
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rare-earth-based chalcogenides and their derivatives: an encouraging IR nonlinear optical material candidate
Ping
Feng†
abcd,
Jia-Xiang
Zhang†
abe,
Mao-Yin
Ran
abe,
Xin-Tao
Wu
 abd,
Hua
Lin
*abd and
Qi-Long
Zhu
*abdf
abd,
Hua
Lin
*abd and
Qi-Long
Zhu
*abdf
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: linhua@fjirsm.ac.cn; qlzhu@fjirsm.ac.cn
bFujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fujian 350108, China
cCollege of Chemistry, Fuzhou University, Fuzhou 350002, China
dFujian College, University of Chinese Academy of Sciences, Fuzhou 350002, China
eUniversity of Chinese Academy of Sciences, Beijing 100049, China
fFujian Key Laboratory of Rare-earth Functional Materials, Fujian Shanhai Collaborative Innovation Center of Rare-earth Functional Materials, Longyan 366300, China
First published on 22nd March 2024
Abstract
With the continuous development of laser technology and the increasing demand for lasers of different frequencies in the infrared (IR) spectrum, research on infrared nonlinear optical (NLO) crystals has garnered growing attention. Currently, the three main commercially available types of borate materials each have their drawbacks, which limit their applications in various areas. Rare-earth (RE)-based chalcogenide compounds, characterized by the unique f-electron configuration, strong positive charges, and high coordination numbers of RE cations, often exhibit distinctive optical responses. In the field of IR-NLO crystals, they have a research history spanning several decades, with increasing interest. However, there is currently no comprehensive review summarizing and analyzing these promising compounds. In this review, we categorize 85 representative examples out of more than 400 non-centrosymmetric (NCS) compounds into four classes based on the connection of different asymmetric building motifs: (1) RE-based chalcogenides containing tetrahedral motifs; (2) RE-based chalcogenides containing lone-pair-electron motifs; (3) RE-based chalcogenides containing [BS3] and [P2Q6] motifs; and (4) RE-based chalcohalides and oxychalcogenides. We provide detailed discussions on their synthesis methods, structures, optical properties, and structure–performance relationships. Finally, we present several favorable suggestions to further explore RE-based chalcogenide compounds. These suggestions aim to approach these compounds from a new perspective in the field of structural chemistry and potentially uncover hidden treasures within the extensive accumulation of previous research.
 Ping Feng | Ping Feng is a joint graduate student at Fujian Institute of Research on the Structure of Matter (FJIRSM) and Fuzhou University (FZU). Her research interests focus on the design and synthesis of RE-based nonlinear optical (NLO) materials for infrared (IR) applications. |
 Jia-Xiang Zhang | Jia-Xiang Zhang is a M.S. candidate at Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS). His research interests focus on the design, synthesis, theory, and structure–property relationships of infrared nonlinear optical (IR NLO) crystals. |
 Mao-Yin Ran | Mao-Yin Ran is a PhD candidate at Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS). He received his Master's degree at Fuzhou University (FZU). His research interest is exploring new heteroanionic nonlinear optical (NLO) materials. |
 Xin-Tao Wu | Xin-Tao Wu is an Academician of Chinese Academy of Sciences. He has published more than 400 peer-reviewed SCI papers and was recognized as the most-cited Chinese author by Elsevier for five consecutive years (2014–2018). He is mainly engaged in structural chemistry and cluster chemistry and has made leading achievements in the fields of sulfur-containing cluster chemistry, rare earth-amino acid cluster chemistry, and supramolecular inorganic assembly chemistry. |
 Hua Lin | Hua Lin is a professor at Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS). He received his PhD degree in Physical Chemistry from FJIRSM-CAS in 2012 and joined Prof. Qi-Long Zhu's group in 2017. He has published more than 120 papers in refereed journals. His research interest currently focuses on the design, synthesis, characterization, and structure–property relationships of thermoelectric (TE) materials and infrared nonlinear optical (IR NLO) crystals. |
 Qi-Long Zhu | Qi-Long Zhu is a professor at Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS). He received his PhD degree in Physical Chemistry from FJIRSM-CAS in 2012. He then joined Prof. Qiang Xu's group at National Institute of Advanced Industrial Science and Technology (AIST, Japan) as a postdoctoral and JSPS (Japan Society for the Promotion of Science) fellow before taking his current faculty position in 2017. He has published more than 170 papers in refereed journals. His group currently focuses on heterogeneous catalysis and nonlinear optics. |
1. Introduction
The laser, an acronym for “Light Amplification by Stimulated Emission of Radiation,” has found extensive applications in the military, technology, medical, and industrial production sectors. The outstanding features of lasers, including excellent collimation, monochromaticity, coherence, and high brightness, make them an indispensable tool.1–5 However, these advantages of lasers also pose limitations to their development. The generation mechanism of lasers dictates that their wavelength is constrained. Second harmonic generation (SHG), which is a nonlinear optical (NLO) effect, enables the expansion of the working wavelengths for lasers.6–20 Extensive research has been carried out in the ultraviolet-visible (UV-vis) spectrum over the past few decades, with a focus on inorganic crystals known for their stability and significant effective frequency-doubling coefficient (deff). Crystals such as KH2PO4 (KDP),21 KTiOPO4 (KTP),22 LiB3O5 (LBO),23 and β-BaB2O4 (β-BBO)24 have been discovered and boast excellent properties.Due to the evolving demands of laser technology, the UV-vis spectrum is no longer sufficient. This has led to a need for research into longer and shorter wavelengths. The study of NLO crystals in the infrared (IR) and deep ultraviolet (DUV) ranges has become crucial in laser technology. Currently, the commercially available IR-NLO crystals primarily consist of AgGaS2,25 AgGaSe2,26 and ZnGeP2.27 However, they all have some unavoidable drawbacks. For instance, AgGaS2 and AgGaSe2 both have a low laser-induced damage threshold (LIDT) and AgGaSe2 is non-phase-matching (NPM) at 1064 nm. Additionally, ZnGeP2 exhibits unfavourable multi-photon absorption. Therefore, there is an urgent demand for superior commercial large crystals, which has prompted research into outstanding candidates for IR-NLO crystals. The traditional method of exploring new IR-NLO crystals involves using powder sample tests to evaluate performance instead of waiting for large-size single crystals. However, this traditional experiment is time consuming. Nowadays, new theoretical calculation techniques are often used to deepen the knowledge of the structure–performance relationship and aid in design, even using machine learning techniques such as SHG-weighted density analysis, the real-space atom-cutting technique, the dipole flexibility model, and partial response functionals.28–38 Through a dual analysis involving both experimental and theoretical calculations, optical testing conditions can be determined for the identification of promising candidates as ideal IR-NLO materials.
To meet the demands of commercial applications, an ideal candidate for an IR-NLO crystal must fulfil several conditions.39–53 Firstly, it should crystallize in a non-centrosymmetric (NCS) space group, which is a prerequisite for being a NLO crystal. Secondly, it should have a large deff value, preferably more than 4 pm V−1 (but even better if it exceeds 8 pm V−1), in order to enhance the conversion efficiency. Additionally, it should have a high LIDT to withstand high power densities of fundamental frequency light waves. The LIDT is positively correlated with the band gap (Eg), which should be at least 3.0 eV, but it is even more desirable for it to be above 3.5 eV. Furthermore, the crystal should possess moderate birefringence (Δn) falling within the range of 0.03–0.10. Excessive birefringence can lead to the walk-off effect, while a small birefringence is not conducive to achieving phase-matching (PM). In addition, the crystal should have a wide optical transparency range that encompasses two significant atmospheric windows: 3–5 μm and 8–12 μm. Finally, the perfect IR-NLO crystal should exhibit stable physical and chemical properties, maintaining stability even when exposed to the air. It should also demonstrate thermal stability, which is advantageous for growing large-sized crystals. Among the various families of materials, chalcogenides show promising potential as excellent candidates for IR-NLO crystals. Extensive research has been conducted on these materials, demonstrating their ability to achieve a superior balance between deff and Eg compared to other families.
Rare-earth (RE)-based chalcogenides, which are a subset of chalcogenides, continue to garner attention from researchers due to the unique properties of lanthanide elements. According to Pearson's hard and soft acid–base theory,54 RE3+ cations are categorized as hard bases. As a result, the bonds formed between these cations and anions exhibit a certain degree of ionic character, similar to alkaline earth metal cations. Additionally, the covalent nature of these bonds is primarily derived from the outer 5d and 6s orbitals. The 4f orbitals situated in the inner shell receive shielding from the outer 5s and 5p electrons, limiting their contribution to bonding. As the atomic number increases, the 4f orbitals become more dispersed, causing these electrons to not fully occupy the inner regions of the 5s and 5p orbitals. This incomplete shielding effect on the nucleus results in the phenomenon known as lanthanide contraction. Consequently, the ionic radii of Sc3+ and Y3+ are similar to those of Lu3+ and Er3+ in the lanthanide series, hence, Y is classified as a member of the RE elements.
Due to the relatively large size of RE3+ cations, they can accommodate higher coordination numbers. Their common coordination numbers are typically less than or equal to 9, approaching the sum of the 6s, 6p, and 5d orbitals, and can even reach up to 12. This complexity in coordination numbers contributes to the diverse geometric configurations of their polyhedra. The combination of the high coordination numbers and moderate positive charge of RE3+ cations enables them to effectively disperse excess negative charges on anionic groups. This dispersion reduces the mutual attraction between anionic groups, presenting an opportunity to obtain low-dimensional anionic framework materials.
So far, numerous reviews on IR-NLO materials have been published. While there are a few examples involving a small amount of RE-based materials,55–67 there has been no systematic overview analyzing and summarizing the fascinating RE-based chalcogenide family. In order to better illustrate the advantages of RE-based chalcogenide compounds and address the challenges in this field, this paper provides detailed explanations for 85 representative examples out of over 400 NCS compounds.68–187 These examples are categorized into four groups based on the various asymmetric building motifs: (1) RE-based chalcogenides containing tetrahedral motifs; (2) RE-based chalcogenides containing lone-pair-electron motifs; (3) RE-based chalcogenides containing [BS3] and [P2Q6] motifs; and (4) RE-based chalcohalides and oxychalcogenides. Finally, the conclusions and perspectives for RE-based chalcogenides and their derivatives are provided for further exploration.
2. RE-based chalcogenides containing various asymmetric building motifs
2.1. RE-based chalcogenides containing tetrahedral motifs
Spontaneous self-aggregation of a single [MQ4] anion disperses negative charges and increases the likelihood of acquiring a high-dimensional framework. RE cations exhibit excellent abilities to disperse negative charges, resulting in the formation of RE–Q bonds when charges are transferred to the anion. This creates favourable conditions for the generation of low-dimensional anionic frameworks. Additionally, the charge transfer optimizes the distribution of electrons in the compound, contributing more effectively to the NLO effects under the synergistic action of the polyhedral [REQn] and tetrahedral [MQ4] asymmetric building motifs. In the La3LiMIVS7 (MIV = Ge and Sn) system, the presence of a closed-ring structure for La–S–MIV reveals the existence of d–p π bonds attributed to the La-5d orbitals and S-3p orbitals. However, the empty 5d orbitals of La3+ lack additional 5d electrons to form d–p back-bonding π bonds. As a result, the formation of extra bonds between RE3+ and Q2− is hindered, limiting the enhancement of induced dipole moment oscillations. On the other hand, the lack of d electrons can be seen as an advantage as it avoids d–d and f–f transitions and enables better absorption of light. Next, the twelve systems containing tetrahedral motifs are introduced.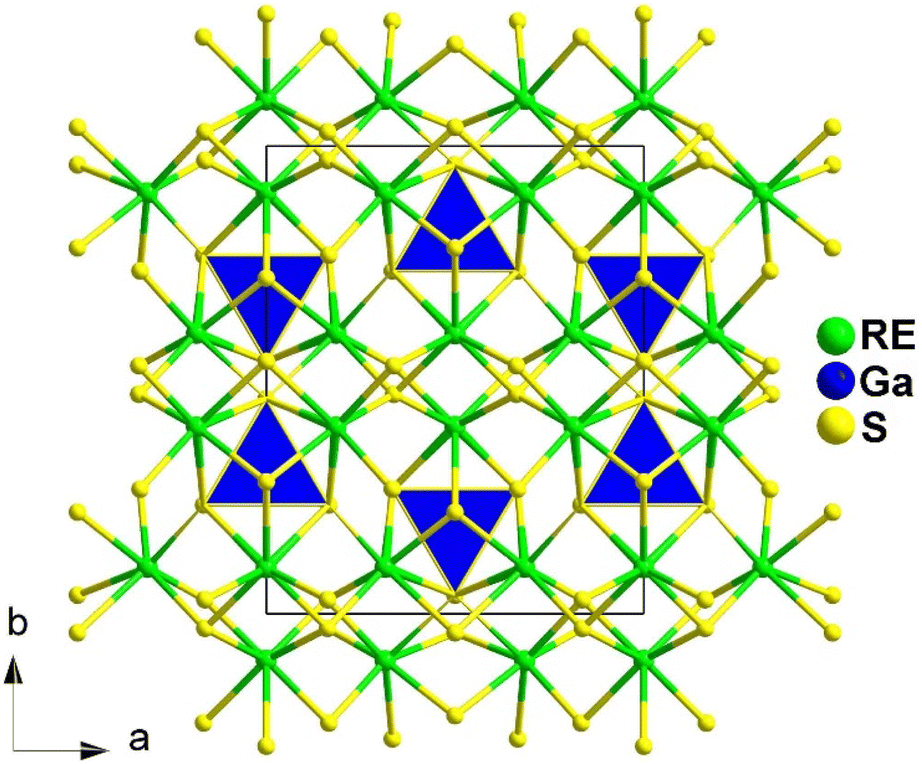 | ||
| Fig. 1 Structures of RE3GaS6 along the c axis with unit cell and discrete [GaS4] polyhedra (blue) outlined. | ||
The results of the SHG test indicate that the [RES7] functional primitives play a key role in the NLO activity, in addition to the [GaS4] tetrahedron. The deff of RE3GaS6 increases as the Shannon ionic radius of the RE3+ ion decreases, namely, in the approximate sequence Dy3GaS6 < Ho3GaS6 ≈ Y3GaS6 < Er3GaS6 when irradiated under a 2100 nm laser. This increase in deff can be attributed to the increasing covalent bond feature of the RE–S bonds. Upon comparing the calculation results for RE3GaS6, it can be concluded that the frontier orbitals of all compounds are primarily influenced by the RE element and S, with very little involvement of Ga. Additionally, the calculated SHG coefficients for these sulfides closely match their experimental deff. Furthermore, the calculated value of Δn, which determines the phase-matchability of IR-NLO materials, is also in line with experimental PM behaviors.
The structure of Eu8Sn4Se20, which belongs to the orthorhombic space group P21212, is displayed in Fig. 2a. The fundamental structural components consist of discrete [Sn4Se14] tetra-nuclear clusters and [Se3] trimers. The coordination environments of the Eu and Sn atoms in the structure are depicted in Fig. 2b and c. It is worth noting that Guo's group, through literature research and detailed structural analysis, pointed out in their report that the coordination geometry of Sn should be more reasonable for four-fold-coordination and six-fold-coordination, contrary to the 4- and 5-coordination mentioned in Yang's article.
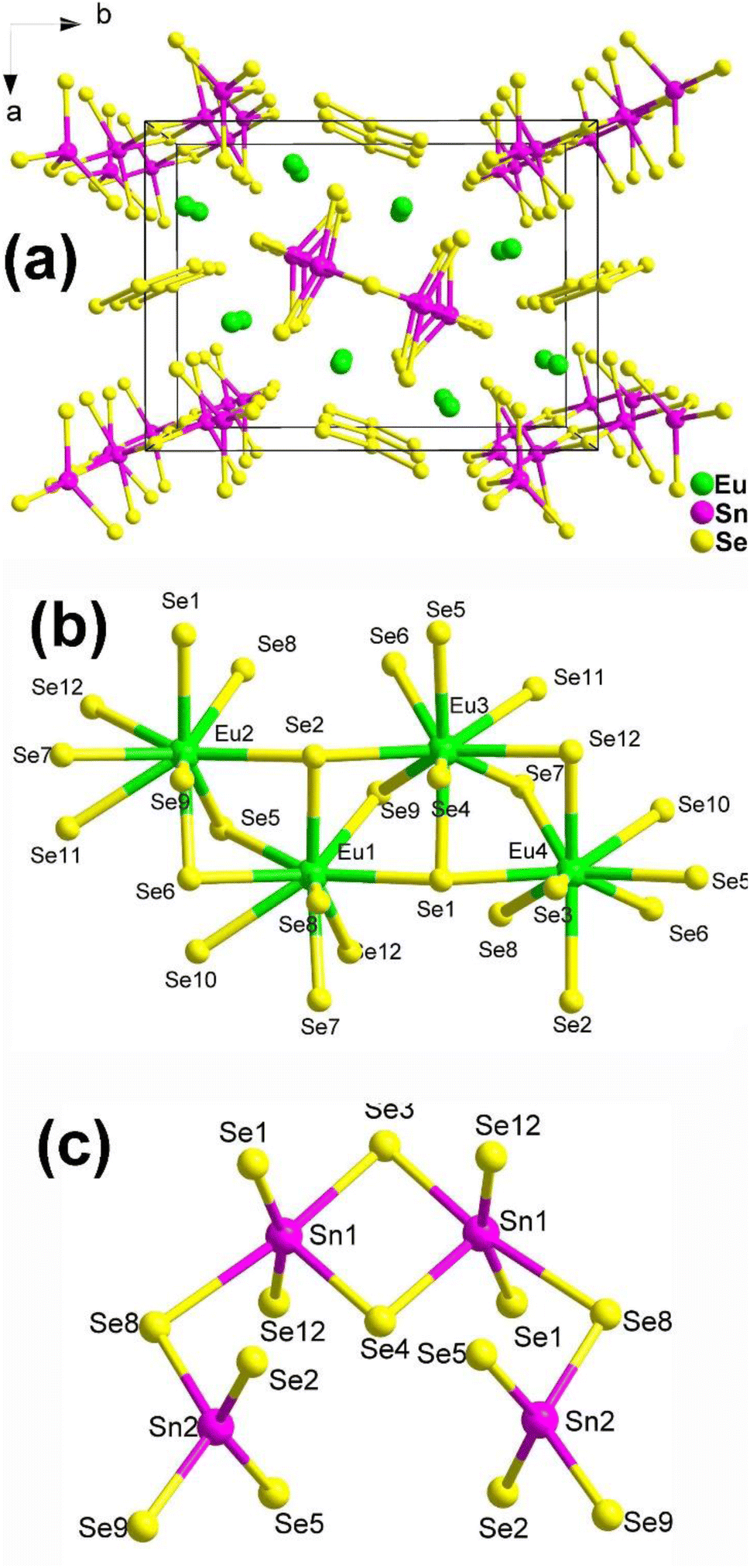 | ||
| Fig. 2 (a) Central projection of Eu8Sn4Se20 along the c axis with the unit cell outlined; (b and c) coordination environment of the crystallographically independent Eu atoms and the discrete [Sn4Se14] tetra-nuclear cluster. | ||
Experimental results reveal that Eu8Sn4Se20 possesses an indirect Eg of 1.33 eV and displays antiferromagnetic-type behaviour. Most significantly, Eu8Sn4Se20 exhibits SHG activity (1 × α-SiO2 in the range of 150–210 μm under 2010 nm laser radiation), although the intensity is not particularly promising. Dipole moment measurements indicate that the majority of the dipole moment vectors of Eu/Se are in opposite directions, resulting in smaller dipole moments. This could be the primary reason for the compound's weak SHG signals.
Although these compounds have the same composition ratio, they crystallize in three different spatial groups represented by the compounds EuCu2SiS4 (space group: P3121), (b) EuCu2GeS4 (space group: P3221), and (c) EuCu2SnS4 (space group: Ama2). Compounds EuCu2SiS4 and EuCu2GeS4 have similar coordination geometries and crystal structures (Fig. 3a and b), while EuCu2SnS4 has completely different ones (Fig. 3c). Similar phenomena are also common in the MIIMI2MIVQ4 family.188–192 Therefore, the description of their structures will only focus on EuCu2SiS4 and EuCu2SnS4.
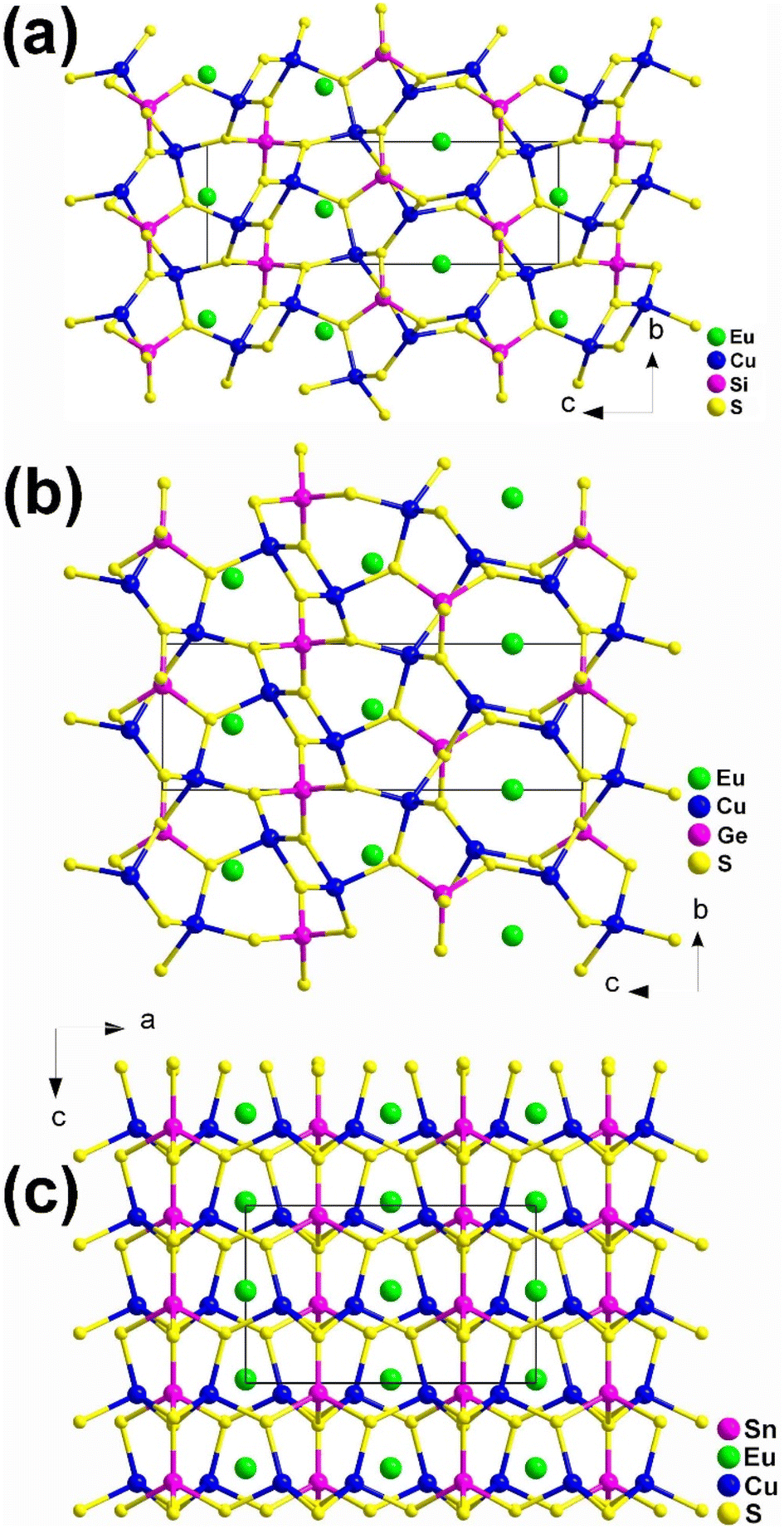 | ||
| Fig. 3 Structures of (a) EuCu2SiS4 (space group: P3121), (b) EuCu2GeS4 (space group: P3221), and (c) EuCu2SnS4 (space group: Ama2) with the unit cell outlined. | ||
In EuCu2SiS4, there is 1 Eu, 1 Cu, 1 Si, and 2 S atoms in the crystallographically unique unit. This structure can be seen as a 3D network formed by [EuS8] bicapped trigonal prisms, with Cu and Si atoms occupying the tetrahedral cavities (Fig. 3a). Each [EuS8] motif links 8 neighboring [EuS8] motifs via sharing vertexes and interconnects with 2, 4, and 2 [CuS4] tetrahedra by sharing corners, edges, and faces, respectively.
For EuCu2SnS4, there is 1 Eu, 1 Cu, 1 Sn, and 3 S atoms in its crystallographically unique units. It also demonstrates a 3D framework made of [EuS8] motifs, with each [EuS8] motif connecting with 4 and 2 neighboring [EuS8] and [CuS4] groups by sharing corners and edges, respectively. It also links 6, 2, and 2 [SnS4] tetrahedra through sharing vertexes, edges, and faces, respectively (Fig. 3c).
Although EuCu2MIVQ4 (MIV = Si, Ge; Q = S, Se) belong to the NCS space groups, no apparent SHG signals were detected under the two most commonly used wavelengths of 1064 nm and 2100 nm. This result is completely different from the previously reported isomorphic compounds of alkaline earth metal groups with strong SHG signals, and the underlying mechanism is currently unclear.
 | ||
| Fig. 4 Two classic structural types in the RE3M1−xMIVQ7 family: (a) La3CuSiS7-type and (b) La3FeGaS7-type structures with the unit cell outlined. | ||
It is worth mentioning that the compounds in this family exhibit significantly different SHG intensities, the underlying reasons for which have not been revealed. In 2015, Chen's research group provided an unprecedented explanation for the unique atomic distribution within this family, which is observed on both the octahedral 2a and tetrahedral 2b sites. This distribution is a direct consequence of the combined considerations of total energy and charge balance. What is even more intriguing is that, in the case of the RE3M1−xM′Q7 family, the atomic distribution predominantly determines the NLO properties. Consequently, members exhibiting a high deff value should adhere to the formula RE3M0.5MIVQ7.
Recently, Wu's group discovered a series of new isomorphic compounds, RE3LiMS7. Compared with the previously reported NPM behavior, these compounds exhibit useful PM features. Among them, La3LiGeS7 and La3LiSnS7 show strong deff values (0.7 and 1.2 × AgGaS2, respectively) and large LIDTs (6.0 and 2.5 × AgGaS2, respectively). Detailed theoretical calculations indicate that the deff values of the RE3LiMS7 family originate from the cooperation of intrinsic dipole moments and d–p delocalized-π-electron-induced dipole oscillations.
EuMIIMIVQ4 (MII = Cd, Hg; MIV = Ge, Sn; Q = S, Se) are isostructural and crystallize in the NCS orthorhombic Ama2 space group. As shown in Fig. 5, a 2D covalent layer of [MIIMIVQ4]2− is made of tetrahedral [MIIQ4] and [MIVQ4] motifs, and further stacks along the a direction with [EuQ8] units occupying the intervals. The geometry of the [EuQ8] units can be viewed as a distorted bicapped trigonal prism.
 | ||
| Fig. 5 Structures of EuMIIMIVQ4 (space group: Ama2) along the c axis with the unit cell outlined. | ||
Owing to the structural traits that arrange the overall NLO-active motifs ([MIIQ4] and [MIVQ4] units) in an orderly manner along a specific direction, all units exhibit strong deff (approximately 2–4 × AgGaS2@2090 nm) along with type-I PM behaviour. Furthermore, theoretical calculations and local dipole moment analyses suggest the significant contribution of distorted [MIIQ4] tetrahedra to the improved deff. These results confirm the intriguing potential applications of these materials as IR NLO crystals. This work also indicates the feasibility of replacing AE elements with RE elements in the exploration of novel IR-NLO candidates.
![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21m. They formed stacked 2D layers of interconnected [GaS4] and [GeS4] tetrahedral functional primitives with La3+ or Eu2+ cations filled into these layers (Fig. 6). However, their functional primitives are linked in different fashions. The 2D [Ga2GeS8]6− layers in La2Ga2GeS8 are formed by wavy 1D [GaS4] chains that are interlinked by individual [GeS4] units through sharing S corners. In contrast, the 2D [Ga2GeS7]4− layers in Eu2Ga2GeS7 are formed by [(GaS4)2] dimers connected via individual [GeS4] units.
21m. They formed stacked 2D layers of interconnected [GaS4] and [GeS4] tetrahedral functional primitives with La3+ or Eu2+ cations filled into these layers (Fig. 6). However, their functional primitives are linked in different fashions. The 2D [Ga2GeS8]6− layers in La2Ga2GeS8 are formed by wavy 1D [GaS4] chains that are interlinked by individual [GeS4] units through sharing S corners. In contrast, the 2D [Ga2GeS7]4− layers in Eu2Ga2GeS7 are formed by [(GaS4)2] dimers connected via individual [GeS4] units.
 | ||
| Fig. 6 Structures of (a) La2Ga2GeS8 and (b) Eu2Ga2GeS7 along the ac plane with the unit cell outlined. | ||
It is worth mentioning that the number of terminal S atoms per formula plays a crucial role in determining the linkage of [GaS4] and [GeS4] functional primitives as well as the aggregation density of the anionic moieties. Theoretical analysis shows that RE cations narrow the Eg but Li does not, and that all components affect the deff through the electronic transitions from the S-3p state to the La/Eu/Li–S, Ga–S, and Ge–S antibonding states. This understanding may provide useful guidance for the further exploration and development of novel IR-NLO materials. Interestingly, the powder Eu2Ga2GeS7 exhibits a large deff (1.6 × AgGaS2@2050 nm) with type-I NPM features, a wide transparent range, and a large theoretical Δn.
According to the report, the Ba2REGaSe5 (RE = Y, Nd, Sm, Gd, Dy, and Er) and Ba2REGaTe5 (RE = Sm and Gd) compounds are isomorphic, with a centrosymmetric triclinic system in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. On the other hand, the Ba2REInSe5 (RE = Y, Nd, Sm, Gd, Dy, and Er), Ba2REGaTe5 (RE = Y, Dy, and Er), and Ba2REInTe5 (RE = Y, Ce, Nd, Sm, Gd, Dy, and Er) compounds belong to an NCS orthorhombic system in the Cmc21 space group. This discussion will focus solely on the NCS compounds.
space group. On the other hand, the Ba2REInSe5 (RE = Y, Nd, Sm, Gd, Dy, and Er), Ba2REGaTe5 (RE = Y, Dy, and Er), and Ba2REInTe5 (RE = Y, Ce, Nd, Sm, Gd, Dy, and Er) compounds belong to an NCS orthorhombic system in the Cmc21 space group. This discussion will focus solely on the NCS compounds.
Fig. 7 shows the structure of the NCS Ba2REMIIIQ5 compounds. The structure consists of 1D [REMIIIQ5]4− chains formed by two types of chains connected through shared Q atoms. These two types of chains are constructed by [REQ6] octahedra and link together through shared edges, whereas [MIIIQ4] tetrahedra connect through shared vertices.
 | ||
| Fig. 7 Structure of Ba2REMIIIQ5 along the bc plane with the unit cell outlined. | ||
Experimental results reveal that the deff value of Ba2InYSe5 is similar to that of AgGaSe2, making it the largest value among the compounds in this family. The Ba2REInSe5 (RE = Gd, Er) compounds exhibit very weak SHG responses, while the Nd, Sm, and Dy compounds show undetectable SHG responses. The detection of weak SHG signals or the failure to detect signals in the compounds of this system may be attributed to the absorption of fundamental and harmonic light by the sample. Similar phenomena have also been observed in other NCS chalcogenides.203–205 On the other hand, it is evident that in the Ba2REInSe5 compounds, the RE metal cation does not impact the crystal structure but does affect the intensity of the deff.
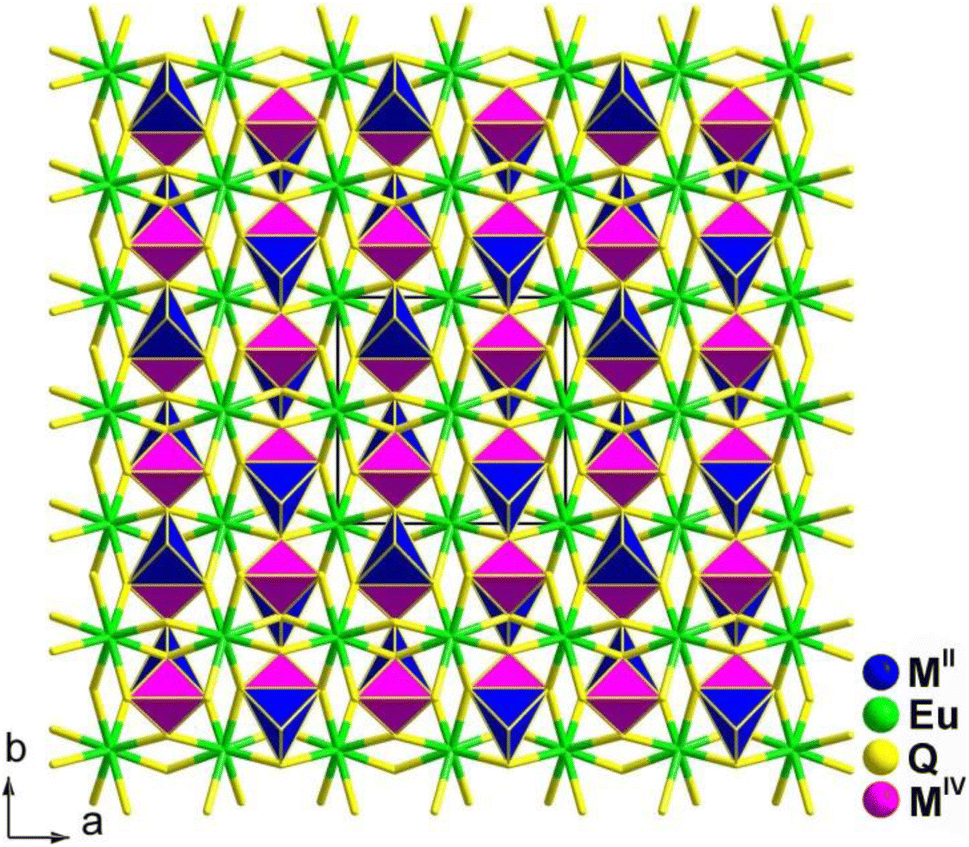
The preparation of the flat needle transparent-brown crystals of Ba4RE2Cd3S10 involved a solid-phase method of elemental mixtures in a KCl/BaCl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) flux at 1173 K. All of them are isostructural and adopt the NCS orthorhombic space group Cmc21 (no. 36). Fig. 8 displays a view of the structure of Ba4RE2Cd3S10 on the bc plane, where the most prominent feature is the 2D complex anionic [RE2Cd3S10]8− layers stacked in an ABABAB fashion along the ab-plane, with discrete Ba2+ cations located in between.
:3) flux at 1173 K. All of them are isostructural and adopt the NCS orthorhombic space group Cmc21 (no. 36). Fig. 8 displays a view of the structure of Ba4RE2Cd3S10 on the bc plane, where the most prominent feature is the 2D complex anionic [RE2Cd3S10]8− layers stacked in an ABABAB fashion along the ab-plane, with discrete Ba2+ cations located in between.
 | ||
| Fig. 8 Structure of Ba4RE2Cd3S10 along the a axis with the unit cell outlined. | ||
Remarkably, Ba4Sm2Cd3S10 exhibits a strong deff (1.8 × AgGaS2) and a significantly higher LIDT (14.3 × AgGaS2). This is the first case of a quaternary AE/RE/TM/Q system possessing an IR-NLO property. Furthermore, the theoretical results for the structure–activity relationships suggest that the combined action of different types of NLO-active motifs (i.e., [RES6] and [CdS4]) contributes to the SHG activity.
LaAEMIII3S7 adopt the P21m space group of the tetragonal system. In the structure, La and AE atoms co-occupy the same position with occupation ratios of 50% and 50%. As shown in Fig. 9, one [MIIIS4] motif is connected with four other [MIIIS4] motifs to generate a [MIII5S16]17− windmill cluster and further interlink together to form a 2D Cairo pentagonal layer along the c direction. Polyhedral [(La/AE)S8] groups filled in the interlayers to bridge neighbouring layers together to build the overall 3D framework.
 | ||
| Fig. 9 Structure of LaAEMIII3S7 along the b axis with the unit cell outlined. | ||
It should be noted that the 2D Cairo pentagonal layer in LaAEAl3S7 contributes to their excellent optical performance. This includes a large deff (0.8–1.1 times that of AgGaS2), a broad optical Eg of 3.76–3.78 eV (the first known cases to achieve the breakthrough of the “3.5 eV wall” among all RE-based IR-NLO chalcogenides), high LIDTs (approximately 9 times that of AgGaS2), and moderate Δn (0.059 for the Ca-compound and 0.077 for the Sr compound at 2000 nm) for PM. These findings offer a new structural guidance route for the development of high-performance IR-NLO chalcogenides.
![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 2m), as well as 17 CS compounds, RE2AE3Ge3S12 and RE2AE3Si3S12, constructed from rigid [GeS4] or [SiS4] tetrahedra.141,142
2m), as well as 17 CS compounds, RE2AE3Ge3S12 and RE2AE3Si3S12, constructed from rigid [GeS4] or [SiS4] tetrahedra.141,142
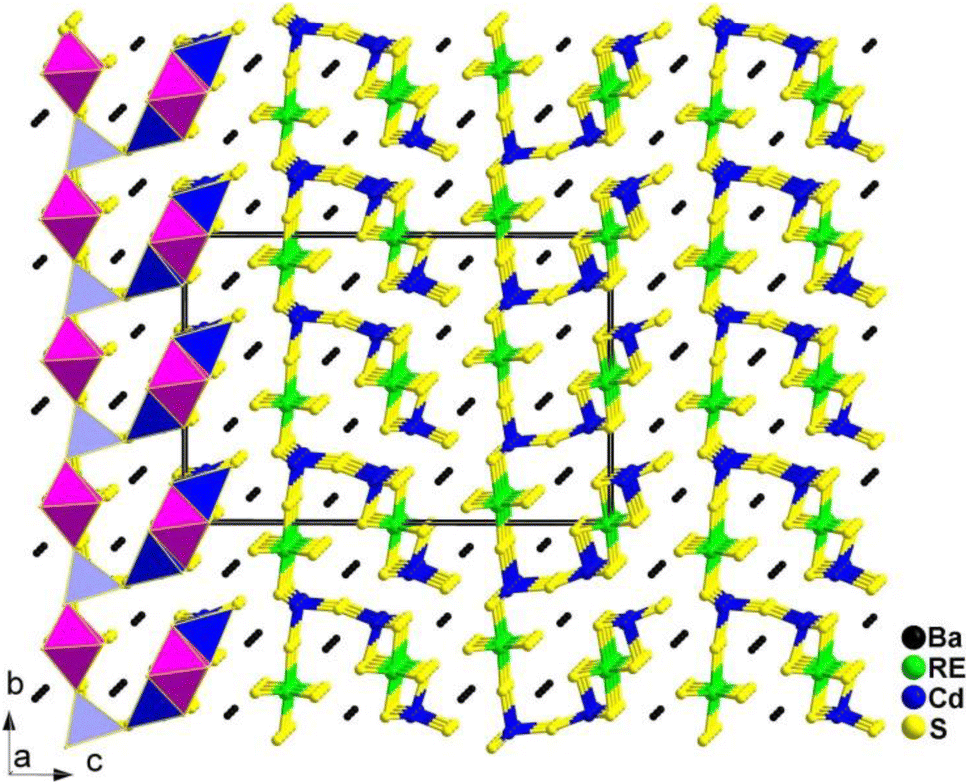
La2Sr3MIV3S12 (MIV = Si, Ge), La2Ca3Sn3S12, and La2Sr3Sn3S12 serve as three typical examples to illustrate the inherent structural features of the family (Fig. 10). The structure of La2Sr3MIV3S12 (MIV = Si, Ge) exhibits isolated [MIVS4] groups, and all the isolated [MIVS4] groups located in the c direction are symmetrical. Additionally, the [LaS8] polyhedra are connected together to generate a 3D framework, while the polyhedral [SrS7] and [SrS9] groups are interconnected to create an isolated triple-chain structure. In the La2Ca3Sn3S12 structure, La and Ca atoms occupy the same positions and Sn atoms are coordinated to 6 S atoms to build the [SnS6] octahedron. The [SnS6] units interconnect with each other to form edge-sharing 1D [(SnS4)]n chains, which are situated within the tunnels encircled by the polyhedral [(La/Ca)S7] and [(La/Ca)S9] motifs. It is worth noting that [SnIVS5] and [SnIVS6] units were only found in the structures of La2Sr3Sn3S12. The [SnS5] and [SnS6] units have no connection, and each forms isolated 1D chains with various linking modes. For instance, [SnS5] units are interlinked together via sharing vertexes to build 1D [(SnS4)]n chains, while [SnS6] units interconnect with each other to generate edge-sharing 1D [(SnS4)]n chains. The arrangement orientation of [SnS5] units in the unit cell demonstrates a nearly uniform arrangement along the ab plane.
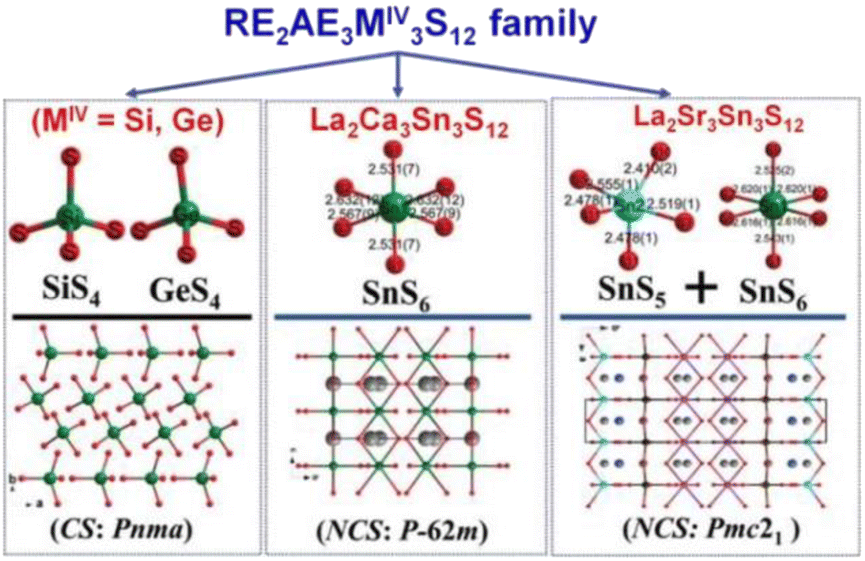 | ||
| Fig. 10 CS-to-NCS structural transformation with varying anionic motifs in the RE2AE3MIV3S12 family. This figure has been adapted from ref. 142 with permission from Wiley-VCH, copyright 2024. | ||
The remarkable CS-to-NCS structural transition (from Pnma to P2m to Pmc21) observed in the RE2AE3MIV3S12 family indicates that the substitution of tetrahedral [GeS4]/[SiS4] motifs with [SnSn] disrupts the original symmetry, resulting in the formation of the necessary NCS structures.
Significantly, the [SnS5] unit exhibits strong polarization anisotropy and hyperpolarizability, leading to significant performance improvements in NPM deff for compounds like La2Ca3Sn3S12 (1.4 × AgGaS2 and Δn = 0.008), compared to the strong PM deff observed in compounds like La2Sr3Sn3S12 (3.0 × AgGaS2 and Δn = 0.086). These findings demonstrate [SnS5] to be a promising “NLO-active motif”.
By incorporating flexible [SnSn] (n = 5 and 6) NLO-active motifs into the crystal structure, this study offers a viable approach for designing novel NCS thiostannates that exhibit very large SHG effects and significant optical anisotropy.
:1:1:4 stoichiometry, these compounds belong to different crystal systems and exhibit different structural symmetries.
Two new RE-based thiosilicates, specifically, RELiSiS4 (Ln = La and Ce), were successfully discovered by aliovalent substitution based on SrCdSiS4 as a parent compound in 2023 by Mao's group. They were synthesized using Li2S, RE2S3, SiS2, and S in a molar ratio of 3:1:3:8 through a high-temperature solid-state method at 1073 K. They are isostructural and belong to the orthorhombic space group Ama2 (no. 40). As shown in Fig. 11a, a 2D covalent layer of [LiSiS4]3− is constructed by tetrahedral [LiS4] and [SiS4] motifs, which further stacks along the a direction with [RES8] units occupying the intervals. Interestingly, this is the first observation of this linking mode in the Li–Si–S system. RELiSiS4 (RE = La and Ce) also exhibit perfectly balanced properties for IR-NLO chalcogenides: the largest deff among all reported thiosilicates (2.0 and 2.1 × AgGaS2@2050 nm for LaLiSiS4 and CeLiSiS4, respectively) with PM ability, high LIDTs (14 and 9 × AgGaS2@1064 nm), large Eg (3.71 and 2.92 eV), wide IR transmission window (0.35–18 μm), and good thermal stability above 973 K. This study not only enriches the structural chemistry of RE-based thiosilicates, but also offers an efficient method to design novel high-performance IR-NLO candidates.
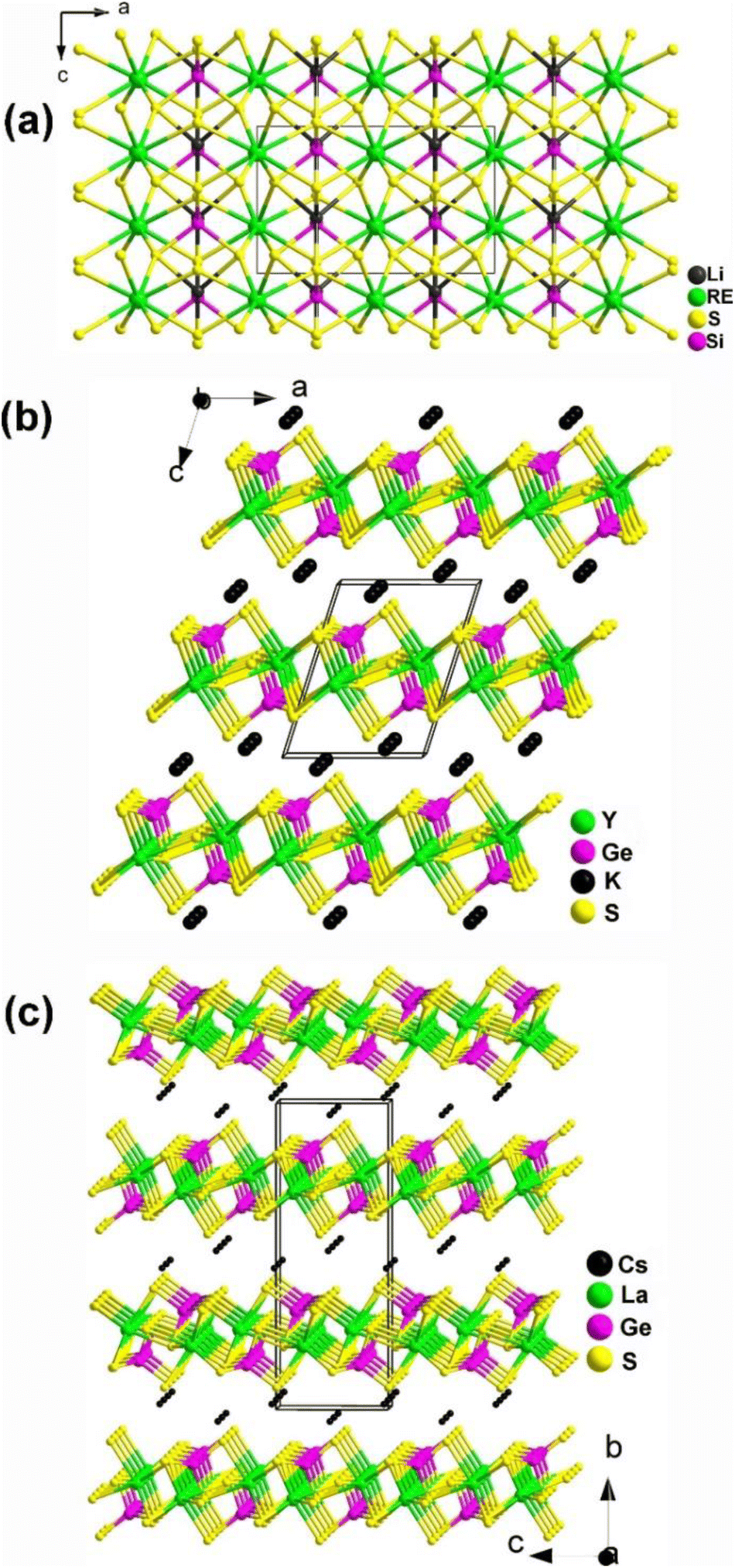 | ||
| Fig. 11 Structures of (a) LiRESiS4 (space group: Ama2), (b) KYGeS4 (space group: P21), and (c) CsLaGeS4 (space group: P212121) with the unit cell outlined. | ||
KREMIVQ4 compounds (RE = Y, La–Nd, Eu–Tb; MIV = Si, Ge; Q = S, Se) belong to the NCS monoclinic space group P21 (no. 4) and are isostructural. Therefore, for the purposes of this description, only the structure of KYGeS4 will be detailed as a representative. This compound was successfully synthesized in 2021 by Mei's group using a charge transfer engineering strategy. Fig. 11b shows that the [YS7] and [GeS4] motifs are interconnected by sharing polyhedral edges, and the [YS7] units are linked to one another through sharing vertexes to generate the infinite 2D [YGeS4]n layers. The interlayer interstices are filled by K+ cations to maintain charge balance. It is noteworthy that both [GeS4] and [YS7] polyhedra are uniformly arranged along the ab plane, which facilitates the positive superposition of microscopic second-order susceptibility, resulting in a strong deff. KYGeS4 overcomes the Eg limitation found in rare earth chalcogenides with the largest Eg (3.15 eV) within this material system, while displaying a significant deff (ca. 1.0 × AgGaS2). Moreover, KYGeS4 is the first RE-based chalcogenide to surpass the “3.0 eV wall” observed in IR-NLO crystals.
Isostructural AREMIVQ4 compounds (RE = La–Nd, Eu–Yb; MIV = Si, Ge; Q = S, Se) were synthesized by Loye and co-workers using a molten alkali halide flux growth method in 2019. All of them adopt the NCS monoclinic space group P212121 (no. 19), and in this discussion, we will focus on the structure of CsLaGeS4 as an example. CsLaGeS4 can be easily obtained by adding Ge to the reaction mixture of La2S3 and S in the CsCl/KCl eutectic flux. In the structure of CsLaGeS4, there are [LaS7] monocapped trigonal prisms and [GeS4] tetrahedrons. These polyhedral [LaS7] motifs and tetrahedral [GeS4] units interlink together through the sharing edge to generate the infinite 2D [LaGeS4]− slabs in the ac plane. The Cs+ cations fill in the gaps between the layers and act as charge balancers. Unfortunately, only CsLaGeS4 was found to be SHG-active, exhibiting nearly half the intensity of α-SiO2 when irradiated with a Nd:YAG 1064 nm laser. This compound also behaves as a semiconductor with an Eg of 3.60 eV based upon UV-vis diffuse reflectance measurements.
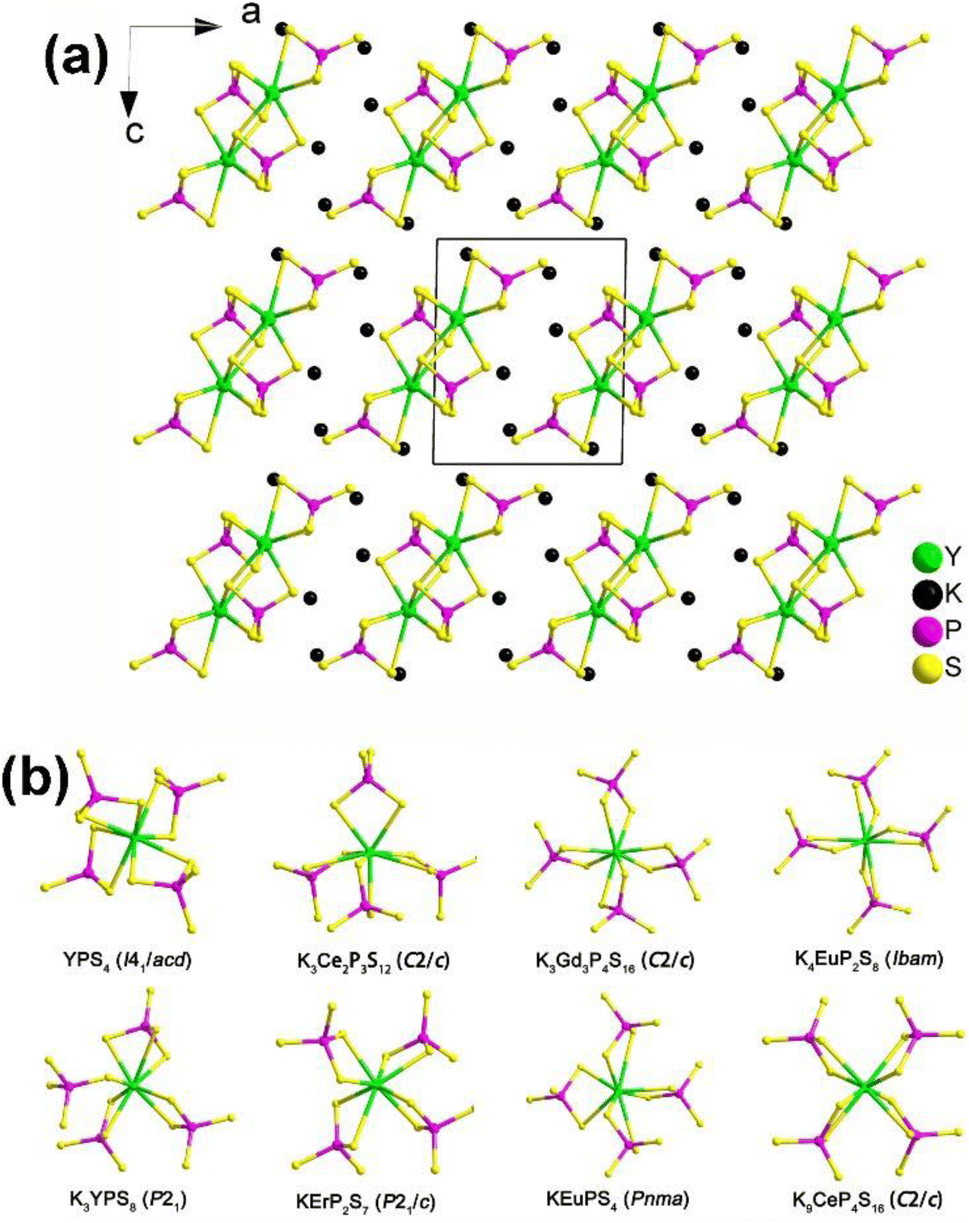 | ||
| Fig. 12 (a) Structure of K3YP2S8 along the ac plane with the unit cell outlined and (b) comparison on the local coordination modes between [RES8] and [PS4] motifs in the reported K/RE/P(V)/S family. | ||
Remarkably, NCS K3REP2S8 displaying strong PM deff (1.1–1.4 × AgGaS2) and large optical anisotropy (Δn = 0.084–0.099) were also proven to be promising IR-NLO candidates. The SHG density and dipole moment calculations indicate that the synergistic effect between the [RES8] and [PS4] units creates an inherent NLO origin, which verifies that RE-based thiophosphates can be regarded as an excellent research system for exploring novel IR-NLO chalcogenides.
2.2. RE-based chalcogenides containing lone-pair-electron motifs
The unique structural characteristics of polyhedra formed by cations that contain a stereochemically active lone pair (SCALP) give rise to a strong local built-in electric field, resulting in a significant dipole moment. As a result, neighbouring polyhedra with a SCALP often connect in opposite orientations, increasing the likelihood of crystallizing in an NCS space group.207–214 The inclusion of highly positively charged RE cations effectively creates a built-in electric field within their immediate surroundings. This takes advantage of the electrostatic interaction between the SCALP electrons and the RE cations to align the initially different orientations, thereby facilitating the macroscopic overlap of the static dipole moments. This alignment enhances the chances of crystallization in an NCS space group. Three types of RE-based chalcogenides containing lone-pair-electron motifs are presented below: RE8Sb2S15 (RE = La, Ce, Pr, and Nd), La2CuSbS5, and RE4MIIISbQ9 (RE = Y, La, Pr, Nd, Sm, Tb, Dy, and Ho; MIII = Ga, In; Q = S, Se). | ||
| Fig. 13 (a) Central projection of RE8Sb2S15 with the unit cell outlined and (b) coordination environment of crystallographically independent RE atoms. | ||
The compound La8Sb2S15 shows an Eg of 2.30 eV and a high effective deff of 1.2 × AgGaS2 (at 74–106 μm under 2050 nm laser irradiation) with NPM behavior. However, no significant deff was observed for Pr8Sb2S15, which may be linked to its poor crystallinity.
Single-crystal XRD result indicated that La2CuSbS5 belongs to the NCS orthorhombic system with the space group Ima2 (no. 46). The structure of La2CuSbS5 made of several distinct units, namely, [LaS10] polyhedra, [CuS4] tetrahedra, and [SbS4] pyramids. Fig. 14a depicts the projection of La2CuSbS5 viewed down the ab plane. The 3D network is constructed from two types of 2D layers (La/Cu/S and La/Sb/S) that are interconnected through shared S–S apexes and edges. The detailed coordination environment of the La atoms is shown in Fig. 14b.
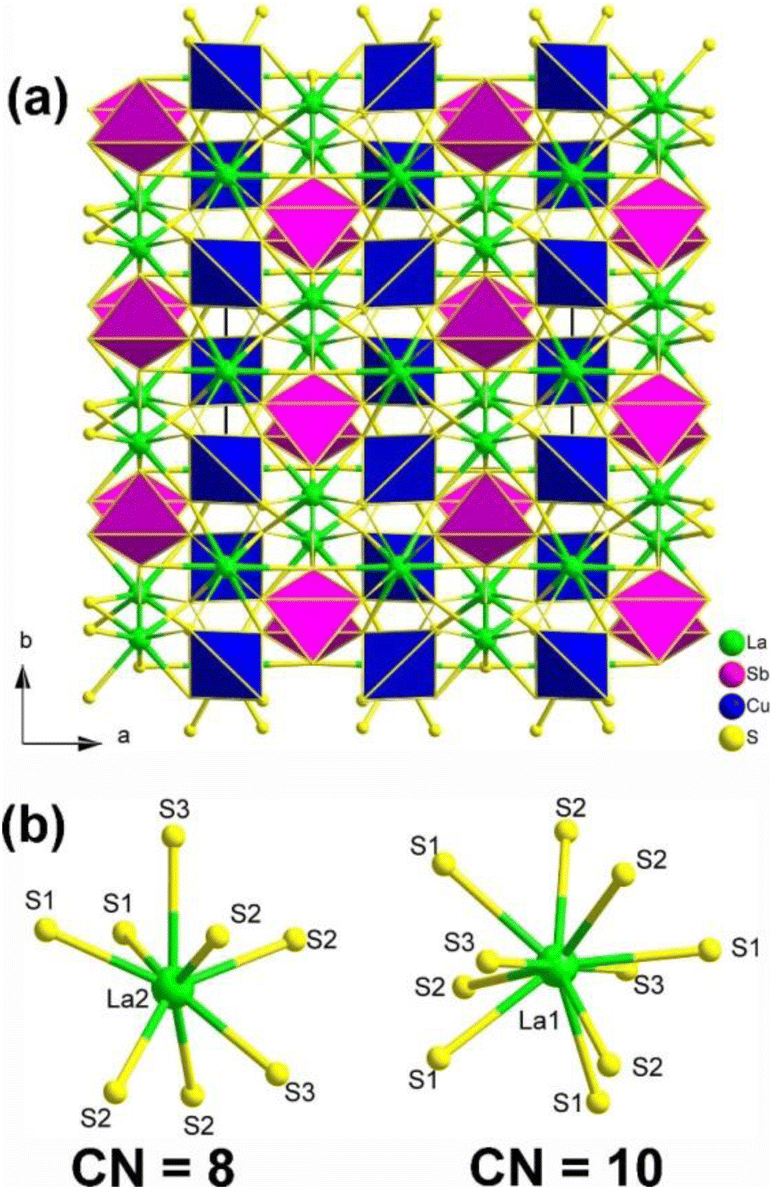 | ||
| Fig. 14 (a) Structure of La2CuSbS5 along the ab plane with the unit cell outlined; (b) coordination environment of crystallographically independent La atoms. | ||
Moreover, La2CuSbS5 can be seen as a result of the stereochemically active lone pair induction (SCALPI) strategy, which is based on the previously known CS La2CuInS5. Investigation into its optical properties showed that La2CuSbS5 displays a suitable PM deff (0.5 × AgGaS2) and a large LIDT (6.7 × AgGaS2), making it a promising candidate for IR-NLO materials. Furthermore, theoretical calculations support the notion that the deff can be attributed to the synergistic effects of the functional primitives, with a particular emphasis on the [SbS4] motifs. These results confirm the practicality of utilizing the SCALPI strategy for the design and exploration of new IR NLO crystals.
 | ||
| Fig. 15 Structures of (a) RE4GaSbS9 and (b) RE4InSbS9 with the unit cell outlined. | ||
The experimental results demonstrate that RE4GaSbS9 and RE4InSbQ9 exhibit different NLO performances. The former is NPM while the latter is PM under the same test conditions. For example, among the Ga analogues, Sm4GaSbS9 shows the strongest deff (3.8 × AgGaS2 in the particle size of 46–74 μm) with NPM features at 2050 nm. Among the In analogues, La4InSbS9 exhibits a strong deff (1.5 × AgGaS2 in the particle size of 150–210 μm) with PM features at 2050 nm.
The theoretical results suggest that the SHG response of RE4GaSbS9 can be attributed to the electronic transitions from the S-3p states in the valence bands to the Sb–S and RE–S antibonding states. On the other hand, the SHG response of RE4InSbQ9 is attributed to the thermal vibrations of the lattice.
2.3. RE-based chalcogenides containing [BS3] and [P2Q6] motifs
The [BS3] motif, with its distinctive planar conjugated π46 electron structure, is a crucial functional unit. Its introduction not only benefits the synergistic enhancement of NLO response and LIDT, but also has a significant impact on birefringence. By anchoring RE cations to the [BS3] unit, the arrangement of the [BS3] unit is optimized. The bonding interaction between them synergistically enhances the contribution to conductive bands, reinforcing the contribution to the empty virtual–hole transition process by the low-lying 4f or 5d orbitals in the rare earth ions and the π* anti-bonding orbitals of the [BS3] unit. For the crossed ethane configuration of the [P2Q6] unit, the 6 exposed Q atoms serve as sites to balance its negative charge through chemical bonding. The high coordination number of rare earth ions precisely matches this unit, allowing one RE cation to bond with one or even multiple [P2Q6] units. This maximizes the functionality of this basic unit and synergistically enhances the NLO effects in conjunction with [REQn] polyhedra. In the following discussion, four systems are introduced: REBS3 (RE = La, Ce, Pr, Nd, Sm, and Tb), Ca2RE(BS3)(SiS4) (RE = La, Ce, and Gd), Eu2P2S6, and KREP2Se6 (RE = Sm, Tb, and Gd).REBS3 (RE = La, Ce, Pr, Nd, Sm, and Tb) are isomorphic and adopt an orthorhombic Pna21 space group. LaBS3 can serve as an example to discuss the crystal structure; as shown in Fig. 16, its structure comprises triangular [BS3]3− units, with each La atom connecting to 9 S atoms to link with the [BS3]3− units. Each [BS3]3− unit is linked to 6 La3+ atoms, ultimately forming the entire 3D network structure.
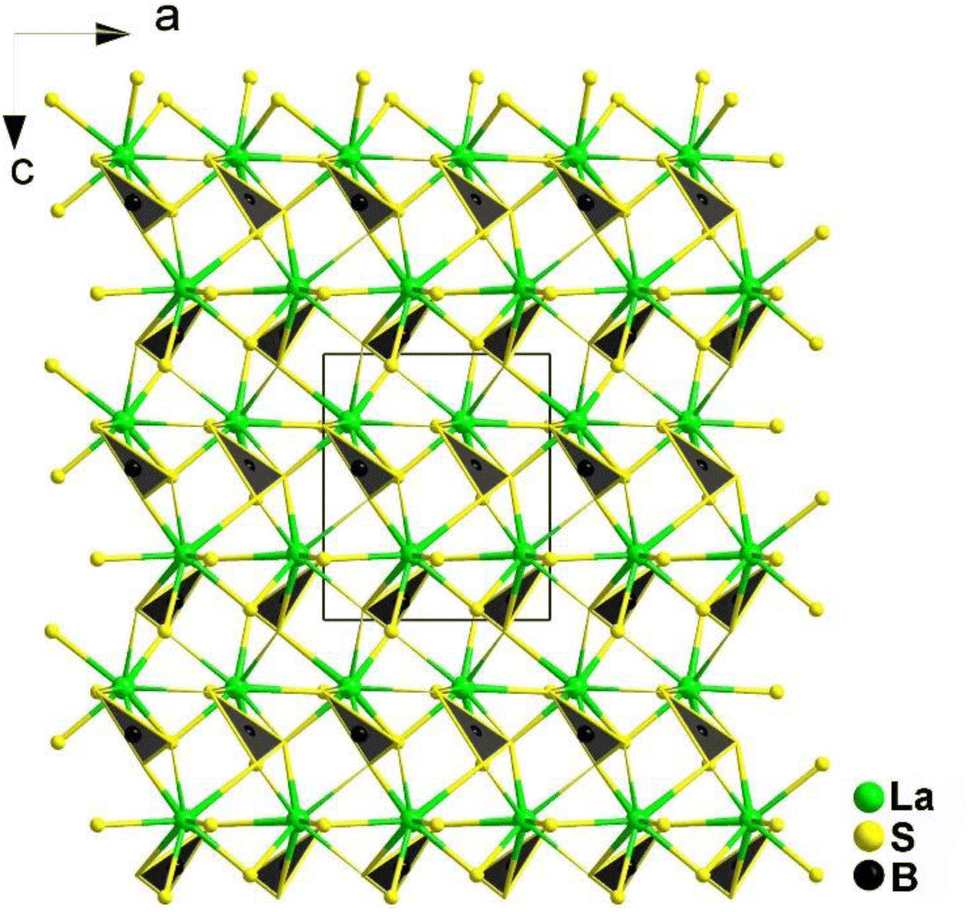 | ||
| Fig. 16 Structure of LaBS3 along the ac plane with the unit cell outlined. | ||
As expected, LaBS3 possesses a large Eg of 3.18 eV, the highest deff among ortho-thioborates (1.2 × AgGaS2 at 2050 nm), an ultra-high LIDT of 14 × AgGaS2, a wide IR transmission range (0.35–25 μm), and exhibits PM behaviour. The results of theoretical calculations indicate that the [LaS9]15− groups and the π-conjugated [BS3]3− anions are the main sources of the SHG effect. In general, this research provides us with a new method to synthesize new NLO materials.
The compounds Ca2RE(BS3)(SiS4) (RE = La, Ce, and Gd) are isostructural and crystallize in the hexagonal P63mc space group. Fig. 17 illustrates the structure of Ca2RE(BS3)(SiS4) along the ab plane. In this structure, the RE3+ and Ca2+ ions occupy the same site, referred to as the M site, with a ratio of 1/3 and 2/3, respectively. Each M site coordinates with 6 S atoms to form an [MS6] polyhedron, which connects with isolated [BS3]3− and [SiS4]4− units along the c axis, resulting in the overall 3D structure of Ca2RE(BS3)(SiS4).
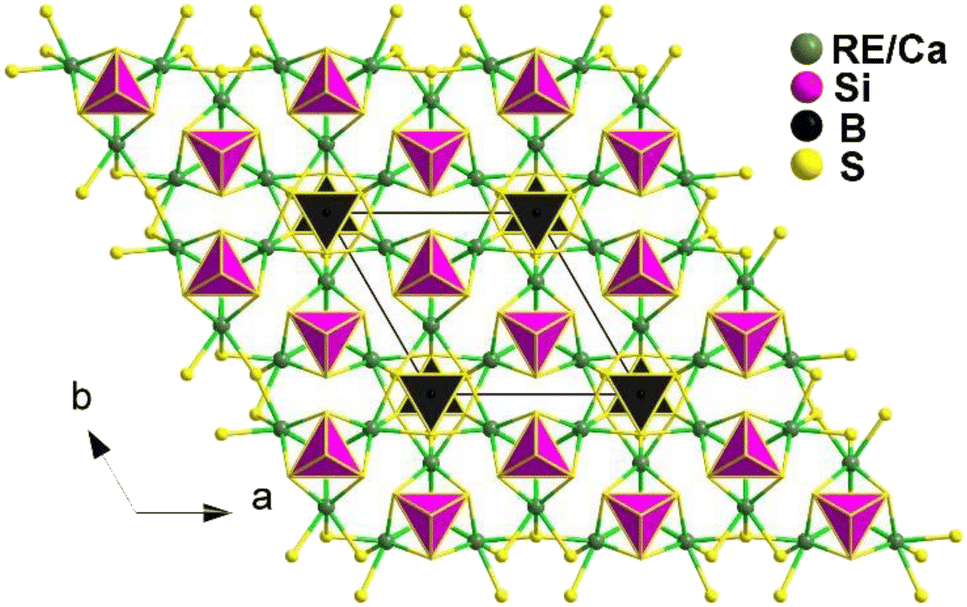 | ||
| Fig. 17 Structure of Ca2RE(BS3)(SiS4) along the ab plane with the unit cell outlined. | ||
Remarkably, all of them exhibit comprehensive properties that make them promising candidates for IR-NLO materials. These properties include strong PM deff at 2050 nm (1.1–1.2 × AgGaS2), high LIDTs (7–10 × AgGaS2), a wide transmission range (0.45–11 μm), high thermal stabilities (>1073 K), and large calculated Δn (0.126–0.149@1064 nm). These properties validate the material design strategy of combining [BS3]3− and [SiS4]4− NLO-active motifs.
Theoretical calculations indicate that the big deff mainly stem from the synergy between the [RES6], [BS3], and [SiS4] groups. These findings not only expand the scope of research on chalcogenides but also offer a straightforward synthetic method for heteroanionic thioborates.
Yellow block crystals of Eu2P2S6 were prepared through a high-temperature solid-phase method between stoichiometric Eu, P2S5, and S at 1233 K. It belongs to the monoclinic Pn space group. The structure of Eu2P2S6 can be viewed as comprising bicapped-triangular-prism [EuS8] and dimeric [P2S6] functional primitives built into a 3D framework via 8:18:13 intergrowth (Fig. 18).
 | ||
| Fig. 18 Structure of Eu2P2S6 along the bc plane with the unit cell outlined. | ||
This first IR-NLO RE-based chalcogenophosphate, Eu2P2S6, displays wonderful comprehensive optical properties, including a moderate PM deff (0.9 × AgGaS2), large LIDT (3.4 × AgGaS2), and broad IR transparency region (0.49–15.4 μm). Based on the results of structural analysis and theoretical studies, the overall IR-NLO properties are attributable to the synergetic effect of [EuS8] and [P2S6] functional primitives. This study not only developed the first RE-based chalcogenophosphate as an advanced NLO candidate, but also provided a representative example for the future discovery of high-performance RE-based IR-NLO chalcogenides.
The first RE-based selenophosphates KREP2Se6 (RE = Sm, Tb, and Gd) belong to the monoclinic P21 space group. They are composed of a 2D [REP2Se6]− layer with K+ ions filling the spaces in between (Fig. 19). The [REP2Se6]− layer is built by layers in which each bicapped-trigonal-prism [RESe8] unit is linked to four [RESe8] units by sharing edges and vertices. These [RESe8] units also connect with 4 [P2Se6]4− units by sharing faces, edges, and corners. The co-vertices are arranged on one side, while the co-edges are arranged on the other.
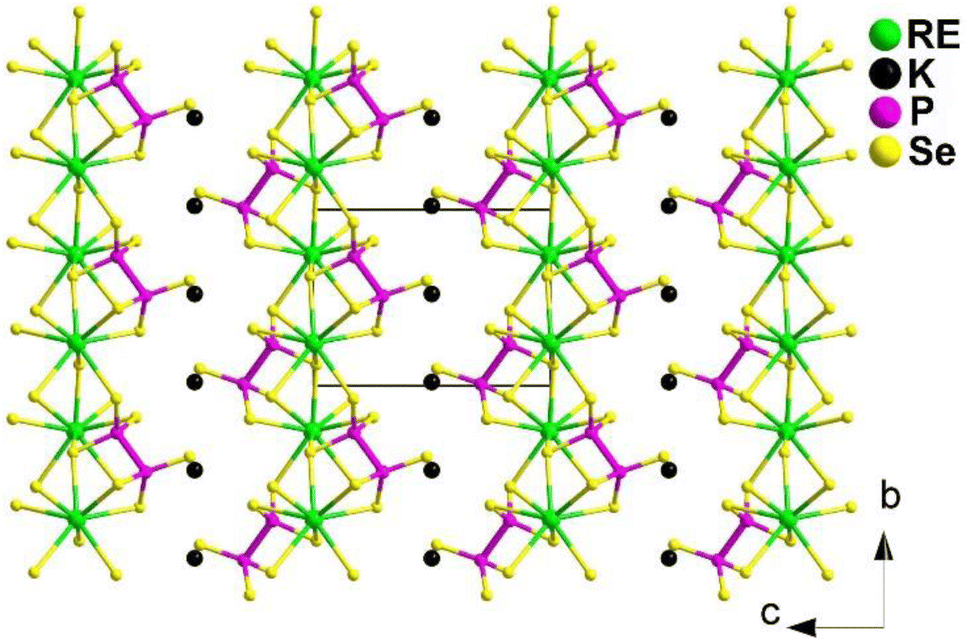 | ||
| Fig. 19 Structure of KREP2Se6 along the bc plane with the unit cell outlined. | ||
As a result, KREP2Se6 (RE = Sm, Tb, and Gd) exhibit a PM deff effect ranging from 0.34 to 1.08 × AgGaS2 at 2100 nm and a LIDT ranging from 1.43 to 4.33 × AgGaS2. The theoretical calculations suggest that the cooperation between [P2Se6] and [RESe8] plays a predominant role in the deff. This research demonstrates that RE-based selenophosphates are a promising type of IR-NLO material. It not only enhances the understanding of crystal chemistry in RE-based chalcophosphates but also expands the range of potential applications for NLO materials.
2.4. RE-based chalcohalides and oxychalcogenides
The emergence of heteroanions has enriched traditional functional units, as the differences in electronegativity result in a greater charge distribution shift within the interior of heteroanionic groups compared to conventional functional units.215–228 This leads to distortions and the display of significant static dipole moments and anisotropy.229–233 Unlike traditional chalcogenides, the presence of heteroanionic groups combines the high polarizability of chalcogenides with the advantages of oxides and halides that have wide Eg. RE-based chalcohalides and oxychalcogenides not only incorporate disparate anions but also feature the presence of salt-inclusion compounds. Below, eleven RE-based chalcohalides and oxychalcogenides are introduced.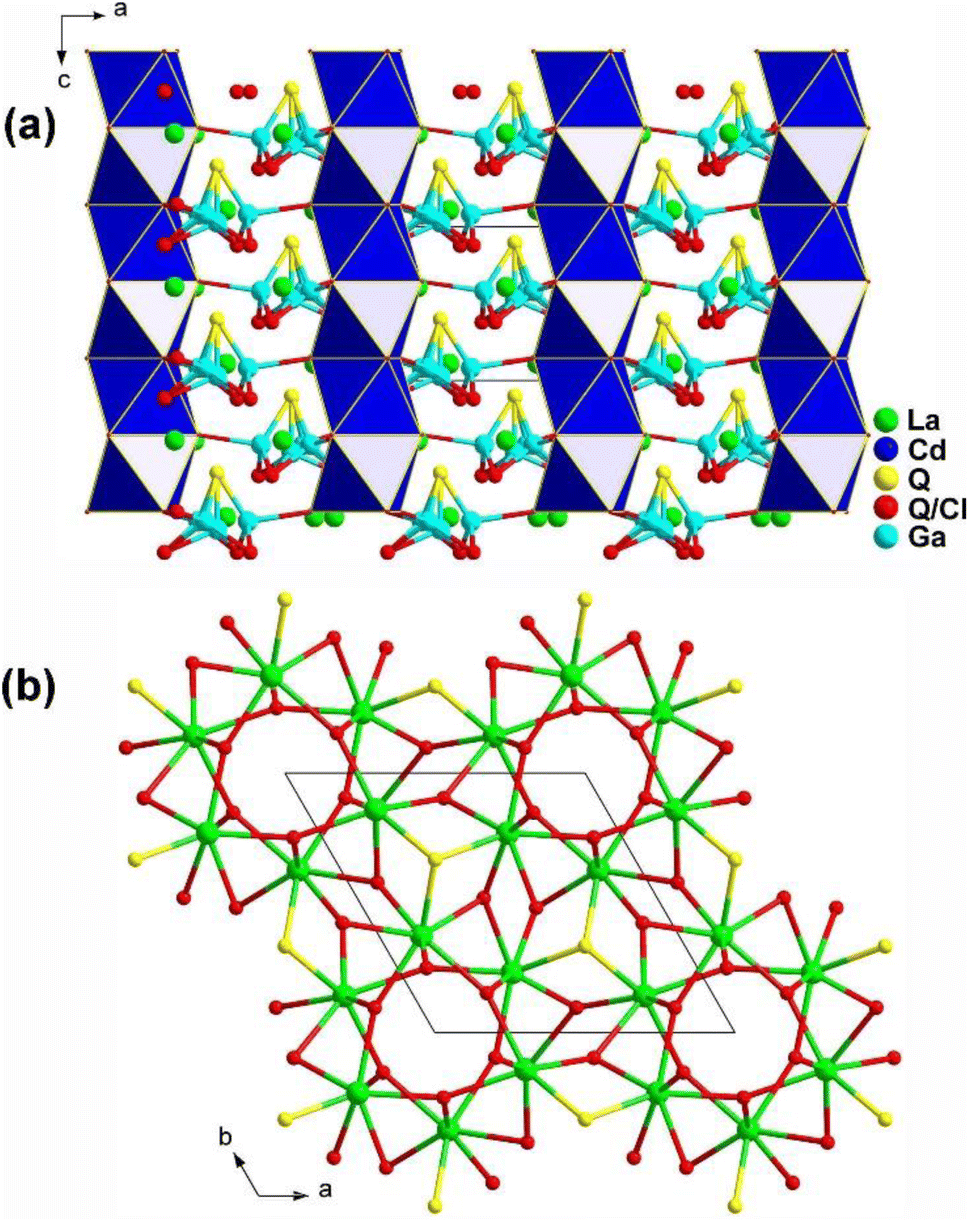 | ||
| Fig. 20 Structure of (a) La6Cd0.75Ga2Q11.5Cl2.5 along the ac plane and (b) 3D La/Q/(Q/Cl) network along the ab plane with the unit cell outlined. | ||
Moreover, the introduction of various heteroanionic functional motifs with varying sizes and electronegativity increases the distortion of structural groups while keeping the structural symmetry. This enhances the polarity of the NLO-active motifs, resulting in a greater deff. As expected, La6Cd0.75Ga2S11.5Cl2.5 exhibits a large LIDT of 18.6 × AgGaS2 @1064 nm and a strong deff of 0.8 × AgGaS2 @43–75 μm under 2050 nm laser irradiation.
The 3D network of RE3AsS5X2 (RE = La, Pr; X = Cl, Br) is composed of [RE1S5X3] bicapped trigonal prisms, [RE2S5X3] bicapped trigonal prisms, [RE3S7] capped trigonal prisms, and SACLP [AsS3] trigonal pyramids, which are interconnected with each other (Fig. 21a). The coordination environment of the crystallographically independent RE atoms in the structure of RE3AsS5X2 is provided in Fig. 21b. The electron localization function results confirmed that the arrangement of the [AsS3] groups contributes to the NCS nature of RE3AsS5X2.
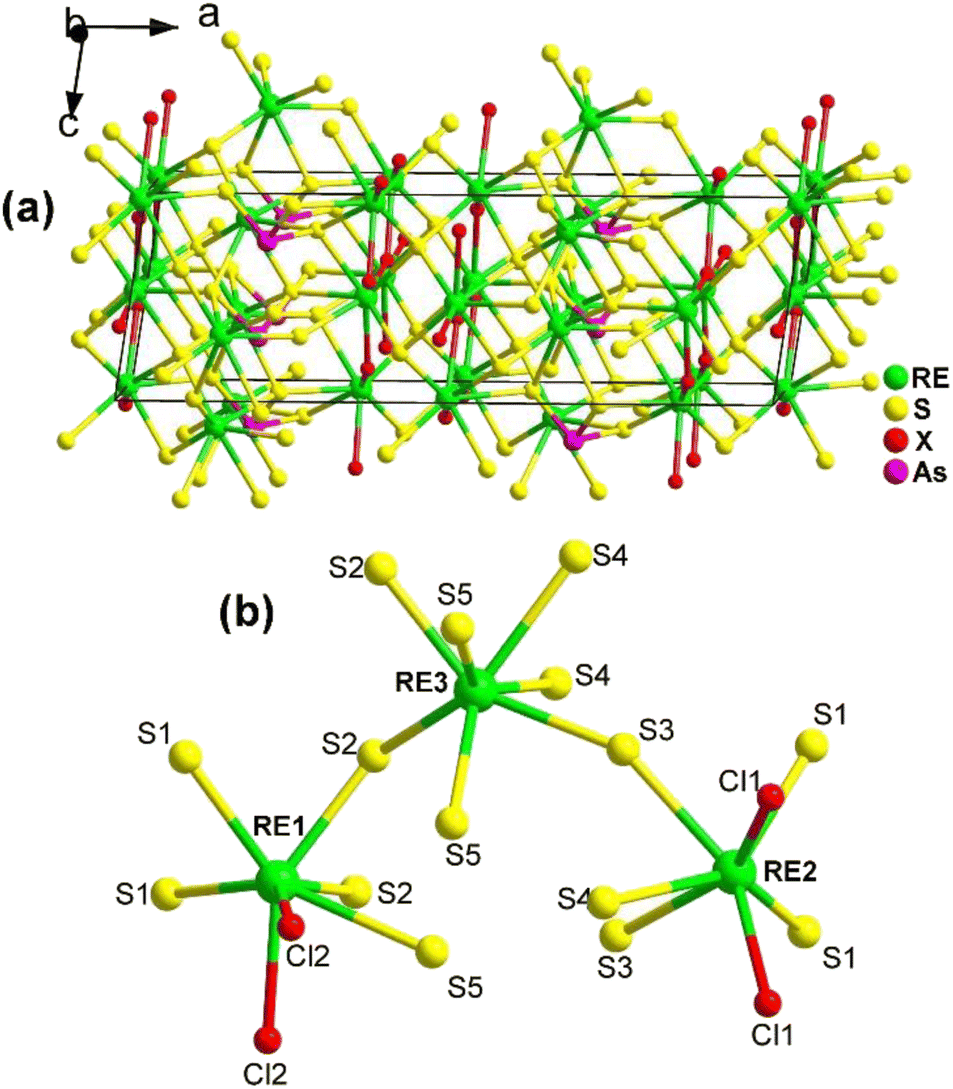 | ||
| Fig. 21 Structure of (a) RE3AsS5X2 along the ac plane with the unit cell outlined and (b) coordination environment of the crystallographically independent RE atoms. | ||
The moderate PM deff (0.23 × AgGaS2 @150–210 μm), easy growth of millimeter-sized crystals, excellent ambient stability, and wide Eg (ca. 2.9 eV) of La3AsS5Br2 suggest its potential applicability as an IR-NLO candidate.
The structure of Eu4.5(B5O9)2SI along the ab plane is displayed in Fig. 22a. It consists of a 3D polyanionic network {(B5O9)3−}∞, which is constructed by linking 3 [BO4] tetrahedra with 2 [BO3] planar triangles through shared O atoms. Along the c axis, there is a tubular accumulation [Eu2(B5O9)]∞ framework built by Eu1 and Eu2 located in the cavities of the 3D network. The cavities of the 3D network are also occupied by I− ions. Fig. 22b shows the coordination environment of the crystallographically independent Eu atoms, which have three different coordination modes: [Eu1O5S2I], [Eu2O5S2I], and [Eu3O4S2].
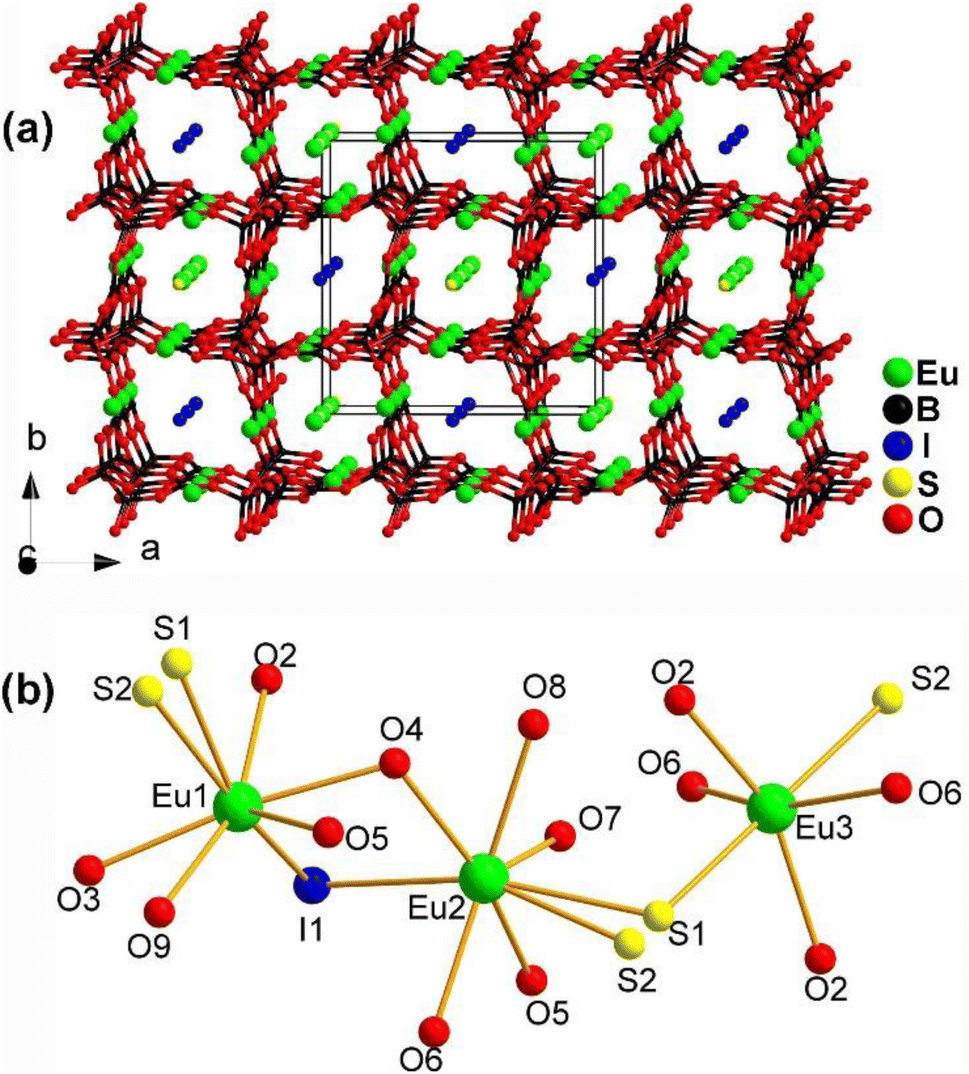 | ||
| Fig. 22 Structure of (a) Eu4.5(B5O9)2SI along the ab plane with the unit cell outlined and (b) coordination environment of the crystallographically independent Eu atoms. | ||
The combination of Eu2+, S2−, I−, and borates allows for the realization of a large deff and a high LIDT. Eu4.5(B5O9)2SI has an Eg of 1.99 eV, a moderate deff of 0.5 × AgGaS2, a high LIDT of 15 × AgGaS2, and PM behaviour. These results indicate that the Eu–S and B–O motifs play an important role in the deff and offer an example of the practicality of this strategy.
(K3I)[REB12(GaS4)3] (RE = Sm, Gd) share the same structure and belong to the hexagonal chiral space group P6322. In the asymmetric unit, there are 1 A, 1 X, 1 RE, 1 Ga, 2 B, and 2 Q positions. The crystal structure consists of two parts: the polycationic [K6I]5+ and the 3D open [REB12(GaS4)3]5− anionic framework. The 3D framework comprises icosahedral [B12], tetrahedral [GaS4], and octahedral [RES6] functional primitives. Within the structure, the [B12] icosahedra are surrounded by 6 [GaS4] tetrahedra and consolidated by 2 [RES6] octahedra, resulting in a 3D honeycomb-like open framework. This framework forms channels occupied by face-sharing [IK6] octahedral chains along the c direction (see Fig. 23).
 | ||
| Fig. 23 Structure of (K3I)[REB12(GaS4)3] with the unit cell outlined along the ab plane. | ||
Due to the significantly low yield for most compounds, the majority of samples in this system were unable to undergo size-dependent SHG measurements. For instance, the deff of (K3I)[SmB12(GaS4)3] is approximately 0.3 times that of KDP when subjected to a 1940 nm laser. The small deff can be attributed to the arrangement of the SHG-active groups in their structures, which hinders the improvement of macroscopic polarizabilities.
[RE3S2Cl2][SbS3] (RE = La, Ce) were obtained from a mixture of CsCl, RE, GaCl3, SbCl3, and S in a ratio of 1:4:1:1:5 using a high-temperature solid-state method at 1073 K. As shown in Fig. 24a, [RE3S2Cl2][SbS3] belongs to the NCS Cc space group, where the 2D layers of [RE3S2Cl2]12+ arranged in the bc plane and the trigonal-pyramid [SbS3] motifs are stacked in an alternating pattern. However, the stacking of [SbS3] motifs in the crystal cell is in-phase and they are correlated with each other through the n sliding planes at (x, 1/4, z). Unfortunately, no NLO performance has been reported for these two compounds.
 | ||
| Fig. 24 Structures of (a) [RE3S2Cl2][SbS3] along the ac plane and (b) [La3OSCl2][SbS3] along the ab plane with unit cell outlined. | ||
Yellow crystals of [La3OSCl2][SbS3] were grown from a mixture of La, LaCl3, Sb, Sb2O3, and S with a ratio of 7:2:1:1:12 at 1223 K. It can be seen as being obtained through partial isovalent anion substitution of the parent structure [La3S2Cl2][SbS3]. Interestingly, [La3OSCl2][SbS3] adopts the hexagonal space group P63mc, which is not isostructural with [La3S2Cl2][SbS3] (monoclinic space group Cc), despite their having similar functional primitives. [La3OSCl2][SbS3] is made of 3D cationic [La3OSCl2]3+ frameworks hosting covalent [SbS3]3− motifs. The 3D cationic [La3OSCl2]3+ are built via the 1D hexagonal columns of corner-sharing [(Cl/S)La3] polyhedra propagating along the 63 axis situated in (0, 0, z) and [OLa3] trigonal pyramids through vertex-sharing. It is worth mentioning that experimental results indicate a moderate deff (ca. 0.7 × AgGaS2@50–100 μm) under a 2050 nm laser, and the calculated Δn value is about 0.27 at 2050 nm, respectively.
YSeBO2 adopts the orthorhombic polar space group Cmc21, and the 3D structure is formed by the connection between [YO3Se4]11− pentagonal bipyramids and [BO3]3− planar triangles by sharing O edges (see Fig. 25). It is worth mentioning that the heteroanionic [YO3Se4]11− functional motif was first discovered in the chalcogenide system. In addition, YSeBO2 displays a weak deff of about 0.2 × KDP (@150–210 μm) with PM features. Theoretical analysis shows that the Se-4p and Y-4d states play a major role in the origin of SHG. The contribution of the [BO3]3− planar triangles did not show any impact due to its unfavorable arrangement.
 | ||
| Fig. 25 Structure of YSeBO2 along the bc plane with the unit cell outlined. | ||
Eu3GeOS4 belongs to the orthorhombic Pca21 space group. As displayed in Fig. 26, neighbouring [EuOS6] mono-capped trigonal prisms are interlinked through sharing faces to form a 3D framework structure, with isolated [GeOS3] tetrahedra occupying the cavities. In other words, without considering the Eu–O/S bonds, the crystal structure of Eu3GeOS4 can also be considered a pseudo-0D structure. It is interesting to note that the coordination mode of the heteroanionic [EuOS6] functional motif was discovered for the first time in oxychalcogenides.
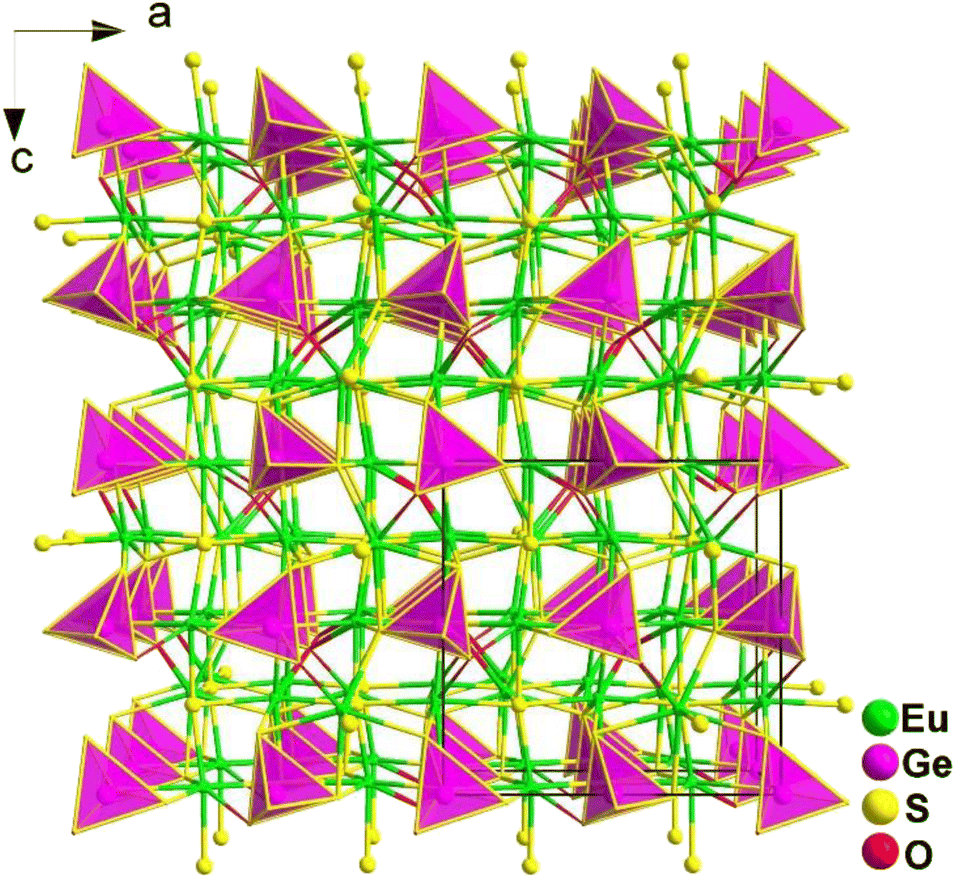 | ||
| Fig. 26 Central projection of Eu3GeOS4 along the ac plane with the unit cell outlined. | ||
Eu3GeOS4 not only shows an obvious deff (0.24 × AgGaS2@2050 nm), but also a large LIDT (8.86 × AgGaS2@1064 nm). Theoretical calculations indicate that the deff is attributed to the synergistic effect of the heteroanionic [EuOS6] and [GeOS3] functional motifs.
The crystals of RE3NbS3O4 were prepared through solid-phase reactions at 1273 K using RE2O3, Nb2O5, B, and S as the raw materials and excess KI as the flux. When charge-balanced RE3+ cations are omitted, the NCS structure of RE3NbS3O4 (RE = Sm, Gd) can be seen as a hexagonal cage formed by 6 Nb atoms encasing 6 RE atoms when viewed along the bc plane (see Fig. 27). Each RE forms two different coordination forms: a [RES4O4] double-capped triangular prism and a [RES5O3] dodecahedron.
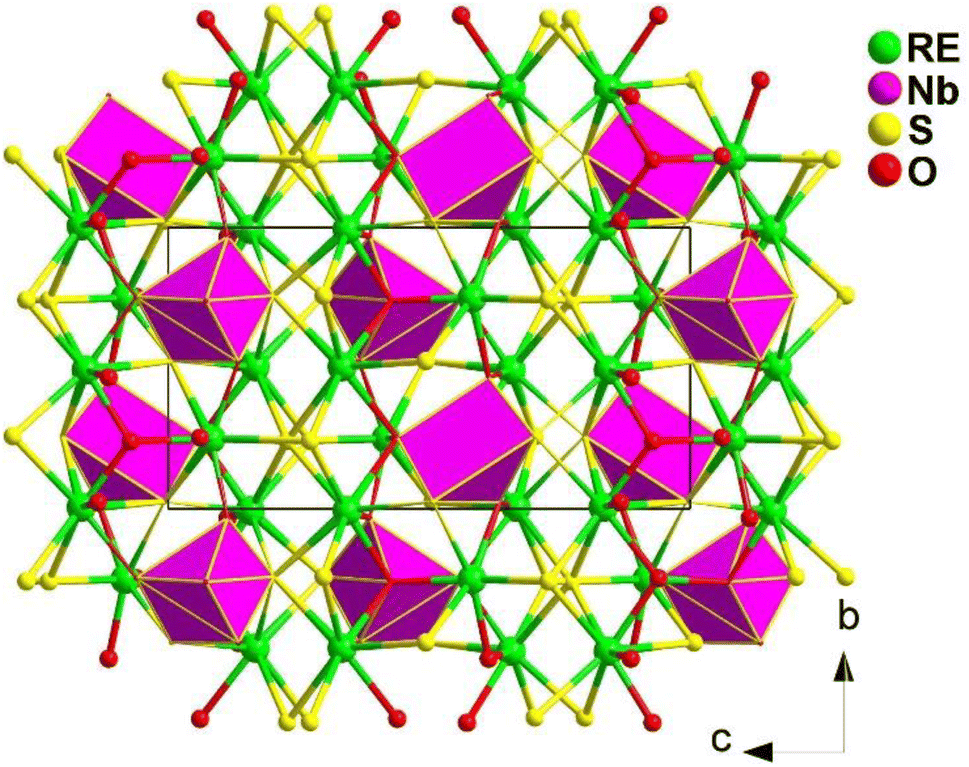 | ||
| Fig. 27 Structure of RE3NbS3O4 along the bc plane with the unit cell outlined. | ||
Sm3NbS3O4 and Gd3NbS3O4 exhibit PM deff of about 0.3 and 0.4 × AgGaS2 under a 2100 nm laser and large LIDTs of 12.5 and 4.5 × AgGaS2 at 1064 nm. The heteroanionic [NbS2O4] unit was first found as a SHG-active motif, which offers a new route for discovering novel IR-NLO oxychalcogenides.
Nd3[Ga3O3][Ge2O7] was discovered using the solid-phase method at 1273 K and adopts the hexagonal space group P2c (no. 190). Its unique structure consists of three different modules: charge-balanced Nd3+ cations, 0D [Ge2O7]6− dimers, and 1D [Ga3O3S3]3− tubular chains (Fig. 28). The Nd3+ cation is coordinated with O/S atoms to form a heteroligand [NdO6S2] polyhedron. Experimental investigations of Nd3[Ga3O3][Ge2O7] demonstrate that it is the first RE-based IR-NLO oxychalcogenide, exhibiting excellent optical performance. This includes a strong deff (ca. 0.8 × AgGaS2@2050 nm), a large Eg (ca. 4.35 eV) corresponding to an ultrahigh LIDT of 23 × AgGaS2@1064 nm, a wide transparent window (0.25–13.7 μm), and good thermal stability (<1200 K).
 | ||
| Fig. 28 Structure of Nd3[Ga3O3S3][Ge2O7] along the ab plane with unit the cell outlined. | ||
Detailed theoretical results indicate that the remarkable deff in Nd3[Ga3O3][Ge2O7] is mainly attributed to the cooperation of heteroanionic [GaO2S2] and [NdO2S6] functional motifs. This research not only expands the possibilities of IR-NLO families with heteroanionic motifs, but also offers a feasible strategy for the development of other high-performance IR-NLO systems.
Eu2MIIGe2OS6 (MII = Mn, Fe, and Co) are isomorphic and grow in a tetragonal NCS P21m space group. As shown in Fig. 29a, two neighboring [GeOS3] tetrahedra build a dimer, [Ge2OS6], by sharing an O atom. Each [Ge2OS6] dimer then interlinks with 4 [MIIS4] tetrahedra by sharing S corners, constructing corrugated 2D [MIIGe2OS6]4− layers along the ab plane. The [EuOS7] bicapped trigonal prisms are located within these 2D interlayers and serve to bridge adjacent [EuOS7] together, forming the 3D network (Fig. 29b).
 | ||
| Fig. 29 Structure of (a) Eu2MIIGe2OS6 along the ac plane and (b) the 2D Eu/O/S network along the ab plane with the unit cell outlined. | ||
As a result, Eu2MIIGe2OS6 (MII = Mn, Fe, and Co) display excellent IR-NLO performance with an Eg of 2.11–2.40 eV, a PM deff of 0.3–0.5 × AgGaS2 at 200–250 μm, and a large LIDT of 2.8–8.3 × AgGaS2. They proposed that these compounds have the potential to replace MII using Cu or other d-block metals and other substitutions within the same family.
All of the REAEGa3S6O oxysulfides belong to the P21m space group of the tetragonal system, and they are isostructural with LaAEGa3O7 and LaAEGa3S7. Fig. 30 presents the unique structure of these compounds, which can be described as consisting of two functional modules: charge-balanced [RE/AE]5+ cations and 2D [Ga3S6O]5− Cairo pentagonal layers. The charge-balanced module is composed of [(RE/AE)S7O] groups that are connected through edge- and face-sharing interactions, forming a 2D [(RE/AE)S7O]n layer. On the other hand, the 2D layers can be seen as a combination of many 5-membered rings, with two [GaS4] and three [GaS3O] units surrounding each ring.
 | ||
| Fig. 30 Structure of LaAEGa3S6O along the ac plane with the unit cell outlined. | ||
In particular, REAEGa3S6O fulfills the property balance requirements (e.g., wide Eg: 3.21–3.27 eV and PM deff: 0.9–1.0 × AgGaS2). This compound shows promise as a potential IR-NLO crystal, as it combines the advantages of LaAEGa3O7 and LaAEGa3S7 through heteroanion-oriented performance engineering. The structure–activity relationships reveal that the heteroanionic [(RE/AE)S7O] and [GaS3O] functional motifs are particularly promising for NLO activity, as they exhibit large cooperation in the origin of SHG. These findings not only provide a clear understanding of the variation in properties from oxide to sulfide to oxysulfide, but also highlight the feasibility of using heteroanion-oriented design to develop novel NLO candidates with well-balanced performances.
3. Conclusions and perspectives
RE-based chalcogenides, with their unique electronic configurations, have been extensively discussed in the field of IR-NLO for several decades, and there has been growing interest in recent years. According to incomplete statistics, there are over 400 compounds with non-centrosymmetric structures among RE-based chalcogenides. However, to date, there has not been a comprehensive review that systematically summarized these compounds. Therefore, this paper provided a summary analysis of their synthesis methods, structures, optical properties, and structure–property relationships. The results of the statistical analysis of these compounds are summarized in Tables 1–4 and Schemes 1–4. Additionally, some key findings have been uncovered:| Compound | [REQn] polyhedra | Space group | E g (eV) | d eff (×AgGaS2) | LIDTb (×AgGaS2) | NPM/PMc | Δnd | Maximum temperaturef | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Experimental value. b Powder sample. c PM = phase-matchability, NPM = nonphase-matchability. d Theoretical value. e N/A = not available. f The maximum temperature of solid-state reaction. | |||||||||
| Dy3GaS6 | [DyS7] | Cmc21 (no. 36) | 2.80 | 0.084 | 14 | PM | 0.068 | 1223 K | 69 |
| Y3GaS6 | [YS7] | Cmc21 (no. 36) | 2.88 | 0.21 | 18 | PM | 0.069 | 1223 K | 69 |
| Ho3GaS6 | [HoS7] | Cmc21 (no. 36) | 3.03 | 0.4 | N/Ae | PM | 0.041 | 1223 K | 70 |
| Er3GaS6 | [ErS7] | Cmc21 (no. 36) | 3.08 | 0.6 | N/A | PM | 0.034 | 1223 K | 70 |
| Eu8Sn4Se20 | [EuSe9] | P21212 (no. 18) | 1.33 | 1 × α-SiO2@150–210 μm | N/A | N/A | 0.16 | 1223 K | 72 |
| EuCu2GeSe4 | [EuSe8] | Ama2 (no. 40) | 1.74 | No signal | N/A | N/A | N/A | 1073 K | 75 |
| EuCu2GeS4 | [EuSe8] | P3221 (no. 154) | 2.32 | No signal | N/A | N/A | N/A | 1073 K | 75 |
| EuCu2SiS4 | [EuS8] | P3121 (no. 152) | 2.36 | No signal | N/A | N/A | N/A | 1073 K | 75 |
| Dy3Si0.85Al0.53S7 | [DyS8] | P63 (no. 173) | 2.03 | 1 × KTP@∼20 μm | N/A | N/A | N/A | 1223 K | 108 |
| Dy6Al2SiS14 | [DyS8] | P63 (no. 173) | 2.22 | 2 × KTP@∼20 μm | N/A | N/A | N/A | 1223 K | 108 |
| Sm3Al0.33SiS7 | [SmS8] | P63 (no. 173) | 2.26 | 0.3 × KDP@∼150 μm | N/A | N/A | N/A | 1223 K | 108 |
| Y6ZnSi2S14 | [YS8] | P63 (no. 173) | 2.38 | 0.84@∼20 μm | N/A | N/A | N/A | 1223 K | 108 |
| La3GaGe0.5S7 | [LaS8] | P63 (no. 173) | 2.54 | 4.8@74–106 μm | N/A | NPM | 0.023 | 1223 K | 82 |
| Sm3GaGe0.5S7 | [SmS8] | P63 (no. 173) | 2.50 | Weak | N/A | N/A | N/A | 1253 K | 82 |
| La3CuGeSe7 | [LaSe8] | P63 (no. 173) | 2.00 | 0.3 × SiO2 | N/A | N/A | N/A | 1073 K | 94 |
| La3LiSnS7 | [LaS8] | P63 (no. 173) | 2.40 | 1.2 | 2.5 | PM | 0.031 | 1073 K | 126 |
| Sm3LiSiS7 | [SmS8] | P63 (no. 173) | 2.83 | 1.5 @88–105 μm | 3.7 | NPM | N/A | 1373 K | 124 |
| La3LiGeS7 | [LaS8] | P63 (no. 173) | 3.02 | 0.7 | 6.0 | PM | 0.025 | 1173 K | 126 |
| La6Ga2GeS14 | [LaS8] | P63 (no. 173) | 2.54 | 4.8@74–106 μm | N/A | NPM | 0.023 | 1223 K | 82 |
| La6In2GeS14 | [LaS8] | P63 (no. 173) | 2.61 | 1.8@74–106 μm | N/A | NPM | 0.007 | 1173 K | 82 |
| Y6Ga2GeS14 | [YS8] | P63 (no. 173) | 2.30 | Weak | 3.0 | NPM | N/A | 1153 K | 130 |
| EuHgGeSe4 | [EuSe8] | Ama2 (no. 40) | 1.97 | 3.1 | N/A | PM | 0.31 | 1173 K | 133 |
| EuHgSnS4 | [EuS8] | Ama2 (no. 40) | 2.14 | 1.77 | N/A | PM | 0.33 | 1173 K | 133 |
| EuCdGeSe4 | [EuSe8] | Ama2 (no. 40) | 2.25 | 3.8 | N/A | PM | 0.18 | 1273 K | 132 |
| EuCdGeS4 | [EuS8] | Ama2 (no. 40) | 2.50 | 2.6 | N/A | PM | 0.18 | 1273 K | 132 |
| Eu2Ga2GeS7 | [EuS6] |
P21m (no. 113) |
2.30 | 1.6@46–74 μm | N/A | NPM | 0.098 | 1123 K | 134 |
| La2Ga2GeS8 | [LaS8] | Cmc21 (no. 36) | 2.78 | weak@46–74 μm | N/A | N/A | 0.077 | 1373 K | 134 |
| Ba2InYSe5 | [YSe6] | Cmc21 (no. 36) | 2.31 | 1 × AgGaSe2 | N/A | N/A | N/A | 1323 K | 135 |
| Ba4Sm2Cd3S10 | [SmS6] | Cmc21 (no. 36) | 2.77 | 1.8@46–74 μm | 14.3 | N/A | 0.044 | 1173 K | 137 |
| LaSrGa3S7 | [LaS8] |
P21m (no. 113) |
2.92 | 1.3 | 5.0 | PM | 0.099 | 1273 K | 139 |
| LaCaGa3S7 | [LaS8] |
P21m (no. 113) |
2.96 | 1.3 | 5.0 | PM | 0.134 | 1273 K | 139 |
| LaCaAl3S7 | [LaS8] |
P21m (no. 113) |
3.76 | 0.8 | 9 | PM | 0.059 | 1473 K | 138 |
| LaSrAl3S7 | [LaS8] |
P21m (no. 113) |
3.78 | 1.1 | 9 | PM | 0.077 | 1473 K | 138 |
| La2Ca3Sn3S12 | [(La/Ca)S7]/[(La/Ca)S9] |
P2m (no. 189) |
1.65 | 1.4@200–250 μm | N/A | NPM | 0.008 | 1073 K | 142 |
| Sm2CaSn3S12 | N/A |
P2m (no. 189) |
1.66 | 1.2@200–250 μm | N/A | NPM | N/A | 1073 K | 142 |
| Gd2Ca3Sn3S12 | N/A |
P2m (no. 189) |
1.63 | 1.0@200–250 μm | N/A | NPM | N/A | 1073 K | 142 |
| La2Sr3Sn3S12 | [(La/Ca)S7]/[(La/Ca)S8] | Pmc21 (no. 26) | 1.68 | 3.0 | N/A | PM | 0.086 | 1073 K | 142 |
| Sm2Sr3Sn3S12 | N/A | Pmc21 (no. 26) | 1.71 | 2.5 | N/A | PM | N/A | 1073 K | 142 |
| Gd2Sr3Sn3S12 | N/A | Pmc21 (no. 26) | 1.51 | 2.6 | N/A | PM | N/A | 1073 K | 142 |
| CeLiSiS4 | [CeS8] | Ama2 (no. 40) | 2.92 | 2.1 | 9 | PM | 0.054 | 1073 K | 143 |
| LaLiSiS4 | [LaS8] | Ama2 (no. 40) | 3.71 | 2.0 | 14 | PM | 0.033 | 1073 K | 143 |
| CsLaGeS4 | [LaS7] | P212121 (no. 19) | 3.60 | 0.5 × α-SiO2 | N/A | N/A | N/A | 1173 K | 146 |
| KYGeS4 | [YS7] | P21 (no. 4) | 3.15 | 1.0 | 10 | PM | 0.12 | 1173 K | 148 |
| K3YP2S8 | [YS8] | P21 (no. 4) | 3.37 | 1.4 | 7.0 | PM | 0.096 | 923 K | 149 |
| K3HoP2S8 | [HoS8] | P21 (no. 4) | N/A | 1.1 | 3.0 | PM | 0.084 | 923 K | 149 |
| K3ErP2S8 | [ErS8] | P21 (no. 4) | N/A | 1.2 | 2.5 | PM | 0.099 | 923 K | 149 |
| Compound | [REQn] polyhedra | Space group | E g (eV) | d eff (×AgGaS2) | LIDTb (×AgGaS2) | NPM/PMc | Δnd | Maximum temperaturef | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Experimental value. b Powder sample. c PM = phase-matchability, NPM = nonphase-matchability. d Theoretical value. e N/A = not available. f The maximum temperature of solid-state reaction. | |||||||||
| Ce8Sb2S15 | [CeS7]/[CeS8] | I41cd (no. 110) | 1.99 | 0.03 | N/A | NPM | N/A | 1223 K | 163 |
| La8Sb2S15 | [LaS7]/[LaS8] | I41cd (no. 110) | 2.30 | 1.2@74–106 μm | N/A | NPM | N/A | 1223 K | 171 |
| La2CuSbS5 | [LaS8]/[LaS10] | Ima2 (no. 46) | 2.06 | 0.5 | 6.7 | PM | 0.11 | 1273 K | 154 |
| La4InSbS9 | [LaS6]/[LaS7] | P41212 (no. 92) | 2.07 | 1.5 | N/A | PM | N/A | 1223 K | 156 |
| Pr4InSbS9 | [PrS6]/[PrS7] | P43212 (no. 96) | 2.09 | No signal | N/A | N/A | N/A | 1223 K | 156 |
| Nd4InSbS9 | [NdS6]/[NdS7] | P43212 (no. 96) | 2.12 | No signal | N/A | N/A | N/A | 1223 K | 156 |
| Sm4InSbS9 | [SmS6]/[SmS7] | P43212 (no. 96) | 2.13 | 0.75@150–210 μm | N/A | PM | N/A | 1223 K | 158 |
| Y4GaSbS9 | [YS6]/[YS7] | Aba2 (no. 41) | 2.06 | 7.5 × α-SiO2@74–106 μm | N/A | NPM | N/A | 1223 K | 159 |
| Sm4GaSbS9 | [SmS6]/[SmS7] | Aba2 (no. 41) | 2.23 | 3.8@46–74 μm | N/A | NPM | N/A | 1223 K | 157 |
| Ho4GaSbS9 | [HoS6]/[HoS7] | Aba2 (no. 41) | 2.25 | 0.25@46–74 μm | N/A | N/A | N/A | 1223 K | 157 |
| Gd4GaSbS9 | [GdS6]/[GdS7] | Aba2 (no. 41) | 2.41 | 0.8@46–74 μm | N/A | NPM | N/A | 1223 K | 157 |
| Tb4GaSbS9 | [TbS6]/[TbS7] | Aba2 (no. 41) | 2.44 | Weak | N/A | N/A | N/A | 1223 K | 157 |
| Compound | [REQn] polyhedra | Space group | E g (eV) | d eff (×AgGaS2) | LIDT (×AgGaS2)b | NPM/PMc | Δnd | Maximum temperaturef | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Experimental value. b Powder sample. c PM = phase-matchability, NPM = nonphase-matchability. d Theoretical value. e N/A = not available. f The maximum temperature of solid-state reaction. | |||||||||
| LaBS3 | [LaS9] | Pna21 (no. 33) | 3.18 | 1.2 | 14 | PM | 0.143 | 1023 K | 167 |
| Ca2La(BS3)(SiS4) | [LaS6] | P63mc (no. 186) | 3.38 | 1.1 | 10 | PM | 0.149 | 1123 K | 169 |
| Ca2Ce(BS3)(SiS4) | [CeS6] | P63mc (no. 186) | 2.65 | 1.2 | 7 | PM | 0.149 | 1123 K | 169 |
| Ca2Gd(BS3)(SiS4) | [GdS6] | P63mc (no. 186) | 3.20 | 1.1 | 8 | PM | 0.126 | 1123 K | 169 |
| Eu2P2S6 | [EuS8] | Pc (no. 7) | 2.54 | 0.9 | 3.4 | PM | 0.074 | 1223 K | 171 |
| KSmP2Se6 | [SmSe8] | P21 (no. 4) | 1.92 | 1.08 | 1.43 | PM | N/A | 1023 K | 172 |
| KTbP2Se6 | [TbSe8] | P21 (no. 4) | 2.46 | 0.35 | 2.29 | PM | N/A | 1023 K | 172 |
| KGdP2Se6 | [GdSe8] | P21 (no. 4) | 2.53 | 0.44 | 4.33 | PM | 0.081 | 1023 K | 172 |
| Compound | [REQn] polyhedra | Space group | E g (eV)a | d eff (×AgGaS2)b | LIDT ( ×AgGaS2)b | NPM/PMc | Δnd | Maximum temperaturef | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Experimental value. b Powder sample. c PM = phase-matchability, NPM = nonphase-matchability. d Theoretical value. e N/A = not available. f The maximum temperature of solid-state reaction. | |||||||||
| La6Cd0.75Ga2Se11.5Cl2.5 | [LaSe7] | P63 (no. 173) | 1.65 | 0.1@43–75 μm | N/A | NPM | N/A | 1173 K | 173 |
| La6Cd0.75Ga2S11.5Cl2.5 | [LaS7] | P63 (no. 173) | 2.28 | 0.8@43–75 μm | 18.6 | NPM | N/A | 1173 K | 173 |
| La3AsS5Br2 | [LaS7] | Cc (no. 9) | 2.90 | 0.23 | N/A | N/A | N/A | 1123 K | 174 |
| Eu4.5(B5O9)2SI | [EuO5S2I]/[EuO5S2I]/[EuO4S2] | Pnn2 (no. 34) | 1.99 | 0.5 | 15 | PM | N/A | 1223 K | 176 |
| (K3I)[SmB12(GaS4)3] | [SmS6] | P6322 (no. 182) | 2.35 | 0.3 × KTP | N/A | N/A | N/A | 1223 K | 177 |
| [La3OSCl2][SbS3] | [(Cl/S)La3] | P63mc (no. 186) | 2.50 | 0.7 | N/A | N/A | 0.269 | 1223 K | 178 |
| YSeBO2 | [YO3Se4]11− | Cmc21 (no. 36) | 3.45 | 0.0054 | N/A | PM | N/A | 1223 K | 180 |
| Eu3GeOS4 | [EuOS6] | Pca21 (no. 29) | 2.05 | 0.24@110–150 μm | 8.86 | NPM | 0.019 | 1148 K | 181 |
| Sm3NbS3O4 | [SmS8] | Pna21 (no. 33) | 2.68 | 0.3 | 12.5 | PM | N/A | 1273 K | 185 |
| Gd3NbS3O4 | [GdS8] | Pna21 (no. 33) | 2.74 | 0.4 | 4.5 | PM | N/A | 1273 K | 185 |
| Nd3[Ga3O3S3][Ge2O7] | [NdO6S2] |
P2c (No. 190) |
4.35 | 0.8 | 23 | PM | 0.091 | 1273 K | 186 |
| Eu2MnGe2OS6 | [EuOS7] |
P21m (no. 113) |
2.40 | 0.3 | 8.3 | PM | 0.13 | 1173 K | 187 |
| Eu2FeGe2OS6 | [EuOS7] |
P21m (no. 113) |
2.11 | 0.3 | 2.8 | PM | N/A | 1173 K | 187 |
| Eu2CoGe2OS6 | [EuOS7] |
P21m (no. 113) |
2.14 | 0.5 | 3.2 | PM | N/A | 1173 K | 187 |
| LaSrGa3S6O | [LaS7O] |
P21m (no. 113) |
3.21 | 1.0 | 14 | PM | 0.138 | 1273 K | 139 |
| LaCaGa3S6O | [LaS7O] |
P21m (no. 113) |
3.27 | 0.9 | 14 | PM | 0.163 | 1273 K | 139 |
(1) Due to the Gd discontinuity effect, there is a slight variation in the ionic radius of lanthanide elements, which leads to differences in their chemical and physical properties. Therefore, based on differences in electronic configuration, as well as physical and chemical properties, the elements preceding Gd are classified as light RE elements or cerium-group elements (La–Eu), while the remaining elements are classified as heavy RE elements (Gd–Lu, Y, and Sc). As shown in Scheme 1, light RE-based chalcogenide compounds have greater appeal than heavy ones. Based on the classification of light and heavy RE elements, an analysis of these compounds reveals that the probability of obtaining NCS compounds through the synthesis of light RE elements is 47.9% (excluding the radioactive Pm element), which is twice the average of heavy RE elements (20.0%). Furthermore, the NLO-active compounds in the light RE elements group account for a remarkable 70.7% of the total, which is also double the percentage found in the heavy group (29.3%). Of course, the price difference between light and heavy RE elements will also impact the synthesis and amount of research into the compounds. However, a more profound reason might be that the synthesis of light RE elements is more conducive to obtaining NCS structures and exhibiting NLO activity. Moreover, by statistically analysing the NCS compounds of each RE element and specifically examining those with NLO activity, it is observed that the top three elements are Eu (88.2%), La (38.2%), and Sm (29.7%), which all belong to the light RE category. Additionally, the percentage of NLO-active materials among Eu-based NCS materials is remarkably high, reaching 88.2%, surpassing the second-ranked La (38.2%). This demonstrates that among the 16 elements (excluding the radioactive Pm), Eu-based compounds stand out as promising candidates for IR-NLO materials with excellent potential.
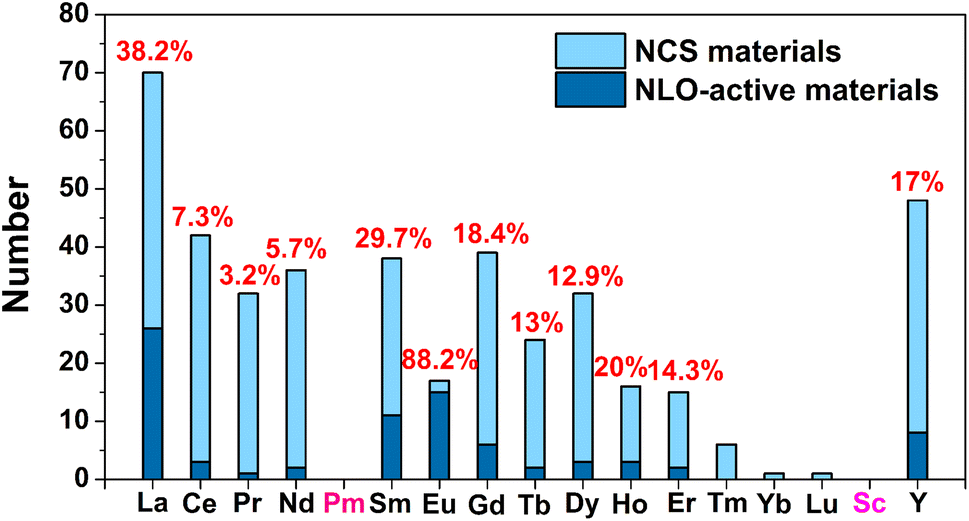 | ||
| Scheme 1 Distribution and proportion of NLO activity in different RE-based NCS materials. | ||
(2) These compounds can be divided into four categories based upon the number of elements: ternary (9, 10.6%), quaternary (62, 72.9%), quinary (13, 15.3%), and hexanary (1, 1.2%). Furthermore, in terms of the dimensionality of the crystal structure, there are 6 (7.1%) 0D compounds, 1 (1.2%) 1D compound, 22 2D (25.9%) compounds, 42 (49.4%) 3D compounds, and 14 (16.5%) MD compounds. Additionally, based on the crystal systems, they can be divided into five categories: monoclinic (12, 14.1%), orthorhombic (31, 36.5%), tetragonal (16, 18.8%), trigonal (2, 2.4%), and hexagonal (24, 28.2%). Analysis of the space group distribution revealed that the top three space groups are P63 (No.173, 15 examples), Cmc21 (No.36, 11 examples), and P21m (no. 113, 10 examples) (see Scheme 2 for details).
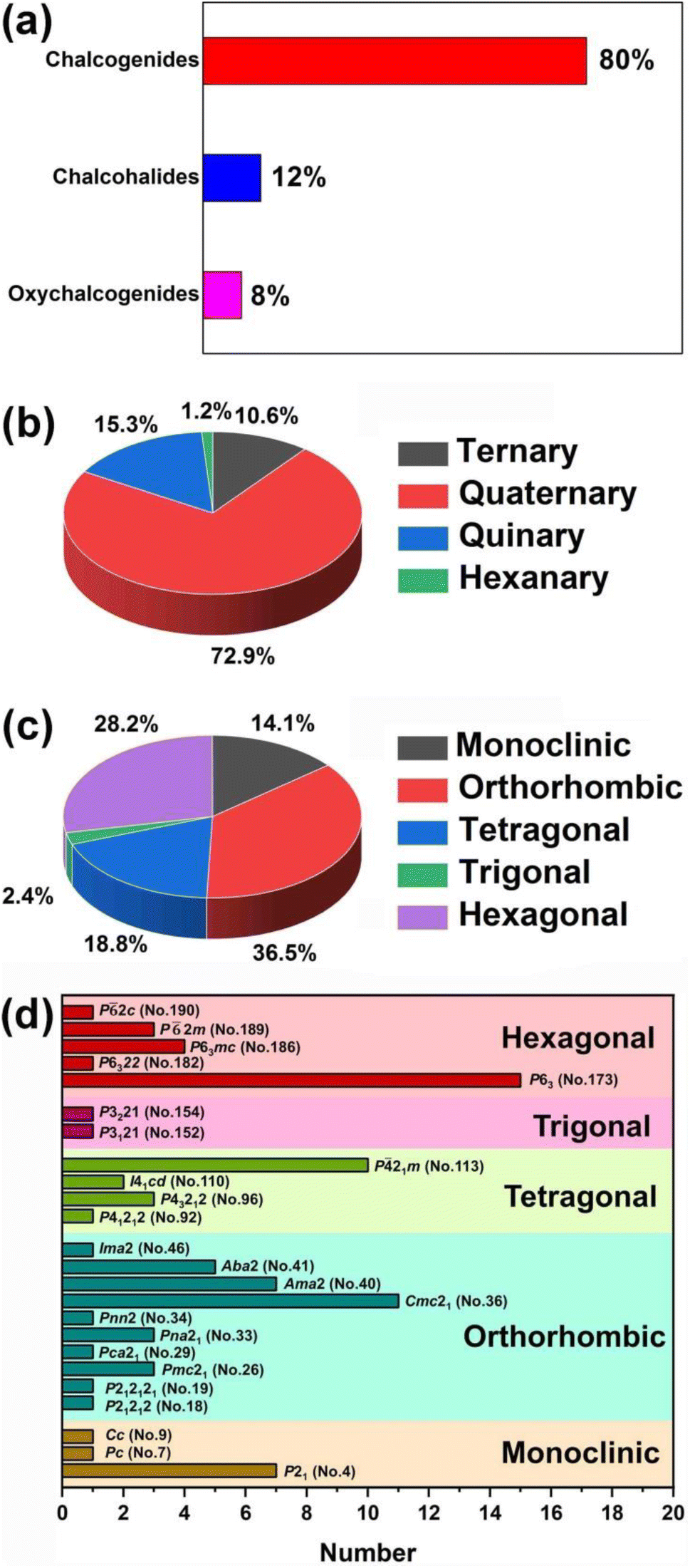 | ||
| Scheme 2 Distributions of RE-based IR-NLO materials based on (a and b) chemical composition, (c) crystal system, and (d) the space groups in five crystal systems. | ||
(3) The RE-based asymmetric building motifs that have been reported currently exhibit two different coordination modes. These include the single-anion coordination mode, which comprises [REQn] (n = 6–10), and the heteroanion coordination mode, which consists mainly of [REQ2O4], [REQ4O3], [REQ6O], [REQ7O], [REQ2O6], [REQ4OX3], and [REQ2O5X] (Scheme 3). It is important to note that the structures of these asymmetric building motifs determine their performance. Consequently, RE-based materials composed of these different asymmetric building motifs exhibit distinct IR-NLO properties. In Scheme 4, a comparison of the deff and Eg of RE-based IR-NLO materials is displayed. La6Ga2GeS14 has the largest deff of 4.8 × AgGaS2 at a particle size of 74–106 μm among all the RE-based chalcogenide IR-NLO materials, but it has a relatively small Eg of 2.54 eV. Nd3[Ga3O3S3][Ge2O7] has the largest Eg of 4.35 eV, while it has a relatively small deff of 0.8 × AgGaS2 at a particle size of 150–210 μm. The “performance-balanced area” indicates the potential materials whose deff and Eg are both higher than those of AgGaS2. In this performance-balanced area, there are 11 PM chalcogenides, but only 1 PM oxychalcogenide and no chalcohalides, which demonstrates the outstanding performance of RE-based chalcogenides. The exploration of oxychalcogenides and chalcohalides should be continued to enable a more in-depth analysis of their structural chemistry and understanding the origin of their properties through additional experimental results.
 | ||
| Scheme 3 Coordination modes of RE-based asymmetric building motifs (CN = coordination number). Green atom: RE; yellow atom: chalcogen (Q); red atom: O; blue atom: halogen (X). | ||
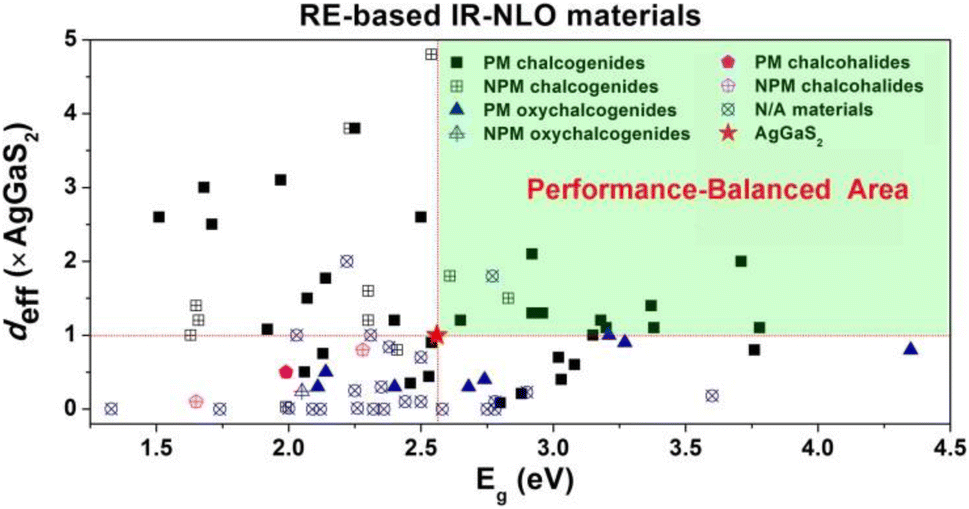 | ||
| Scheme 4 Comparison of the deff (×AgGaS2) and Eg (eV) for selected RE-based materials with IR-NLO properties (the green shaded region represents the performance-balanced area, wherein deff > 1.0 AgGaS2 and Eg > 2.56 eV). | ||
(4) For the synthesis method of RE-based IR-NLO compounds, the common method is high-temperature solid-state synthesis. Additionally, the introduction of RE elements not only involves the use of elemental RE, but also employs the reactive flux and boron–chalcogen mixture methods. The boron–chalcogen mixture method uses boron and RE oxides to remove oxygen in situ from rare earth oxides during the reaction, allowing the participation of RE elements. The emergence of this method enriches the selection of reaction pathways.
Currently, out of over 400 RE-based compounds, fewer than 100 demonstrate NLO properties. With decades of accumulated knowledge, our predecessors have laid a substantial foundation for us. Now is the opportune moment to strategically delve into synthesizing and exploring the underlying mechanisms, cultivating the soil to harvest successful outcomes. For this, the following outlook is provided:
(1) Further investigation into the mechanisms of RE-based polyhedra is needed. Currently, a significant question remains unanswered in the realm of RE-based compounds: why do only certain compounds of RE elements exhibit nonlinear effects in isomeric compounds? The differences among RE cations go beyond simple considerations of atomic radius; the underlying reasons should be attributed to variations in their electronic structures. Previous researchers have explored and synthesized numerous compounds in this field. In-depth studies of the electronic structures of different cations can help to uncover the answer to this question. Additionally, the contribution of the 5d and 4f empty orbitals of RE cations plays a crucial role in the conductive bands. By delving into the electronic structures of various anions and selectively substituting RE cations and anions with different 4f and 5d electronic structures based on their distinct structures, blind synthesis of RE-based compounds can be avoided, and hidden treasures within the accumulated knowledge can be uncovered. In contemporary research, the continual progress in theoretical calculations and machine learning plays a pivotal role in the exploration and design of novel materials. When synthesizing RE-based chalcogenides, the substitutability of RE sites with various RE elements is commonly considered, yet synthesis endeavours are often time-consuming. Consequently, upon successfully synthesizing a particular compound, it becomes important to validate, through theoretical calculations, the generalizability of the synthesis method to other RE elements. Additionally, when investigating isostructural compounds, a judicious approach involves preliminary theoretical analyses to identify RE elements that may exhibit superior performance, guiding subsequent synthesis explorations.
(2) According to Pauling's third and fourth rules, in systems containing more than one type of cation, polyhedra with high oxidation states and low coordination numbers tend to be connected through sharing corners or remain unconnected. The two functional motifs [PS4] and [SiS4], which are beneficial in terms of Eg, can be taken as examples. Due to the high positive charge and low coordination of the central cations, self-aggregation into high-dimensional frameworks becomes challenging under electrostatic rules. Therefore, a common approach for stabilization is to connect them with other polyhedra possessing central cations with lower positive charges and higher coordination. When alkali earth metals or alkali metals bond with these functional motifs, the resulting bonds are primarily ionic in nature. One characteristic of ionic bonds is isotropy, which makes it difficult to restrict the orientation of functional motifs and thus facilitates the formation of a thermodynamically stable centrosymmetric phase. Additionally, the isotropic nature of these bonds leads to low optical anisotropy. The bonding between REn+ and S2− is complex, primarily due to the complexity of their respective electronic structures. Furthermore, as different REn+ engage in bonding, the variability in their individual electronic structures contributes to distinct electron density of states within the system. As a result, this diversity leads to varying responses to external light stimuli. The presence of high-coordination RE cations acts as a binder between polyhedra with high oxidation states and coordination numbers. In these systems, RE cations not only play a role as a structural binder, but also contribute to providing nonlinear optical responses. Therefore, re-examining the structures of different compounds in the database reveals that introducing RE cations to connect previously disordered functional units may have a rejuvenating effect of “old wood meets spring”, enhancing performance while adjusting the structure.
(3) The growth of large crystals is necessary. Currently, the major RE-based compounds are tested in the powder state, but the real working state of crystal materials is the single-crystal state. Consequently, in order to satisfy the demands of test accuracy, further test results are needed in the large single-crystal state, which is also a prerequisite for evaluating the commercialization of NLO materials.
Data availability
There is no experimental or computational data associated with this article.Author contributions
Ping Feng: investigation, writing – original draft. Jia-Xiang Zhang: investigation, writing – original draft. Mao-Yin Ran: investigation, formal analysis. Xin-Tao Wu: conceptualization, formal analysis. Hua Lin: supervision, conceptualization, writing – review & editing. Qi-Long Zhu: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (22175175), Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (2021ZR118), and the Natural Science Foundation of Fujian Province (2022L3092 and 2023H0041).Notes and references
- D. F. Voss, Science, 1987, 237, 573 CrossRef CAS PubMed
.
- J. C. Whitehead, Nature, 1998, 393, 430 CrossRef CAS
- S. W. Eaton, A. Fu, A. B. Wong, C.-Z. Ning and P. Yang, Nat. Rev. Mater., 2016, 1, 16028 CrossRef CAS
- N. T. Otterstrom, R. O. Behunin, E. A. Kittlaus, Z. Wang and P. T. Rakich, Science, 2018, 360, 1113 CrossRef CAS PubMed
- N. Ahn, C. Livache, V. Pinchetti and V. I. Klimov, Chem. Rev., 2023, 123, 8251 CrossRef CAS PubMed
-
T. Schneider, Nonlinear Optics in Telecommunications, Springer Science & Business Media, 2004 Search PubMed
-
D. N. Nikogosyan, Nonlinear Optical Crystals: A Complete Survey, Springer, New York, 1st edn, 2005 Search PubMed
-
M. G. Papadopoulos, A. J. Sadlej and J. Leszczynski, Non-linear optical properties of matter, Springer, 2006 Search PubMed
-
C. T. Chen, T. Sasaki, R. K. Li, Y. C. Wu, Z. S. Lin and Y. Mori, Nonlinear Optical Borate Crystals: Principals and Applications, John Wiley & Sons, 2012 Search PubMed
-
A. Newell, Nonlinear Optics, CRC Press, 2018 Search PubMed
- P. S. Halasyamani and J. M. Rondinelli, Nat. Commun., 2018, 9, 2972 CrossRef PubMed
- M. Mutailipu, M. Zhang, H. Z. Yang and L. S. Pan, Acc. Chem. Res., 2019, 52, 791 CrossRef CAS PubMed
- M. Mutailipu and S. Pan, Angew. Chem., Int. Ed., 2020, 59, 20302 CrossRef CAS PubMed
- Y. C. Liu, Y. G. Shen, S. G. Zhao and J. H. Luo, Coord. Chem. Rev., 2020, 407, 213152 CrossRef CAS
- M. Mutailipu, K. R. Poeppelmeier and L. S. Pan, Chem. Rev., 2021, 121, 1130 CrossRef CAS PubMed
- J. Chen, C.-L. Hu, F. Kong and J.-G. Mao, Acc. Chem. Res., 2021, 54, 2775 CrossRef CAS PubMed
- X. H. Meng, W. L. Yin and M. J. Xia, Coord. Chem. Rev., 2021, 439, 213916 CrossRef CAS
- H. Fan, N. Ye and M. Luo, Acc. Chem. Res., 2023, 56, 3099 CrossRef CAS PubMed
- X. Liu, Y.-C. Yang, M.-Y. Li, L. Chen and L.-M. Wu, Chem. Soc. Rev., 2023, 52, 8699 RSC
- J.-X. Zhang, P. Feng, M.-Y. Ran, X.-T. Wu, H. Lin and Q.-L. Zhu, Coord. Chem. Rev., 2024, 505, 215617 CrossRef
- R. C. Eckardt, H. Masuda, Y. X. Fan and R. L. Byer, IEEE J. Quantum Electron., 1990, 26, 922 CrossRef CAS
- M. E. Hagerman and K. R. Poeppelmeier, Chem. Mater., 1995, 7, 602 CrossRef CAS
- Y. N. Xia, C. T. Chen, D. Y. Tang and B. C. Wu, Adv. Mater., 1995, 7, 79 CrossRef CAS
- C. T. Chen, B. C. Wu, A. D. Jiang and G. M. You, Sci. Sin., Ser. B, 1985, 28, 235 Search PubMed
- A. Harasaki and K. Kato, Jpn. J. Appl. Phys., 1997, 36, 700 CrossRef CAS
- G. C. Catella, L. R. Shiozawa, J. R. Hietanen, R. C. Eckardt, R. K. Route, R. S. Feigelson, D. G. Cooper and C. L. Marquardt, Appl. Opt., 1993, 32, 3948 CrossRef CAS PubMed
- G. D. Boyd, E. Buehler and F.
G. Storz, Appl. Phys. Lett., 1971, 18, 301 CrossRef CAS
- L. Kang, D. M. Ramo, Z. Lin, P. D. Bristowe, J. Qin and C. Chen, J. Mater. Chem. C, 2013, 1, 7363–7370 RSC
- R. He, H. Huang, L. Kang, W. Yao, X. Jiang, Z. Lin, J. Qin and C. Chen, Appl. Phys. Lett., 2013, 102, 231904 CrossRef
- Z. Lin, X. Jiang, L. Kang, P. Gong, S. Luo and M. H. Lee, J. Phys. D: Appl. Phys., 2014, 47, 253001 CrossRef
- F. Liang, L. Kang, Z. Lin and Y. Wu, Cryst. Growth Des., 2017, 17, 2254 CrossRef CAS
- G. Peng, Y. Yang, Y. H. Tang, M. Luo, T. Yan, Y. Zhou, C. Lin, Z. Lin and N. Ye, Chem. Commun., 2017, 53, 9398 RSC
- C. Hu, B. Zhang, B.-H. Lei, S. Pan and Z. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 26413 CrossRef CAS PubMed
- Z. Yang, B.-H. Lei, W. Zhang and S. Pan, Chem. Mater., 2019, 31, 2807 CrossRef CAS
- J. Yu, B. Zhang, X. Zhang, Y. Wang, K. Wu and M.-H. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 45023 CrossRef CAS PubMed
- B.-W. Liu, X.-M. Jiang, S.-M. Pei, W.-F. Chen, L.-Q. Yang and G.-C. Guo, Mater. Horiz., 2021, 8, 3394 RSC
- H. Li, J. Min, Z. Yang, Z. Wang, S. Pan and A. R. Oganov, Angew. Chem., Int. Ed., 2021, 60, 10791 CrossRef CAS PubMed
- L. Xiong, L.-M. Wu and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 25063 CrossRef CAS PubMed
- L. Kang, M. L. Zhou, J. Y Yao, Z. S. Lin, Y. C Wu and C. T. Chen, J. Am. Chem. Soc., 2015, 137, 13049 CrossRef CAS PubMed
- F. Liang, L. Kang, Z. S. Lin, Y. C. Wu and C. T. Chen, Coord. Chem. Rev., 2017, 333, 57 CrossRef CAS
- J.-R. Xiao, S.-H. Yang, F. Feng, H.-G. Xue and S.-P. Guo, Coord. Chem. Rev., 2017, 347, 23 CrossRef CAS
- M.-M. Chen, H.-G. Xue and S.-P. Guo, Coord. Chem. Rev., 2018, 368, 115 CrossRef CAS
- P. Gong, F. Liang, L. Kang, X. Chen, J. Qin, Y. Wu and Z. Lin, Coord. Chem. Rev., 2019, 380, 83 CrossRef CAS
- L. Kang, F. Liang, X. Jiang, Z. Lin and C. Chen, Acc. Chem. Res., 2020, 53, 209 CrossRef CAS PubMed
- K. Wu, Y. Yang and L. Gao, Coord. Chem. Rev., 2020, 418, 213380 CrossRef CAS
- L. Gao, J. Huang, S. Guo, Z. Yang and S. Pan, Coord. Chem. Rev., 2020, 421, 213379 CrossRef CAS
- G. Zou and K. M. Ok, Chem. Sci., 2020, 11, 5404 RSC
- J. Dang, D. Mei, Y. Wu and Z. Lin, Coord. Chem. Rev., 2021, 431, 213692 CrossRef CAS
- F. Hou, D. Mei, M. Xia and Y. Wu, Coord. Chem. Rev., 2021, 444, 214038 CrossRef CAS
- C. X. Li, X. H. Meng, Z. Li and J. Y. Yao, Coord. Chem. Rev., 2022, 453, 214328 CrossRef CAS
- X. L. Chen and K. M. Ok, Chem. Sci., 2022, 13, 3942 RSC
- H.-D. Yang, M.-Y. Ran, W.-B. Wei, X.-T. Wu, H. Lin and Q.-L. Zhu, Mater. Today Phys., 2023, 35, 101127 CrossRef CAS
- M.-Y. Ran, A.-Y. Wang, W.-B. Wei, X.-T. Wu, H. Lin and Q.-L. Zhu, Coord. Chem. Rev., 2023, 481, 215059 CrossRef CAS
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS
- I. Chung and M. G. Kanatzidis, Chem. Mater., 2014, 26, 849 CrossRef CAS
- C. L. Hu and J. G. Mao, Coord. Chem. Rev., 2015, 288, 1 CrossRef CAS
- S.-P. Guo, Y. Chi and G.-C. Guo, Coord. Chem. Rev., 2017, 335, 44 CrossRef CAS
- K. Wu and S. L. Pan, Coord. Chem. Rev., 2018, 377, 191 CrossRef CAS
- Y. Y. Li, W. J. Wang, H. Wang, H. Lin and L. M. Wu, Cryst. Growth Des., 2019, 19, 4172 CrossRef CAS
- J. Zhao, D. Mei, W. Wang, Y. Wu and D. Xue, J. Rare Earths, 2021, 39, 1455 CrossRef CAS
- Y. F. Shi, W. B. Wei, X. T. Wu, H. Lin and Q. L. Zhu, Dalton Trans., 2021, 50, 4112 RSC
- H. Chen, W.-B. Wei, H. Lin and X.-T. Wu, Coord. Chem. Rev., 2021, 448, 214154 CrossRef CAS
- H.-D. Yang, M.-Y. Ran, W.-B. Wei, X.-T. Wu, H. Lin and Q.-L. Zhu, Chem.–Asian J., 2021, 16, 3299 CrossRef CAS PubMed
- H. Chen, M.-Y. Ran, W.-B. Wei, X.-T. Wu, H. Lin and Q.-L. Zhu, Coord. Chem. Rev., 2022, 470, 214706 CrossRef CAS
- Z.-X. Chen, W. Liu and S.-P. Guo, Coord. Chem. Rev., 2023, 474, 214870 CrossRef CAS
- J. J. Xu and K. Wu, Coord. Chem. Rev., 2023, 486, 215139 CrossRef
- P.-F. Li, J.-G. Mao and F. Kong, Mater. Today Phys., 2023, 37, 101197 CrossRef CAS
- A. M. Lozac’h, S. Jaulmes and M. Guittard, C. R. Acad. Sci., Ser. C, 1971, 272, 1123 Search PubMed
- M. J. Zhang, B. X. Li, B. W. Liu, Y. H. Fan, X. G. Li, H. Y. Zeng and G. C. Guo, Dalton Trans., 2013, 42, 14223 RSC
- H. Y. Chen, Y. Y. Zhang, Y. F. Yang and S. P. Guo, J. Alloys Compd., 2021, 868, 1873 Search PubMed
- C. R. Evenson IV and P. K. Dorhout, Z. Anorg. Allg. Chem., 2001, 627, 2178 CrossRef
- S. H. Yang, X. H. Li, W. D. Yao, Q. T. Xu and S. P. Guo, J. Solid State Chem., 2020, 288, 1095 Search PubMed
- J. Llanos, C. Mujica, V. Sánchez and O. Peña, J. Solid State Chem., 2003, 173, 78 CrossRef CAS
- J. A. Aitken, J. W. Lekse, J.-L. Yao and R. Quinones, J. Solid State Chem., 2009, 182, 141 CrossRef CAS
- Z. D. Sun, Y. Chi and S. P. Guo, J. Solid State Chem., 2019, 269, 225 CrossRef CAS
- F. Q. Huang and J. A. Ibers, Acta Crystallogr., Sect. C, 1999, 55, 1210 CrossRef
- L. D. Gulay, O. S. Lychmanyuk, J. Stepien-Damm, A. Pietraszko and I. D. Olekseyuk, J. Alloys Compd., 2005, 402, 201 CrossRef CAS
- I. Hartenbach, A. C. Mueller and T. Schleid, Z. Anorg. Allg. Chem., 2006, 632, 2147 CrossRef
- L. D. Gulay, O. S. Lychmanyuk, I. D. Olekseyuk, M. Daszkiewicz, J. Stepien-Damm and A. Pietraszko, J. Alloys Compd., 2007, 431, 185 CrossRef CAS
- M. Daszkiewicz, L. D. Gulay, A. Pietraszko and V. Y. Shemet, J. Solid State Chem., 2007, 180, 2053 CrossRef CAS
- I. Hartenbach, T. Nilges and T. Schleid, Z. Anorg. Allg. Chem., 2007, 633, 24452 Search PubMed
- Y. F. Shi, Y. K. Chen, M. C. Chen, L. M. Wu, H. Lin, L. J. Zhou and L. Chen, Chem. Mater., 2015, 27, 1876 CrossRef CAS
- A. Michelet and J. Flahaut, C. R. Acad. Sci. Ser. C, 1969, 269, 1203 CAS
- G. Perez, M. Darriet-Duale and P. Hagenmuller, J. Solid State Chem., 1970, 2, 42 CrossRef CAS
- G. Perez and M. Darriet, C. R. Acad, Sci. Ser. C, 1970, 270, 420 CAS
- M. Guittard and M. Julienpo, Bull. Soc. Chim. Fr., 1970, 7, 2467 Search PubMed
- G. Collin and J. Flahaut, C. R. Acad. Sci. Ser. C., 1970, 270, 488 CAS
- Y. Q. Zhou, A. K. Iyer, A. O. Oliynyk, M. Heyberger, Y. X. Lin, Y. Qiu and A. Mar, J. Solid State Chem., 2019, 278, 120914 CrossRef CAS
- M. A. Agaev, V. O. Aliev, A. B. Agaev and E. V. Magerramov, Inorg. Mater., 1994, 30, 1423 Search PubMed
- S. J. Hwu, C. K. Bucher, J. D. Carpenter and S. P. Taylor, Inorg. Chem., 1995, 34, 1979 CrossRef CAS
- R. L. Gitzendanner, C. M. Spencer, F. J. DiSalvo, M. A. Pell and J. A. Ibers, J. Solid State Chem., 1996, 131, 399 CrossRef
- S. H. Lin, J. G. Mao, G. C. Guo and J. S. Huang, J. Alloys Compd., 1997, 252, 8 CrossRef
- Y. T. Yang and J. A. Ibers, J. Solid State Chem., 2000, 155, 433 CrossRef CAS
- K. M. Poduska, F. J. DiSalvo, K. Min and P. S. Halasyamani, J. Alloys Compd., 2002, 335, L5 CrossRef CAS
- I. Hartenbach and T. Schleid, J. Solid State Chem., 2003, 171, 382 CrossRef CAS
- L. D. Gulay, I. D. Olekseyuk, M. Wolcryz and J. Stepien-Damm, Z. Anorg. Allg. Chem., 2005, 631, 1919 CrossRef CAS
- L. D. Gulay and I. D. Olekseyuk, J. Alloys Compd., 2005, 388, 274 CrossRef CAS
- L. B. Wu and F. Q. Huang, Z. Kristallogr.–New Cryst. Struct., 2005, 220, 307 CAS
- L. D. Gulay, O. S. Lychmanyuk, M. Wolcyrz, A. Pietraszko and I. D. Olekseyuk, J. Alloys Compd., 2006, 425, 159 CrossRef CAS
- L. D. Gulay, O. S. Lychmanyuk, I. D. Olekseyuk and A. Pietraszko, J. Alloys Compd., 2006, 422, 203 CrossRef CAS
- M. R. Huch, L. D. Gulay and I. D. Ekseyuk, J. Alloys Compd., 2006, 424, 114 CrossRef CAS
- L. D. Gulay, M. Daszkiewicz, M. R. Huch and A. Pietraszko, Acta Crystallogr., Sect. E, 2007, 63, I187 CrossRef CAS
- O. S. Lychmanyuk, L. D. Gulay, I. D. Olekseyuk, J. Stepien-Damm, M. Daszkiewicz and A. Pietraszko, Pol. J. Chem., 2007, 81, 353 CAS
- M. Daszkiewicz, L. D. Gulay, O. S. Lychmanyuk and A. Pietraszko, J. Alloys Compd., 2008, 460, 201 CrossRef CAS
- H. Y. Zeng, F. K. Zheng, G. C. Guo and J. S. Huang, J. Alloys Compd., 2008, 458, 1239 Search PubMed
- M. Daszkiewicz, L. D. Gulay, O. S. Lychmanyuk and A. Pietraszko, J. Alloys Compd., 2009, 467, 168 CrossRef CAS
- M. Daszkiewicz, L. D. Gulay and O. S. Lychmanyuk, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 126 CrossRef CAS PubMed
- S. P. Guo, G. C. Guo, M. S. Wang, J. P. Zou, G. Xu, G. J. Wang, X. F. Long and J. S. Huang, Inorg. Chem., 2009, 48, 7059 CrossRef CAS PubMed
- S. P. Guo, G. C. Guo and J. S. Huang, Sci. China, Ser. B: Chem., 2009, 52, 16095 Search PubMed
- O. M. Strok, M. Daszkiewicz, L. D. Gulay and D. Kaczorowski, J. Alloys Compd., 2010, 493, 47 CrossRef CAS
- B. W. Rudyk, S. S. Stoyko and A. Mar, J. Solid State Chem., 2013, 208, 78 CrossRef CAS
- B. W. Liu, H. Y. Zeng, Z. Y. Zhao, F. K. Zheng, G. C. Guo and J. S. Huang, Chin. J. Struct. Chem., 2013, 32, 1537 CAS
- B. W. Rudyk, S. S. Stoyko, A. O. Oliynyk and A. Mar, J. Solid State Chem., 2014, 210, 79 CrossRef CAS
- W. L. Yin, Y. G. Shi, B. Kang, J. G. Deng, J. Y. Yao and Y. C. Wu, J. Solid State Chem., 2014, 213, 87 CrossRef CAS
- X. Zhang, W. Chen, D. Mei, C. Zheng, F. Liao, Y. Li, J. Lin and F. Huang, J. Alloys Compd., 2014, 610, 617 CrossRef
- M. Daszkiewicz, O. V. Marchuk, L. D. Gulay and D. Kaczorowski, J. Alloys Compd., 2014, 610, 258 CrossRef CAS
- M. Daszkiewicz, Y. O. Pashynska, O. V. Marchuk, L. D. Gulay and D. Kaczorowski, J. Alloys Compd., 2014, 616, 243 CrossRef CAS
- H. J. Zhao, Synthesis, Crystal and Electronic Structure, and Optical Property of the Quaternary Selenide: La3Sb0.33SiSe7, Z. Anorg. Allg. Chem., 2015, 641, 917 CrossRef CAS
- O. Strok, M. Daszkiewicz and L. Gulay, Chem. Met. Alloys, 2015, 8, 16 CrossRef CAS
- M. Daszkiewicz, Y. O. Pashynska, O. V. Marchuk, L. D. Gulay and D. Kaczorowski, J. Alloys Compd., 2015, 647, 445 CrossRef CAS
- A. K. Iyer, B. W. Rudyk, X. S. Lin, H. Singh, A. Z. Sharma, C. R. Wiebe and A. Mar, J. Solid State Chem., 2015, 229, 150 CrossRef CAS
- A. Choudhury and P. K. Dorhout, Inorg. Chem., 2015, 54, 1055 CrossRef CAS PubMed
- A. K. Iyer, W. L. Yin, E. J. Lee, X. S. Lin and A. Mar, J. Solid State Chem., 2017, 250, 14 CrossRef CAS
- N. Zhen, L. Y. Nian, G. M. Li, K. W and S. L. P, Crystals, 2016, 6, 121 CrossRef
- Y. Zhou, A. K. Iyer, A. O. Oliynyk, M. Heyberger, Y. Lin, Y. Qiu and A. Mar, J. Solid State Chem., 2019, 278, 120914 CrossRef CAS
- Y. Yang, Y. Chu, B. B. Zhang, K. Wu and S. L. Pan, Chem. Mater., 2021, 33, 4225 CrossRef CAS
- G. Akopov, N. W. Hewage, P. Yox, G. Viswanathan, S. J. Lee, L. P. Hulsebosch, S. D. Cady, A. L. Paterson, F. A. Perras, W. Q. Xu, K. Wu, Y. Mudryk and K. Kovnir, Chem. Sci., 2021, 12, 14718 RSC
- L. H. Gao, X. W. Wu, J. J. Xu, X. Y. Tian, B. B. Zhang and K. Wu, J. Alloys Compd., 2022, 900, 163535 CrossRef CAS
- A. J. Craig, J. B. Cho, S. H. Shin, S. H. Ha, S. S. Stoyko, J. I. Jang and J. A. Aitken, J. Alloys Compd., 2022, 910, 164855 CrossRef CAS
- Y. Wang, Y. F. Shi, Y. Lin, Z. Chen and L. T. Li, Inorg. Chem. Commun., 2023, 153, 110829 CrossRef CAS
- J.-X. Zhang, M.-Y. Ran, X.-T. Wu, H. Lin and Q.-L. Zhu, Inorg. Chem. Front., 2023, 10, 5244 RSC
- W. Xing, N. Wang, Y. Guo, Z. Li, J. Tang, K. Kang, W. Yin, Z. Lin, J. Yao and B. Kang, Dalton Trans., 2019, 48, 17620 RSC
- W. Xing, C. Tang, N. Wang, C. Li, Z. Li, J. Wu, Z. Lin, J. Yao, W. Yin and B. Kang, Inorg. Chem., 2020, 59, 18452 CrossRef CAS PubMed
- M. C. Chen, P. Li, L. J. Zhou, L. H. Li and L. Chen, Inorg. Chem., 2011, 50, 12402 CrossRef CAS PubMed
- W. Yin, K. Feng, W. Wang, Y. Shi, W. Hao, J. Yao and Y. Wu, Inorg. Chem., 2012, 51, 6860 CrossRef CAS PubMed
- W. Yin, W. Wang, L. Bai, K. Feng, Y. Shi, W. Hao, J. Yao and Y. Wu, Inorg. Chem., 2012, 51, 11736 CrossRef CAS PubMed
- Q. G. Yue, S. H. Zhou, B. Li, X. T. Wu, H. Lin and Q. L. Zhu, Inorg. Chem., 2022, 61, 1797 CrossRef CAS PubMed
- A. Mazurier, S. Jaulmes and M. Guittard, Acta Crystallogr., Sect. C, 1987, 43, 1859 CrossRef
- J. J. Xu, K. Wu, Y. Xiao, B. B. Zhang, H. H. Yu and J. H. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 37967 CrossRef CAS PubMed
- J. J. Xu, K. Wu, B. B. Zhang, H. H. Yu and J. H. Zhang, Inorg. Chem. Front., 2023, 10, 2045 RSC
- H. Y. Zeng, Z. Y. Zhao, S. P. Guo, F. K. Zheng, G. C. Guo and J. S. Huang, J. Alloys Compd., 2012, 514, 135 CrossRef CAS
- J. Xu, Y. Xiao, K. Wu, B. Zhang, D. Lu, H. Yu and H. Zhang, Small, 2024, 20, 2306577 CrossRef CAS PubMed
- Y. X. Han, C. L. Hu, B. X. Li and J. G. Mao, Mater. Today Phys., 2023, 31, 100987 CrossRef CAS
- P. Wu and J. A. Ibers, J. Solid State Chem., 1993, 107, 347 CrossRef CAS
- M. Usman, M. D. Smith, G. Morrison, V. V. Klepov, W. Zhang, P. S. Halasyamani and H. C. Zur Loye, Inorg. Chem., 2019, 58, 8541 CrossRef CAS PubMed
- A. K. Gray, J. M. Knaust, B. C. Chan, L. A. Polyakova and P. K. Dorhout, Z. Kristallogr.–New Cryst. Struct., 2005, 220, 293 CAS
- S. P. Guo, H. Y. Zeng, G. C. Guo, J. P. Zou, G. Xu and J. S. Huang, Chin. J. Struct. Chem., 2008, 27, 1543 CAS
- D. J. Mei, W. Z. Cao, N. Z. Wang, X. X. Jiang, J. Zhao, W. K. Wang, J. H. Dang, S. Y. Zhang, Y. D. Wu and P. H. Rao, Mater. Horiz., 2021, 8, 2330 RSC
- X. Tian, Y. Xiao, B. Zhang, D. Yang and K. Wu, Mater. Today Phys., 2022, 28, 100885 CrossRef CAS
- A. Assoud, K. M. Kleinke and H. Kleinke, Chem. Mater., 2006, 18, 1041 CrossRef CAS
- H. J. Zhao and L. J. Zhou, Eur. J. Inorg. Chem., 2015, 6, 964 CrossRef
- G. G. Guseinov, F. K. Mamedov, I. R. Amiraslanov and K. S. Mamedov, Z. Kristallogr., 1981, 26, 831 CAS
- H. J. Zhao and X. A. Zhong, J. Solid State Chem., 2017, 251, 65 CrossRef CAS
- H. Lin, Y. Y. Li, M. Y. Li, Z. Ma, L. M. Wu, X. T. Wu and Q. L. Zhu, J. Mater. Chem. C, 2019, 7, 4638 RSC
- A. M. Kussainova, L. G. Akselrud, N.-T. Suen, L. Voss, S. Stoyko and S. Bobev, J. Solid State Chem., 2016, 233, 269 CrossRef CAS
- H. J. Zhao, Y. F. Zhang and L. Chen, J. Am. Chem. Soc., 2012, 134, 1993 CrossRef CAS PubMed
- M. C. Chen, L. H. Li, Y. B. Chen and L. Chen, J. Am. Chem. Soc., 2011, 133, 4617 CrossRef CAS PubMed
- H. J. Zhao, Z. Anorg. Allg. Chem., 2015, 642, 56 CrossRef
- Y. Wang, X. C. Zou, X. Feng, Y. F. Shi and L. M. W, J. Solid State Chem., 2017, 245, 110 CrossRef CAS
- H. J. Zhao, J. Solid State Chem., 2016, 237, 99 CrossRef CAS
- Y. Wang, H. J. Zhao, Y.-F. Shi, P. F. Liu, X. C. Zou and Y. R. Ren, Chin. J. Struct. Chem., 2017, 36, 1465 CAS
- W. Yin, A. K. Iyer, C. Li, J. Yao and A. Mar, J. Alloys Compd., 2017, 710, 424 CrossRef CAS
- J. Hunger, M. Borna and R. Kniep, J. Solid State Chem., 2010, 183, 702 CrossRef CAS
- M. Borna, J. Hunger and R. Kniep, Z. Kristallogr.–New Cryst. Struct., 2010, 225, 223 CAS
- M. Borna, J. Hunger and R. Kniep, Z. Kristallogr.–New Cryst. Struct., 2010, 225, 225 CAS
- M. Borna, J. Hunger, A. Ormeci, D. Zahn, U. Burkhardt, W. C. Cabrera, R. C. Gil and R. Kniep, J. Solid State Chem., 2011, 184, 296 CrossRef CAS
- L. S. Breton, G. Morrison, M. R. Lacroix, P. S. Halasyamani and H. C. Zur Loye, Chem. Commun., 2022, 58, 7992 RSC
- Y. X. Han, C. L. Hu, Z. Fang, Q. Q. Chen, B. X. Li, Y. Lin and J. G. Mao, J. Mater. Chem. C, 2022, 10, 12556 RSC
- Y. X. Han, C. L. Hu and J. G. Mao, Small, 2024, 20, 2305828 CrossRef CAS PubMed
- W. Brockner and R. Becker, Z. Naturforsch. A, 1987, 42, 511 CrossRef CAS
- X. Huang, S. H. Yang, X. H. Li, W. Liu and S. P. Guo, Angew. Chem., Int. Ed., 2022, 61, e202206791 CrossRef CAS PubMed
- Z. X. Chen, C. Y. Zhao, X. H. Li, W. D. Yao, W. Liu and S. P. Guo, Small, 2023, 19, 2206910 CrossRef CAS PubMed
- F. Lin, M. Luo, R. Wang, X. Che and F. Huang, CrystEngComm, 2021, 23, 2133 RSC
- A. Cicirello, A. Swindle and J. Wang, CrystEngComm, 2023, 25, 6354 RSC
- D. H. Kang, F. Ledderboge, H. Kleinke and T. Schleid, Z. Anorg. Allg. Chem., 2015, 641, 322 CrossRef CAS
- Y. Chi, H. G. Xue and S. P. Guo, Inorg. Chem., 2020, 59, 1547 CrossRef CAS PubMed
- S. P. Guo, G. C. Guo, M. S. Wang, J. P. Zou, H. Y. Zeng, L. Z. Cai and J. S. Huang, Chem. Commun., 2009, 29, 4366 RSC
- H. J. Zhao, P. F. Liu and L. M. Wu, Dalton Trans., 2021, 50, 2075 RSC
- H. J. Zhao, H. D. Yang, P. F. Liu and H. Lin, Cryst. Growth Des., 2022, 22, 1437 CrossRef CAS
- Z. T. Lu, W. J. Fan, Z. Q. Wang, N. Gu, Z. H. Yue, H. G. Xue and S. P. Guo, Inorg. Chem., 2020, 59, 7905 CrossRef CAS PubMed
- M. Yang, W. D. Yao, W. Liu and S. P. Guo, Chem. Commun., 2023, 59, 3894 RSC
- C. Boyer-Candalen, A. Meerschaut and P. Palvadeau, Mater. Res. Bull., 2000, 35, 1593 CrossRef CAS
- H. Kabbour, L. Cario, C. Deudon and A. Meerschaut, Acta Crystallogr., Sect. E, 2003, 59, i101 CrossRef CAS
- S. Altmannshofer and D. Johrendt, Z. Anorg. Allg. Chem., 2008, 634, 1361 CrossRef CAS
- X. Lian, Z. T. Lu, W. D. Yao, S. H. Yang, W. Liu, R. L. Tang and S. P. Guo, Inorg. Chem., 2021, 60, 10885 CrossRef CAS PubMed
- M. Y. Ran, S. H. Zhou, W. B. Wei, B. X. Li, X. T. Wu, H. Lin and Q. L. Zhu, Small, 2023, 19, 2300248 CrossRef CAS PubMed
- N. Zhang, X. Huang, W. D. Yao, Y. Chen, Z. R. Pan, B. Li, W. Liu and S. P. Guo, Inorg. Chem., 2023, 62, 16299 CrossRef CAS PubMed
- D. Mei, W. Yin, L. Bai, Z. Lin, J. Yao, P. Fu and Y. Wu, Dalton Trans., 2011, 40, 3610 RSC
- K. Wu, Z. Yang and S. Pan, Chem. Mater., 2016, 28, 2795 CrossRef CAS
- H. Chen, P. Liu, B. Li, H. Lin, L. Wu and X.-T. Wu, Dalton Trans., 2018, 47, 429 RSC
- Y. Yang, K. Wu, X. Wu, B. Zhang and L. Gao, J. Mater. Chem. C, 2020, 8, 1762 RSC
- G. Li, Z. Yang, J. Li and S. L. Pan, Chem. Commun., 2020, 56, 11565 RSC
- D. Desaintg, P. Laruelle and J. Flahaut, C. R. Acad. Sci., Ser. C, 1968, 267, 1029 Search PubMed
- M. Patrie and M. Guittard, C. R. Acad. Sci., Ser. C, 1969, 268, 1136 CAS
- Y. Zhang, D. Mei, Y. Yang, W. Cao, Y. Wu, J. Lu and Z. Lin, J. Mater. Chem. C, 2019, 7, 8556 RSC
- Y. Guo, F. Liang, W. Yin, Z. Li, X. Luo, Z.-S. Lin, J. Yao, A. Mar and Y. Wu, Chem. Mater., 2019, 31, 3034 CrossRef CAS
- Y.-J. Lin, B.-W. Liu, R. Ye, X.-M. Jiang, L.-Q. Yang, H.-Y. Zeng and G.-C. Guo, J. Mater. Chem. C, 2019, 7, 4459 RSC
- M. Yan, Z.-D. Sun, W.-D. Yao, W. Zhou, W. Liu and S.-P. Guo, Inorg. Chem. Front., 2020, 7, 2451 RSC
- H.-D. Yang, M.-Y. Ran, S.-H. Zhou, X.-T. Wu, H. Lin and Q.-L. Zhu, Chem. Sci., 2022, 13, 10725 RSC
- Y.-N. Li, Z.-X. Chen, W.-D. Yao, R.-L. Tang and S.-P. Guo, J. Mater. Chem. C, 2021, 9, 8659 RSC
- Q. Q. Liu, X. Liu, L. M. Wu and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202205587 CrossRef CAS PubMed
- M.-Y. Ran, S.-H. Zhou, W.-B. Wei, B.-X. Li, X.-T. Wu, H. Lin and Q.-L. Zhu, Small, 2024, 20, 2304563 CrossRef CAS PubMed
- H. Lin, Y. Liu, L. J. Zhou, H. J. Zhao and L. Chen, Inorg. Chem., 2016, 55, 4470 CrossRef CAS PubMed
- H. Lin, H. Chen, Y. J. Zheng, J. S. Yu and L. M. Wu, Dalton Trans., 2016, 45, 17606 RSC
- M.-Y. Ran, Z. Ma, X.-T. Wu, H. Lin and Q.-L. Zhu, Inorg. Chem. Front., 2021, 8, 4838 RSC
- Y. T. Yang and J. A. Ibers, J. Solid State Chem., 2000, 149, 384 CrossRef CAS
- M. C. Chen, L. M. Wu, H. Lin, L. J. Zhou and L. Chen, J. Am. Chem. Soc., 2012, 134, 6058 CrossRef CAS PubMed
- M.-Y. Li, B. Li, H. Lin, Z. Ma, L.-M. Wu, X.-T. Wu and Q.-L. Zhu, Chem. Mater., 2019, 31, 6268 CrossRef CAS
- M.-M. Chen, Z. Ma, B.-X. Li, W.-B. Wei, X.-T. Wu, H. Lin and Q.-L. Zhu, J. Mater. Chem. C, 2021, 9, 1156 RSC
- Y. Xiao, M. M. Chen, Y. Y. Shen, P. F. Liu, H. Lin and Y. Liu, Inorg. Chem. Front., 2021, 8, 2835 RSC
- M. Yan, H.-G. Xue and S.-P. Guo, Cryst. Growth Des., 2021, 21, 698 CrossRef CAS
- C. Liu, S.-H. Zhou, Y. Xiao, C. Zhang, H. Lin and Y. Liu, J. Mater. Chem. C, 2021, 9, 15407 RSC
- X.-H. Li, Z.-H. Shi, M. Yang, W. Liu and S.-P. Guo, Angew. Chem., Int. Ed., 2022, 61, e202115871 CrossRef CAS PubMed
- M.-M. Chen, S.-H. Zhou, W. Wei, M.-Y. Ran, B. Li, X.-T. Wu, H. Lin and Q.-L. Zhu, ACS Mater. Lett., 2022, 4, 1264 CrossRef CAS
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Adv. Mater., 2019, 31, 1805295 CrossRef PubMed
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef PubMed
- Y. Pan, S.-P. Guo, B.-W. Liu, H.-G. Xue and G.-C. Guo, Coord. Chem. Rev., 2018, 374, 464 CrossRef CAS
- R. Wang, F. Liang, F. Wang, Y. Guo, X. Zhang, Y. Xiao, K. Bu, Z. Lin, J. Yao, T. Zhai and F. Q. Huang, Angew. Chem., Int. Ed., 2019, 58, 8078 CrossRef CAS PubMed
- M. Y. Ran, Z. J. Ma, H. Chen, B. X. Li, X. T. Wu, H. Lin and Q. L. Zhu, Chem. Mater., 2020, 32, 5890 CrossRef CAS
- H. Chen, Y. Y. Li, B. X. Li, P. F. Liu, H. Lin, Q. L. Zhu and X. T. Wu, Chem. Mater., 2020, 32, 8012 CrossRef CAS
- H. Yan, Y. Matsushita, K. Yamaura and Y. Tsujimoto, Angew. Chem., Int. Ed., 2021, 60, 26561 CrossRef CAS PubMed
- Y.-F. Shi, Z. Ma, B.-X. Li, X. Wu, H. Lin and Q.-L. Zhu, Mater. Chem. Front., 2022, 6, 3054 RSC
- Y. F. Shi, S. H. Zhou, B. Li, Y. Liu, X. T. Wu, H. Lin and Q. L. Zhu, Inorg. Chem., 2023, 62, 464 CrossRef CAS PubMed
- Q.-G. Yue, W.-B. Wei, H. Chen, X.-T. Wu, H. Lin and Q.-L. Zhu, Dalton Trans., 2020, 49, 14338 RSC
- M.-Y. Ran, S.-H. Zhou, B.-X. Li, W.-B. Wei, X.-T. Wu, H. Lin and Q.-L. Zhu, Chem. Mater., 2022, 34, 3853 CrossRef CAS
- H.-D. Yang, S.-H. Zhou, M.-Y. Ran, X.-T. Wu, H. Lin and Q.-L. Zhu, Inorg. Chem., 2022, 61, 15711 CrossRef CAS PubMed
- J. Wang, Y. Cheng, H. Wu, Z. Hu, J. Wang, Y. Wu and H. Yu, Angew. Chem., Int. Ed., 2022, 61, e202201616 CrossRef CAS PubMed
- H.-D. Yang, S.-H. Zhou, M.-Y. Ran, X.-T. Wu, H. Lin and Q.-L. Zhu, Inorg. Chem. Front., 2023, 10, 2030 RSC
- Y. F. Shi, X. F. Li, Y. X. Zhang, H. Lin, Z. J. Ma, L. M. Wu, X. T. Wu and Q. L. Zhu, Inorg. Chem., 2019, 58, 6588 CrossRef CAS PubMed
- M. Y. Li, Y. X. Zhang, H. Lin, Z. J. Ma, X. T. Wu and Q. L. Zhu, Dalton Trans., 2019, 48, 17588 RSC
- S.-H. Zhou, M.-Y. Ran, W.-B. Wei, A.-Y. Wang, X.-T. Wu, H. Lin and Q.-L. Zhu, Inorg. Chem. Front., 2023, 10, 5997 RSC
- X. Chen, S.-H. Zhou, C. Zhang, H. Lin and Y. Liu, Chem. Commun., 2023, 59, 12124 RSC
- Y.-F. Shi, S.-H. Zhou, P.-F. Liu, X.-T. Wu, H. Lin and Q.-L. Zhu, Inorg. Chem. Front., 2023, 10, 4425 RSC
- S. P. Guo, C. Yang, B. W. Liu and G. C. Guo, Dalton Trans., 2016, 45, 10459 RSC
- Z. D. Sun, Y. Chi, H. G. Xue and S. P. Guo, Inorg. Chem. Front., 2017, 4, 1841 RSC
- S. S. Han, W. D. Yao, S. X. Yu, Y. L. Sun, A. H. Gong and S. P. Guo, Inorg. Chem., 2021, 60, 3375 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |