
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA-directed formation of plasmonic core–satellite nanostructures for quantification of hepatitis C viral RNA†
Siddhant
Jaitpal‡
 ab,
Ka Wai
Ng‡
ab,
Angela Michelle
San Juan
ab,
Cecilia
Martinez
a,
Christian
Phillips
a,
Sayantan
Tripathy
ab and
Samuel
Mabbott
*ab
ab,
Ka Wai
Ng‡
ab,
Angela Michelle
San Juan
ab,
Cecilia
Martinez
a,
Christian
Phillips
a,
Sayantan
Tripathy
ab and
Samuel
Mabbott
*ab
aDepartment of Biomedical Engineering, Texas A&M University, 600 Discovery Drive, College Station, TX 77840-3006, USA. E-mail: smabbott@tamu.edu
bCenter for Remote Health Technologies & Systems, Texas A&M Engineering Experiment Station, 600 Discovery Drive, College Station, TX 77840-3006, USA
First published on 22nd April 2024
Abstract
Hepatitis C virus (HCV) continues to be a significant public health challenge, affecting an estimated 71 million people globally and posing risks of severe liver diseases. Despite advancements in treatments, diagnostic limitations hinder the global elimination efforts targeted by 2030. This study introduces an innovative diagnostic approach, integrating catalytic hairpin assembly (CHA) with plasmonic core–satellite gold nanoparticle (AuNP) assemblies, to enable sensitive and specific detection of HCV RNA. We optimized the stoichiometry of DNA hairpins to form highly stable three-way junctions (3WJs), minimizing non-specific reactions in an enzyme-free, isothermal amplification process. The resulting dual-transduction biosensor combines colorimetric and surface-enhanced Raman spectroscopy (SERS) techniques, utilizing the Raman reporter malachite green isothiocyanate (MGITC) for signal generation. Our system targets a conserved 23-nucleotide sequence within the HCV 5′-UTR, essential for RNA replication, facilitating pan-genotypic HCV detection that complements direct-acting antiviral strategies. We evaluated the biosensor's efficacy using fluorescence spectroscopy, native PAGE, AFM, and TEM. Findings indicate that the 60 nm core AuNPs surrounded by 20 nm satellite AuNPs achieved a ten-fold increase in sensitivity over the 10 nm satellites, detecting HCV RNA concentrations as low as 1.706 fM. This sensitivity is crucial, given the extremely low viral loads present during early infection stages. Our research demonstrates the promise of enzyme-free molecular biosensors for HCV, with the potential to provide cost-efficient, rapid, point-of-care testing, although further sensitivity enhancements are needed to address the challenges of early-stage detection.
Introduction
Global burden and challenges of hepatitis C
Despite the existence of curative treatments, hepatitis C virus (HCV) remains a global public health threat. An estimated 71 million infected individuals worldwide risk developing severe liver disease and hepatocellular carcinoma.1 While the World Health Organization targeted HCV elimination by 2030,2 current diagnosis and treatment rates appear inadequate. Direct-acting antivirals can clear over 95% of chronic HCV cases,3 yet only 18% of the infected population is diagnosed.4 Studies predict 80% of high-income countries will fail the 2030 target,5 with 67% still falling short if given until 2050.6 This outlook seems even more ominous in resource-limited settings.7 Expanding point-of-care screening will prove critical to attaining elimination goals by enabling access for vulnerable groups frequently beyond the reach of conventional laboratory testing. The early detection and treatment of individuals infected with HCV is critical to reducing transmission. This situation highlights an urgent need for developing and distributing cost-effective, user-friendly, and rapid point-of-care tests.Conventional diagnostic approaches for HCV
Currently, the predominant clinical diagnostic techniques utilized for HCV detection are serological tests for identifying antibodies developed against the virus, and HCV-RNA in serum.8 As the most common first-line screening method, clinical laboratories frequently employ anti-HCV enzyme immunoassays to detect antibodies signaling exposure to the hepatitis C virus. However, these assays can produce false negatives during the initial acute phase of infection (0–10 days post-exposure) when the antibody response may not yet be substantial. This issue is especially problematic in cases of HIV co-infection.9,10 The medical community regards nucleic acid amplification tests (NAATs) as the definitive method for diagnosing active HCV infection. Popular nucleic acid detection methods encompass PCR variants, like RT-qPCR and nested PCR. However, performing NAATs remains restricted to centralized laboratories due to prohibitive costs for equipment and intensive staff training demands, creating barriers to widespread access. Isothermal amplification offers a more affordable alternative to PCR, needing minimal instrumentation amenable to point-of-care use. Enzyme-assisted isothermal methods include nucleic acid sequence amplification (NASBA),11,12 loop-mediated isothermal amplification (LAMP),13–15 rolling circle amplification (RCA),16,17 and recombinase polymerase amplification (RPA).18,19 However, dependence on enzyme catalysis introduces supply, cost, and technical hurdles that need to be addressed for decentralized adoption. Enzyme-free techniques present a promising solution, instead utilizing the inherent self-assembly of nucleic acids – such as Watson–Crick base pairing and nucleoside modifications – to construct nanostructures in a bottom-up approach.20 This avoids instability while retaining the adaptability underpinning amplification reactions.Enzyme-free, isothermal molecular biosensors using nucleic acid amplification for detection of HCV
Molecular biosensors often use enzyme-free toehold-mediated strand displacement as the main mechanism for amplifying test signal output21–23 In a typical approach, the spontaneous hybridization between two complementary sequences results in the displacement of a previously combined strand. The rate of TMSD reactions is governed by sequence composition and length of the toehold. Catalytic hairpin assembly (CHA) is one approach that utilizes enzyme-free amplification through the TMSD mechanism.24,25 Typically, a CHA reaction is executed using two complementary DNA strands prepared as meta-stable hairpin structures. Spontaneous hybridization between the two DNA strands is purposely prohibited, as the complementary domains are enclosed within the hairpin stems. When a target sequence is present, the two hairpins are configured to sequentially unfold based on their rationally designed toehold regions, leading to the rapid formation of stable DNA complexes. The forward reaction and amplification are achieved through recycling of the molecular target which catalyzes further complex formations. CHA has shown vast versatility in biosensor development through successful integration with multiple analytical signal transduction modalities. These have included fluorescence,26,27 chemiluminescence,28,29 electrochemical,30,31 SERS,32,33 and colorimetry,34,35 resulting in sensing platforms that are both highly sensitive and precise.Plasmonic core–satellite nanostructures as SERS and colorimetric biosensors
Surface-enhanced Raman spectroscopy (SERS) offers ultrasensitive detection down to single molecules by exploiting plasmonic nanostructures to amplify electromagnetic fields and Raman signals. Rapid, non-destructive SERS maintains compatibility with diverse transduction mechanisms and portable formats applicable across settings from basic research to clinical diagnostics.36 Biomolecular detection using SERS is typically achieved using nanoparticles composed of noble metals whose surface is co-functionalized with Raman-active dyes and biorecognition elements, including aptamers or oligonucleotides. Significant increases in SERS intensity and detection sensitivity can be achieved by exploiting areas of high electric field intensity (hot spots), generated between the point of tangency at the interface of two or more plasmonic nanoparticles.37 Furthermore the grouping of the nanoparticles in close proximity, causes a red shift in the surface plasmon resonance (SPR) wavelength and a visible color change in colloidal solutions.38 Different morphologies of gold nanoparticles (AuNPs) have been explored for SERS-based detection.39 Core–satellite nanoparticles have demonstrated significant potential for both SERS and colorimetric detection. This is primarily attributed to the precise control achievable over the spatial arrangement and closeness of the core and satellite nanoparticles. Such control allows for a more refined manipulation of their plasmonic properties.40 By integrating structural alterations, like varying spacer lengths, with diverse Raman-active molecules and different sizes of core–satellite nanoparticles, a wide array of configurations can be generated. These configurations are well-suited for highly sensitive and specific optical biosensing applications.In our research, we employ isothermal CHA amplification resulting in the generation of stable three-way junctions (3WJs) as a mechanism to induce the formation of rigid core–satellite assemblies. We then evaluate the optical signal transduction properties of these assemblies, which serve as a quantifiable measure for detecting hepatitis C virus RNA (HCV-RNA). Our CHA mechanism is designed to precisely target the 5′-untranslated region (5′-UTR) of the HCV-RNA sequence, a region that is preserved across approximately 90% of HCV genotypes.41 Specifically, our target is a 23-nt sequence, spanning positions 21 to 43, that is believed to play a role in RNA replication, due to it being situated directly adjacent to the internal ribosomal entry site.42–44 Moreover, the target is well-suited for our antisense oligonucleotide strategy as it lacks substantial secondary structures, which would necessitate additional processing to unfold before targeting. Our pan-genotypic approach to HCV detection also augurs well with the strategies of direct-acting antiviral treatments.45
Materials and methods
Chemicals
All DNA oligonucleotides were purchased from Integrated DNA Technologies. The sequences and their modifications is shown in Table S1.† Sodium chloride, sodium phosphate monobasic, sodium phosphate dibasic, potassium chloride, magnesium chloride, nuclease-free water, HEPES, poly-L-lysine hydrobromide (MW 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000, PLL), and Tween 20 were purchased from Sigma-Aldrich. Citrate-capped AuNPs (10 nm, 20 nm, and 60 nm, all supplied with an OD of 10) were purchased from Cytodiagnostics, nuclease-free water was used in all experiments and to prepare solutions. Acrylamide/bis-acrylamide solution (29:1), 10× Tris–borate–EDTA (TBE buffer), ammonium persulfate (APS), N,N,N′,N′-tetramethylethylenediamine (TEMED), SYBR Gold staining and Invitrogen ultra-low range DNA ladder (10–300 bp) were purchased from Thermo Fisher Scientific.
000–30000, PLL), and Tween 20 were purchased from Sigma-Aldrich. Citrate-capped AuNPs (10 nm, 20 nm, and 60 nm, all supplied with an OD of 10) were purchased from Cytodiagnostics, nuclease-free water was used in all experiments and to prepare solutions. Acrylamide/bis-acrylamide solution (29:1), 10× Tris–borate–EDTA (TBE buffer), ammonium persulfate (APS), N,N,N′,N′-tetramethylethylenediamine (TEMED), SYBR Gold staining and Invitrogen ultra-low range DNA ladder (10–300 bp) were purchased from Thermo Fisher Scientific.
Instruments used for characterization and sample measurements
The hydrodynamic dynamic radius and the ζ – potential of the AuNPs and DNA hairpin-modified AuNPs were measured using the Malvern Zetasizer at 25 °C. UV-Vis spectrophotometry was performed on a Tecan Infinite M Nano plate reader. A Nanodrop spectrophotometer was used to measure DNA concentration. SERS measurements were performed using a Wasatch Photonics benchtop fiber optic Raman spectrometer equipped with a 785 nm laser, spectra were collected using an 10 s integration time, 25 mW laser power, and optimal z distance of 11 mm across a spectral range of 260–2000 cm−1. All SERS samples measured were fixed an OD of 1. A BIORAD Mini-Protean system electrophoresis apparatus and Typhoon FLA 9500 biomolecular imager system were used for running and imaging the native PAGE gels. Imaging of the AuNPs and core–satellite assemblies was carried out using a JEOL 1200 EX transmission electron microscope (TEM). Atomic force microscopy (AFM) was performed using a Dimension Icon ScanAsyst AFM.AFM imaging and analysis
To prepare DNA oligonucleotide samples for AFM imaging, 3 μL of NiCl2 (10 mM) was deposited onto a freshly cleaved mica substrate and left to adsorb for 5 min at room temperature, before it was washed three times with a 1× TAE/Mg buffer (40 mM Tris, 20 mM acetic acid, 2 mM EDTA, 12.5 mM MgCl2, pH = 7.5).46 10 μL of the sample either consisting of 1 μM each hairpin (H), H1, H2, and H3 (negative control) or 1 μM each of H1, H2 and H3 with 100 nM HCV target (positive control) was deposited onto the mica surface and left to adsorb for 3 min. The AFM measurements across the mica surface were performed in air-tapping mode. Drive frequencies were modulated between 5 and 500 kHz. After flattening the images, a protocol adapted from Lysetska et al. was used to determine the DNA contour length within ImageJ.4715% native PAGE
For the preparation of the 15% polyacrylamide gel plates, a base solution was created by mixing equal volumes of acrylamide/bis-acrylamide (29:1) with 2× TBE. To this base solution, we added APS at a concentration of 0.01% of the base volume and TEMED at a concentration of 0.001% of the base volume. For all gel experiments the H1, H2, H3, and HCV RNA, were used at a final concentration of 1 μM. To generate the hairpin structures, 10 μM of hairpin-forming oligonucleotide sequences were heated to 95 °C for 5 minutes, then allowed to cool to RT over a period of 2 hours. We employed the same method previously established to confirm the formation of DNA complexes during the refinement of the CHA amplification protocol. During this optimization, we adjusted the component volumes to obtain the desired concentration in the final CHA reaction mixture. The reaction buffer for this final mixture was composed of 20 mM Tris (with a pH of 7.6), 2 mM EDTA, 200 mM HEPES, 400 mM NaCl, 10 mM MgCl2, and 100 mM KCl. After conducting the CHA reaction for we added glycerol to the reaction. This addition increased the glycerol concentration to 10% of the total mixture volume. Consequently, the overall volume of the reaction mixture was adjusted to 12 μL. This final volume was then ready for loading into the wells of a polyacrylamide gel for analysis.
Fluorescence spectroscopy for optimization of hairpin concentrations
To study the interactions between the hairpins and evaluate the reaction kinetics of the CHA mechanism used to form the stable 3WJs, we carried out fluorescence quenching experiments. Since fluorescein (FAM) was used as the fluorescent component in all experiments the excitation and emission wavelengths were set at 495 nm and 520 nm, respectively. The gain settings were normalized to the highest expected signal intensity. For CHA mechanism validation studies, the hairpins featured either a 5′-FAM modification, a 3′-quencher modification, or both. To determine the ideal concentrations of the hairpins required to detect the lowest concentrations of HCV-RNA target H1 was kept at a constant concentration of 500 nM. While H2 and H3 concentrations were varied from 50 to 500 nM. HCV-RNA target was tested at serial dilutions with concentrations of 10 nM, 2 nM, 400 pM, 80 pM, and 16 pM.To determine the optimal signal-to-noise ratio (SNR), we first tested varying concentrations of H2 while maintaining both FAM-H1-quencher and H3-quencher at a fixed concentration of 500 nM. To optimize the H3-quencher concentrations we carried out two sets of experiments. In the first set of experiments, FAM-H1-quencher was incubated with the aforementioned concentrations of HCV RNA. Following a 2 hour incubation, the reaction volume was adjusted from 15 μL to 30 μL with the addition of CHA reaction buffer and water. In the second set, the procedure was mirrored with the addition of H3-quencher after the initial 2 hour period, adjusting the H3-quencher concentration to fall within the 50–500 nM range. Control measurements included FAM-H1-quencher alone and in combination with either H2 or H3-quencher. These controls were essential to distinguish specific CHA-induced fluorescence from non-specific signals. In all experiments fluorescence measurements were captured every 5 minutes over a span of 4 hours, separate wells were employed for each measurement to prevent photobleaching. The total reaction volume for these measurements was maintained at 30 μL and a 1× final concentration of the CHA reaction buffer was kept consistent throughout. An integration time of 1 ms was used for all fluorescence readings to ensure consistent data capture.
Preparation of AuNPs for CHA mediated formation of core–satellite assemblies
Spherical AuNPs were functionalized with monothiolated hairpin DNA using a modified pH-assisted conjugation approach.48 In a typical modification approach, 60 nm AuNP core particles (OD 10) were mixed with monothiolated H1 (500 nM), in the presence of Tween 20 (0.01% w/v) and trisodium citrate (30 mM, pH 3.0). The mixture was allowed to incubate for 30 min, after which the hairpin-functionalized AuNPs were isolated by centrifuging them at 4000 rpm for 30 minutes at 4 °C. The AuNP cores were then resuspended in phosphate-buffered saline containing 0.01% (w/v) of Tween 20 (PBST). The 10 and 20 nm AuNP satellite particles were each coated with monothiolated H2 (50 nM) and H3 (300 nM), using the previously outlined approach. The functionalized satellite AuNPs were washed and isolated by ultracentrifugation two times at 6000 rpm for 30 min at 4 °C in PBST. Samples were stored at 4 °C in PBS buffer (0.01 M, pH 7.4) until further use. To enable SERS activity, the hairpin functionalized satellite AuNPs were modified with malachite green isothiocyanate dye (MGITC) using a ratio of approximately 1000 dye molecules per particle.49 This was achieved by agitating the mixture for 2 hours at RT. After mixing, the SERS-active, hairpin-functionalized AuNP satellites were centrifuged at a speed of 6000 rpm for 30 min at 4 °C. Subsequently, the supernatant was discarded, and the nanoparticles were reconstituted in PBS and stored at a temperature of 4 °C.Formation of the core–satellite assemblies in the presence of CHA
The 3WJ-CHA mechanism was performed using the H1-functionalized 60 nm AuNP cores and either 10 or 20 nm H2 and H3-functionalized satellites, SERS-active AuNP satellites. Before mixing the core and satellite particles they were separately resuspended in 1× CHA buffer (pH 7.2). The target HCV-RNA was then introduced to the core AuNPs and allowed to incubate for 2 h at 37 °C. Afterward, the satellite AuNPs were mixed with the core AuNPs, and the CHA reaction was allowed to proceed while the mixture was kept at 37 °C. Before mixing, UV-Vis measurements were used to confirm that the ratio of satellite-to-core nanoparticles was 100:1. For SERS measurements 20 μL of the reaction solution was transferred to a 384 well microplate for Raman interrogation. The acquired spectra were smoothed and baseline corrected in MATLAB prior to analysis.50 MGITC vibrational modes observed at 1171 cm−1, 1368 cm−1, and 1617 cm−1 were used to create standard curves for evaluating the relationship between HCV-RNA concentration and SERS signal intensity.51,52
For colorimetric quantification, the extracted CHA reaction was treated with PLL to improve the stability of the core–satellite assemblies prior to analysis.53 The core–satellite assemblies were incubated in an aqueous solution containing 8 mg per mL PLL and 10 mM MgCl2 for 1 h. The stabilized particles were centrifuged at 4000 rpm for 30 min after which they were resuspended in the same PLL/MgCl2 solution. We hypothesized that plasmonic coupling within the core–satellite assemblies would display a relationship in response to HCV-RNA concentration, resulting in a measurable red shift in the UV-visible spectrum. To quantify this shift, we compared the absorbance ratios at 600 nm and 528 nm. Control experiments were also performed, which involved taking corresponding measurements in the absence of HCV RNA to serve as a baseline for comparison.
Results and discussion
Designing the 3WJ-CHA amplification mechanism
The CHA mechanism displayed in Fig. 1 is facilitated using three rationally designed, meta-stable hairpin DNA sequences (H1, H2, and H3) in which amplification is catalyzed via the target HCV-RNA sequence which gets recycled upon the formation of a stable 3WJ. A total of 9 complementary domains were chosen to make up the nucleotide sequences of the hairpins. The mechanism can be broken down into three integral steps. The catalytic hairpin assembly (CHA) mechanism for detecting HCV-RNA begins with the introduction of the HCV-RNA target strand (TS). This target strand contains sequences complementary to regions on hairpin H1. Specifically, the toehold domains marked as 1* and 1 on TS and H1, respectively, initiate hybridization, leading to the formation of an intermediate complex, denoted as IC1. The single-stranded overhangs on IC1, labeled as domains 4–5–3*–2*, then interact with hairpin H2. This interaction utilizes the toehold-mediated strand displacement to open H2, resulting in a second intermediate complex, IC2. Subsequently, a stable three-way junction (3WJ) is established. The formation of this 3WJ is driven by the overhangs on IC2, labeled as domains 4–9–1, which now engage with hairpin H3. The interaction with H3 allows for the hybridization and subsequent displacement of the TS, freeing it to participate in further rounds of CHA amplification. This series of hybridization and displacement events is the basis for the amplification of the signal, which correlates with the presence and quantity of the HCV-RNA target.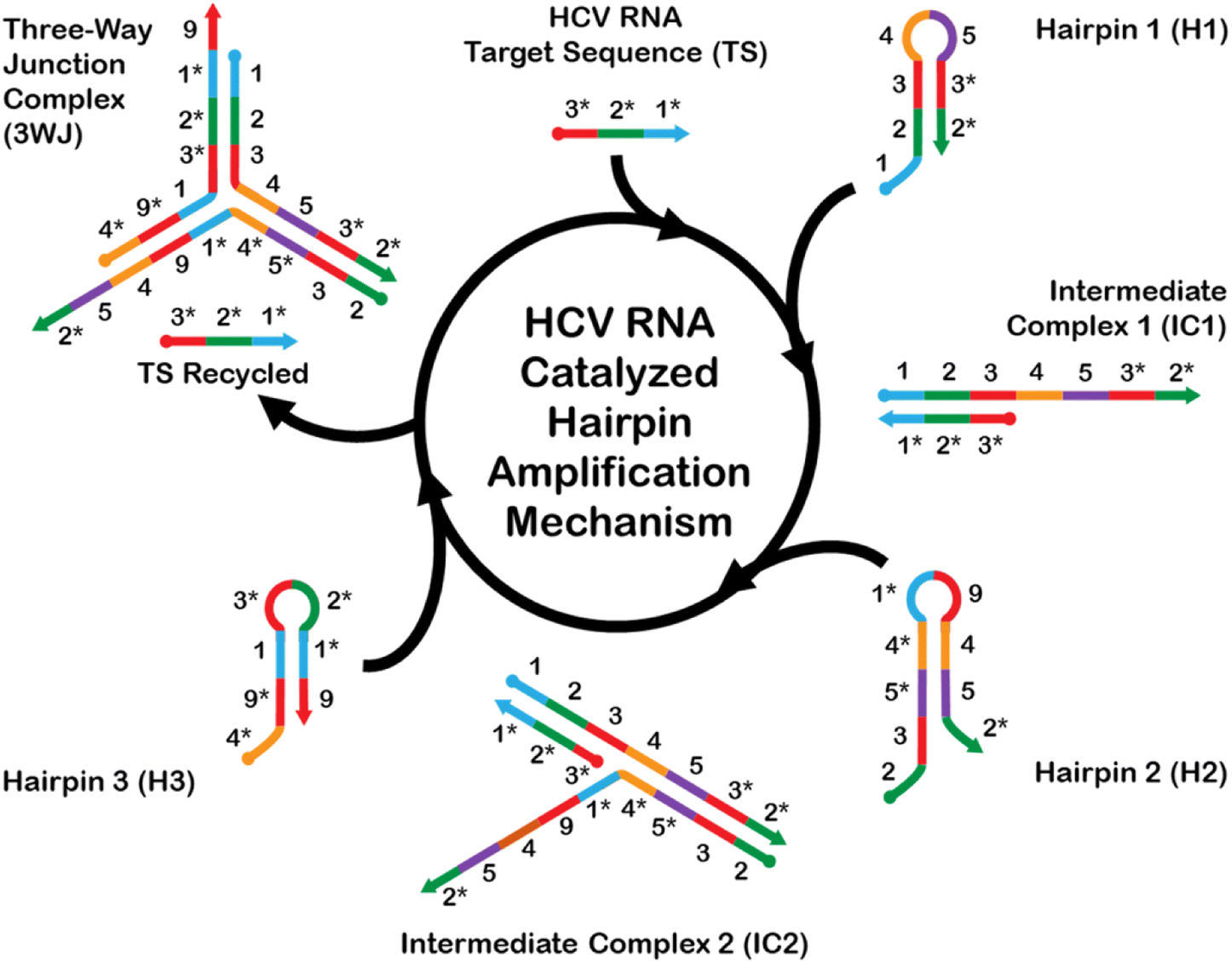 | ||
| Fig. 1 HCV-RNA catalyzed hairpin amplification (CHA) mechanism. This schematic illustrates the cyclic process of HCV-RNA target sequence (TS) recognition and amplification through a series of hairpin structures and intermediate complexes. The diagram is color-coded according to the 9 complementary domains and annotated to depict the step-by-step formation and transformation of CHA complexes. | ||
The complete list of sequences used for the CHA amplification mechanism is provided in Table S1.† H1 and H3 each contain one toehold region of 8 nts each, while sequence H2 consists of two toehold sequences, consisting of 15 nts at the 5′-end and 8 nts at the 3′-end. The longer toehold at the 5′-end of H2 was purposely chosen to facilitate hybridization to the 4 complementary domains of H1. Stem sequences were purposely designed to have the highest CG content (greater than 55% within all hairpins), and to be longer in length than any exposed toeholds. This measure was implemented to minimize the likelihood of unintended leakage and the nonspecific opening of hairpins in scenarios where the HCV-RNA target is not present. However, it is important to acknowledge that fully eliminating these risks is unfeasible. The forward propagation of the CHA amplification mechanism using the devised sequences was evaluated using NUPACK nucleic acid design and analysis software.
Validating the 3WJ-CHA amplification mechanism using native-PAGE and AFM
The image of the gel shown in Fig. 2 confirms the generation of the stable 3WJ, and the respective intermediates including the tentative identification of leakage products which inevitably form when employing CHA approaches.54–57 The dotted boxes displayed on the image are color coordinated to the hairpins, target sequence, intermediates, and 3-way junction represented above the image. Major leakage products are highlighted by a grey box and their tentative assignments are written in blue on the gel. Lanes 1, 2, and 3 represent the individual bands observed for H1, H2, and H3, respectively. Lane 4 shows the bands observed when H1 and H3 are mixed, the presence of a single band at a position representative of H1 and H3 and a very light band at higher molecular weight indicates there is little cross-hybridization. Similarly, only a small amount of cross-hybridization is observed when H1 is combined with H2 in lane 5. When H2 is combined with H3 (lane 10) we do observe slight formation of a much higher molecular weight complex, which may be due to the hybridization between adjacent regions 4, 9, and 1* on H2 to regions 4*, 9*, and 1 on H3. However, since it is located at the top of the gel we hypothesize that the complex involves more than one of each sequence. When H2 and TS are mixed (lane 6) we also observed a second band at higher molecular weight which we tentatively assigned to hybridization between the toehold domain in regions, 2 and 3 of H2 with regions 2* and 3* on the TS. No hybridization was observed upon mixing of H3 with TS (lane 7). The successful formation of the IC1 complex was confirmed in lanes 8 and 9 where H1 and TS (lane 8) and H1, H3 and TS (lane 9) were mixed. Lane 11 confirms the formation of the H2 + TS complexes in the presence of H2, H3, and TS, in the absence of H1, while in lane 12 we confirm the formation of the IC2 complex upon mixing H1, H2, and TS. Lane 13 displays a band corresponding to the 3WJ when all hairpins are mixed. However, with the introduction of the TS in lane 14, there is a noticeable intensification of the 3WJ band, along with a significant decrease in the intensity of the bands for H2 and H3. This suggests that the presence of the HCV-RNA target facilitates more efficient assembly of the 3WJ through TS recycling.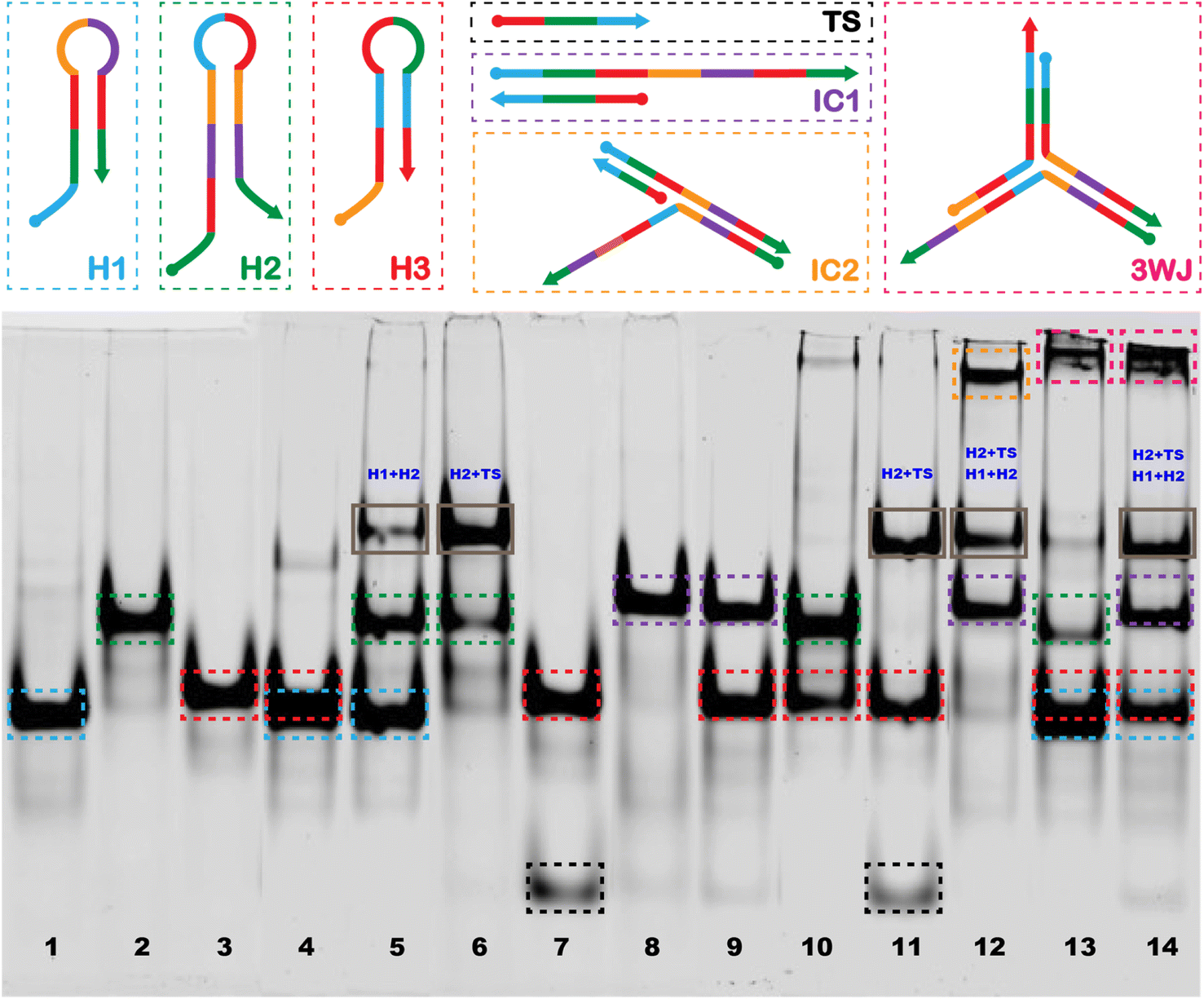 | ||
| Fig. 2 15% native PAGE image highlighting the tentative assignment of CHA products. We prepared a range of sample mixtures for analysis: lane 1 contains H1; lane 2 has H2; lane 3 features H3; lane 4 includes a mixture of H1 and H3; lane 5 combines H1 and H2; lane 6 is composed of H2 with HCV RNA; lane 7 has H3 with HCV RNA; lane 8 includes H1 with HCV RNA; lane 9 combines H1, H3, and HCV RNA; lane 10 is a mix of H2 and H3; lane 11 features H2, H3, and HCV RNA; lane 12 contains H1, H2, and HCV RNA; lane 13 includes all three hairpins H1, H2, and H3; and lane 14 has the full set: H1, H2, H3, and HCV RNA. Colored dotted boxes on the gel correspond to the tentative products formed, as depicted in the schematic. | ||
Our methodology mirrored that of Lysetska et al.,47 who employed contour length measurements obtained from AFM imaging to determine the length of DNA immobilized on a mica surface. AFM images depicted in Fig. 3, panels (A) through (D), illustrate the varying extent of complex formation, corresponding to the mixtures detailed in lanes 8 (H1 and TS), 12 (H1, H2, and TS), 13 (H1, H2, H3), and 14 (H1, H2, H3 and TS), of Fig. 2 respectively. Contour length measurements were compared with end-to-end distances of DNA hairpins of different lengths based on a method previously reported by Shlyakhtenko et al.58 Based on the estimation that the average base pair was ∼0.34 nm in length. When H1 and TS were combined (Fig. 3A and the red line in Fig. 3E) many fragments are observed to be below 10 nm, which suggests that a significant percentage of H1 is associated with TS to form the IC1 complex, which should be ∼7.82 nm in length (0.34 nm × 23). When H1, H2 and TS are combined (from Fig. 3B, and the green line in Fig. 3E); a triplex consisting of 46 nt is expected to form which should approximate a length of 15.64 nm (0.34 nm × 46). In our case, most of the fragments are under 16 nm which can be tentatively assigned to the formation of products that include populations of both H1–HCV RNA duplexes (IC1) and H1–H2–HCV RNA triplexes (IC2). When all the hairpins are combined in the absence of HCV RNA (Fig. 3C, and the black line in Fig. 3E) we expected based on the comparative gel electrophoresis data (Fig. 2, lane 5 for H1 + H2 and lane 13 for all hairpins) that H1–H2 which is observed very faintly in lane 13 and 3WJ complexes would be formed. The H1–H2 complex is 32 nt and has an estimated length of 10.88 nm (0.34 nm × 32), while the end-to-end length of the complementary sequences within the 3WJ is estimated to be between 48 and 56 nt thus has a length of between 16.34 nm (0.34 nm × 48) and 20 nm (0.34 nm × 56). Consequently, as seen in Fig. 3E (black line); a high percentage of the product population has sizes greater than 11 nm which indicates the dynamic nature of these reactions where hairpins are attempting to reach steady state by forming stable complexes. Finally, in the presence of HCV RNA in Fig. 3D and E (blue line), we see most fragments having an average size between 16 and 20 nm which is consistent with the anticipated size of the 3WJ. A magnified AFM image of the 3WJ can be seen in Fig. S1.†
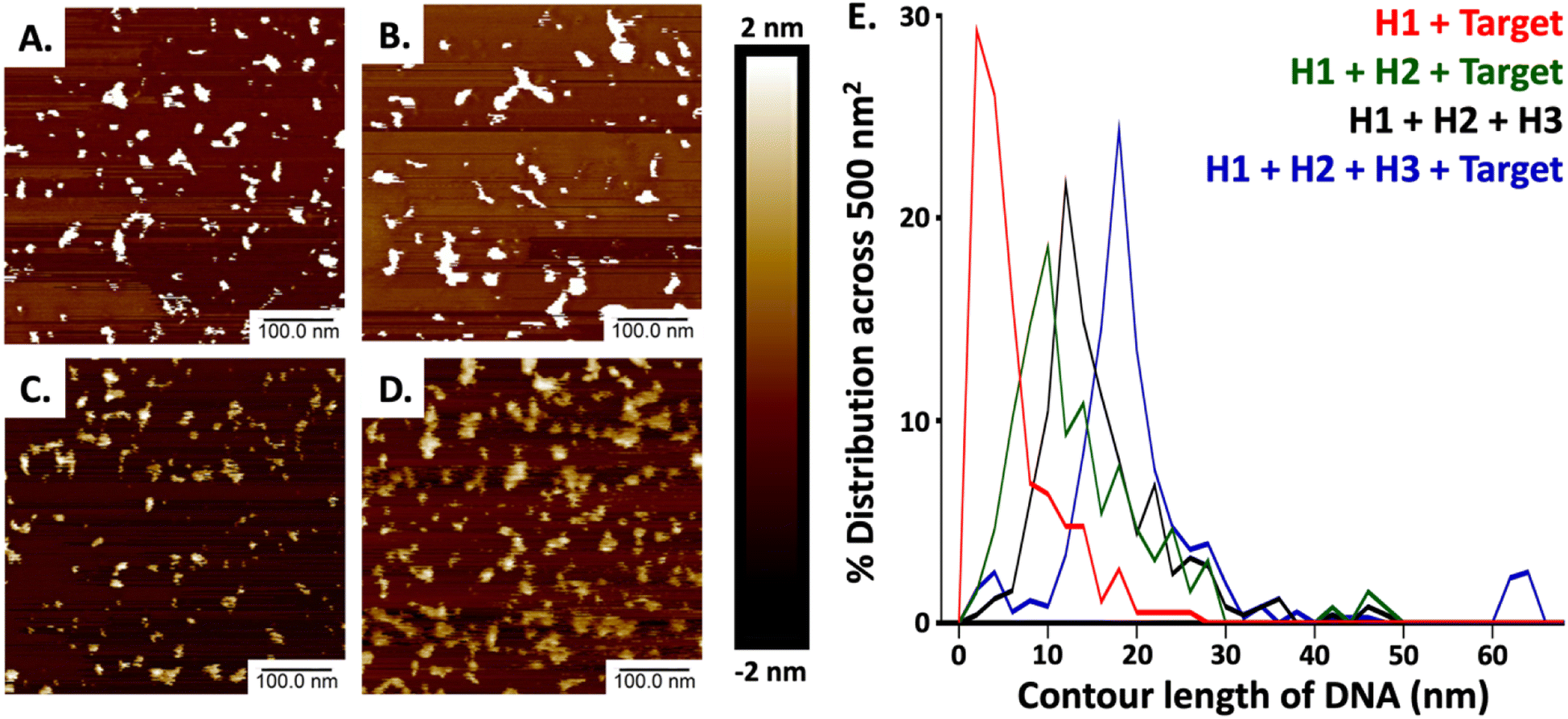 | ||
| Fig. 3 AFM analysis of the 3WJ CHA reaction. All AFM measurements were collected in height mode across an area of 500 × 500 nm2 using a Z range of 4 nm using hairpin concentrations (H1, H2, H3) of 1 μM and an HCV-RNA concentration of 10 nM. (A) AFM image for H1 incubated with HCV-RNA. (B) AFM image for H1 incubated with H2, and HCV RNA. (C) AFM image for H1 incubated with H2, and H3. (D) AFM image for H1 incubated with H2, H3, and HCV-RNA. (E) The plot of contour length distributions acquired from the AFM images. | ||
Optimization of the hairpin concentrations using fluorophore-quencher studies
The gel and AFM experiments demonstrate that the CHA mechanism effectively uses the target TS to form the desired 3WJ complex. Nevertheless, the detection of leakage in the cycling process, which H2 initiates, prompts concerns regarding the potential reduction in the sensitivity of our HCV-RNA quantification method. Consequently, we investigated how variations in H2 concentration would influence the detection of HCV-RNA using fluorophore-quencher assays. We kept the concentrations of our FAM-H1-quencher sequence and the H3 sequence constant at 500 nM and 250 nM, respectively, while adjusting H2 concentrations from 25 to 250 nM and the TS from 40 pM to 25 nM. We maintained high levels of H1 and H3 to enhance TS recognition and ensure the maximal conversion of intermediate complexes to the stable 3WJ. Our findings, illustrated in Fig. S2,† indicated that higher H2 concentrations (125 nM or 250 nM) made it challenging to differentiate between various TS concentrations. However, a concentration of 50 nM for H2 allowed for the most distinct differentiation between TS concentrations after approximately 210 minutes of reaction time, especially in distinguishing between the control (no TS present) and the lowest TS concentration of 40 pM. Fig. 4A presents the correlation between TS concentration and fluorescence intensity at H2 concentrations of 25 nM and 50 nM. While the 25 nM H2 concentration also showed clear separation from the control at the lowest TS concentration, there was negligible difference in fluorescence intensity for TS concentrations ranging from 5 nM to 200 pM. Therefore, we proceeded with 50 nM H2 for subsequent experiments.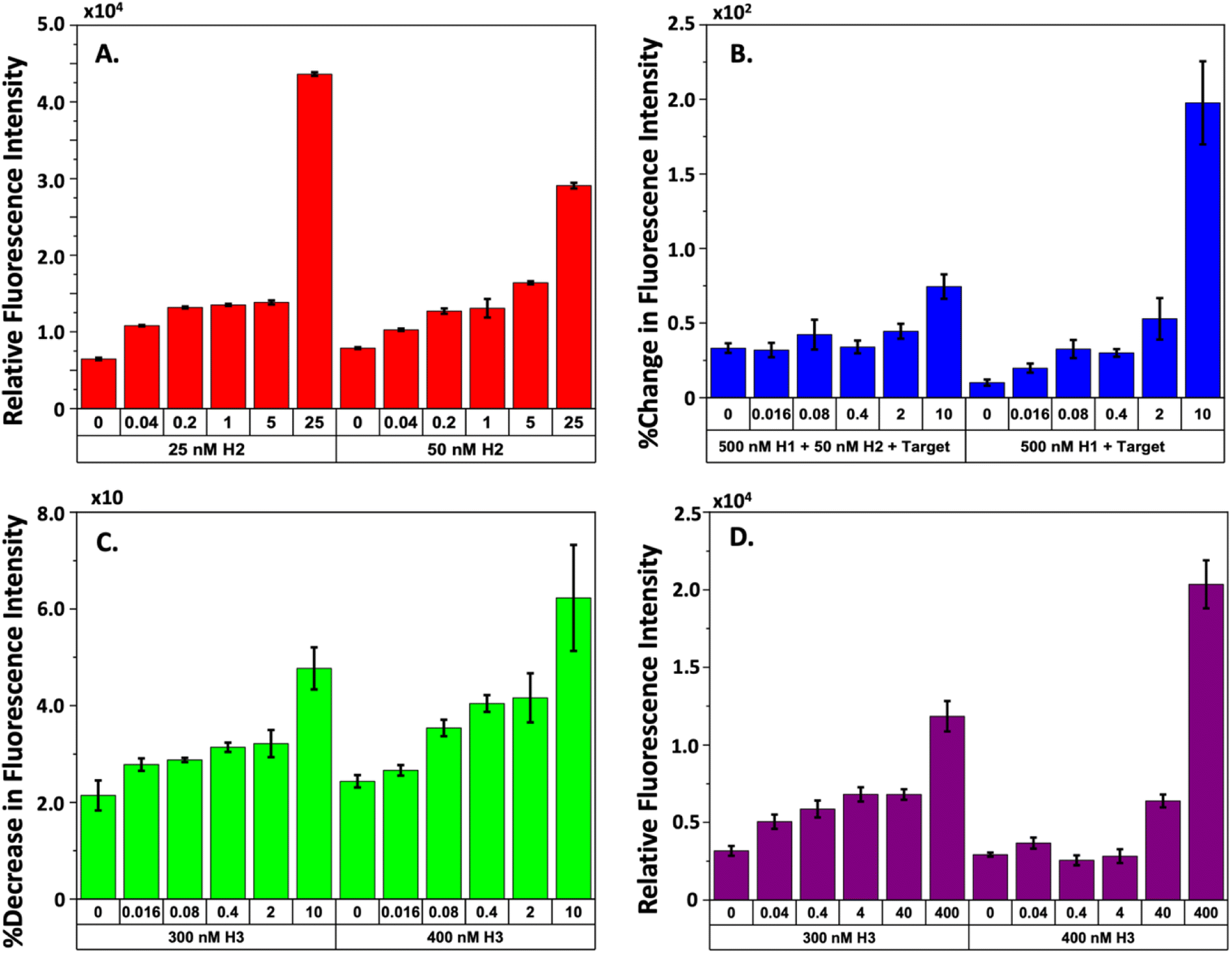 | ||
| Fig. 4 Refinement of 3WJ CHA reaction conditions. (A) The plot shows the fluorescence intensity of the 3WJ CHA reaction at two H2 concentrations (25 & 50 nM), in the presence of 500 nM FAM-H1-quencher and H3 across varying HCV RNA concentrations (0.04–25 nM, with three replicates per concentration). (B) Displays the percentage increase in fluorescence intensity for 3WJ CHA products in two scenarios: group 1 (500 nM H1, 50 nM H2, plus the target sequence), and group 2 (500 nM H1 with the target sequence). This comparison aims to assess the role of H2 in stabilizing intermediate structures at HCV RNA concentrations (16 pM–10 nM, with three replicates per concentration). (C) Depicts the percentage reduction in fluorescence intensity, signaling the completion of the 3WJ CHA reaction and the recycling of the target, under two H3-quencher concentrations (300 & 400 nM) for HCV RNA concentrations (16 pM–10 nM, with three replicates per concentration). (D) The plot illustrates the decline in fluorescence intensity of the 3WJ CHA reaction after a 6 hour incubation period using 500 nM FAM-H1-quencher and 50 nM H2, followed by the addition of H3-quencher (300 & 400 nM). Measurements were taken after 1 hour for low-range HCV RNA concentrations (0.04–400 pM, with three replicates per concentration). | ||
Observing interactions between our H2 and H3 sequences, we further explored the optimal H3 concentration to enhance the CHA mechanism and improve the sensitivity of TS detection. In these experiments, we held the concentrations of FAM-H1-quencher and H2 steady at 500 nM and 50 nM, respectively, while the concentration of H3-quencher ranged from 50 to 500 nM and the TS varied from 16 pM to 10 nM. Introducing a quencher to H3 enabled confirmation that the TS was being effectively recycled. We also posited that the successful formation of the 3WJ would be indicated by a decrease in fluorescence intensity, due to the proximity of the quencher on H3 to the FAM on H1. We established two experimental groups to test this hypothesis. Group 1 consisted of reactions with FAM-H1-quencher, H2, and TS, while group 2 included FAM-H1-quencher and TS without H2. For both groups, the CHA mechanism was allowed to run for 2 hours, after which H3-quencher was introduced, and the reaction continued for an additional 2 hours. We anticipated that a stable intermediate would form in the reactions of group 1, leading to a marked increase in fluorescence in contrast to group 2, followed by a rapid decrease in fluorescence upon the addition of H3-quencher.
Fig. S3–S5† present the raw time-series fluorescence data from our experiments that varied H3 concentrations. Fig. 4B illustrates the relationship between increasing TS concentrations and fluorescence intensity for both experimental groups. A striking difference between the two groups is the significantly higher fluorescence observed in group 2 with a 10 nM TS compared to group 1. Upon adding the H3-quencher, as depicted in Fig. 4C, we noted a fluorescence reduction of approximately 2–6% across all TS levels when 300 nM and 400 nM concentrations of H3 were introduced. This decrease in fluorescence was attributed to the formation of 3WJs because of H3 binding to the IC2 complex. At all concentrations of H3-quencher tested the addition of 300 and 400 nM led to the most marked decrease in fluorescence intensity. Notably, the decrease was more pronounced at higher TS concentrations. For TS levels ranging from 16 pM to 2 nM, the fluorescence signal correlation was almost linear, especially at H3-quencher concentrations of 300 or 400 nM. While other H3-quencher concentrations (500, 200, 100, and 50 nM, as shown in Fig. S3–S5†) also resulted in a drop in fluorescence intensity, they did not provide as clear a differentiation as the 300 nM and 400 nM concentrations. In summary, all concentrations of H3-quencher tested facilitated target recycling within the CHA process. Yet, the most effective discrimination of HCV RNA was achieved with quencher concentrations of 300 nM and 400 nM, suggesting a stoichiometric regulation within the system. Determining the optimal H3 concentration for the most sensitive detection of HCV-RNA, we varied the TS concentration from 0.04 to 400 pM in a focused study. Additionally, to align with key findings from seminal research on CHA mechanisms for viral RNA detection, we extended the incubation period of 500 nM FAM-H1-quencher with 50 nM H2 and the TS to 6 hours, as suggested by established literature.24,25 Post incubation, H3-quencher was introduced, and fluorescence intensity was recorded for an hour. The observed signal reduction for the two refined H3 concentrations is depicted in Fig. S6† and encapsulated in Fig. 4D, which shows a more linear correlation of fluorescence intensity with TS concentration at 300 nM H3, unlike the 400 nM H3 concentration, which presented negligible correlation.
These fluorophore-quencher experiments underscore the crucial influence that the stoichiometry of the hairpins has in crafting an effective CHA mechanism, especially when considering different incubation times. Fig. 4C and D indicate that fine-tuning hairpin concentrations is vital for detecting analytes over a broad dynamic range. Our research illustrates that the design and concentration of hairpins must be meticulously considered to maximize signal-to-noise ratios and mitigate leakage. These factors are paramount in the development of isothermal amplification mechanisms for biosensing applications, regardless of the method of signal transduction employed.
Colorimetric detection of HCV-RNA though 3WJ-CHA triggered AuNP core–satellite assembly
Following the successful validation and stoichiometric optimization of our three-way junction catalytic hairpin assembly (3WJ-CHA) mechanism, we shifted our focus to integrating hairpins with gold nanoparticles (AuNPs) to develop a colorimetric assay for quantifying HCV-RNA. Fig. 5A illustrates the sequential formation of a core–satellite structure through the 3WJ-CHA mechanism for the optical detection of the HCV-RNA target sequence (TS). Although not depicted in the figure, each hairpin sequence includes an 18-carbon spacer between the terminal monothiol and the sequence, enhancing hybridization efficiency by providing greater flexibility. Fig. S7A–C† suggests successful surface functionalization of the AuNPs, as evidenced by an increased hydrodynamic radius, despite minor signs of aggregation in the 10 nm and 20 nm AuNPs after labeling with 50 nM of H2. The subsequent interaction of the modified AuNPs with the TS under 3WJ-CHA conditions led to a further increase in hydrodynamic radius, as shown in Fig. S7D and E,† signaling the assembly of satellite structures. Moreover, the introduction of various TS concentrations (ranging from 100 fM to 10 nM) to the hairpin-AuNP complex resulted in an immediate color change in the colloidal mixture, as documented in Fig. S8.† To stabilize the core–satellite assemblies, poly-L-lysine (PLL) was added, ensuring the integrity of the structures.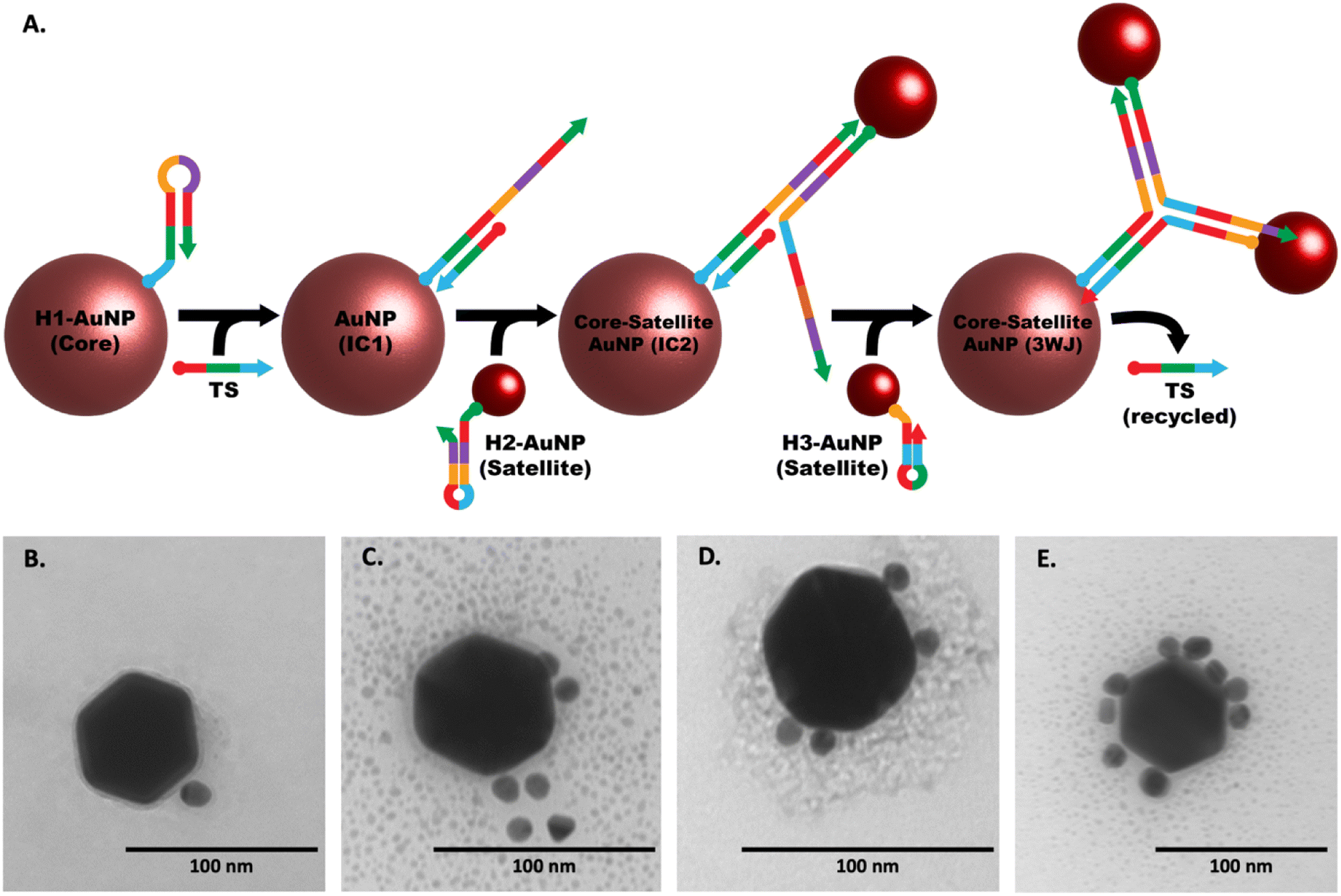 | ||
| Fig. 5 Visualization of core–satellite assembly via 3WJ-CHA. (A) The schematic details the sequential stages of core–satellite assembly formation driven by the 3WJ-CHA mechanism. (B–E) TEM images sequentially illustrate the core–satellite assemblies ranging from 60–10 nm in size, capturing the progressive stages of the reaction, with (B) showing the initial stage and (E) showing the final stage. | ||
Confirmation of the CHA-triggered core–satellite assembly formation was captured using TEM. The TEM images displayed in Fig. 5(B)–(E) were acquired at different stages of the CHA reaction and show the grouping of the smaller H2 and H3 modified-satellite AuNPs around the central core H1-modified AuNP. The speckled contrast surrounding the core–satellite structures can be attributed to the formation of the PLL protective layer, which serves as a stabilizing agent. To ascertain the performance of the core–satellite nanoparticles for colorimetric sensing, we evaluated their extinction spectra both with and without the TS present. Plots shown in Fig. 6A and B contain data representative of the 60 nm core, 10 nm satellite nano-assemblies, where (A) shows the normalized absorbance spectra collected in the presence of varying TS concentrations and (B) summarizes the observed red-shift in the absorbance spectrum by ratioing the measured intensities at 600 nm to 528 nm (λmax of combined AuNPs in the absence of TS). Fig. 6C and D are representative of the 60 nm core, and 20 nm satellite nano-assemblies, where (C) shows the measured absorbance relative to TS concentration and (D) represents the ratioed intensities. Upon target addition, the plasmonic resonance peak of the newly formed core–satellite assemblies shifts to a new maximum at 600 nm (indicated by the cyan line in Fig. 6A and C) and broadens as the target concentration increases. To examine the aggregation-induced peak shift in greater detail, we used the absorbance ratio at 528 nm and 600 nm (A600/A528) as a measure of AuNP aggregation. As depicted in Fig. 6B and D, this ratio proportionally increases with the HCV-RNA TS concentration for both sizes of core–satellites, across a broad dynamic range. However, while we do observe some concentration-dependent relationship, the colorimetric analysis only provides a semi-quantitative insight into HCV-RNA concentrations due to the overlap in error bars.
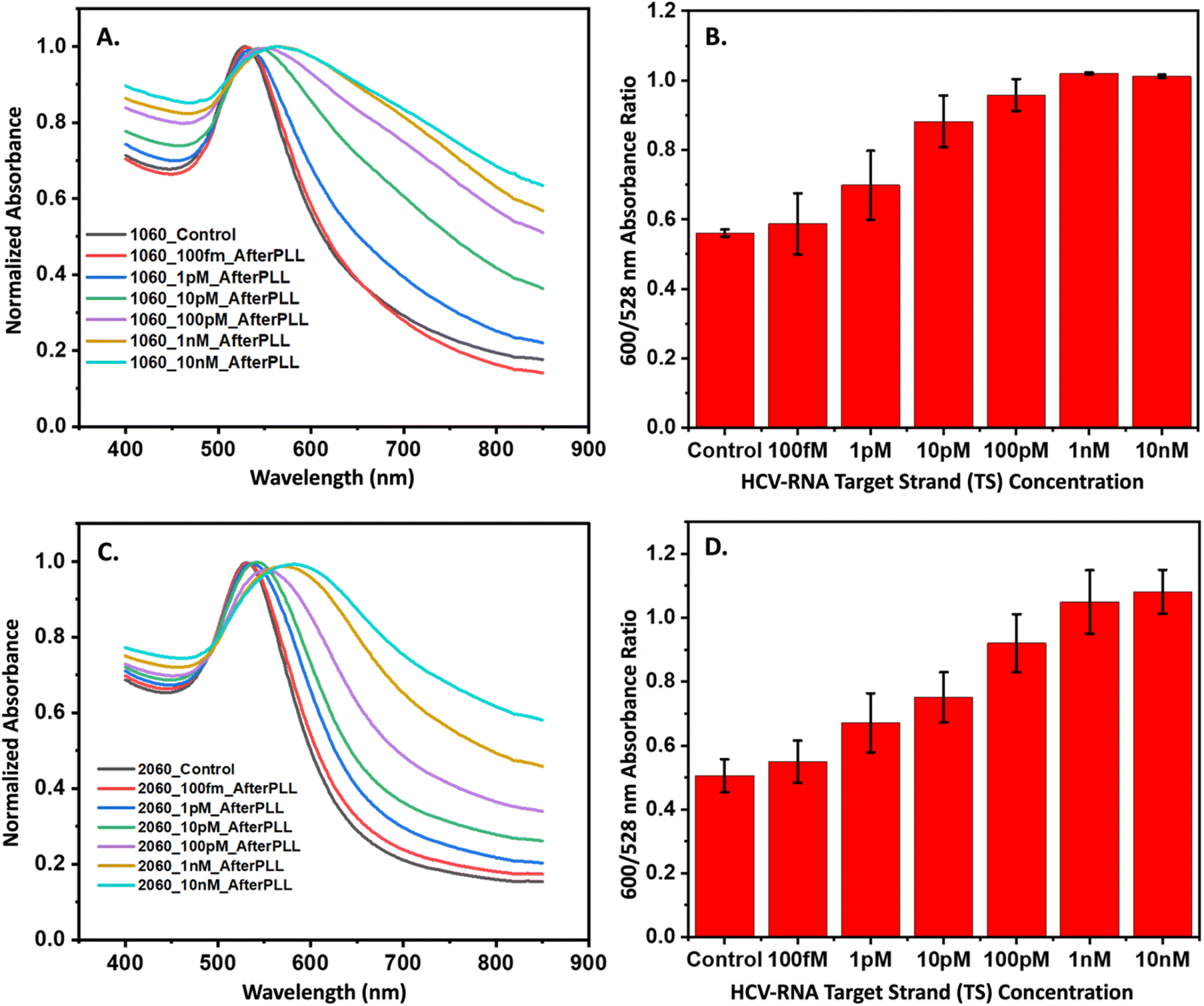 | ||
| Fig. 6 Colorimetric detection of the HCV-RNA using the 3WJ CHA to facilitate the formation of core–satellite assemblies. (A) shows the absorbance spectra for 60–10 nm core–satellite assemblies following incubation with HCV RNA (100 fM–10 nM in 10× increments). A red-shift and peak broadening can be observed when higher concentrations of HCV-RNA are present. (B) The red-shift is emphasized when the ratio of absorbance intensities at 600 nm to 528 nm are plotted for 60–10 core–satellite assemblies. (C) Shows the absorbance spectra for 60–20 nm core–satellite assemblies following incubation with HCV RNA (100 fM–10 nM in 10× HCV-RNA are present. (D) A similar red-shift is emphasized when the ratio of increments). A red-shift and peak broadening can be observed when higher concentrations of absorbance intensities at 600 nm to 528 nm are plotted for 60–20 core–satellite assemblies. | ||
Quantitative analysis of HCV-RNA concentration using SERS
As an extension of the colorimetric concentration studies of HCV-RNA we decided to modify the hairpin-functionalized satellite AuNPs with MGITC to enable the generation of a SERS signal. Since we had confirmed the CHA-catalyzed formation of the core–satellite nanostructures using DLS and TEM, and had further confirmed a semi-quantitative relationship between HCV-RNA TS concentration and colorimetric shifts we hypothesized that the SERS could be used to more sensitively quantify HCV-RNA. The results of the SERS experiments for the two combinations of core–satellite nanostructures (60–10 nm and 60–20 nm) are shown in Fig. 7. Average SERS spectra collected (n = 3) for each TS concentration are shown in Fig. 7A and B, for the 60–10 nm and 60–20 nm core–satellite combinations, respectively.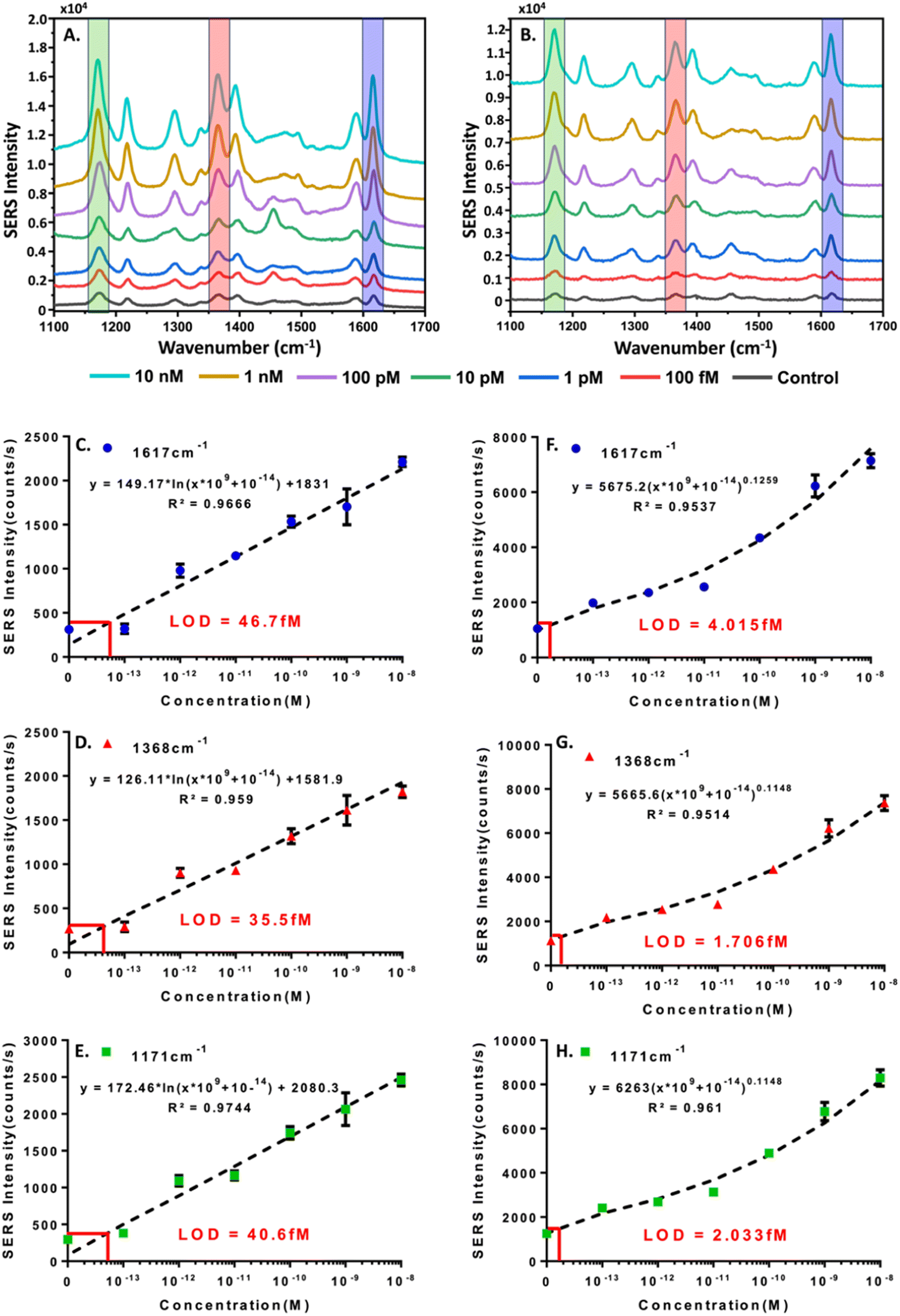 | ||
| Fig. 7 SERS quantification of the HCV-RNA using the 3WJ CHA to facilitate the formation of core–satellite assemblies. The SERS intensity of vibrational modes at 1171, 1368, and 1617 cm−1 were analyzed to generate standard curves to relating to HCV-RNA concentrations. (A and B) shows the baseline corrected SERS spectra for the different concentrations (100 fM–10 nM) of HCV-RNA for 60–10 & 60–20 nm core–satellite assemblies respectively. The spectra displayed represent the averages of n = 3 independent samples. (C–E) displays the standard curves for LOD determination based on the SERS intensities of the three vibrational modes using the 60–10 nm core–satellite assemblies. (F–H) displays the standard curves for LOD determination based on the SERS intensities of the three vibrational modes using the 60–20 nm core–satellite assemblies. | ||
Standard curves were generated for the 60–10 nm (Fig. 7C–E) and 60–20 nm (Fig. 7F–H) core–satellites using the intensities of the MGITC vibrational peaks positioned at 1617 (N–Ph ring and C–C stretching), 1368 (N–Ph ring stretching) & 1171 cm−1 (ν9 benzene in-plane mode).51,52 For the standard curves related to the 60–10 nm assembly, a logarithmic best-fit equation was derived to determine the LOD using the calculated limit of blank (LOB),59 while a power best-fit equation was used for the 60–20 nm assembly. For the 60–10 nm assembly LODs of 46.7 fM, 35.5 fM, and 40.6 fM were obtained for the peaks situated at 1617, 1368, and 1171 cm−1 respectively. For the 60–20 LODs of 4.015 fM, 1.706 fM, and 2.033 fM were obtained for the vibrational modes at 1617, 1368, and 1171 cm−1 respectively. While both core–satellite configurations are capable of detecting HCV-RNA at femtomolar levels, the 60–20 nm assemblies were approximately 10 times more sensitive. The increased sensitivity of the 60–20 nm nanostructures could be a result of several factors including optimal interparticle spacing between the 60 nm core and 20 nm satellites resulting in the creation of ‘hot spots’, an LSPR closest to the 785 nm excitation wavelength resulting in better matching and stronger plasmonic enhancement or the fact that the 20 nm satellites have a greater surface area for MGITC attachment compared to the 10 nm satellites, which can contribute to the generation of a stronger signal. Regardless of core–satellite configuration used, it can be concluded that SERS provides a much more sensitive methodology for the quantification of the HCV-RNA TS in contrast to the colorimetric approach.
Discussion and conclusion
In our research, we have pioneered a novel method that employs catalytic hairpin assembly (CHA) to construct SERS-active core–satellite assemblies anchored by a highly stable three-way junction. This method is designed for the specific detection of a 23-nucleotide segment within the 5′-UTR of HCV RNA. Our findings underscore the significance of fine-tuning the stoichiometric balance of the hairpins, which is crucial for minimizing non-specific reactions and improving the overall effectiveness of non-enzymatic, isothermal amplification techniques. The employment of a 3WJ-CHA-based strategy to initiate the formation of core–satellite structures has yielded a novel dual-transduction technique that leverages both colorimetric and SERS modalities to detect and quantify HCV-RNA target sequences. Methods like the one we have developed here hold promise as powerful, enzyme-independent, and cost-effective tools for molecular sensing. Improving the sensitivity of this method is crucial, especially because HCV RNA can be present at very low concentrations, ranging from 0.1 aM to 6.8 fM, during the early stages of infection.60 Currently, commercial gold-standard HCV NAAT assays such as the COBAS AmpliPrep/COBAS TaqMan HCV Test v2.0 (Roche Molecular Systems) and the Abbott RealTime HCV Assay (Abbott Molecular) have limits of detection (LODs) of 40–50 copies per mL (0.07–0.08 aM).61,62 Future enhancements in sensitivity could be achieved by engineering cascades of multiple DNA hairpin arrangements, potentially resulting in greater signal amplification. Furthermore, optimizing nanoparticle substrates to augment SERS-related electromagnetic enhancements presents an additional path to strengthen the detection signal.Data availability
Data to be made available upon request.Author contributions
Siddhant Jaitpal: conceptualization, methodology, investigation, validation, writing the manuscript. Ka Wai Ng: conceptualization, methodology, investigation, validation, writing the manuscript. Angela Michelle San Juan: methodology, validation, formal analysis, data curation, writing – review & editing. Cecilia Martinez: methodology and validation. Christian Phillips: methodology and validation. Sayantan Tripathy: writing – review & editing. Samuel Mabbott: conceptualization, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support from Texas A&M University (TAMU) and Texas Engineering Experiment Station (TEES) start-up funds and the National Science Foundation (NSF) (award numbers: 1648451 and 2022805).References
- C. Stasi, C. Silvestri and F. Voller, Update on Hepatitis C Epidemiology: Unaware and Untreated Infected Population Could Be the Key to Elimination, SN Compr. Clin. Med., 2020, 2(12), 2808–2815, DOI:10.1007/s42399-020-00588-3.
- R. K. Dhiman and M. Premkumar, Hepatitis C Virus Elimination by 2030: Conquering Mount Improbable, Clin. Liver Dis., 2021, 16(6), 254–261, DOI:10.1002/cld.978 . PMID: 33489098; PMCID: PMC7805299..
- D. S. Fierer and D. L. Wyles, Re-treatment of Hepatitis C Infection After Multiple Failures of Direct-Acting Antiviral Therapy, Open Forum Infect. Dis., 2020, 7(4), ofaa095, DOI:10.1093/ofid/ofaa095 . PMID: 32296728; PMCID: PMC7148001..
- A. C. Spaulding, E. J. Anderson, M. A. Khan, C. A. Taborda-Vidarte and J. A. Phillips, HIV and HCV in U.S. Prisons and Jails: The Correctional Facility as a Bellwether Over Time for the Community's Infections, AIDS Rev., 2017, 19(3), 134–147 CrossRef PubMed.
- J. D. Stanaway, A. D. Flaxman and M. Naghavi, et al., The global burden of viral hepatitis from 1990 to 2013: findings from the Global Burden of Disease Study 2013, Lancet, 2016, 388(10049), 1081–1088, DOI:10.1016/S0140-6736(16)30579-7.
- A. L. Cox, M. H. El-Sayed, J. H. Kao, J. V. Lazarus, M. Lemoine, A. S. Lok and F. Zoulim, Progress towards elimination goals for viral hepatitis, Nat. Rev. Gastroenterol. Hepatol., 2020, 17(9), 533–542, DOI:10.1038/s41575-020-0332-6 . PMID: 32704164; PMCID: PMC7376316..
- M. J. Akiyama, N. Kronfli and J. Cabezas, et al., The role of low-income and middle-income country prisons in eliminating hepatitis C, Lancet Public Health, 2022, 7(7), e578–e579, DOI:10.1016/S2468-2667(22)00119-0.
- US Preventive Services Task Force, D. K. Owens and K. W. Davidson, et al., Screening for Hepatitis C Virus Infection in Adolescents and Adults: US Preventive Services Task Force Recommendation Statement, JAMA, 2020, 323(10), 970–975, DOI:10.1001/jama.2020.1123.
- E. C. Thomson, E. Nastouli, J. Main, P. Karayiannis, J. Eliahoo, D. Muir and M. O. McClure, Delayed anti-HCV antibody response in HIV-positive men acutely infected with HCV, AIDS, 2009, 23(1), 89–93, DOI:10.1097/QAD.0b013e32831940a3 . PMID: 19050390; PMCID: PMC2646374.
- L. E. Taylor, T. Swan and K. H. Mayer, HIV coinfection with hepatitis C virus: evolving epidemiology and treatment paradigms, Clin. Infect. Dis., 2012, 55(suppl. 1), S33–S42, DOI:10.1093/cid/cis367 . PMID: 22715212; PMCID: PMC3491862..
- K. Pardee, A. A. Green and M. K. Takahashi, et al., Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components, Cell, 2016, 165(5), 1255–1266, DOI:10.1016/j.cell.2016.04.059.
- Q. Wu, C. Suo, T. Brown, T. Wang, S. A. Teichmann and A. R. Bassett, INSIGHT: a population-scale COVID-19 testing strategy combining point-of-care diagnosis with centralized high-throughput sequencing, Sci. Adv., 2021, 7(7), eabe5054, DOI:10.1126/sciadv.abe5054.
- H. M. Pham, C. Nakajima, K. Ohashi and M. Onuma, Loop-mediated isothermal amplification for rapid detection of Newcastle disease virus, J. Clin. Microbiol., 2005, 43(4), 1646–1650, DOI:10.1128/JCM.43.4.1646-1650.2005 . PMID: 15814979; PMCID: PMC1081312..
- X. Huang, G. Tang, N. Ismail and X. Wang, Developing RT-LAMP assays for rapid diagnosis of SARS-CoV-2 in saliva, EBioMedicine, 2022, 75, 103736, DOI:10.1016/j.ebiom.2021.103736.
- W. Witkowska McConnell, C. Davis, S. R. Sabir, A. Garrett, A. Bradley-Stewart, P. Jajesniak, J. Reboud, G. Xu, Z. Yang, R. Gunson, E. C. Thomson and J. M. Cooper, Paper microfluidic implementation of loop mediated isothermal amplification for early diagnosis of hepatitis C virus, Nat. Commun., 2021, 12(1), 6994, DOI:10.1038/s41467-021-27076-z . PMID: 34848705; PMCID: PMC8632961..
- B. Wang, S. J. Potter, Y. Lin, A. L. Cunningham, D. E. Dwyer, Y. Su, X. Ma, Y. Hou and N. K. Saksena, Rapid and sensitive detection of severe acute respiratory syndrome coronavirus by rolling circle amplification, J. Clin. Microbiol., 2005, 43(5), 2339–2344, DOI:10.1128/JCM.43.5.2339-2344.2005 . PMID: 15872263; PMCID: PMC1153787..
- H. Takahashi, M. Ohkawachi and K. Horio, et al., RNase H-assisted RNA-primed rolling circle amplification for targeted RNA sequence detection, Sci. Rep., 2018, 8(1), 7770, DOI:10.1038/s41598-018-26132.
- F. Shen, E. K. Davydova, W. Du, J. E. Kreutz, O. Piepenburg and R. F. Ismagilov, Digital isothermal quantification of nucleic acids via simultaneous chemical initiation of recombinase polymerase amplification reactions on SlipChip, Anal. Chem., 2011, 83(9), 3533–3540, DOI:10.1021/ac200247e . PMID: 21476587; PMCID: PMC3101872..
- C. T. Chia, A. T. Bender, L. Lillis, B. P. Sullivan, C. D. Martin, W. Burke, C. Landis, D. S. Boyle and J. D. Posner, Rapid detection of hepatitis C virus using recombinase polymerase amplification, PLoS One, 2022, 17(10), e0276582, DOI:10.1371/journal.pone.0276582 . PMID: 36282844; PMCID: PMC9595512.
- J. K. Jung, C. M. Archuleta, K. K. Alam and J. B. Lucks, Programming cell-free biosensors with DNA strand displacement circuits, Nat. Chem. Biol., 2022, 18(4), 385–393, DOI:10.1038/s41589-021-00962-9 . PMID: 35177837; PMCID: PMC8964419..
- A. A. Green, P. A. Silver, J. J. Collins and P. Yin, Toehold switches: de novo-designed regulators of gene expression, Cell, 2014, 159(4), 925–939, DOI:10.1016/j.cell.2014.10.002.
- F. C. Simmel, B. Yurke and H. R. Singh, Principles and Applications of Nucleic Acid Strand Displacement Reactions, Chem. Rev., 2019, 119(10), 6326–6369, DOI:10.1021/acs.chemrev.8b00580 . PMID: 30714375..
- S. Li, L. Zhu, S. Lin and W. Xu, Toehold-mediated biosensors: types, mechanisms and biosensing strategies, Biosens. Bioelectron., 2023, 220, 114922, DOI:10.1016/j.bios.2022.114922 . PMID: 36423392..
- P. Yin, H. M. Choi, C. R. Calvert and N. A. Pierce, Programming biomolecular self-assembly pathways, Nature, 2008, 451(7176), 318–322, DOI:10.1038/nature06451.
- B. Li, A. D. Ellington and X. Chen, Rational, modular adaptation of enzyme-free DNA circuits to multiple detection methods, Nucleic Acids Res., 2011, 39(16), e110, DOI:10.1093/nar/gkr504 . PMID: 21693555; PMCID: PMC3167626..
- F. Jin and D. Xu, A fluorescent microarray platform based on catalytic hairpin assembly for microRNAs detection, Anal. Chim. Acta, 2021, 1173, 338666, DOI:10.1016/j.aca.2021.338666 . PMID: 34172148..
- J. Zhou, Q. Lin, Z. Huang, H. Xiong, B. Yang, H. Chen and J. Kong, Aptamer-Initiated Catalytic Hairpin Assembly Fluorescence Assay for Universal, Sensitive Exosome Detection, Anal. Chem., 2022, 94(15), 5723–5728, DOI:10.1021/acs.analchem.2c00231 . PMID: 35377617..
- J. Y. Do, J. Y. Jeong and C. A. Hong, Catalytic hairpin DNA assembly-based chemiluminescent assay for the detection of short SARS-CoV-2 target cDNA, Talanta, 2021, 233, 122505, DOI:10.1016/j.talanta.2021.122505 . PMID: 34215120; PMCID: PMC8124025..
- J. Chen, H. Qiu, M. Zhang, T. Gu, S. Shao, Y. Huang and S. Zhao, Hairpin assembly-triggered cyclic activation of a DNA machine for label-free and ultrasensitive chemiluminescence detection of DNA, Biosens. Bioelectron., 2015, 68, 550–555, DOI:10.1016/j.bios.2015.01.054 . PMID: 25638797..
- Y. Zhang, Y. Yan, W. Chen, W. Cheng, S. Li, X. Ding, D. Li, H. Wang, H. Ju and S. Ding, A simple electrochemical biosensor for highly sensitive and specific detection of microRNA based on mismatched catalytic hairpin assembly, Biosens. Bioelectron., 2015, 68, 343–349, DOI:10.1016/j.bios.2015.01.026 . PMID: 25603399..
- Y. Zhang, X. Zhang, B. Situ, Y. Wu, S. Luo, L. Zheng and Y. Qiu, Rapid electrochemical biosensor for sensitive profiling of exosomal microRNA based on multifunctional DNA tetrahedron assisted catalytic hairpin assembly, Biosens. Bioelectron., 2021, 183, 113205, DOI:10.1016/j.bios.2021.113205 . Erratum in: Biosens. Bioelectron., 2022, 205, 113989. PMID: 33813210..
- J. Chen, Y. Wu, C. Fu, H. Cao, X. Tan, W. Shi and Z. Wu, Ratiometric SERS biosensor for sensitive and reproducible detection of microRNA based on mismatched catalytic hairpin assembly, Biosens. Bioelectron., 2019, 143, 111619, DOI:10.1016/j.bios.2019.111619 . PMID: 31454694..
- C. Liu, C. Chen, S. Li, H. Dong, W. Dai, T. Xu, Y. Liu, F. Yang and X. Zhang, Target-Triggered Catalytic Hairpin Assembly-Induced Core-Satellite Nanostructures for High-Sensitive "Off-to-On" SERS Detection of Intracellular MicroRNA, Anal. Chem., 2018, 90(17), 10591–10599, DOI:10.1021/acs.analchem.8b02819 . PMID: 30058321..
- C. Ma, W. Wang, Z. Li, L. Cao and Q. Wang, Simple colorimetric DNA detection based on hairpin assembly reaction and target-catalytic circuits for signal amplification, Anal. Biochem., 2012, 429(2), 99–102, DOI:10.1016/j.ab.2012.07.009 . PMID: 22800654..
- H. He, J. Dai, Z. Duan, Y. Meng, C. Zhou, Y. Long, B. Zheng, J. Du, Y. Guo and D. Xiao, Target-catalyzed autonomous assembly of dendrimer-like DNA nanostructures for enzyme-free and signal amplified colorimetric nucleic acids detection, Biosens. Bioelectron., 2016, 86, 985–989, DOI:10.1016/j.bios.2016.07.045 . PMID: 27498325..
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J. F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J. M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L. L. Tay, K. G. Thomas, Z. Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, Present and Future of Surface-Enhanced Raman Scattering, ACS Nano, 2020, 14(1), 28–117, DOI:10.1021/acsnano.9b04224 . PMID: 31478375; PMCID: PMC6990571..
- M. Culha, D. Stokes, L. R. Allain and T. Vo-Dinh, Surface-enhanced Raman scattering substrate based on a self-assembled monolayer for use in gene diagnostics, Anal. Chem., 2003, 75(22), 6196–6201, DOI:10.1021/ac0346003 . PMID: 14616001..
- C. M. Niemeyer, Nanoparticles, Proteins, and Nucleic Acids: Biotechnology Meets Materials Science, Angew Chem. Int. Ed. Engl., 2001, 40(22), 4128–4158, DOI:10.1002/1521-3773(20011119)40:22<4128::AID-ANIE4128>3.0.CO;2-S . PMID: 29712109..
- F. Tian, F. Bonnier, A. Casey, A. E. Shanahan and H. J. Byrne, Surface enhanced Raman scattering with gold nanoparticles: effect of particle shape, Anal. Methods, 2014, 6(22), 9116–9123, 10.1039/c4ay02112f.
- Q. Gu, J. Zhu, G. J. Weng, J. J. Li and J. W. Zhao, Core-satellite nanostructures and their biomedical applications, Mikrochim. Acta, 2022, 189(12), 470, DOI:10.1007/s00604-022-05559-0.
- J. Bukh, R. H. Purcell and R. H. Miller, Sequence analysis of the 5' noncoding region of hepatitis C virus, Proc. Natl. Acad. Sci. U. S. A., 1992, 89(11), 4942–4946, DOI:10.1073/pnas.89.11.4942.
- S. T. Shi and M. M. C. Lai, HCV 5′ and 3′UTR: When Translation Meets Replication, in Hepatitis C Viruses: Genomes and Molecular Biology, ed. S. L. Tan, Horizon Bioscience, Norfolk (UK), 2006, ch. 2. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1624/\ Search PubMed.
- D. M. Mauger, M. Golden, D. Yamane, S. Williford, S. M. Lemon, D. P. Martin and K. M. Weeks, Functionally conserved architecture of hepatitis C virus RNA genomes, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(12), 3692–3697, DOI:10.1073/pnas.1416266112 . PMID: 25775547; PMCID: PMC4378395..
- A. J. Lyons, J. R. Lytle, J. Gomez and H. D. Robertson, Hepatitis C virus internal ribosome entry site RNA contains a tertiary structural element in a functional domain of stem-loop II, Nucleic Acids Res., 2001, 29(12), 2535–2541, DOI:10.1093/nar/29.12.2535 . PMID: 11410661; PMCID: PMC55737..
- M. A. Martinez and S. Franco, Therapy Implications of Hepatitis C Virus Genetic Diversity, Viruses, 2020, 13(1), 41, DOI:10.3390/v13010041.
- L. S. Shlyakhtenko, V. N. Potaman, R. R. Sinden, A. A. Gall and Y. L. Lyubchenko, Structure and dynamics of three-way DNA junctions: atomic force microscopy studies, Nucleic Acids Res., 2000, 28(18), 3472–3477, DOI:10.1093/nar/28.18.3472.
- M. Lysetska, A. Knoll, D. Boehringer, T. Hey, G. Krauss and G. Krausch, UV light-damaged DNA and its interaction with human replication protein A: an atomic force microscopy study, Nucleic Acids Res., 2002, 30(12), 2686–2691, DOI:10.1093/nar/gkf378.
- X. Zhang, M. R. Servos and J. Liu, Instantaneous and quantitative functionalization of gold nanoparticles with thiolated DNA using a pH-assisted and surfactant-free route, J. Am. Chem. Soc., 2012, 134(17), 7266–7269, DOI:10.1021/ja3014055.
- M. Schechinger, H. Marks, S. Mabbott, M. Choudhury and G. Cote, A SERS approach for rapid detection of microRNA-17 in the picomolar range, Analyst, 2019, 144(13), 4033–4044, 10.1039/c9an00653b.
- K. Gracie, E. Correa, S. Mabbott, J. A. Dougan, D. Graham, R. Goodacre and K. Faulds, Simultaneous Detection and Quantification of Three Bacterial Meningitis Pathogens by SERS, Chem. Sci., 2014, 5, 1030–1040, 10.1039/C3SC52875H.
- H. A. Kashmery, D. G. Thompson, R. Dondi, S. Mabbott, D. Graham, A. W. Clark and G. A. Burley, SERS enhancement of silver nanoparticles prepared by a template-directed triazole ligand strategy, Chem. Commun., 2015, 51(65), 13028–13031, 10.1039/C5CC02883C.
- S. Mabbott, S. C. Fernandes, M. Schechinger, G. L. Cote, K. Faulds, C. R. Mace and D. Graham, Detection of cardiovascular disease associated miR-29a using paper-based microfluidics and surface enhanced Raman scattering, Analyst, 2020, 145, 983–991, 10.1039/C9AN01748H.
- L. Y. Chou, F. Song and W. C. Chan, Engineering the Structure and Properties of DNA-Nanoparticle Superstructures Using Polyvalent Counterions, J. Am. Chem. Soc., 2016, 138(13), 4565–4572, DOI:10.1021/jacs.6b00751.
- N. Srinivas, J. Parkin, G. Seelig, E. Winfree and D. Soloveichik, Enzyme-free nucleic acid dynamical systems, Science, 2017, 358(6369), eaal2052, DOI:10.1126/science.aal2052.
- N. Srinivas, T. E. Ouldridge and P. Sulc, et al., On the biophysics and kinetics of toehold-mediated DNA strand displacement, Nucleic Acids Res., 2013, 41(22), 10641–10658, DOI:10.1093/nar/gkt801.
- X. Chen, N. Briggs, J. R. McLain and A. D. Ellington, Stacking nonenzymatic circuits for high signal gain, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5386–5391, DOI:10.1073/pnas.1222807110.
- Y. S. Jiang, S. Bhadra, B. Li and A. D. Ellington, Mismatches improve the performance of strand-displacement nucleic acid circuits, Angew Chem. Int. Ed. Engl., 2014, 53(7), 1845–1848, DOI:10.1002/anie.201307418.
- L. S. Shlyakhtenko, V. N. Potaman, R. R. Sinden, A. A. Gall and Y. L. Lyubchenko, Structure and dynamics of three-way DNA junctions: atomic force microscopy studies, Nucleic Acids Res., 2000, 28(18), 3472–3477, DOI:10.1093/nar/28.18.3472 . PMID: 10982865; PMCID: PMC110733..
- D. A. Armbruster and T. Pry, Limit of blank, limit of detection and limit of quantitation, Clin. Biochem. Rev., 2008, 29(suppl. 1), S49–S52 Search PubMed . PMID: 18852857; PMCID: PMC2556583..
- B. Hajarizadeh, J. Grebely, T. Applegate, G. V. Matthews, J. Amin, K. Petoumenos, M. Hellard, W. Rawlinson, A. Lloyd, J. Kaldor, G. J. Dore and ATAHC Study Group, Dynamics of HCV RNA levels during acute hepatitis C virus infection, J. Med. Virol., 2014, 86(10), 1722–1729, DOI:10.1002/jmv.24010 . PMID: 25042465; PMCID: PMC4276420..
- Roche Molecular Systems, COBAS AmpliPrep/COBAS TaqMan HCV Test, https://www.accessdata.fda.gov/cdrh_docs/pdf15/P150015c.pdf, accessed April 2024 Search PubMed.
- Abbott Molecular, Abbott RealTime HCV Assay, https://www.accessdata.fda.gov/cdrh_docs/pdf10/P100017b.pdf, accessed April 2024 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00891j |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |