
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow synthesis and post-synthetic conversion of single-crystalline covalent organic frameworks†
Michael
Traxler
 and
William R.
Dichtel
*
and
William R.
Dichtel
*
Department of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. E-mail: wdichtel@northwestern.edu
First published on 9th April 2024
Abstract
The synthesis and scale-up of high quality covalent organic frameworks (COFs) remains a challenge due to slow kinetics of the reversible bond formation and the need for precise control of reaction conditions. Here we report the rapid synthesis of faceted single crystals of two-dimensional (2D) COFs using a continuous flow reaction process. Two imine linked materials were polymerized to the hexagonal CF-TAPB-DMPDA and the rhombic CF-TAPPy-PDA COF, respectively. The reaction conditions were optimized to produce single crystals of micrometer size, which notably formed when the reaction was cooling to room temperature. This indicated a growth mechanism consistent with the fusion of smaller COF particles. The optimized conditions were used to demonstrate the scalability of the continuous approach by synthesizing high quality, faceted COFs at a rate of more than 1 g h−1. The materials showed high crystallinity and porosity with surface areas exceeding 2000 m2 g−1. Additionally, the versatility of the continuous flow reaction approach was demonstrated on a post-synthetic single crystal to single crystal demethylation of CF-TAPB-DMPDA to afford a hydroxyl functionalized COF CF-TAPB-DHPDA. Throughout the modification process, the material maintained its hexagonal morphology, crystallinity, and porosity. This work reports the first example of synthesizing and post-synthetically modifying imine linked COF single crystals in continuous flow and will prove a first step towards scaling high quality COFs to industrial levels.
Introduction
Covalent organic frameworks (COFs) have attracted increasing interest since the first report in the mid-2000s, owing to their unique characteristics compared to traditional macromolecular architectures.1–3 With their high porosity, absorption in the visible light range or crystallinity, the organic framework materials have been introduced to wide set of applications ranging from gas storage4 and separation5,6 to photocatalytic applications.7–9 Alongside the exploration of the application space of COF materials, a broader range of synthetic conditions and fundamental understanding of 2D polymerization has emerged over the last two decades.10–15 Solvothermal batch synthesis still remains the most broadly applied synthetic strategy.16–18 However, major drawbacks of this method include extended reaction times, batch dependent quality, low scalability and limited control in nucleation and growth of the framework material.19In contrast to batch synthesis, continuous flow approaches offer enhanced efficiency, precise control over reaction parameters, and the ability to achieve consistent and reproducible outcomes in materials quality over an extended timeframe.20,21 Multiple successful attempts to synthesize COFs in continuous flow have been reported in recent years, dating back to 2016, when COF-LZU1 was synthesized by Zhao and coworkers.22 Thereafter, continuous flow approaches have been mainly used in the preparation of COF films23–25 and fibers.26 However, the latest development in the continuous flow synthesis of COFs is fueled by the demand to increase their production beyond the small academic laboratory scale. By ensuring a consistent and ample production a scalable synthesis opens a pathway to use these framework materials in their plethora of possible applications.27 A high-pressure homogenization approach for the synthesis of various crystalline porous polymeric materials has shown the potential in scalability in COF manufacturing.28 Nevertheless, the reported materials have low material quality, small particle sizes, and undefined morphologies. These features result in low performance in a suite of applications, where higher crystallinity of the materials has proven superior to polycrystalline alternatives.5,29,30 Recently, high quality 3D-COF fibers with micrometer particle sizes were realized in a continuous microfluidic synthesis approach with reaction times of just a few seconds showing promise of this strategy in the scaling of COF preparation.31
A viable strategy to obtain high quality COFs is to perform reactions under solvothermal reactions that allow the formation of stable colloids of the framework material.32,33 Using this strategy micrometer sized COF particles have been obtained in significantly reduced reaction times moving from multi-day synthesis to several minutes.5,34–36 The benzoic acid catalyzed polymerization relies on the use of benzonitrile as a solvent and modulation with aniline, which enables efficient error correction necessary to form single crystalline materials.
Here, we further advance this reaction concept and develop an efficient and continuous synthesis of single-crystalline COFs. The flow approach is demonstrated on the two different COF systems, denoted CF-TAPB-DMPDA and CF-TAPPy-PDA, which crystallize in hexagonal and square planar topologies, respectively (Fig. 1a and c). Additionally, the continuous flow synthetic approach was used in the post-synthetic modification of COFs, which was demonstrated previously by the reduction of nitro side groups to amines in a keto-enamine COF.37 Here, we demonstrate the continuous single crystal to single crystal demethylation of CF-TAPB-DMPDA to obtain CF-TAPB-DHPDA.38,39 The strong Lewis acid used for the reaction can be safely used in the PSM, since only low quantities of the reagent are in the reactive zone in the continuous flow approach. Using this strategy, a TAPB-DHPDA COF with previously unreported crystallite size was obtained. These findings indicate the promise of flow conditions to enable the continuous manufacture of high quality COFs in potentially large quantities.
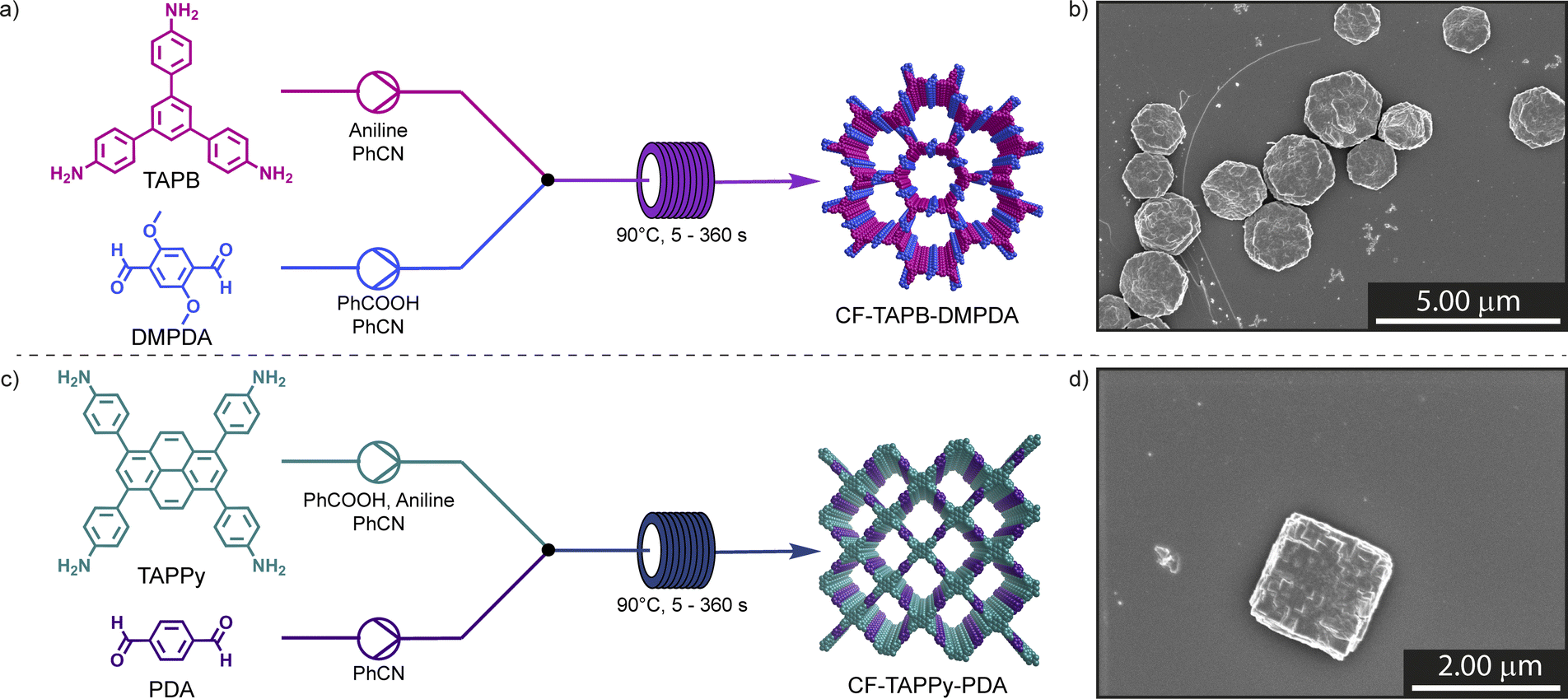 | ||
| Fig. 1 Continuous flow synthesis of single-crystalline CF-TAPB-DMPDA (a) and CF-TAPPy-PDA (c) covalent organic frameworks. Scanning electron microscopy (SEM) images of the hexagonal CF-TAPB-DMPDA COF (b) and the rhombic CF-TAPPy-PDA COF (d). | ||
Results and discussion
The continuous flow synthesis was investigated on an in-lab flow setup, where precursor solutions, containing the aldehyde and amine linkers necessary to form the imine-based framework, are combined in a Y-mixing unit (Scheme S1 and Fig. S1†). This setup is affordable, accessible, and can be implemented easily in most research laboratories.40 For transferring the batch reaction conditions towards a continuous flow approach, several design criteria, such as temperature, catalyst solubility or material quality, were considered. Firstly, the precursor solutions containing linkers, catalyst and modulator must remain in solution throughout the flow reaction to prevent clogging of the reactor. To accommodate for the high catalyst loadings necessary to efficiently form COF single crystals the two precursor solutions are heated prior to the reaction. Secondly, the solutions are combined in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixing ratio, where components with low solubility are dissolved in the high flow portion. Furthermore, the temperature was set to 90 °C, slightly below the temperature limits of the tubing and connectors, and the concentration was set to 8 mM with respect to the polyfunctional amine monomer. To prevent an imine pre-equilibrium all amine compounds were delivered in the opposite solution than the aldehyde linker.41 Finally, the success of the reaction was determined in part by scanning electron microscopy (SEM), where aliquots of the collected sample were held at room temperature for 60 min, diluted with methanol, and imaged after drop-casting onto a silicon-wafer. The flow setup was equipped with a 0.5 mL reactor defined by a length of 10′′ of a tubing with an inner diameter of 1/16′′. The hexagonal framework (CF-TAPB-DMPDA) was synthesized by combing a reaction mixture containing the aldehyde linker 2,5-dimethoxyterephtalaldehyde (DMPDA) and benzoic acid in benzonitrile with a solution 1,3,5-tris(4-aminophenyl)benzene (TAPB) and aniline as modulator of in the same solvent (Fig. 1a, Section C1 in the ESI†).
:1 mixing ratio, where components with low solubility are dissolved in the high flow portion. Furthermore, the temperature was set to 90 °C, slightly below the temperature limits of the tubing and connectors, and the concentration was set to 8 mM with respect to the polyfunctional amine monomer. To prevent an imine pre-equilibrium all amine compounds were delivered in the opposite solution than the aldehyde linker.41 Finally, the success of the reaction was determined in part by scanning electron microscopy (SEM), where aliquots of the collected sample were held at room temperature for 60 min, diluted with methanol, and imaged after drop-casting onto a silicon-wafer. The flow setup was equipped with a 0.5 mL reactor defined by a length of 10′′ of a tubing with an inner diameter of 1/16′′. The hexagonal framework (CF-TAPB-DMPDA) was synthesized by combing a reaction mixture containing the aldehyde linker 2,5-dimethoxyterephtalaldehyde (DMPDA) and benzoic acid in benzonitrile with a solution 1,3,5-tris(4-aminophenyl)benzene (TAPB) and aniline as modulator of in the same solvent (Fig. 1a, Section C1 in the ESI†).
Initial screening was performed by changing the benzoic acid catalyst (19.2 equiv. & 38.4 equiv. with respect to aldehyde functional groups) and aniline modulator (0.8 equiv. & 1.6 equiv.) aliquots in a reaction with a residence time of 1 min. All tested reaction conditions yielded stable dark red colloids solutions after cooling to room temperature for 60 min (Fig. S4†). The crystallinity of the material was confirmed by capillary powder X-ray diffractometry (PXRD) (Fig. S5†). However, for the evaluation of the COF particles in the micrometer range we used SEM imaging, since the Scherrer equation, conventionally used for determining particle sizes in COFs, is not applicable within the obtained size regime.42 SEM imaging revealed that the conditions with the 1.6 equiv. of aniline and 38.4 equiv. of benzoic acid loadings resulted in uniform, hexagonal single-crystalline COF with a mean particle size of 1.4 ± 0.4 μm (Fig. 2a and S11†). The importance of the catalyst and modulator loadings was demonstrated by the smaller particle size of 0.8 ± 0.2 μm, when either the acid or aniline loading is reduced. When both variables are changed simultaneously even smaller particles are obtained. Moreover, the impact on the reaction time was investigated, by changing the overall flow rate and thus the residence time in the reactor tubing ranging from 5 s to 6 min (360 mL h−1 to 5 mL h−1). Such short reaction times are hardly feasible using a batch synthesis strategy, due to the inherent limitations of mass and heat flow. Dark red colloids of CF-TAPB-DMPDA were formed in all cases and the same trend in catalyst and modulator concentrations holds true over the tested residence timeframe (Fig. S6–S10†). The average particle size of the hexagonal COF single crystals is with 0.9 ± 0.2 μm slightly smaller at a residence time of 5 s. At longer reaction times the average particle size stays nearly the same, however, the dispersity increased.
 | ||
| Fig. 2 SEM images of Screening of CF-TAPB-DMPDA (a) and CF-TAPPy-PDA (b) COF reaction condition screenings with 60 s residence time. SEM images of CF-TAPB-DMPDA COF particle formation at time points after leaving the reactor tubing to ambient conditions (c). | ||
Intrigued by the observation that the COF reaction mixture turns from a clear solution to a colloidal suspension over the course of 60 min, we probed the particle formation mechanism of the CF-TAPB-DMPDA COF to determine whether the growth is occurring within the tubing, or upon cooling to room temperature. Therefore, 0.5 mL of COF colloid solution was collected in a glass vial after a residence time of 5 s at 90 °C in the continuous flow tubing. This solution was thereafter kept at 25 °C and over the course of the colloidal formation process aliquots of this solution were quenched in methanol and imaged using SEM (Fig. 2c, S12 and S13†). Initially, the material consists of small undefined particles with a size of ∼100 nm up to 5 min after leaving the continuous flow tubing. At around 7.5 min these small particles start to fuse together to form bigger hexagonally shaped particles in the micrometer range. Over the course of 30 min the transformation of the small particulates into the final COF material continues, resulting in faceted hexagonal particles. On the one hand, these findings highlight the importance of the cooling step at ambient condition to form high quality single-crystalline COF materials. On the other hand, the delayed crystallization outside of the tubing holds potential for solution processing of the small particulate solution before it crystallizes into high quality COF material.
Cooling the colloids at different temperatures had a significant impact on the formation of the single-crystalline COFs. When CF-TAPB-DMPDA was transferred to the freezer at −20 °C, polycrystalline framework material was obtained. We attribute these colloidal particles to cooling below the freezing point of the benzonitrile solvent, which impedes their fusion into single-crystalline crystals. Stepwise cooling to 60 °C for 30 minutes followed to room temperature for another 30 minutes results in less defined spherical COF material, while direct cooling in an ice bath affords hexagons with a similar quality as obtained by ambient cooling (Fig. S14†).
For the synthesis of the square planar CF-TAPPy-PDA framework the reaction conditions were modified, due to the lower solubility of the 1,3,6,8-tetrakis(4-aminophenyl)pyrene (TAPPy) linker to prevent its precipitation during the reaction. Therefore, acid catalyst, TAPPy linker as well as the aniline modulator were dissolved in benzonitrile, while the second stream consisted of terephtalaldehyde (PDA) in benzonitrile (Fig. 1c, Section C4 in the ESI†). Similarly, different reaction mixtures were screened for the formation of the framework with different aliquots of benzoic acid catalyst (19.2 equiv. & 38.4 equiv.) and aniline modulator (0.8 equiv. & 1.6 equiv.) at a residence time of 1 min in the reactor tubing after combination of the two reaction mixtures. After PXRD confirmed the formation of the targeted CF-TAPPy-PDA framework, the crystal morphology of the dark red colloids was investigated by SEM. Compared to CF-TAPB-DMPDA the images revealed small agglomerating particulates with a particle size of ∼100 nm at an aniline loading of 0.8 equiv. for both catalyst loadings (Fig. 2b). Slowing down the reaction by the addition of double the amount of modulator resulted in bigger more defined still agglomerating rhombic particles. Therefore, the aniline amount in the reaction mixture was further increased to 2.0 equiv., which resulted in the formation of faceted squared particles with an average diameter of 1.9 ± 0.4 μm for the reaction with 38.4 equiv. benzoic acid, while the lower acid concentration resulted in less defined agglomerated rhombic particles. Reaction time screening from 5 s to 360 s revealed the maintenance of faceted squared particles with the optimized conditions of 2.0 equiv. aniline and 38.4 equiv. benzoic acid (Fig. S16–S21†). Interestingly, the conditions with 1.6 equiv. of modulator yielded squared particles at lower reaction times, while a decrease in modulator loading to 0.8 equiv. was leading not only to poor materials quality, but also resulted in clogging of the reactor tubing at longer residence times. Judging from these results, it became clear that both a high catalyst and modulator loading are necessary to give single-crystalline COFs in the probed timeframe.
Scale-up of 2D COFs under continuous flow
After finding suitable and robust reaction conditions for each COF system, the scalability of the production of high-quality single-crystalline COFs was investigated. To produce a higher amount of COF colloid solution one syringe pump was exchanged with a peristaltic pump (Fig. S2, Section C3 in the ESI†). Using this setup, the production of CF-TAPB-DMPDA COF was scaled to a production of >250 mL in one run with a pumping speed of 360 mL h−1, while taking aliquots for SEM imaging to ensure the high quality of the COFs throughout the process. After cooling down to room temperature, the red colloids were precipitated from the reaction solution using brine and methanol, filtered, extracted with methanol, and finally activated using a supercritical CO2 dryer (Fig. 3a and S3†). Using this setup that remains inexpensive, a continuous production of >1 g h−1 single-crystalline COF is obtained. Compared to the highest yielding reaction conditions reported for the batch synthesis of 2D COF single crystals the flow synthesis approach offers a 2–3-fold increase in framework production rate and similar to the recent reports of 3D COF synthesis in microfluidic systems.5,31 PXRD confirmed the high crystallinity of the yellow CF-TAPB-DMPDA COF with a sharp diffraction at 2.78 2θ degree angles corresponding to the (100) facet of a primitive hexagonal lattice (Fig. 3b and S22†). Additional higher order diffractions are as well in full agreement with the simulated diffractogram of the hexagonal eclipsed structure of this COF. The high quality of the COF was moreover confirmed using nitrogen sorption measurements, which confirm the permanent porosity of the mesoporous COF (Fig. 3c). The internal surface area of the CF-COF was determined at 2133 m2 g−1 using the Brunauer–Emmett–Teller (BET) theory. The pore size distribution analysis from non-local density functional theory (NLDFT) proved a narrow pore distribution around 3.2 nm, which is in full agreement with the expected pore diameter of the COF in hexagonal eclipsed structure. The chemical integrity of the formed COF was investigated using 13C CP-MAS solid state nuclear magnetic resonance (NMR) and Fourier-transform infrared spectroscopy (FT-IR). Besides the peaks in the aromatic region of the NMR spectrum, the carbon of the methyl group of the DMPDA linker can be assigned to the signal at a chemical shift of 53 ppm (Fig. 3d). The IR-spectrum reveals the expected disappearance of the vibrations of the monomers, while the imine stretch at around 1620 cm−1 is obtained (Fig. S24†).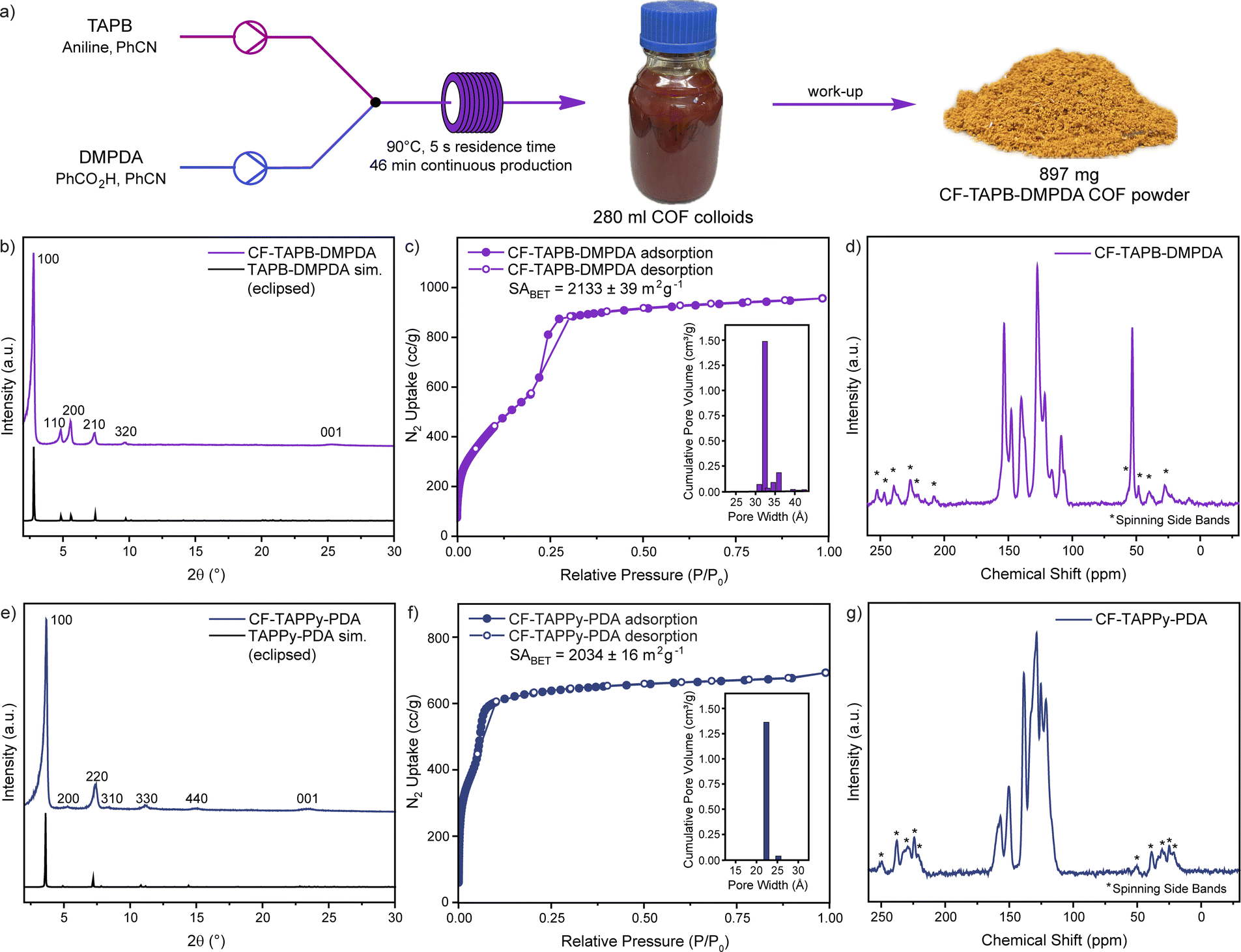 | ||
| Fig. 3 Scaled-up continuous production of CF-TAPB-DMPDA (a). PXRD (b), nitrogen isotherms with pore size distribution in the inlet (c) and 13C solid state NMR analysis (d) of scaled-up CF-TAPB-DMPDA. PXRD (e), nitrogen isotherms with pore size distribution in the inlet (f) and 13C solid state NMR analysis (g) of scaled-up CF-TAPPy-PDA. | ||
Similarly, the scalability of the CF-TAPPy-PDA COF was demonstrated using the same setup with the adapted reaction mixtures (Section C5 in the ESI†). With a pumping speed of 360 mL h−1 for this COF also a production rate of >1 g h−1 was obtained for the isolated COF powder. PXRD confirmed the square planar structure of the COF, with the diffraction of the (100) facet at 3.66 2θ degree angles and additional higher order diffractions that are in full agreement with the simulated eclipsed structure (Fig. 3e and S23†). Nitrogen sorption experiments revealed a BET surface area as high as 2034 m2 g−1 and a pore size distribution centered at 2.2 nm (Fig. 3f). The formation of the framework was further confirmed by the expected aromatic peaks in the 13C solid state NMR (Fig. 3g), and the imine stretch in the FT-IR (Fig. S25†).
Continuous flow single-crystal to single-crystal post-synthetic modification
Post-synthetic modification (PSM) has proven to be a valuable tool for accessing high quality samples of otherwise difficult to prepare COFs.43 The conversion of side groups or linkages as well as the exchange of linkers has significantly expanded synthetic strategies in the formation of framework materials. After confirming the synthesis and scalability of a continuous formation of COFs, a PSM demethylation strategy was developed for a single crystal to single-crystal transformation of CF-TAPB-DMPDA. The deprotection results in the COF with free hydroxyl groups CF-TAPB-DHPDA, which has not been reported as a single-crystalline material previously. For the conversion crude CF-TAPB-DMPDA is synthesized, and solvent exchanged with methanol, followed by hexane and chlorobenzene. Finally, the COF was suspended in anhydrous o-dichlorobenzene to give a yellow suspension. This suspension was pumped with a peristaltic pump and combined with a solution of boron tribromide in the same solvent. Immediately after those streams are mixed the color of the COF particles turns black and this suspension is pumped at 90 °C with a residence time of 6 minutes. The solution is quenched at exiting of the heating bath by a stream of triethylamine/methanol (1:5) and the demethylated COF is collected as a dark red solid, which was washed with methanol, before extraction in the same solvent and drying using supercritical CO2 (Section F in the ESI†). The PXRD of the CF-TAPB-DHPDA COF exhibits a strong diffraction of the (100) facet at 2.78 2θ degree angles, and additional higher order diffractions, that are unchanged in position to the methylated COF (Fig. 4d and S32†). This confirms that the hexagonal structure of the framework is unchanged during the demethylation step. Moreover, SEM imaging shows that the hexagonal morphology of the COF single crystals is maintained during the post-synthetic modification step (Fig. 4b and S33†). FT-IR spectroscopy reveals that the imine vibration of the demethylated framework is unchanged, therefore confirming the structural integrity of the material (Fig. S34†). Furthermore, characteristic vibration bands for C–O–C motifs of CF-TAPB-DMPDA at 1288 and 1040 cm−1 disappear in the demethylation step. The 13C solid-state NMR of the collected powder confirms the nearly complete disappearance of the intense signal of the methoxy carbon, comparable to the demethylation efficiency reported for a protocol using batch conditions (Fig. S35†).38 Nitrogen porosimetry revealed that the COF is still highly porous with a BET surface area of 1994 m2 g−1 and narrow calculated pore size distribution around 3.2 nm (Fig. 4e). One unique feature of the TAPB-DHPDA COF is that it reversibly changes color in the presence of solvents or solvent vapor. This behavior is enabled through the tautomerization of the diiminol-form present in dry and apolar environments to the iminol/cis-ketoenamine form in the presence of polar solvents, especially water.44 To investigate if the post-synthetically modified COF (CF-TAPB-DHPDA) also shows this feature, both the methylated COF CF-TAPB-DMPDA and its demethylated analogue CF-TAPB-DHPDA were investigated before and after making a slurry with water. While the change in color upon adding water to the methoxy functionalized COF is from yellow to orange, the hydroxyl functionalized COF changes from a bright orange solid to a black slurry (Fig. 4c). Diffuse reflectance spectroscopy (DRS) confirmed this change in optical absorbance, where the absorption onset for the hydroxyl functionalized COF is drastically red shifted from approximately 575 nm (2.2 eV) to 690 nm (1.8 eV) (Fig. 4f). This provides evidence that the material produced over a two-step continuous flow production could be used in previously proposed humidity sensing applications.
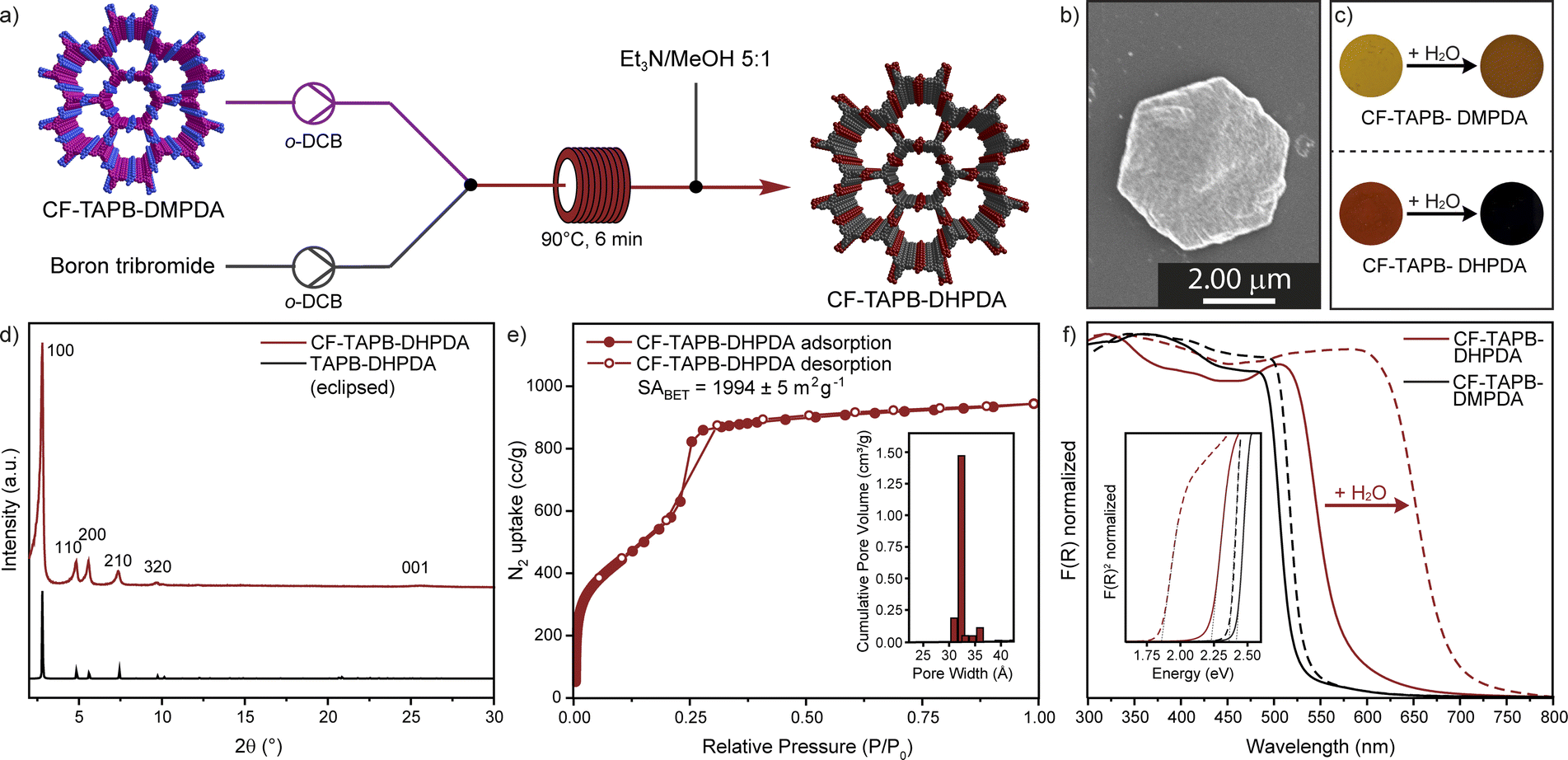 | ||
| Fig. 4 Continuous flow post-synthetic single crystal to single crystal conversion of CF-TAPB-DMPDA (a). Scanning electron microscopy image (b). Optical image of methylated and demethylated COF before and after dispersion in water (c). PXRD (d) and nitrogen isotherms with pore size distribution in the inlet of CF-TAPB-DHPDA(e). UV-vis spectroscopy with Tauc plot in the inlet of methylated and demethylated COF before and after dispersion in water (f). | ||
Conclusions
In this study we prepared single-crystalline imine-linked 2D COFs using a flow approach. The continuous process was optimized to afford single crystals in the low micrometer regime for both a hexagonal COF (CF-TAPB-DMPDA) and a rhombic COF (CF-TAPPy-PDA). We found that the residence time in the reactor can be lowered to 5 s followed by a cooling phase at ambient conditions during which smaller nanometer sized particles are fusing to the final product. This unexpected behavior at these low reaction times gives new insights in the formation of 2D COF single crystals and offers the possibility for solution processing before the final crystallization step. Additionally, the scalability of the continuous reaction was demonstrated with a productivity of more than one gram of high-quality single crystals per hour. The COF materials were fully characterized and exhibit high crystallinity and porosity. Besides the direct synthesis of COF single crystals, the post-synthetic demethylation of CF-TAPB-DMPDA was performed in continuous flow. Here, the material maintained its single-crystalline morphology as well as permanent porosity, while establishing the solvent dependent optical properties of the CF-TAPB-DHPDA COF. Ultimately, this work demonstrates the use of continuous flow methods for the synthesis, scaling, and post-synthetic modification of single-crystalline COFs and will open a pathway for further exploration of this class of porous 2D materials towards industrial implementation.Data availability
All the experimental data related to this study are available from the corresponding author upon reasonable request.Author contributions
M. T. designed and carried out the flow experiment as well as the characterizations of the materials. M. T. and W. R. D wrote the manuscript. W. R. D. managed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was sponsored by the Army Research Office under Grant Number W911NF-23-1-0306. This work was partially funded by the Trienens Institute for Sustainability and Energy at Northwestern University. This work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-2308691). This work made use of the IMSERC Crystallography facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), and Northwestern University. This work made use of the IMSERC NMR facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), International Institute of Nanotechnology, and Northwestern University.Notes and references
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- A. Natraj, W. Ji, J. Xin, I. Castano, D. W. Burke, A. M. Evans, M. J. Strauss, M. Ateia, L. S. Hamachi, N. C. Gianneschi, Z. A. Alothman, J. Sun, K. Yusuf and W. R. Dichtel, J. Am. Chem. Soc., 2022, 144, 19813–19824 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, R. P. Sivasankaran, M. Y. Ye, A. Brückner, J. Schmidt and A. Thomas, Chem. Sci., 2019, 10, 8316–8322 RSC.
- M. Traxler, S. Gisbertz, P. Pachfule, J. Schmidt, J. Roeser, S. Reischauer, J. Rabeah, B. Pieber and A. Thomas, Angew. Chem., Int. Ed., 2022, 61, e202117738 CrossRef CAS PubMed.
- A. M. Evans, M. J. Strauss, A. R. Corcos, Z. Hirani, W. Ji, L. S. Hamachi, X. Aguilar-Enriquez, A. D. Chavez, B. J. Smith and W. R. Dichtel, Chem. Rev., 2022, 122, 442–564 CrossRef CAS PubMed.
- J. Maschita, T. Banerjee, G. Savasci, F. Haase, C. Ochsenfeld and B. V. Lotsch, Angew. Chem., Int. Ed., 2020, 59, 15750–15758 CrossRef CAS PubMed.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed.
- W. Zhao, P. Yan, H. Yang, M. Bahri, A. M. James, H. Chen, L. Liu, B. Li, Z. Pang, R. Clowes, N. D. Browning, J. W. Ward, Y. Wu and A. I. Cooper, Nat. Synth., 2022, 1, 87–95 CrossRef.
- X. Wang, J. Yang, X. Shi, Z. Zhang, C. Yin, Y. Wang, X. Wang, J. Yang, X. Shi, Z. Zhang, C. Yin and Y. Wang, Small, 2022, 18, 2107108 CrossRef CAS PubMed.
- N. L. Campbell, R. Clowes, L. K. Ritchie and A. I. Cooper, Chem. Mater., 2009, 21, 204–206 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1171 CrossRef PubMed.
- A. Acharjya, P. Pachfule, J. Roeser, F. J. Schmitt and A. Thomas, Angew. Chem., Int. Ed., 2019, 58, 14865–14870 CrossRef CAS PubMed.
- J. Feng, Y. J. Zhang, S. H. Ma, C. Yang, Z. P. Wang, S. Y. Ding, Y. Li and W. Wang, J. Am. Chem. Soc., 2022, 144, 6594–6603 CrossRef CAS PubMed.
- J. Hu, Z. Huang and Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202306999 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, ACS Macro Lett., 2020, 9, 123–133 CrossRef CAS PubMed.
- Y. Peng, W. K. Wong, Z. Hu, Y. Cheng, D. Yuan, S. A. Khan and D. Zhao, Chem. Mater., 2016, 28, 5095–5101 CrossRef CAS.
- R. P. Bisbey, C. R. DeBlase, B. J. Smith and W. R. Dichtel, J. Am. Chem. Soc., 2016, 138, 11433–11436 CrossRef CAS PubMed.
- N. Contreras-Pereda, D. Rodriguez-San-Miguel, C. Franco, S. Sevim, J. P. Vale, E. Solano, W. K. Fong, A. Del Giudice, L. Galantini, R. Pfattner, S. Pane, T. S. Mayor, D. Ruiz-Molina and J. Puigmarti-Luis, Adv. Mater., 2021, 33, e2101777 CrossRef PubMed.
- Y. Yang, C. Schäfer and K. Börjesson, Chem, 2022, 8, 2217–2227 CAS.
- D. Rodriguez-San-Miguel, A. Abrishamkar, J. A. Navarro, R. Rodriguez-Trujillo, D. B. Amabilino, R. Mas-Balleste, F. Zamora and J. Puigmarti-Luis, Chem. Commun., 2016, 52, 9212–9215 RSC.
- H. Vardhan, G. Rummer, A. Deng and S. Ma, Membranes, 2023, 13, 696 CrossRef CAS PubMed.
- X. Liu, A. Wang, C. Wang, J. Li, Z. Zhang, A. M. Al-Enizi, A. Nafady, F. Shui, Z. You, B. Li, Y. Wen and S. Ma, Nat. Commun., 2023, 14, 7022 CrossRef CAS PubMed.
- T. Ma, L. Wei, L. Liang, S. Yin, L. Xu, J. Niu, H. Xue, X. Wang, J. Sun, Y.-B. Zhang and W. Wang, Nat. Commun., 2020, 11, 6128 CrossRef CAS PubMed.
- B. Mishra, A. Alam, B. Kumbhakar, D. Díaz Díaz and P. Pachfule, Cryst. Growth Des., 2023, 23, 4701–4719 CrossRef CAS.
- M. Mattera, A. T. Ngo, J. P. Vale, C. Franco, S. Sevim, M. Guix, R. Matheu, T. Sotto Mayor, S. Pané and J. Puigmartí-Luis, Chem. Mater., 2024, 36, 959–967 CrossRef CAS.
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 58–65 CrossRef CAS PubMed.
- R. L. Li, N. C. Flanders, A. M. Evans, W. Ji, I. Castano, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Chem. Sci., 2019, 10, 3796–3801 RSC.
- L. Peng, Q. Guo, C. Song, S. Ghosh, H. Xu, L. Wang, D. Hu, L. Shi, L. Zhao, Q. Li, T. Sakurai, H. Yan, S. Seki, Y. Liu and D. Wei, Nat. Commun., 2021, 12, 5077 CrossRef CAS PubMed.
- C. Kang, K. Yang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, Y. Wang, J. Jiang, X. Zou, Z. Huang and D. Zhao, Nat. Commun., 2022, 13, 1370 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- V. Singh, S. Jang, N. K. Vishwakarma and D.-P. Kim, NPG Asia Mater., 2018, 10, e456 CrossRef.
- S. J. Lyle, T. M. Osborn Popp, P. J. Waller, X. Pei, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 11253–11258 CrossRef CAS PubMed.
- S. Kandambeth, V. Venkatesh, D. B. Shinde, S. Kumari, A. Halder, S. Verma and R. Banerjee, Nat. Commun., 2015, 6, 6786 CrossRef CAS PubMed.
- J. Britton and T. F. Jamison, Nat. Protoc., 2017, 12, 2423–2446 CrossRef CAS PubMed.
- W. Ji, L. S. Hamachi, A. Natraj, N. C. Flanders, R. L. Li, L. X. Chen and W. R. Dichtel, Chem. Sci., 2021, 12, 16014–16022 RSC.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- S. Jhulki, A. M. Evans, X.-L. Hao, M. W. Cooper, C. H. Feriante, J. Leisen, H. Li, D. Lam, M. C. Hersam, S. Barlow, J.-L. Brédas, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 783–791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and instrumentation, experimental procedures, additional analysis (PDF). See DOI: https://doi.org/10.1039/d4sc01128g |
| This journal is © The Royal Society of Chemistry 2024 |