
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural engineering of atomic catalysts for electrocatalysis
Tianmi
Tang
,
Xue
Bai
,
Zhenlu
Wang
 and
Jingqi
Guan
*
and
Jingqi
Guan
*
Institute of Physical Chemistry, College of Chemistry, Jilin University, Changchun 130021, PR China. E-mail: guanjq@jlu.edu.cn
First published on 5th March 2024
Abstract
As a burgeoning category of heterogeneous catalysts, atomic catalysts have been extensively researched in the field of electrocatalysis. To satisfy different electrocatalytic reactions, single-atom catalysts (SACs), diatomic catalysts (DACs) and triatomic catalysts (TACs) have been successfully designed and synthesized, in which microenvironment structure regulation is the core to achieve high-efficiency catalytic activity and selectivity. In this review, the effect of the geometric and electronic structure of metal active centers on catalytic performance is systematically introduced, including substrates, central metal atoms, and the coordination environment. Then theoretical understanding of atomic catalysts for electrocatalysis is innovatively discussed, including synergistic effects, defect coupled spin state change and crystal field distortion spin state change. In addition, we propose the challenges to optimize atomic catalysts for electrocatalysis applications, including controlled synthesis, increasing the density of active sites, enhancing intrinsic activity, and improving the stability. Moreover, the structure–function relationships of atomic catalysts in the CO2 reduction reaction, nitrogen reduction reaction, oxygen reduction reaction, hydrogen evolution reaction, and oxygen evolution reaction are highlighted. To facilitate the development of high-performance atomic catalysts, several technical challenges and research orientations are put forward.
1. Introduction
More than 80% of modern chemical industrial processes involve catalytic reactions, which have greatly promoted the development of modern industry.1 Therein, the wide application of electrocatalysis technology reduces the industrial dependence on fossil fuels and alleviates the environmental crisis, for instance, CO2 can be converted into chemicals with high added value through the electrocatalytic CO2 reduction reaction (CO2RR).2,3 Fuel cells and water splitting are new energy conversion technologies, in which no carbon-containing compounds are released and the core reactions are the oxygen reduction reaction (ORR), oxygen evolution reaction (OER) and hydrogen evolution reaction (HER).4–7 Ammonia synthesis by the Haber–Bosch process requires high temperature and high pressure, which can be achieved by the electrocatalytic nitrogen reduction reaction (NRR) at normal temperature and normal pressure. Moreover, the electrocatalytic NRR can be used to treat wastewater and reduce nitrogen pollution.8 However, the application of electrocatalysis to industry not only requires high catalytic activity and specific selectivity, but also needs to ensure economic benefit and high catalytic stability. The commonly used electrocatalysts are Pt, Au, Ru, Ir and other precious metals, but their small reserves and high price hinder the large-scale commercial applications.3,9 The development of efficient and economical transition metal-based catalysts is expected to promote the development of industrial electrocatalysis.By reducing the size of metal particles of metal-based catalysts from bulk to nanoscale, and further to the single-atom level, atomic dispersion of metal species is achieved. In 1998, Basset et al. modified Sn atoms around Pt atoms by surface organometallic chemistry (SOMC), showing nearly 100% selectivity for isobutane dehydrogenation.10 In addition, they used the Si–O bond on the surface of silicon oxide to fix single Zr atoms, which can be used for low-temperature hydrogenolysis of alkanes.11 In 2011, the concept of “single atom catalysis” was first proposed by Zhang et al.12 Single-atom catalysts (SACs) are a new catalytic family. The uniformly dispersed isolated metal atoms on the substrate have well-defined active centers and 100% atom utilization.13
As a new family of catalysts, isolated transition metal atoms in SACs anchored to specific substrates act as active sites. In addition to the central metal, the surrounding coordination environment affects the electronic structure and geometric configuration of single metal sites, and the catalytic activity of SACs is related to the strong force between metal centers and the substrate. Therefore, the electrocatalytic activity can be effectively improved by selecting appropriate central metal atoms and adjusting the local microenvironment of SACs.14,15 Diatomic catalysts (DACs) have isolated pairs of metal atoms as the active sites, in which pairs of the same metal are called homonuclear DACs, while pairs of different metals are called heteronuclear DACs. In catalytic reactions, the metal pairs synergistically participate in the reactions. DACs are an extension of SACs, which not only have the advantages of SACs, but also introduce another degree of freedom for adsorption regulation of reaction intermediates, providing more possibilities for adsorption. At the same time, the interaction between bimetal sites can effectively regulate the activation energy and adsorption energy of reaction intermediates.16 By further adjusting the number of central metal atoms, isolated active sites with three or four metal atoms called triatomic catalysts (TACs) or tetratomic catalysts can be obtained. The main advantage of such catalysts is the synergistic effect between multi-nuclear metals, providing a variety of reaction intermediates and transition states in catalytic reactions, searching for the optimal reaction path and the fastest reaction rate, which are more prominent for some complex multi-electron transfer processes.17,18 At present, there are relatively few reports about the application of multi-atomic catalysts in electrocatalysis, although the research on SACs and DACs has achieved great progress. The study of catalysts at an atomic level opens a door to explore the relationship between activity and structure.
With the deepening of research, the electrocatalytic activity of atomic catalysts can be improved by adjusting the coordination structure of metal sites. Müllen et al. synthesized a P and N double-doped Fe single-atom catalyst (P/Fe–N–C) by introducing P atoms into the second coordination layer of Fe–N4, which showed OER/ORR bifunctional activity. Theoretical calculations showed that P doping can adjust the *OOH/*O adsorption energy at the Fe–N4 site and accelerate the reactions.19 Liu et al. used a controlled “precursor preselection” strategy to synthesize a class of Cu and Co DACs with different coordination shells.20 When Cu and Co metals bond to S, they have lower valence states, which are conducive to the ORR. However, when S is in the second shell and is coordinated with N, the metal atoms show an intermediate equivalent state, which is conducive to the OER. Additionally, when S is doped on the base surface it is far away from the metal, which is in a high valence state and is favourable for the HER.
In addition to SACs and DACs, the synthesis and characterization of TACs can be experimentally realized. However, TACs are still in the development stage, since the more central the metal atoms, the greater the probability of metal agglomeration, and the greater the challenge of synthesis. TACs have great potential to catalyse the conversion of CO2 to C2 and C2+ products due to multi-metal site synergistic catalysis. Using theoretical calculations, Dou et al. demonstrated that spatially confined three-centered metal atoms can simultaneously fix a few molecules of carbon dioxide and have multiple reaction paths, which are spatially and electronically conducive to the coupling of C–C and C2–C to generate C2 and C3 products with high added-value.21 On M3@NG, the reaction steps of CO2 → *COOH → *CO → *CHO → *CH are similar to those on copper catalysts, and the speed control step is *CO → *CHO. Then *CH can be converted into *CH2 or *CH–CO, and the C–C coupling process of the latter can be carried out on the middle spatially confined trinuclear metal to form C2 or continue coupling to produce C3. This provides a new idea for designing novel electrocatalysts for CO2 reduction to C2+ products. Zhang et al. reported a trinuclear nickel catalyst (TNC-Ni), where the interaction among the three nickel sites plays a key role in charge accumulation and O–O bond formation during water oxidation.22
About 297 million tons of nitrogen are annually converted by biological nitrogen fixation, in which dimeric enzyme complexes (FeMo, FeV, or FeFe active sites) play a catalytic role, effectively reducing the reaction energy barrier. However, the slow kinetics limits high yields, and the stability and recyclability of catalysts are challenging.23–26 Inspired by natural nitrogen fixation, Ma et al. proposed a heteronuclear FeV@C2N diatomic electrocatalyst, which effectively inhibited competitive HER and had good NRR activity and selectivity.27 Dey et al. reported the synthesis of a trinuclear Ni complex (Ni3S8) using PhOH as a proton source, which selectively reduces N2 to N2H4.28
In this review, we summarize the recent progress of atomic catalysts in the electrocatalysis of the HER, OER, ORR, CO2RR and NRR, and a main development timeline of SACs, DACs and TACs is shown in Fig. 1. The regulation strategies of metal active sites are discussed, including reasonable design of substrates, regulating the electronic structure of central metal atoms and the coordination environment. Furthermore, the influencing factors of atomic catalysts for electrocatalysis are theoretically analysed and the strategy of optimizing the atomic catalyst is proposed. Finally, we put forward the current challenges and opportunities for future development of atomic catalysts to guide the reasonable design of multi-atomic catalysts in the future.
 | ||
| Fig. 1 History of catalysts with different nuclear numbers for electrocatalysis. Pt1/FeOx.12 Pt–MoS2. Reproduced with permission.29 Copyright 2015, The Royal Society of Chemistry. FeTPyP-Co. Reproduced with permission.30 Copyright 2016, American Chemical Society. Ru3/CN. Reproduced with permission.31 Copyright 2017, American Chemical Society. Co–N5/HNPCSs. Reproduced with permission.32 Copyright 2018, American Chemical Society. Ni/Fe–N–C. Reproduced with permission.33 Copyright 2019, Wiley-VCH. Se/Te-doped C catalysts. Reproduced with permission.34 Copyright 2020, Wiley-VCH. TNC-Cu. Reproduced with permission.35 Copyright 2021, American Chemical Society. RuCoNx/C. Reproduced with permission.36 Copyright 2022, American Chemical Society. Homonuclear and heteronuclear DACs. Reproduced with permission.37 Copyright 2023, American Chemical Society. CoMn–N/C. Reproduced with permission.38 Copyright 2023, Wiley-VCH GmbH. | ||
2. Reasonable control of the geometric and electronic structure of metal active sites
Geometric configuration and electronic structure are crucial for metal active sites towards advanced electrocatalysis.39 The geometric configuration reflects the coordination environment between the active metal and the surrounding atoms, and the electronic structure of the active site is closely related to the coordination environment. When the distance between two metal atoms is less than 1.0 nm, the two metal atoms will interact and affect the distribution of electrons.40 The selection and modification of the substrate also play an important role in the geometric configuration and electronic structure of central metal atoms, and the selection of special position on the substrate is beneficial to the formation of a unique electronic structure. The active center is composed of metal and coordination atoms, and the interaction between metal and metal/non-metal will cause charge rearrangement, and therefore, reasonable selection of the number and type of metal is very important to design catalysts with different electrocatalytic activities.41 In this section, we first introduce the rational design of atomic catalysts with different numbers of metal nuclei through defect engineering and spatial limiting, and focus on how to regulate the central metal atoms and the coordination environment.2.1. Reasonable design of substrates
 | ||
| Fig. 2 (a–c) High angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) images of A-Ni@DG. (d–f) Illustrations of three different types of catalytic active sites corresponding to a single Ni atom supported on (d) perfect hexagons, (e) D5775, and (f) di-vacancy. (g and h) Energy profiles of three configurations (d–f) for the HER (g) and OER (h). Reproduced with permission.45 Copyright 2018, Elsevier. | ||
Xu et al. reported a carbon-supported defect-anchored Pt SAC.46 The ORR on the Pt SAC is a four-electron reaction with a high half-wave potential (0.835 V vs. RHE). Theoretical calculations showed that the Pt–C4 site with two carbon vacancies is the active center of the ORR. Wang et al. constructed defect sites on TiO2 nanosheets to stabilize single-atom Au sites, which can improve catalytic activity by reducing the energy barrier and competitive adsorption at isolated Au atomic sites.47 Chen et al. prepared a catalyst with a copper atomic pair (Cu01–Cu1x+) that was stabilized by Te surface defects on Pd10Te3 alloy nanowires. High FE (FECO = 92%) was shown for the CO2RR, and the HER was almost completely suppressed.48 Theoretical calculations showed that the Cu01–Cu1x+ site can promote CO2 activation by the “diatomic activation bimolecular mechanism”. Yao et al. reported a locally distributed atomic Pt–Co nitrocarbon-based catalyst (A-CoPt-NC) with 267 times higher mass ORR activity than commercial Pt/C.49 DFT calculations showed that the high ORR activity is due to the synergistic interaction between Pt–Co atom pairs and the surrounding defective coordination environment, and the electron distribution of the Pt–Co active center is asymmetrical.
The introduction of nonmetallic heteroatoms such as P or S into the carbon carrier will cause the change of charge density of active sites due to the mismatch between the heteroatoms and the outer orbitals of carbon, thus regulating the electrocatalytic performance.50,51 Deng et al. synthesized an N,P codoped FeNi-NPC catalyst with diatomic Fe–Ni sites, where P does not coordinate with the diatomic Fe–Ni site.52 By adjusting the microenvironment and designing the structure, the catalytic OER/ORR activities of FeNi-NPC were effectively improved. Zou et al. synthesized a S-doped Mo single atom/carbon multichannel fiber catalyst (MoSA/CMF-S) using template assisted electrospinning.53 The catalyst catalyzed the NRR to generate NH3 with a yield of 46.6 μg h−1 mgcat−1 and FE of 28.9% at −0.2 V. Theoretical calculations showed that the catalytic active site of MoSA/CMF-S is Mo1N1C2–S4, where S does not coordinate with a Mo atom. Bader analysis showed that S doping makes the charge of the Mo atom rearrange. By comparing the charge of Mo in Mo1N1C2–S4 and Mo1N1C2, the charge of the Mo atom in the former increases more than that in the latter, and a charge accumulates between S and Mo in the Mo1N1C2–S4. In addition, HER kinetics was slow at the Mo1N1C2–S4, indicating that S doping effectively inhibited the HER. The adsorption energy of N2 on the side-on (–0.87 eV) and end-on (–0.82 eV) orientations of a single Mo atom at the active site was calculated. The adsorption energy of N2 at the Mo1N1C2–S4 site was −0.88 eV, which is stronger than that at Mo1N1C2 (−0.79 eV). In addition, the speed control step at the Mo1N1C2–S4 (0.58 eV) is easier to overcome than that at Mo1N1C2 (0.81 eV) (Fig. 3). In short, the introduction of S rearranges the electrons of single-atom Mo, which is conducive to the adsorption of N2 and effectively inhibits HER activity.
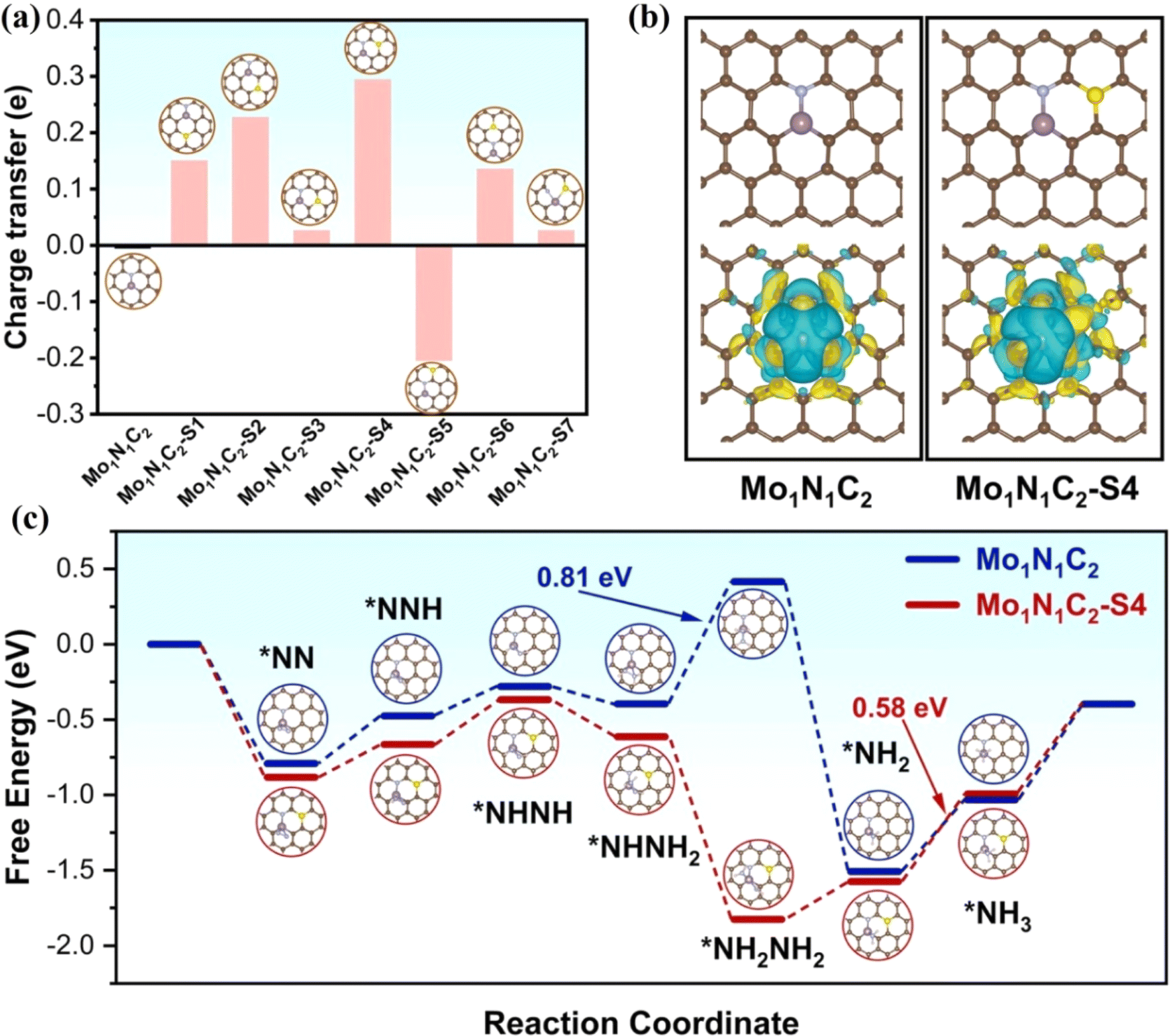 | ||
| Fig. 3 (a) Bader charge variations of the Mo atom in different S-doped samples. (b) Optimized geometries and contour plots of the charge density differences. (c) Gibbs free energy diagram of enzymatic NRR. Reproduced with permission.53 Copyright 2022, Elsevier B.V. | ||
Li et al. used cage encapsulation and pyrolysis strategies to obtain a single-atom iron catalyst with good ORR activity, exhibiting a half-wave potential of 0.9 V in 0.1 M KOH and good stability.61 DFT calculations showed that the good ORR activity is attributable to the formation of single-atom Fe, which coordinates with four N atoms and is axially adsorbed with O2 (Fig. 4a). Li et al. packaged a W precursor into a MOF and pyrolyzed it to obtain a W single-atom catalyst with a local structure of W1N1C3.62 The catalyst showed good HER activity with an overpotential of 85 mV at 10 mA cm−2 (η10) in 0.1 M KOH. Huang et al. prepared an ultra-stable bimetallic phthalocyanine COF (Fig. 4b).63 The overlap stacking mode of metal phthalocyanine units is conducive to electron transfer and improves the FE of CO2 conversion to CO (FE = 97%). Niu et al. used encapsulation and pyrolysis strategies to construct a series of diatomic catalysts, including homo-nuclear (Fe2, Co2, Ni2, Cu2, Mn2 and Pd2) and hetero-nuclear (Fe–Cu, Fe–Ni, Cu–Mn and Cu–Co) DACs.37 After pyrolysis, the molecular skeleton of macrocyclic complexes can be preserved (Fig. 4c). Taking Fe–Cu DAC as an example, it showed good ORR activity (E1/2 = 0.793 V) and stability. Besides the above two atomic confinement methods, Zhang et al. designed a click-limiting strategy to disperse cobalt-coordinated porphyrin units onto the substrate to obtain a Co–N–C electrocatalyst, which showed excellent OER/ORR activities (ΔE = 0.79 V) (Fig. 4d–f).64
 | ||
| Fig. 4 (a) Schematic illustration of the formation of Fe-ISAs/CN. (b) Top views of a graphical representation of the eclipsed stacking of CuPcF8-CoNPc-COF. (c) Schematic illustration of the synthesis of DACs via a macrocyclic precursor-mediated encapsulation–pyrolysis process. (d–f) Different confinement strategies for fabricating M–N–C SACs. (a) Reproduced with permission.61 Copyright 2017, Wiley-VCH. (b) Reproduced with permission.63 Copyright 2021, American Chemical Society. (c) Reproduced with permission.37 Copyright 2023, American Chemical Society. (d–f) Reproduced with permission.64 Copyright 2022, American Association for the Advancement of Science. | ||
2.2. Regulating the electronic structure of central metal atoms
The electronic structure of central metal atoms plays an important role in optimizing the physical and chemical properties of catalysts. The central metal atom determines the energy level of the d orbital, the number of d electrons and the electron configuration. The distance between metal centers and the combination of metal centers are important factors affecting chemical reactions.65–67 Single, double and triple metal atoms with isolation are obtained by adjusting the distance of metal centers, namely SACs, DACs and TACs, respectively (Fig. 5). The charge distribution between the active sites is optimized by adjusting metal combination, thereby changing the electronic properties of the metal atoms. The electronic structure of the central metal atom determines the atomic charge distribution and Fermi level, thus influencing the adsorption/desorption energy of reaction intermediates.68 In this section, we will discuss how to adjust the electronic structure of the central metal atoms of SACs, DACs and TACs. | ||
| Fig. 5 (a–c) Typical structures of SACs, DACs and TACs. | ||
Hydrogen production by electrolysis of water is a green and sustainable method, which is expected to replace fossil fuels in the future. Precious metal platinum-, palladium-, and iridium-based catalysts play an important role in water electrolysis. However, Pt, Pd and Ir have small reserves and high costs, which are obstacles in large-scale electrocatalytic hydrogen production.69 At present, ideal electrocatalysts can be developed by two strategies, one is to prepare SACs with fewer precious metals, and the other is to develop non-precious metal catalysts.70,71 In the HER, there are three basic steps: Tafel, Heyrovsky, and Volmer, where the Volmer step is the fastest and Tafel is the rate-determining step. The formation and desorption processes of the reaction intermediate H* affect the reaction mechanism and reaction rate. According to Sabatier's principle, when the hydrogen adsorption free energy GH* is close to 0, the HER is the most favourable to occur. Under alkaline conditions, the activity of the HER is also affected by the kinetic energy barrier of water splitting and the energy barrier of hydroxyl desorption.72
For the HER, Pt-based catalysts are the most active electrocatalysts. Lee et al. loaded Pt onto low-oxide tungsten (Pt SA/WO3−x), which greatly improved the utilization of Pt, and demonstrated that Pt SA/WO3−x was significantly superior to WO3−x loaded with Pt nanoparticles (Pt NP/WO3−x).73 At an overpotential of 50 mV, the mass activity of Pt SA/WO3−x was 10.7 times that of Pt NP/WO3−x. In practical applications, pH has a great effect on HER activity. Single-atom Pt catalysts show good performance under different pH values. Tan et al. modified Pt atoms into nanoporous Co0.85Se (Pt/np-Co0.85Se), and the catalyst showed excellent HER activity under neutral conditions, with an initial potential close to 0 and a Tafel slope of 35 mV dec−1.74 Operando XAS and theoretical calculations showed that Pt in Pt/np-Co0.85Se can not only regulate the surface state of the active center, but also decrease the ΔG of the HER, optimize the hydrogen adsorption/desorption energy, and promote thermodynamics and kinetics.
Compared with acidic conditions, the HER rate and apparent activity of catalysts is slower under alkaline conditions, and thus it is relatively difficult to design atomic catalysts that can catalyse the HER under alkaline conditions. The π-electron hybridization at the edge of the carrier graphene is used to influence the electronic state of the Pt 5d orbital. Amal et al. used edge-rich vertical graphene to anchor Pt, and Pt coordinated with the graphene edge showed excellent HER activity with a turnover frequency (TOF) up to 22.6 s−1 at η = 150 mV.75
The anchoring of single-atom platinum to a suitable carrier can facilitate the HER due to the optimization of metal–carrier interaction and water adsorption. Jiang et al. anchored single-atom Pt to α-MoC1−x@C (PtSA/α-MoC1−x@C), which showed high activity and stability for the pH-universal HER.76 The η10 of PtSA/α-MoC1−x@C under alkaline and acidic conditions is 21 mV and 12 mV, respectively. X-ray photoelectron spectrometry (XPS) and XAS analysis indicated that Pt in PtSA/α-MoC1−x@C is present in a low state, which is caused by electron migration from Mo to Pt. Experimental results showed that platinum atoms are monodispersed in the α-MoC1−x@C lattice. DFT analysis showed that the ΔG of hydrogen adsorption decreases significantly (0.60 eV → 0.21 eV) with the construction of the Pt unit point under acidic conditions, which is favourable for hydrogen adsorption/desorption. Under neutral and alkaline conditions, α-MoC1−x-Pt is beneficial to water adsorption, promotes the capture of reactants, and accelerates the HER. In addition, the hydroxide adsorption free energy is reduced on α-MoC1−x–Pt, and the interaction between α-MoC1−x and Pt atoms optimizes the adsorption/desorption energy of reaction intermediates. Thus, PtSA/α-MoC1−x@C showed pH-universal HER activity (Fig. 6).
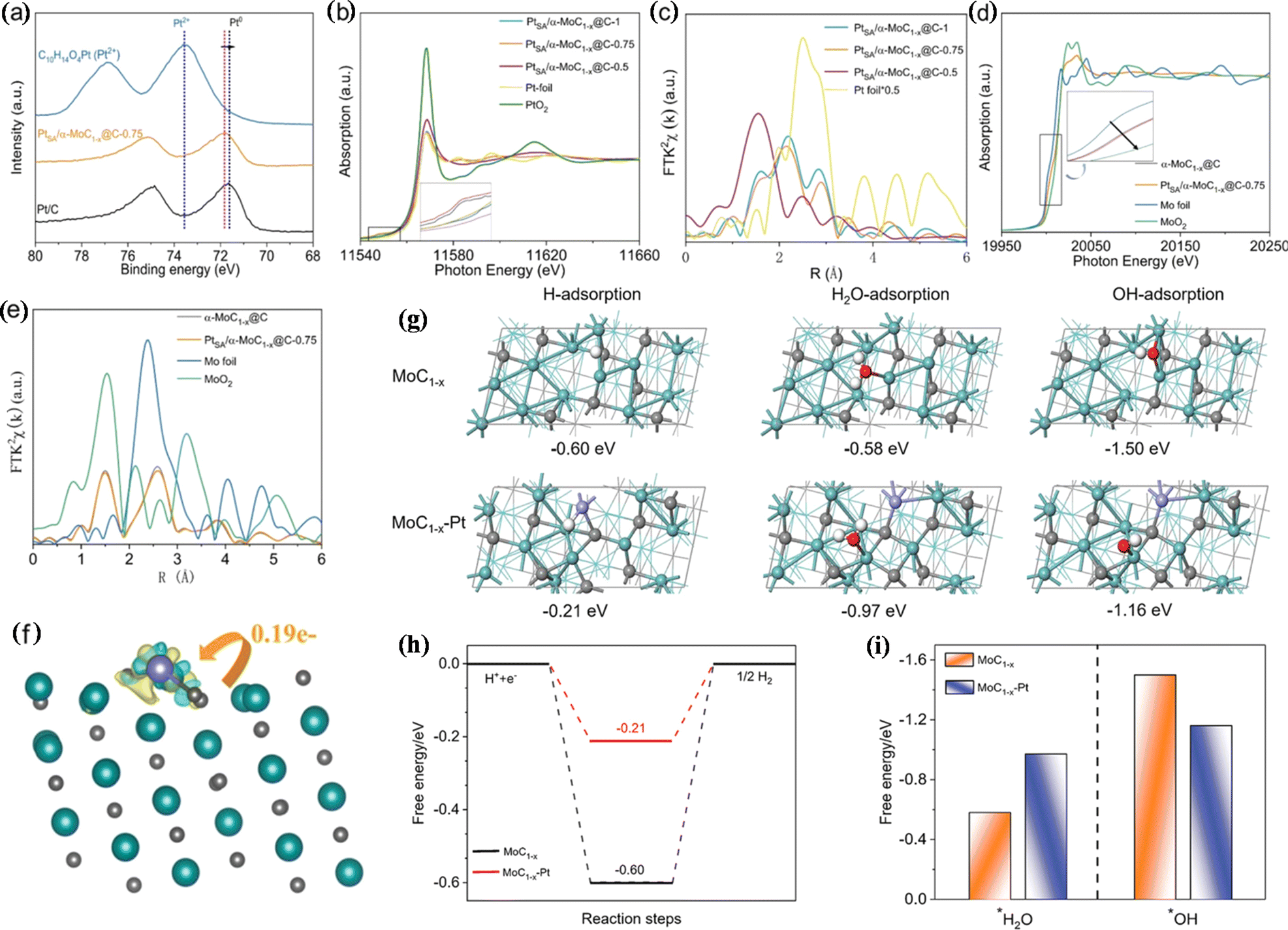 | ||
| Fig. 6 (a) Pt 4f XPS spectra. (b) Pt L3-edge XANES spectra. (c) Fourier transform X-ray absorption fine structure (FT-EXAFS) curves. (d) Mo K-edge X-ray absorption near edge structure (XANES) spectra. (e) FT-EXAFS curves of the Mo K-edge. (f) Differential charge density for Pt1/α-MoC1−x. (g) Optimized adsorption geometries. (h) Gibbs free energy diagram. (i) Calculated adsorption free energies. Reproduced with permission.76 Copyright 2021, Wiley-VCH GmbH, Weinheim. | ||
In addition to the precious metal Pt, transition metal cobalt and nickel-based catalysts are used to catalyse the HER in acidic environments.77–80 Studies have shown that transition metal carbides have a low energy barrier in hydrogen adsorption, and the density of states near the Fermi level is similar to that of precious metals. Nickel carbide (NiC) shows stability in a wide pH range. By introducing N into NiC, the electronic structure of Ni can be adjusted to improve the HER activity. In addition, the conductivity of the catalyst can be improved by reducing its size. Sun et al. prepared hybrid Ni–C–N nanosheets with thickness smaller than 2 nm.81 Ni–C–N is chemically stable and has metal conductivity, which showed good HER activity in 0.5 M H2SO4, 1.0 M KOH and 1.0 M PBS (pH = 7). Yu et al. prepared nitrogen-doped porous nanofibers with Fe–N5 sites (Fe SA/PNCNFs-0.1) by a wet impregnation method.79 The catalyst showed a high catalytic HER activity with an η10 of 44.3 mV and Tafel slope of 45.4 mV dec−1. However, there are still some issues in transition metal catalysts catalysing the HER at full pH, because the process will cause the change of proton concentration. To make water splitting more energy-saving, the ideal state should show excellent activity at any pH.
The OER is a complex four-electron process whose reactivity determines the efficiency of hydrogen production by water electrolysis.82 At present, the most advanced OER electrocatalysts are IrO2 and RuO2, but they have poor stability in alkaline media.83 SACs have attracted much attention because of their unique electronic structure.84 By anchoring precious metals to supports, SACs can be prepared to reduce the use of precious metals while ensuring high OER activity. Huang et al. prepared single-atom Rh-doped CuO nanowires on a copper foam (Rh SAC-CuO NAs/CF) using a cation exchange strategy, which exhibited an η10 of 357 mV for the OER.85 Theoretical calculations showed that the introduction of Rh atoms into the CuO lattice can effectively reduce the reaction energy barrier and accelerate the kinetics. Gu et al. synthesized a single-atom iridium-doped nickel phosphide catalyst (IrSA-Ni2P) using a top-down phosphating strategy, which showed excellent OER activity with an η10 of 149 mV.86 Simulated results of IrSA-Ni12P5 and IrSA-Ni2P nanospheres showed that iridium atoms replace the outermost nickel and occupy the Ni sites in the NixPy (Ni2P and Ni12P5) matrix (Fig. 7).
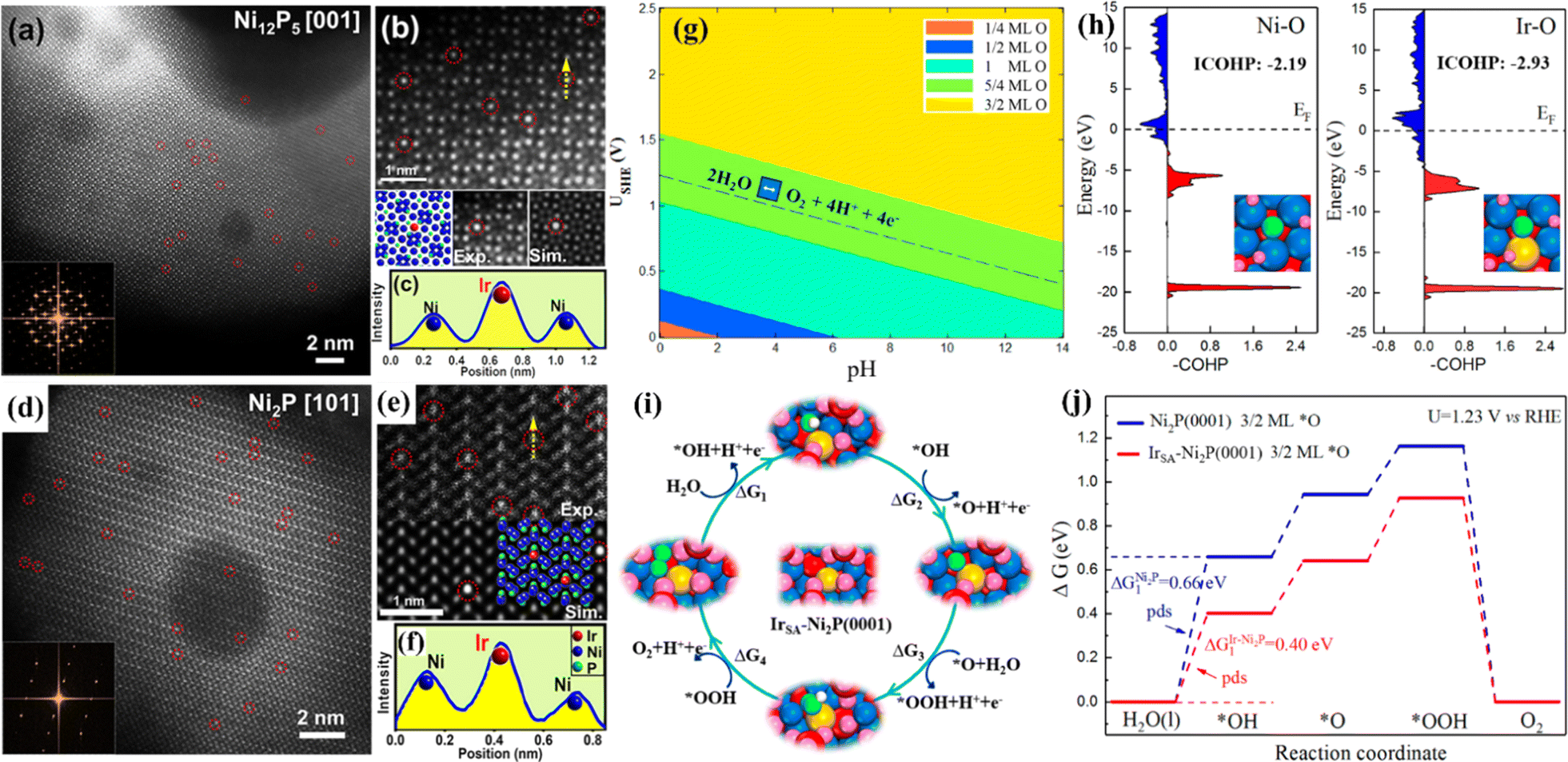 | ||
| Fig. 7 (a and d) HAADF-STEM images and corresponding FFT (inset). (b and e) Experimental and simulated HAADF-STEM images. (c and f) Intensity profile of the yellow line in (b and e) with the matrix Ni column and the decorated Ir atom. (g) Surface Pourbaix diagram of a single Ir atom doped Ni2P (0001) surface (IrSA-Ni2P (0001)). (h) Projected crystal orbital Hamilton populations (COHP) between chemisorbed *O and active metal centers on Ni2P (0001) and IrSA-Ni2P (0001). (i) The optimized OER intermediates on IrSA-Ni2P (0001). (j) Free energy diagrams. Reproduced with permission.86 Copyright 2021, American Chemical Society. | ||
In addition, the abundant and inexpensive transition metals Mn, Fe, Co and Ni are often used as active centers of SACs for the OER.87 Kibria et al. synthesized a cobalt-anchoring nitrogen-rich carbon single-atom catalyst (Co SAC) with a Co loading content of 10.6 wt% using a macromolecular assisted strategy.88 The Co SAC had an OER η10 of 351 mV under alkaline conditions and a mass activity of 2209 mA at 1.65 V, and showed tolerance for up to 300 h. According to operando XAS analysis, the pyridine nitrogen in a cobalt phthalocyanine tetramer (CoPc) with melem (CoMM) was suitable for the OER and had good reproducibility. Close Co–Co atomic spacing (∼3.85 A) and eg packing can increase OER activity. In addition, theoretical calculations showed that electron-deficient Co–O coordination intermediates are easily formed to accelerate the reaction kinetics, and the Co–N4 coordination structure can effectively reduce the reaction energy barrier and increase the OER activity (Fig. 8). According to the Sabatier principle, a good catalyst should optimize the adsorption energy of reaction intermediates. The OER involves adsorption and desorption of three reaction intermediates (*OH, *O and *OOH). Therefore, for a catalyst with high OER activity, the binding strength of the three intermediates to the active site should be moderate. Therefore, regulating the electronic structure and coordination environment of the active sites plays an important role in improving OER activity.89–91
 | ||
| Fig. 8 (a) Schematics of operando XAS analysis of CoMM. (b) Chemical state change of CoMM during the OER. (c) OER free energy profile of pyridinic-nitrogen–cobalt and pyrrolic-nitrogen–cobalt single-atom models. (d) Projected density of states (PDOS) on the Co atom for both pyridinic-nitrogen–cobalt and pyrrolic-nitrogen–cobalt single-atom models. (e) Top view and (f) side view of *OH. (g) Top view and (h) side view of *O. Reproduced with permission.88 Copyright 2023, American Chemical Society. | ||
The ORR plays an important role in energy storage and chemical production, such as in fuel cells, metal air batteries and hydrogen peroxide synthesis. However, these devices are faced with the problems of low ORR efficiency and slow dynamics.92–96 According to the volcanic curve (Fig. 9a), metals with slightly lower oxygen binding energy than platinum have higher oxygen reduction rate, and transition metals Fe, Co, Ni and Cu exhibit weaker binding energy of reaction intermediates. Anchoring them in the form of single atoms into conductive supports can promote the inherent ORR activity.97,98 Zhang et al. synthesized an iron-doped mesoporous nitrogen-doped carbon material (Fe SAC-MIL101-T).99 When the pyrolysis temperature was 1000 °C, the ORR activity was the best, and the half-wave potential was 0.94 V. The excellent ORR activity was attributed to the high dispersion of FeNx active centers and abundant pore structure, which was conducive to mass transfer and improved the utilization rate of FeNx active sites.
 | ||
| Fig. 9 (a) Trends in oxygen reduction activity plotted as a function of the oxygen binding energy. (b) The normalized K-edge XANES spectra and (c) FT-EXAFS spectra. (d) FT-EXAFS fitting curve of Co–N–C in R space and (e) wavelet transform (WT) for the k3-weighted EXAFS signals. (f) ΔG diagram. (g) Kinetic energy barriers. (h) ΔG diagram of the two-electron transfer ORR pathway. (i) PDOS in Co–N5 near the Fermi level (Ef). (j) PDOS in Co–N4 near Ef. Reproduced with permission.97 (a) Copyright 2004, American Chemical Society. (b–j) Reproduced with permission.105 Copyright 2021, The Royal Society of Chemistry. | ||
The strong binding of Co-based SACs with oxygen can promote a two-electron process in the ORR, which is conducive to the generation of H2O2. Co-based SACs with Co–N4 structure like cobalt phthalocyanine and cobalt porphyrin are conducive to the 4e− transfer process. However, when the cobalt is coordinated with five nitrogen atoms (Co–N5), it shows high selectivity of H2O2.100–104 Shao et al. used a coordination engineering strategy to prepare a Co single-atom catalyst with Co–N5 sites (Co SAC), which had a good selectivity for H2O2 in the ORR.105 The Co SAC demonstrated the two-electron process of the ORR in seawater with fast kinetics and chloride ion tolerance. The FE of H2O2 in 0.5 M NaCl solution with pH = 7 is as high as 95.6%. Theoretical calculation results showed that the ORR barrier of the two-electron pathway on the Co–N5 is lower than that of the four-electron transfer process, and the Co–N5 is more conducive to the activation and reduction of *O2 than Co–N4 (Fig. 9b–j).
In the CO2RR, most metal-based SACs can reduce CO2 to CO, and Sn- and Bi-based SACs can convert CO2 to HCOOH, while Cu-based catalysts with moderate adsorption strength of CO2 can reduce CO2 to C1, C2 and C2+ products.106–113 Cu is the only metal reported to be able to convert CO2 into C2 and C2+ products with high added value.14,114 Chen et al. studied a series of TM-phthalocyanine (TM-Pcs, TM = Fe, Co, Ni, Cu, and Zn) crystal molecular catalysts with TM-N4 active sites for the CO2RR and revealed that the binding strength of the metal to reaction intermediates determines the activity of the CO2RR.115
Shen et al. synthesized a series of M–N–C SACs (M = Fe, CO, Ni and Cu) by a leaching method.116 The FE of CO on Ni–N–C reached up to 97%, and the activity of the CO2RR was affected by the d-orbital electron configuration of the central metal ion. As shown in Fig. 10a–h, during the activation process, CO2 interacts with the active site, and electrons are transferred from CO2 to the active center metal, and then fed back to the π orbital of CO2. Ni–N–C has higher electron density around Ni than other M–N–C because the outermost d orbital of Ni2+ is empty, which is conducive to the transfer of electrons from the C atom in the CO2 molecule to the outermost orbital of the Ni atom and promotes the progress of the CO2RR. Theoretical calculations showed that the rate-determining step of Ni–N–C is the formation of the *COOH intermediate, and the energy barrier of the rate-determining step on Fe–N–C, Co–N–C and Cu–N–C in the CO2RR is greater than that in the HER, which is not conducive to the progress of the CO2RR (Fig. 10i and j). Crystal field theory showed that the outermost space orbital of Ni2+ in the Ni–N–C is beneficial to the transfer of electrons from CO2 to the active site Ni, effectively activating CO2 molecules adsorbed on the surface.
 | ||
| Fig. 10 Schematics of d-orbital electronic configurations of central metals in (a) Fe–N–C, (b) Co–N–C, (c) Ni–N–C, and (d) Cu–N–C and their electronic interaction with CO2 molecules. Side-view differential charge density maps of (e) Fe–N–C, (f) Co–N–C, (g) Ni–N–C, and (h) Cu–N–C. The blue and red regions represent net electron accumulation and depletion, respectively. Reaction pathways and free energy diagrams of (i) the CO2RR and (j) HER over M–N–C SACs at 0 V versus RHE. Reproduced with permission.116 Copyright 2023, American Chemical Society. | ||
Cu-based catalysts can induce CO* dimerization in the CO2RR to obtain C2 and C2+ products. The activity and selectivity can be improved by modifying the surface/interface of catalysts, such as adjusting the morphology, ligand modification, vacancy fabrication and doping. Han et al. applied a site-specific underpotential electrochemical deposition (UPD) strategy to treat single-atom Cu-anchoring Ag2S/Ag nanowires.117 Cu/Ag(S) was synthesized through single atom Cu occupying the defect position after sulfur removal. The FE of C2+ on Cu/Ag(S) is significantly higher than that on the Ag(S) catalyst without single-atom Cu, and Cu/Ag(S) showed the maximum FE (17%) at 1.4 V. Theoretical calculations showed that the interaction between Cu and Ag on the surface defects causes the d-band center to shift down, reduces the energy barrier of *CO formation (∼0.5 eV), promotes the adsorption of reaction intermediates, and improves the selectivity of C2+.
Ammonia (NH3) is an important energy carrier and an important raw material for the synthesis of agricultural fertilizers. At present, NH3 is industrially produced by the Haber–Bosch method, which needs high temperature and pressure. The conditions are harsh and the energy input is not in line with the trend of green and low-carbon development. The electrocatalytic NRR has attracted much attention because of its low energy consumption and mild reaction conditions. However, the NRR is a six-electron-proton transfer process with complex intermediates and slow kinetics. In addition, side reactions can occur through a two-electron pathway to diazene (N2H2) and four-electron pathway to hydrazine (N2H4). Therefore, in recent years, the development of SACs with high activity and selectivity for the electrocatalytic NRR has been on the rise.118,119
Since the active metal center of the natural nitrogenase is Mo, it has been inferred that single-atom Mo catalysts might be efficient for the NRR. Xin et al. synthesized a single-atom Mo-anchoring nitrogen-doped porous carbon material (SA-Mo/NPC), showing an FENH3 of 14.6 ± 1.6%.120 NH3 yields reached up to 34.0 ± 3.6 μg h−1 mgcat.−1, and SA-Mo/NPC was well tolerated in the NRR (Fig. 11a–g). Zhang et al. simulated a monatomic Mo-anchoring C9N4 catalyst (Mo@C9N4) for the NRR.121 The results showed that the Mo atom is the active center of the NRR, and N2 can be efficiently converted to NH3 on the surface of Mo@C9N4. By comparing the remote and alternate pathways, the energy barrier (0.40 V) of the rate control step of *N2 → *N2H in the remote pathway is lower than that (0.70 V) of the rate control step of *NNH → *NHNH in the alternate pathway. Through the analysis of the bond length and charge changes in each step of the NRR, it was concluded that single-atom Mo serves as both the reservoir and the electron transfer transmitter between the intermediate *NxHy and the carrier C9N4 in the NRR process (Fig. 11h–k).
 | ||
| Fig. 11 (a) Illustration of SA-Mo/NPC. (b) Mo EDS mapping. (c) HAADF-STEM image. (d) LSV curves. (e) NH3 yield rate and FE at each given potential. (f) NH3 yield rates with different Mo loadings. (g) Chronoamperometric curve and FE stability. (h) Mo–N bond length during the NRR process. (i) Charge variation of a single Mo atom during the NRR process. Charge variation of the three moieties (C9N4, single Mo atom and the adsorbed NxHy species) along (j) distal and (k) alternating pathways. (a–g) Reproduced with permission.120 Copyright 2019, Wiley-VCH. (h–k) Reproduced with permission.121 Copyright 2020, Elsevier B.V. | ||
2.2.2.1. Homonuclear DACs. Homonuclear DACs can be obtained by increasing metal loadings. Compared with SACs, the active sites of DACs increase due to the increase in the metal loadings.122 In addition, the interaction between homonuclear metals can regulate the electronic structure of active centers, which is conducive to reducing the reaction energy barrier. Zhou et al. constructed a diatomic Ag/graphene with AgN3–AgN3 structure (Ag2–G), which showed good activity in catalysing the conversion of CO2 to CO, with an FE of up to 93.4%.123 Theoretical calculations showed that the AgN3–AgN3 active site has a lower energy barrier of *COOH formation than single-site Ag1–G. Wang et al. reported a diatomic Pd catalyst (Pd2 DAC) for the CO2RR, exhibiting an FECO of 98.2%.124 DFT calculations unveiled that the enhanced activity of the CO2RR is due to the electron transfer between Pd and Pd, optimizing the adsorption strength of the reaction intermediate CO*. Quan et al. synthesized a nitrogen-coordinated Fe DAC (Fe2NPC) using Fe2(CO)9 as the Fe source, exhibiting an FECO of 96.0%.125 XAS characterization showed that the Fe2 dimer was coordinated with six N atoms (Fe2N6) (Fig. 12a–c). Theoretical calculations showed that CO2 → *CO2 is an exothermic process (−0.03 eV) at Fe2N6, while an energy barrier of +0.59 eV needs to be overcome at Fe–N4. In addition, another CO2 can be adsorbed at the Fe2N6 site to form a simultaneous adsorption of *CO and *CO2, which is subsequently converted into a *CO and *CO configuration, and the desorption of CO requires only 0.20 eV. The maximum energy barrier of CO2 to CO at the Fe2N6 site is 0.90 eV (Fig. 12d). The desorption temperature of CO2 at the Fe2N6 site is also higher than that of Fe–N4 (Fig. 12e). The synergistic effect of the Fe2 dimer is conducive to the adsorption of CO2 and the desorption of CO.
 | ||
| Fig. 12 (a) FT-EXAFS spectra at the Fe K-edge. (b) Corresponding EXAFS R-space fitting curves and (c) WT of Fe2NPC. (d) Calculated free energy. (e) CO2-TPD curves. Reproduced with permission.125 Copyright 2022, American Chemical Society. | ||
2.2.2.2. Heteronuclear DACs. The synergistic effect of bimetallic active sites can break the linear proportional relationship of single atomic sites, which is conducive to catalysing complex multi-electron reactions and improving the stability.126 The electron coupling of adjacent metal sites can optimize the adsorption energy and adsorption mode of reaction intermediates, reduce the energy barrier and accelerate the reaction kinetics. Due to the unique electronic structure of DACs, great progress has been made in electrocatalytic reactions such as the HER, OER, ORR, CO2RR and NRR.9,127
The development of high-performance DACs is expected to replace platinum for the HER. Fan et al. prepared a W1Mo1-NG DAC with W–O–Mo–O–C structure by combining a hydrothermal method and chemical vapor deposition (CVD) technology.128 The W and Mo diatomic structure is the active center of the HER and has high stability. Theoretical calculations showed that the electron delocalization of the W–O–Mo–O–C configuration provided the best adsorption strength for H and accelerated the kinetics of the HER. Hu et al. used atomic layer deposition to anchor Pt to Ni and N co-doped carbon (PtNi-NC), exhibiting an η10 of 30 mV for the HER in an acidic medium.129 XAS characterization indicated the presence of a Pt–Ni dimer in the PtNi-NC. DFT calculations showed that Pt–Ni is beneficial to regulate adsorption energy and increase catalytic activity by regulating the local electronic structure and charge. Liu et al. studied cobalt-based DACs (Co–M DACs) for the HER and found that the activity of DACs was significantly better than that of a single-atom Co catalyst.130 Theoretical calculations showed that OH is more easily adsorbed on the CoFe DAC, promoting hydrogen evolution under alkaline conditions.
The ORR and OER are the core reactions of various energy conversion devices, and their main challenge is the complex four-electron transfer process with a high overpotential. Fu et al. used a plasma engineering method to immobilize an Fe–Co dimer onto mesoporous nitrogen-doped carbon nanotubes (Fe, Co SAs-PNCF) with metal loading of up to 9.8 wt%.131 Fe, Co SAs-PNCF showed good ORR activity (Eonset = 1.04 V and E1/2 = 0.93 V). XAS analysis revealed that the active center of the Fe, Co SAs-PNCF catalyst is the N3–Fe–Co–N3 structure. In situ XAS and Raman spectroscopy were used to study the changes of oxidation states of Fe and Co and the bond length of the N3–Fe–Co–N3 structure during the ORR. The Raman peak intensities of CoOOH/FeOOH and Fe(OH)2/Co(OH)2 changed with increasing bias, and oxygen intermediates on the catalyst surface increased continuously, indicating that Fe/Co species are the active sites (Fig. 13a–c). In situ XAS results showed that the oxidation states of Fe and Co become smaller and Fe/Co–N bonds become longer during the ORR process at lower voltages (Fig. 13d–f), indicating that the N3–Fe–Co–N3 structure is the active center of the ORR. Theoretical calculations were further used to reveal the mechanism of the ORR. The limiting potential (UL) of Fe, Co SAs–V (0.89 V) is higher than that of Fe, Co SAs (0.81 V) and NCF (0.278 V). The spin orbit of Fe, Co SAs-V is closer to the Fermi level, and the transfer of electrons between metal and oxygen atoms facilitates electron accumulation and accelerates the ORR process (Fig. 13g–i).
 | ||
| Fig. 13 Potential-dependent in situ Raman spectra during the ORR process in (a) alkaline and (b) acidic solutions. (c) Normalized Raman intensities of the stretching mode of OOH at 730 cm−1. (d) In situ XAS spectra recorded at the Fe K-edge. (e) Fitted average oxidation states of Fe from XANES spectra. (f) Lengths of the Fe–N bond obtained from the FT-EXAFS spectra. (g) Optimized structures of OH*, O*, and OOH* intermediates on the Fe, Co SAs-PNCF surfaces. (h) Free energy diagram. (i) Electronic density of states (DOS). Reproduced with permission.131 Copyright 2021, Elsevier Ltd. | ||
Hu et al. synthesized a cobalt–iron diatomic catalyst using in situ electrochemistry by introducing iron onto a single-atom Co catalyst.132 The dimerized Co–Fe structure is the active site of the OER. Lou et al. synthesized a nitrogen-doped carbon material (a-NiCo/NC) anchored by Ni–Co sites using a multi-step template strategy, in which Ni and Co are dispersed on hollow prisms of the carbon.133 DFT calculations showed that the strong interaction between Ni and Co sites optimizes the electronic structure, reduces the reaction energy barrier, and increases the OER activity. Wu et al. obtained a diatomic catalyst (FeCo-NC) by calcining an Fe and Co co-doped zeolite imidazole skeleton (ZIF-8).134 FeCo-NC showed good bifunctional catalytic activity, exhibiting E1/2 of 0.877 V in the ORR and Ej=10 of 1.579 V in the OER. The increase in the activity of the ORR and OER can be attributed to the adjustable electronic structure of FeCo–N6. In addition, the fractional micropore–mesopore structure of the catalyst can increase the specific surface area and facilitate mass transfer.
Compared with SACs, DACs provide a variety of adsorption sites for the CO2RR, and the synergistic effect between diatomic sites can improve the selectivity of CO2 conversion to CO and regulate the electronic structure to promote the subsequent reaction. The CO selectivity on the diatomic Ni–Fe catalyst synthesized by Zhao et al. was as high as 98% at −0.7 V, and the electron interaction between Ni and Fe is beneficial to COOH* formation and CO desorption.33 Wu et al. synthesized a diatomic catalyst with Co–Cu heterodiatomic pairs, which has an FE of 99.1% for CO generation and FECO of more than 95% at high current densities.135 The introduction of the second metal can effectively regulate the electronic structure and promote the formation of intermediate *COOH. He et al. prepared a nitrogen-doped carbon matrix catalyst with Fe–Cu diatomic sites, and the CO FE in the CO2RR was as high as 99.2%.136 The synergistic effect of Fe–Cu accelerates the charge transfer, and effectively regulates the position of the d-band center, reducing the reaction energy barrier. He et al. prepared a Ni/Cu–N–C catalyst with Ni–Cu double sites, in which an N4Ni/CuN4 site structure was formed (Fig. 14a–c).137 The catalyst had an FECO of 99.2% at −0.79 V. Theoretical calculation results showed that the adjustment of electronic structure promoted by the two-site interaction can effectively narrow the band gap, promote charge transfer, and reduce the reaction energy barrier, which is conducive to the generation of CO (Fig. 14d–f).
 | ||
| Fig. 14 (a) Ni K-edge FT-EXAFS curves. (b) Ni K-edge EXAFS fitting curves of Ni/Cu–N–C in the R space. (c) HOMO and LUMO spin electron densities of the proposed Ni/Cu–N–C and Ni–N–C models. (d) Optimized catalytic models and reaction pathways. (e) Free energy profiles of the CO2RR. (f) Difference in limiting potentials for the CO2RR and HER. Reproduced with permission.137 Copyright 2021, American Chemical Society. | ||
At present, C1 products are mainly reported on DACs in the catalytic CO2RR, and further in-depth products have been rarely reported. The mechanism of multi-electron reaction to produce methane and methanol on DACs for the CO2RR was explored, demonstrating the way of C–C coupling to obtain C2 products. Lu et al. constructed Fe/Co SACs and found that CO was the only product in the CO2RR.138 With the help of CO intermediates, the limiting potential on the FeCo-NC DAC to reduce CO2 to CH3OH and CH4 is as low as −0.64 V. Under applied voltage, FeCo-NC is more likely to generate CH3OH and CH4, since the two-site center facilitates spin polarization and electron transfer. *CO tends to adsorb the bridge position of the dual-sites on either side, and C–C coupling is not easy to occur since greater excitation energy is required. In addition, the increase of CO concentration at the dual-sites will cause poisoning.139
 | ||
| Fig. 15 (a) HAADF-STEM image of Ru3/CN. (b) Corresponding intensity maps (left), models (right) and (c) intensity profiles obtained in areas labeled 1, 2, and 3 in (a). (d) Ru K-edge XANES spectra. (e) EXAFS Fourier transformed (FT) k2-weighted χ(k) function spectra. (f) Wavelet transforms for the k2-weighted Ru K-edge EXAFS signals for the high-coordination shells in Ru3/CN. The colors in the contour figures indicate the moduli of the Morlet wavelet transform. (g) Comparison of the q-space magnitudes for FEFF-calculated k2-weighted EXAFS paths. (h) Corresponding EXAFS R-space fitting curve for Ru3/CN. (i) Schematic model of Ru3/CN. Reproduced with permission.31 Copyright 2017, American Chemical Society. | ||
To study the synergistic effect between three nuclear metal atoms, Wang et al. systematically simulated the catalytic performance of single atoms, dual atoms and triple atoms in the electrocatalytic NRR by theoretical calculations.144 The results showed that the catalytic activity of the TAC was generally superior to that of the SAC and DAC since the reduction of adsorption energy of the NH2 intermediate effectively promotes the rate control step of *NH2 hydrogenation. Due to the relatively low loading capacity of single/double/triple site catalysts, there are few active sites which affect the catalytic activity. Jiang et al. used density functional theory to construct a triatomic Fe3-GDY/Gra catalyst with a theoretical mass loading of up to 35.8 wt%, showing excellent activity in the theoretical NRR at a potential of −0.37 V.145 High loading provides more active sites to activate N2 more easily. Zhou et al. theoretically demonstrated that a trinuclear catalyst anchored on nitrogen-doped carbon can effectively convert CO2 into C2 and C2+ products in the CO2RR.21 The trinuclear active sites can adsorb multiple CO2 at the same time, which is conducive to C–C coupling.
2.3. Regulating the coordination environment of central metal sites
Electrocatalytic active sites are formed by bonding between central metal atoms and the surrounding coordination atoms. Therefore, adjusting the bonding mode can tune the electronic structure of central metal atoms, including the valence and spin states and the electron cloud density near the Fermi level. The target active structure is obtained by adjusting the number, type and configuration of coordination atoms, which is conducive to understanding the relationship between coordination configuration and catalytic performance.146,147Among them, the M–N4 structure is the most studied active site, which is like that of metal porphyrins and metal phthalocyanines, but the chemical environment after loading on the carbon carrier is obviously different from that of metal porphyrins/metal phthalocyanines. M–N4 sites showed good activity for many electrocatalytic reactions. Guo et al. synthesized single-atom Co and Ni catalysts through three steps of impregnation–carbonation–acidification.152 The single-atom Co catalyst showed excellent catalytic activity and stability for the OER/ORR, and the reversible oxygen overpotential was 0.75 V. It was assembled as a cathode into a zinc–air battery with a capacity of 530.17 mA h gzn−1. Theoretical calculation results showed that the OH* hydrogenation barrier increases when there are clusters in the catalyst, which is not conducive to the promotion of ORR/OER activity.
Xie et al. synthesized a Ni–N4–C catalyst using a topochemical transformation strategy, which effectively avoided the agglomeration of Ni metal.153 The FE of converting CO2 to CO was up to 99%. Theoretical calculations showed that the Ni–N4 site effectively reduces the generation energy of COOH* and accelerates charge transfer to promote the conversion of CO2 to CO (Fig. 16a–e). The distance between different M–Nx coordination structures affects the selectivity of the CO2RR. Zheng et al. adjusted the Cu–Nx structure by adjusting pyrolysis temperature and Cu doping amount.154 Experimental and theoretical results showed that Cu–Nx can promote the conversion of CO2 to CH4 when the concentration of Cu is less than 2.4%mol, while when the concentration is increased to 4.9%mol, CO2 can be transformed into C2H4. Two CO intermediates can be coupled to produce C2H4 at adjacent Cu–N2 sites (Fig. 16f).
 | ||
| Fig. 16 (a) Schematic illustration of the topochemical transformation strategy. (b) FT of the Ni K-edge EXAFS oscillations of Ni-doped g-C3N4. (c) FT of the Ni K-edge EXAFS oscillations of Ni–N4–C. (d) Calculated free energy diagram. (e) Difference in limiting potentials for CO2 reduction and H2 evolution. (f) Schematic of the procedure to synthesize Cu–N–C–T catalysts. (a–e) Reproduced with permission.153 Copyright 2017, American Chemical Society. (f) Reproduced with permission.154 Copyright 2020, American Chemical Society. | ||
Fu et al. used in situ XAS to reveal the relationship between the ORR and the configuration of active sites.155 They studied the dynamic changes of the Mn–N4 configuration during the ORR process. Under applied voltage, Mn–N4 was transformed into Mn–N3C, and then further transformed into the Mn–N2C2 configuration. The valence state of Mn increased from +3.0 to +3.8 and then decreased to +3.2. Therefore, Mn–N3C and Mn–N2C2 configurations really regulated the adsorption energy barrier and adsorption mode of the reaction intermediates in the ORR process. Wang et al. synthesized nitrogen-doped graphene with highly dispersed FeN5 active sites, which achieved 97.0% FE in converting CO2 to CO.156 Theoretical calculations showed that five N coordination depleted the electron density of Fe 3d, effectively promoting the formation of CO. Jiang et al. prepared single-atom Ni–Nx–C with different N coordination numbers by a post-synthetic metal substitution method.157 The FE on the Ni–N3–C catalyst was 95.6%, which is significantly higher than that of the Ni–N4–C catalyst. DFT calculations showed that the Ni–N3 site was conducive to COOH* production and accelerated CO production. Wang et al. reasonably designed a single-atom W catalyst with a W1N1C3 structure, showing an η10 of 85 mV in 0.1 M KOH solution for the HER.62
In DACs, the M1M2-N6 coordination structure is usually formed. Zhao et al. anchored diatomic Ni–Fe sites on carbon nitride, which showed an FECO of up to 90% and CO2RR stability.158 Theoretical calculations revealed that the NiN3–FeN3 active site reduced the formation energy of the COOH* intermediate and the desorption energy of CO. In addition, other M1M2–Nx coordination structures also play an important role in the CO2RR. Lu et al. anchored diatomic Ni2–N3 sites onto carbon nanotubes (CNTs), on which the energy barrier of the *COOH intermediate could be more significantly reduced than on Ni–N3, Ni–N4 and Ni2–N4 sites.159 The energy barrier of the rate-limiting step of *CO desorption is 0.76 eV, which is conducive to the conversion of CO2 to CO. Zhong et al. reported three coordination modes of Ni2–N7, Ni2–N5C2 and Ni2–N3C4, among which Ni2–N3C4 showed excellent activity in the CO2RR.160 DFT calculations showed that a suitable coordination environment regulated the electronic structure of Ni2 and reduced the energy barrier of COOH*.
The Fe–Nx structure has been considered to be an effective active site for the ORR.161–163 However, a two-electron transfer process will inevitably occur, generating a small amount of H2O2, which attacks N ligands, resulting in the dissolution of Fe and inactivation. To inhibit the two-electron ORR process, it is needed to regulate the Gibbs free energy of *OOH to prevent the spontaneous generation of H2O2.164,165 By introducing a second metal, the electronic structure of Fe–N4 can be effectively adjusted to optimize the adsorption free energy of reaction intermediates.166 Cao et al. synthesized a diatomic catalyst (Fe–Mn–N–C) with FeN4–MnN3 sites.167 There is a defect around FeN4–MnN3 and the Fe–Mn bond has a strain effect, leading to a significant change of the space charge density around Fe and Mn. The rate-limiting step of the ORR on FeN4 is OH* → H2O, while it changes to O* → OH* after the introduction of MnN3. The intermediate OOH* is adsorbed at the Fe–Mn bridging position, which is conducive to O–O dissociation, and hydrogen peroxide generation by 2e− can be completely inhibited. The FeN4–MnN3 configuration decreased the d-band center of FeN4 compared with FeN4, and weakened the binding strength, thus reducing the adsorption energy of oxygen-containing intermediates (Fig. 17).
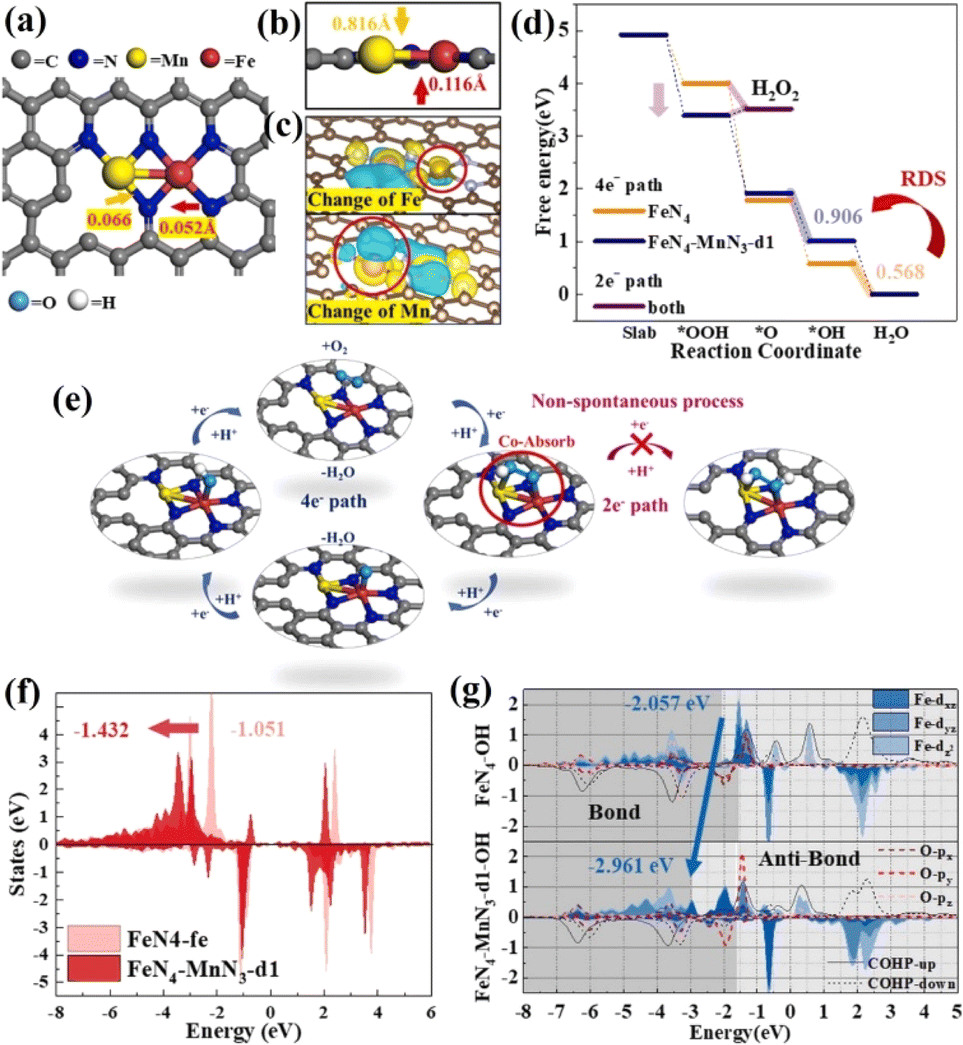 | ||
| Fig. 17 (a) FeN4–MnN3-d1 structure (d1 referring to the number of defect being 1). (b) Side view of the FeN4–MnN3 structure. (c) Charge density difference diagrams of FeN4–MnN3-d1. (d) Oxygen-free energy diagrams of FeN4 and FeN4–MnN3-d1. (e) Illustrations of the ORR process on the FeN4–MnN3-d1 structure. (f) The d-band changes of Fe in FeN4 and FeN4–MnN3-d1. (g) PDOS of Fe in the dxz, dyz and dz2 orbitals and the bonding and anti-bonding state information obtained by COHP. Reproduced with permission.167 Copyright 2020, Elsevier B.V. | ||
 | ||
| Fig. 18 (a) Schematic atomic interface model of S-Cu-ISA/SNC. (b) Schematic of the in situ electrochemical cell set-up. (c) Cu K-edge XANES spectra of S-Cu-ISA/SNC at various potentials during the ORR. (d) Differential Δμ XANES spectra. (e) Current density as a function of potential for S-Cu-ISA/SNC (left) and the average oxidation number of Cu species in S-Cu-ISA/SNC as a function of potential (right). (f) k3-weighted FT-EXAFS. (g) The proposed ORR mechanism for the S-Cu-ISA/SNC. (h) ηORR as a function of ΔGO* on different Cu-centered moieties. (i) Free-energy diagram. Reproduced with permission.169 Copyright 2020, Nature. | ||
P and N belong to the same group V, but the electronegativity of P is weaker than that of N, and therefore, the adsorption activity of reaction intermediates can be regulated by introducing P in the common tetragonal configuration. Wang et al. synthesized a single-atom Co catalyst (Co-SA/P-in situ) with a Co1–P1N3 coordination structure, showing an η10 of 98 mV for the HER.170 The bond lengths of Co–P and Co–N were 2.18 Å and 1.95 Å, respectively. In situ XAS results showed that the valence state of Co increased from +1.53 to +1.78 during the HER, and the bond lengths of Co–P and Co–N became longer, indicating that Co1–P1N3 was the active center. DFT calculations showed that P and N doping promoted the charge transfer, and effectively improved the catalytic activity of the HER. Chen et al. found that a N- and P-codoped single-atom Fe catalyst (Fe–N/P–C) can efficiently catalyze the CO2RR, and the FECO can reach 98%.171 DFT calculations showed that the introduction of P reduces the oxidation state of Fe, optimizes the adsorption energy of the *CO intermediate, and improves the catalytic activity.
B is an electron-deficient atom, and when B and N co-coordinate with the metal, B shares electrons with the electron-rich N to regulate the electronic structure around the metal. Xin et al. synthesized a single-atom Ni catalyst (Ni–B/N–C) with a Ni–B1N3 coordination structure, exhibiting a half-wave potential of 0.87 V for the ORR.174 Theoretical calculation results show that the introduction of B can adjust the local coordination environment of Ni–B1N3, reduce the adsorption energy barrier of reaction intermediates, and accelerate the ORR process.
The electronegativity of O is greater than that of N, and O and N can form quad-coordination with the metal in the plane and quintuple coordination outside the plane. Compared to other non-metallic coordination atoms, no additional oxygen source is introduced during the synthesis process due to the presence of oxygen in most carbon precursors. Chen et al. successfully introduced single-atom Mn–N3O1 into a 3D graphene framework (Mn/C–NO), optimized the position of the Mn d-band center by regulating the geometry of local coordination, making it conducive to the adsorption and desorption of reaction intermediates, and accelerating the kinetics of the ORR.175 Chen et al. used a sacrificial template method to controllably anchor Ni–N4–O sites on nitrogen-doped porous graphite carbon (NiSA-N-PGC).172 The highest FE of NiSA-N-PGC in the catalytic conversion of CO2 to CO was 97.2%. In situ infrared spectroscopy was used to study the reaction intermediates adsorbed on the catalyst surface, demonstrating that *COOH and *CO were the main intermediates in the CO2RR process. With the increase of potential, the peak strength of *CO increased, while the peak strength of *COOH decreased, indicating that *COOH → *CO accelerated, and the activity of the CO2RR increased. Theoretical calculations revealed the promotion effect of the axial Ni–O coordination on the CO2RR. In the CO2RR, the conversion step of CO2 to CO is CO2 → *COOH → *CO → CO. The oxidation state of Ni in Ni–N4–O is +1.12 e, which is higher than that in Ni–N4 due to the transfer of electrons from Ni to the axial O. Therefore, the Ni 3d orbitals in the former exhibit higher DOS near the Fermi level than the latter. The coordination of axial O with high electronegativity enhances the electron delocalization of the active center metal Ni, reduces the reaction energy barrier in the CO2RR, and facilitates the adsorption and desorption of reaction intermediates (Fig. 19a–d).
 | ||
| Fig. 19 (a) Potential-dependent operando attenuated total reflection surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) spectra of NiSA(0.3)-N-PGC. (b) Proposed reaction steps of electrocatalytic reduction of CO2 to CO over the Ni–N4–O site. (c) PDOS calculated using DFT for Ni 3d. (d) Reaction paths and free energy diagrams. (e) Operando XANES spectra recorded at the Ni K-edge of Ni DSC. (f) Least-squares curve-fitting analysis of operando EXAFS spectra at the Ni K-edge. (g) Comparison between the Ni K-edge XANES experimental spectra (solid lines) and the theoretical spectra. (h) Projected crystal orbital Hamilton population (pCOHP) between the metal center and the COOH* intermediate. (i) Electron density difference plot of the *COOH intermediate adsorption structure and the Bader charge analysis. (a–d) Reproduced with permission.172 Copyright 2022, American Chemical Society. (e–i) Reproduced with permission.176 Copyright 2021, American Chemical Society. | ||
In DACs, the electron effect between two adjacent metal atoms can provide a variety of absorbable sites and accelerate electrocatalytic reactions. Hou et al. synthesized an atomically dispersed indium–nickel diatomic catalyst (InNi DS/NC), in which an oxygen atom is axially bridged between In and Ni.173 XAS characterization showed that the coordination configuration was O–In–N6–Ni, and the FE of CO generation was up to 96.7%. In situ characterizations and theoretical calculations showed that the interaction between the In–N6–Ni site and bridging oxygen reduced the energy barrier of the *COOH intermediate and inhibited the competitive HER. In addition, the electrolyte can provide an oxygen atom axially between the two core metals and affect the activity and efficiency of the catalyst. Yao et al. synthesized a binuclear Ni cluster precursor and then anchored it to a nitrogen-doped carbon material to obtain Ni2/NC with binuclear Ni2–N6 sites.176 The FE of catalytic conversion of CO2 to CO was greater than 94%. Operando XAS analysis and theoretical calculation results showed that the Ni2–N6 in the electrolyte first adsorbed an oxygen-containing group to form an O–Ni2–N6 structure during the initial reaction, and then the CO2 adsorbed on the O–Ni2–N6 structure was rapidly activated, and then the CO* intermediate was obtained through two proton coupling processes. Finally, CO* was desorbed on the O–Ni2–N6 structure. The occupancy rate of Ni–COOH* antibonding states at O–Ni2–N6 is lower than that at Ni–N4, indicating stronger adsorption of intermediates at the former. In addition, the positive charge (0.23 e) at O–Ni2–N6 was higher than that at Ni–N4 (0.18 e), indicating that the O–Ni2–N6 site was more conducive to the activation of CO2 conversion to COOH* (Fig. 19e–i).
3. Theoretical understanding of atomic catalysts for electrocatalysis
Electrocatalytic reactions usually take place at the interface or surface of catalysts, and structural variations affect the adsorption and the desorption. On the one hand, well-designed engineering of the geometry or electronic structure produces synergistic effects that can enhance catalytic activity, stability and selectivity. On the other hand, the changes in the electronic structure affect the charge density and the degree of d-band filling in the spin state, which modulates the binding model, leading to a decrease in the reaction energy and an increase in the catalytic activity.68,177,1783.1. Synergistic effect
Modulating the coordination environment and electron distribution of atomic catalysts is an effective but still challenging strategy to improve the electrocatalytic performance.179 Many works have shown the activity of DACs and TACs is higher than that of SACs due to a synergistic effect.68 The synergistic effect between polynuclear metal atoms can regulate the local electronic structure, promote the transfer of electrons, optimize the adsorption and desorption properties, and reduce the overall reaction barrier, thus improving the electrocatalytic performance.41 Theoretical calculations showed that the synergistic action of neighboring metals optimizes the d-band center positions of the metal centers and balances the free energies of the oxygen intermediates, thus increasing the oxygen electrocatalytic activity. Bu et al. reported a homogeneously conceived FeCo-DACs/NC catalyst, in which the synergistic interaction between Fe and Co exerted a significant effect on the reaction barrier.180 The tiny band gap of FeCo-DACs/NC increased the electronic conductivity and accelerated the electron transport. Meanwhile, the synergistic interaction between Fe and Co made the metal d-band center of FeCo-DACs/NC lower than the Fermi energy level and the energy level position closer to the 2p orbital of O than that of the corresponding SACs (Fig. 20a and b). Lee et al. reported a more efficient Fe2–N–C electrocatalyst than Fe1–N–C. The diatomic Fe orbital coupling led to the reduction of orbital energy levels and electron delocalization, which facilitated *CO desorption. The orbital coupling between the dual-site Fe atoms reduced the energy gap between the antibonding and bonding states in *CO adsorption (Fig. 20c–e).181 Li et al. reported the construction of a bimetallic catalyst by introducing single-atom Co into the second ligand shell layer of the Fe center.182 The addition of the Co atom to the d-band center in FeCo-NC (−0.98 eV) brought it closer to the Fermi energy level than Fe-NC (−1.61 eV), indicating that fewer electrons populate the antibonding orbitals of the active center, thus enhancing the phase hundred interaction between the active and adsorbed substances, and facilitating the subsequent substrate reaction (Fig. 20f). | ||
| Fig. 20 (a) Proposed architecture of the Fe–Co metal pair in FeCo-DACs/NC. (b) DOS patterns of FeCo1, Co-SACs/NC, and Fe-SACs/NC. (c) Bare surface with a range from −0.01e Å−3(blue) to +0.04e Å−3 (red) and (d) *CO adsorption with an isosurface level of 0.002e Å−3. (e) PDOS of Fe–N4 and N3–Fe–N–Co. (f) Schematic illustration of orbital interaction between Fe-3d (dz2 and dxz/dyz) and adsorbed CO (5σ and 2π*) for Fe1–N4–C and Fe2–N6–C-o. (a and b) Reproduced with permission.180 Copyright 2022, Wiley-VCH GmbH. (c and e) Reproduced with permission.181 Copyright 2022, American Chemical Society. (f) Reproduced with permission.182 Copyright 2023, Wiley-VCH GmbH. | ||
3.2. Defect coupled spin state change
Defects are prevalent in heterogeneous materials, and can be used as active sites to fabricate specific catalysts or to promote catalytic performance. Defect engineering is an effective way to modulate the surface physical/chemical properties of catalytic materials, and the presence of defects will significantly change the electronic structure and chemical properties, leading to the formation of new physicochemical properties or strong synergistic effects, and thus optimizing the catalytic performance.183 The change of defect-coupled spin states has gradually attracted more and more attention. Yang et al. reported that the charge redistribution of the attached exfoliated monolayer of iron phthalocyanine (FePc) could be effectively regulated using a defective graphene substrate with 585 defects.184 The transfer of electrons from the 585 defects to FePc formed an electron-rich region on the Fe atoms, and the high-density electrons further elevated the d-band center of the Fe atoms. Obviously, this adjustment of the electronic structure of Fe atoms favoured the adsorption and reaction of O2 molecules (Fig. 21a–c). Nitrogen-doped carbon-coupled FeNi3 intermetallic compounds (FeNi3@NC) with excellent OER and ORR properties were reported by Mu et al.185 The defects in FeNi3@NC, including impurities, dopants, vacancies, and interstitial defects, can significantly change the chemical properties and electronic structure, promoting the formation of new physicochemical properties or strong synergistic effects. Mai et al. utilized the coupling effect of edge defects of coordinated bismuth (Bi) and sulfur to improve the selectivity of the CO2RR and inhibit the competing HER.186 DFT calculations showed that sulfur tends to bind to the Bi edge defects, reduces the ligand-unsaturated Bi sites (*H adsorption sites), and modulates the charge state of the neighboring Bi sites to improve the *OCHO adsorption (Fig. 21d and e). Wang et al. utilized a seeded decarboxylate-induced defect strategy to improve the intrinsic activity of the Co SAC by increasing the defect density.43 DFT calculations confirmed that in the defect-rich Co SAC, the defect structure formed near the Co–N4 site likely influences the charge redistribution, which can greatly improve the intrinsic catalytic performance and reduce the free energy of OH adsorption on the Co–N4 site, thus facilitating the electrochemical reaction. | ||
| Fig. 21 (a) The top views of optimized FePc/DG-585 (FePc/G or FePc/NG) based hybrid interfaces. (b) The side views of the 3D charge density difference plot for the interfaces between the DG-585 (G or NG) sheet and FePc layer. (c) The schematic of a probable ORR electrocatalytic mechanism of FePc/DG-585 for the ORR. (d) The correlation between the adsorption energy (ΔG*OCHO) and p-band center of active Bi atoms. (e) The formation energies. (a–c) Reproduced with permission.184 Copyright 2020, Elsevier B.V. (d and e) Reproduced with permission.186 Copyright 2023, Wiley-VCH GmbH. | ||
3.3. Crystal field distortion spin state change
Although some previous discussions have demonstrated the important role of the coordination field of M–N–C catalysts in improving the catalytic performance, the mechanism is still unclear. Breaking the D4h symmetry of the FeN4 active center provides a new way to improve the activity of Fe–N–C catalysts.187 Zhang et al. achieved the modulation of the electronic spin state of the Fe active center in SAC-Fe–N–C and the modulation of the electronic spin state of the FeN4 active center from the low-spin state of FeN5 to the high-spin states of FeN4 and FeN3, based on ligand-field theory by converting the defect-rich pyrrole-type N-coordinated FeNx sites, thus allowing the electrons to easily penetrate the antibonding π-orbitals of oxygen.188 Fe–N4–HS has a three-dimensional electronic structure of t2g and eg, which promotes the kinetics of the ORR significantly. The asymmetric arrangement of the d-orbital electrons of Fe in the spin channel indicates spin polarization. Utilizing the activity-enhancing effect of the high spin state of Fe(III), the designed Fe–N4-HS catalyst exhibited higher ORR activity than commercial Pt/C catalysts (Fig. 22a). Ling et al. showed the first crystal-field deformation of the FeN4 portion achieved by the introduction of sulfur–oxygen-polar functional groups into Fe–N–C catalysts, and revealed that the disruption of FeN4's D4h symmetry leads to the rearrangement of Fe 3d electrons and increases the spin moment of the Fe center.187 The effective spin-state manipulation optimized the adsorption energy of ORR intermediates, which significantly improved the intrinsic ORR activity of the Fe–N–C catalysts (Fig. 22b–d). Ruan et al. remodelled the metal-centered coordination of the Fe centers by altering the crystal field of the Fe–N portion through the introduction of sacrificial bonds.162 Selective breaking of the sacrificial Fe–S bond resulted in electron-withdrawing sulfur oxides in the Fe center. The reduced electron localization around the Fe center gave the catalyst excellent ORR activity and remarkable stability, which facilitated the desorption of ORR intermediates. | ||
| Fig. 22 (a) Schematic illustration of bond formation between the Fe–N–C and the adsorbates (left), and schematic illustration between the d-band centers and adsorption–desorption abilities of ORR intermediates (right). (b–d) Schematic diagrams of breaking FeN4 square-planar coordination by introducing polar XO2 (X = S, Se, and Te) groups. (a) Reproduced with permission.188 Copyright 2022, Elsevier Ltd. (b–d) Reproduced with permission.187 Copyright 2022, Wiley-VCH GmbH. | ||
3.4. Stress-induced spin state change
Changes in catalyst geometry induce strain effects that affect the spin polarization of active centers and the changes of charge density. Zhang et al. succeeded in introducing strain effects on the active C–C bond near the edge graphite nitrogen, which increased the proper spin polarization and charge density of the carbon active center, and kinetically favoured the adsorption of O2 and the activation of O-containing intermediates.189 The spin-polarization strategy allows the construction of highly curved carbon-based multinuclear metal electrocatalysts with improved ORR performance by introducing strain effects. Dong et al. designed magical concave Pt–Zn nanocubes with high-index faceted platinum skins, which produced a strain effect different from that of the ordinary cubes.190 The length of the Pt–Pt bond was significantly shortened, which reduced oxygen-containing intermediates' adsorption energy on the platinum surface. Jiang et al. proposed a spin-state transition strategy modulated by axial Fe–O–Ti ligands to enhance the ORR activity of Fe centers.191 Theoretical calculations showed that the Fe–O–Ti ligand in FeN3O–O–Ti induces a low to medium spin-state transition to optimize the adsorption of O2 on the FeN3O. The magnetic moment of Fe increased from 1.51 μB (FeN3O) to 3.52 μB (FeN3O–O–Ti). The low to moderate spin transition provides better Eg filling for Fe, which favors O2 affinity. FeN3O–O–Ti has suitable O2 adsorption energy (1.84 eV) and ICOHP value (1.88 eV), meaning a moderate binding strength of O2. It was therefore inferred that FeN3O–O–Ti may have convincing ORR activity.4. Challenges for optimizing atomic catalysts
4.1. Controlled synthesis
Accurate and controllable synthesis techniques play an important role in the fabrication of atomic catalysts. The active sites determine the catalytic activity and selectivity. Uniform active sites are conducive to the study of the relationship between structure and performance. At present, the common synthesis strategies for atomic catalysts include high temperature annealing and encapsulation.13,66,192,193 Random dispersion is suitable for low metal loading, while high metal loading might lead to metal agglomeration since metal atoms migrate at high temperatures. The encapsulation strategy can satisfy the need to obtain DACs with definite structure, but the size of the selected precursor must match the carrier cage size. Using the steric hindrance of the carrier to inhibit atomic migration, Qu et al. constructed a diatomic catalyst with Fe2, Cu2 and Ir2 sites by using the interface encapsulation strategy.194 To prevent thermal migration during synthesis, the interface encapsulation carrier ZIF-8 with polydopamine was used, and the bimetallic precursor was anchored into the hollow nanocages (Fig. 23a–d). Bu et al. adopted a “pre-constrained metal twins” strategy to encapsulate an FeCo dimer into ZIF-8 in situ for the creation of adjacent Fe–N4 and Co–N4 geometric configurations (Fig. 23e).180 Theoretical calculations showed that the interaction between Fe and Co optimizes the d-band center of the catalyst, optimizes the adsorption energy of oxygen intermediates, and improves the catalytic activity of the ORR/OER.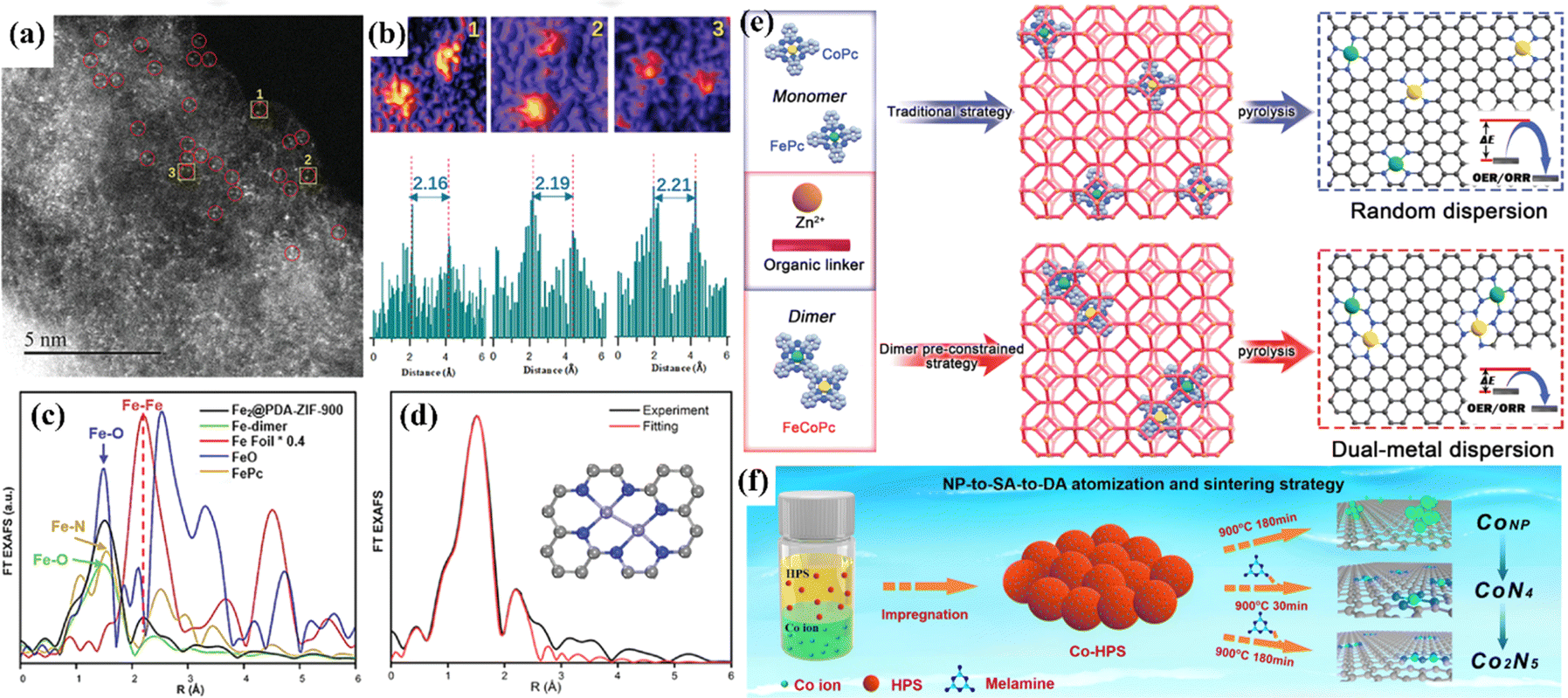 | ||
| Fig. 23 (a) HAADF-STEM image. (b) Intensity profiles highlighted in yellow in (a). (c) Fe K edge FT EXAFS spectra. (d) FT EXAFS fitting spectrum at R space. (e) Schematic illustration of the traditional strategy and “pre-constrained metal twins” strategy for fabricating DACs. (f) Schematic diagram of the NP-to-SA-to-DA atomization and sintering strategy. (a–d) Reproduced with permission.194 Copyright 2022, Wiley-VC. (e) Reproduced with permission.180 Copyright 2022, Wiley-VC. (f) Reproduced with permission.195 Copyright 2023, Nature. | ||
The major challenge of controlled synthesis is that the dynamic evolution of metal atoms and ligands during the synthesis is still not clear. To date, there have been some reports revealing this complex process. Huang et al. designed an atomization and sintering strategy to transfer Co nanoparticles to single-atom CoN4 and then to diatomic Co2N5.195 In this process, the nitrogen source played the role of stripping and fixing the metal atoms, and the Co2N5 sites were acquired by thermal migration spontaneous sintering (Fig. 23f). Wu et al. used in situ XAS to investigate the structural changes of the CoN4 site during synthesis.196In situ XAS was used to monitor the conversion of inactive Co–OH/O to the active CoN4 site. The conversion could not be completed below 700 °C, and the best activity was obtained at 900 °C. However, due to the limitation of advanced characterization techniques, it is still very difficult to dynamically study the whole process of the formation of active sites.
4.2. Increasing the density of active sites
For typical synthesis of SACs, it is usual to reduce the metal load on a high specific surface substrate to gain atomic dispersion and prevent aggregation, but due to the low metal load, there are few metal active sites. The number of catalytic active sites can be effectively increased by increasing the metal loading, but the probability of metal agglomeration increases gradually with the increase of metal loading. At present, the commonly used methods to increase metal loading and reduce agglomeration are to stabilize single metal atoms by introducing heteroatoms and using covalent coordination between the metal and nonmetal. Choi et al. loaded 5 wt% Pt in atomically dispersed form on a high sulfur doped zeolite template carbon by a wet impregnation method.197 The rich sulfur doping and 3D carbon structure provided a stable loading environment.Compared to SACs, the density of active sites in DACs and TACs can be enhanced. For instance, the loading capacity of iron in the monatomic iron catalyst (Fe–N4–C) and the positive catalyst of diatomic iron (Fe2–N6–C-o) synthesized by Han et al. was 0.155 and 0.313 wt%, respectively.181 More than 80% of the iron was identified as double metal sites by calculation, and the mean distance of Fe–Fe was 2.37 ± 0.31 Å. The Fe and Ni content in diatomic catalyst NiFe-DASC synthesized by Chen et al. using the pyrolysis strategy was 3.24 and 4.05 wt% respectively, which is twice the metal load of the Fe/Ni-SAC synthesized by the same method.198 The HAADF-STEM images showed that the Ni and Fe atoms in the NiFe DASC are evenly dispersed, with a distance of about 0.24 nm between the Ni and Fe (Fig. 24a–g). Using the large number of periodic N–H functional groups in polymeric carbon nitride (PCN) as a metal coordination site, the metal can replace the H atom and can be fixed by N, avoiding agglomeration. Lu et al. used stepwise ion exchange and ligand removal strategies to synthesize low-coordination bimetallic atoms, where the content of Cu was up to 18.3 wt%.199
 | ||
| Fig. 24 (a) Schematic illustration of NiFe-DASC synthesis. (b) Dark-field scanning TEM and AFM (inset) images. (c) TEM image and (d) EDS elemental mapping of NiFe-DASC. (e) HAADF-STEM image of NiFe-DASC. (f) Intensity profiles of the three sites in (e). (g) Electronic energy loss spectroscopy (EELS) of NiFe-DASC. Reproduced with permission.198 Copyright 2021, Nature. | ||
4.3. Enhancing intrinsic activity
To improve the inherent electrocatalytic activity of catalysts, the central metal atom and the coordination environment can be adjusted. Selecting an appropriate central metal atom as the active center can significantly improve the electrocatalytic activity and effectively adjust the product selectivity. For example, Fe-pyridine/pyrrole-N4 and Mn-pyrrole-N4 sites can catalyse the ORR, while the Co-pyrrole-N4 site can catalyse the OER efficiently.200 Cu is the only metal species that can convert CO2 to C2+ in the CO2RR.201 The coordination environment includes a coordination atom, base atom and axial ligand. Unsaturated central metal atoms interact with ligands to regulate the metallic energy band, spin state, and charge redistribution to enhance the inherent activity. The heteroatoms on the base plane can regulate the electronic structure of the active site through long-range delocalization. In addition, the interaction of the active site with reactants and reaction intermediates during electrocatalysis can also effectively regulate its activity. Therefore, the inherent activity of the central metal atoms can be changed by adjusting the coordination environment. Zhang et al. synthesized a series of (Mn, Fe, Co, Ni, Cu or Zn) framework porphyrin (POF) materials, and found that the ORR activity of Co-POF was superior to that of Fe-POF.202 Lu et al. constructed an Fe–N2S2 active site, and found that the activity of the CO2RR was significantly optimized compared to that on an Fe–N4 porphyrin.203 On the Fe–N4 porphyrin, the reduction to HCOOH is favourable at −0.70 V, while on the Fe–N2S2 porphyrin, CO2 can be converted to HCOOH and CH3OH at −0.38 V and −0.40 V, respectively (Fig. 25). The introduction of para-S atoms elongates the bond length of Fe–N, causing electrons to gather in the Fe–S bond, and the interaction between Fe and S is enhanced. Moreover, additional Fe orbitals in the Fe–N2S2 porphyrin can effectively regulate the adsorption/desorption energy of reaction intermediates and accelerate the CO2RR. | ||
| Fig. 25 (a) The schematic of elementary steps of the CO2RR. (b) CO2 and H binding energies. (c) Volcano plot. (d) Bader charge difference of C and O atoms of the CO molecule and Fe atom before and after CO binding. (e) Charge density difference of CO binding. Reproduced with permission.203 Copyright 2021, Wiley-VCH GmbH. | ||
In addition, adjacent active sites can regulate each other to promote the inherent electrocatalytic activity. Yu et al. demonstrated that the catalytic ORR activity was enhanced when the distance between the Fe–N4 active sites was in the subnanometer level. When the distance between the two sites was less than 1.2 nm, the strong interaction between them altered the electronic structure and improved the inherent ORR activity. The activity is optimal when the distance is about 0.7 nm.204 Qiao et al. demonstrated theoretically and experimentally that when the distance between Ni–N4 and Cu–N4 is about 5.3 Å, the electronic structure of the active site can be effectively regulated to improve the selectivity and activity of the CO2RR.205
4.4. Improving the stability
For atomic active sites, due to the high surface energy of metal atoms and the unsaturated coordination environment, the isolated metal sites will partially agglomerate under working conditions, resulting in a decrease in catalytic activity. The stability of active sites can be improved by enhancing the interaction between isolated metals and ligands and carriers. The electronegativity of the B atom is less than that of the N atom, which can effectively enhance the electron density of the Ni–N4 center and strengthen the binding ability of the Ni site with reaction intermediates. Xin et al. synthesized a SAC (Ni–B/N–C) with Ni–B1N3 sites, which had a higher E1/2 (0.87 V) than Ni–N–C (0.76 V) in the ORR, and was more stable than most Fe–N–C and Co–N–C catalysts.174 In addition, based on the degradation mechanism of Fe–N–C SACs, some strategies have been proposed, such as the introduction of a co-catalyst and free radical scavenger in the active sites to effectively reduce the degradation. Sun et al. anchored CeO2 NPs around Fe–N4 active sites by an organic gas doping method. CeO2 NPs were used to eliminate the ˙OH and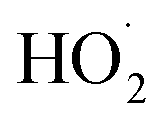 radical generated at the Fe–N4 site (Fig. 26).206 When the Fe-NC/Scaad-CeO2 catalyst was assembled into a fuel cell, the attenuation of peak power density was reduced from 69% to 28%.
radical generated at the Fe–N4 site (Fig. 26).206 When the Fe-NC/Scaad-CeO2 catalyst was assembled into a fuel cell, the attenuation of peak power density was reduced from 69% to 28%.
 | ||
| Fig. 26 Schematic illustration of the radical elimination behaviour. Reproduced with permission.206 Copyright 2023, Wiley-VCH GmbH. | ||
In addition, the active sites can be reconstructed under applied voltage, and the structure is restored upon removal of applied voltage, which is also considered to be a stable catalyst. Fontecave et al. reported nitrogen-doped graphene with CuN4 sites (Cu–N–C), which reduced CO2 to ethanol with an FE of 55%.207 Under the applied voltage, the isolated CuN4 transformed into copper clusters, and Cu–N–C recovered after the voltage was removed. Operando XAS showed that the transformed copper clusters were the active structure. During electrolysis, the reconstruction of CuN4 in Cu–N–C was reversible.
5. Conclusions and outlook
In the past decade, SACs have made great progress in the field of electrocatalysis due to their unique geometry and electronic structures, which are expected to replace commercial precious metal-based catalysts. DACs with the interaction between metal dimers are also emerging and show excellent electrocatalytic activity, which can solve the limitation caused by SACs to a certain extent. To better suit multi-electron complex reactions, TACs have been developed, which provide more possibilities for the adsorption of intermediates, and are conducive to better study the relationship between the structure and electrocatalytic activity. In this review, we summarize the progress of SACs, DACs and TACs in electrocatalysis systematically. By using defect engineering and an atomic limiting strategy to design precursors, the number of central metal atoms and the coordination environment can be regulated for specific electrocatalytic reactions. Up to now, SACs and DACs designed by electronic regulation have made good progress in electrocatalysis. As a rising star, TACs are in the initial stage of development. In general, electrocatalysts with different nuclear numbers and uniform active sites have broad application prospects, but some problems still need to be solved.(i) Controlled synthesis is still one of the biggest obstacles for atomic catalysts. At present, most of the atomic catalysts are synthesized by pyrolysis at high temperatures. The randomness of the coordination environment of the obtained catalysts is relatively large, and metal migration and agglomeration will inevitably occur. In addition, for DACs and TACs, active sites may possess multiple structures. By encapsulating precursors into MOFs, COFs and other materials with pore structure, DACs and TACs can be synthesized through high-temperature pyrolysis. However, this method needs to ensure that the size of the precursor and the pores is compatible, which brings certain obstacles to the accurate control of the coordination environment. To solve the issue of migration and agglomeration of metal atoms during high temperature pyrolysis, Joule thermal synthesis technology with ultra-fast heating and cooling (millisecond level) processes has been developed. However, in the current synthesis technology, single-atom sites are unavoidably formed in DACs and TACs, resulting in the reduction of the target effective active sites. In addition, the atomic distance and configuration between active sites cannot be guaranteed to be consistent. The ratio of the target effective active sites to other structural sites plays a crucial role in determining catalytic activity. Therefore, it is pivotal to develop new and operable synthesis techniques to achieve precise regulation of the coordination environment and to better explain the relationship between structure and mechanism.
(ii) Atomic catalysts synthesized by controllable technology provide a good platform for studying the structure–activity relationships and reaction mechanisms. In catalytic processes, reaction intermediates adsorbed at active sites are dynamically evolved. In addition, the structures of some catalysts change under applied voltage, and therefore it is necessary to in situ detect the changes of active sites and reaction intermediates in catalytic reactions. At present, some in situ techniques have been developed, such as in situ XAS, IR, Raman, and TEM, but there are still issues in time accuracy and atomic resolution. For example, it is difficult to distinguish coordination atoms with similar atomic numbers and similar bonds (e.g. Co–N and Co–O). Therefore, advanced characterization techniques are needed to deeply understand the dynamic evolution of active sites during electrolysis.
(iii) At present, DFT calculations have been successfully applied to guide the selection of atomic catalysts. However, screening the coordination environment of metal center atoms is limited by computational cost and time, and traditional DFT calculations are not suitable for such a huge working system. With the development of computers, the first-principles high-throughput screening method will guide the experimental design of more excellent catalysts. In addition, the high-throughput method can also be used to screen the structures of reaction intermediates in catalytic reactions, and the combination with in situ characterizations will further promote the study of dynamic mechanisms. However, these calculations are carried out in an ideal environment, and the external conditions and internal environment in the electrocatalysis process are very complicated, which makes experimental results deviate from theoretical simulation to a certain extent, and a relatively large model is required to fully simulate the electrocatalytic environment. Therefore, new methods need to be developed in a timely manner.
(iv) Industrial grade catalysts need to have high activity and stability. Although atomic catalysts have made good progress, they are still in the basic research stage. At present, theoretical calculations of high-throughput screening can provide guidance for high-performance catalysts. Catalysts synthesized in the laboratory are in the gram scale, whereas the kilogram scale is commercially required. Therefore, catalyst amplification synthesis is also a key for commercial applications, since the mass-synthesized catalysts need to maintain high activity and stability. Moreover, the test of catalyst stability in the laboratory usually takes several hundred hours, but it falls far short of commercial requirements, since it'll take at least a few months. Therefore, more synthetic methods should be developed to synthesize catalysts that can effectively stabilize the active sites in corrosive electrolytes and reduce economic losses caused by frequent catalyst replacement.
In short, although atomic catalysts have made good progress in the field of electrocatalysis, there are still some issues to be addressed. However, we believe that with the development of new synthesis methods, characterization techniques and computing theories, atomic catalysts will play a significant role in the field of energy storage and conversion in the future.
Author contributions
All of the authors contributed to the literature search, writing and editing of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22075099) and the Natural Science Foundation of Jilin Province (No. 20220101051JC).Notes and references
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem, 2018, 2, 65–81 CrossRef CAS
.
- L. Xu, P. Trogadas and M.-O. Coppens, Adv. Energy Mater., 2023, 13, 2302974 CrossRef CAS
- L. Xiao, L. Qi, J. Sun, A. Husile, S. Zhang, Z. Wang and J. Guan, Nano Energy, 2024, 120, 109155 CrossRef CAS
- Y.-X. Zhao, J.-H. Wen, P. Li, P.-F. Zhang, S.-N. Wang, D.-C. Li, J.-M. Dou, Y.-W. Li, H.-Y. Ma and L. Xu, Angew. Chem., Int. Ed., 2023, 62, e202216950 CrossRef CAS PubMed
- J. Chang, Q. Zhang, J. Yu, W. Jing, S. Wang, G. Yin, G. I. N. Waterhouse and S. Lu, Adv. Sci., 2023, 10, 2301656 CrossRef CAS
- J. Quílez-Bermejo, S. García-Dalí, A. Daouli, A. Zitolo, R. L. S. Canevesi, M. Emo, M. T. Izquierdo, M. Badawi, A. Celzard and V. Fierro, Adv. Funct. Mater., 2023, 33, 2300405 CrossRef
- T. Sun, W. Zang, J. Sun, C. Li, J. Fan, E. Liu and J. Wang, Adv. Funct. Mater., 2023, 33, 2301526 CrossRef CAS
- S. Chen, Y. Gao, W. Wang, O. V. Prezhdo and L. Xu, ACS Nano, 2023, 17, 1522–1532 CrossRef CAS PubMed
- Y. Gao, B. Liu and D. Wang, Adv. Mater., 2023, 35, 2209654 CrossRef CAS PubMed
- F. Humblot, J. P. Candy, F. Le Peltier, B. Didillon and J. M. Basset, J. Catal., 1998, 179, 459–468 CrossRef CAS
- J. Corker, F. Lefebvre, C. Lécuyer, V. Dufaud, F. Quignard, A. Choplin, J. Evans and J.-M. Basset, Science, 1996, 271, 966–969 CrossRef CAS
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS
- X. Bai, J. Han, X. Niu and J. Guan, Nano Res., 2023, 16, 10796–10802 CrossRef CAS
- T. Tang, Z. Wang and J. Guan, Coord. Chem. Rev., 2023, 492, 215288 CrossRef CAS
- L. Sun, V. Reddu and X. Wang, Chem. Soc. Rev., 2022, 51, 8923–8956 RSC
- B. Zhang, Y. Chen, J. Wang, H. Pan and W. Sun, Adv. Funct. Mater., 2022, 32, 2202227 CrossRef CAS
- Y. Zhou, R. Lu, X. Tao, Z. Qiu, G. Chen, J. Yang, Y. Zhao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2023, 145, 3647–3655 CrossRef CAS PubMed
- Y. Chen, J. Mao, H. Zhou, L. Xing, S. Qiao, J. Yuan, B. Mei, Z. Wei, S. Zhao, Y. Tang and C. Liu, Adv. Funct. Mater., 2023, 2311664 Search PubMed
- W. Pei, S. Zhou, J. Zhao, X. Xu, Y. Du and S. X. Dou, Nano Energy, 2020, 76, 105049 CrossRef CAS
- Q.-F. Chen, Y. Xiao, R.-Z. Liao and M.-T. Zhang, CCS Chem., 2022, 5, 245–256 CrossRef
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330, 192–196 CrossRef CAS PubMed
- R. L. Robson, R. R. Eady, T. H. Richardson, R. W. Miller, M. Hawkins and J. R. Postgate, Nature, 1986, 322, 388–390 CrossRef CAS
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed
- J. Rittle and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 4243–4248 CrossRef CAS PubMed
- Z. Wei, J. He, Y. Yang, Z. Xia, Y. Feng and J. Ma, J. Energy Chem., 2021, 53, 303–308 CrossRef CAS
- P. Saha, S. Amanullah and A. Dey, J. Am. Chem. Soc., 2020, 142, 17312–17317 CrossRef CAS PubMed
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC
- B. Wurster, D. Grumelli, D. Hötger, R. Gutzler and K. Kern, J. Am. Chem. Soc., 2016, 138, 3623–3626 CrossRef CAS PubMed
- S. Ji, Y. Chen, Q. Fu, Y. Chen, J. Dong, W. Chen, Z. Li, Y. Wang, L. Gu, W. He, C. Chen, Q. Peng, Y. Huang, X. Duan, D. Wang, C. Draxl and Y. Li, J. Am. Chem. Soc., 2017, 139, 9795–9798 CrossRef CAS PubMed
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed
- Y. Yang, L. Zhang, Z. Hu, Y. Zheng, C. Tang, P. Chen, R. Wang, K. Qiu, J. Mao, T. Ling and S.-Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 4525–4531 CrossRef CAS PubMed
- Q.-F. Chen, Z.-Y. Cheng, R.-Z. Liao and M.-T. Zhang, J. Am. Chem. Soc., 2021, 143, 19761–19768 CrossRef CAS PubMed
- M. Liu, H. Chun, T.-C. Yang, S. J. Hong, C.-M. Yang, B. Han and L. Y. S. Lee, ACS Nano, 2022, 16, 10657–10666 CrossRef CAS PubMed
- Y.-X. Zhang, S. Zhang, H. Huang, X. Liu, B. Li, Y. Lee, X. Wang, Y. Bai, M. Sun, Y. Wu, S. Gong, X. Liu, Z. Zhuang, T. Tan and Z. Niu, J. Am. Chem. Soc., 2023, 145, 4819–4827 CrossRef CAS PubMed
- X. Yan, D. Liu, P. Guo, Y. He, X. Wang, Z. Li, H. Pan, D. Sun, F. Fang and R. Wu, Adv. Mater., 2023, 35, 2210975 CrossRef CAS PubMed
- X. Xie, Z. Zhai, L. Peng, J. Zhang, L. Shang and T. Zhang, Sci. Bull., 2023, 68, 2862–2875 CrossRef CAS PubMed
- Y. Chen, J. Lin, B. Jia, X. Wang, S. Jiang and T. Ma, Adv. Mater., 2022, 34, 2201796 CrossRef CAS PubMed
- L. Li, K. Yuan and Y. Chen, Acc. Mater. Res., 2022, 3, 584–596 CrossRef CAS
- Y. Zhang, L. Guo, L. Tao, Y. Lu and S. Wang, Small Methods, 2019, 3, 1800406 CrossRef
- S. Yuan, J. Zhang, L. Hu, J. Li, S. Li, Y. Gao, Q. Zhang, L. Gu, W. Yang, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 21685–21690 CrossRef CAS PubMed
- R. Jiang, Z. Qiao, H. Xu and D. Cao, Chin. J. Catal., 2023, 48, 224–234 CrossRef CAS
- L. Zhang, Y. Jia, G. Gao, X. Yan, N. Chen, J. Chen, M. T. Soo, B. Wood, D. Yang, A. Du and X. Yao, Chem, 2018, 4, 285–297 CAS
- J. Liu, M. Jiao, B. Mei, Y. Tong, Y. Li, M. Ruan, P. Song, G. Sun, L. Jiang, Y. Wang, Z. Jiang, L. Gu, Z. Zhou and W. Xu, Angew Chem. Int. Ed. Engl., 2019, 58, 1163–1167 CrossRef CAS PubMed
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1705369 CrossRef
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed
- L. Zhang, J. M. T. A. Fischer, Y. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu, L. Zhuang, M. Hankel, D. J. Searles, K. Huang, S. Feng, C. L. Brown and X. Yao, J. Am. Chem. Soc., 2018, 140, 10757–10763 CrossRef CAS PubMed
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed
- X. Bai, J. Han, S. Chen, X. Niu and J. Guan, Chin. J. Catal., 2023, 54, 212–219 CrossRef CAS
- Z. Wang, C. Li, Y. Liu, Y. Wu, S. Zhang and C. Deng, J. Energy Chem., 2023, 83, 264–274 CrossRef CAS
- L. Li, W. Yu, W. Gong, H. Wang, C.-L. Chiang, Y. Lin, J. Zhao, L. Zhang, J.-M. Lee and G. Zou, Appl. Catal., B, 2023, 321, 122038 CrossRef CAS
- J. Han and J. Guan, Chin. J. Catal., 2023, 49, 1–4 CrossRef CAS
- Q. Sun, C. Jia, Y. Zhao and C. Zhao, Chin. J. Catal., 2022, 43, 1547–1597 CrossRef CAS
- J. Li, L. Zhang, K. Doyle-Davis, R. Li and X. Sun, Carbon Energy, 2020, 2, 488–520 CrossRef CAS
- J. Han, X. Meng, L. Lu, J. Bian, Z. Li and C. Sun, Adv. Funct. Mater., 2019, 29, 1808872 CrossRef CAS
- V. Hasija, S. Patial, P. Raizada, A. Aslam Parwaz Khan, A. M. Asiri, Q. Van Le, V.-H. Nguyen and P. Singh, Coord. Chem. Rev., 2022, 452, 214298 CrossRef CAS
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS
- L. Xiao, Z. Wang and J. Guan, Adv. Funct. Mater., 2023, 2310195 Search PubMed
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W.-C. Cheong, J. Mao, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1800396 CrossRef PubMed
- Y. Yue, P. Cai, K. Xu, H. Li, H. Chen, H.-C. Zhou and N. Huang, J. Am. Chem. Soc., 2021, 143, 18052–18060 CrossRef CAS
- C.-X. Zhao, J.-N. Liu, J. Wang, C. Wang, X. Guo, X.-Y. Li, X. Chen, L. Song, B.-Q. Li and Q. Zhang, Sci. Adv., 2022, 8, eabn5091 CrossRef CAS PubMed
- W.-H. Li, J. Yang and D. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213318 CrossRef CAS PubMed
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS
- H. Zhang, X. Jin, J.-M. Lee and X. Wang, ACS Nano, 2022, 16, 17572–17592 CrossRef CAS PubMed
- P. Zhu, X. Xiong, D. Wang and Y. Li, Adv. Energy Mater., 2023, 13, 2300884 CrossRef CAS
- L. Xiao, Z. Wang and J. Guan, Chem. Sci., 2023, 14, 12850–12868 RSC
- T. Tang, Z. Wang and J. Guan, Chin. J. Catal., 2022, 43, 636–678 CrossRef CAS
- T. Tang, S. Li, J. Sun, Z. Wang and J. Guan, Nano Res., 2022, 15, 8714–8750 CrossRef CAS
- Y. Yang, Y. Yu, J. Li, Q. Chen, Y. Du, P. Rao, R. Li, C. Jia, Z. Kang, P. Deng, Y. Shen and X. Tian, Nano-Micro Lett., 2021, 13, 160 CrossRef CAS
- J. Park, S. Lee, H.-E. Kim, A. Cho, S. Kim, Y. Ye, J. W. Han, H. Lee, J. H. Jang and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 16038–16042 CrossRef CAS PubMed
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y.-R. Lu, T.-S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2019, 10, 1743 CrossRef PubMed
- C. Tsounis, B. Subhash, P. V. Kumar, N. M. Bedford, Y. Zhao, J. Shenoy, Z. Ma, D. Zhang, C. Y. Toe, S. Cheong, R. D. Tilley, X. Lu, L. Dai, Z. Han and R. Amal, Adv. Funct. Mater., 2022, 32, 2203067 CrossRef CAS
- W. Wang, Y. Wu, Y. Lin, J. Yao, X. Wu, C. Wu, X. Zuo, Q. Yang, B. Ge, L. Yang, G. Li, S. Chou, W. Li and Y. Jiang, Adv. Funct. Mater., 2022, 32, 2108464 CrossRef CAS
- H. Yan, C. Tian, L. Wang, A. Wu, M. Meng, L. Zhao and H. Fu, Angew. Chem., Int. Ed., 2015, 54, 6325–6329 CrossRef CAS PubMed
- Q. Jin, N. Liu, C. Dai, R. Xu, B. Wu, G. Yu, B. Chen and Y. Du, Adv. Energy Mater., 2020, 10, 2000291 CrossRef CAS
- M. Li, J. Yu, Q. Liu, J. Liu, R. Chen, J. Zhu, R. Li and J. Wang, ACS Sustain. Chem. Eng., 2022, 10, 13505–13513 CrossRef CAS
- J. Wang, X. Ge, L. Shao, J. Zhang, D. Peng, G. Zou, H. Hou, W. Deng, S. Xu, X. Ji and W. Zhang, Mater. Today Energy, 2020, 17, 100436 CrossRef
- J. Yin, Q. Fan, Y. Li, F. Cheng, P. Zhou, P. Xi and S. Sun, J. Am. Chem. Soc., 2016, 138, 14546–14549 CrossRef CAS PubMed
- J. Han and J. Guan, Nano Res., 2023, 16, 1913–1966 CrossRef CAS
- S. Chen, T. Zhang, J. Han, H. Qi, S. Jiao, C. Hou and J. Guan, Nano Res. Energy, 2024, 3, e9120106 CrossRef
- T. Tang, Z. Duan, D. Baimanov, X. Bai, X. Liu, L. Wang, Z. Wang and J. Guan, Nano Res., 2023, 16, 2218–2223 CrossRef CAS
- H. Xu, T. Liu, S. Bai, L. Li, Y. Zhu, J. Wang, S. Yang, Y. Li, Q. Shao and X. Huang, Nano Lett., 2020, 20, 5482–5489 CrossRef CAS
- Q. Wang, Z. Zhang, C. Cai, M. Wang, Z. L. Zhao, M. Li, X. Huang, S. Han, H. Zhou, Z. Feng, L. Li, J. Li, H. Xu, J. S. Francisco and M. Gu, J. Am. Chem. Soc., 2021, 143, 13605–13615 CrossRef CAS PubMed
- X. Bai, Y. Wang, J. Han, X. Niu and J. Guan, Appl. Catal., B, 2023, 337, 122966 CrossRef CAS
- P. Kumar, K. Kannimuthu, A. S. Zeraati, S. Roy, X. Wang, X. Wang, S. Samanta, K. A. Miller, M. Molina, D. Trivedi, J. Abed, M. A. Campos Mata, H. Al-Mahayni, J. Baltrusaitis, G. Shimizu, Y. A. Wu, A. Seifitokaldani, E. H. Sargent, P. M. Ajayan, J. Hu and M. G. Kibria, J. Am. Chem. Soc., 2023, 145, 8052–8063 CrossRef CAS PubMed
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC
- C. Zhang, Y.-C. Wang, B. An, R. Huang, C. Wang, Z. Zhou and W. Lin, Adv. Mater., 2017, 29, 1604556 CrossRef PubMed
- Q. Liu, X. Liu, L. Zheng and J. Shui, Angew. Chem., Int. Ed., 2018, 57, 1204–1208 CrossRef CAS PubMed
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J.-Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed
- X. Han, X. Ling, Y. Wang, T. Ma, C. Zhong, W. Hu and Y. Deng, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS PubMed
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
- C.-X. Zhao, B.-Q. Li, J.-N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4448–4463 CrossRef CAS PubMed
- X. Xie, L. Peng, H. Yang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Mater., 2021, 33, 2101038 CrossRef CAS PubMed
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS PubMed
- M. Kunitski, N. Eicke, P. Huber, J. Köhler, S. Zeller, J. Voigtsberger, N. Schlott, K. Henrichs, H. Sann, F. Trinter, L. P. H. Schmidt, A. Kalinin, M. S. Schöffler, T. Jahnke, M. Lein and R. Dörner, Nat. Commun., 2019, 10, 1 CrossRef CAS PubMed
- C. Wang, D. Wang, S. Liu, P. Jiang, Z. Lin, P. Xu, K. Yang, J. Lu, H. Tong, L. Hu, W. Zhang and Q. Chen, J. Catal., 2020, 389, 150–156 CrossRef CAS
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed
- Y. Wang, R. Shi, L. Shang, G. I. N. Waterhouse, J. Zhao, Q. Zhang, L. Gu and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 13057–13062 CrossRef CAS PubMed
- Q. Zhao, Y. Wang, W.-H. Lai, F. Xiao, Y. Lyu, C. Liao and M. Shao, Energy Environ. Sci., 2021, 14, 5444–5456 RSC
- Y. Zhao, J. Liang, C. Wang, J. Ma and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1702524 CrossRef
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, 1808135 CrossRef PubMed
- Z. Jiang, T. Wang, J. Pei, H. Shang, D. Zhou, H. Li, J. Dong, Y. Wang, R. Cao, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2020, 13, 2856–2863 RSC
- S. Kusama, T. Saito, H. Hashiba, A. Sakai and S. Yotsuhashi, ACS Catal., 2017, 7, 8382–8385 CrossRef CAS
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed
- T. Wang, L. Xu, Z. Chen, L. Guo, Y. Zhang, R. Li and T. Peng, Appl. Catal., B, 2021, 291, 120128 CrossRef CAS
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef
- T. Tang, Z. Wang and J. Guan, Acta Phys.-Chim. Sin., 2022, 39, 2208033 Search PubMed
- Q. Chang, Y. Liu, J.-H. Lee, D. Ologunagba, S. Hwang, Z. Xie, S. Kattel, J. H. Lee and J. G. Chen, J. Am. Chem. Soc., 2022, 144, 16131–16138 CrossRef CAS PubMed
- J. Wang, Y.-C. Huang, Y. Wang, H. Deng, Y. Shi, D. Wei, M. Li, C.-L. Dong, H. Jin, S. S. Mao and S. Shen, ACS Catal., 2023, 13, 2374–2385 CrossRef CAS
- Z. Ma, T. Wan, D. Zhang, J. A. Yuwono, C. Tsounis, J. Jiang, Y.-H. Chou, X. Lu, P. V. Kumar, Y. H. Ng, D. Chu, C. Y. Toe, Z. Han and R. Amal, ACS Nano, 2023, 17, 2387–2398 CrossRef CAS PubMed
- H. Li, F. Pan, C. Qin, T. Wang and K.-J. Chen, Adv. Energy Mater., 2023, 13, 2301378 CrossRef CAS
- M. Humayun, M. Israr, A. Khan and M. Bououdina, Nano Energy, 2023, 113, 108570 CrossRef CAS
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lü, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS PubMed
- Z. Xue, X. Zhang, J. Qin and R. Liu, J. Energy Chem., 2021, 57, 443–450 CrossRef CAS
- J. Han and J. Guan, Chin. J. Catal., 2023, 47, 1–31 CrossRef CAS
- Y. Li, C. Chen, R. Cao, Z. Pan, H. He and K. Zhou, Appl. Catal., B, 2020, 268, 118747 CrossRef CAS
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu, Y. Wang, Q. Zhang, L. Gu, L. Liu, W. Song, J. Liu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 13388–13393 CrossRef CAS PubMed
- X. Zhao, K. Zhao, Y. Liu, Y. Su, S. Chen, H. Yu and X. Quan, ACS Catal., 2022, 12, 11412–11420 CrossRef CAS
- T. Tang, Y. Wang, J. Han, Q. Zhang, X. Bai, X. Niu, Z. Wang and J. Guan, Chin. J. Catal., 2023, 46, 48–55 CrossRef CAS
- Y. Ying, X. Luo, J. Qiao and H. Huang, Adv. Funct. Mater., 2021, 31, 2007423 CrossRef CAS
- Y. Yang, Y. Qian, H. Li, Z. Zhang, Y. Mu, D. Do, B. Zhou, J. Dong, W. Yan, Y. Qin, L. Fang, R. Feng, J. Zhou, P. Zhang, J. Dong, G. Yu, Y. Liu, X. Zhang and X. Fan, Sci. Adv., 2020, 6, eaba6586 CrossRef CAS PubMed
- Y. Da, Z. Tian, R. Jiang, Y. Liu, X. Lian, S. Xi, Y. Shi, Y. Wang, H. Lu, B. Cui, J. Zhang, X. Han, W. Chen and W. Hu, Sci. China Mater., 2023, 66, 1389–1397 CrossRef CAS
- M. Jiao, Z. Chen, N. Wang and L. Liu, Appl. Catal., B, 2023, 324, 122244 CrossRef CAS
- M. Jiang, F. Wang, F. Yang, H. He, J. Yang, W. Zhang, J. Luo, J. Zhang and C. Fu, Nano Energy, 2022, 93, 106793 CrossRef CAS
- L. Bai, C.-S. Hsu, D. T. L. Alexander, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2019, 141, 14190–14199 CrossRef CAS PubMed
- Z. Pei, X. F. Lu, H. Zhang, Y. Li, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2022, 61, e202207537 CrossRef CAS PubMed
- Y. He, X. Yang, Y. Li, L. Liu, S. Guo, C. Shu, F. Liu, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2022, 12, 1216–1227 CrossRef CAS
- J.-d. Yi, X. Gao, H. Zhou, W. Chen and Y. Wu, Angew. Chem., Int. Ed., 2022, 61, e202212329 CrossRef CAS PubMed
- M. Feng, X. Wu, H. Cheng, Z. Fan, X. Li, F. Cui, S. Fan, Y. Dai, G. Lei and G. He, J. Mater. Chem. A, 2021, 9, 23817–23827 RSC
- H. Cheng, X. Wu, M. Feng, X. Li, G. Lei, Z. Fan, D. Pan, F. Cui and G. He, ACS Catal., 2021, 11, 12673–12681 CrossRef CAS
- S. Cao, S. Zhou, H. Chen, S. Wei, S. Liu, X. Lin, X. Chen, Z. Wang, W. Guo and X. Lu, Energy Environ. Mater., 2021, 6, e12287 CrossRef
- W. Yang, Z. Jia, B. Zhou, L. Chen, X. Ding, L. Jiao, H. Zheng, Z. Gao, Q. Wang and H. Li, ACS Catal., 2023, 13, 9695–9705 CrossRef CAS
- D. Fang, P. Sun, S. Huang, Y. Shang, X. Li, D. Yan, Y. V. Lim, C.-Y. Su, B.-J. Su, J.-Y. Juang and H. Y. Yang, ACS Mater. Lett., 2022, 4, 1–10 CrossRef CAS
- R. Wang, L.-Z. Dong, J.-W. Shi, M. Zhang, S.-L. Li, Y.-Q. Lan and J. Liu, ACS Catal., 2024, 14, 741–750 CrossRef CAS
- D.-S. Huang, H.-L. Zhu, Z.-H. Zhao, J.-R. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 8444–8450 CrossRef CAS
- M. Kato, M. Muto, N. Matsubara, Y. Uemura, Y. Wakisaka, T. Yoneuchi, D. Matsumura, T. Ishihara, T. Tokushima, S.-i. Noro, S. Takakusagi, K. Asakura and I. Yagi, ACS Appl. Energy Mater., 2018, 1, 2358–2364 CrossRef CAS
- W. J. Li, Z. X. Lou, H. Y. Yuan, H. G. Yang and H. F. Wang, J. Mater. Chem. A, 2022, 10, 16106–16114 RSC
- Z. W. Chen, L. X. Chen, M. Jiang, D. Chen, Z. L. Wang, X. Yao, C. V. Singh and Q. Jiang, J. Mater. Chem. A, 2020, 8, 15086–15093 RSC
- X. Fu, X. Zhao, T.-B. Lu, M. Yuan and M. Wang, Angew. Chem., Int. Ed., 2023, 62, e202219242 CrossRef CAS PubMed
- Y. Zhang, J. Yang, R. Ge, J. Zhang, J. M. Cairney, Y. Li, M. Zhu, S. Li and W. Li, Coord. Chem. Rev., 2022, 461, 214493 CrossRef CAS
- G. Bae, S. Han, H.-S. Oh and C. H. Choi, Angew. Chem., Int. Ed., 2023, 62, e202219227 CrossRef CAS PubMed
- Y. Wang, D. Wang and Y. Li, Adv. Mater., 2021, 33, 2008151 CrossRef CAS PubMed
- P. Zhang, K. Chen, J. Li, M. Wang, M. Li, Y. Liu and Y. Pan, Adv. Mater., 2023, 35, 2303243 CrossRef CAS PubMed
- Y. Zhu, J. Sokolowski, X. Song, Y. He, Y. Mei and G. Wu, Adv. Energy Mater., 2020, 10, 1902844 CrossRef CAS
- D. Ji, L. Fan, L. Li, S. Peng, D. Yu, J. Song, S. Ramakrishna and S. Guo, Adv. Mater., 2019, 31, 1808267 CrossRef PubMed
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS
- M. Tong, F. Sun, G. Xing, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2023, 62, e202314933 CrossRef CAS PubMed
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed
- X.-M. Liang, H.-J. Wang, C. Zhang, D.-C. Zhong and T.-B. Lu, Appl. Catal., B, 2023, 322, 122073 CrossRef CAS
- Y.-N. Gong, C.-Y. Cao, W.-J. Shi, J.-H. Zhang, J.-H. Deng, T.-B. Lu and D.-C. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202215187 CrossRef CAS PubMed
- X. Yang, Y. Wang, G. Zhang, L. Du, L. Yang, M. Markiewicz, J.-y. Choi, R. Chenitz and S. Sun, Appl. Catal., B, 2020, 264, 118523 CrossRef
- L. Yu, Y. Li and Y. Ruan, Angew. Chem., Int. Ed., 2021, 60, 25296–25301 CrossRef CAS PubMed
- S. H. Lee, J. Kim, D. Y. Chung, J. M. Yoo, H. S. Lee, M. J. Kim, B. S. Mun, S. G. Kwon, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2019, 141, 2035–2045 CrossRef CAS PubMed
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS
- M. Ferrandon, X. Wang, A. J. Kropf, D. J. Myers, G. Wu, C. M. Johnston and P. Zelenay, Electrochim. Acta, 2013, 110, 282–291 CrossRef CAS
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed
- S. Huang, Z. Qiao, P. Sun, K. Qiao, K. Pei, L. Yang, H. Xu, S. Wang, Y. Huang, Y. Yan and D. Cao, Appl. Catal., B, 2022, 317, 121770 CrossRef CAS
- J. Zhang, M. Zhang, Y. Zeng, J. Chen, L. Qiu, H. Zhou, C. Sun, Y. Yu, C. Zhu and Z. Zhu, Small, 2019, 15, 1900307 CrossRef PubMed
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed
- J. Wan, Z. Zhao, H. Shang, B. Peng, W. Chen, J. Pei, L. Zheng, J. Dong, R. Cao, R. Sarangi, Z. Jiang, D. Zhou, Z. Zhuang, J. Zhang, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 8431–8439 CrossRef CAS PubMed
- K. Li, S. Zhang, X. Zhang, S. Liu, H. Jiang, T. Jiang, C. Shen, Y. Yu and W. Chen, Nano Lett., 2022, 22, 1557–1565 CrossRef CAS PubMed
- M. Huang, B. Deng, X. Zhao, Z. Zhang, F. Li, K. Li, Z. Cui, L. Kong, J. Lu, F. Dong, L. Zhang and P. Chen, ACS Nano, 2022, 16, 2110–2119 CrossRef CAS PubMed
- Z. Fan, R. Luo, Y. Zhang, B. Zhang, P. Zhai, Y. Zhang, C. Wang, J. Gao, W. Zhou, L. Sun and J. Hou, Angew. Chem., Int. Ed., 2023, 62, e202216326 CrossRef CAS PubMed
- F. Wang, R. Zhang, Y. Zhang, Y. Li, J. Zhang, W. Yuan, H. Liu, F. Wang and H. L. Xin, Adv. Funct. Mater., 2023, 33, 2213863 CrossRef CAS
- Y. Yang, K. Mao, S. Gao, H. Huang, G. Xia, Z. Lin, P. Jiang, C. Wang, H. Wang and Q. Chen, Adv. Mater., 2018, 30, 1801732 CrossRef PubMed
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang, B. Pang, D. Wu, L. Cao, L. Wang, T. Liu, Y. Li, H. Sheng, M. Zhu and T. Yao, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS PubMed
- X. Li, S. Mitchell, Y. Fang, J. Li, J. Perez-Ramirez and J. Lu, Nat. Rev. Chem, 2023, 7, 754–767 CrossRef CAS PubMed
- D.-C. Zhong, Y.-N. Gong, C. Zhang and T.-B. Lu, Chem. Soc. Rev., 2023, 52, 3170–3214 RSC
- Z. Pei, H. Zhang, Y. Guo, D. Luan, X. Gu and X. W. Lou, Adv. Mater., 2023, 2306047 CrossRef PubMed
- M. Liu, N. Li, S. Cao, X. Wang, X. Lu, L. Kong, Y. Xu and X.-H. Bu, Adv. Mater., 2022, 34, 2107421 CrossRef CAS PubMed
- Y. Wang, B. J. Park, V. K. Paidi, R. Huang, Y. Lee, K.-J. Noh, K.-S. Lee and J. W. Han, ACS Energy Lett., 2022, 7, 640–649 CrossRef CAS
- X. Yi, H. Yang, X. Yang, X. Li, C. Yan, J. Zhang, L. Chen, J. Dong, J. Qin, G. Zhang, J. Wang, W. Li, Z. Zhou, G. Wu and X. Li, Adv. Funct. Mater., 2023, 2309728 Search PubMed
- B. Zhang, J. Hou, Y. Wu, S. Cao, Z. Li, X. Nie, Z. Gao and L. Sun, Adv. Energy Mater., 2019, 9, 1803693 CrossRef
- X. Yu, S. Lai, S. Xin, S. Chen, X. Zhang, X. She, T. Zhan, X. Zhao and D. Yang, Appl. Catal., B, 2021, 280, 119437 CrossRef CAS
- D. Chen, J. Zhu, X. Mu, R. Cheng, W. Li, S. Liu, Z. Pu, C. Lin and S. Mu, Appl. Catal., B, 2020, 268, 118729 CrossRef CAS
- L. Lv, R. Lu, J. Zhu, R. Yu, W. Zhang, E. Cui, X. Chen, Y. Dai, L. Cui, J. Li, L. Zhou, W. Chen, Z. Wang and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202303117 CrossRef CAS PubMed
- R. Wang, L. Zhang, J. Shan, Y. Yang, J.-F. Lee, T.-Y. Chen, J. Mao, Y. Zhao, L. Yang, Z. Hu and T. Ling, Adv. Sci., 2022, 9, 2203917 CrossRef CAS PubMed
- D. Xue, P. Yuan, S. Jiang, Y. Wei, Y. Zhou, C.-L. Dong, W. Yan, S. Mu and J.-N. Zhang, Nano Energy, 2023, 105, 108020 CrossRef CAS
- Y. Wang, W. Cheng, P. Yuan, G. Yang, S. Mu, J. Liang, H. Xia, K. Guo, M. Liu, S. Zhao, G. Qu, B.-A. Lu, Y. Hu, J. Hu and J.-N. Zhang, Adv. Sci., 2021, 8, 2102915 CrossRef CAS PubMed
- M. Liu, B.-A. Lu, G. Yang, P. Yuan, H. Xia, Y. Wang, K. Guo, S. Zhao, J. Liu, Y. Yu, W. Yan, C.-L. Dong, J.-N. Zhang and S. Mu, Adv. Sci., 2022, 9, 2200147 CrossRef CAS PubMed
- Y. Liu, X. Liu, Z. Lv, R. Liu, L. Li, J. Wang, W. Yang, X. Jiang, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2022, 61, e202117617 CrossRef CAS PubMed
- K. Maiti, S. Maiti, M. T. Curnan, H. J. Kim and J. W. Han, Adv. Energy Mater., 2021, 11, 2101670 CrossRef CAS
- A. Pedersen, J. Barrio, A. Li, R. Jervis, D. J. L. Brett, M. M. Titirici and I. E. L. Stephens, Adv. Energy Mater., 2022, 12, 2102715 CrossRef CAS
- K. Leng, J. Zhang, Y. Wang, D. Li, L. Bai, J. Shi, X. Li, L. Zheng, J. Bai and Y. Qu, Adv. Funct. Mater., 2022, 32, 2205637 CrossRef CAS
- X. Wang, L. Xu, C. Li, C. Zhang, H. Yao, R. Xu, P. Cui, X. Zheng, M. Gu, J. Lee, H. Jiang and M. Huang, Nat. Commun., 2023, 14, 7210 CrossRef CAS PubMed
- Y. He, Q. Shi, W. Shan, X. Li, A. J. Kropf, E. C. Wegener, J. Wright, S. Karakalos, D. Su, D. A. Cullen, G. Wang, D. J. Myers and G. Wu, Angew. Chem., Int. Ed., 2021, 60, 9516–9526 CrossRef CAS PubMed
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed
- Z. Zeng, L. Y. Gan, H. Bin Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang, W. Cai, Z. Zhang, Y. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef CAS PubMed
- X. Hai, Y. Zheng, Q. Yu, N. Guo, S. Xi, X. Zhao, S. Mitchell, X. Luo, V. Tulus, M. Wang, X. Sheng, L. Ren, X. Long, J. Li, P. He, H. Lin, Y. Cui, X. Peng, J. Shi, J. Wu, C. Zhang, R. Zou, G. Guillén-Gosálbez, J. Pérez-Ramírez, M. J. Koh, Y. Zhu, J. Li and J. Lu, Nature, 2023, 622, 754–760 CrossRef CAS PubMed
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS
- S. H. Lee, J. C. Lin, M. Farmand, A. T. Landers, J. T. Feaster, J. E. Avilés Acosta, J. W. Beeman, Y. Ye, J. Yano, A. Mehta, R. C. Davis, T. F. Jaramillo, C. Hahn and W. S. Drisdell, J. Am. Chem. Soc., 2021, 143, 588–592 CrossRef CAS PubMed
- C.-X. Zhao, B.-Q. Li, J.-N. Liu, J.-Q. Huang and Q. Zhang, Chin. Chem. Lett., 2019, 30, 911–914 CrossRef CAS
- S. Cao, S. Wei, X. Wei, S. Zhou, H. Chen, Y. Hu, Z. Wang, S. Liu, W. Guo and X. Lu, Small, 2021, 17, 2100949 CrossRef CAS PubMed
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS
- D. Yao, C. Tang, X. Zhi, B. Johannessen, A. Slattery, S. Chern and S.-Z. Qiao, Adv. Mater., 2022, 35, 2209386 CrossRef PubMed
- X. Cheng, X. Jiang, S. Yin, L. Ji, Y. Yan, G. Li, R. Huang, C. Wang, H. Liao, Y. Jiang and S. Sun, Angew. Chem., Int. Ed., 2023, 62, e202306166 CrossRef CAS PubMed
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |