
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrophage-engaging peptidic bispecific antibodies (pBsAbs) for immunotherapy via a facile bioconjugation strategy†
Chihao
Shao
 a,
Bo
Tang
a,
Jacky C. H.
Chu
b,
Kwai Man
Lau
a,
Wai-Ting
Wong
a,
Chi-Ming
Che
bc,
William C. S.
Tai
*a,
Wing-Tak
Wong
*a and
Clarence T. T.
Wong
*a
a,
Bo
Tang
a,
Jacky C. H.
Chu
b,
Kwai Man
Lau
a,
Wai-Ting
Wong
a,
Chi-Ming
Che
bc,
William C. S.
Tai
*a,
Wing-Tak
Wong
*a and
Clarence T. T.
Wong
*a
aDepartment of Applied Biology and Chemical Technology and State Key Laboratory of Chemical Biology and Drug Discovery, Hong Kong Polytechnic University, Kowloon, Hong Kong, China. E-mail: william-cs.tai@polyu.edu.hk; w.t.wong@polyu.edu.hk; clarence-tt.wong@polyu.edu.hk
bLaboratory for Synthetic Chemistry and Chemical Biology Limited, Units 1503-1511, 15/F, Building 17W, Hong Kong Science Park, New Territories, Hong Kong, China
cDepartment of Chemistry, State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China
First published on 30th April 2024
Abstract
Bispecific antibodies are artificial molecules that fuse two different antigen-binding sites of monoclonal antibodies into one single entity. They have emerged as a promising next-generation anticancer treatment. Despite the fascinating applications of bispecific antibodies, the design and production of bispecific antibodies remain tedious and challenging, leading to a long R&D process and high production costs. We herein report an unprecedented strategy to cyclise and conjugate tumour-targeting peptides on the surface of a monoclonal antibody to form a novel type of bispecific antibody, namely the peptidic bispecific antibody (pBsAb). Such design combines the merits of highly specific monoclonal antibodies and serum-stable cyclic peptides that endows an additional tumour-targeting ability to the monoclonal antibody for binding with two different antigens. Our results show that the novel pBsAb, which comprises EGFR-binding cyclic peptides and an anti-SIRP-α monoclonal antibody, could serve as a macrophage-engaging bispecific antibody to initiate enhanced macrophage–cancer cell interaction and block the “don't eat me” signal between CD47-SIRP-α, as well as promoting antibody-dependent cellular phagocytosis and 3D cell spheroid infiltration. These findings give rise to a new type of bispecific antibody and a new platform for the rapid generation of new bispecific antibodies for research and potential therapeutic uses.
Introduction
The concept of bispecific antibodies was first introduced in 1960 by Nisonoff and co-workers.1,2 They are designed to recognise two or more different epitopes or antigens on single or multiple target cells. This dual-targeting mechanism enables the bispecific antibody to achieve therapeutic effects unachievable by conventional monoclonal antibodies. One of the mechanisms is the recruitment of immune cells to cancer cells: the bispecific can bind to a tumour-specific antigen with one arm and an immune cell marker such as CD3 on T-cells with the other arm.3 In this way, the bispecific can effectively bring immune cells into direct contact with cancer cells, promoting their destruction. Another mechanism involves simultaneously blocking two signalling pathways on target cells, offering a synergistic therapeutic effect by inhibiting multiple disease-related targets. This dual binding capacity of bispecific antibodies opens avenues for innovative treatments in oncology, autoimmune diseases, and beyond.4,5 Hence, they are regarded as the next generation of antibody-based therapeutic agents with the potential to improve clinical efficacy and safety.6,7 Bispecific antibodies can be classified into IgG-like and non-IgG-like structures.8 IgG-like subtypes maintain the basic structure of conventional IgG, typically incorporating two Fab arms and an Fc region. This structure imparts a longer half-life and stability, similar to natural IgG antibodies, and enables them to engage the immune system effectively. In contrast, non-IgG-like subtypes deviate from this traditional antibody structure. They are often smaller, lacking the Fc region, which can result in shorter half-lives but potentially improved tissue penetration (Fig. 1).9,10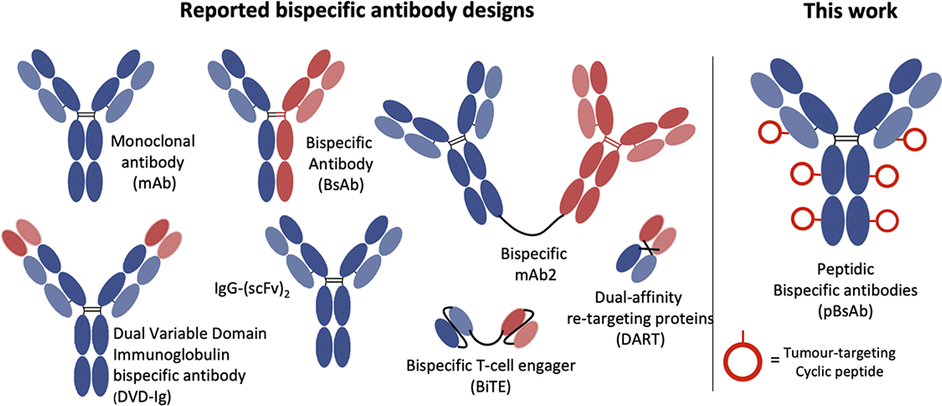 | ||
| Fig. 1 Schematic structures of the monoclonal antibody, different IgG-like, non-IgG-like bispecific antibodies, and our novel peptidic bispecific antibody (pBsAb). | ||
These bispecific antibodies have been shown to significantly improve survival chances of cancer patients compared to monoclonal antibodies.11 Blinatumomab, which is the first T-cell-engaging bispecific antibody approved by the FDA in 2015, can target both CD19 on B-lineage leukemia and CD3 on T cells.12,13 Such an engaging mechanism redirects the immune cells to engage and destroy the target cancer cells. There is another T cell engager that binds to both PD-1 and CTLA4 currently in clinical trials.14 There were eight approved bispecific anti-bodies on the market by the end of 2022.15 To date, there are more than 150 bispecific antibodies at different stages of clinical trials.16 Despite the aforementioned advantages and potential applications, the production of bispecific antibodies remains challenging, tedious, and expensive due to the extra requirement in design considerations, extensive purification steps, and more complicated quality control.17–19 Thus, there is a strong demand for a robust platform that can rapidly and efficiently generate novel bispecifics for future research and therapeutic development.
The use of biological approaches to generate bispecific antibodies was started two decades ago20–22 such as the knobs-into-holes strategy,23 strand-exchange engineered domain (SEED) CH3 heterodimers approach,24 orthogonal Fab interface,25 and CorssMab design.26 Recently, chemical approaches27–35 to develop bispecific antibodies has recently emerged, which shows advantages in the bispecific antibody production. In 2010, Doppalapudi et al. introduced a novel technology for creating bispecific antibodies, termed bispecific CovX-Bodies, which are generated by fusing pharmacophore peptide heterodimers to a scaffold antibody that effectively targets both vascular endothelial growth factor and angiopoietin-2.28 Chudasama and co-workers reported a simple plug-and-play approach to facilitate the generation of bispecific molecules.29 In 2023, they developed the bio-orthogonal approach to generate Synthetic Antibodies (SynAbs).30 In this literature, we report a new strategy that allows efficient design and generation of a new type of bispecific antibody, namely the peptidic bispecific antibody (pBsAb) (Fig. 1). This novel pBsAb is composed of a monoclonal antibody that covalently conjugated with tumour-targeting cyclic peptides, which can be readily constructed using our recently reported facile peptide cyclisation and protein conjugation reaction (Fig. 2A).36,37 The key compound for this reaction is the bifunctional linker 1 that consists of a dibromomethyl benzene (DBMB) unit for peptide cyclisation with a bis-cysteine-containing peptide,38–41 and an ortho-phthalaldehyde (OPA) for phthalaldehyde-amine capture (PAC) protein conjugation.42–45 With the extraordinary stability in serum, cyclic peptides are expected to resist enzymatic degradation, which could significantly enhance the half-life of the molecules.46 With such an idea in mind, we used such a method to construct a novel pBsAb using this robust approach.
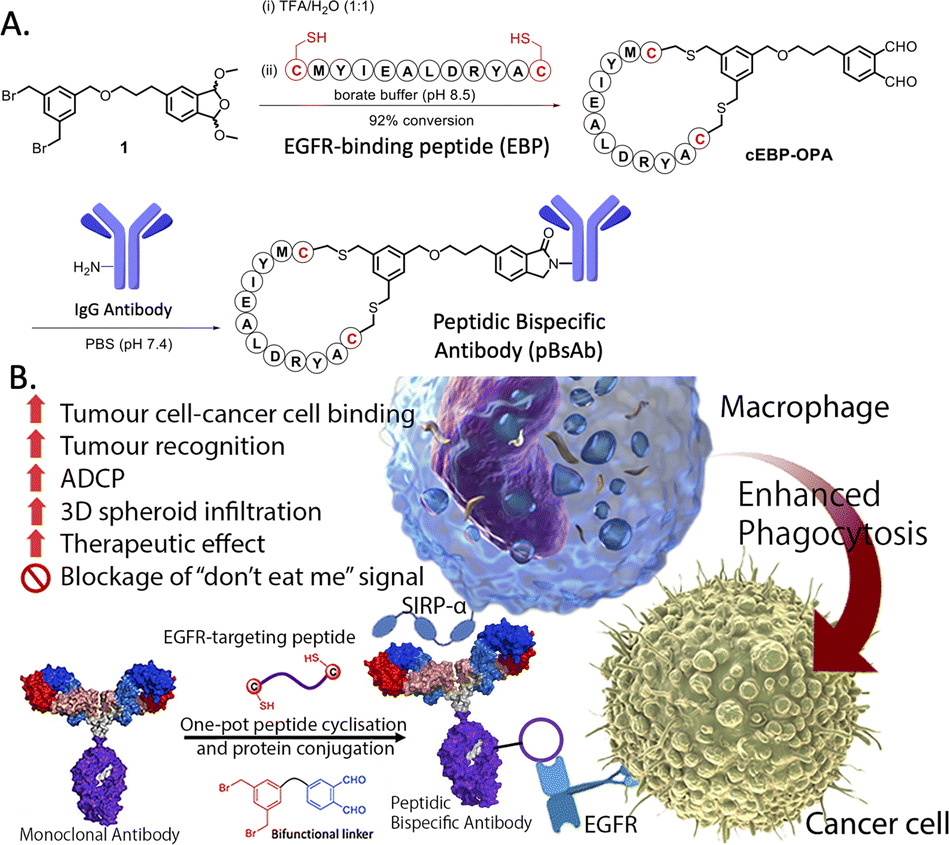 | ||
| Fig. 2 (A) Scheme of one-pot peptide cyclisation and (B) anti-body conjugation and schematic representation of the mechanism of novel EGFR × SIRP-α macrophage-engaging pBsAb. | ||
Results and discussion
First, a proof-of-concept experiment was performed by cyclising and conjugating EGFR-binding peptides (EBP) (AcNH-CMYIEALDRYAC-CONH2) to a nonspecific human IgG that did not have specificity against tumour cells. To this end, the linear EBP was cyclised with a bifunctional linker via site-specific dialkylation of the sulfhydryl side chains of two cysteine residues in borate buffer (pH 8.5, 1 mM) for 1 h at room temperature (Fig. 2).36 The resulting ortho-phthalaldehyde-functionalised cyclic EBP peptide (labelled as cEBP-OPA) was formed successfully, as characterised by high-resolution electrospray ionisation (ESI) mass spectrometry (Fig. S1†). The cEBP-OPA (1 mM) was then further reacted with the non-specific human IgG antibody (50 μM) in phosphate-buffered saline (PBS) (pH 7.4) via PAC reaction for 30 min at room temperature. After removing the excess reagents using the Zeba Spin Desalting Column (40K cut-off), the cyclic peptide-conjugated antibody cEBP-IgG was obtained. The SDS-PAGE result showed the increased molecular weight of the light chain and heavy chain of the antibody (Fig. S2†). The selective binding ability of cEBP-IgG was then investigated by confocal microscopy, using EGFR-positive HT29 human colorectal adenocarcinoma cells and EGFR-negative HeLa human cervical carcinoma cells. Alexa Fluor 647 conjugated rabbit anti-human secondary antibody (Abcam, USA) was used to visualise the cEGP-IgG. The in vitro data showed that the cEBP-IgG was able to bind to the EGFR-overexpressed HT29 cells but not the HeLa cells. In comparison, the native IgG without peptide modification could not bind to any cell surface (Fig. S3†). The findings of this study indicate that the conjugation of cyclic peptides onto non-specific IgG antibodies can confer an additional tumour-targeting function on the antibody.With the plausible preliminary data, we further use this strategy to design a novel macrophage-engaging bispecific antibody for immunotherapy. Macrophages are known to be an important immune modulator capable of performing phagocytosis for cancer elimination and effective infiltration into solid tumours.47 For immunotherapy, the macrophages may have a reduced risk of cytokine release syndrome and T-cell exhaustion. SIRP-α (CD172a) is a macrophage receptor that negatively controls the effector functions of cytotoxicity. When SIRP-α on the macrophage binds to CD47 on the cancer cells, the “don't eat me” signal is activated, thereby inhibiting the macrophage-mediated cancer cell phagocytosis. This mechanism is often exploited by cancer cells to avoid immune destruction. Consequently, targeting the SIRP-α-CD47 axis has emerged as a promising therapeutic strategy in cancer immunotherapy, aiming to block this interaction and thereby enhancing the immune system's ability to eliminate tumour cells.48,49 Therefore, SIRP-α could be one of the possible targets for de-signing the novel bispecific antibody. To link cancer cells to the macrophage, cyclic EPB will be conjugated to the anti-SIRP-α monoclonal antibody as EGFR is known to be overexpressed in many cancer cells.43,50 Having such design, we expect that after the construction of the novel pBsAb, the EBP can target overexpressed EGFR on the cancer cell surface while an anti-SIRP-α monoclonal antibody was used to target SIRP-α on macrophages. This approach could bring macrophages and cancer cells in proximity, improving cancer cell recognition, and initiating antibody-dependent cellular phagocytosis (ADCP) (Fig. 2B). Furthermore, it is anticipated that the pBsAb design can block the SIRP-α “don't eat me” checkpoint signal, thus preventing the inactivation of macrophage-mediated phagocytosis by CD47 on cancer cells.
To this end, a conjugation reaction between the cyclic peptide and antibody was optimised to determine the optimal conditions with the best bioactivities. First, different groups of pBsAbs were produced by conjugating different amounts of cEBP-OPA with a fixed amount of the anti-SIRP-α mAb (100 nM) in different mole ratios (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10:1, 20:1, and 50:1) according to the aforementioned procedure. After purification with the Zeba Spin Desalting Column (40K cut off) to remove excess peptides, pBsAbs were characterised by SDS-PAGE (Fig. S4†) and ESI mass spectrometry (Fig. 3). The results showed that the pBsAb prepared by adding 20 equivalents of cEBP-OPA resulted in an average of 9.6 peptide-to-antibody ratio. Then a detailed target binding study of the novel pBsAb was performed to determine the binding affinities against EGFR and SIRP-α using enzyme-linked immunosorbent assay (ELISA). Our data showed that the pBsAb prepared with 20 equivalents of peptides to antibody gave the best binding affinity against EGFR compared to the other conditions (Fig. S5†), while 50-fold excess of cEBP-OPA led to denaturation and precipitation of the antibody. Size exclusion chromatography analysis was also conducted and showed no significant aggregation for the pBsAb after overnight incubation in PBS (Fig. S6†).
:1, 10:1, 20:1, and 50:1) according to the aforementioned procedure. After purification with the Zeba Spin Desalting Column (40K cut off) to remove excess peptides, pBsAbs were characterised by SDS-PAGE (Fig. S4†) and ESI mass spectrometry (Fig. 3). The results showed that the pBsAb prepared by adding 20 equivalents of cEBP-OPA resulted in an average of 9.6 peptide-to-antibody ratio. Then a detailed target binding study of the novel pBsAb was performed to determine the binding affinities against EGFR and SIRP-α using enzyme-linked immunosorbent assay (ELISA). Our data showed that the pBsAb prepared with 20 equivalents of peptides to antibody gave the best binding affinity against EGFR compared to the other conditions (Fig. S5†), while 50-fold excess of cEBP-OPA led to denaturation and precipitation of the antibody. Size exclusion chromatography analysis was also conducted and showed no significant aggregation for the pBsAb after overnight incubation in PBS (Fig. S6†).
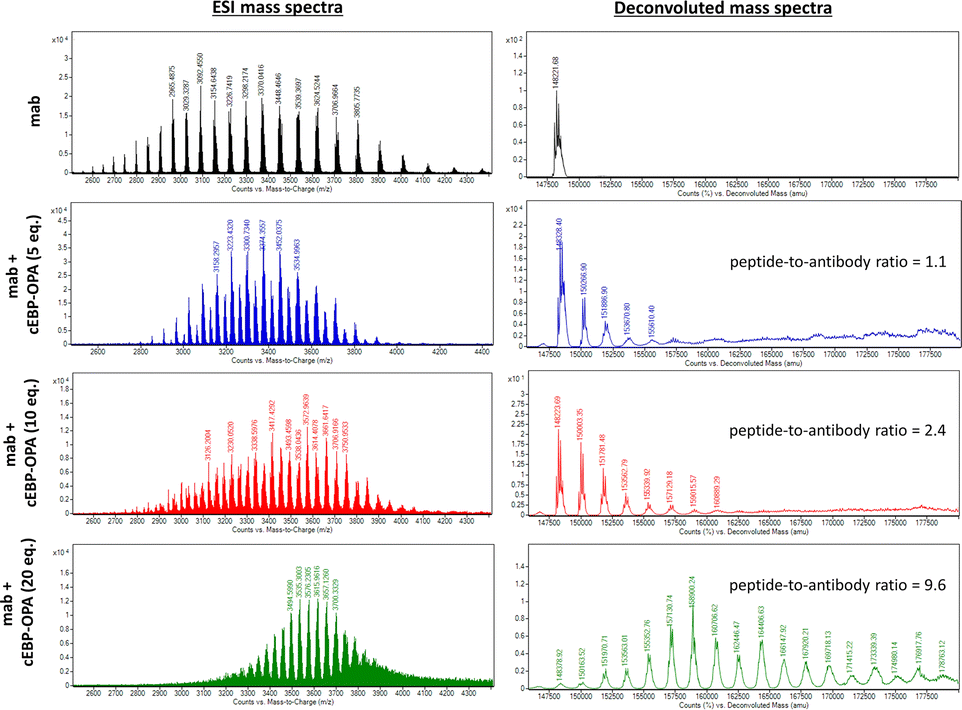 | ||
| Fig. 3 ESI mass analysis of the mAb and pBsAb prepared with different equivalents (5, 10, and 20) of cEBP-OPA. The data revealed that an average of 1.1 cyclic peptides was conjugated on the antibody when 5 equivalents of cEBP-OPA were added. An average of 2.4 cyclic peptides were conjugated on the antibody when 10 equivalents of cEBP-OPA were used, while the addition of 20 equivalents of cEBP-OPA resulted in a 9.6 peptide-to-antibody ratio. | ||
After optimising the conjugation conditions, the binding ability of the pBsAb against EGFR was shown in a dose-dependent manner with an EC50 value of 2.6 nM (Fig. 4A). In contrast, the monoclonal antibody (mAb) demonstrated no binding activity against EGFR by the ELISA binding assay. Our data, on the other hand, showed that the chemical modification of the pBsAb did not significantly interfere with its SIRP-α binding ability (EC50 = 0.12 nM) when compared to the native mAb (EC50 = 0.05 nM) (Fig. 4B). Furthermore, the protein–protein interaction between CD47 and SIRP-α was found to be inhibited in the presence of the pBsAb in our ELISA experiment, showing the blockage of the interaction of CD47-SIRP-α (Fig. S7 and S11†). In addition, we challenged the pBsAb with a serum stability experiment to demonstrate the binding of the two receptors after overnight incubation in serum. The data showed that the binding affinities against EGFR and SIRP-α did not decrease over time (Fig. S8†). All these results indicated that an extra antigen-binding ability against EGFR could be incorporated into the monoclonal antibody to generate a novel bispecific antibody by our facile reaction. It is noteworthy that although heterogeneous conjugated products could be produced using our approach, our data confirmed that neither Fab binding activity was significantly affected.
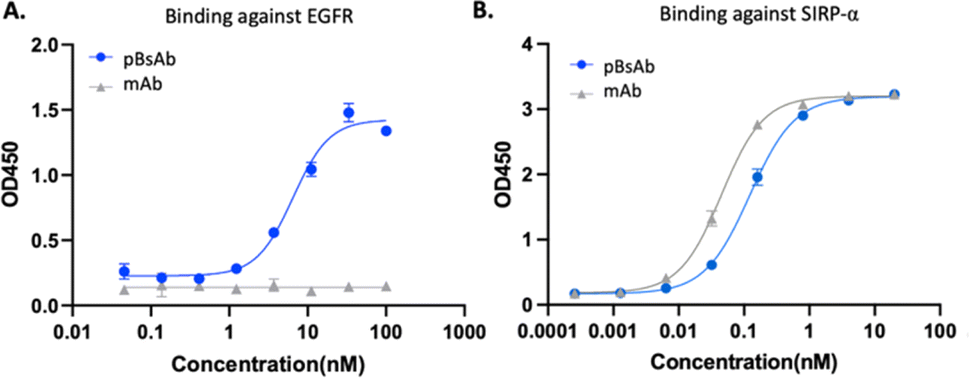 | ||
| Fig. 4 ELISA binding assay of the pBsAb and mAb against (A) EGFR and (B) SIRP-α. Data are expressed as the mean value ± standard error of the mean (SEM) of three independent experiments, each performed in triplicate. | ||
With the confirmed bispecificity of EGFR × SIRP-α pBsAb, the in vitro target binding experiment was further studied by confocal microscopy. The SIRP-α-positive RAW264.7 murine macrophage cells, the EGFR-positive A549 human lung carcinoma cells and HT29 human colorectal adenocarcinoma cells, as well as the EGFR-negative HeLa human cervical carcinoma cells were incubated with the anti-SIRP-α mAb or pBsAb (10 nM) for 30 min at 4 °C, followed by Alexa 488-labelled secondary antibody for 30 min. As shown in Fig. 5A the pBsAb showed strong fluorescence on the surface of the RAW264.7, demonstrating that both antibodies could bind with the SIRP-α on the RAW264.7 cell surface. For the binding with the EGFR, the fluorescence signal could only be observed on the EGFR-positive A549 and HT29 cells, but not the EGFR-negative HeLa cells, while the anti-SIRP-α mAb could not bind with these cells. Thus, it illustrated that our novel pBsAb could selectively bind with both EGFR and SIRP-α proteins on the cell surface. Similar results were also obtained by flow cytometry (Fig. 5B). Upon incubation with pBsAb (20 nM), a significant 15-fold difference in fluorescence intensity could be observed between the EGFR-positive A549 cells and the EGFR-negative HeLa cells, while incubation with mAb showed low fluorescence intensity in these two cells. On the other hand, incubation of both anti-SIRP-α mAb and pBsAb showed similar fluorescence intensity in RAW264.7 cells. These results were consistent with the ELISA binding assay, which confirmed that under an in vitro environment, the antibody could gain extra targeting ability against EGFR on cancer cells via our novel approach.
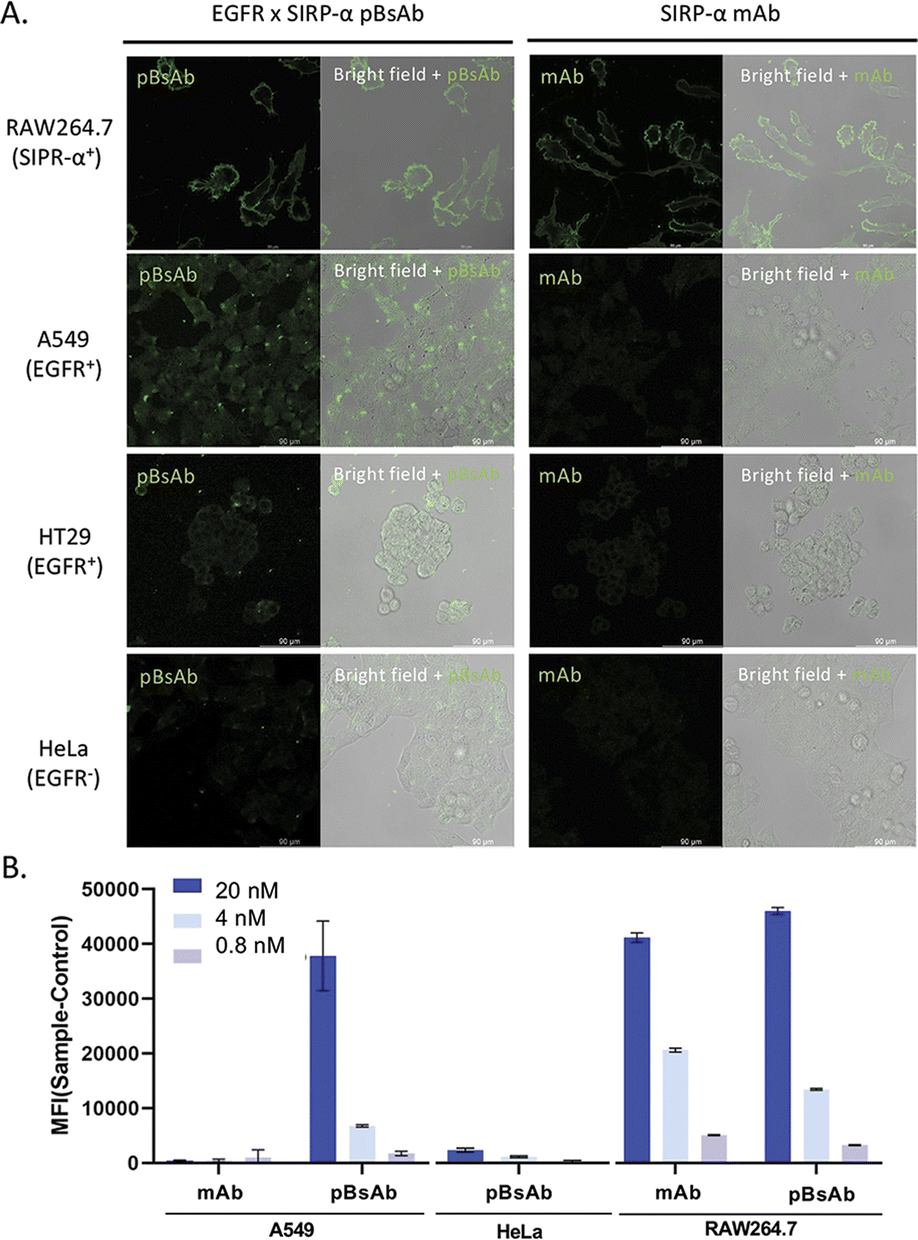 | ||
| Fig. 5 (A) Confocal microscopic images of RAW264.7, A549, HT29, and HeLa cells upon incubation with the mAb and pBsAb (10 nM), followed by Alexa 488-labelled secondary antibody. (B) Flow cytometric data of RAW264.7, A549, and HeLa cells treated with the mAb or pBsAb at different concentrations. Data are expressed as the mean value ± SEM of three independent experiments. | ||
Encouraged by the bispecificities of the pBsAb against EGFR and SIRP-α, we further investigated the macrophage-engaging property of the pBsAb. The initiation of the macrophage–cancer cell interaction by the pBsAb was studied by the cell–cell adhesion assay.51 In brief, A549 cells (2 × 105 cells) were seeded in a 12-well plate and cultured in the medium until 90–95% confluency was reached. Then carboxyfluorescein succinimidyl ester (CFSE)-labelled RAW264.7 macrophages (2 × 105 cells) were added into each well in the presence of 20 nM of mAb, pBsAb, or PBS as the control. After incubation for 10 min, the cells were washed extensively with PBS. The number of green labelled RAW264.7 cells that remained adhered at the bottom was quantified using ImageJ. As shown in Fig. 6, a higher number of macrophages could be detected in the presence of the pBsAb, indicating enhanced macrophage–cancer cell adhesion compared to the mAb and PBS negative control. Additionally, we verified that similar outcomes are achievable with the human monocyte cell line THP-1, following its differentiation into macrophages. This demonstrates the cross-reactivity of our pBsAb against human SIRP-α, further validating our findings (Fig. S10†).
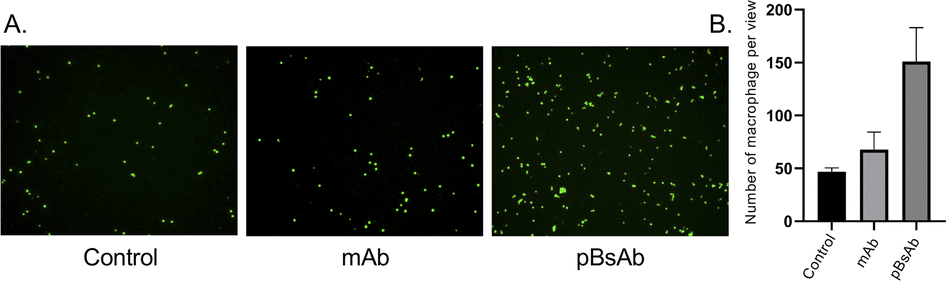 | ||
| Fig. 6 (A) Fluorescence microscopy images of the cell–cell adhesion experiment illustrating the adhesion of RAW264.7 macrophages (green) to the monolayer of EGFR-positive A549 cells in the presence of different antibodies at 20 nM or PBS. (B) Bar chart of the quantified number of macrophages adhering to A549 cells. | ||
After confirming the macrophage-engaging property of our pBsAb, we further investigated its antibody-dependent cellular phagocytosis (ADCP) activity by confocal microscopy and flow cytometry.52,53 As shown in Fig. 7A and B, upon treatment with pBsAb (20 nM) in the co-culture of RAW264.7 macrophages (prelabelled with CFSE) and A549, HT29, and HeLa cancer cells (prelabelled with CellTracker Red CMPTX) for 24 h, intense red fluorescence originating from the A549 cells was observed in the cytoplasm of RAW264.7 macrophages. This indicated that phagocytosis occurred to engulf the red-labelled cancer cells. On the other hand, incubation with the anti-SIRP-α mAb resulted in a much lesser phagocytic activity (Fig. 7A and S9†). In addition, we also quantified ADCP activity using flow cytometry. After pre-staining the macrophages and cancer cells, they were co-cultured in a U-bottom 96-well plate at 1:1 ratio (1 × 104 cells) in the presence or absence of the pBsAb or anti-SIRP-α mAb (20 nM) for 2 h at 37 °C. The percentage of phagocytosis was calculated as the population of phagocytic macrophages among the total macrophages. The quadrant analysis clearly showed that in the presence of the pBsAb, the percentage of double-positive phagocytic macrophages against EGFR-positive A549 and HT29 cells increased by 2.0 and 2.5-fold, respectively, compared to the treatment with mAb control. However, for the EGFR-negative HeLa cells, treatment with the pBsAb or mAb showed a similar percentage of phagocytic macrophages, indicating there was no observable enhancement of phagocytotic activity (Fig. 7C). These results clearly illustrated that the ADCP activity was enhanced by our novel EGFR × SIRP-α pBsAb.
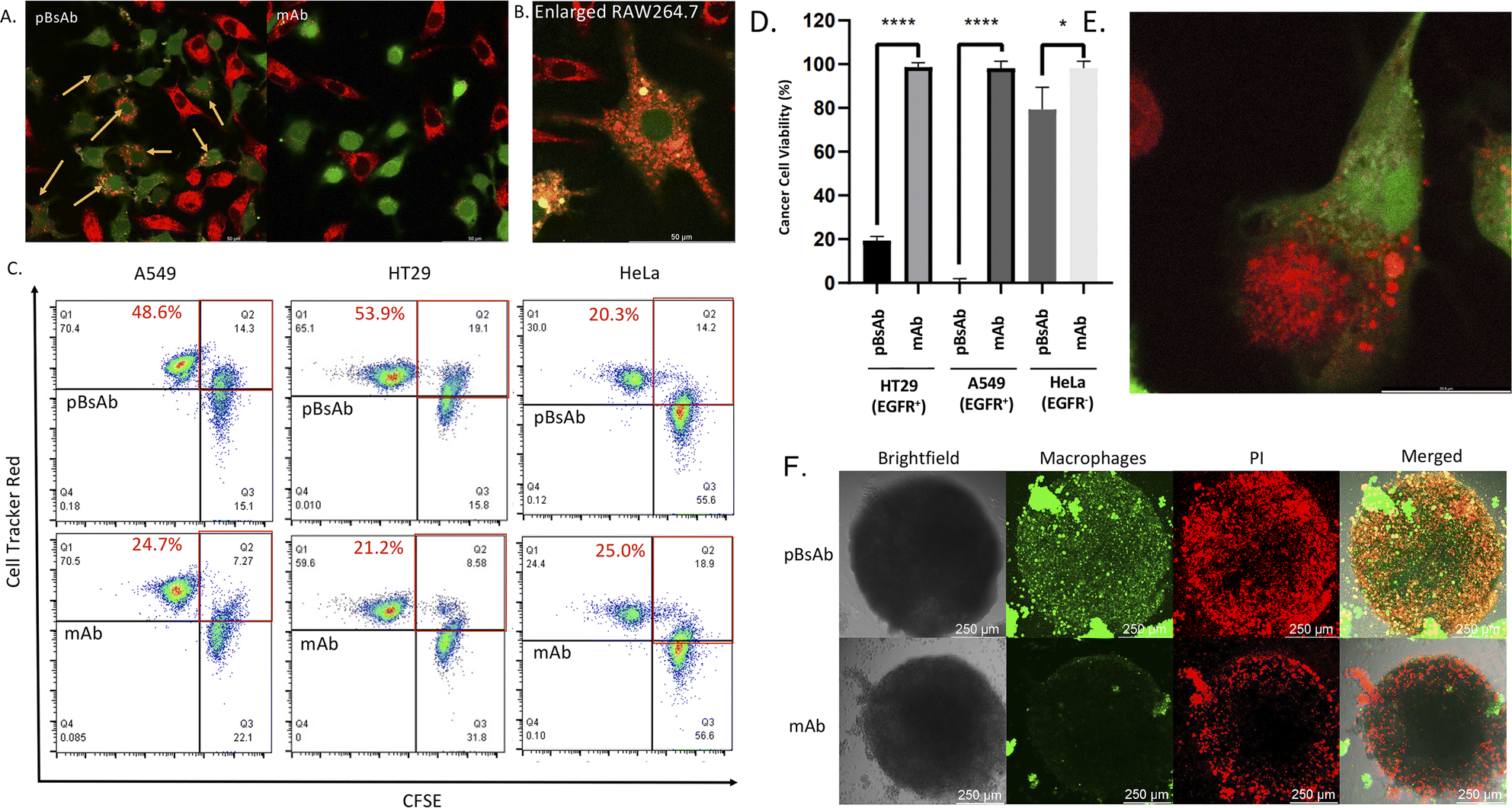 | ||
| Fig. 7 (A) Confocal microscopic images of the co-cultured RAW264.7 macrophages (green) and A549 cells (red) treated with the pBsAb or anti-SIRP-α mAb (20 nM) for 24 h. The yellow arrow shows the phagocytic macrophages. (B) Enlarged confocal microscopic image of the phagocytic macrophages (green) treated with the pBsAb (20 nM) for 24 h. (C) Flow cytometric quadrant analysis of the co-cultured RAW264.7 macrophages (green) and A549, HT29, and HeLa cells (red) treated with the pBsAb (20 nM) or anti-SIRP-α mAb (20 nM) for 2 h. The red squares show the percentage of phagocytotic macrophages under different conditions. (D) Cancer cell viability assay of RAW264.7 macrophages against EGFR-overexpressed (A549 and HT29) and EGFR-negative cells (HeLa) upon treatment with 50 nM of pBsAb and anti-SIRP-α mAb. Data are expressed as the mean value ± SEM of three independent experiments. ****P < 0.0001 compared to the corresponding control of mAb, * no significant difference. (E) Confocal image of the phagocytosis process. The macrophage (green) was engulfing the HT29 (red) in the presence of the pBsAb. (F) Confocal Z-stack maximum projection microscopic images of HT29 cancer cell spheroids co-cultured with RAW264.7 macrophages (green) upon treatment with the pBsAb or anti-SIRP-α mAb (50 nM) for 24 h. The dead cells were stained with PI (red). | ||
The ADCP anti-cancer activity of RAW264.7 macrophages was further determined by using a CellTiter-Glo Luminescent Cell Viability Kit (Promega, USA) to determine the viability of cancer cells.54 As shown in Fig. 7D, pBsAb treatment could significantly reduce the number of cancer cells, which indicated that the pBsAb enhanced the anticancer effect against EGFR-overexpressed cancer cells, compared to the unmodified anti-SIRP-α mAb at 50 nM. We also confirmed that the EGFR-binding peptide itself did not contribute to any cytotoxicity effect (Fig. S12†). Lastly, we further investigated the macrophage infiltration and ADCP anti-tumour effect in a three-dimensional HT29 spheroid model. The three-dimensional tumour spheroid model is known to resemble the tumour situation in vivo more closely than the monolayer setting.55 As shown in Fig. 7F, the Z-stack maximum projection confocal images revealed that the number of macrophages (green) inside the core of the spheroid treated with the pBsAb was significantly higher than that treated with the anti-SIRP-α mAb.
In addition, an intensified red-fluorescent PI signal was observed in the core of the spheroid treated with the pBsAb. This set of data further supports our hypothesis that, in the presence of the pBsAb, macrophages can effectively bind to and infiltrate the solid tumour core, leading to an enhanced anti-cancer effect. Collectively, these results suggest that our innovative and facile approach for generating this novel type of pBsAb has the potential to become a preferred method in developing bispecific antibodies, ultimately improving cancer recognition and immunotherapeutic efficacy.
Conclusions
In this study, a novel and robust chemical method was developed for generating a new type of bispecific antibody, namely the peptidic bispecific antibody (pBsAb), from a monoclonal antibody. The EGFR × SIRP-α pBsAb was derived from an anti-SIRP-α monoclonal antibody by conjugating EGFR-targeting cyclic peptides on the protein surface using a robust one-pot peptide cyclisation and antibody conjugation reaction. The results demonstrated that the pBsAb was capable of binding both the EGFR-overexpressed cancer cells and SIRP-α-expressed macrophages, thereby initiating macrophage–cancer cell interaction, which enhanced EGFR-targeting antibody-dependent cellular phagocytosis. The successful establishment of this novel platform provides a means of rapid production of bispecific antibodies for immunotherapy at a lower cost.Data availability
All our data has been placed in the main text and the ESI.†Author contributions
Conceptualisation: C. T. T. W., W. C. S. T., W. T. W.; funding acquisition: C. T. T. W.; resources: C. T. T. W., C. M. C., W. T. W.; methodology: C. T. T. W., C. S., B. T., K. M. L., J. C. H. C., W.-T. W.; sample preparation and data acquisition: C. S., B. T.; data analysis: C. S., B. T., K. M. L., J. C. H. C., W.-T. W.; data interpretation: C. S., B. T., K. M. L., J. C. H. C., W.-T. W.; visualisation: C. T. T. W., C. S., B. T.; supervision: C. T. T. W., W. T. W., C. M. C.; writing – original draft: C. T. T. W., C. S.; writing – review and editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the General Research Fund from the Hong Kong Research Grant Council (Grant No. 15303321), Hong Kong Innovation and Technology Commission (ITS/013/22FP), and Research Impact Fund (R5008-22). We also acknowledge the funding support from “Laboratory for Synthetic Chemistry and Chemical Biology” under the Health@InnoHK Program launched by the Innovation and Technology Commission, The Government of HKSAR, China.Notes and references
- A. Nisonoff, F. C. Wissler and L. N. Lipman, Science, 1960, 132, 1770–1771 CrossRef CAS PubMed
.
- A. Nisonoff and M. M. Rivers, Arch. Biochem. Biophys., 1961, 93, 460–462 CrossRef CAS PubMed
- H. R. Robinson, J. Qi, E. M. Cook, C. Nichols, E. L. Dadashian, C. Underbayev, S. E. M. Herman, N. S. Saba, K. Keyvanfar, C. Sun, I. E. Ahn, S. Baskar, C. Rader and A. Wiestner, Blood, 2018, 132, 521–532 CrossRef CAS PubMed
- N. Gera, Expert Opin. Biol. Ther., 2022, 22, 945–949 CrossRef CAS
- A. Krishnamurthy and A. Jimeno, Pharmacol. Ther., 2018, 185, 122–134 CrossRef CAS PubMed
- J. Ma, Y. Mo, M. Tang, J. Shen, Y. Qi, W. Zhao, Y. Huang, Y. Xu and C. Qian, Front. Immunol., 2021, 12, 626616 CrossRef CAS PubMed
- A. F. Labrijn, M. L. Janmaat, J. M. Reichert and P. W. H. I. Parren, Nat. Rev. Drug Discovery, 2019, 18, 585–608 CrossRef CAS PubMed
- C. Rader, Curr. Opin. Biotechnol., 2019, I65, 9–16 Search PubMed
- K. Runcie, D. R. Budman, V. John and N. Seetharamu, Mol. Med., 2018, 24, 50 Search PubMed
- J. Kang, T. Sun and Y. Zhang, Front. Immunol., 2022, 13, 1020003 CrossRef CAS PubMed
- S. E. Sedykh, V. V. Prinz, V. N. Buneva and G. A. Nevinsky, Drug Des., Dev. Ther., 2018, 12, 195–208 CrossRef CAS PubMed
- N. Bumma, N. Papadantonakis and A. S. Advani, Future Oncol., 2015, 11, 1729–1739 CrossRef CAS PubMed
- H. Kantarjian, A. Stein, N. Gökbuget, A. K. Fielding, A. C. Schuh, J.-M. Ribera, A. Wei, H. Dombret, R. Foà, R. Bassan, Ö. Arslan, M. A. Sanz, J. Bergeron, F. Demirkan, E. Lech-Maranda, A. Rambaldi, X. Thomas, H.-A. Horst, M. Brüggemann, W. Klapper, B. L. Wood, A. Fleishman, D. Nagorsen, C. Holland, Z. Zimmerman and M. S. Topp, N. Engl. J. Med., 2017, 376, 836–847 CrossRef CAS PubMed
- A. Berezhnoy, B. J. Sumrow, K. Stahl and K. Shah,
et al.
, Cell Rep. Med., 2020, 1, 100163 CrossRef CAS PubMed
- A. Esfandiari, S. Cassidy and R. M. Webster, Nat. Rev. Drug Discovery, 2022, 21, 411–412 CrossRef CAS PubMed
- M. S. Kinch, Z. Kraft and T. Schwartz, Drug Discovery Today, 2023, 28, 103462 CrossRef CAS PubMed
- A. Thakur, M. Huang and L. G. Lum, Blood Rev., 2018, 32, 339–347 CrossRef CAS PubMed
- S. W. Chen and W. Zhang, Antibody Ther., 2021, 4, 73–88 CrossRef CAS PubMed
- D. J. Underwood, J. Bettencourt and Z. Jawad, Expert Opin. Biol. Ther., 2022, 22, 1043–1065 CrossRef CAS
- U. Brinkmann and R. E. Kontermann, mAbs, 2017, 9, 182–212 CrossRef CAS PubMed
- Y. Chen, J. Park, X. Liu, Y. Hu, T. Wang, K. McFarland and M. J. Betenbaugh, Antibodies, 2019, 8, 43 CrossRef PubMed
- Y. W. Iwasaki, K. Tharakaraman, V. Subramanian, A. Khongmanee, A. Hatas, E. Fleischer, T. T. Rurak, P. Ngok-ngam, P. Tit-oon, M. Ruchirawat, J. Satayavivad, M. Fuangthong and R. Sasisekharan, Front. Immunol., 2022, 13, 1063002 CrossRef CAS PubMed
- A. M. Merchant, Z. Zhu, J. Q. Yuan, A. Goddard, C. W. Adams, L. G. Presta and P. Carter, Nat. Biotechnol., 1998, 16, 677–681 CrossRef CAS PubMed
- J. H. Davis, C. Aperlo, Y. Li, E. Kurosawa, Y. Lan, K.-M. Lo and J. S. Huston, Protein Eng., Des. Sel., 2010, 23, 195–202 CrossRef CAS PubMed
- S. M. Lewis, X. Wu, A. Pustilnik, A. Sereno, F. Huang, H. L. Rick, G. Guntas, A. Leaver-Fay, E. M. Smith, C. Ho, C. Hansen-Estruch, A. K. Chamberlain, S. M. Truhlar, E. M. Conner, S. Atwell, B. Kuhlman and S. J. Demarest, Nat. Biotechnol., 2014, 32, 191–198 CrossRef CAS PubMed
- W. Schaefer, J. T. Regula, M. Bähner, J. Schanzer, R. Croasdale, H. Dürr, C. Gassner, G. Georges, H. Kettenberger, S. Imhof-Jung, M. Schwaiger, K. G. Stubenrauch, C. Sustmann, M. Thomas, W. Scheuer and C. Klein, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11187–11192 CrossRef CAS PubMed
- P. Szijj and V. Chudasama, Nat. Rev. Chem., 2021, 5, 78–92 CrossRef CAS PubMed
- V. R. Doppalapudi, J. Huang, D. Liu, P. Jin, B. Liu, L. Li, J. Desharnais, C. Hagen, N. J. Levin, M. J. Shields, M. Parish, R. E. Murphy, J. Del Rosario, B. D. Oates, J.-Y. Lai, M. J. Matin, Z. Ainekulu, A. Bhat, C. W. Bradshaw, G. Woodnutt, R. A. Lerner and R. W. Lappe, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 22611–22616 CrossRef CAS PubMed
- A. Maruani, P. A. Szijj, C. Bahou, J. C. F. Nogueira, S. Caddick, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2020, 31, 520–529 CrossRef CAS PubMed
- F. Thoreau, P. A. Szijj, M. K. Greene, L. N. C. Rochet, I. A. Thanasi, J. K. Blayney, A. Maruani, J. R. Baker, C. J. Scott and V. Chudasama, ACS Cent. Sci., 2023, 9, 476 CrossRef CAS PubMed
- H. Khalili, A. Godwin, J. Choi, R. Lever, P. T. Khaw and S. Brocchini, Bioconjugate Chem., 2013, 24, 1870–1882 CrossRef CAS PubMed
- J. M. Scheer, W. Sandoval, J. M. Elliott, L. Shao, E. Luis, S. C. Lewin-Koh, G. Schaefer and R. Vandlen, PLoS One, 2012, 7, e51817 CrossRef CAS PubMed
- C. H. Kim, J. Y. Axup, A. Dubrovska, S. A. Kazane, B. A. Hutchins, E. D. Wold, C. C. Smider and P. G. Schultz, J. Am. Chem. Soc., 2012, 134, 9918–9921 CrossRef CAS PubMed
- E. Walseng, C. G. Nelson, J. Qi, A. R. Nanna, W. R. Roush, R. K. Goswami, S. C. Sinha, T. R. Burke Jr and C. Rader, J. Biol. Chem., 2016, 291, 19661–19673 CrossRef CAS PubMed
- A. Ueda, M. Umetsu, T. Nakanishi, K. Hashikami, H. Nakazawa, S. Hattori, R. Asano and I. Kumagai, Int. J. Mol. Sci., 2020, 21, 71129 Search PubMed
- J. Xiong, J. C. H. Chu, W. P. Fong, C. T. T. Wong and D. K. P. Ng, J. Am. Chem. Soc., 2022, 144, 10647–10658 CrossRef CAS
- J. C. H. Chu, C. Shao, S. Y. Y. Ha, W. P. Fong, C. T. T. Wong and D. K. P. Ng, Biomater. Sci., 2021, 9, 7832–7837 RSC
- D. E. Streefkerk, M. Schmidt, J. H. Ippel, T. M. Hackeng, T. Nuijens, P. Timmerman and J. H. van Maarseveen, Org. Lett., 2019, 21, 2095–2100 CrossRef CAS PubMed
- S. Chen, D. Bertoldo, A. Angelini, F. Pojer and C. Heinis, Angew. Chem., Int. Ed., 2014, 53, 1602–1606 CrossRef CAS PubMed
- J. C. H. Chu, W. P. Fong, C. T. T. Wong and D. K. P. Ng, J. Med. Chem., 2021, 64, 2064–2076 CrossRef CAS PubMed
- J. C. H. Chu, M. L. Chin, C. T. T. Wong, M. Hiu, P. C. Lo and D. K. P. Ng, Adv. Ther., 2020, 4, 2000204 CrossRef
- C. L. Tung, C. T. T. Wong, E. Y. Fung and X. Li, Org. Lett., 2016, 18, 2600–2603 CrossRef CAS
- Y. Zhang, Q. Zhang, C. T. T. Wong and X. Li, J. Am. Chem. Soc., 2019, 141, 12274–12279 CrossRef CAS
- C. H. P. Cheung, T. H. Chong, T. Wei, H. Liu and X. Li, Angew. Chem., Int. Ed., 2023, 135, e202217150 CrossRef
- C. T. T. Wong, J. C. H. Chu, S. Y. Y. Ha, R. C. H. Wong, G. Dai, T. T. Kwong, C. H. Wong and D. K. P. Ng, Org. Lett., 2020, 22, 7098–7102 CrossRef CAS PubMed
- H. Zhang and S. Chen, RSC Chem. Biol., 2022, 3, 18–31 RSC
- J. L. Guerriero, Trends Mol. Med., 2018, 24, 472–489 CrossRef CAS PubMed
- A. Alausa, K. A. Lawal, O. A. Babatunde, E. N. O. Obiwulu, O. C. Oladokun, O. S. Fadahunsi, U. O. Celestine, E. U. Moses, I. R. Akaniro and P. I. Adegbola, Pharmacol. Res., 2022, 181, 106264 CrossRef CAS PubMed
- N. Dizman and E. I. Buchbinder, Cancers, 2021, 13, 6229 CrossRef CAS PubMed
- S. Sigismund, D. Avanzato and L. Lanzetti, Mol. Oncol., 2018, 12, 3–20 CrossRef PubMed
- A. Zepeda-Moreno, I. Taubert, I. Hellwig, V. Hoang, L. Pietsch and V. K. Lakshmanan, Cell Adhes. Migr., 2011, 5, 215–219 CrossRef PubMed
- M. E. Ackerman, B. Moldt, R. T. Wyatt, A.-S. Dugast, E. McAndrew, S. Tsoukas, S. Jost, C. T. Berger, G. Sciaranghella, Q. Liu, D. J. Irvine, D. R. Burton and G. Alter, J. Immunol. Methods, 2011, 366, 8–19 CrossRef CAS PubMed
- C. Xu, H. Wu, Y. Liu, F. Li, R. K. Manne and H. K. Lin, STAR Protoc., 2022, 4, 101940 CrossRef PubMed
- Y. Yang, X. Sun, J. Xu, C. Cui, H. Safari Yazd, X. Pan, Y. Zhu, X. Chen, X. Li, J. Li and W. Tan, ACS Nano, 2020, 14, 9562–9571 CrossRef CAS PubMed
- J. Caleb and Y. Teng, Front. Mol. Biosci., 2020, 7, 33 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00851k |
| This journal is © The Royal Society of Chemistry 2024 |