
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation on the substrate specificity and N-substitution tolerance of PseF in catalytic transformation of pseudaminic acids to CMP-Pse derivatives†
Xing
Guo‡
ac,
Yan Chu
Cheung‡
b,
Can
Li
a,
Han
Liu
*a,
Pengfei
Li
*c,
Sheng
Chen
*b and
Xuechen
Li
 *ad
*ad
aDepartment of Chemistry, State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, P. R. China. E-mail: liuhan@hku.hk; xuechenl@hku.hk
bState Key Lab of Chemical Biology and Drug Discovery and the Department of Food Science and Nutrition, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, P. R. China. E-mail: sheng.chen@polyu.edu.hk
cDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong Province, P. R. China. E-mail: lipf@sustech.edu.cn
dLaboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Ocean University China, Qingdao 266237, People's Republic of China
First published on 21st March 2024
Abstract
Pseudaminic acid (Pse) belongs to a class of bacterial non-2-ulosonic acids, and has been implicated in bacterial infection and immune evasion. Various Pse structures with diverse N-substitutions have been identified in pathogenic bacterial strains like Pseudomonas aeruginosa, Campylobacter jejuni, and Acinetobacter baumannii. In this study, we successfully synthesized three new Pse species, including Pse5Ac7Fo, Pse5Ac7(3RHb) and Pse7Fo5(3RHb) using chemical methods. Furthermore, we investigated the substrate specificity of cytidine 5′-monophosphate (CMP)-Pse synthetase (PseF), resulting in the production of N-modified CMP-Pse derivatives (CMP-Pses). It was found that PseF was promiscuous with the Pse substrate and could tolerate different modifications at the two nitrogen atoms. This study provides valuable insights into the incorporation of variable N-substitutions in the Pse biosynthetic pathway.
Introduction
Bacterial non-2-ulosonic acids, including pseudaminic acid (Pse), legionaminic acid (Leg), acinetaminic acid (Aci), fusaminic acid, and their epimers, have been identified from pathogenic bacteria, and play crucial roles in bacterial morphology, bacterial pathogenicity, immune escape, and other related processes.1–6 Pse derivatives present as key components of surface glycans in various clinical pathogens, which are widely distributed in lipopolysaccharides (LPS),7 capsular polysaccharides (CPS),8,9 exopolysaccharides (EPS),10 pili,11 and flagella.12 Unlike the case of sialic acids in mammalian cells, Pse species bear variable N5/N7-substitutions and are connected to other sugars through both α and β linkages (Fig. 1). The fascinating structures and less discovered biological functions of these compounds make them attractive subjects for investigation in the field of synthetic glycobiology.13–15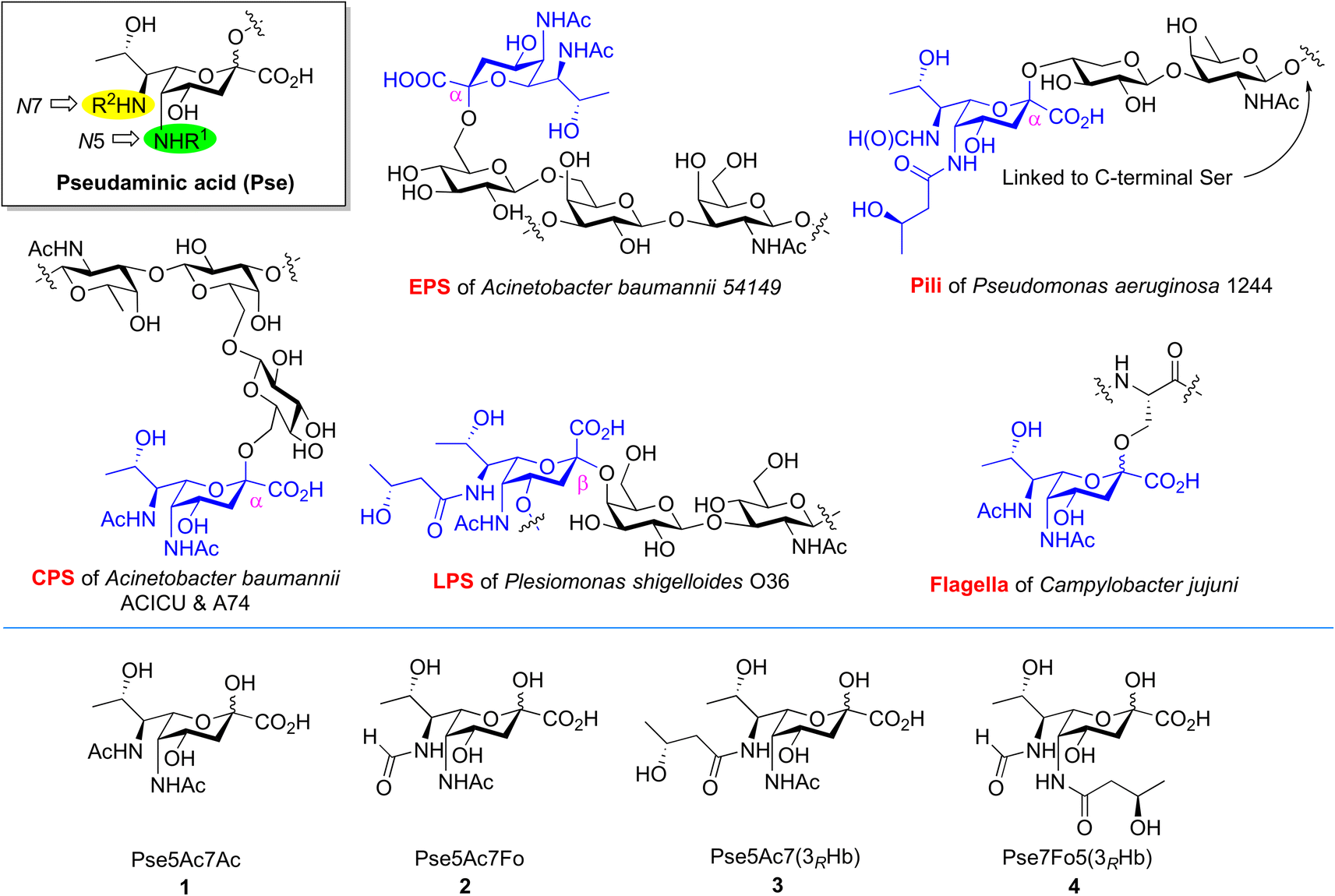 | ||
| Fig. 1 Occurrence of Pse in bacterial glycans bearing different N5/N7-substitutions and structures of Pse species selected for synthesis and enzymatic transformation studies. | ||
The biosynthetic pathway and associated enzymes for Pse5Ac7Ac 1 have been elucidated clearly by Logan et al., including PseB, PseC, PseH, PseG and PseI (Fig. 2a).16 Based on this, Fascione et al. developed the enzymatic synthetic approach of Pse5Ac7Ac 1 and CMP-Pse5Ac7Ac 7,17 and realized the enzymatic glycosylation of CMP-Pse5Ac7Ac by promiscuous sialyltransferases18 and Pse-specific glycosyltransferase KpsS1.19 The identification of novel formyltransferase PseJ20 and amidine synthase PseA21 filled in the map of the biosynthetic pathway of Pse5Ac7Fo and Pse5Ac7Am, respectively. However, unlike sialic acid, which can be produced via industrial scale fermentation, the variable N-substitutions of Pse species and largely unknown enzymes for N-modifications make the biosynthetic approach more challenging, especially for large-scale in vitro biosynthesis.
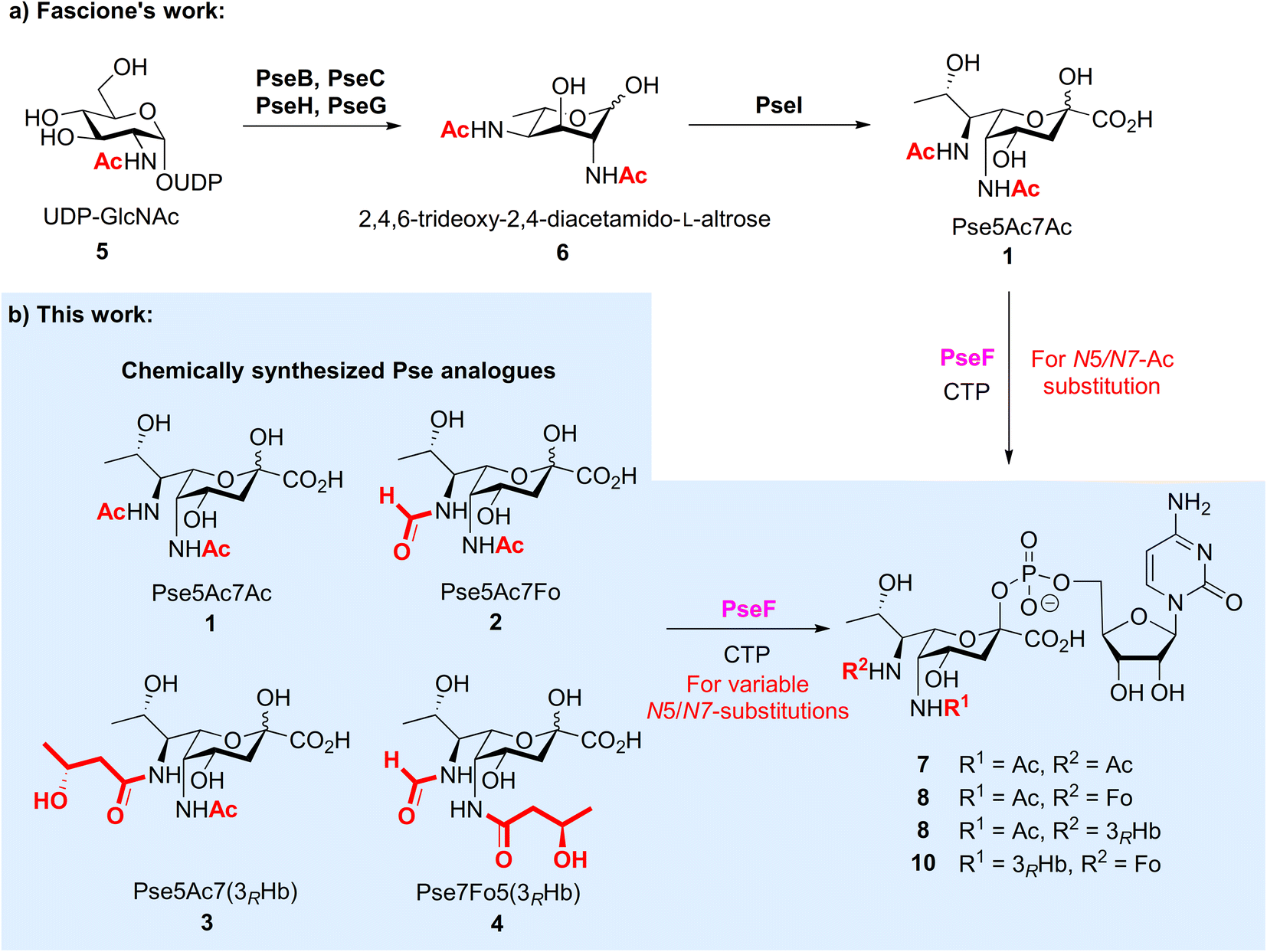 | ||
| Fig. 2 Enzymatic and chemoenzymatic syntheses of CMP-Pse bearing variable N5/N7-substitutions. (a) Fascione's enzymatic synthesis of CMP-Pse. (b) Chemoenzymatic synthesis of N5/N7-modified CMP-Pse in this work. | ||
To investigate the biological significance of Pse at a molecular level, we started a research program to chemically synthesize Pse and its derivatives,22,23 with which we develop probes24 and explore their applications as synthetic antibacterial vaccines.25 On the other forefront, we aim to use synthetic Pse derivatives to study the biosynthesis and biological function of Pse. It is noted that, after the biosynthesis of Pse, the pseudaminic acid cytidylyltransferase PseF is responsible for converting Pse into corresponding cytidine 5′-monophosphate (CMP) derivatives,16,26 which is then transferred to other glycans to form Pse glycosides. At this stage, the Pse-specific glycosyltransferase has just been identified.19 Furthermore, natural Pse carries diverse N-acylations at two nitrogen atoms, which makes the process more complicated. The promiscuity and specificity of PseF and the glycosyltransferase, as well as the order of N-acylation and transglycosylation are open questions. To this end, we started to answer the question related to PseF by using N-modified Pse derivatives, which were only available via chemical synthesis at this stage. Given that the two amino groups in Pse derivatives are naturally modified by variable N-substitutions, in this study, we first developed chemical synthetic approaches to prepare three new naturally existing Pse derivatives 2–4 selected from bacterial glycans that were modified with acetyl, formyl and (R)-3-hydroxybutyryl on N5/N7 sites, respectively, followed by the enzymatic studies of PseF with Pse derivatives 1–4 to form the corresponding Pse-CMP compounds 7–10 (Fig. 2b). We showed for the first time that PseF was well compatible with Pse derivatives in vitro, which indicates the variable N-substitution installation most likely occurs before the formation of pseudaminosides in the biosynthetic pathway. Deeper understanding of the Pse biosynthetic pathway may accelerate the development of novel inhibitors to fight pathogenic Gram-negative bacteria.
Results and discussion
Chemical synthesis of Pse5Ac7Fo, Pse5Ac7(3RHb) and Pse7Fo5(3RHb)
In 2017, we have reported the de novo total synthesis of Pse5Ac7Ac 1 from the 5N-Troc/7N-Cbz thioglycoside donor.22 The orthogonal protections installed on N5 and N7 sites during this de novo approach perfectly fit our needs in the synthesis of Pse derivatives bearing different N5/N7-substitutions. Herein, starting from the 5N-Troc/7N-Cbz S-adamantyl thioglycoside donor 11 developed in 2019,23 we were able to complete the synthesis of Pse5Ac7Fo 2, Pse5Ac7(3RHb) 3 and Pse7Fo5(3RHb) 4, respectively.As illustrated in Scheme 1, the synthesis of Pse5Ac7Fo 2 started from the β-selective glycosylation of donor 11 with benzyl alcohol. The benzyl group installed at the glycosidic site of 12 served as a protecting group during the following synthetic steps. N5-Troc was then transformed to N5-Ac via Zn mediated orthogonal deprotection and N-acetylation. During the modification of N7, to avoid the O8-to-N7 acetyl transfer when removing Cbz, the O4/O8-acetyl groups in 13 were first removed by treating with 5 mM MeONa in MeOH. The obtained intermediate 14 containing methyl ester generated by partial isopropyl/methyl exchange was used in the following transformations, since both ester groups would be hydrolyzed at the end stage. Next, the selective hydrogenolysis of Cbz without removing glycosidic OBn was achieved using the pyrimidine deactivated Pd/C catalyst (see ESI† for details) after extensive screening. Several nitrogen-containing compounds have been developed as deactivators to regulate the activity of the Pd/C catalyst.27–31 In our case, other deactivators (e.g., ammonium acetate, tertiary amine, ethylenediamine, pyridine, 2,2′-bipyridine) gave inferior chemoselectivity. After N7-formylation, the isopropyl/methyl ester in 15 was hydrolyzed by LiOH, and Pse5Ac7Fo 2 was obtained in 79% yield after Bn removal. We also tried to avoid the methyl ester contamination by directly removing N7-Cbz with O8-Ac intact, since the O8-to-N7 acyl transfer could be partially suppressed by shortening the reaction time. Unfortunately, product 16 was obtained in only 25% yield, which was inferior to the above 3-step process (50% for 15 from 13). This established approach was also used to synthesize Pse5Ac7(3RHb) 3, where symmetric anhydride 17 was used for N7-acylation instead of formic anhydride.
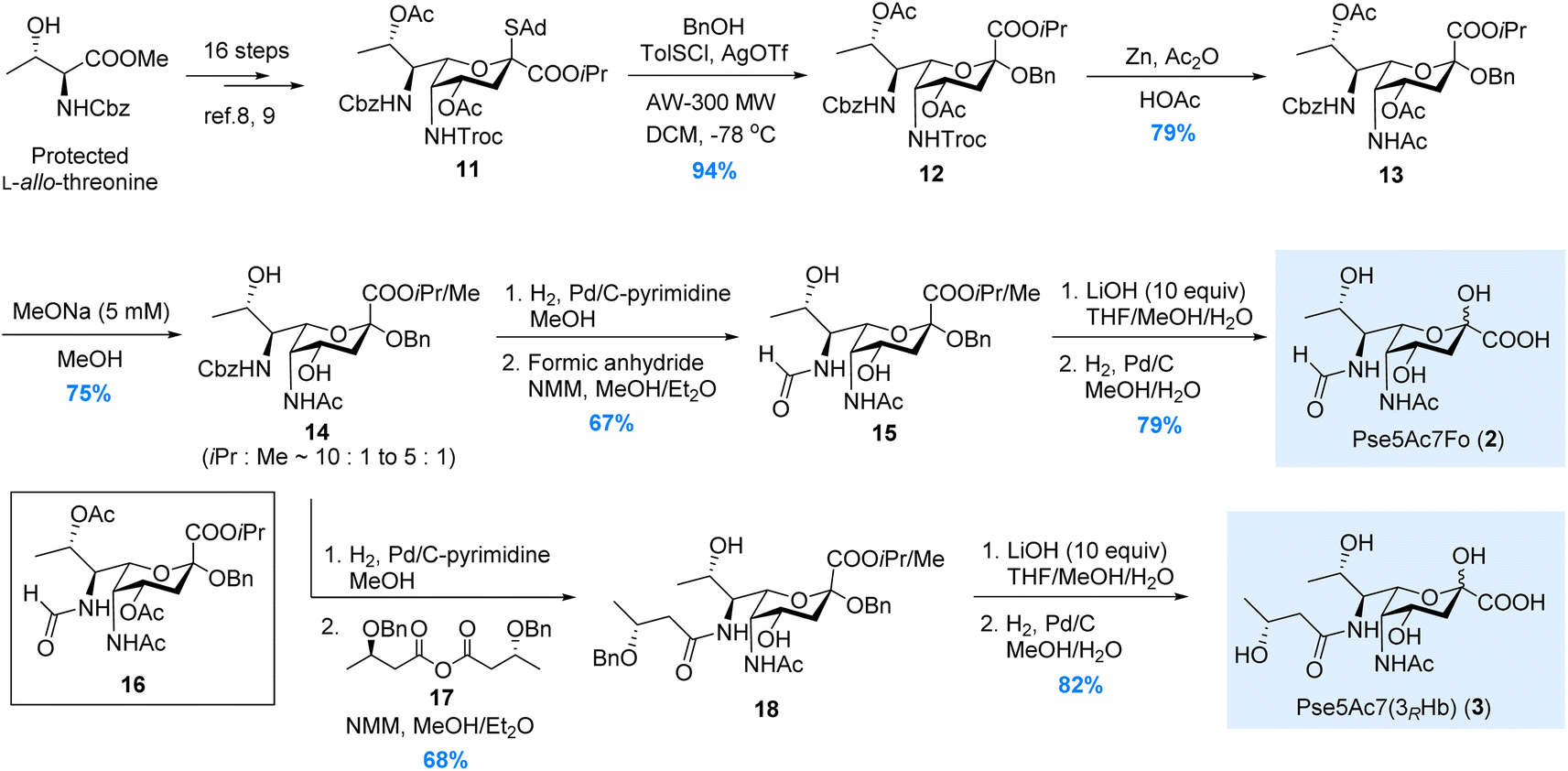 | ||
| Scheme 1 Synthesis of the Pse5Ac7Fo and Pse5Ac7(3RHb). | ||
For the synthesis of Pse7Fo5(3RHb) 4, the approach we developed during the total synthesis of the Pseudomonas aeruginosa 1244 pilin trisaccharide22 was adopted, where the O4-to-N5 acyl transfer was used to install the N5-(R)-3-hydroxybutyryl group. As illustrated in Scheme 2, the two acetyl groups in thioglycoside 11 were removed under acidic conditions to avoid destroying the Troc group. Two (R)-3-hydroxybutyryl groups were installed on the O4/O8 hydroxyl groups in 19, followed by β-selective glycosylation of 20 with benzyl alcohol. Intermediate 21 was subjected to the TBAF mediated Troc removal conditions developed by Fukase.32 The key carbamoyl fluoride intermediate proposed by Coudert et al.33 was hydrolyzed in situ by the trace amount of H2O to release the amino group for acyl transfer. The addition of acetic acid to partially buffer the basicity of the fluoride anion significantly suppressed side reactions, and mild heating ensured full conversion in 48 hours. To install the N7-formyl group, the Cbz was selectively removed by hydrogenolysis, and the amino group was formylated immediately to minimize O8-to-N7 acyl transfer. The 3-step process used in the synthesis of 2 and 3 was abandoned here due to significant side reactions during MeONa treatment. Intermediate 23, isolated in moderate 31% yield, was then subjected to saponification and hydrogenolysis to give rise to Pse7Fo5(3RHb) 4 in 76% yield.
 | ||
| Scheme 2 Synthesis of the Pse7Fo5(3RHb). | ||
PseF catalyzed enzymatic synthesis of N5/N7-modified CMP-Pse
To evaluate the substrate tolerance of PseF, we designed the single step catalytic activity assay using purified PseF and synthetic N-substituted Pse derivatives 1–4. The full-length pseF gene was cloned from Acinetobacter baumannii clinical isolates and the recombinant PseF protein containing a His tag was expressed in E. coli BL21 cells and purified via a nickel column (see ESI† for details).With the recombinant PseF protein in hand, we validated its catalytic activity in the reported model reaction. According to the conditions suggested by Logan et al.16 and Fascione et al.,17 we observed smooth formation of CMP-Pse5Ac7Ac 7 by treating synthetic Pse5Ac7Ac 1 and CTP with PseF. In contrast, no conversion was observed when sialic acid was tested as the substrate, which showcased the Pse-specificity of the enzyme. In order to explore the substrate tolerance of PseF towards other N-substituted Pse derivatives in a quantitative way, we first conducted quantitative kinetic analysis of this enzymatic transformation using a HILIC column. As depicted in Fig. 3, a rapid enzymatic reaction rate was revealed. The conversion of Pse5Ac7Ac 1 to CMP-Pse5Ac7Ac 7 reached up to 66% within a 30 minutes timeframe. Further time-dependent data are presented in the ESI (Fig. S2).†
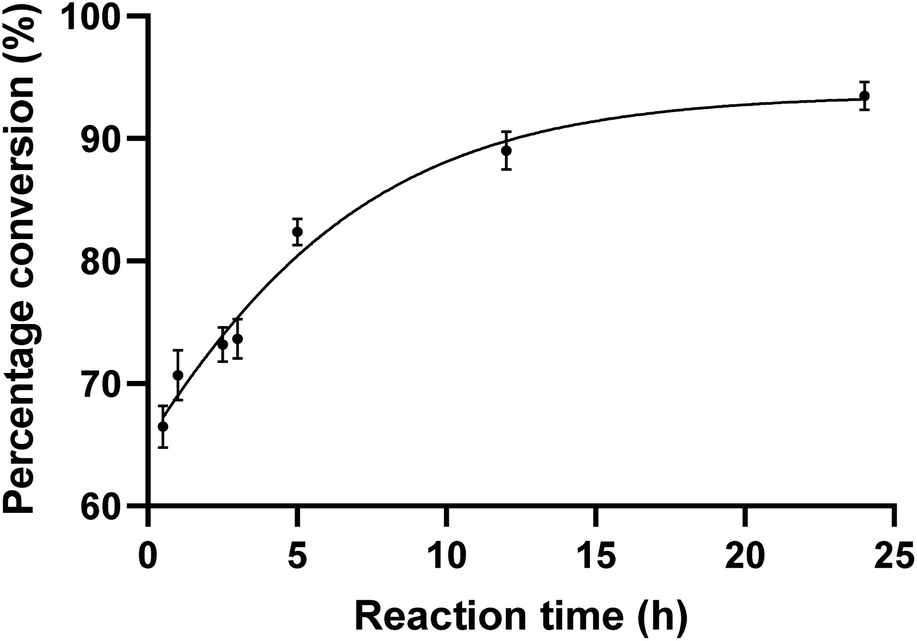 | ||
| Fig. 3 CMP-Pse5Ac7Ac percentage conversion (%) at varying reaction times. PseF (3.8 μM) was assayed using Pse5Ac7Ac (0.9 mM), CTP (2 mM) as substrates in the presence of MgCl2 (20 mM) and sodium phosphate (30 mM, pH 7.4) at 37 °C for 0.5, 1, 2.5, 3, 5, 12, and 24 h, respectively. The error bars were derived from three parallel replicates of each experiment. | ||
To investigate the influence of metal ions on the activity of PseF, we conducted initial experiments (Fig. 4). PseF was subjected to assays using Pse5Ac7Ac 1 (0.9 mM) and CTP (2 mM) as substrates in the presence of various metal cations or EDTA (20 mM) as a metal ion sequestering agent. The reactions were carried out in a sodium phosphate buffer (30 mM, pH 7.4) for a duration of 5 minutes. The obtained results clearly demonstrated the requirement of metal cations for the enzymatic activity of PseF, as no activity was observed in the presence of EDTA. Among the Mn2+, Ca2+, Zn2+, Ni2+, Co2+, Na+ and Mg2+, only Ca2+ and Na+ gave obviously lower efficiency. In contrast, Zn2+ and Mg2+ gave 49% and 47% conversion, respectively. To keep the consistency with reported conditions,16,17 Mg2+ was chosen as the metal ion in the following assays. Mg2+ is known to be involved in the enzymatic generation of sugar-UDP34 and sugar-CMP35 species under physiological conditions (intracellular c[Mg2+] ∼1.5 mM36). According to Pohl's report, the strong binding of Mg2+ to UTP led to the Mg2+-UTP complex serving as the active substrate of the uridylyltransferase enzymes in the sugar-UDP synthesis.37 In the PseF catalyzed Pse-CMP synthesis, a similar role of Mg2+ can be proposed. The pH and temperature profiles of PseF are described in Fig. S3 and S4.† Moreover, to identify a suitable concentration of CTP, we measured the reaction rates under fixed c(Pse5Ac7Ac) concentration with varied c(CTP). As illustrated in Fig. S5,†c(CTP) higher than Km(CTP) (0.6295 ± 0.065 mM) should be used to ensure rapid conversion.
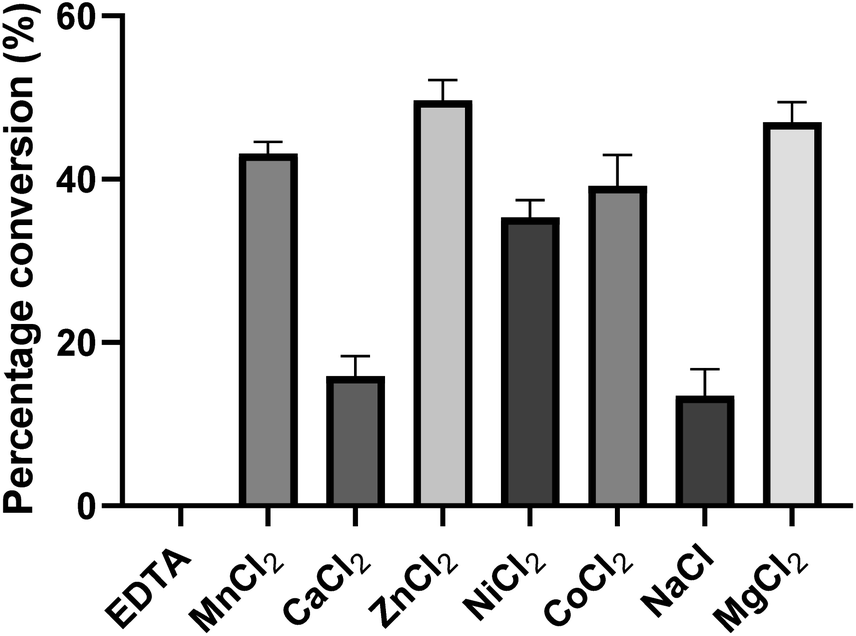 | ||
| Fig. 4 Metal ion effects of PseF. PseF (2.27 μM) was assayed using Pse5Ac7Ac (0.9 mM), CTP (2 mM) as substrates in the presence of a metal cation or EDTA (20 mM), and sodium phosphate (30 mM, pH 7.4) at 37 °C for 5 minutes. Note: 5 minutes is a suitable time to compare their differences. The error bars were derived from three parallel replicates of each experiment. | ||
With the optimized conditions (0.9 mM Pse analogue, 0.1 mg mL−1 PseF, 6 mM CTP, 20 mM MgCl2, pH 7.4, 37 °C) in hand, we tested the N-substitution tolerance of PseF using our synthetic Pse derivatives 1–4. We extended the incubation time to 24 hours in each case to maximize the conversion, followed by isolation of the four CMP-Pse derivatives 7–10 through a Bio-Gel P-2 resin column. The isolated yields of CMP-Pse5Ac7Ac 7, CMP-Pse5Ac7Fo 8, CMP-Pse5Ac7(3RHb) 9 and CMP-Pse7Fo5(3RHb) 10 were 72%, 72%, 70% and 24%, respectively. For compounds 7–9, 1.1–1.2 mg products could be obtained from 2.3–2.7 μmol scale transformations. This is the first report to show that PseF is compatible with other N-substituted Pse derivatives in vitro, in addition to Pse5Ac7Ac. This result indicates that PseF is a promiscuous enzyme with the Pse substrate, showing higher tolerance to N7-substitutions than N5. Also, the installation of variable N-substitution most likely occurs before the formation of CMP-Pse and pseudaminosides in the biosynthetic pathway.
To gain a comprehensive understanding of PseF's characteristics and to elucidate the underlying reasons for the relatively low yield of CMP-Pse7Fo5(3RHb), measurement of PseF catalytic kinetics was conducted. To determine the apparent kinetic parameters for PseF activity, we selected a time point of 15 seconds based on Fig. S2,† where the percentage conversion of CMP-Pse5Ac7Ac was 7% using 0.9 mM of Pse. This early time point was chosen to mimic the initial velocity and facilitate the calculation of PseF apparent kinetic parameters. Once the predetermined time was reached, the reaction samples were immediately quenched by MeCN and subsequently subjected to UPLC analysis on a HILIC column. A high concentration of CTP (2 mM) giving rapid conversion at high c(Pse) could lead to a narrow time window (shorter than 15 s) for initial velocity measurement, which was difficult for us to achieve reliable accuracy. Thus, PseF was assayed using a fixed moderate concentration of CTP (0.8 mM) and varying Pse concentrations (ranging from 0.2 to 1.8 mM) in the presence of MgCl2 (20 mM) and sodium phosphate (30 mM, pH 7.4) for 15 seconds (Fig. 5). The corresponding Michaelis–Menten best-fit values are summarized in Table 1. It is clear that Pse7Fo5(3RHb) 4 showed around one quarter of kcat value and one fifth of kcat/Km value than other Pse derivatives, which demonstrated that the lower yield of CMP-Pse7Fo5(3RHb) 10 originated from inferior conversion. This kinetic discrimination of N5-substituted Pse is also consistent with our observations in metabolic labelling studies, where the N2-azidoacetyl modified probe that was metabolically transformed into N5-modified Pse gave much lower labelling efficiency compared to the N4-modified probe.24
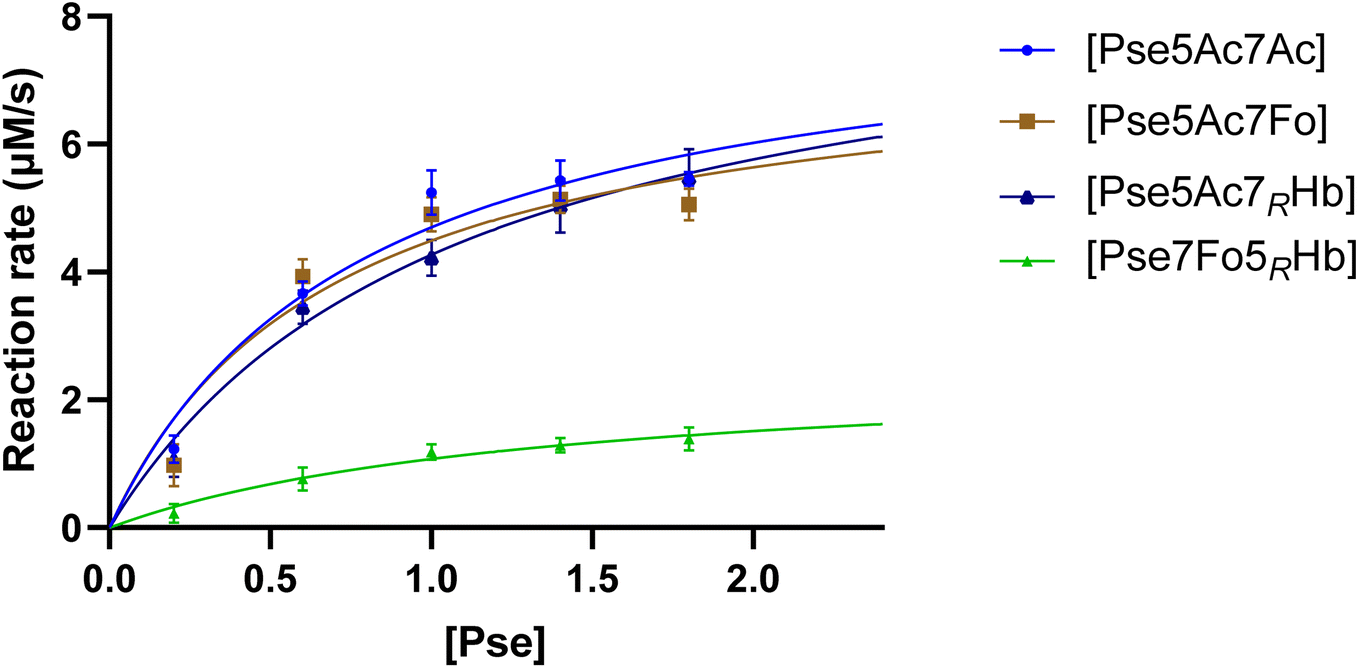 | ||
| Fig. 5 PseF kinetic assay results at varying Pse concentrations. PseF (2.27 μM) was assayed using a fixed concentration of CTP (0.8 mM) and varying Pse concentrations (ranging from 0.2 to 1.8 mM) in the presence of MgCl2 (20 mM) and sodium phosphate (30 mM, pH 7.4) at 37 °C for 15 seconds. The error bars were derived from three parallel replicates of each experiment. | ||
| Pse | V max (μM s−1) | K m (mM) | k cat (s−1) | k cat/Km (s−1 mM−1) |
|---|---|---|---|---|
| a Reactions were performed with 0.8 mM CTP and 2.27 μM PseF in 20 mM MgCl2, 30 mM sodium phosphate (pH 7.4) at 37 °C for 15 seconds. b Estimated conversion was determined by UV absorption via UPLC analysis on a HILIC column. | ||||
| Pse5Ac7Ac | 8.377 ± 0.816 | 0.783 ± 0.185 | 3.690 ± 0.359 | 4.710 |
| Pse5Ac7Fo | 7.575 ± 0.865 | 0.688 ± 0.202 | 3.337 ± 0.381 | 4.853 |
| Pse5Ac7(3RHb) | 8.863 ± 0.936 | 1.077 ± 0.239 | 3.904 ± 0.412 | 3.624 |
| Pse7Fo5(3RHb) | 2.543 ± 0.524 | 1.367 ± 0.536 | 1.120 ± 0.231 | 0.819 |
Conclusions
In summary, we finished the de novo total synthesis of three new N-substituted Pse derivatives, including Pse5Ac7Fo, Pse5Ac7(3RHb) and Pse7Fo5(3RHb), from orthogonally protected Pse thioglycoside donor 11. The synthetic strategies we demonstrated can be useful for the chemical synthesis of other bacterial non-2-ulosonic acids, since the variation of N-substitutions is widely observed in this family of molecules. Furthermore, we probed the enzymatic substrate specificity of PseF by measuring the enzymatic kinetics of the transformation of our synthetic Pse derivatives to corresponding CMP-Pse products. To our delight, the results proved that PseF could specifically recognize a series of N-substituted Pse derivatives, but not sialic acid. Thus, PseF could be regarded as a universal Pse cytidylyltransferase that is compatible with variable N-substitution. More critically, this is a piece of evidence that variable N-substitutions of Pse species in bacterial glycans are possibly decorated before the biosynthetic incorporation of Pse units into polysaccharides. This also indicates that the CMP-Pse compounds with variable N-substitution may be recognized by the same pseudaminyltransferase, which may reduce the difficulty of in vitro experiments to search for putative pseudaminyltransferases in future.By gaining a more comprehensive understanding of the Pse biosynthetic pathway, researchers can potentially develop novel inhibitors to target pathogenic Gram-negative bacteria.38,39 This could lead to innovative therapeutic approaches in combating drug-resistant infections and improving the overall efficacy of antibiotic treatments. Moreover, this study may contribute to the ongoing exploration of bacterial glycosylation and its implications in human health and diseases by guiding the design of molecular probes with better labelling efficiency.
Experimental section
All experimental details are provided in the ESI† of this article.Data availability
The data that support the findings, including the details of the synthesis, protein expression, enzymatic reactions, and copies of NMR spectra, are available in the ESI† of this article.Author contributions
X. Li, S. Chen and P. Li conceived the project. X. Guo, Y. C. Cheung and C. Li did the experiments. X. Guo, H. Liu, S. Chen and X. Li analyzed the data. H. Liu and X. Li wrote the manuscript with the assistance of other contributors.Conflicts of interest
The authors declare no conflict interest.Acknowledgements
The work was supported by Health and Medical Research Fund (20190802), Collaborative Research Fund (C7003-20G), and General Research Fund (17312822).References
- M. Zunk and M. J. Kiefel, RSC Adv., 2014, 4, 3413 RSC.
- N. D. McDonald and E. F. Boyd, Trends Microbiol., 2021, 29, 142 CrossRef CAS PubMed.
- S. M. Logan, J. F. Kelly, P. Thibault, C. P. Ewing and P. Guerry, Mol. Microbiol., 2002, 46, 587 CrossRef CAS PubMed.
- F. Liu, A. J. Aubry, I. C. Schoenhofen, S. M. Logan and M. E. Tanner, ChemBioChem, 2009, 10, 1317 CrossRef CAS PubMed.
- H. N. Stephenson, D. C. Mills, H. Jones, E. Milioris, A. Copland, N. Dorrell, B. W. Wren, P. R. Crocker, D. Escors and M. Bajaj-Elliott, J. Infect. Dis., 2014, 210, 1487 CrossRef CAS PubMed.
- I.-M. Lee, F.-L. Yang, T.-L. Chen, K.-S. Liao, C.-T. Ren, N.-T. Lin, Y.-P. Chang, C.-Y. Wu and S.-H. Wu, J. Am. Chem. Soc., 2018, 140, 8639 CrossRef CAS PubMed.
- M. Kaszowska, K. Stojkovic, T. Niedziela and C. Lugowski, Carbohydr. Res., 2016, 434, 1 CrossRef CAS PubMed.
- S. N. Senchenkova, A. S. Shashkov, M. M. Shneider, N. P. Arbatsky, A. V. Popova, K. A. Miroshnikov, N. V. Volozhantsev and Y. A. Knirel, Carbohydr. Res., 2014, 391, 89 CrossRef CAS PubMed.
- J. J. Kenyon, A. M. Marzaioli, R. M. Hall and C. De Castro, Glycobiology, 2014, 24, 554 CrossRef CAS PubMed.
- I.-M. Lee, I.-F. Tu, F.-L. Yang, T.-P. Ko, J.-H. Liao, N.-T. Lin, C.-Y. Wu, C.-T. Ren, A. H.-J. Wang, C.-M. Chang, K.-F. Huang and S.-H. Wu, Sci. Rep., 2017, 7, 42711 CrossRef CAS PubMed.
- P. Castric, F. J. Cassels and R. W. Carlson, J. Biol. Chem., 2001, 276, 26479 CrossRef CAS PubMed.
- P. Thibault, S. M. Logan, J. F. Kelly, J. R. Brisson, C. P. Ewing, T. J. Trust and P. Guerry, J. Biol. Chem., 2001, 276, 34862 CrossRef CAS PubMed.
- E. K. P. Flack, H. S. Chidwick, M. Best, G. H. Thomas and M. A. Fascione, ChemBioChem, 2020, 21, 1397 CrossRef CAS PubMed.
- K. Pradhan and S. S. Kulkarni, Eur. J. Org Chem., 2020, 44, 6819 CrossRef.
- X. Guo, P. Li, H. Liu and X. Li, Org. Chem. Front., 2023, 10, 3429 RSC.
- I. C. Schoenhofen, D. J. McNally, J.-R. Brisson and S. M. Logan, Glycobiology, 2006, 16, 8C CrossRef CAS PubMed.
- H. S. Chidwick, E. K. P. Flack, T. Keenan, J. Walton, G. H. Thomas and M. A. Fascione, Sci. Rep., 2021, 11, 4756 CrossRef CAS PubMed.
- E. K. P. Flack, H. S. Chidwick, G. Guchhait, T. Keenan, D. Budhadev, K. Huang, P. Both, J. Mas Pons, H. Ledru, S. Rui, G. P. Stafford, J. G. Shaw, M. C. Galan, S. Flitsch, G. H. Thomas and M. A. Fascione, ACS Catal., 2020, 10, 9986 CrossRef CAS.
- A. J. Walklett, E. K. P. Flack, H. S. Chidwick, N. E. Hatton, T. Keenan, D. Budhadev, J. Walton, G. H. Thomas and M. A. Fascione, Angew. Chem., Int. Ed., 2024, e202318523 Search PubMed.
- J. M. Reimer, I. Harb, O. G. Ovchinnikova, J. Jiang, C. Whitfield and T. M. Schmeing, ACS Chem. Biol., 2018, 13, 3161 CrossRef CAS PubMed.
- C. P. Ewing, E. Andreishcheva and P. Guerry, J. Bacteriol., 2009, 191, 7086 CrossRef CAS PubMed.
- H. Liu, Y. Zhang, R. Wei, G. Andolina and X. Li, J. Am. Chem. Soc., 2017, 139, 13420 CrossRef CAS PubMed.
- R. Wei, H. Liu, A. H. Tang, R. J. Payne and X. Li, Org. Lett., 2019, 21, 3584 CrossRef CAS PubMed.
- G. Andolina, R. Wei, H. Liu, Q. Zhang, X. Yang, H. Cao, S. Chen, A. Yan, X. D. Li and X. Li, ACS Chem. Biol., 2018, 13, 3030 CrossRef CAS PubMed.
- R. Wei, X. Yang, H. Liu, T. Wei, S. Chen and X. Li, ACS Cent. Sci., 2021, 7, 1535 CrossRef CAS PubMed.
- S. U. H. Wahid, Adv. Appl. Bioinf. Chem., 2017, 10, 79 Search PubMed.
- H. Sajiki and K. Hirota, Tetrahedron, 1998, 54, 13981 CrossRef CAS.
- H. Sajiki, H. Kuno and K. Hirota, Tetrahedron Lett., 1998, 39, 7127 CrossRef CAS.
- H. Sajiki and K. Hirota, Chem. Pharm. Bull., 2003, 51, 320 CrossRef CAS PubMed.
- H. Sajiki, K. Hattori and K. Hirota, J. Org. Chem., 1998, 63, 7990 CrossRef CAS.
- M. B.-U. Surfraz, M. Akhtar and R. K. Allemann, Tetrahedron Lett., 2004, 45, 1223 CrossRef CAS.
- C. Y. Huang, N. Wang, K. Fujiki, Y. Otsuka, M. Akamatsu, Y. Fujimoto and K. Fukase, J. Carbohydr. Chem., 2010, 29, 289 CrossRef CAS.
- U. Jacquemard, V. Bénéteau, M. Lefoix, S. Routier, J.-Y. Mérour and G. Coudert, Tetrahedron, 2004, 60, 10039 CrossRef CAS.
- R. M. Mizanur, C. J. Zea and N. L. Pohl, J. Am. Chem. Soc., 2004, 126, 15993 CrossRef CAS PubMed.
- R. M. Mizanur and N. L. Pohl, Appl. Microbiol. Biotechnol., 2008, 80, 757 CrossRef CAS PubMed.
- L. Csernoch, J. C. Bernengo, P. Szentesi and V. Jacquemond, Biophys. J., 1998, 75, 957 CrossRef CAS PubMed.
- C. J. Zea, G. Camci-Unal and N. L. Pohl, Chem. Cent. J., 2008, 2, 15 CrossRef PubMed.
- R. Ménard, I. C. Schoenhofen, L. Tao, A. Aubry, P. Bouchard, C. W. Reid, P. Lachance, S. M. Twine, K. M. Fulton, Q. Cui, H. Hogues, E. O. Purisima, T. Sulea and S. M. Logan, Antimicrob. Agents Chemother., 2014, 58, 7430 CrossRef PubMed.
- D. J. McNally, I. C. Schoenhofen, R. S. Houliston, N. H. Khieu, D. M. Whitefield, S. M. Logan, H. C. Jarrell and J.-R. Brisson, ChemMedChem, 2008, 3, 55 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00758a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |