
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-component dicarbofunctionalization of allylamines via nucleopalladation pathway: unlocking vicinal and geminal selectivity†
Nityananda
Ballav
,
Shib Nath
Saha
,
Shailesh
Yadav
and
Mahiuddin
Baidya
 *
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: mbaidya@iitm.ac.in
First published on 23rd February 2024
Abstract
A palladium(II)-catalyzed vicinal as well as geminal selective dicarbofunctionalization of allylamine embedded in a removable picolinamide auxiliary is developed by exploiting a nucleopalladation-triggered intermolecular three-component coupling reaction. The vicinal selectivity was accomplished by engaging allylamine, indoles, and aryl or styrenyl halides through a Pd(II)/Pd(IV) reaction manifold, while the two-fold coupling of allylamine and indoles via a Pd(II)/Pd(0) reaction modality resulted in geminal selectivity. The protocol is operationally simple, scalable, and offers two distinct types of products bearing functionalized tryptamine and bisindolyl frameworks in very high to excellent yields. The reaction features a wide substrate generality and also remains effective in the presence of various medicinally relevant scaffolds. Notably, this work represents the first example of nucleopalladation-guided intermolecular dicarbofunctionalization of allylamines.
Introduction
Aliphatic amines are ubiquitous substructures found in a wide array of pharmaceutical compounds, natural products, and organic materials.1 They serve as pivotal building blocks for the production of other important functional groups and value-added molecules.1,2 Remarkably, a substantial proportion of top-selling small-molecule drugs are derivatives of aliphatic amines.1a,2 Consequently, the pursuit of innovative and efficient methods en route to functionally enriched aliphatic amines has become a longstanding focus within the synthetic chemistry community.3 One rewarding route could be the catalytic three-component dicarbofunctionalization of alkenyl amines that potentially allows the installation of two new carbon–carbon bonds across the olefin functionality.4 Specifically, the dicarbofunctionalization of a simple allylamine is significant owing to its ready availability while the olefin motif is unactivated and potentially vicinal as well as geminal dicarbofunctionalizations leading to sp3-dense products are feasible (Scheme 1a). In 2018, Zhao et al. exploited Ni(COD)2 as a catalyst and showcased difunctionalization of N-pyrimidyl allyl amine with aryl boronic acids and diverse organohalides where substrate-dependent vicinal and distal selectivities were observed (Scheme 1b).5 In this direction, the Engle group also reported Ni(COD)2 catalyzed vicinal dicarbofunctionalization of N-benzoyl allylamines with aryl boronates and aryl iodides (Scheme 1c).6 However, the operational complexity of these protocols is noteworthy due to the extreme sensitivity of the low valent Ni(COD)2 catalyst, requiring execution in a glovebox and handling in an utmost inert atmosphere. Meanwhile, the dicarbofunctionalization of allylamines with geminal selectivity still remains elusive. Thus, developing an operationally simple catalytic strategy that is tunable for both vicinal as well as geminal dicarbofunctionalizations of allylamines is highly desirable.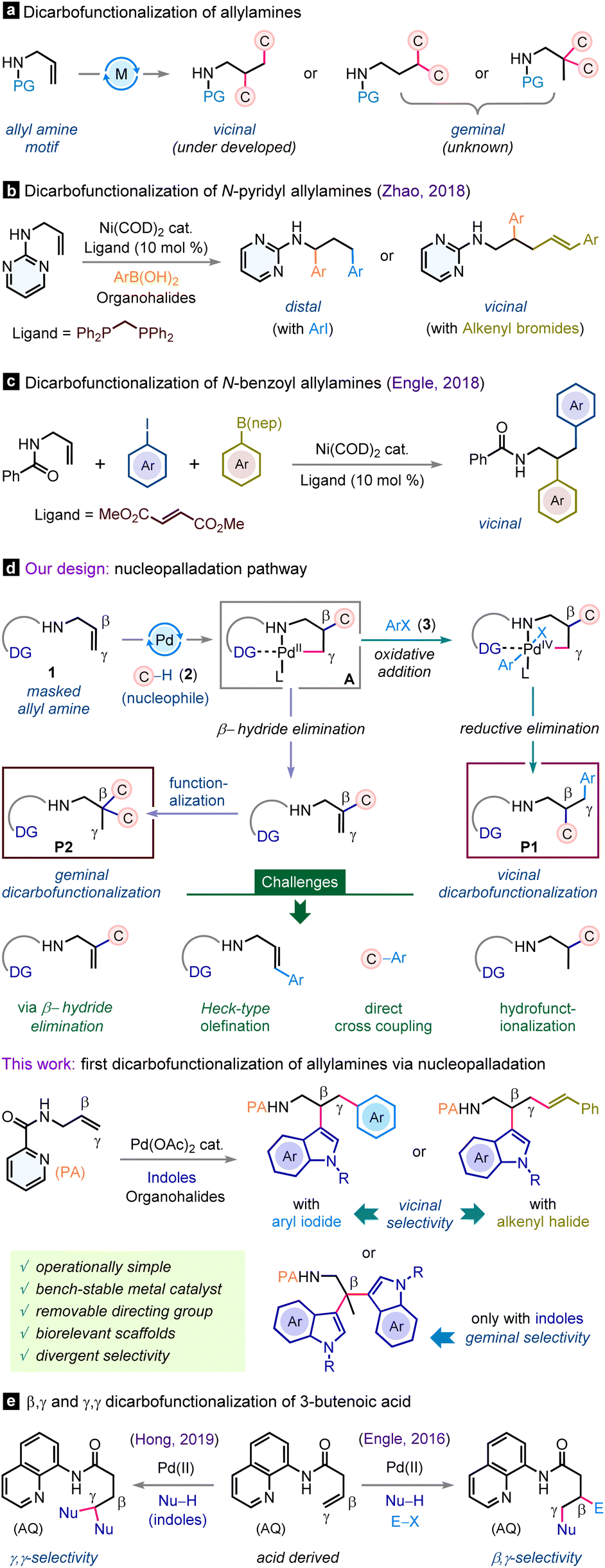 | ||
| Scheme 1 Regioselective dicarbofunctionalization of allylamines. | ||
The nucleopalladation methodology is fundamental, capitalizing on the π-activation of alkenes with palladium metal and concomitant nucleophilic addition-triggered functionalization event.7 Despite its versatile synthetic portfolio, to our surprise, heretofore, this approach has not been explored for intermolecular three-component dicarbofunctionalization of alkenyl amines. With our continuous interest in allylamine functionalization strategies,8 we surmise that the nucleopalladation blueprint may be effective for the rapid dicarbofunctionalization of allylamines (Scheme 1d). Strategically, allylamine (1) can be tethered to a suitable directing group, facilitating its binding with the palladium catalyst and expediting olefin π-activation through coordination. This scenario is expected to favor nucleophilic addition to generate the pivotal palladacycle intermediate A. We expect a β-regioselective attack of nucleophile 2, forming a more stable 5,5-palladacycle intermediate. The intermediate A can partake in the subsequent cross-coupling reaction with suitable carbon electrophile 3 to produce vicinal dicarbofunctionalized product P1 (Scheme 1d). Alternatively, by fine-tuning the reaction conditions, the palladacycle intermediate A can be pushed forward towards β-hydride elimination to regenerate the allylamine motif, which can then undergo further functionalization with the carbon nucleophile, enabling access to the geminal dicarbofunctionalized product P2. However, at the outset, we foresaw several challenges as the progress of the reaction can be prematurely terminated. One potential issue is the susceptibility of the key palladacycle intermediate to facile protodemetalation reactions or a β-hydride elimination event, which may result in undesired mono-functionalization9 rather than the intended difunctionalization (Scheme 1d). Also, the direct cross-coupling10 between nucleophiles and aryl halides or Heck-type functionalization11 of an alkene with aryl iodides in the presence of a palladium catalyst is a renowned protocol and may hamper the reaction outcome.
Herein, we report the first nucleopalladation-guided three-component regioselective dicarbofunctionalization of allylamine bearing a removable picolinamide (PA) auxiliary with inexpensive indole heterocycles and aryl or alkenyl halides. This new protocol is operationally simple, accommodates a wide range of substrates including valuable pharmacophore scaffolds, and has the provision for accessing both vicinal and geminal dicarbofunctionalizations selectively. Notably, our investigations into the dicarbofunctionalization of allylamines complement the prior works of the Engle and Hong groups on dicarbofunctionalization of 3-butenoic amides bearing an 8-aminoquinoline directing group, albeit with different selectivity outcomes (Scheme 1e).12 In our study, nucleopalladation commences at the β-center of allylamine, in contrast to the nucleophilic attack occurring at the γ-center of the 3-butenoic amide in Engle's study. Similarly, while the Hong group accomplished γ,γ-dicarbofunctionalization, we achieved selective β,β-dicarbofunctionalization. This distinct selectivity can be attributed to the formation of a stable 5,5-palladacycle intermediate, as indicated in the preceding section.
Results and discussion
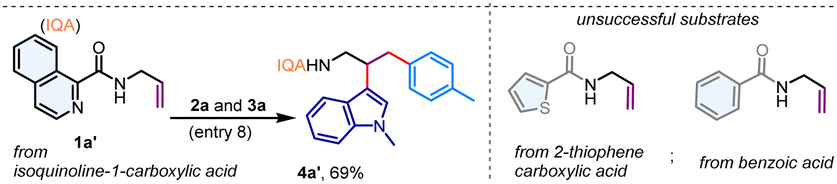
The picolinamide directing group, initially introduced by Daugulis in 2005, has exhibited outstanding directing capabilities in a variety of metal-catalyzed transformations involving both aromatic and aliphatic amines.13 Inspired by these findings, we considered the N-allyl amide 1a, prepared from 2-picolinic acid and allylamine, as a model substrate. Our initial focus was on the three-component coupling of 1a with indole 2a and aryl iodide 3a, potentially yielding a valuable tryptamine derivative (Table 1). Gratifyingly, when a combination of 1a, 2a, and 3a was subjected to a Pd(OAc)2 catalyst in hexafluoroisopropanol (HFIP) solvent with K3PO4 (1.0 equiv.) at 80 °C, we observed the formation of the vicinal dicarbofunctionalization product 4a in a 56% yield (Table 1, entry 1). A comparable yield of 4a was achieved with K2HPO4; however, the reaction yield significantly decreased with K2CO3, and KOAc failed to promote the reaction (entries 2–3). Interestingly, the use of a combination of K3PO4 and K2HPO4 resulted in an improved yield of 69% for 4a (entries 4–5). Exploration of other aprotic solvents such as acetonitrile, trifluorotoluene (TFT), and 1,2-dichloroethane (DCE), as well as polar protic solvents including trifluoroethanol (TFE), methanol, and ethanol, did not yield any difunctionalization product (entries 6–7). Notably, the addition of H2O (10 equiv.) resulted in a remarkably clean reaction, and the desired vicinal difunctionalization product 4a was isolated in a 78% yield (entry 8). Performing the reaction at either lower (60 °C) or higher (100 °C) temperatures produced inferior results (entry 9). Control experiments indicated that the reaction was unproductive in the absence of the Pd(OAc)2 catalyst (entry 10). Additionally, other catalysts such as Co(OAc)2 and Ni(OAc)2 were incapable of promoting this dicarbofunctionalization reaction (entry 11). Investigation into other directing groups revealed that the N-allyl amide derived from isoquinoline-1-carboxylic acid (1a′) also effectively participated in this reaction, yielding 4a′ in a 69% yield. However, such difunctionalization proved unsuccessful with N-allyl amides of 2-thiophenecarboxylic acid as well as benzoic acid, indicating that an effective bidentate coordination is critical for this reaction (Table 1, below).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield of 4a (%)b |
| a Reaction conditions: 1a (0.25 mmol), 2a (1.1 equiv.), 3a (4.0 equiv.), Pd(OAc)2 (5 mol%), K3PO4 (0.5 equiv.), K2HPO4 (1.0 equiv.), H2O (10.0 equiv.), HFIP (1.5 M), 80 °C for 24 h (open air operation). b Isolated yields were provided. c Without H2O additive. | ||
| 1c | Only with K3PO4 (1.0 equiv.) | 56 |
| 2c | Only with K2HPO4 (1.0 equiv.) | 52 |
| 3c | With K2CO3/KOAc instead of phosphates | 38/− |
| 4c | K3PO4 (1.0 equiv.) and K2HPO4 (1.0 equiv.) | 62 |
| 5c | K3PO4 (0.5 equiv.) and K2HPO4 (1.0 equiv.) | 69 |
| 6c | CH3CN, TFT, or DCE instead of HFIP | — |
| 7c | TFE, MeOH, or EtOH instead of HFIP | — |
| 8 | None | 78 |
| 9 | At 60 °C/100 °C | 65/70 |
| 10 | Without Pd(OAc)2 | — |
| 11 | With Co(OAc)2/Ni(OAc)2 instead of Pd(OAc)2 | — |
|
||
After identifying the optimal reaction conditions (Table 1, entry 8), we aimed to explore the substrate diversity in this three-component vicinal dicarbofunctionalization reaction (Scheme 2). Delightfully, the reaction exhibited uniformity across a broad spectrum of structurally diverse aromatic iodides encompassing both electron-donating and electron-withdrawing groups, and desired dicarbofunctionalized products 4b–j were obtained in consistently high yields. Disubstituted aromatic iodide (4m) and 2-naphthyl iodide (4n) were also effective, and commonly used protecting groups, for example, allyl (4k) and benzyl (4l), were well-tolerated. To advance the synthetic utility further, an array of pharmacophore-coupled aryl iodides were explored where substrates featuring bio-relevant motifs like isoxepac (4q), gemfibrozil (4r), ibuprofen (4s), naproxen (4t), and valproic acid (4u) produced the desired products in good to excellent yields. Furthermore, the protocol proved equally efficacious with aryl iodides linked to saturated fatty acid motifs such as lauric acid and palmitic acid, offering 4o and 4p in 73% and 70% yields, respectively.
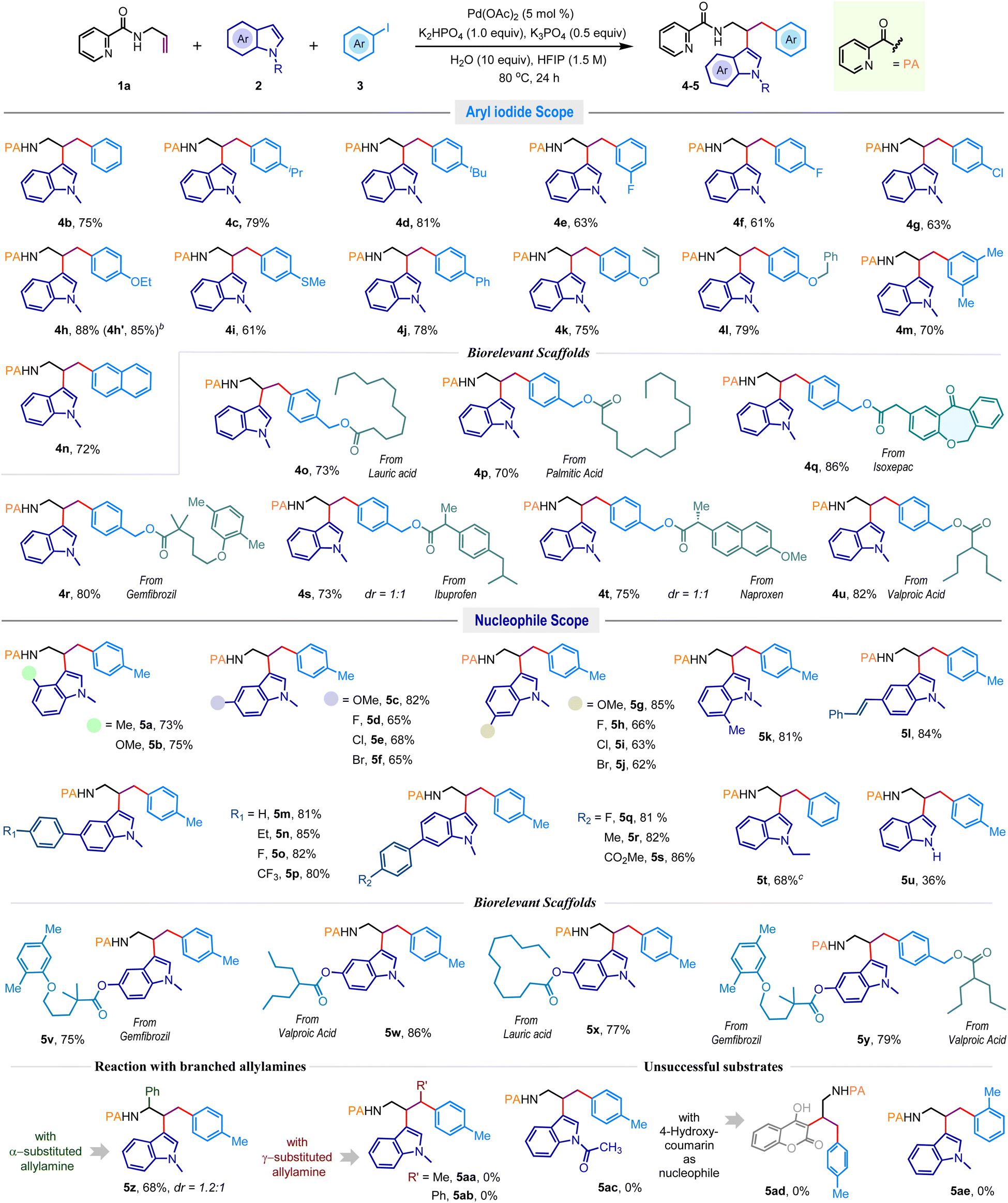 | ||
| Scheme 2 Substrate scope in the vicinal dicarbofunctionalization reactiona. aReaction conditions: 1a (0.25 mmol), 2 (1.1 equiv.), 3 (4.0 equiv.) for 24 h. bIQA was used as a directing group. cIodobenzene (4.0 equiv.) was used. Isolated yields were provided. | ||
Next, we evaluated the reaction competence with different indoles (Scheme 2). N-methylindoles featuring alkyl, alkoxy, and halogen functionalities at the C4, C5, C6, and C7 positions yielded dicarbofunctionalized products 5a–k in high yields. Generally, electron-rich indoles exhibited higher reactivity compared to their electron-deficient counterparts. Halogen functionalities such as fluorine (5d, 5h), chlorine (5e, 5i), and bromine (5f, 5j) remained unaffected under the reaction conditions. The presence of a reactive olefin functionality in the indole did not impede the reaction outcome, producing 5l in 84% yield. Indoles with aryl substitutions at the C5 and C6 positions smoothly produced 5m–p and 5q–s in excellent yields. The N-ethylindole also delivered the desired product 5t in 68% yield; however, the difunctionalization was sluggish with N-unsubstituted indole, giving 5u in 36% yield. Of note, indoles encompassing lauric acid, gemfibrozil, and valproic acid frameworks were also suitable substrates for this reaction and produced high-value amines 5v–x in very high yields. More importantly, the reaction was also fruitful when both indole and aryl iodide were adorned with bioactive motifs. For example, the reaction of 1a with gemfibrozil derived indole and valproic acid derived aryl iodide coupling partners gave densely functionalized amine 5y in 79% isolated yields (Scheme 2). Examination of branched allylamines showed that α-substitution is tolerable, offering 5z in 68% yield, while γ-substitution (5aa–5ab) dramatically hampers the progress of the reaction. Also, the difunctionalization was unsuccessful with less reactive N-acylindole (5ac) and 4-hydroxycoumarin (5ad) nucleophiles, as well as the sterically demanding 2-iodotoluene (5ae) electrophile (Scheme 2, below).
Pleasingly, the same catalytic system is also effective for styrenyl iodide 6a, and vicinal dicarbofunctionalized products bearing a bishomoallylic amine motif (7a–h) were obtained in very high yields (Scheme 3). The reaction was also amenable with styrenyl bromide and TIPS-protected alkynyl bromide where desired products 7f and 7i were isolated in 75% and 46% yields, respectively. However, under the standard reaction conditions, we observed the decomposition of (iodoethynyl)benzene without the formation of the desired product 7j. When the reaction was performed with alkyl iodide, for example, 2-iodobutane, the vicinal difunctionalization did not take place; on the contrary, we surprisingly noticed the formation of geminal difunctionalized product 8a, featuring a valuable bisindole moiety, albeit in poor yield. This finding prompted us to tune the catalytic reaction conditions further to materialize the geminal dicarbofunctionalization of the allylamine motif, which was not heretofore reported. We have realized that such difunctionalization most likely arises through the functionalization of the regenerated olefin as depicted in Scheme 3, below (for an alternative mechanism, see ESI, page S15†).9b,12b
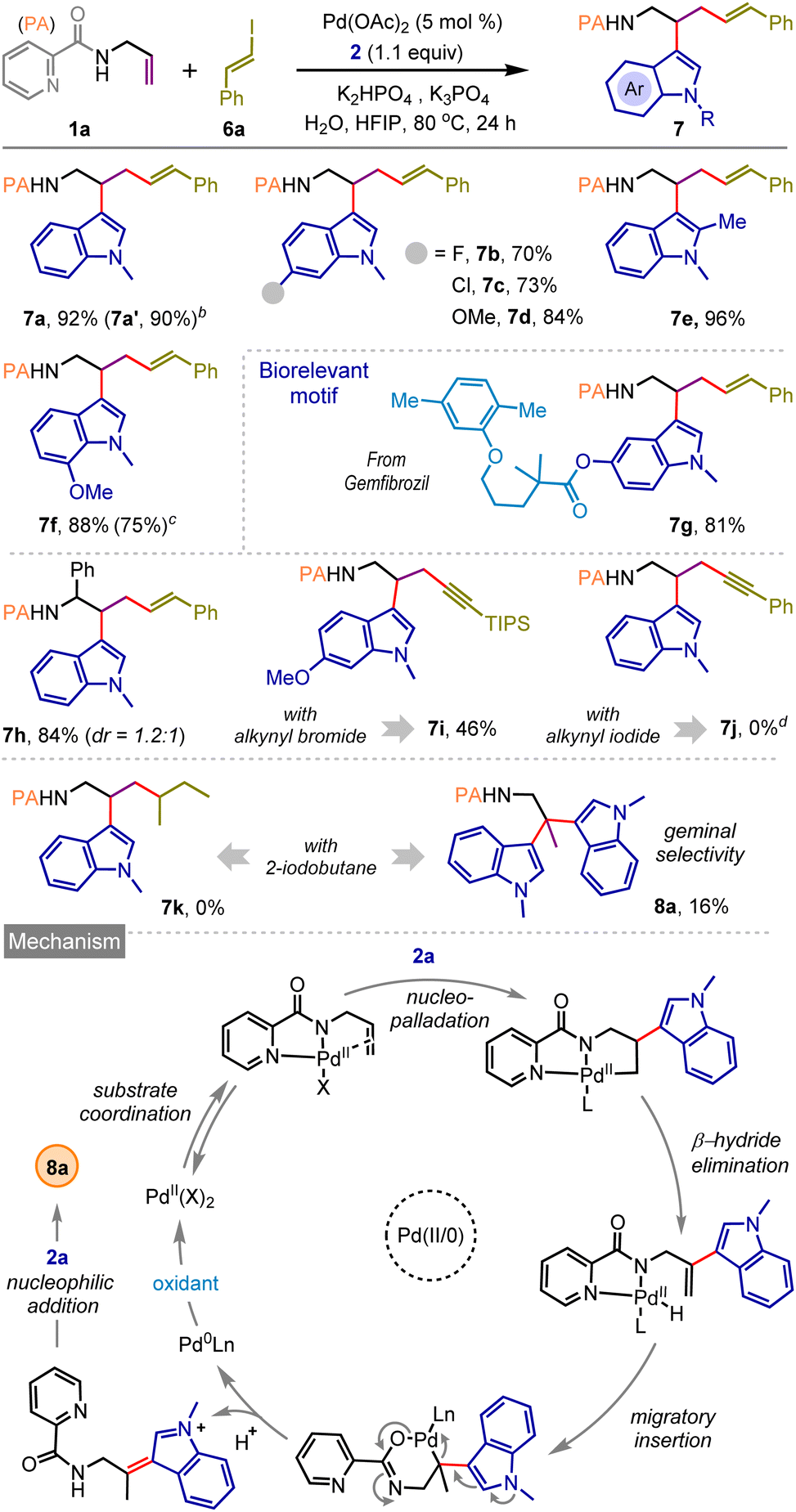 | ||
| Scheme 3 Exploration of three-component dicarbofunctionalization with styrenyl, alkynyl, and alkyl halidesa. aReaction conditions: 1a (0.25 mmol), 2 (1.1 equiv.), 6a (4.0 equiv.), Pd(OAc)2 (5 mol%), K3PO4 (0.5 equiv.), K2HPO4 (1.0 equiv.), H2O (10.0 equiv.), HFIP (1.5 M), 80 °C for 24 h. bIQA was used as a directing group. cStyrenyl bromide (4.0 equiv.) was used. dStarting materials decomposed. | ||
Accordingly, additional oxidants were introduced under the catalytic conditions. Delightfully, the geminal dicarbofunctionalization proceeded smoothly when a mixture of 1a and 2a was treated with the Pd(OAc)2 catalyst in HFIP (0.5 M) solvent in the presence of K3PO4 (1.0 equiv.), 1,4-benzoquinone (BQ, 0.2 equiv.), and MnO2 (3.0 equiv.) to deliver desired product 8a in 92% yield (Scheme 4; see ESI page S9 for optimization details†). The protocol is quite general, enabling the construction of a small library of bis-indolyl14 molecules (8b–8o) with electronically diverse functional groups and substitution patterns (Scheme 4). Compound 8j was crystallized, and single-crystal X-ray analysis unequivocally confirmed the product structure and the selectivity of geminal dicarbofunctionalization.
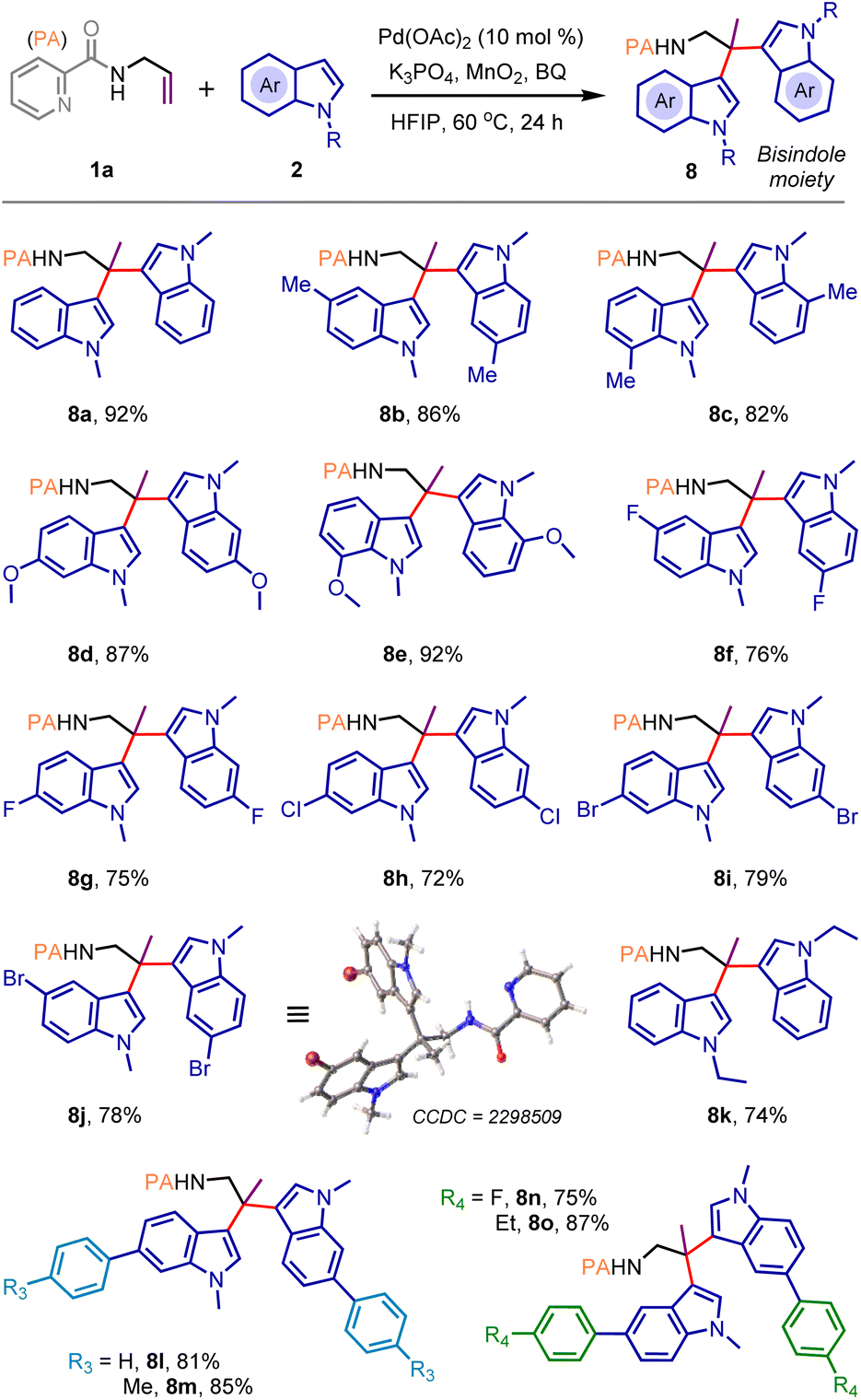 | ||
| Scheme 4 Substrates scope in the geminal dicarbofunctionalization reactiona. aReaction conditions: 1a (0.25 mmol), 2 (2.5 equiv.), Pd(OAc)2 (10 mol%), K3PO4 (1.0 equiv.), MnO2 (3.0 equiv.), 1, 4-benzoquinone (BQ) (0.2 equiv.), HFIP (0.5 M), 60 °C for 24 h (open air operation). | ||
To showcase its synthetic utility, we performed gram–scale reactions. The efficacy of the small-scale reaction was comparable to scale-up synthesis, delivering vicinal functionalization product 7e and geminal functionalization product 8a in 87% and 82% yields, respectively (Scheme 5a). The picolinamide directing group was also removed to access tryptamine analog 9 in 97% yield (Scheme 5b). The product diversification has also been elaborated through picolinamide-directed regioselective alkenylation of 3a, providing functionalized indole 10 in 62% yield (Scheme 5c). In addition, the amino alkynylation of the internal olefin was also accomplished to construct pyrrolidine heterocycle 12 from 7e in synthetically useful yield (Scheme 5d).15
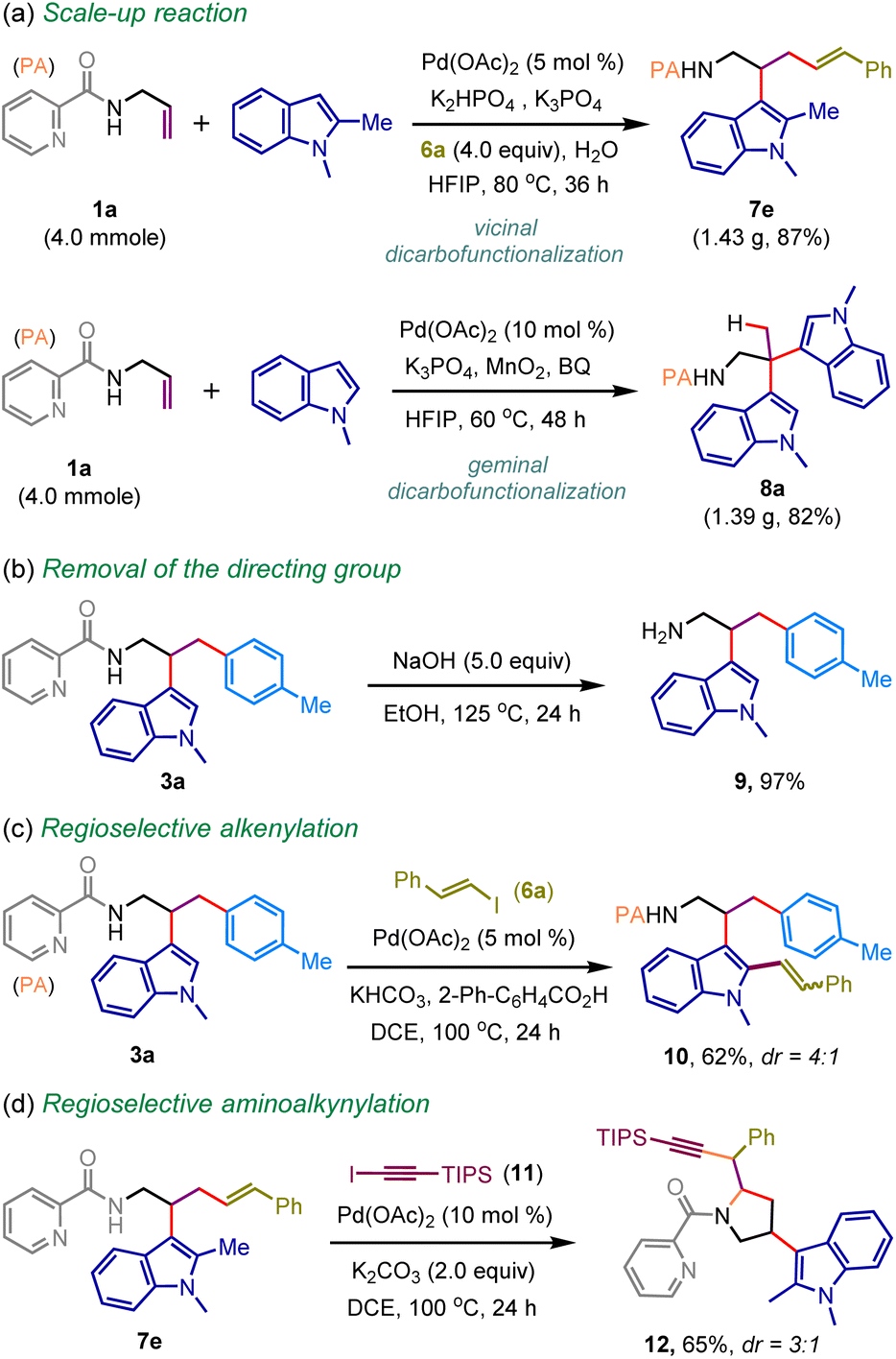 | ||
| Scheme 5 Scale-up and synthetic application. | ||
Conclusions
In summary, we have delineated a palladium(II)-catalyzed dicarbofunctionalization of allylamine, showcasing both vicinal and geminal regioselectivity. This approach utilizes a removable picolinamide auxiliary to facilitate nucleopalladation-triggered intermolecular three-component coupling reactions. The vicinal selectivity is achieved through a Pd(II)/Pd(IV) reaction manifold involving allylamine, indoles, and aryl or styrenyl halides. Conversely, geminal selectivity is realized via a two-fold coupling of allylamine and indoles employing a Pd(II)/Pd(0) reaction modality. The protocol is characterized by its operational simplicity, scalability, substrate generality including diversification of substrates containing pharmacophore scaffolds, and the generation of two distinct types of products featuring functionalized tryptamine and bisindolyl frameworks, all obtained in very high to excellent yields. Importantly, this work represents a pioneering instance of nucleopalladation-guided dicarbofunctionalization of allylamines.Data availability
General information, experimental procedures, characterization data for all new compounds, and NMR spectra are in the ESI.† Data for the crystal structure reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition number CCDC 2298509.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge SERB, India (CRG/2023/001052) for the financial support. N.B. acknowledges CSIR for fellowship, and S.N.S. acknowledges the PMRF fellowship from MHRD, Government of India. We also thank the Department of Chemistry, SAIF, and IIT-Madras for providing instrumental facilities.Notes and references
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (b) A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, 2004 Search PubMed.
- (a) A. Hager, N. Vrielink, D. Hager, J. Lefranc and D. Trauner, Nat. Prod. Rep., 2016, 33, 491–522 RSC; (b) N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- (a) A. Das and J. Waser, Tetrahedron, 2022, 128, 133135 CrossRef CAS; (b) L. Zhu, X. Meng, L. Xie, Q. Shen, W. Li, L. Zhang and C. Wang, Org. Chem. Front., 2022, 9, 3068–3074 RSC; (c) L. Xie, S. Wang, L. Zhang, L. Zhao, C. Luo, L. Mu, X. Wang and C. Wang, Nat. Commun., 2021, 12, 6280 CrossRef CAS PubMed; (d) A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed; (e) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed; (f) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413–2423 CrossRef CAS PubMed; (g) C. Lepori and J. Hannedouche, Synthesis, 2016, 48, 1158–1167 Search PubMed; (h) J. W. Xu, Z. Z. Zhang, W. H. Rao and B. F. Shi, J. Am. Chem. Soc., 2016, 138, 10750–10753 CrossRef CAS PubMed; (i) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed; (j) P. X. Ling, S. L. Fang, X. S. Yin, K. Chen, B. Z. Sun and B. F. Shi, Chem.–An Euro. J., 2015, 21, 17503–17507 CrossRef CAS PubMed; (k) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J. Q. Yu, Nat. Chem., 2014, 6, 146–150 CrossRef CAS PubMed; (l) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675–703 CrossRef PubMed.
- (a) T. Kang, O. Apolinar and K. M. Engle, Synthesis, 2023, 56, 1–15 CrossRef; (b) J. Han, R. He and C. Wang, Chem Catal., 2023, 3, 100690 CrossRef CAS; (c) O. Apolinar, T. Kang, T. M. Alturaifi, P. G. Bedekar, C. Z. Rubel, J. Derosa, B. B. Sanchez, Q. N. Wong, E. J. Sturgell, J. S. Chen, S. R. Wisniewski, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2022, 144, 19337–19343 CrossRef CAS PubMed; (d) S. Wang, C. Luo, L. Zhao, J. Zhao, L. Zhang, B. Zhu and C. Wang, Cell Rep. Phys. Sci., 2021, 2, 100574 CrossRef CAS; (e) V. T. Tran, Z. Q. Li, T. J. Gallagher, J. Derosa, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7029–7034 CrossRef CAS PubMed; (f) O. Apolinar, V. T. Tran, N. Kim, M. A. Schmidt, J. Derosa and K. M. Engle, ACS Catal., 2020, 10, 14234–14239 CrossRef CAS.
- W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600–607 RSC.
- J. Derosa, R. Kleinmans, V. T. Tran, M. K. Karunananda, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 17878–17883 CrossRef CAS PubMed.
- (a) S. Giofrè, L. Molteni and E. M. Beccalli, Eur. J. Org Chem., 2023, 26, e202200976 CrossRef; (b) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed.
- (a) C. K. Giri, S. Dana and M. Baidya, Chem. Commun., 2021, 57, 10536–10539 RSC; (b) N. Ballav, S. Dana and M. Baidya, Org. Lett., 2022, 24, 9228–9232 CrossRef CAS PubMed; (c) N. Ballav, S. N. Saha, S. Yadav and M. Baidya, ChemRxiv, Preprint, 2024, DOI:10.26434/chemrxiv-2024-77qnb.
- (a) Q. Liu, Z. Zhou, Z. Huang and Y. Zhao, J. Org. Chem., 2023, 88, 15350–15357 CrossRef CAS PubMed; (b) K. S. Yang, J. A. Gurak, Z. Liu and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 14705–14712 CrossRef CAS PubMed.
- (a) P. Kumar, P. J. Nagtilak and M. Kapur, New J. Chem., 2021, 45, 13692–13746 RSC; (b) N. Lebrasseur and I. Larrosa, Adv. Heterocycl. Chem., 2012, 105, 309–351 CrossRef CAS; (c) L. Joucla, N. Batail and L. Djakovitch, Adv. Synth. Catal., 2010, 352, 2929–2936 CrossRef CAS.
- R. Parella and S. A. Babu, J. Org. Chem., 2017, 82, 6550–6567 CrossRef CAS PubMed.
- (a) Z. Liu, T. Zeng, K. S. Yang and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 15122–15125 CrossRef CAS PubMed; (b) J. Jeon, H. Ryu, C. Lee, D. Cho, M. H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 10048–10059 CrossRef CAS PubMed.
- (a) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; (b) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- (a) M. Xu, R. Peng, Q. Min, S. Hui, X. Chen, G. Yang and S. Qin, Eur. J. Med. Chem., 2022, 243, 114748 CrossRef CAS PubMed; (b) A. Palmieri and M. Petrini, Synthesis, 2019, 51, 829–841 CrossRef CAS; (c) S. Imran, M. Taha and N. Ismail, Curr. Med. Chem., 2015, 22, 4412–4433 CrossRef CAS PubMed; (d) M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Chem. Rev., 2010, 110, 2250–2293 CrossRef CAS PubMed; (e) S. Safe, S. Papineni and S. Chintharlapalli, Cancer Lett., 2008, 269, 326–338 CrossRef CAS PubMed.
- N. Müller, B. S. Schreib, S. U. Leutenegger and E. M. Carreira, Angew. Chem., Int. Ed., 2022, 61, e202204535 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2298509. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00607k |
| This journal is © The Royal Society of Chemistry 2024 |