Friščić and D. Cinčić, Nat. Commun., 2019, 10, 61 CrossRef CAS PubMed.
- J. M. Hoffmann, A. K. Sadhoe and T. J. Mooibroek, Synthesis, 2020, 52, 521–528 CrossRef CAS.
- B. Lin, H. Liu, I. Karki, E. C. Vik, M. D. Smith, P. J. Pellechia and K. D. Shimizu, Angew. Chem., Int. Ed., 2023, 62, e202304960 CrossRef CAS PubMed.
- T. J. Mooibroek, CrystEngComm, 2017, 19, 4485–4488 RSC.
- J. Han, D. Yang, X. Jin, Y. Jiang, M. Liu and P. Duan, Angew. Chem., Int. Ed., 2019, 58, 7013–7019 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145–10243 CrossRef CAS PubMed.
- F. Wang, C. Shen, F. Gan, G. Zhang and H. Qiu, CCS Chem., 2023, 5, 1592–1601 CrossRef CAS.
- H. Wang, H. K. Bisoyi, B. X. Li, M. E. McConney, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2020, 59, 2684–2687 CrossRef CAS PubMed.
- X. Lin, B.-H. Kou, J. Cao, P. Weng, X. Yan, Z. Li and Y.-B. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202205914 CrossRef CAS PubMed.
- R. L. Sutar, E. Engelage, R. Stoll and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 6806–6810 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- J. Cao, X. Yan, W. He, X. Li, Z. Li, Y. Mo, M. Liu and Y.-B. Jiang, J. Am. Chem. Soc., 2017, 139, 6605–6610 CrossRef CAS PubMed.
- C. Raju, S. Kunnikuruvan and K. M. Sureshan, Angew. Chem., Int. Ed., 2022, 61, e202210453 CrossRef CAS PubMed.
- L. de Azevedo Santos, T. A. Hamlin, T. C. Ramalho and F. M. Bickelhaupt, Phys. Chem. Chem. Phys., 2021, 23, 13842–13852 RSC.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- J. D. Tovar, Acc. Chem. Res., 2013, 46, 1527–1537 CrossRef CAS PubMed.
- P. Moreau, F. Anizon, M. Sancelme, M. Prudhomme, C. Bailly, C. Carrasco, M. Ollier, D. Severe, J. F. Riou, D. Fabbro, T. Meyer and A. M. Aubertin, J. Med. Chem., 1998, 41, 1631–1640 CrossRef CAS PubMed.
- Z. Genene, W. Mammo, E. Wang and M. R. Andersson, Adv. Mater., 2019, 31, 1807275 CrossRef PubMed.
- B. M. Boyle, O. Heinz, G. M. Miyake and Y. Ding, Macromolecules, 2019, 52, 3426–3434 CrossRef CAS PubMed.
- (a) S. V. Bhosale, M. A. Kobaisi, R. W. Jadhav, P. P. Morajkar, L. A. Jones and S. George, Chem. Soc. Rev., 2021, 50, 9845–9998 RSC; (b) S. Chen, P. Slattum, C. Wang and L. Zang, Chem. Rev., 2015, 115, 11967–11998 CrossRef CAS PubMed.
- (a) S. K. Keshri, S. Kumar, K. Mandal and P. Mukhopadhyay, Chem.–Euro. J., 2017, 23, 11802–11809 CrossRef CAS PubMed; (b) K. Mandal, D. Bansal, Y. Kumar, Rustam, J. Shukla and P. Mukhopadhyay, Chem.–Euro. J., 2020, 26, 10607–10619 CrossRef CAS PubMed; (c) S. Kumar, J. Shukla, K. Mandal, Y. Kumar, R. Prakash, P. Ram and P. Mukhopadhyay, Chem. Sci., 2019, 10, 6482–6493 RSC.
- S. S. Parikh, G. Walcher, G. D. Jones, G. Slupphaug, H. E. Krokan, G. M. Blackburn and J. A. Tainer, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 5083–5088 CrossRef CAS PubMed.
- S. An, L. Gao, A. Hao and P. Xing, ACS Nano, 2021, 15, 20192–20202 CrossRef CAS PubMed.
- I. Hisaki, T. Sasaki, N. Tohnai and M. Miyata, Chem.–Euro. J., 2012, 18, 10066–10073 CrossRef CAS PubMed.
- Z. Wang, Y. Li, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2021, 60, 3138–3147 CrossRef CAS PubMed.
- Y. Wang, D. Niu, G. Ouyang and M. Liu, Nat. Commun., 2022, 13, 1710 CrossRef CAS PubMed.
- N. Ousaka, M. Itakura, A. Nagasaka, M. Ito, T. Hattori, D. Taura, T. Ikai and E. Yashima, J. Am. Chem. Soc., 2021, 143, 4346–4358 CrossRef CAS PubMed.
- M. Bursch, L. Kunze, A. M. Vibhute, A. Hansen, K. M. Sureshan, P. G. Jones, S. Grimme and D. B. Werz, Chem.–Euro. J., 2020, 27, 4627–4639 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. P. Walsh, J. A. Barclay, C. S. Begg, J. Xuan, N. T. Johnson, J. C. Cole and M. O. Kitching, JACS Au, 2022, 2, 2235–2250 CrossRef CAS PubMed.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092–6107 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2309600, 2309601, 2309599, 2309598, 2309602. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00554f |
| This journal is © The Royal Society of Chemistry 2024 |