
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu site differentiation in tetracopper(I) sulfide clusters enables biomimetic N2O reduction†
Pinar
Alayoglu
a,
Suresh C.
Rathnayaka
 a,
Tieyan
Chang
b,
SuYin Grass
Wang
b,
Yu-Sheng
Chen
b and
Neal P.
Mankad
*a
a,
Tieyan
Chang
b,
SuYin Grass
Wang
b,
Yu-Sheng
Chen
b and
Neal P.
Mankad
*a
aDepartment of Chemistry, University of Illinois at Chicago, Chicago, IL 60607, USA. E-mail: npm@uic.edu
bChemMatCARS, The University of Chicago, Argonne, IL 60439, USA
First published on 31st July 2024
Abstract
Copper clusters feature prominently in both metalloenzymes and synthetic nanoclusters that mediate catalytic redox transformations of gaseous small molecules. Such reactions are critical to biological energy conversion and are expected to be crucial parts of renewable energy economies. However, the precise roles of individual metal atoms within clusters are difficult to elucidate, particularly for cluster systems that are dynamic under operating conditions. Here, we present a metal site-specific analysis of synthetic Cu4(μ4-S) clusters that mimic the CuZ active site of the nitrous oxide reductase enzyme. Leveraging the ability to obtain structural snapshots of both inactive and active forms of the synthetic model system, we analyzed both states using resonant X-ray diffraction anomalous fine structure (DAFS), a technique that enables X-ray absorption profiles of individual metal sites within a cluster to be extracted independently. Using DAFS, we found that a change in cluster geometry between the inactive and active states is correlated to Cu site differentiation that is presumably required for efficient activation of N2O gas. More precisely, we hypothesize that the Cuδ+⋯Cuδ− pairs produced upon site differentiation are poised for N2O activation, as supported by computational modeling. These results provide an unprecedented level of detail on the roles of individual metal sites within the synthetic cluster system and how those roles interplay with cluster geometry to impact the reactivity function. We expect this fundamental knowledge to inform understanding of metal clusters in settings ranging from (bio)molecular to nanocluster to extended solid systems involved in energy conversion.
Introduction
Numerous critical reactions in biology rely on multiproton/multielectron redox transformations that serve both to provide biosynthetic building blocks and to store and transduce cellular energy. These reactions are commonly catalyzed by metalloenzymes whose catalytic active sites feature multinuclear metal clusters capable of managing the orchestrated delivery of multiple proton and electron equivalents to/from an activated substrate.1 Examples of biological reactions catalyzed by enzymatic metal clusters include the hydrogen evolution (HER), hydrogen oxidation (HOR),2 oxygen evolution (OER),3 oxygen reduction (ORR),4 carbon dioxide reduction (CO2RR),5 carbon monoxide oxidation,6 and nitrogen reduction (N2RR)7 reactions, among others. Synthetic analogues of many of these reactions are thought to be requirements for supporting a renewable energy economy but often depend on precious metal catalysts such as platinum or iridium.8 Thus, detailed knowledge about the operating mechanisms of bioinorganic metal clusters composed of only earth-abundant metals is of urgent value. The pinnacle of understanding would include information on how individual metal sites within a given cluster contribute to its overall behavior.One fascinating case study is the tetranuclear CuZ site of nitrous oxide reductase (N2OR),9 which catalyzes the 2H+/2e− conversion of nitrous oxide (N2O) to N2 and H2O during the bacterial denitrification branch of the global nitrogen cycle.10 Nitrous oxide remediation is an emerging societal problem, because anthropogenic N2O emissions have a pronounced greenhouse effect (∼300× that of CO2) and are projected to be the leading cause of ozone layer depletion in the next century.11 The metal coordination chemistry of N2O is underdeveloped due to its inability to bind strongly to transition metal sites in an end-on fashion, in some cases being outcompeted even by N2 as a ligand.12 The first crystallographically characterized complex with an N2O ligand was reported only in 2011,13 and just a small number of examples have emerged since.14–17 Because rational design of synthetic catalysts for N2O fixation is thus elusive, it will be informative to learn the details by which the CuZ site is capable of efficient N2O reduction at ambient conditions on 1012 g per y scale.18 Furthermore, gaining detailed knowledge about the CuZ cluster has ramifications on fundamental understanding of extended Cu-based materials renowned for CO2RR19 and other important processes20,21via the “cluster-surface analogy”.22–25
Especially given the experimental difficulties in studying complicated and dynamic metalloenzymes like N2OR directly,26–28 synthetic model clusters can play an important role in elucidating relevant reaction pathways.29–32 However, even for well-defined synthetic Cu–S clusters, there are obstacles toward gaining detailed knowledge of chemical behavior. First, N2OR activity of CuZ (and the synthetic mimics discussed herein) is correlated to cluster reduction to a closed-shell, diamagnetic 4CuI state,28,33 thus limiting applicability of spectroscopic and physical tools (e.g. EPR, Mössbauer, SQUID) commonly employed to study synthetic clusters experimentally. Second, CuZ is a homometallic cluster active in a homovalent redox state, making deconvolution of the roles of individual Cu sites a challenge.
We have contributed synthetic compounds mimicking the Cu4(μ4-S) structural core of CuZ34–36 and have observed stoichiometric N2O reactivity in some of the cases,37–39 thus opening an opportunity for systematic studies. Here, we report a comparative study of two closely related Cu4(μ4-S) clusters to probe factors controlling relative reactivity towards 2H+/2e− reduction of N2O. Using resonant X-ray diffraction anomalous fine structure (DAFS) measurements,40–43 we have determined that N2O reductase activity in the model clusters is correlated to Cu site differentiation upon hydrogen-bond induced structural distortion. Calibrated by these measurements, quantum chemical calculations were used to simulate N2O activation on a site-specific basis. Collectively, the study addresses – on a metal atom-by-atom basis – how the cluster's geometric and electronic parameters enable multimetallic cooperativity during biomimetic N2O reduction.
Results and discussion
Yam originally discovered in 1993 that addition of sulfide sources to dicopper(I) complex [Cu2(dppm)2]2+ induces assembly of tetracopper(I) sulfide complex [Cu4(μ4-S)(dppm)4]2+ [1, dppm = Ph2PCH2PPh2].44 Although the composition of the active CuZ site in N2OR is under debate,26–28 we recognized that 1 shares both the 4Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1S stoichiometry and 4CuI redox state of the active form of CuZ favored by Solomon and Moura (Fig. 1a) based on many years of spectroscopic and enzymological investigations.26,28,33 In 2014, we prepared a new derivative, [Cu4(μ4-S)(dppa)4]2+ [2, dppa = Ph2PNHPPh2], by addition of Na2S to [Cu2(dppa)2(CH3CN)2]2+ (3).34 In 2020, we reported that exposure of 2 to N2O (1 atm) in the presence of a sacrificial reductant (CoCp2) in a hydrogen bond acceptor solvent (MeOH or acetone) produces stoichiometric quantities of N2 and H2O.39 Crucially, activity towards N2O conversion was not observed in the absence of reductant, in alternative solvents incapable of accepting hydrogen bonds, or when compound 2 with –NH bridgeheads was replaced by compound 1 with –CH2 bridgeheads. Multiple X-ray crystal structures of 2 have exhibited both –NH⋯solvent hydrogen bonds and highly distorted Cu4(μ4-S) cores with unsymmetrical Cu⋯Cu distances.34,39,45 The nature of this distortion bears resemblance to the unsymmetrical structure of the Cu4(μ4-S) core of CuZ in the native enzyme.9,46,47 In contrast, the structure of complex 1 that lacks the ability to hydrogen bond shows a fairly symmetrical, square pyramidal Cu4(μ4-S) core.44
:1S stoichiometry and 4CuI redox state of the active form of CuZ favored by Solomon and Moura (Fig. 1a) based on many years of spectroscopic and enzymological investigations.26,28,33 In 2014, we prepared a new derivative, [Cu4(μ4-S)(dppa)4]2+ [2, dppa = Ph2PNHPPh2], by addition of Na2S to [Cu2(dppa)2(CH3CN)2]2+ (3).34 In 2020, we reported that exposure of 2 to N2O (1 atm) in the presence of a sacrificial reductant (CoCp2) in a hydrogen bond acceptor solvent (MeOH or acetone) produces stoichiometric quantities of N2 and H2O.39 Crucially, activity towards N2O conversion was not observed in the absence of reductant, in alternative solvents incapable of accepting hydrogen bonds, or when compound 2 with –NH bridgeheads was replaced by compound 1 with –CH2 bridgeheads. Multiple X-ray crystal structures of 2 have exhibited both –NH⋯solvent hydrogen bonds and highly distorted Cu4(μ4-S) cores with unsymmetrical Cu⋯Cu distances.34,39,45 The nature of this distortion bears resemblance to the unsymmetrical structure of the Cu4(μ4-S) core of CuZ in the native enzyme.9,46,47 In contrast, the structure of complex 1 that lacks the ability to hydrogen bond shows a fairly symmetrical, square pyramidal Cu4(μ4-S) core.44
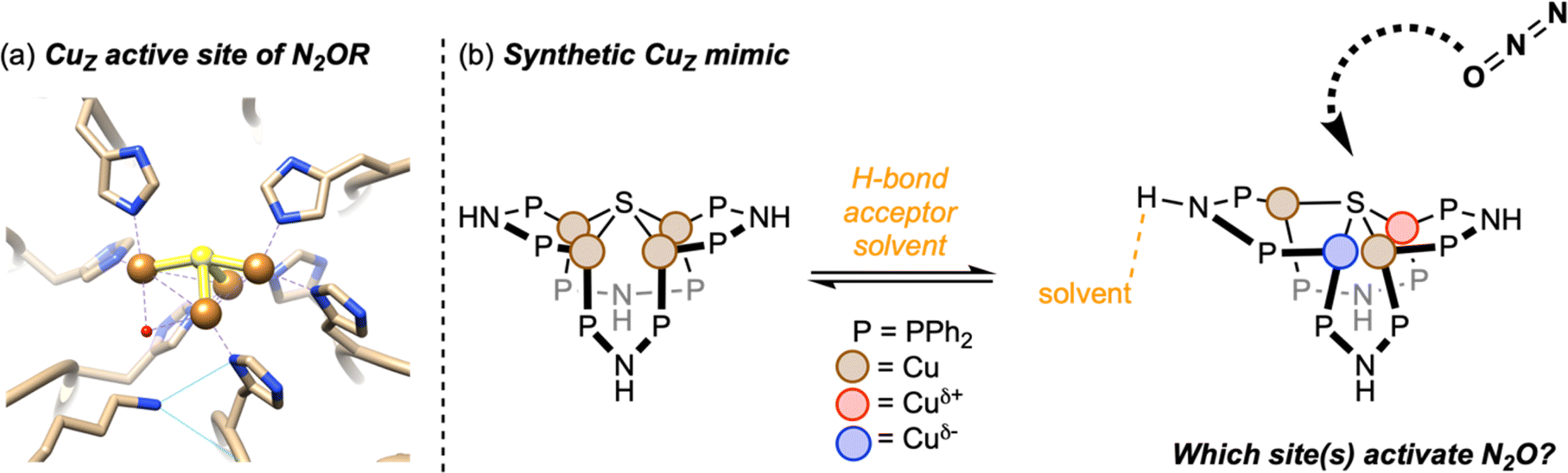 | ||
| Fig. 1 (a) The Cu4(μ4-S) active site of nitrous oxide reductase from Paracoccus denitrificans (PDB ID 1FWX, image reproduced from Rathnayaka et al.9); (b) schematic of the synthetic CuZ mimic in this study. | ||
Based on these observations, we hypothesize that the Cu4(μ4-S) core is dynamic in solution, with the symmetrical (inactive) form being the ground state and the distorted (active) form being a strained geometry that is stabilized by hydrogen bonding in the secondary coordination sphere (Fig. 1b). Variable-temperature 31P NMR spectroscopy provide support for this hypothesis. The room-temperature 31P NMR spectrum of 1 shows a singlet at 14.6 ppm corresponding to the eight P-atoms in the cluster that are chemically and magnetically equivalent on the NMR timescale. Over the temperature range 244–315 K, the chemical shift of this peak was found to vary by <0.3 ppm, and it remained as a sharp singlet with no detectable line broadening (Fig. S1†). These observations are consistent with the solution structure of 1 matching the symmetrical solid-state structure characterized by X-ray crystallography, as noted by Yam previously.44 On the other hand, for complex 2 in methanol-d4, decoalescence of the 31P NMR signal at 36.1 ppm was resolved in high-quality spectra even at room temperature. Over the temperature range 230–276 K, more than two inequivalent P-atom environments were resolved, with each chemical shift having different temperature dependences (Fig. S2†). These observations are consistent with 2 having access in solution to unsymmetrical conformations akin to the solid-state structures characterized by X-ray crystallography, such that these conformers can be frozen out at low temperatures. Previously reported34 solvent dependence of the 31P NMR spectroscopy of 2 indicates that these conformational changes are correlated to hydrogen bonding interactions with solvent molecules.
The main goal of this study was to compare electronic structures of crystalline forms of 1 and 2, which provide structural snapshots of solution-phase inactive and active forms of the synthetic CuZ mimic, respectively. We were particularly interested in elucidating Cu site differentiation in unsymmetrical 2 and determining how that might enable its biomimetic activity towards N2O (Fig. 1b). To achieve this, we used resonant X-ray diffraction anomalous fine structure (DAFS), a technique that exploits crystallographic refinement at multiple wavelengths near the metal K-edge to resolve different X-ray absorption profiles of individual metal sites within homometallic clusters.40,41,43,48–50 These resonant X-ray absorption energies, in turn, are proportional to each metal site's effective nuclear charge (Zeff) in a manner akin to more traditional X-ray absorption near edge structure (XANES). As with other synthetic systems analyzed by DAFS that commonly crystallize in centrosymmetric space groups,40,42,48,50 the real component of anomalous scattering (f′) is plotted against incident X-ray energy for each crystallographically independent metal site to facilitate DAFS analysis. The overall negative peak shapes obtained in f′ vs. E plots have three distinct regions: the falling edge, the intricate in-edge features, and the rising edge. In line with previous DAFS literature on highly covalent cluster complexes,40,48 our previous work calibrating DAFS at the Cu K-edge indicates that the peak shape observed at the in-edge region is sensitive to coordination geometry, the peak width (i.e., difference between falling and rising edges) is sensitive to metal–ligand covalency, and the rising edge position is the best reporter of Zeff.51,52 Complexes 1 and 2 were analyzed by DAFS, as was dicopper(I) complex 3 as a reference point. The ability to correlate DAFS data for clusters 1 and 2 to their reactivity behavior is a unique aspect of this study without precedent in the DAFS literature.
Single crystals of 1 suitable for DAFS were obtained for its PF6− salt as an acetone solvate. As in previously reported structures,44 the cationic portion of 1 was found to reside on a crystallographic special position, causing only two of the four Cu sites to be crystallographically unique within the asymmetric unit. The Cu4(μ4-S) core features four coplanar Cu sites slightly distorted from an idealized square shape according to the Cu⋯Cu distances [2.8581(7) and 3.1072(7) Å], with effectively identical Cu–S distances [2.2650(4) and 2.2659(4) Å] across the cluster. The DAFS profiles for the two unique Cu sites are nearly identical (Fig. 2a), with indistinguishable falling and rising edge positions and with matching, symmetrical doublet in-edge shapes corresponding to each Cu site having four nearest-neighbor atoms (CuP2S).51 These data indicate that the individual Cu sites in 1 experience effectively identical environments in terms of metal–ligand covalency, coordination geometry, and Zeff. In other words, there is no Cu site differentiation in the Cu4(μ4-S) cluster that is unreactive towards N2O.
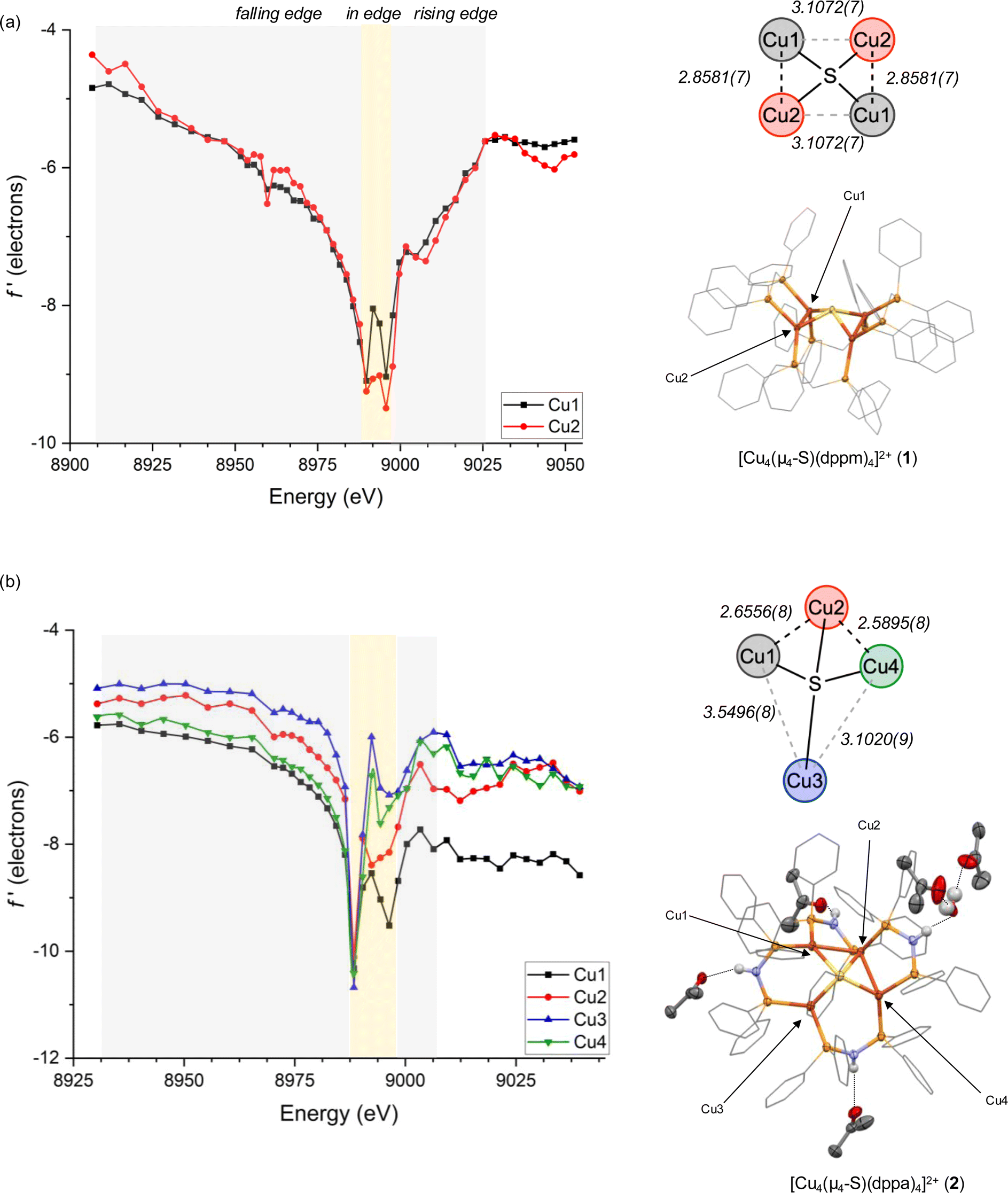 | ||
| Fig. 2 DAFS data and X-ray crystal structures for (a) [Cu4S(dppm)4][PF6]2 (1) and (b) [Cu4S(dppa)4][PF6]2 (2). Bond distances are given in Å. Anions, C–H hydrogens, and co-crystallized solvent molecules not involved in hydrogen bonding are omitted from X-ray structures for clarity. Phenyl carbons are shown as wireframes, and all other non-hydrogen atoms are shown as thermal ellipsoids (50% probability). | ||
Single crystals of 2 were obtained for its PF6− salt as a mixed acetone/H2O solvate, here with all four Cu sites being crystallographically unique. The structure features three of the –NH groups donating hydrogen bonds to acetone molecules and one participating in a hydrogen bonding network with multiple H2O and acetone molecules. Nonetheless, the distorted Cu4(μ4-S) core bears similarities to previously characterized MeOH and acetone solvates34,39,45 and to the Cu4(μ4-S) core of native CuZ.9,46,47 Three of the Cu sites (Cu1, Cu2, Cu4) are spaced closely together in a bent arrangement with Cu1⋯Cu2 distances and Cu2⋯Cu4 distances of 2.6556(8) and 2.5895(8) Å, respectively, and at a Cu1⋯Cu2⋯Cu4 angle of 109.20(2)°. A fourth Cu site (Cu3) is displaced further away with Cu3⋯Cu1 and Cu3⋯Cu4 distances of 3.5496(8) and 3.1020(9) Å, respectively. Once again, the Cu–S distances are nearly identical across the cluster, ranging from 2.222(1) to 2.269(1) Å. In this case, all four Cu sites are well resolved by DAFS and have distinctive f′ vs. E profiles (Fig. 2b). In other words, there is clearly Cu site differentiation in the Cu4(μ4-S) cluster that is reactive towards N2O.
Within the DAFS plots for complex 2, the Cu1 and Cu4 sites are nearly superimposed in the falling edge region (8930–8988 eV), while the Cu2 and Cu3 sites are blue shifted in this region by ∼4 and ∼8 eV, respectively. In the rising edge region (8996–9039 eV), the Cu1 is the bluest shifted and Cu4 the reddest shifted, with ∼10 eV between them. The Cu2 and Cu3 sites fall between Cu1 and Cu4 in the rising edge region, with red shifts of ∼5 eV and ∼7–8 eV, respectively, compared to Cu1. As will be discussed further below, these rising edge positions initially indicate that the Cu sites experience relative Zeff in the order Cu1 > Cu2 > Cu3 > Cu4 before correcting for other factors (especially coordination number). Additionally, because the Cu1 site exhibits a measurably broader peak than the other comparable Cu sites, it is likely that Cu1 experiences a particularly large degree of covalent bonding with its neighboring atoms.
This relative covalency may be influenced by orbital overlap that, in turn, is heavily impacted by coordination geometry. This motivated us to examine the in-edge DAFS region (8988–8996 eV) more closely as it relates to each Cu site's local environment. The four Cu sites each give unsymmetrical doublet shapes at the in-edge region, with major features at 8988 eV and minor features at slightly higher energy. Although the major features are nearly invariant across the series, the minor feature is most prominent for Cu1 and least so for Cu3. These distinct in-edge peak shapes are reflective of the different local geometries surrounding the four Cu sites (Fig. 3a). The Cu3 site is displaced far away from the other three Cu atoms, giving it only three neighboring atoms (P2S). As such, its peak shape closely matches those of three-coordinate Cu sites we previously characterized by DAFS (Fig. S9†).51 The Cu2 site, on the other hand, has five neighboring atoms (Cu2P2S) and a correspondingly different in-edge shape. The Cu1 site in 2 has four nearest neighbor atoms (CuP2S) in a seesaw arrangement (τ4 = 0.43), giving an in-edge peak shape by DAFS that closely matches those of the two seesaw Cu sites in 1 (τ4 = 0.44 and 0.44) (the τ4 geometric index ranges from 0 for idealized square planar centers to 1 for idealized tetrahedral centers53). These seesaw-shaped Cu sites also give DAFS responses closely resembling that of a trigonal pyramidal Cu site in 3 (τ4 = 0.44, Fig. S7†), albeit significantly blue shifted (Fig. 3b). We tentatively attribute this blueshift to a nuclearity effect, i.e., that higher nuclearity clusters tend to have blue-shifted f′ features. The origin of this nuclearity effect is unclear at this time, but we hypothesize that it may be induced by the electrostatic fields of the other CuI ions in the cluster that are outside bonding range but within reasonable Coulombic range. Similar trends were observed for trinuclear Cu clusters previously,51 suggesting that the nuclearity effect may be systematic. Lastly, the Cu4 site also has four neighboring atoms (CuP2S) but is distorted towards a square-planar arrangement (τ4 = 0.25) quite dissimilar from Cu1 (Fig. 3a), as reflected in its unique in-edge shape. Overall, it is clear that coordination number and geometry impact the DAFS responses of the Cu sites in this study, in agreement with previous Cu K-edge DAFS experiments.51 Hence, in accord with those previous calibration measurements that indicated a ∼3.5 eV blue shift per coordination number increase, correction factors of −3.5 eV and +3.5 eV are applied to the rising edge positions of Cu2 and Cu3, respectively, to facilitate direct comparisons to the various four-coordinate Cu sites in the global analysis below. A similar correction was applied previously to a five-coordinate Cu complex for direct comparison of its DAFS response to related four-coordinate complexes.52
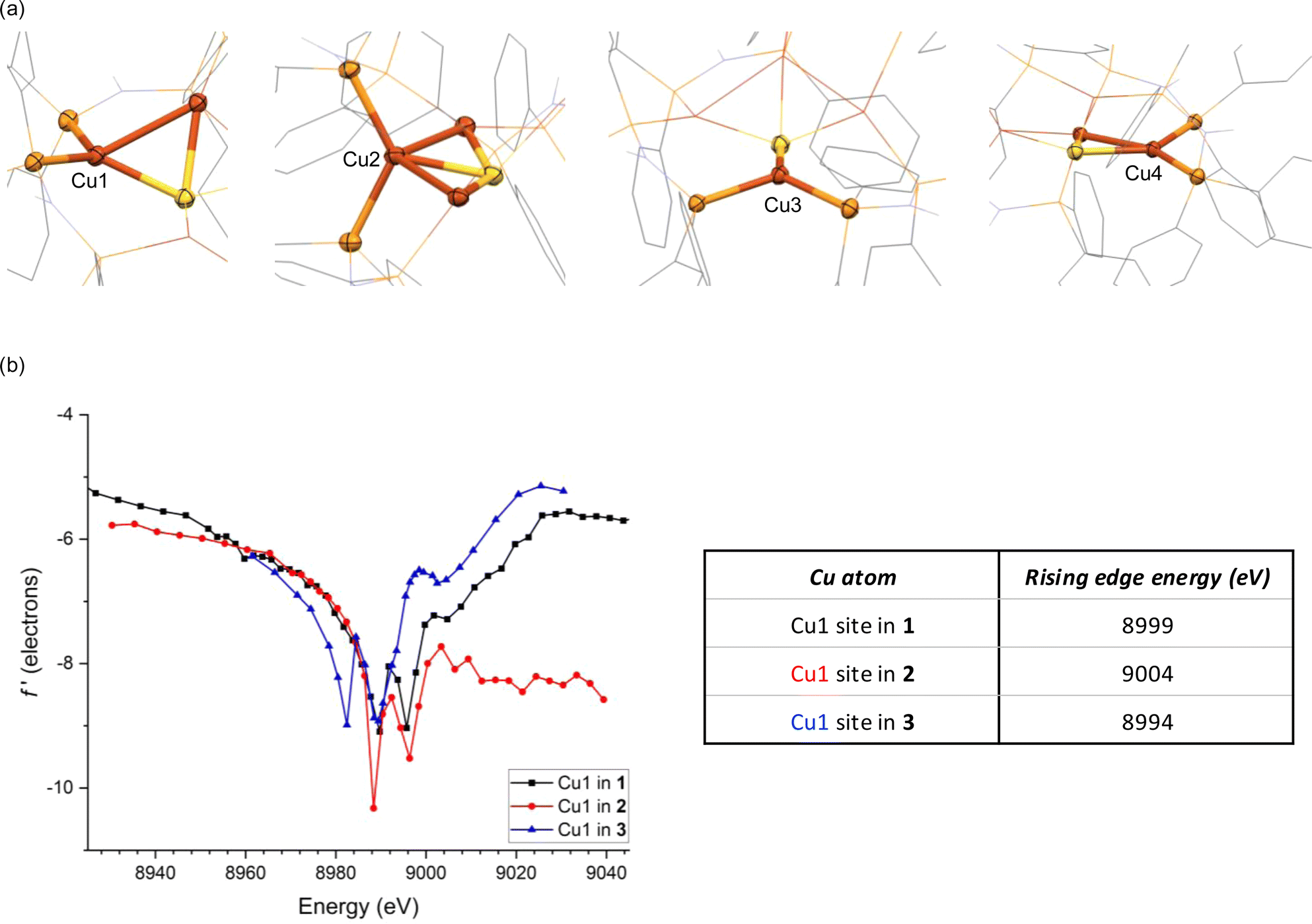 | ||
| Fig. 3 (a) Nearest neighbor atoms surrounding each Cu site in 1; (b) DAFS data for the Cu1 sites in 1, 2, and [Cu2(dppa)2][PF6]2 (3). Rising edge energies were estimated using a linear regression protocol described in ESI.† | ||
A previously used52 linear fitting procedure (Fig. S10†) was employed to estimate the rising edge position of each Cu site, enabling semi-quantitative comparisons across the series (Fig. 4a). For the dicopper(I) complex 3, the two Cu sites have rising edge positions (f′ = −7.5) of 8994 and 8995 eV. The two crystallographically unique Cu sites in 1 are blue shifted compared to 3, with rising edge positions of 8999 and 9000 eV. This measurable blue shift of 4–6 eV even upon introduction of an electron-rich S2− ligand is indicative of the effect of cluster nuclearity on Zeff. Accordingly, all four Cu sites in tetracopper(I) sulfide complex 2 are also blue shifted compared to its dicopper(I) analogue 3. When comparing the two tetranuclear complexes and applying corrections for coordination number as described above, it is evident that distortion from symmetrical 1 to unsymmetrical 2 causes site differentiation, with the Cu1 and Cu3 sites in 2 showing blue shifts and Cu2 and Cu4 showing red shifts compared to the baseline provided by 1. In other words, the geometric distortion taken on by complex 2 leads to polarization, with half the Cu sites taking on more positive charge character and half the Cu sites taking on less positive charge character compared to the relaxed geometry of 1. The most dramatic change is seen for the Cu1 site in 2, which shows a blue shift of 4–5 eV compared to the Cu sites in 1. While this polarization phenomenon is clearly demonstrated by the DAFS analysis, it would have been difficult to predict a priori since it is influenced by subtleties of the coordination environments in an otherwise homovalent (all cuprous) cluster. For example, the Cu1 site in 2 apparently has a relatively higher Zeff than the other three sites due to an interplay of its distance to other Cu(I) cations and its covalency dictated by coordination geometry.
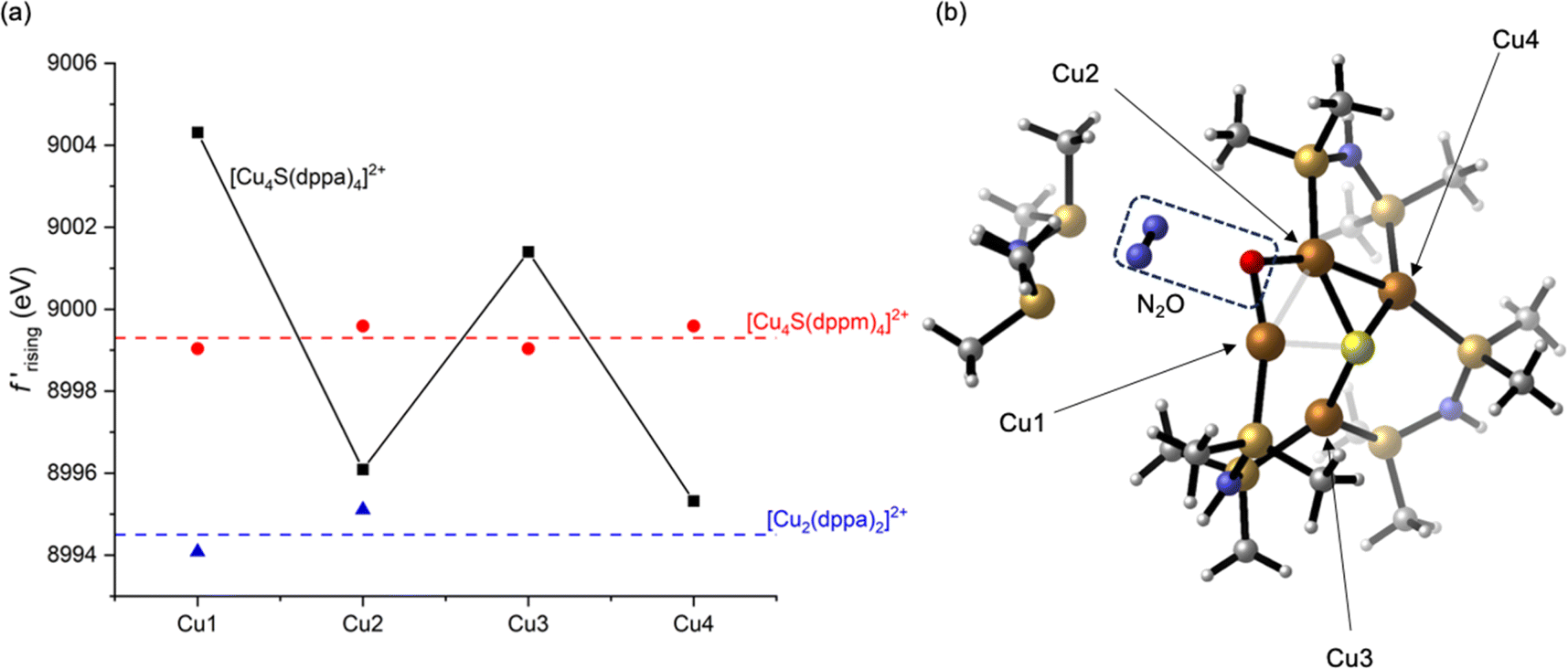 | ||
| Fig. 4 (a) Rising-edge energies (interpolated at f′ = −7.5, see ESI†) for all crystallographically independent Cu sites in this study; (b) a computational model for N2O activation along the Cu1⋯Cu2 edge of 2. For rising-edge energies, correction factors of −3.5 and +3.5 eV were applied to the Cu2 and Cu3 sites in 2, respectively, to correct for coordination number as described in the text. | ||
As a validation of the experimental analysis, we computed atomic partial charges54 at the B3LYP//LANL2TZ/6-31+G(d,p) level of DFT with implicit MeOH solvation, as well as one explicit MeOH molecule hydrogen-bound to 2. The resulting model for 2 indicated that the Cu1 site is, indeed, predicted to bear more positive charge than the other three Cu sites (Table S3†). For example, the calculated Hirshfeld charge for Cu1 is +0.27e−, compared to +0.14–0.16e− for the other three Cu sites. As expected from the preceding DAFS analysis, the calculated Hirshfeld charges for 1 at the same level of theory are uniform across the four Cu sites (+0.16e−) and fall between Cu1 and the other three Cu sites of 2. It should be noted that previously published, lower-level calculations provided Cu partial charges for 2 that are inconsistent with the DAFS data and the higher-level calculations presented here,39 although they were still indicative of distortion-induced charge polarization.
Having established that Cu site differentiation in 2 is correlated to enhanced reactivity, we used computational modeling to probe which Cu atom(s) within the cluster activate the N2O molecule. Recognizing that site differentiation results in polarized (i.e., Cuδ+⋯Cuδ−) dicopper pairs, we explored N2O binding along each of the four cluster edges of 2. In none of the four cases were we able to locate adducts of the cluster with intact N2O, either as minima or saddle points on the potential energy surfaces. However, stationary points after N–O bond cleavage were located for each case. The relevance of N2O activation processes along the Cu1⋯Cu3 and Cu3⋯Cu4 cluster edges were ruled out since they resulted in P–O and S–O bond formation, respectively (Fig. S11 and S12†). Neither of these are consistent with experimental results, which indicate quantitative formation of H2O from N2O.39 Furthermore, these are the two longest cluster edges [experimental: 3.5496(8) and 3.1020(9) Å, respectively], which may inhibit dicopper(I) cooperativity. Neither of the two shorter edges, Cu1⋯Cu2 and Cu2⋯Cu4 [experimental: 2.6556(8) and 2.5895(8) Å, respectively] can be ruled out definitively. N2O activation along these dicopper(I) edges resulted in formation of μ2-O ligands along with liberation of N2 (Fig. 4b and S13†), with the Cu1⋯Cu2 case indicating dissociation of a dppa ligand that was also observed experimentally. However, given that the preceding DAFS and computational data indicate that Cu1 is the most dramatically differentiated in 2 and that the Cu1⋯Cu2 edge is more polarized than the Cu2⋯Cu4 edge, we favor the relevance of computed N2O activation along Cu1⋯Cu2 edge (Fig. 4b) to experimental N2O activation behavior. Upon formation of the μ2-O ligand, orchestrated deliveries of 2H+ from the dppa groups and 2e− from the stoichiometric reductant would deliver H2O.
Conclusions
Deconvoluting contributions of individual metal sites to the overall behavior of metal clusters during small-molecule redox transformations is experimentally challenging but provides basic knowledge about catalytic systems crucial to both biology and energy science. In this study, a synthetic model system that mimics structural and functional aspects of the CuZ active site of nitrous oxide reductase was analyzed using DAFS, a technique that allows for the X-ray absorption profiles of individual metal sites within a cluster to be probed experimentally. Specifically, compounds 1 and 2 provide structural snapshots of active and inactive conformations, respectively, of synthetic Cu4(μ4-S) clusters. Whereas the individual Cu sites within inactive 1 were found to be nearly indistinguishable, site differentiation was evident from DAFS analysis of active 2. This site differentiation, and the corresponding charge polarization (i.e., Cuδ+⋯Cuδ−) along dicopper cluster edges, is correlated with N2O reactivity and likely enables cooperative N2O activation in situ. This concept of cooperative reactivity of localized Cuδ+⋯Cuδ− pairs may have relevance to other Cu clusters ranging from molecular species to nanoclusters and extended solids that catalyze critical reactions like CO2RR.Data availability
Crystallographic data for compounds 1–3 have been deposited at the CCDC under 2293203–2293205. Experimental section, supporting data, and computational output coordinates have been uploaded as part of the ESI.†Author contributions
P. A. analysed and interpreted data, carried out computations, and prepared the manuscript. S. C. R. prepared samples and collected data. T. C. and S. G. W. assisted with data collection. Y.-S. C. supervised and procured funding. N. P. M. supervised, procured funding, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by NIH (R35 GM140850 to N. P. M.). P. A. was supported by a Moriarty Graduate Fellowship from the UIC Department of Chemistry. NSF's ChemMatCARS Sector 15 is supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1834750. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.Notes and references
- A. C. Ghosh, C. Duboc and M. Gennari, Coord. Chem. Rev., 2021, 428, 213606 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- J. P. Collman, N. K. Devaraj, R. A. Decréau, Y. Yang, Y.-L. Yan, W. Ebina, T. A. Eberspacher and C. E. D. Chidsey, Science, 2007, 315, 1565–1568 CrossRef CAS PubMed.
- J. H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- H. Dobbek, L. Gremer, R. Kiefersauer, R. Huber and O. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15971–15976 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, 769 CrossRef PubMed.
- S. C. Rathnayaka and N. P. Mankad, Coord. Chem. Rev., 2021, 429, 213718 CrossRef CAS PubMed.
- S. R. Pauleta, M. S. P. Carepo and I. Moura, Coord. Chem. Rev., 2019, 387, 436–449 CrossRef CAS.
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS PubMed.
- J. N. Armor and H. Taube, J. Am. Chem. Soc., 1969, 91, 6874–6876 CrossRef CAS.
- N. A. Piro, M. F. Lichterman, W. H. Harman and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 2108–2111 CrossRef CAS PubMed.
- V. Zhuravlev and P. J. Malinowski, Angew. Chem., Int. Ed., 2018, 57, 11697–11700 CrossRef CAS PubMed.
- C. C. Mokhtarzadeh, C. Chan, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2019, 141, 15003–15007 CrossRef CAS PubMed.
- M. R. Gyton, B. Leforestier and A. B. Chaplin, Angew. Chem., Int. Ed., 2019, 58, 15295–15298 CrossRef CAS PubMed.
- B. M. Puerta Lombardi, C. Gendy, B. S. Gelfand, G. M. Bernard, R. E. Wasylishen, H. M. Tuononen and R. Roesler, Angew. Chem., Int. Ed., 2021, 60, 7077–7081 CrossRef CAS PubMed.
- N. Lehnert, B. W. Musselman and L. C. Seefeldt, Chem. Soc. Rev., 2021, 50, 3640–3646 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- D. K. Pappas, A. Martini, M. Dyballa, K. Kvande, S. Teketel, K. A. Lomachenko, R. Baran, P. Glatzel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef CAS PubMed.
- E. L. Muetterties, T. N. Rhodin, E. Band, C. F. Brucker and W. R. Pretzer, Chem. Rev., 1979, 79, 91–137 CrossRef CAS.
- B. C. Gates, Angew Chem. Int. Ed. Engl., 1993, 32, 228–229 CrossRef.
- J. L. Peltier, M. Soleilhavoup, D. Martin, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2020, 142, 16479–16485 CrossRef CAS PubMed.
- A. W. Beamer and J. A. Buss, J. Am. Chem. Soc., 2023, 145, 12911–12919 CrossRef CAS PubMed.
- E. M. Johnston, S. Dell'Acqua, S. Ramos, S. R. Pauleta, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 614–617 CrossRef CAS PubMed.
- A. Pomowski, W. G. Zumft, P. M. H. Kroneck and O. Einsle, Nature, 2011, 477, 234–237 CrossRef CAS PubMed.
- E. M. Johnston, C. Carreira, S. Dell'Acqua, S. G. Dey, S. R. Pauleta, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2017, 139, 4462–4476 CrossRef CAS PubMed.
- I. Bar-Nahum, A. K. Gupta, S. M. Huber, M. Z. Ertem, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2009, 131, 2812–2814 CrossRef CAS PubMed.
- C. Esmieu, M. Orio, S. Torelli, L. Le Pape, J. Pécaut, C. Lebrun and S. Ménage, Chem. Sci., 2014, 5, 4774–4784 RSC.
- S. Bagherzadeh and N. P. Mankad, Chem. Commun., 2018, 54, 1097–1100 RSC.
- Y. Liu, S. Chatterjee, G. E. I. Cutsail, S. Peredkov, S. K. Gupta, S. Dechert, S. DeBeer and F. Meyer, J. Am. Chem. Soc., 2023, 145, 18477–18486 CrossRef CAS PubMed.
- S. Ghosh, S. I. Gorelsky, P. Chen, I. Cabrito, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 15708–15709 CrossRef CAS PubMed.
- B. J. Johnson, S. V. Lindeman and N. P. Mankad, Inorg. Chem., 2014, 53, 10611–10619 CrossRef CAS PubMed.
- B. J. Johnson, W. E. Antholine, S. V. Lindeman and N. P. Mankad, Chem. Commun., 2015, 51, 11860–11863 RSC.
- S. C. Rathnayaka, C. W. Hsu, B. J. Johnson, S. J. Iniguez and N. P. Mankad, Inorg. Chem., 2020, 59, 6496–6507 CrossRef CAS PubMed.
- B. J. Johnson, W. E. Antholine, S. V. Lindeman, M. J. Graham and N. P. Mankad, J. Am. Chem. Soc., 2016, 138, 13107–13110 CrossRef CAS PubMed.
- S. C. Rathnayaka, S. M. Islam, I. M. Dimucci, S. N. MacMillan, K. M. Lancaster and N. P. Mankad, Chem. Sci., 2020, 11, 3441–3447 RSC.
- C.-W. W. Hsu, S. C. Rathnayaka, S. M. Islam, S. N. MacMillan and N. P. Mankad, Angew. Chem., Int. Ed., 2020, 59, 627–631 CrossRef CAS PubMed.
- A. K. Bartholomew, J. J. Teesdale, R. H. Sánchez, B. J. Malbrecht, C. E. Juda, G. Ménard, W. Bu, D. A. Iovan, A. A. Mikhailine, S. L. Zheng, R. Sarangi, S. Y. G. Wang, Y. S. Chen and T. A. Betley, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15836–15841 CrossRef CAS PubMed.
- O. Einsle, S. L. A. Andrade, H. Dobbek, J. Meyer and D. C. Rees, J. Am. Chem. Soc., 2007, 129, 2210–2211 CrossRef CAS PubMed.
- G. Wu, Y. Zhang, L. Ribaud, P. Coppens, C. Wilson, B. B. Iversen and F. K. Larsen, Inorg. Chem., 1998, 37, 6078–6083 CrossRef CAS PubMed.
- O. Einsle and D. C. Rees, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed.
- V. W. W. Yam, W. K. Lee and T. F. Lai, J. Chem. Soc. Chem. Commun., 1993, 4, 1571–1573 RSC.
- C.-H. Lam, W. K. Tang and V. W.-W. Yam, Inorg. Chem., 2023, 62, 1942–1949 CrossRef CAS PubMed.
- T. Haltia, K. Brown, M. Tegoni, C. Cambillau, M. Saraste, K. Mattila and K. Djinovic-Carugo, Biochem. J., 2003, 369, 77–88 CrossRef CAS PubMed.
- K. Brown, K. Djinovic-Carugo, T. Haltia, I. Cabrito, M. Saraste, J. J. G. Moura, I. Moura, M. Tegoni and C. Cambillau, J. Biol. Chem., 2000, 275, 41133–41136 CrossRef CAS PubMed.
- A. K. Bartholomew, R. A. Musgrave, K. J. Anderton, C. E. Juda, Y. Dong, W. Bu, S.-Y. Wang, Y.-S. Chen and T. A. Betley, Chem. Sci., 2021, 12, 15739–15749 RSC.
- T. Spatzal, J. Schlesier, E.-M. Burger, D. Sippel, L. Zhang, S. L. A. Andrade, D. C. Rees and O. Einsle, Nat. Commun., 2016, 7, 10902 CrossRef CAS PubMed.
- R. Hernández Sánchez, A. M. Champsaur, B. Choi, S. G. Wang, W. Bu, X. Roy, Y. S. Chen, M. L. Steigerwald, C. Nuckolls and D. W. Paley, Angew. Chem., Int. Ed., 2018, 57, 13815–13820 CrossRef PubMed.
- P. Alayoglu, T. Chang, M. V. Lorenzo Ocampo, L. J. Murray, Y.-S. Chen and N. P. Mankad, Inorg. Chem., 2023, 62, 15267–15276 CrossRef CAS PubMed.
- P. Alayoglu, T. Chang, C. Yan, Y.-S. Chen and N. Mankad, Angew. Chem., Int. Ed., 2023, 62, e202313744 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- M. Cho, N. Sylvetsky, S. Eshafi, G. Santra, I. Efremenko and J. M. L. Martin, ChemPhysChem, 2020, 21, 688–696 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, additional data plots, and computational output. CCDC 2293203–2293205. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00701h |
| This journal is © The Royal Society of Chemistry 2024 |