
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in CO2 reduction with renewable reductants under hydrothermal conditions: towards efficient and net carbon benefit CO2 conversion
Zien
Tang
a,
Xu
Liu
a,
Yang
Yang
*a and
Fangming
Jin
 *abc
*abc
aSchool of Environmental Science and Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: fmjin@sjtu.edu.cn; yangyang0120@sjtu.edu.cn
bShanghai Key Laboratory of Hydrogen Science, Center of Hydrogen Science, Shanghai Jiao Tong University, Shanghai 200240, P. R. China
cShanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, P. R. China
First published on 28th May 2024
Abstract
The ever-growing atmospheric CO2 concentration threatening the environmental sustainability of humankind makes the reduction of CO2 to chemicals or fuels an ideal solution. Two priorities are anticipated for the conversion technology, high efficiency and net carbon benefit, to ensure the mitigation of the CO2 problem both promptly and sustainably. Until now, catalytic hydrogenation or solar/electro-chemical CO2 conversion have achieved CO2 reduction promisingly while, to some extent, compromising to fulfill the two rules, and thus alternative approaches for CO2 reduction are necessary. Natural geochemical processes as abiotic CO2 reductions give hints for efficient CO2 reduction by building hydrothermal reaction systems, and this type of reaction atmosphere provides room for introducing renewable substances as reductants, which offers the possibility to achieve CO2 reduction with net carbon benefit. While the progress in CO2 reduction has been abundantly summarized, reviews on hydrothermal CO2 reduction are relatively scarce and, more importantly, few have focused on CO2 reduction with renewable reductants with the consideration of both scale of efficiency and sustainability. This review provides a fundamental and critical review of metal, biomass and polymer waste as reducing agents for hydrothermal CO2 reduction. Various products including formic acid, methanol, methane and multi-carbon chemicals can be formed, and effects of operational parameters such as temperature, batch holding time, pH value and water filing as well as detailed reaction mechanisms are illustrated. Particularly, the critical roles of high temperature and pressure water as reaction promotor and catalyst in hydrothermal CO2 conversion are discussed at the mechanistic level. More importantly, this review compares hydrothermal CO2 reduction with other methods such as catalytic hydrogenation and photo/electrocatalysis, evaluating their efficiency and potential for net carbon benefit. The aim of this review is to promote the understanding of CO2 activation under a hydrothermal environment and provide insights into the efficient and sustainable strategy of hydrothermal CO2 conversion for future fundamental research and industrial applications.
 Zien Tang | Zien Tang received his master's degree in 2024 from the School of Environmental Science and Engineering at Shanghai Jiao Tong University, under the guidance of Professor Fangming Jin. His research interests focus on the hydrothermal co-conversion of lignin and CO2. |
 Xu Liu | Xu Liu is a doctoral candidate at the School of Environmental Science and Engineering, Shanghai Jiao Tong University under the supervision of Prof. Fangming Jin. His research focuses on cobalt based catalytic CO2 reduction under hydrothermal conditions and the corresponding density functional theory (DFT) calculations. |
 Yang Yang | Yang Yang obtained her PhD in 2019 from Shanghai Jiao Tong University, China, under the guidance of Prof. Fangming Jin. She is currently an Assistant Researcher at the School of Environmental Science and Engineering at Shanghai Jiao Tong University. Previously, she was a postdoctoral fellow in the same group (2019–2023) and was supported by the Postdoctoral Innovation Talents Support Program. Her research focuses on the conversion of CO2 and biomass under hydrothermal conditions, aiming to develop sustainable and efficient technologies for CO2 reduction. |
 Fangming Jin | Fangming Jin received her PhD in Earth Engineering from Tohoku University in 1999. She was a postdoctoral fellow and later an Assistant Professor and Associate Professor at Tohoku University until 2007. She became a Professor at Tongji University in 2007 and was honored as a Changjiang Scholars Program Distinguished Professor. Since 2011, she has been a Distinguished Professor at the School of Environmental Science and Engineering, Shanghai Jiao Tong University. Her research focuses on mimicking the natural hydrothermal environment for highly efficient conversion of biomass/waste and CO2 to amend the carbon cycle. |
1 Introduction
The world is currently facing an energy crisis since the demand for energy continuously grows to support the rapid development of the society. This has resulted in a heavy reliance on carbon-intensive energy sources, leading to increasing levels of carbon dioxide (CO2) emissions. Given that CO2 is a major greenhouse gas, extensive emissions of CO2 will bring a series of negative impacts including global warming, ocean acidification, melting of polar ice caps, and other environmental problems.1–3 As the world grapples with challenges related to climate change and energy security, the importance of utilizing CO2 as a carbon resource is increasingly recognized for its potential to mitigate climate change and promote sustainable development. In this context, CO2 can be viewed as a valuable resource that can be utilized in various ways such as in the production of fuels, chemicals, and materials,4,5 which can reduce greenhouse gas emissions, create economic opportunities, and promote the transition to a low-carbon economy.When fully considering the utilization of CO2 as a carbon source to build an environmentally benign and sustainable society for the future, the underlying technologies are expected to achieve two targets: net carbon benefit and high efficiency, which ideally require no extra fossil fuel sourced energy during the whole CO2 reduction process and simultaneously an efficient conversion process. This consideration aligns with the growing consensus that sustainability should be integrated into the performance metrics of conversion systems.6 In recent years, various catalytic methods, including catalytic hydrogenation,7,8 photo-catalysis,9,10 electrocatalysis,11,12 the recently arisen photo-thermal catalysis,13,14 and photo-electro catalysis,15,16 have been applied to convert CO2 into high-value-added products. During these processes, carbon-containing compounds like formic acid, methanol, methane, acetic acid, ethylene, and even long-chain alkanes are formed, which can serve as chemical stocks or fuels depending on the circumstances.17–19 However, bottlenecks in enhancing the efficiency of photo-catalysis or large consumption of high-quality electricity during the electro-catalytic CO2 reduction process and the necessity of delicately prepared catalysts restrict their applications. For catalytic CO2 hydrogenation, hydrogen is an essential raw material, yet it has complex systematic issues in its preparation, storage, and transportation processes, which rely heavily on fossil fuels as the energy supplier.20 Consequently, to meet the undeniable requirements of net carbon benefit and highly efficient CO2 reduction, alternative approaches that balance the paradox are anticipated.
Natural geochemical processes that regulate the carbon cycle, including the transformation of CO2 over geological timescales, could be instrumental in counteracting anthropogenic CO2 emissions. In the absence of solar irradiation and electrical power, Earth's natural hydrothermal reduction environments, enhanced by the significant catalytic properties of bedrock minerals, can abiotically reduce CO2 into long-chain hydrocarbons and organic compounds like carboxylic acids.21 This process is also postulated to have facilitated the origin of life and the formation of most natural petroleum deposits.22,23 Mimicking this natural phenomenon, efficient CO2 reduction could be achieved using artificially constructed hydrothermal reaction systems.24 Compared to solar/electro-catalytic CO2 reduction or catalytic CO2 hydrogenation, under hydrothermal conditions, high-temperature water creates an environment that facilitates the utilization of various hydrogen sources, such as metals, biomass, and even organic waste, for CO2 reduction, demonstrating a flexibility in selecting reductant feedstock which eliminates the need for gaseous hydrogen or high-quality electricity. Additionally, owing to the vigorousness of the hydrothermal reaction, highly efficient and fast CO2 reduction could be achieved, providing prospects for large scale application. Further advances in hydrothermal CO2 reduction lie in the direct reduction of bicarbonate or carbonate (HCO3− or CO32−), since CO2 generally needs to be captured from either the atmosphere or point sources by alkaline absorbents before utilization, and this process transforms CO2 into HCO3− and/or CO32− (the final products are pH dependent), while for solar/electro-catalytic CO2 reduction or catalytic CO2 hydrogenation, generally only gaseous CO2 can be directly reduced. However, central to this geochemically inspired approach is the selection of appropriate reductants. The ideal reductants should be renewable hydrogen sources or regenerable through renewable energy, ensuring that the CO2 reduction process aligns with the principles of sustainability.
A series of pioneering studies on CO2 reduction by building hydrothermal reaction systems have been reported, in which various zero-valent metals such as Fe, Zn, Mn have been used as reductants for in situ hydrogen production.25–51 While CO2 was successfully reduced to C1 or C2+ products, the zero-valent metals were oxidized to the corresponding metal oxides, leading to the metals being discarded after one use. To cope with this issue, the regeneration of metals using renewable energy becomes necessary. When Fe was used as the reductant to hydrothermally convert CO2, the corresponding oxidized high valence metal could be reduced to zero valence by biomass or biomass derivatives with the formation of organic acids, which means no metal consumption during those processes, and both CO2 and the biomass can be converted to high value-added products.48,51–55 Furthermore, in the systematic work by A. Steinfeld,56 the heat obtained from concentrated solar radiation was utilized to drive multiple cycles of metal oxide redox pairs, including Zn/ZnO, potentially offering Zn as another reductant for CO2 conversion. This research substantiates the proposal that the reduction of CO2 can be driven by the redox reactions of zero-valent metals (Fe or Zn), underscoring a critical mechanism by which these metals facilitate the transformation of CO2 into various reduced forms without being consumed.
Intriguingly, biomass and bio-related organic compounds containing highly active and reductive functional groups (–OH, –CHO, –NH2) can produce hydrogen in hydrothermal environments, rendering biochemicals as a viable hydrogen donor for CO2 reduction. Since biomass stores solar energy, using it to drive CO2 reduction offers great potential in achieving net-zero emission reaction systems. Moreover, as CO2 is naturally fixed through photosynthesis in biomass, additional CO2 reduction with biomass allows double CO2 fixation. Following this concept, wastes such as polyvinyl chloride (PVC) have the potential to generate reductive functional groups such as –OH if processed properly, offering another potential and economic reductant for CO2 reduction. Thus, the employment of regenerable metals, biomass, or organic waste as renewable reductants for CO2 reduction under hydrothermal conditions becomes a plausible solution for efficient and net carbon benefit CO2 reduction (the notion of CO2 reduction with renewable reductants under hydrothermal conditions is illustrated in Fig. 1).
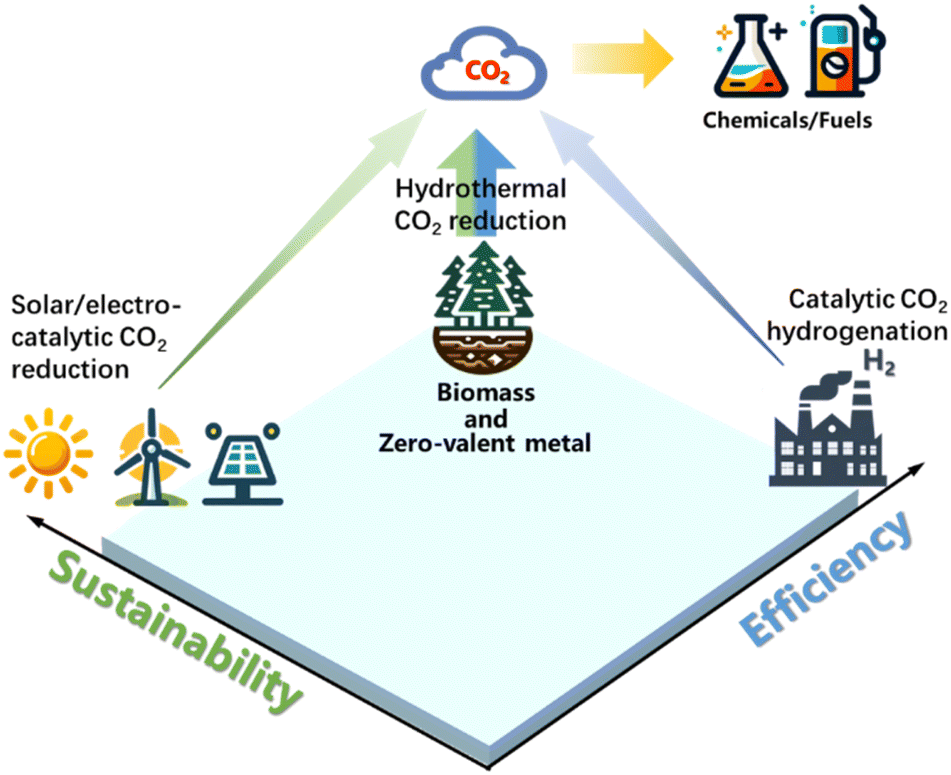 | ||
| Fig. 1 Comparison of CO2 reduction by renewable reductants under hydrothermal conditions with solar/electro-catalytic CO2 reduction or catalytic CO2 hydrogenation. | ||
Despite the technologies for CO2 reduction being readily summarized, hydrothermal CO2 reduction is relatively new with scarce reviews in this field, and more importantly, few discuss this issue with consideration for both efficiency and sustainability. Starting with the advantages and geologic origin of a hydrothermal environment for CO2 reduction, this minireview systematically summarizes recent advances in hydrothermal CO2 reduction using metals (Fe or Zn), biomass, and polymer wastes, discussing operational factors such as temperature, reaction time, pH value, and pressure. The superiority of CO2 reduction under hydrothermal conditions is illustrated at a mechanistic level, revealing the catalytic influence of hydrothermal conditions based on the intrinsic characteristics of the hydrothermal environment, with a specific section emphasizing the promoting function of water molecules. For the primary purpose of advancing CO2 reduction with net carbon benefit and high efficiency, this review compares the summarized approaches with reported methods such as catalytic hydrogenation or photo/electric catalyzed CO2 reduction, analyzing the degree of carbon mitigation, and therefore provides insights on the efficient and sustainable strategy of hydrothermal CO2 conversion for future fundamental research and industrial applications.
2 The origin of life under hydrothermal conditions and insights on CO2 reduction
High temperature and high pressure water (HTHP water) in hydrothermal conditions is recognized as an important factor in various geological processes, such as hydrothermal circulation, ore deposition, and rock alteration,57 occurring in both sub-aerial and sub-seafloor environments and profoundly affecting the physicochemical properties of rocks and fluids. At elevated temperature and pressure, HTHP water exhibits as good solubility for organics and gases as many nonpolar organic solvents due to its lower dielectric constant (ε), which decreases from about 80 at ambient condition to 5 at the critical point58–60 (374 °C and 22.1 MPa). Another significant factor is the ionic product of HTHP water (Kw = [H+]·[OH−]; [H+] = [OH−]). For instance, at ambient conditions, Kw is 10−14 (mol kg−1),2 but it can sharply rise to a maximum of 10−11 at around 250 °C.61 This increase in the concentration of H+ or OH− ions can facilitate acid/base-catalyzed reactions, which can significantly accelerate reactions, such as the dehydration of carbohydrates and alcohols. The diffusion rate of supercritical water is reduced to nearly 1/100 that of liquid water. The weak interaction of hydrogen bonds among water molecules in the HTHP water phase eliminates the mass transfer resistance between interfaces.62 Consequently, the efficient heat transfer and rapid mass diffusion resulting from the high diffusion rate and low viscosity of HTHP water promote reaction rates, hopefully contributing to invoke the reductive capacity of metals or organic hydrogen sources for CO2 reduction.59,63Due to its unique properties, HTHP water has been proposed as a possible setting for the origin of life on Earth. Hydrothermal vents are fissures in the Earth's surface, often found along mid-ocean ridges, where superheated water, gases (CH4, CO2, H2S and H2), minerals (pyrrhotite, sphalerite, pyrite and copper-iron sulfide) and metals (mainly Fe, Co and Mg) are expelled from the ocean floor.64 The presence of those mineral surfaces in hydrothermal environments enhances the stability and reactivity of organic compounds and provides catalytic surfaces for prebiotic reactions.65 Furthermore, the hydrothermal environment can protect organic molecules from destructive radiation and other environmental stresses, providing a dynamic and energy-rich environment for the emergence and evolution of early life forms.66
Inspired by the theory of the origin of deep-sea life, a series of studies on CO2 conversion under hydrothermal conditions was conducted. Fiebig et al.67 provided definitive evidence that CH4 is generated from mixed aliquots of limestone and mantle-derived CO2 in volcanic hydrothermal systems. It was found that CH4 generation highly depends on the availabilities of Fe2+, CO2 and H2O and on reaction kinetics. Furthermore, He et al. demonstrated that the formation of Fe–OH during hydrothermal CO2 processing can enhance the adsorption of the siderite-derived CO intermediate on Co, facilitating efficient C–C coupling in which C24+ long chain hydrocarbons were synthesized under 300 °C and 30 MPa hydrothermal conditions.25 These findings support the abiogenic hypothesis for petroleum deposits, propose a method for nonnoble metal-catalyzed CO2 conversion to petroleum fuel, and contribute to a better understanding of the emergence of life. Amino acids, such as glycine, can also be synthesized from simple inorganic chemicals like HCHO and NH3,68 which suggests the huge potential of the hydrothermal environment for CO2 reduction.
Considering the unique characteristics of HTHP water, the influence of hydrothermal conditions on CO2 conversion can be summarized as follows: (1) temperature and reaction time. Temperature directly affects the ion product and dielectric constant of high-temperature water (HTW). At 350 °C, hydrothermal products like formic acid primarily decompose into H2 and CH4.69 This finding underscores the importance of selecting appropriate temperature and time ranges for reducing CO2. Furthermore, different reductants such as metals or biomass require different reaction temperatures to stimulate hydrogen production. For most metal-based reactions, the ideal temperature and reaction time are around 250 °C and 2 h, respectively. Biomass, however, requires a higher temperature of 300 °C or a longer time of 3 h, probably owing to the lower reducing capacity of biomass-derived functional groups compared to metal compounds. (2) Water filling and pressure. Water filling affects the reaction pressure, which significantly impacts CO2 dissolution and hydrogen partial pressure. Higher pressure promotes higher reduction efficiency by enhancing CO2 activation and facilitating the reaction direction via H2 consumption.30 (3) Solution pH and concentration of CO2, HCO3− or CO32−. Solution pH affects CO2 reduction because an acid or alkaline environment can convert CO2 to HCO3− or CO32−, and different carbon sources lead to different products such as methanol, methane, formic acid and multi-carbon products. Furthermore, a volcano-type relationship generally exists between the carbon source concentration and product yields, with a proper carbon source concentration favoring reduction efficiency while decreasing or increasing the concentration inhibits efficient reduction.
When applying hydrothermal technology for CO2 reduction at lab scale, autoclave reactors, which can be heated to 200–400 °C and hold pressure from 1 atm to 300 atm, are generally adopted. According to their different heating systems, three sets of apparatus are used, such as (1) six-batch autoclaves and Parr reactors, which are heated through resistance wire; (2) SUS 316 tubular reactors, which are used in a salt bath and (3) electromagnetic induction reactors heated by the electromagnetic effect (Fig. 2). Due to the different heating rates and stirring modes provided by the different apparatus, each reactor is applied in different scenarios. For instance, six-batch autoclaves and Parr reactors are the most used reactors, as the Parr reactor offers a larger capacity, allowing exploration of industrial-scale processes with real-time monitoring of temperature and pressure. On the other hand, when rapid heating rates are needed, SUS 316 tubular reactors are used; they are commonly chosen when biomass serves as the reductant. The electromagnetic induction reactor has a larger volume and, under the influence of a magnetic field, effective CO2 reduction to long-chain hydrocarbons or multi-carbon products is achieved.
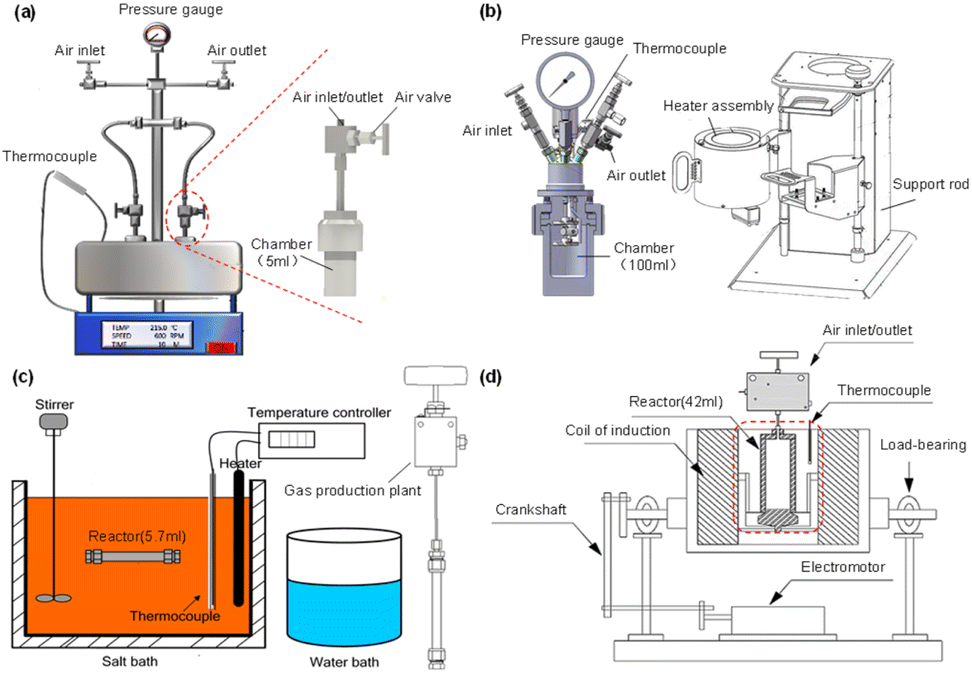 | ||
| Fig. 2 The fundamental devices used in hydrothermal CO2 conversion. (a) Six-batch autoclaves, (b) Parr reactor, (c) SUS 316 tubular reactor, (d) electromagnetic induction reactor. | ||
3 Hydrothermal CO2 reduction with zero-valent metal/metal oxide redox cycles
The strategy for CO2 reduction under hydrothermal conditions using zero-valent metal (M0)/metal oxide (MOx) redox cycles involves the production of formic acid and hydrogen via the oxidation of the M0 in the first step. In the second step, MOx is reduced with solar energy or by a suitable biomass derivative such as glycerin or glucose, resulting in the production of organic acids like formic, acetic, and lactic acids, as shown in Fig. 3. In this context, the following section summarizes CO2 hydrothermal reduction with Fe or Zn as the reductant, focusing on the reaction characteristics, parameters, and mechanism.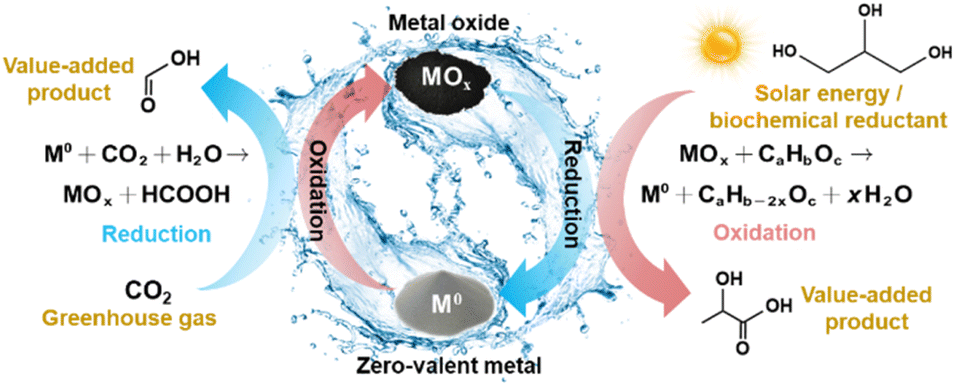 | ||
| Fig. 3 Hydrothermal CO2 reduction with zero-valent metal/metal oxide redox cycles. | ||
3.1 Hydrothermal CO2 reduction with Fe
Using Fe powder as the reductant in a hydrothermal environment, Nakamichi Yamasaki commenced CO2 reduction at the beginning of this century.70 He developed a continuous flow system operating at 200 °C and 2.0 MPa of constant CO2 flow in a hydrothermal environment. When processing carbon steel scraps without Ni powder, no organic compounds were detected. However, with the addition of Ni powder, formic acid formation was observed. Extending the reaction time to 72 hours, He et al.53 used Fe powder as a reductant at 200 °C to successfully reduce CO2 to formic and acetic acids. During this process, Fe powder acts both as a reductant and a catalyst for CO2 reduction. As illustrated in Fig. 4a, the reaction begins with Fe powder reacting with water to generate H2, while dissolved CO2 molecules are adsorbed on the surface of Fe. Under the influence of hydrogen, CO2 is reduced to intermediate A, which subsequently hydrolyzes to form formic acid. Intermediate A continues to react with hydrogen, forming intermediate B, and, eventually, intermediates A and B react to form intermediate C, hydrolyzing into acetic acid. Additionally, phenol can be formed with Fe powder at 200 °C, 1.4 MPa CO2, and 120 h of hydrothermal conditions with a yield of 1.21%.71 The reaction mechanism, shown in Fig. 4b, involves CO2 adsorption and binding to the Fe surface. Concurrently, Fe reacts with water to produce H2 and Fe2+. On the Fe powder surface, activated CO2 forms formaldehyde, eventually transforming into phenol under the action of hydrogen. This process includes two simple reaction types: oxidative coupling and rearrangement reactions. The generation of H2 is the rate-determining step in accelerating the reaction, and water is essential as a hydrogen source. Once hydrogen is produced from the reaction of Fe with water, the reaction proceeds rapidly, and the yield of phenol increases in acidic media.72 Wu et al.55 initially utilized NaHCO3 to replace CO2 in the reaction, due to the fact that, under high-temperature and high-pressure conditions, CO2 readily dissolves in alkaline aqueous solutions, which leads to an equilibrium involving CO2, H2O, HCO3−, CO32−, and OH− ions, similar to conditions in NaHCO3 aqueous solutions. No hydrogen was generated in the absence of NaHCO3, while the hydrogen yield linearly increased with the addition of NaHCO3, indicating that NaHCO3 can effectively enhance hydrogen production from HTHP water. XRD and Raman results indicated no change in Fe valence in the absence of NaHCO3; however, Fe was oxidized to Fe3O4 in the presence of NaHCO3, accompanied by the formation of hydrogen gas. Furthermore, with an increase in NaHCO3 concentration, an increase in Fe3O4 crystallization was observed. Thus, hydrogen was produced from the reaction of H2O with Fe enhanced by NaHCO3. For formate yield from hydrothermal NaHCO3 conversion, the amount of Fe significantly influenced the yield. The formate yield increased from 1.6% to 10.7% when raising the Fe amount from 2 to 8 mmol under the conditions of 1 mmol NaHCO3, 2 mL H2O, 573 K, 2 h time and 35% water filling.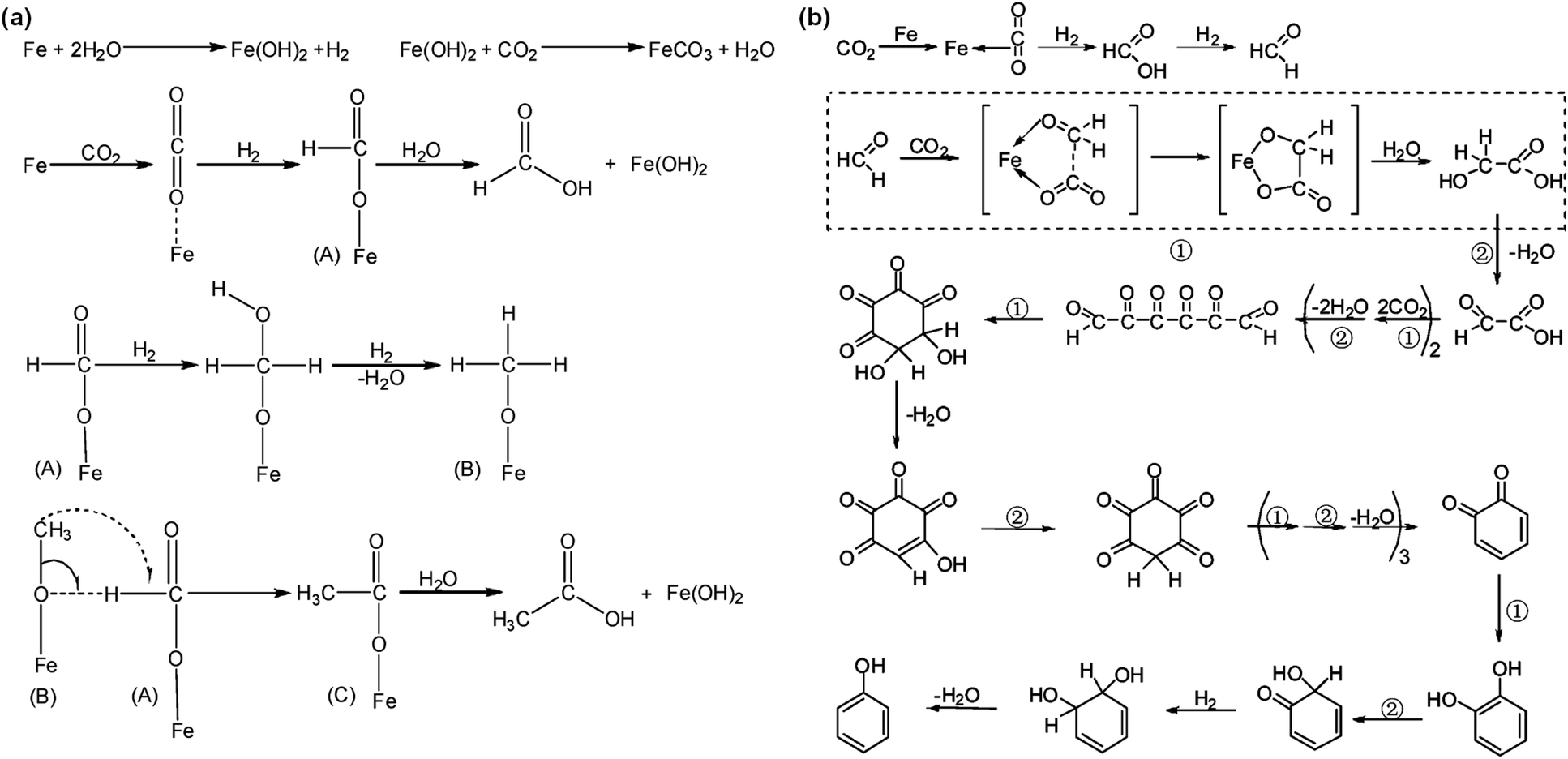 | ||
| Fig. 4 (a) Proposed mechanism for the formation of formic acid and acetic acid in the presence of Fe powder and CO2 under hydrothermal conditions.53 This figure has been reproduced from ref. 53 with permission from American Chemical Society, copyright 2010. (b) Proposed mechanism of phenol formation72 (① refers to the oxidative coupling reactions, and ② refers to the rearrangement reactions). This figure has been reproduced from ref. 72 with permission from American Chemical Society, copyright 2007. | ||
To enhance the productivity of CO2 reduction with Fe under hydrothermal conditions for possible application of the reaction, heterogeneous catalysts were added. The impact of Ni addition was demonstrated, and the yield of formic acid increased from 1.16% to 5.4% with incorporation of Ni.55 The Ni/Fe ratio significantly influences the yield of formic acid. As the Ni/Fe ratio increased from 1/2 to 1/1, the yield of formic acid rose from 9.1% to 15.6%. However, a higher Ni/Fe ratio resulted in a slight decrease in yield, as more Ni sites facilitated the conversion of formic acid to CH4. Additionally, Chen et al.69 found that temperatures exceeding 350 °C, particularly under supercritical water conditions, significantly accelerated the further reduction of formic acid to methane. Like Ni, Cu exhibited both positive and negative effects on formic acid formation. With Cu additions of less than 12 mmol, there was an increase in the yield of formic acid. However, when Cu exceeded 12 mmol, a sharp decrease in the yield of formic acid occurred, as excessive Cu led to the decomposition of formic acid. The highest formic acid yield, 71.3%, was achieved under the conditions of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu = 1:1 for 2 h at 573 K.48 In summary, with Ni or Cu as the catalyst, an appropriate amount is essential to maximize the yield of formic acid.
:Cu = 1:1 for 2 h at 573 K.48 In summary, with Ni or Cu as the catalyst, an appropriate amount is essential to maximize the yield of formic acid.
A significant breakthrough in CO2 hydrothermal reduction using Fe as the reductant occurred upon proper adjustment of the Fe amount. Duo et al.52 found that the amount of Fe was a decisive factor for CO2 reduction. Under the conditions of 1 mmol per L NaHCO3, 300 °C and 2 h, formate yield was dramatically increased from 12% to a peak of 91% as Fe loading was increased from 4 mmol to 24 mmol. Integrating XRD, XPS characterization analysis, and kinetic experimental results, a self-catalytic reaction mechanism was proposed, incorporating a short induction and a rapid formation phase. Initially, in the first 5–10 minutes, hydrogen and Fe3O4 with more oxygen vacancies (Fe3O4−x) were formed. Subsequently, this oxygen vacancy containing Fe3O4−x served as a catalyst to convert HCO3−, leading to a sharp increase in formate yield during the 10-30 minute period. Kinetic modelling indicates that the activation energy for the formate formation from HCO3− reduction is 28 kJ mol−1,74 much lower than the 80 kJ mol−1 reported in previous studies on conventional hydrogenation of CO2 using pincer-supported Ni hydrides as a catalyst,75 demonstrating the supportive contribution of the self-catalytic reaction mechanism. Further support for the self-catalytic mechanism was obtained from the XPS analysis for both recovered and commercially available Fe3O4. As shown in Fig. 5a, the Fe2+/Fe3+ ratio of the recovered Fe3O4 was 0.89:0.11, which is higher than the theoretical ratio of Fe3O4 (0.33:0.67), indicating in situ reduction of Fe3+ into Fe2+ on the surface of Fe3O4. This result was likely caused by the hydrogen formation from Fe oxidation, enhanced by the presence of NaHCO3.
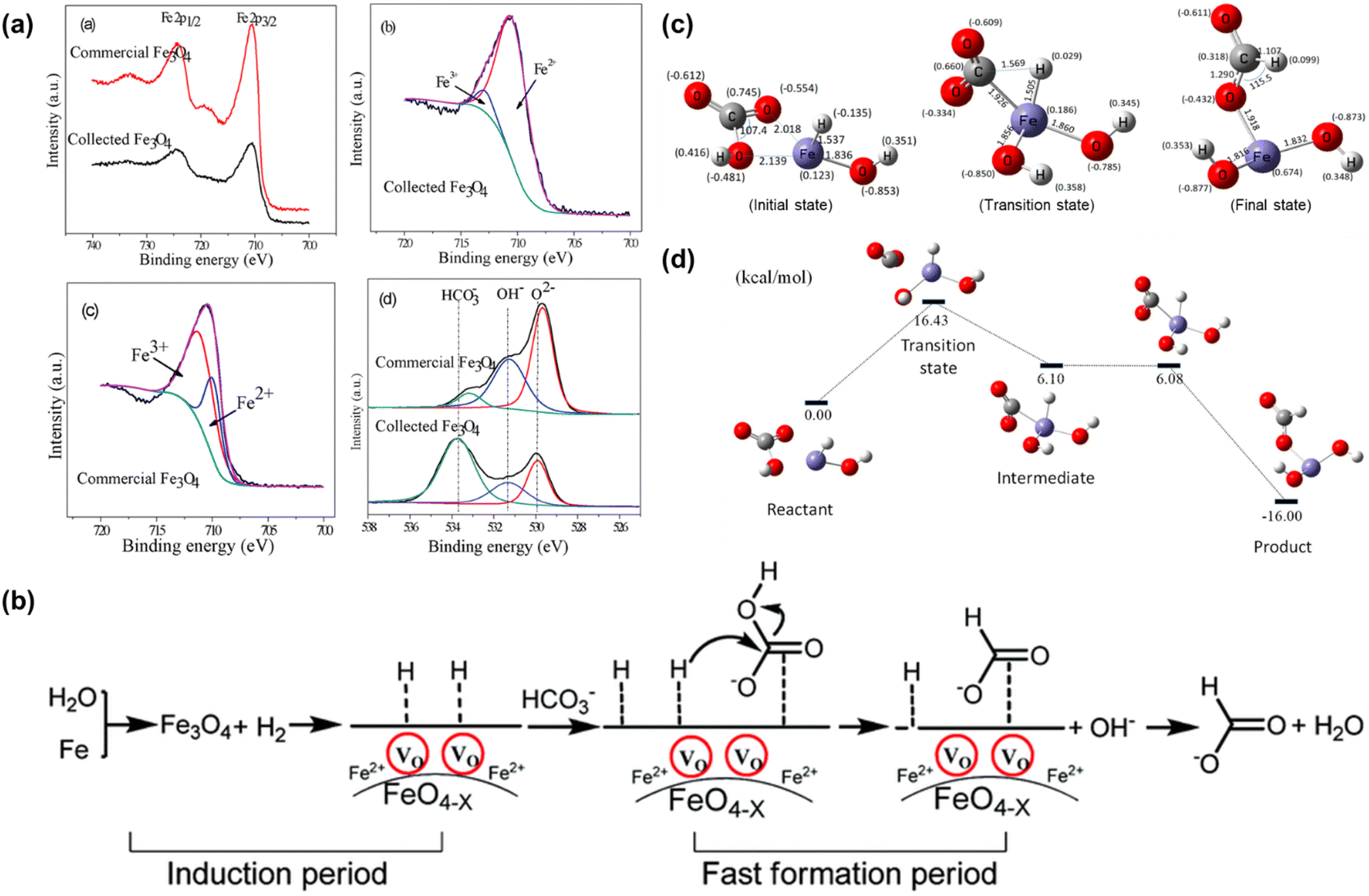 | ||
| Fig. 5 (a) X-ray photoelectron spectra of collected and commercial Fe3O4; (b) proposed mechanism of NaHCO3 reduction into formate with Fe.52 This figure has been reproduced from ref. 52 with permission from The Royal Society of Chemistry, copyright 2016. (c) Optimized geometric parameters of the initial, transition and final states of HCO3− reduction with Fe. (d) Potential energy diagram for HCOO− production from HCO3− reduction with Fe.73 | ||
Based on the experimental results, a possible mechanism for the reduction of NaHCO3 to formate by Fe is proposed in Fig. 5b. Initially, Fe undergoes oxidation to Fe3O4 in water in the presence of HCO3− to form a great amount of hydrogen which simultaneously in situ reduced Fe3O4, creating more active sites on the surface of Fe3O4−x. Subsequently, the generated hydrogen and HCO3− become activated upon adsorption on the Fe3O4−x surface. Following this, the activated H on the Fe3O4−x surface attacks C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, resulting in the release of the hydroxyl group from HCO3−. Ultimately, this process leads to the formation of formate accompanied by H2O. Furthermore, DFT calculations suggest that the Fe–Hδ− species is formed via the dissociation of water73 (Fig. 5c and d). The Mulliken charge of H in the iron hydride was −0.135 in the HCO3− hydrogenation process, wherein a C–H bond forms in a transition state through the attack of Hδ− on Cδ+. Ultimately, a HCOO− species was generated with an energy barrier of 16.43 kcal mol−1.
O, resulting in the release of the hydroxyl group from HCO3−. Ultimately, this process leads to the formation of formate accompanied by H2O. Furthermore, DFT calculations suggest that the Fe–Hδ− species is formed via the dissociation of water73 (Fig. 5c and d). The Mulliken charge of H in the iron hydride was −0.135 in the HCO3− hydrogenation process, wherein a C–H bond forms in a transition state through the attack of Hδ− on Cδ+. Ultimately, a HCOO− species was generated with an energy barrier of 16.43 kcal mol−1.
In addition to formic acid production, C2 and C2+ products were also synthesized from hydrothermal CO2 reduction using Fe as the reductant., With RANEY®-Ni instead of Ni powder, the resulting in situ RANEY®-Ni/Fe3O4 acted as a bifunctional catalyst, facilitating the production of acetate.34 A mechanistic study reveals that Fe3O4 is active for the formation of in situ hydrogen from water and in synthesizing formate, while the RANEY®-Ni site enhances the intermediate CO and its adsorption, leading to further hydrogenation to methylene. Subsequently, acetate was produced through the combination of formate and methylene on the RANEY®-Ni surface.
Moreover, long-chain hydrocarbons (up to C24) were for the first time synthesized using Fe as the reductant and Co as the catalyst in a hydrothermal environment.25 At 300 °C, the in situ formed Fe–OH, confirmed by operando infrared spectroscopy, enhanced CO adsorption on the Co site, thereby favoring further reduction to long-chain hydrocarbons (as shown in Fig. 6). It should be noted that under these hydrothermal conditions, the traditional issue of water-induced deactivation of Co can be inhibited by bicarbonate-assisted CoOx reduction, leading to the formation of honeycomb-native Co nanosheets as a new motif. Accordingly, the Co0 promoter responsible for synthesizing long-chain hydrocarbons is extraordinarily stable when coupled with Fe–OH formation.
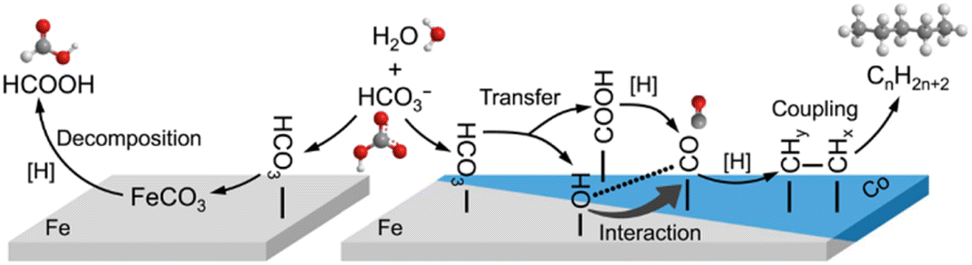 | ||
| Fig. 6 Proposed hydrothermal reduction mechanism of NaHCO3 into long-chain hydrocarbons.25 | ||
3.2 Hydrothermal CO2 reduction with Zn
When the reductant switched from Fe to Zn, methanol can be directly generated from the hydrothermal conversion of CO2, with HCl playing a crucial role. Huo et al.28 found an increase in methanol yield from 0.6% to 4.2% as the HCl concentration increased from 0.5 M to 2 M, and a maximum methanol yield of 11.4% was achieved at 350 °C for 3 h using 70 mmol Cu, 60 mmol Zn, and 2.0 M HCl. XRD analysis confirmed that Zn was oxidized to ZnO, while Cu maintained its valence. The proposed mechanism suggests that CO2 is adsorbed on ZnO and subsequently undergoes a stepwise hydrogenation process to form methoxide species. Simultaneously, atomic hydrogen generated from water decomposition is supplied by spillover from Cu. Ultimately, methanol is formed through the hydrolysis of methoxide groups on ZnO, accompanied by the formation of H2O as a by-product.It is noteworthy that with the exclusive use of Zn for hydrothermal NaHCO3 reduction, over 80% of formate was generated.76 XRD results (Fig. 7a) revealed that almost all Zn underwent rapid oxidation to ZnO within 10 minutes. Additionally, the particle size of the resultant ZnO was found to be smaller than that of the original Zn powder. Thus, HCO3− can be effectively and selectively reduced to formate using solely Zn, without requiring a catalyst. Furthermore, the infrared spectrum indicated that Zn–Hδ− is an active intermediate resulting from the interaction between Zn and H2O, as indicated in Fig. 7b. DFT calculations further confirmed that the Zn–Hδ− species is produced via the reaction of Zn with H2O.44 Specifically, it is in the form of H–Zn⋯O–H, attributable to the chemisorption of H2, which drives the production of formic acid. Subsequently, Hδ− attacks the carbon center Cδ+ of HCO3−, resulting in a bond formation with carbon in a transition state. Finally, the OH− species dissociates, leading to the formation of HCOO− through an SN2-like mechanism (Fig. 7c). Owing to the reversible nature of ZnO reduction and Zn oxidation, Zn–Hδ− formation occurs via the oxidation of Zn in water followed by hydrogen adsorption on ZnO. During the initial stages of the reaction, this in situ generated hydrogen adsorbs on ZnO surfaces and at the Zn/ZnO interface, where oxygen vacancies play a key role in hydrogen activation. The activated hydrogen subsequently nucleophilically attacks bicarbonate ions, with Hδ− added to the carbon of CO, resulting in the formation of formate. This process, in which the autocatalytic mechanism at the Zn/ZnO interface is critical, is pivotal for the effective conversion of HCO3− into formate (as shown in Fig. 7d).77
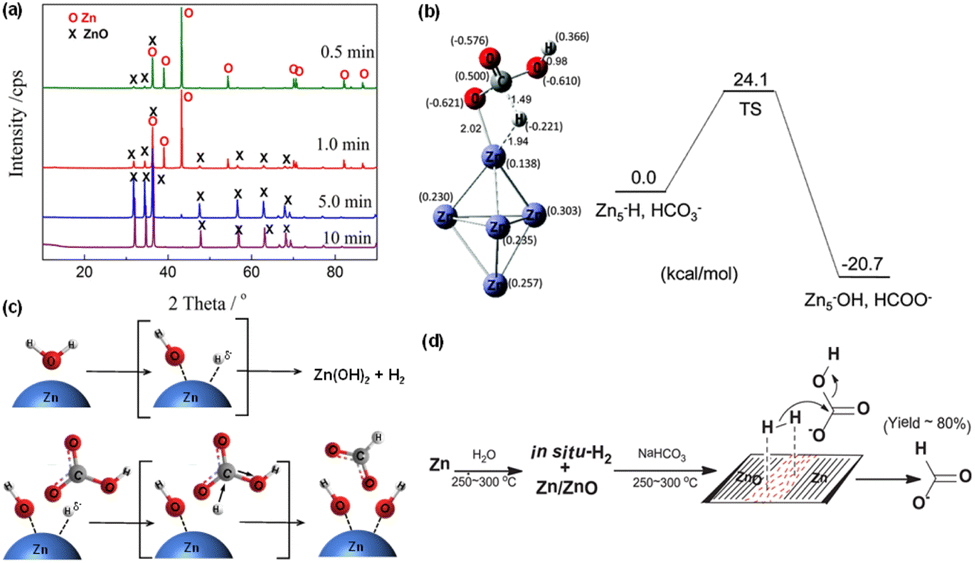 | ||
| Fig. 7 (a) XRD patterns of Zn after NaHCO3 hydrothermal reduction for different reaction times,76 (b) the geometries of the transition state in reaction of Zn–H + HCO3− → Zn5–OH + HCOO−, (c) proposed SN2-like mechanism for the formation of HCOO−.44 This figure has been reproduced from ref. 44 with permission from The Royal Society of Chemistry, copyright 2016. (d) Proposed autocatalytic mechanism of Zn/ZnO formed in water splitting for the conversion of CO2 to formic acid with Zn.77 This figure has been reproduced from ref. 77 with permission from Elsevier, copyright 2017. | ||
Wang et al.78 investigated the catalytic activity of various metals (Cu, Fe, Sn) in the hydrothermal reduction of CO2 using Zn powder. Copper demonstrated the highest catalytic activity for formic acid production, achieving a yield of up to 61%. Zhong et al.79 explored a method for converting bicarbonate into formate using nickel (Ni) and zinc/zinc oxide (Zn/ZnO). The process involves Ni playing a key role in enhancing the formation of oxygen vacancies at the Zn/ZnO interface. The study showed an impressive 81% yield of formate from bicarbonate at 225 °C, representing a significant improvement over the previous research without catalysts, which required 300 °C as the reaction temperature.
Liu et al.80 found that formic acid could also be converted to methanol with Zn as the reductant and Cu as the catalyst. The highest methanol yield was 32% at 300 °C for 5 h utilizing 6.5 mmol Cu and 12 mmol Zn. In this reaction, H2 was derived from both formic acid decomposition (HCOOH = CO2 + H2) and water splitting, coupled with Zn oxidation to ZnO. Subsequently, the newly generated H2 dissociated into H(a) on the surface of Cu/ZnO and H(a) interacted with oxygen atoms in ZnO to produce H2O, resulting in the formation of an oxygen vacancy in ZnO1−x. Following this, CO2 adsorbed on the oxygen vacancies of ZnO1−x, and residual H(a) attacked the C–O bond near Zn to form ZnO–C–O. Therefore, the catalytic cycle concluded with the regeneration of Cu/ZnO and the formation of CH3OH.
4 Hydrothermal CO2 reduction with biomass or polymer wastes
Biomass is defined as all organic substances originating from plants, encompassing algae, trees, and crops. The composition of biomass differs based on its source but typically includes a wide range of organic compounds like lignin, proteins, carbohydrates (such as cellulose, starch, hemicellulose), and lipids. These compounds are characterized by functional groups such as hydroxyl (–OH), aldehyde (–CHO), or amino (–NH2) groups. This section commences with a comprehensive discussion of the hydrothermal reduction of CO2 by carbohydrates and their derivatives, and then transitions to examining protein-containing biomass such as microalgae and finally lignin. Ultimately, the scope of reductants expands from biomass to polymer wastes, which share similar functional groups like hydroxyl (–OH), with the aim of broadening the range of reducing agents to more economically viable sources.4.1 Hydrothermal reduction of CO2 by hydroxyl containing alcohols
Studies of CO2 reduction with carbohydrates as reductants began with the examination of hydroxyl containing chemicals, given that carbohydrates are characterized by their polyhydroxy structure, and methanol as the simplest alcohol was selected as a representative. Wang et al.35 found that formate can be successfully produced through NaHCO3 reduction with methanol. Thermodynamic analysis showed that this process is favorable even at 453 K (eqn (1)).| CH3OH + 2HCO3− → HCOOH + 2HCOO− + H2O | (1) |
| ΔrHθ (298 K) = −74.7 kJ mol−1; ΔrGθ (298 K) = −32.1 kJ mol−1 |
| ΔrHθ (453 K) = −49.7 kJ mol−1; ΔrGθ (453 K) = −15.8 kJ mol−1 |
A formate yield of 68% was achieved in hydrothermal NaHCO3 reduction with methanol at 180 °C using a Pd0.5Cu0.5/C catalyst. 13C-qNMR analysis was conducted to distinguish and quantify formate production from NaHCO3 and CH3OH. Approximately 55% of the formate originated from HCO3− reduction, while the remaining 45% resulted from CH3OH oxidation. The superiority of Pd0.5Cu0.5/C catalyst was concluded not only due to its cost-effective characteristic of combining Pd and Cu but also to its role in enhancing hydrogen production and subsequent formate generation by altering the electronic structure of Pd by Cu. XPS analysis confirmed synergistic interactions between Pd and Cu. Specifically, the Pd 3d peaks in the alloy exhibited a lower binding energy compared to those in Pd/C, indicating electron transfer from Cu to Pd. This electron shift modifies the electronic structure of Pd, reducing the hydrogen binding energy on its surface, thereby enhancing catalytic hydrogen generation, as shown in Fig. 8a. Furthermore, the altered Pd d-band center, distanced from the Fermi level due to Cu integration, lowers the formate adsorption energy, thus facilitating its production and selectivity in HCO3− reduction.
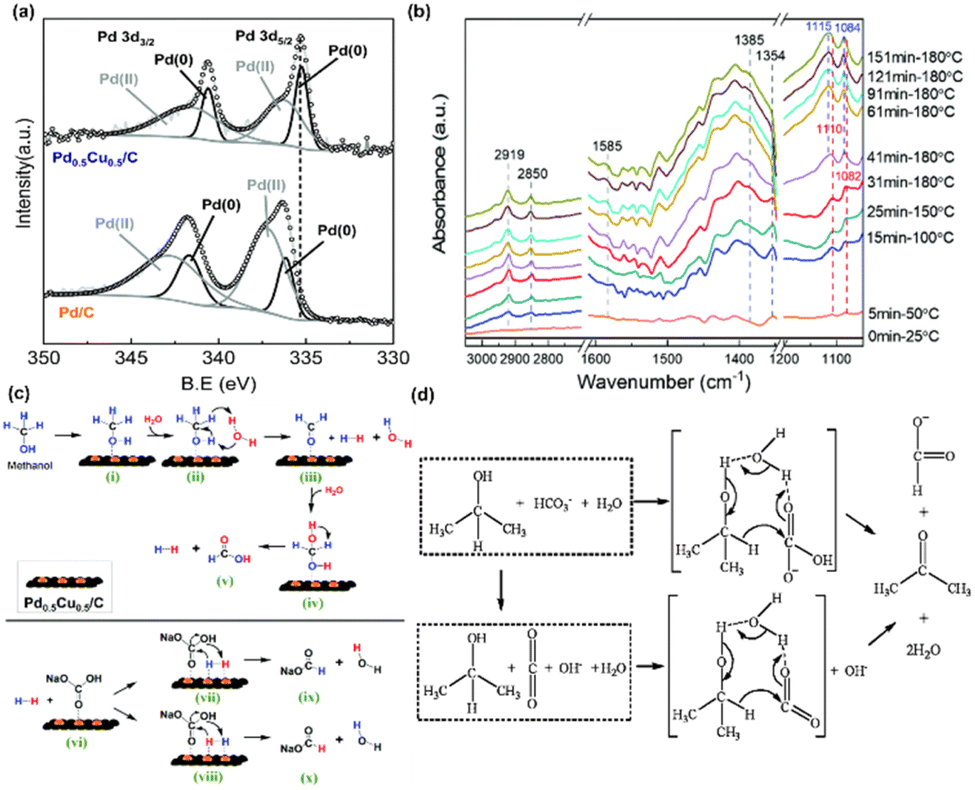 | ||
| Fig. 8 (a) XPS spectra of Pd 3d in Pd0.5Cu0.5/C and Pd/C, (b) operando ATR-FTIR subtraction spectra recorded during NaHCO3 reduction with methanol, (c) reaction mechanism of HCO3− reduction into formate with methanol in water on the Pd0.5Cu0.5/C catalyst.35 This figure has been reproduced from ref. 35 with permission from The Royal Society of Chemistry, copyright 2021. (d) Possible mechanism for hydrogen-transfer reduction of NaHCO3 with isopropanol.81 This figure has been reproduced from ref. 81 with permission from The Royal Society of Chemistry, copyright 2011. | ||
Operando hydrothermal ATR-FTIR studies, as detailed in Fig. 8b, revealed the transformation of reactants during the Pd0.5Cu0.5/C-catalyzed reaction. An increase in temperature and reaction time was found to enhance formate production. A blue shift observed in the peaks of the methanol C–O bond at 1110 and 1082 cm−1 suggested its oxidation to formaldehyde. This implies a mechanism in which CH3OH oxidizes to HCHO, generating H2, and then HCHO reacts with H2O to produce H2 and HCOOH. The adsorption of H2 and HCO3− on Pd0.5Cu0.5/C results in formate formation through a nucleophilic attack on the CO bond and C–OH bond cleavage. The potential reaction mechanism proposed for methanol and bicarbonate reaction under hydrothermal conditions via Pd0.5Cu0.5/C catalyst is illustrated in Fig. 8c. Furthermore, the study explored the reactivity of ethanol, 1-propanol, and 1-butanol in reducing NaHCO3 to formate, testing the general feasibility of alcohols. These alcohols demonstrated activity comparable to methanol while being converted into their respective acids. Interestingly, the reactivity of these alcohols was found to increase marginally with the elongation of their carbon chain.
In addition to examining hydroxyl groups on terminal carbon atoms, isopropanol, the simplest secondary alcohol, was also chosen as a reductant for hydrothermal CO2 reduction, with its operational parameters and reaction mechanism systematically investigated.81 The formate yield was observed to gradually increase with the increase in CO2 quantity, temperature, and reaction time. Conditions of 300 °C and 240 minutes were determined to be optimal. However, a moderate NaOH concentration was necessary to produce formate, as the base aids in improving the enthalpy of CO2 reduction, while excess base can result in CO2 conversion to Na2CO3. Generally, a formate yield of approximately 70% was achieved under conditions of 300 °C, 0.25 mol per L isopropanol, 1.0 MPa CO2, and 0.75 mol per L NaOH. Drawing on the traditional Meerwein–Ponndorf–Verley (MPV) hydrogen-transfer reduction mechanism, which involves a cyclic transition state and the catalytic role of water molecules, a potential mechanism is proposed, as shown in Fig. 8d. Initially, two hydrogen bonds form between three molecules (isopropanol, H2O, and HCO3− or CO2 resulting from the decomposition of NaHCO3), rendering the carbonyl-carbon of HCO3− or CO2 and the hydride of isopropanol more electropositive. Subsequently, the hydride from isopropanol attacks the carbonyl-carbon of HCO3− or CO2, leading to the formation of a cyclic transition state. Ultimately, following the intramolecular hydride shift, formate and acetone are produced, with the recovery of water molecules.
Beyond methanol and isopropanol, hexanol and glycerin, which possess multiple hydroxyl groups, were also utilized for hydrothermal CO2 reduction. Yang et al.40 found that operating parameters significantly impacted CO2 conversion. Specifically, the formic acid yield linearly increased with the initial CO2 loading, and a similar trend was observed for water filling, reaction time, and temperature. However, an optimal pH value was necessary to produce formic acid. The yield of formic acid dramatically increased from a pH of 6.5 and peaked at a pH of 7.5, but excessive alkalinity could result in the conversion of CO2 to Na2CO3.81 During this process, a maximum yield of 76% for formic acid was obtained.
Under alkaline hydrothermal conditions, the reaction of glycerol with NaHCO3 primarily yields pyruvate, glycolate, lactate, formate, and acetate. It is noteworthy that glycolate was initially identified in reactions involving other polyols with NaHCO3. In these reactions, formate tends to yield CO2 through decarboxylation in the liquid phase. Wang82 combined the results of previous research32,83–85 and outlined three main competitive pathways for glycerol transformation: reduction of HCO3− and simultaneous conversion of glycerol (the lower pathway in Fig. 9), self-degradation of glycerol in alkaline media to lactate (the middle pathway in Fig. 9), and reverse aldol condensation yielding glycolate, formate, and lactate (the upper pathway in Fig. 9). For the simultaneous reduction of HCO3− and conversion of glycerol, Shen32,86 further proposed a potential transfer hydrogenation mechanism where glycerol first undergoes dehydration to produce 2-hydroxypropanone, followed by keto–enol tautomerization to acetol. In the second step, potential hydrogen bonding between three molecules (acetol, water, and CO2) enhances the electropositivity of the carbonyl carbon on CO2 and the hydride ion on acetol. The hydride ion then attacks the carbonyl carbon, forming a cyclic transition state and ultimately yielding pyruvaldehyde and formate with the generation of a water molecule.
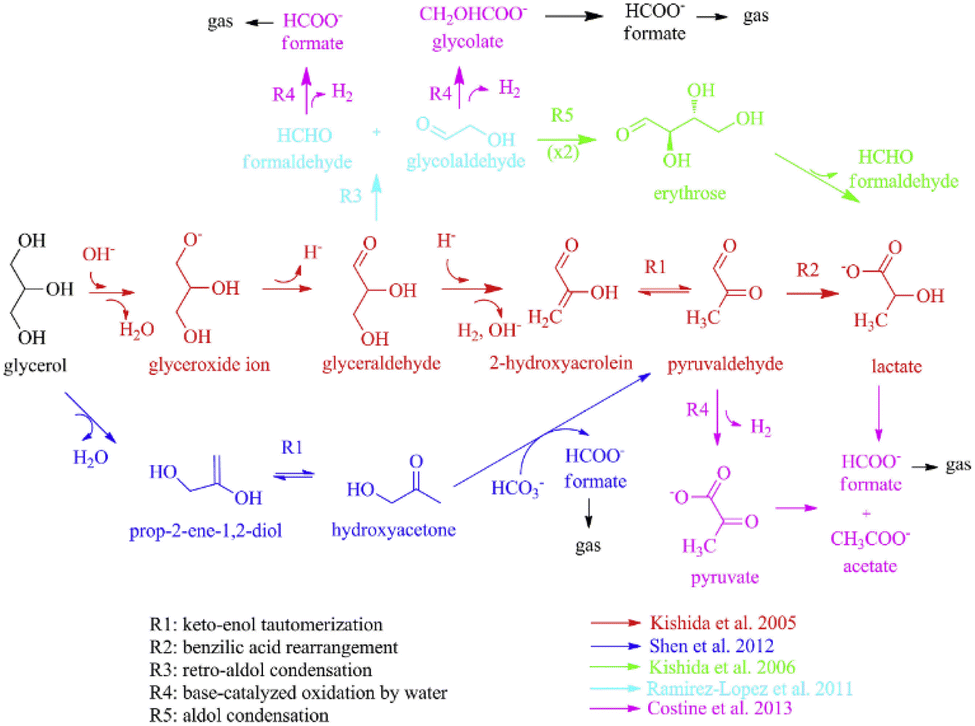 | ||
| Fig. 9 Possible reaction mechanisms of hydrothermal reduction of HCO3− by glycerol.82 This figure has been reproduced from ref. 82 with permission from Elsevier, copyright 2016. | ||
Catalysts are crucial in the glycerol-driven reduction of bicarbonate. In the reduction of bicarbonate using Ru/ZrO2 catalysts and employing biodiesel waste glycerol as a reductant, a maximum formate yield of 25.1% was achieved, which is a significant increase compared to the mere 4.2% yield observed in the absence of catalysts.87 The ruthenium component in the catalyst not only facilitates glycerol dehydrogenation but also enhances the hydrogenation of HCO3−. When employing the Pd/AC catalyst in the bicarbonate reduction with glycerol, the conversion of glycerol to lactate at 240 °C results in yields up to 55%, while simultaneously producing approximately 30% formate.88 The reaction encompasses two potential hydrogen transfer pathways: glycerol dehydrogenation forming H2 gas, which then hydrogenates bicarbonate, or direct transfer of hydrogen from glycerol to bicarbonate. Both pathways are catalyzed on the active sites of the Pd catalysts. For direct hydrogen transfer, firstly, the hydrogen donor dehydrogenates to form a palladium hydride, which then transfers the hydride to the hydrogen acceptor; secondly, the hydrogen donor and acceptor co-adsorb onto the Pd surface, enabling direct hydrogen transfer on the catalyst surface without forming hydrides.
Jaedeuk Park89 devised and evaluated a two-pot/two-step transfer hydrogenation method (2P2S) for CO2 reduction with glycerol. This method represents a comprehensive conversion process utilizing two reactors. The 2P2S method not only enhanced the yields of lactate and formate but also streamlined product purification and separation, ultimately producing lactate and formate or methyl formate (MF) while reducing the consumption of sulfuric acid (H2SO4) and potassium sulfate (K2SO4) waste, offering an environmentally friendly production route. Techno-economic and life cycle assessments reveal that the utilization of CO2 is crucial for attaining significant economic benefits and minimizing climate change impact. Case studies demonstrate the lowest minimum product selling price (MPSP) of $1.25 per kg LA and the highest net present value (NPV) of $33.6 million, showing the economic competitiveness of the process. Moreover, the process significantly reduces material costs, as CO2 utilization in the lactate and methyl formate production pathways reduces sulfuric acid usage and generates internal potassium bicarbonate (KHCO3) through recycling.
4.2 Hydrothermal reduction of CO2 by carbohydrates
As glucose is the primary product of lignocellulosic biomass hydrolysis, the reduction of CO2 with glucose was systematically investigated. Yang et al.90 achieved 54% formate yield under conditions of 0.125 mol per L glucose and 3 mol per L NaHCO3 at 300 °C, employing a two-step reaction strategy. They proposed that the aldehyde group of glucose reduces HCO3− to HCOO−via a direct hydrogen transfer mechanism concurrently with the oxidation of glucose to gluconic acid. Subsequently, gluconic acid undergoes direct α-cleavage, resulting in the formation of formate and pentose (C5 aldose). Further reactions lead to a gradual reduction in the carbon number of pentose, followed by a mechanism identical to the aforementioned process, culminating in the production of formate. As depicted in Fig. 10, this process results in a total of twelve formate and formic acid molecules originating from the reduction of NaHCO3 by glucose (pathway I). Conversely, side reaction pathways (pathway II) also play a role in this reaction. Under hydrothermal conditions, the produced pentose (C5 aldose) may undergo dehydration, keto–enol tautomerism, retro-aldol reactions, and benzilic acid rearrangement due to the long carbon chain, and eventually converts to products such as glyceraldehyde or acetaldehyde, which reduce NaHCO3 to yield formate with themselves oxidized to lactic acid and acetic acid. Through this pathway, only two molecules of NaHCO3 can be reduced. To optimally minimize the influence of side reactions, Yang et al.90 developed a two-step reaction strategy focusing on cutting the carbon chain of glucose or cellulose, which could minimize the side reactions of multi-carbon aldoses. With this strategy, as high as 74% formate yield of NaHCO3 reduction with direct use of cellulose was achieved.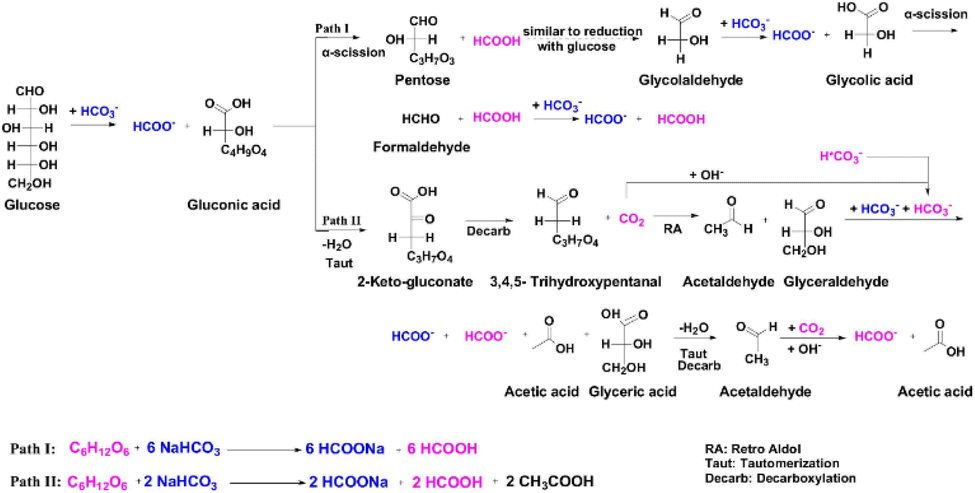 | ||
| Fig. 10 Proposed reaction pathways of NaHCO3 reduction with glucose.90 | ||
Support for the direct hydrogen transfer mechanism was provided by operando hydrothermal diffuse reflectance infrared Fourier transform spectroscopy (OH-DRIFTS). Signals of formaldehyde and glycolic acid indicated glycolaldehyde (the intermediate of glucose) oxidation, which aids NaHCO3 reduction, in assistance with the above proposed reaction pathway. The ν(C–H) signal of formate exhibited a blue shift, indicative of an elevated C–H bond energy. This enhanced C–H bond energy, likely caused by another functional group linked to the C atom, is attributed to the inductive effect dispersing the electron cloud of the central C atom, suggesting orthoformate formation, which forms formate through loss of H2O. This shift, absent in NaHCO3 reduction with gaseous H2, directly proves NaHCO3 reduction via hydrogen transfer between the aldehyde group of glucose and NaHCO3.
Andérez-Fernández and colleagues91 achieved formate yields of 65% for glucose and 35% for fructose at 300 °C, with 0.05 mol per L reactants and 0.5 mol per L NaHCO3. They elucidated the reasons behind the yield disparity, attributing it to the decomposition of glucose into three C2 molecules (such as glyoxal) and fructose into two C3 molecules (like glyceraldehyde and pyruvic acid). It was discovered that under hydrothermal conditions, sugars initially undergo retro-aldol reactions or dehydration, forming smaller compounds like 5-hydroxymethylfurfural (5-HMF) and furfural. These intermediates can also reduce NaHCO3, yielding a formate production rate of 15%, and through a series of reactions, including dehydration, keto–enol tautomerism, retro-aldol reactions, and benzilic acid rearrangement, they eventually convert to products such as lactic acid, acetic acid, and formaldehyde. In the early stages of the reaction, formic acid is directly produced from formaldehyde via dehydrogenation. As the reaction progresses, 13C-NMR analysis detects formate from NaHCO3 reduction gradually replacing the formic acid from organic matter. The proposed reaction mechanism includes hydrogen transfer and redox reactions between formaldehyde and NaHCO3. Under hydrothermal conditions, formaldehyde may decompose through Cannizzaro reaction or dehydrogenation, generating formic acid and hydrogen, with the latter reacting with NaHCO3 to form formate. This mechanism also involves a collection of reversible reactions during which formic acid and formaldehyde decompose into CO, CO2, and H2 at high temperatures, leading to a decrease in the total formate yield.92
Subsequently, a continuous flow reactor with the capacity to process 1.2 liters of bicarbonate solution per hour was developed. At shorter reaction times, the primary mechanism involves the decomposition of glucose into formic acid and other by-products, while at longer reaction times, a higher yield of formate was produced from bicarbonate reduction. These results suggest that the reduction pathway of bicarbonate occurs through the oxidation of by-products rather than the direct oxidation of glucose itself, requiring a longer reaction time to achieve a higher proportion of formate produced from bicarbonate reduction. It is hypothesized that glucose decomposes into glyceraldehyde and glycolaldehyde, which then further break down into formaldehyde, a crucial intermediate in the reaction with bicarbonate to produce formate. Other final products include acetic acid and lactic acid, alternative products of the decompositions of glyceraldehyde and glycolaldehyde.93
Using a Pd/AC and Pt/AC bimetallic catalyst in ethanol–water solvents at room temperature, Ding et al. obtained a 46% yield of formate and 21% yield of sorbitol by coupling glucose dehydrogenation with CO2 hydrogenation. The key intermediate, ethyl carbonate, formed in basic aqueous ethanol, is crucial for the hydrogenation reaction. The reaction yields are significantly influenced by the initial pH and the amounts of glucose, ammonium carbonate, and KOH additives; the optimal pH for maximizing formate yield is around 11.73.95 Oh et al. further found that the addition of alcohols significantly enhanced the catalytic activity.94 A 50% 2-propanol solution exhibited the highest formic acid yield of 30.5%. Alkali metal carbonates showed superior catalytic activity compared to bicarbonates, with larger alkali metal cations effectively increasing the yield due to higher salt solubility. The presence of large alkali metal cations or alcohols enhanced reactivity by increasing the interaction between reactants and the metal surface, attributed to the water structure-breaking effect, as illustrated in Fig. 11. This was particularly crucial for glucose anions and bicarbonate, indicating a stronger hydration sphere for anions than neutral molecules. By exploiting the dual effect of large alkali metal cations and alcohols, the carbohydrate sources could be extended to galactose and lactose.
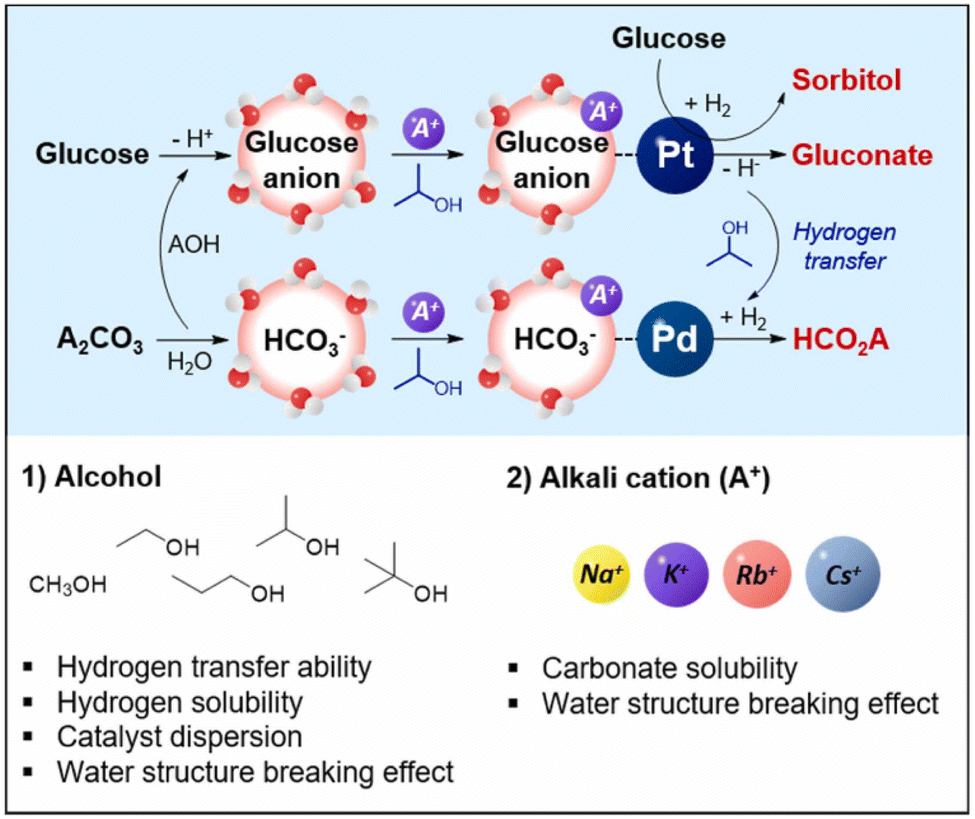 | ||
| Fig. 11 The proposed reaction mechanism for simultaneous transformation of glucose and alkali carbonate.94 This figure has been reproduced from ref. 94 with permission from Elsevier, copyright 2022. | ||
4.3 Hydrothermal reduction of CO2 by microalgae or lignin
In addition to carbohydrate biomass, microalgae are regarded as a third generation of biomass energy due to advantages such as rapid growth rate, high photosynthetic efficiency, and no competition for food resources. In contrast to carbohydrate biomass, the chemical compositions of microalgae include abundant lipid and protein. The former is comprised of glycerol and carboxylic acid, for which the reduction of CO2 is summarized with emphasis on the reductive capacity of hydroxyl group, and the latter is characterized by the amino group, which is also a reductive functional group in addition to the hydroxyl and aldehyde groups. However, few studies have recognized and concentrated on the utilization of the amino group in biomass, particularly in driving CO2 reduction. In this section, we summarize the hydrothermal reduction of CO2 with amino-containing model compounds and actual microalgae biomass.Yang et al.39 conducted pioneering research on the feasibility of hydrothermal CO2 reduction using Spirulina, a commonly known microalga. Comparative experiments demonstrated that the sole conversion of Spirulina yielded over 15 complex products, primarily including organic acids (acetic acid, formic acid, and propionic acid), chain amides (N-ethyl-acetamide, N-methyl-acetamide, and acetamide), N-substituted 2,5-diketopiperazines (N-ethyl-2,5-diketopiperazine and N-methyl-2,5-diketopiperazine), and lactams (N-ethyl-2-pyrrolidinone, N-methyl-2-pyrrolidinone, 2-pyrrolidinone, 2-piperidinone, and 2-oxoazepane). However, the distribution and selectivity of products altered when Spirulina reacted with NaHCO3, and the yields of organic acids and lactams increased obviously, with a sharp decrease in the production of chain amides and diketopiperazines, and, combined with 13C-NMR analysis, the production of formate from the reduction of NaHCO3 was confirmed. Systematic research on the reaction parameters indicated that Spirulina conversion was enhanced by increasing NaHCO3 concentration. Elevating the temperature and extending the reaction time can enhance the conversion of both Spirulina and NaHCO3. In this instance, 325 °C and 2 h represented the optimal conditions for formate production. The addition of NaOH augmented formate production from NaHCO3, while its effect on the conversion of Spirulina was limited. After acknowledging the possible reaction mechanism, the range of reductant substrates was expanded from Spirulina to Chlorella and microalgae residue, demonstrating the general applicability of the reaction protocol.
Amino acid is easily obtained from protein-rich microalgae biomass. Li et al.96 chose L-alanine as the model chemical to verify the feasibility of hydrothermal CO2 reduction by amino acid. In the presence of NaHCO3, there was a significant increase in formate yield (9.3%), a finding also corroborated by 13C-NMR analysis with NaH13CO3 as the reactant. A screening of active metal catalysts (Cu, Co, Ni, Ru, Pd, and Pt) and suitable supports (SiO2, CeO2, ZrO2, ZnO, and gamma-Al2O3) for hydrothermal NaHCO3 reduction with L-alanine was conducted, where Pd/γ-Al2O3 exhibited higher activity, achieving a formate yield of 49.0% under conditions of 0.8 mol per L L-alanine and 0.8 mol per L NaHCO3 at 325 °C for 2 hours with 0.08 g of 5% Pd/γ-Al2O3.
As depicted in Fig. 12a and b, OH-DRIFTS was also utilized to ascertain the reaction mechanism of amino groups reducing NaHCO3. The peaks at 3500–3300 cm−1, exhibiting a growing negative variation pattern, were attributed to the –NH2 group of L-alanine, indicating continuous consumption of –NH2 in L-alanine during the reaction.97 Conversely, the NH4+ signal in the range of 3000–2800 cm−1 exhibited a nearly unchanged state after 5 minutes, while this peak tended to intensify due to the hydrolysis of amido at a rapid rate, implying the continuous consumption of NH4+ over time. The signal at 1260 cm−1 was assigned to hydroxylamine (R–NHOH). Given that hydroxylamine is the product of –NH2 oxidation, this signal provided direct evidence of NaHCO3 reduction via –NH2 oxidation. It is noteworthy that the R–NHOH peaks displayed an increasing and then decreasing trend, suggesting that R–NHOH was an intermediate in the –NH2 oxidation process. Additionally, absorbances of N–O and NO2− were observed at 1642 cm−1 and 1570 cm−1, respectively. Based on these results, the proposed mechanism of amino acid reduction of NaHCO3 to formate is as follows (Fig. 12c). –NH2 is first oxidized into hydroxylamine by releasing one molecule of H2, and then hydroxylamine forms two molecules of H2 by oxidizing to R–NO and R–NO2. Subsequently, NO2− interacts with NH4+ (hydrolyzed from the amino acid) to generate N2 and H2O, with the concurrent formation of alcohols from the amino acid. Meanwhile, NaHCO3 hydrogenation occurs on the catalyst surface, where *H is adsorbed on the Pd site and HCO3− is adsorbed on the support γ-Al2O3, leading to the reduction of NaHCO3 to formate.51
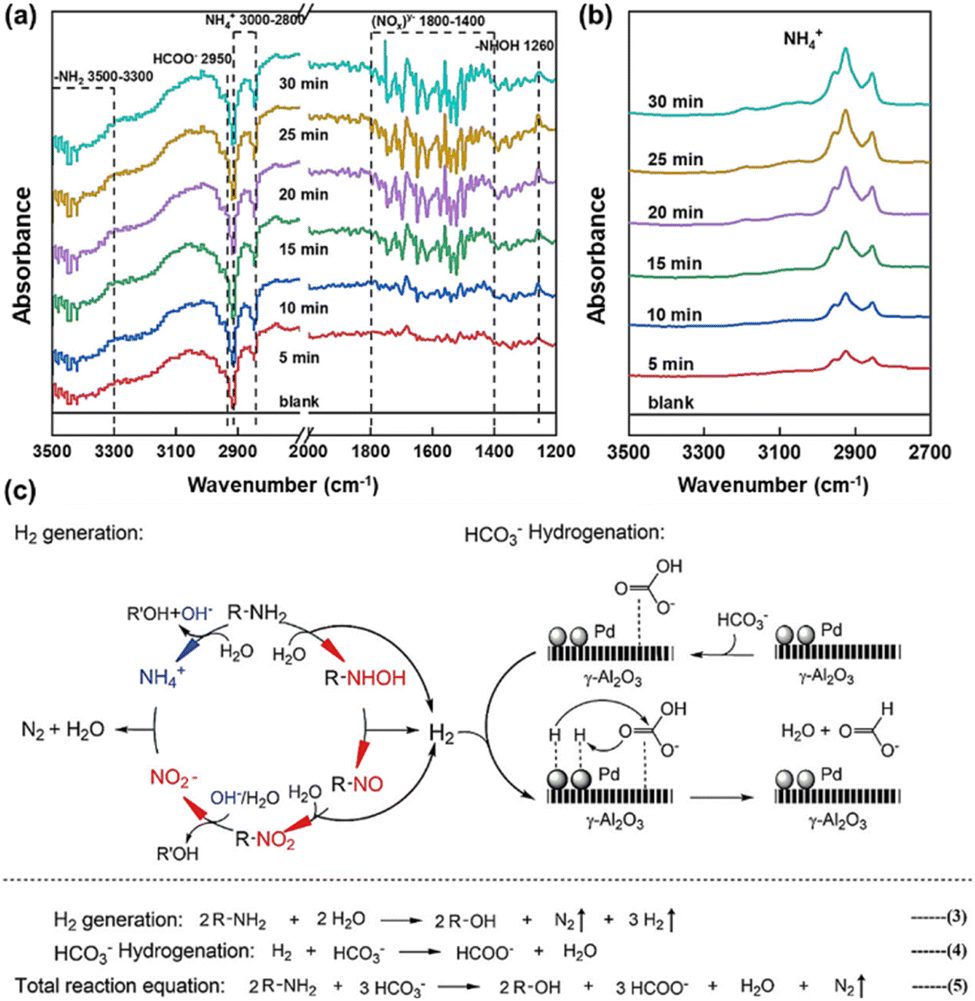 | ||
| Fig. 12 OH-DRIFTS spectra of L-alanine reaction with NaHCO3 (a) or without NaHCO3 (b) (only the absorbance signal for NH4+ was displayed in spectra (b)), (c) proposed reaction mechanism of NaHCO3 hydrothermal reduction with L-alanine.96 This figure has been reproduced from ref. 96 with permission from American Chemical Society, copyright 2021. | ||
The reduction efficiency of aromatic ring-containing lignin has also garnered attention. Andérez-Fernández91 investigated the efficacy of CO2 reduction using various lignin model compounds. Under the conditions of 300 °C for 180 minutes, phenol yielded only 2% formic acid yield. In contrast, higher formic acid yields were observed with resorcinol and catechol solutions (19% and 9%, respectively). The highest yield of 51% was achieved with a vanillin solution, followed by a 24% yield with guaiacol. These results suggest that aromatic compounds possessing ortho or para hydroxyl groups exhibit greater CO2 reduction activity. Oxidation by-products from lignin derivatives suggest that the reaction may proceed via a ring-opening mechanism. Vanillin is hypothesized to undergo Cannizzaro disproportionation to vanillic acid and vanillic alcohol, with the latter being involved in CO2 reduction. Aromatic compounds in high-temperature water may be oxidized to quinone groups, which are prone to ring opening. The proposed CO2 reduction mechanism involves oxidation through a cyclic transition state, wherein the hydrogen atom to be transferred is located on the hydroxyl group.
Apart from biomass, recent advancements have highlighted the potential of biomass polymer waste for CO2 reduction. Specifically, a study98 demonstrated that when 5.0 g per L sugarcane bagasse solution reacted with 42.0 g per L NaHCO3 at 250 °C for 3 h, a maximum formate yield of 10% was achieved. Experiments using various bases, including NaOH and phosphate buffer, indicated that alkaline conditions favor biomass waste oxidation to acids while impeding the conversion of CO2, dissolved as bicarbonates/carbonates. The addition of NaHCO3 serves not only as a secondary carbon source, converted to formate, but also as an oxidant, thereby enhancing the conversion of biomass wastes into organic acids.
4.4 Hydrothermal reduction of CO2 by polymer wastes
Plastics have revolutionized modern life due to their low cost and utility in a wide range of applications. Regrettably, the accumulation of artificial polymers in landfills and environment has led to a global pollution crisis,99 which in turn has accelerated research on the conversion of plastic waste into value-added chemical products.100,101 Recently, Ma et al. developed a process for the co-utilization of waste plastics and CO2. They designed a triple-reaction coupling involving CO2 hydrogenation, methanolysis of polyethylene terephthalate (PET), and hydrogenation of dimethyl terephthalate (DMT), effectively degrading PET and utilizing CO2. As shown in Fig. 13a, CO2 as a solvent precursor overcomes the thermodynamic limits of the reactions. CO2 is hydrogenated to methanol, which then decomposes PET into DMT and ethylene glycol (EG). Subsequently, DMT is hydrogenated into higher-value chemicals such as dimethyl 1,4-cyclohexanedicarboxylate (DMCD) and para-xylene (PX). This coupled reaction significantly improves the yield of CO2 hydrogenation or PET methanolysis by 2 to 7 times compared to uncoupled reactions. In addition, with tandem catalysis, a promotion of the synergistic coupling of CO2 hydrogenation and PET methanolysis was achieved to produce valuable chemicals like cyclohexanedicarboxylate over Cu4Fe1Cr1 catalyst.102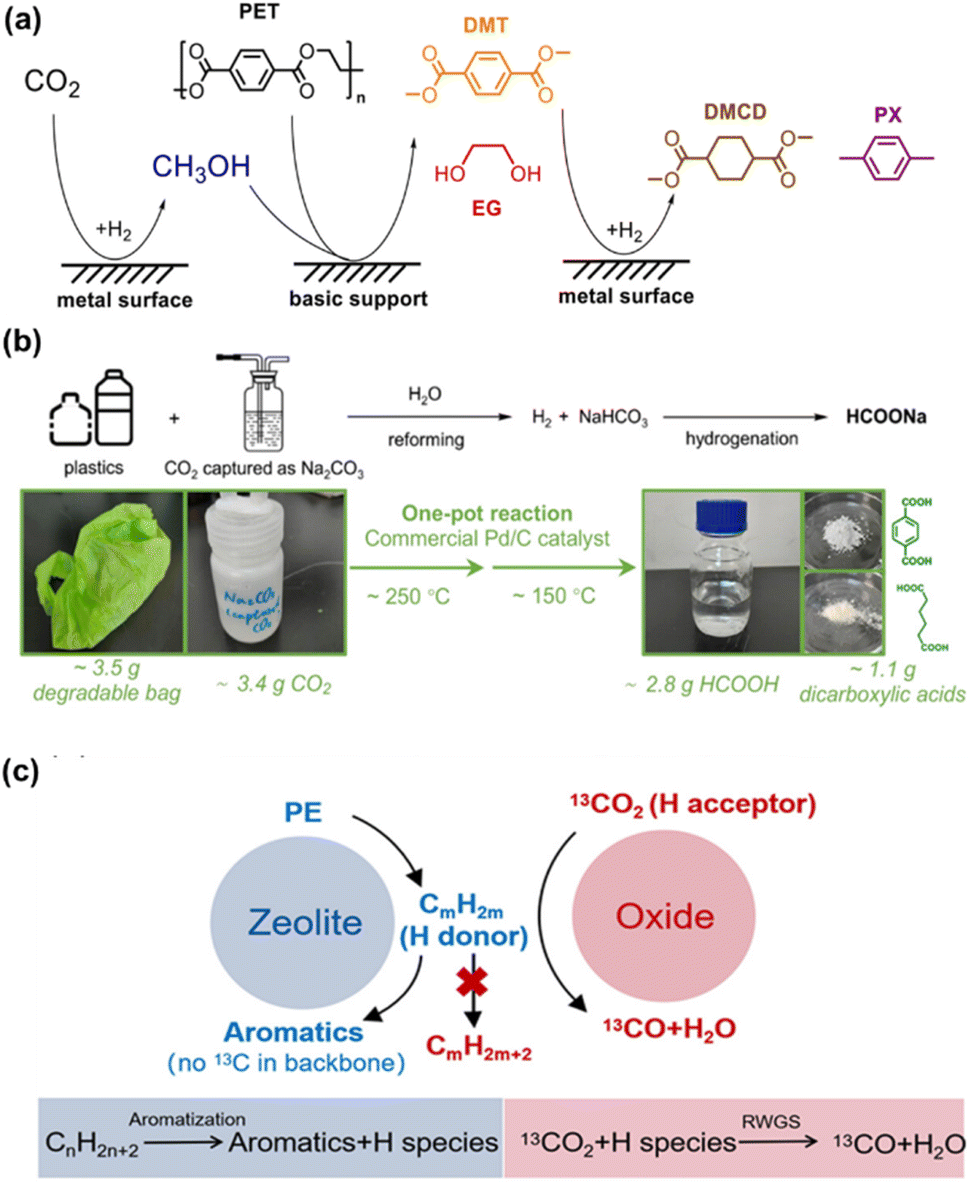 | ||
| Fig. 13 (a) The scheme of CO2 hydrogenation and PET degradation dual-promoted reaction system.102 This figure has been reproduced from ref. 102 with permission from Wiley-VCH GmbH, copyright 2022. (b) Plastic and CO2 in one pot, two-step catalytic conversion process.103 This figure has been reproduced from ref. 103 with permission from American Chemical Society, copyright 2024. (c) Proposed mechanism for the coupling reaction of CO2 with PE over composite catalyst.104 | ||
Under hydrothermal conditions, a one-pot, two-step catalytic method using commercial Pd/C catalysts enabled the conversion of polyesters like polyglycolic acid (PGA), polyethylene terephthalate (PET), and poly(butylene adipate-co-terephthalate) (PBAT) with sodium carbonate into sodium formate, without external hydrogen. As shown in Fig. 13b, at 250–270 °C, polyesters undergo aqueous modification with Pd/C catalysts, breaking down into smaller molecules. Subsequently, at 150 °C, the hydrogen generated in the first step is used for the hydrogenation of NaHCO3. This technique effectively resolves the contradictions in reaction temperature and pressure between plastic reforming and NaHCO3 hydrogenation, achieving a 79% yield of HCOONa.103 Additionally, CO2 facilitates the conversion of polyethylene (PE) into aromatics, achieving synergistic resource utilization, as shown in Fig. 13c. Utilizing Cu–Fe3O4 and Zn/ZSM-5 tandem catalysts at temperatures below 400 °C, an efficient conversion with 64.0% aromatic selectivity was achieved. This process not only exceeds the 50% theoretical limit of aromatic selectivity in PE aromatization but also, through 13C isotope studies, confirms CO2's role in the reverse water–gas shift (RWGS) process. CO2, acting as a hydrogen scavenger and converting to CO, significantly reduces hydrogen transfer reactions in olefinic intermediates, decreasing the fraction of light alkanes and enhancing the formation of aromatic compounds. Notably, CO2 does not participate in extending the carbon chain of aromatic compounds.104
Furthermore, the synergistic transformation of PVC plastics and CO2 was achieved even without the use of any external metal-based catalysts. 13C-NMR analysis was performed on the liquid sample derived from the reaction of NaH13CO3 and PVC. As depicted in Fig. 14a, the labeling experiment using NaH13CO3 revealed a strong signal attributed to H13COO− at a chemical shift of 171.1 ppm in the 13C-NMR spectrum, confirming that formate originated from NaHCO3.105 Research on reaction characteristics indicated that higher temperatures (over 300 °C) and longer times (more than 8 hours) can lead to formate decomposition, and higher NaHCO3 concentrations (more than 4 mmol) can cause an insufficient amount of PVC as the reductant. Under conditions of 125 mg PVC, 300 °C, 8 hours, 1 mmol NaHCO3, and 2 mmol NaOH, approximately 16% formate yield was achieved.
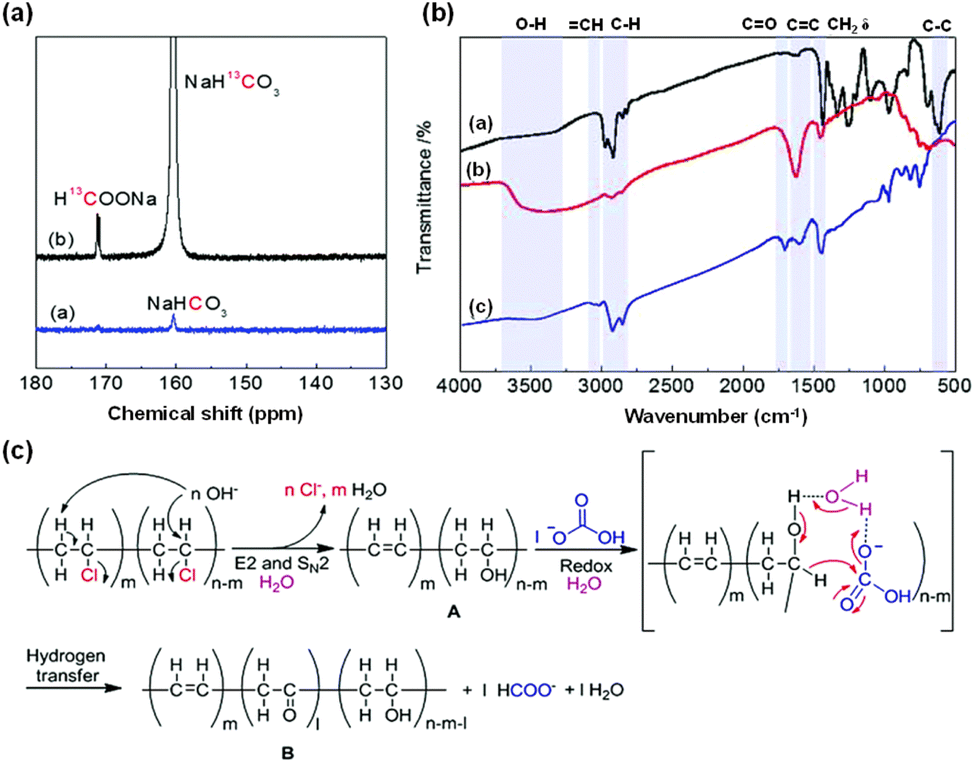 | ||
| Fig. 14 (a) 13C-NMR spectra of liquid samples for the reactions of PVC with NaHCO3 or NaH13CO3, (b) FT-IR spectra of PVC before and after the reaction under different conditions ((b-a) pure PVC, (b-b) solid product for the reaction of PVC + NaOH, (b-c) solid product for the reaction of PVC + NaOH + NaHCO3), (c) proposed mechanism of HCO3− reduction to formate using PVC under hydrothermal conditions.105 This figure has been reproduced from ref. 105 with permission from The Royal Society of Chemistry, copyright 2020. | ||
FT-IR analysis was also utilized to investigate the reduction of bicarbonate to formate using PVC under various reaction conditions. In Fig. 14b, absorption signals of CC and –OH stretching vibrations were observed at 1621 cm−1 and 3200–3700 cm−1, respectively, while the absorption peak of the C–Cl stretching vibration disappeared following the reaction between PVC and NaOH. Collectively, these results indicate that the OH− in water attacks the β-H and α-C of PVC, resulting in the formation of CC, CC–H, and CH–OH groups. Conversely, after the reaction of PVC with NaHCO3 and NaOH, in contrast to the reaction of PVC with NaOH alone, the absorption peak of the CO stretching vibration (1701 cm−1) appeared, and the peak of the –OH stretching vibration decreased significantly with the addition of NaHCO3, implying the formation of the CH–OH group through an SN2 mechanism and its further conversion to CO in the presence of NaHCO3. Overall, the proposed reaction mechanism of NaHCO3 for the hydrothermal reduction of NaHCO3 to formate with PVC is depicted in Fig. 14c. Initially, dichlorination of PVC occurs under hydrothermal conditions, forming intermediate A by the elimination of OH− attacking on the β-H and the substitution of OH− attacking on the α-C; then, the in situ formed CH–OH is oxidized to CO by HCO3−, while HCO3− is reduced to formate.
Additionally, hydrothermal CO2 reduction using ethylene propylene diene monomer (EPDM) as a model compound for sulfur-containing rubber was conducted. The results showed that formic acid was the main product, and carboxylic acids with 2–4 carbon atoms were also formed.45 It should be noted that a higher temperature of 400 °C was required, and, upon increasing the temperature to 450 °C, C5–C6 products were detected, during which a 20% CO2 reduction efficiency was achieved. From the cracking of EPDM, straight-chain n-alkanes with more than nine carbon atoms were formed.
5 Roles of water in hydrothermal CO2 reduction
The general feasibility of reductants for CO2 reduction under hydrothermal conditions, ranging from metals to biomass and even plastics, indicates the unique property of HTHP water in invoking the reductive power. The commonly recognized advantages of HTHP water include: (1) the excellent solubility of organics and gases; (2) the increase in the concentration of H+/OH− ions which facilitates acid/base-catalyzed reactions; (3) the significantly reduced diffusion rate of supercritical water to nearly 1/100 that of liquid water; and (4) the weak interaction of hydrogen bonds among water molecules, which reduces mass transfer resistance at the interface, leading to efficient heat transfer, rapid mass diffusion, and an enhanced reaction rate. However, the mechanistic study of CO2 hydrothermal reduction, particularly through in situ observations with FTIR, suggests that, in addition to these advantages, a potentially significant yet often overlooked homogeneous catalytic function of HTHP water might exist.In hydrothermal CO2 reduction with methanol, the role of water was investigated carefully.35 Wang et al. discovered that water is crucial for the initial step of methanol activation, as no H2 and HCHO were detected in the reaction with only methanol in the absence of water. Furthermore, the generation of H2 from methanol, both with and without H2O, was analyzed through DFT calculations. As depicted in Fig. 15a–c, the added H2O molecule functioned as a proton acceptor, forming a deformed H3O-cation-like conformation with the transition state hydrogen atom from the methanol molecule. This significantly reduced the activation energy required for ionic water dissociation. Consequently, the energy barrier for H2 formation from methanol was lowered from 378 kJ mol−1 without H2O to 220 kJ mol−1 with H2O present (Fig. 15d). Additionally, an isotopic tracing reaction with D2O substituting H2O was conducted, with NaH13CO3 as the carbon source to differentiate formate from NaHCO3 reduction. As illustrated in Fig. 15e and f, a peak corresponding to D13COO− was distinctly observed in the D-NMR spectrum, consisting of two parallel peaks. The 13C-NMR image revealed that the NaH13CO3− derived formate contained three parallel peaks attributed to the resonance effect between 13C and D. The COSY spectrum of the liquid sample post-reaction between NaH13CO3 and methanol in D2O is also depicted in Fig. 15g. These findings confirmed that the formate derived from NaHCO3 reduction incorporated D from D2O, and further analysis demonstrated that the final formate product consisted of nearly equal amounts of H13COO− and D13COO−. Thus, the catalytic role of H2O in methanol activation is substantiated and is in perfect alignment with the DFT calculations.
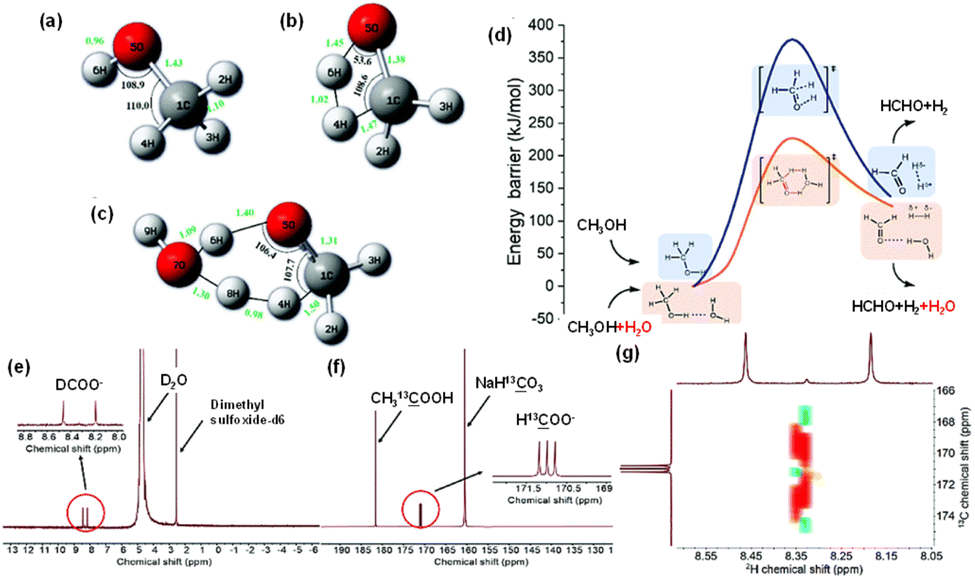 | ||
| Fig. 15 Transition states (a–c) and energy profiles (d) for methanol dissociation into H2 with or without H2O ((a) methanol molecule, (b) the transition state without H2O, (c) the transition state with H2O, (d) the profile with H2O as the promoter is depicted in orange, and the profile without H2O is depicted in blue); (e) D-NMR, (f) 13C-NMR and (g) COSY spectrum of the liquid sample after NaH13CO3 reaction with methanol in D2O.35 This figure has been reproduced from ref. 35 with permission from The Royal Society of Chemistry, copyright 2021. | ||
When PVC was used as a reductant, the reduction of NaHCO3 to formate did not occur in the absence of water,105 and high-temperature water is hypothesized to play a critical role in this process. Initially, hydrogen bonds are presumed to form among three molecules: dechlorinated PVC, H2O, and HCO3−. This interaction is believed to increase the reactivity of the O–H bond in dechlorinated PVC and the C–O bond in HCO3−. Subsequently, the hydride ion from dechlorinated PVC is postulated to attack the carbonyl carbon of HCO3−, leading to a cyclic transition state. Finally, upon hydrogen transfer, formate and the CO group are generated, and water molecules are recovered.
Furthermore, in Yang et al.'s study on CO2 reduction with glucose, the OH-DRIFTS spectra of NaHCO3 reduction with glycolaldehyde, a model compound of carbohydrates, revealed the presence of hydrated aldehyde groups in formaldehyde and glycolaldehyde which helped NaHCO3 reduction through a hydrogen transfer mechanism, and a water molecule was recovered after formate generation. This result directly proved that water assists the hydrothermal reaction as a homogeneous catalyst. Specifically, when a –CHO group becomes hydrated, the reductive hydrogen attached to the central carbon becomes more nucleophilic, enabling it to reduce electron-deficient substances more easily.106
Consequently, when molecular water hydrates the –CHO group to form –CH(OH)2, the reductive hydrogen of the aldehyde group becomes sufficiently active to directly attack NaHCO3 molecules through hydrogen transfer. As shown in Fig. 16, a six-member ring transition state is formed with HCO3−, leading to the formation of a C–H bond between the highly activated hydrogen and HCO3− through nucleophilic addition to a CO bond. Thereafter, HCO3− is reduced to orthoformate, which then releases H2O to form formate to complete the catalytic cycle.
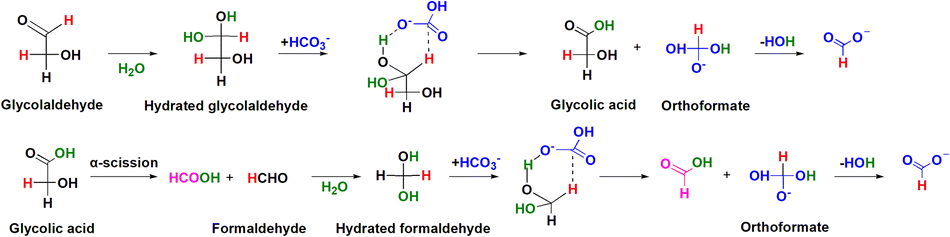 | ||
| Fig. 16 Catalytic role of water in hydrogen transfer for NaHCO3 reduction with glycolaldehyde. (Reactant NaHCO3/its reduced HCOO− and glycolaldehyde formed HCOOH are depicted in blue and violet, respectively. Reductive hydrogen in glycolaldehyde is exhibited in red, and H2O catalyzing the reaction is shown in green.)107 | ||
These results suggest that hydrothermal water can accelerate the reaction either by forming a bridge inside a molecule or between molecules or by hydrating reactants for higher activity. Indeed, the promoting role of water in reactions has been documented in several cases, such as by altering reaction pathways through the blocking of active sites or by forming intermediates or products with substrates via hydroxyl groups from water, thus creating alternative reaction paths.108 The summarized catalytic function of HTHP water here may further expand the understanding of water's role in enhancing reactions and extend the scope of hydrothermal chemistry.
6 The assessment of carbon emission budget and efficiency of CO2 reduction with renewable reductants under hydrothermal conditions
The summarized research demonstrated that multiple value-added chemicals or fuels can be produced from CO2 hydrothermal conversion, as depicted in Fig. 17a, indicating the potential of applying the technology for large scale CO2 conversion. To advance CO2 reduction in meeting the requirements of net carbon benefit and high efficiency, a comparison of the summarized approaches from the reported methods of catalytic hydrogenation and photo/electro-catalyzed CO2 reduction was performed. Fig. 17b demonstrates the efficiencies of various CO2 catalytic methods for formic acid production. Iron powder, used for the hydrothermal reduction of CO2 to formic acid, exhibits the highest efficiency. Additionally, compared to photo/electro-catalysis and hydrogenation catalysis, biomass and metal induced reduction of CO2 not only offer the advantage of high efficiency but also simplicity and cost-effectiveness due to the absence of complex catalyst preparation. Fig. 17c displays the potential greenhouse gas (GHG) emissions throughout the lifecycle of different CO2 catalytic methods. While photo/electro-catalysis aims for green and sustainable development, its low efficiency, the need for catalyst preparation, and the high energy demand of the entire system do not confer an advantage in CO2 emission reduction. In the hydrogenation catalysis process, the sources of CO2 and H2 and the boundaries considered in the life cycle assessment (LCA) significantly impact GHG emissions. For CO2 reduction via biomass and regenerable metals, the system's lower dependency on electricity, the use of sodium hydroxide for effective CO2 absorption, and the use of biomass or solar energy as the energy input contribute to its potential as a carbon-neutral technology. Moreover, implementing low-carbon energy sources and waste heat recovery methods can significantly reduce greenhouse gas emissions.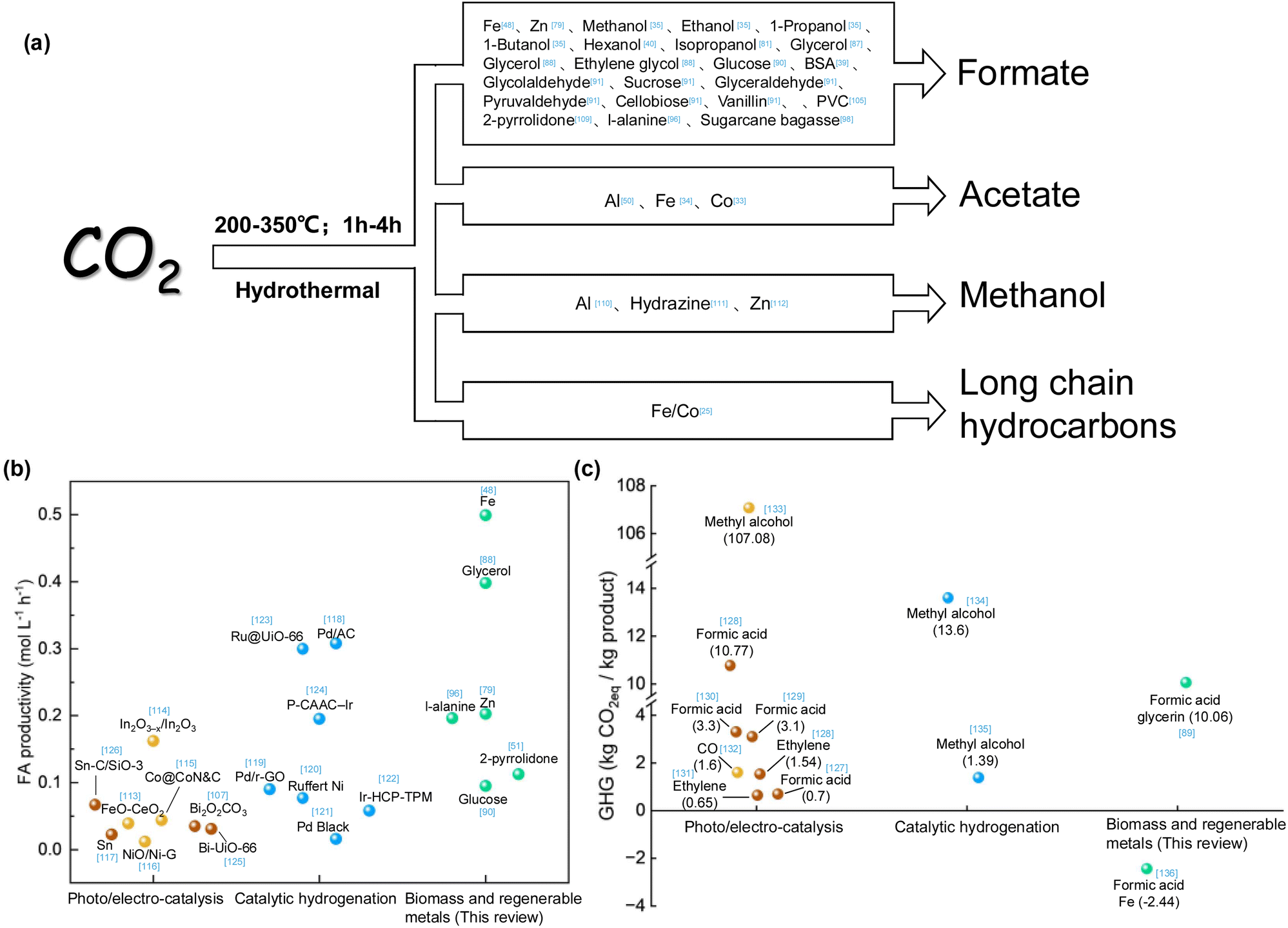 | ||
| Fig. 17 (a) Main products that can be obtained by hydrothermal CO2 conversion,33–35,39,40,48,50,79,81,87,88,90,91,96,98,105,109–112 (b) comparison of formic acid production efficiency by different CO2 catalytic methods,48,51,79,88,90,96,107,113–126 (c) comparison of greenhouse gas emissions by different CO2 catalytic methods89,127–136 (yellow balls represent photocatalysis, brown balls represent electrocatalysis, blue balls represent catalytic hydrogenation, green balls represent biomass and regenerable metals). | ||
7 Challenges, prospects and conclusions
As an indispensable part in dealing with the threat of GHG emissions, CO2 conversion into fuels or chemicals is considered a revolutionary technology for thoroughly coping with the issue and generating sustainable carbon sources without using the conserved fossil sources on Earth. Still far from practical implementation, CO2 conversion approaches face formidable challenges in terms of renewable reductive sources, easy handling reaction systems, and readily accessible high efficiency. Specifically, (1) catalytic CO2 hydrogenation exhibits the highest reaction efficiency, while the need for gaseous hydrogen compromises the net carbon benefit since industrial hydrogen production relies heavily on fossil fuels as the feedstock; moreover, the storage and transportation of high pressure gas also require large amounts of energy input;137 (2) solar or electro induced CO2 reduction could entirely or partly use solar energy as the energy input which promises the high prospect of achieving net carbon benefit; however, the efficiency of solar reactions is relatively poor now, and the competitive reaction of hydrogen evolution in electrochemical CO2 reduction interferes with reaction selectivity; and (3) for solar-/electro-CO2 reduction, delicate prepared catalysts or electrodes are inevitable, leading to difficulties in implementing the technology practically. Therefore, from the perspective of practical applications, it is urgent to develop efficient strategies to use renewable reductants and operable and scalable methods in CO2 conversion to achieve the dual goals of efficient and net carbon benefit CO2 reduction.In this review, we comprehensively summarize the recent achievements of hydrothermal CO2 reduction with renewable reductants, starting with the origin and advantages of hydrothermal technology and the feasibility of certain metals, biomass, and organic waste as renewable reductants for CO2 conversion. Moreover, we focus on the reaction characteristics and mechanisms to discuss whether the summarized technologies can overcome the existing challenges of CO2 conversion. Specifically, we first classify the renewable reductants for efficient CO2 reduction, including the metals Zn9,41,51,56,77,80 and Fe,29,32,34 biomasses as carbohydrates, microalgae and lignin, and organic solid waste as plastics. More importantly, the intrinsic advantages of hydrothermal CO2 reduction are represented, for example, the self-catalytic process of metal induced CO2 reduction, the high reducing capacity of an organic hydrogen source catalyzed by the hydrothermal environment, and the easily controlled reaction process by regulating the hydrothermal reaction settings. Although significant progress has been made in hydrothermal CO2 reduction with renewable reductants, particularly by comparing the hydrothermal technology with existing approaches, and the advantages of hydrothermal conversion are demonstrated to satisfy the dual goals of high efficiency and net carbon benefit, the implementation of this method is still in the early stages and full of challenges. Thus, the following perspectives should be taken into consideration in future research.
First, for metal-initiated CO2 reduction, although high efficiency can be achieved readily even without catalysts, the limited products species should be noted. Currently, the manufacture of C1 products such as formic acid, methanol, and methane is sufficiently ready, while for multi-carbon products, such as hydrocarbons and multi-carbon acids, only certain reaction settings can achieve the target. Thus, it is urgent to screen and synthesize proper catalysts to break the bottleneck. Indeed, primary studies have shown that Co25 and Ni are two feasible core metals for catalyzing carbon–carbon coupling in CO2 hydrothermal reduction, and they have been demonstrated for hydrocarbon and acetic acid production. Moreover, due to the rigid reductive environment of hydrothermal reduction, the oxidation and consequent deactivation of Co or Ni based catalysts are avoided, which are the primary problems affecting the use of these two catalysts in other catalytic processes. However, other products have been scarcely observed, calling for further effort in exploiting more proper catalysts. Moreover, the abovementioned catalysts functioned with the assistance of in situ generated reductant oxides such as Fe3O4, while ZnO could not help the catalytic process,25,77 suggesting that the design of proper catalysts should consider the reductant as well, that is, the added catalysts and the reductant constitute a catalytic system. The reductant oxide could be described as a catalyst support, which could alter the core catalyst morphology as a motif or accommodate the catalyst in the porous structure. Thus, the coordination of the reductant oxide and the core catalyst is important for the design of the catalytic system.
Second, when biomass or organic waste is used as the reductant the reducing efficiency is limited compared to that of metals since it is derived from reductive functional groups; thus, how to increase the reducing efficiency of biomass or organic waste is one obstacle to cope with. As summarized above, the hydroxyl (–OH), aldehyde (–CHO), and amino (NH2–) groups are the main reductive groups of organic reductants. With the assistance of the hydrothermal environment, the reductive capacity of functional groups can be invoked sufficiently, but the ultimate reducing performance also relies on the chemical structure of the organic reductants. For instance, for carbohydrates, the aldehyde group is the main reductive source, but the exposure of the aldehyde group is difficult to control. Taking glucose as an example, if dehydration or isomerization of glucose happens, the aldehyde group will be consumed or concealed in CO2 reduction, which will severely compromise the reduction efficiency. Thus, it is urgent to design optimal reaction procedures or catalysts to conserve the reductive functional groups. Yang et al.117 used a two-step reaction strategy to enhance glycolaldehyde production from glucose, as it is a two-carbon aldose that thoroughly cuts off the possibility of dehydration or isomerization reaction, and, therefore, almost two-fold reduction efficiency was achieved compared to the one-step reaction. For biomass using amino groups as the reductive force, such as in microalgae, the dehydration reaction of the amino group with a carboxylic group to form amide is the main reaction consuming amino groups. However, with proper modulation of the solution pH, the dehydration reaction can be sufficiently avoided.39 Waste plastics such as PVC reduce CO2 with hydroxyl groups.101 Although this group can be preserved well under hydrothermal conditions, its reducing capacity is relatively poor compared to those of other functional groups such as aldehyde or amino. Thus, delicately prepared catalysts such as Cu based catalysts are needed to enhance the reducing efficiency.9 Consequently, to fully utilize the reductive power of functional groups in organic reductants, the general reaction mechanisms should be illustrated clearly with acknowledgment of the possible side reactions that might compromise the functional groups, and optimal reaction procedures, parameters or catalysts must be designed accordingly.
Third, for long-term development, natural biomass is better for use when biomass serves as the reductant. However, natural biomass is very complicated, with not only varied species, but also various chemical compositions (such as carbohydrates, proteins, lipids, lignin, etc.). The synergetic effects of these chemical compositions will inevitably affect both the selectivity and the efficiency of CO2 hydrothermal reduction. Thus, it is necessary to study whether and how the different chemical compositions of biomass will influence CO2 reduction. Taking the reaction of microalgae with CO2 as an example, protein was found to be the main reductant, while carbohydrate and lipid reduced less CO2. In the meantime, reactions between protein and carbohydrate were observed, like the Maillard reaction. However, the reaction between protein and carbohydrate is highly dependent on the solution pH, suggesting it can be partly avoided by proper alkalinity adjustment. On the other hand, it should be noted that the direct use of natural biomass for CO2 reduction is difficult for achieving high selectivity or efficiency as the compositions of biomass are too complicated. With sufficient acknowledgement of the reaction characteristics of different reductants, certain approaches can be suggested to achieve the target with the basic principle that it is important to produce active intermediates in the first step and then achieve CO2 reduction in the second step. Specifically, when carbohydrate is used, cutting the carbon chain to smaller aldoses is one feasible solution to ensure the reducing capacity, which can be accomplished by acid catalyzed pretreatment or the physical process of ball milling. When protein abundant biomass is used, such as microalgae, the hydrolysis of microalgae in the first step is important. After eliminating the products of the Maillard reaction, much higher CO2 reduction efficiency can be expected with the remaining amino acids. For lignin, it is found that the reactivity is hardly influenced by other chemical compositions, while its degradation pathway is important for the CO2 reduction and can be modulated by proper catalysts such as Pd or Ru based catalysts.
Fourth, although great progress has been made in the development of hydrothermal CO2 with renewable reductants, the above analysis suggests that improving the reduction efficiency or producing the target product are still great challenges. Therefore, in-depth understanding of the mechanism of competitive reactions in the hydrothermal environment is desperately needed. The high temperature and high-pressure reaction environment in a sealed reaction box makes it difficult to detect the reaction mechanism. The continuous and rapid development of DFT calculations combined with in situ characterization techniques, such as in situ synchrotron radiation spectroscopy, NMR, Raman and FTIR spectroscopy and chromatography, will undoubtedly accelerate the progress of understanding the reaction mechanism. On one hand, DFT calculations can be used to deeply reveal the transformative behaviors of the reductants under hydrothermal conditions, which will help to understand the possible catalytic function of hydrothermal water and provide guidance for the design of highly selective catalysts or reaction parameters. On the other hand, advanced operando characterization techniques can provide real-time analysis of key intermediates during the reaction, which is conducive to deducing the rate-determining step of the reaction and optimizing the reaction by designing rational procedures such as multi-step reactions. Therefore, the combination of theoretical calculations and advanced in situ characterization techniques will help to understand hydrothermal reactions basically and profoundly and thus assist in achieving industrial-level CO2 hydrothermal reduction with renewable reductants.
Fifth, to meet the requirement of practically implementing CO2 conversion approaches, the advantages of the summarized methods of hydrothermal CO2 reduction with renewable reductants include the operable nature of hydrothermal reactions, the storage and transportation convenience of metals or biomass, and the direct use of bicarbonate, the alkaline product after CO2 capture, as the carbon source. However, the main conundrum to consider is the high temperature and high pressure of hydrothermal reactions. For industrial applications, temperatures around 200 °C can be provided readily, while higher temperatures approaching 300 °C could cause safety issues. Thus, decreasing the temperature of hydrothermal CO2 reduction to around 200 °C is a necessary consideration. In this aspect, Wang et al.35 achieved CO2 reduction with methanol at 180 °C with Pd–Cu catalysts, and Ni and Cu were found to decrease the reduction temperature of CO2 with Fe or Zn as the reductant to around 200 °C. More recently, we found that with Co–Pd catalysts, glucose could reduce CO2 at around 200 °C as well. These results indicate that, with rationally designed catalysts, the reaction temperature can be reduced sufficiently. By investigating the catalytic mechanism in the above research, the basic thought in designing the target catalyst is to enhance the hydrogenation of CO2. Thus, normally noble metals such as Pd, Pt, and Ru can be considered as incorporated components in catalysts, and, with the proper added components, the catalytic effect of the core element can be further enhanced. Indeed, our previous research concerning biomass conversion under hydrothermal conditions has inspired a facility for processing organic waste at ca. 200 °C with the capacity of 100 kg per day. Thus, a lower reaction temperature approaching 200 °C indicates the possibility of applying the technology practically.
Sixth, for industrial applications, the energy consumption and economic benefit of the process should be considered. For hydrothermal reactions, it is generally acknowledged that water must be heated for the reaction and thus the energy consumption is abundant. As a matter of fact, according to the thermodynamic study of the above summarized research, CO2 hydrothermal reduction with renewable reductants is always an exothermic process, since the reductants are oxidized as energy donors, which indicates that if the heat preservation of reactors is supplied, once the reaction is started, no more energy is required. More intriguingly, when metals are used as the reductants, the exuded heat of metal oxidation is so large that extra energy might be recovered if the reactors are designed properly. For the financial budget of hydrothermal technology, since special facilities are required for hydrothermal reactions, the expense of the technology is postulated to be large. However, when the oxidation of reductants such as biomass into chemicals or fuels is considered for calculating the economic benefit, the vision might be different. Taking Fe as the reductant for CO2 conversion as an example, when its recovery is achieved with glycerol reducing Fe3O4, it is determined that the economic benefit of one cycle can reach $2.2 for 1 mol CO2, indicating vast potential in achieving economic benefit from CO2 hydrothermal reduction. When hydrothermal technology is applied in practice, the up-front investment might be large, but for the long term, the financial benefit is relatively promising due to its fast reaction speed and the feasibility in processing different substrates, as is the case in our facilities for processing organic waste.
Seventh, the most acknowledgeable application of hydrothermal technology is its use in reforming biomass for hydrogen production under supercritical hydrothermal conditions, and recently aqueous phase reforming of biomass under subcritical conditions has been developed, which suggests that hydrogen can first be produced from biomass hydrothermal treatment, and then CO2 can be hydrogenated in the second step through the process of catalytic hydrogenation. This consideration also aligns with the practical plant built by Li et al.,135,138 where they first use solar energy to achieve electrolysis of water, and then CO2 is reduced through a thermal catalytic process. This way, the practical obstacles in harnessing direct CO2 solar-/electro-reduction can be avoided, and the need for green hydrogen in CO2 hydrogenation can be achieved. However, when fully considering CO2 hydrothermal reduction, we assume that direct CO2 hydrothermal conversion with reductants in a one-pot reaction is superior, with the following advantages: (1) from the macroscopic view, when CO2 and reductants are present in the same reaction environment, the reductive power and energy held in the reductant can be exploited directly for CO2 reduction without the need for mass or energy transfer, which can naturally lead to less efficiency lost, and (2) from the mechanistic view, CO2 hydrothermal reduction rarely follows the pathway of first hydrogen generation and consequent CO2 hydrogenation. In contrast, it is reduced directly with in situ hydrogen from either the hydrogen spillover from the catalyst surface (as in the case with metals as the reductants) or direct hydrogen transfer from the reductant (as in the case with organic reductants), which have proved to be far more efficient than CO2 hydrogenation. Thus, owing to the improvement in mass and energy transfer under hydrothermal conditions, one-pot CO2 hydrothermal reduction is postulated to be more efficient.
In summary, although progress is still needed in CO2 hydrothermal reduction, such as in the broadness of product species, given the improvement in reducing efficiency and stable rational hydrothermal catalysts, the proposed strategy holds promising potential for industrial application due to the native advantages of hydrothermal reactions and the sustainability of the reductants. It is believed that in the foreseeable future, efficient and net carbon benefit CO2 hydrothermal reduction will make a great breakthrough through systematic mechanistic research and continuous optimization of catalysts and reaction procedures and parameters. Once the concept of hydrothermal CO2 reduction with renewable reductants can be applied in large-scale industrial application, the global warming and energy crises will be greatly alleviated, and a low-carbon economy can be envisioned.
Author contributions
Zien Tang: conceptualization, methodology, writing – original draft; Xu Liu: data curation, visualization; Yang Yang: software, writing – review & editing, project administration, supervision; Fangming Jin: investigation, resources, validation, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support of the National Natural Science Foundation of China (No. 21978170 & 22108171), the Natural Science Foundation of Shanghai (No. 23ZR1435200), and Shanghai Key Laboratory of Hydrogen Science & Center of Hydrogen Science, Shanghai Jiao Tong University, China.References
- M. I. Alam, R. Cheula, G. Moroni, L. Nardi and M. Maestri, Catal. Sci. Technol., 2021, 11, 6601–6629 RSC.
- E. Koohestanian and F. Shahraki, J. Environ. Chem. Eng., 2021, 9, 105777 CrossRef CAS.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- T. Kumar and S. Eswari, Energy Fuels, 2023, 37, 3570–3589 CrossRef CAS.
- H. Salehizadeh, N. Yan and R. Farnood, Chem. Eng. J., 2020, 390, 124584 CrossRef CAS.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y. M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B. M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C. J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef PubMed.
- G. Hasrack, M. C. Bacariza, C. Henriques and P. Da Costa, Catalysts, 2022, 12, 36 CrossRef CAS.
- S. S. Xu, S. Chansai, S. J. Xu, C. E. Stere, Y. L. Jiao, S. H. Yang, C. Hardacre and X. L. Fan, ACS Catal., 2020, 10, 12828–12840 CrossRef CAS.
- X. He, M. Liu, Z. Liang, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, Z. Zheng and B. Huang, J. Solid State Chem., 2021, 298, 122113 CrossRef CAS.
- L. L. Ling, W. Yang, P. Yan, M. Wang and H. L. Jiang, Angew Chem. Int. Ed. Engl., 2022, 61, e202116396 CrossRef CAS PubMed.
- S. F. Hung, A. Xu, X. Wang, F. Li, S. H. Hsu, Y. Li, J. Wicks, E. G. Cervantes, A. S. Rasouli, Y. C. Li, M. Luo, D. H. Nam, N. Wang, T. Peng, Y. Yan, G. Lee and E. H. Sargent, Nat. Commun., 2022, 13, 819 CrossRef CAS PubMed.
- J. B. Jiang, A. J. Matula, J. R. Swierk, N. Romano, Y. S. Wu, V. S. Batista, R. H. Crabtree, J. S. Lindsey, H. L. Wang and G. W. Brudvig, ACS Catal., 2018, 8, 10131–10136 CrossRef CAS.
- X. Ding, X. Liu, J. Cheng, L. Kong and Y. Guo, Catal. Sci. Technol., 2022, 12, 4740–4752 RSC.
- S. Wang, A. A. Tountas, W. Pan, J. Zhao, L. He, W. Sun, D. Yang and G. A. Ozin, Small, 2021, 17, e2007025 CrossRef PubMed.
- N. Dhabarde, J. Selvaraj, A. Yuda, A. Kumar and V. R. Subramanian, Int. J. Hydrogen Energy, 2022, 47, 30908–30936 CrossRef CAS.
- S. Pan, J. Li, Z. Wen, R. Lu, Q. Zhang, H. Jin, L. Zhang, Y. Chen and S. Wang, Adv. Energy Mater., 2021, 12, 2004002 CrossRef.
- Z. H. Liu, K. Wang, Y. Chen, T. W. Tan and J. Nielsen, Nat. Catal., 2020, 3, 274–288 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS PubMed.
- O. Faye, J. Szpunar and U. Eduok, Int. J. Hydrogen Energy, 2022, 47, 13771–13802 CrossRef CAS.
- T. M. McCollom and J. S. Seewald, Chem. Rev., 2007, 107, 382–401 CrossRef CAS PubMed.
- W. Martin, J. Baross, D. Kelley and M. J. Russell, Nat. Rev. Microbiol., 2008, 6, 805–814 CrossRef CAS PubMed.
- G. Proskurowski, M. D. Lilley, J. S. Seewald, G. L. Fruh-Green, E. J. Olson, J. E. Lupton, S. P. Sylva and D. S. Kelley, Science, 2008, 319, 604–607 CrossRef CAS PubMed.
- S. Navarro-Jaen, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem, 2021, 5, 564–579 CrossRef CAS PubMed.
- D. He, X. Wang, Y. Yang, R. He, H. Zhong, Y. Wang, B. Han and F. Jin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2115059118 CrossRef CAS PubMed.
- R. He, B. Hu, H. Zhong, F. Jin, J. Fan, Y. H. Hu and Z. Jing, Chem. Commun., 2019, 55, 1056–1059 RSC.
- B. Y. Hu, Z. Z. Jing, J. J. Fan, G. D. Yao and F. M. Jin, Catal. Today, 2016, 263, 128–135 CrossRef CAS.
- Z. B. Huo, M. B. Hu, X. Zeng, J. Yun and F. M. Jin, Catal. Today, 2012, 194, 25–29 CrossRef CAS.
- C. L. Jiang, H. Zhong, G. D. Yao, J. Duo and F. M. Jin, Int. J. Hydrogen Energy, 2017, 42, 17476–17487 CrossRef CAS.
- X. Liu, H. Zhong, C. Wang, D. He and F. Jin, Energy Sci. Eng., 2022, 10, 1601–1613 CrossRef CAS.
- Z. Ni, H. Zhong, Y. Yang, G. Yao, B. Jin and F. Jin, ACS Sustainable Chem. Eng., 2019, 7, 5827–5834 CrossRef CAS.
- Z. Shen, Y. Zhang and F. Jin, RSC Adv., 2012, 2, 797–801 RSC.
- X. Wang, Y. Yang, T. Wang, H. Zhong, J. Cheng and F. Jin, ACS Sustainable Chem. Eng., 2021, 9, 1203–1212 CrossRef CAS.
- X. G. Wang, Y. Yang, H. Zhong, R. T. He, J. Cheng and F. M. Jin, Catal. Today, 2020, 350, 136–141 CrossRef CAS.
- X. G. Wang, Y. Yang, H. Zhong, T. F. Wang, J. Cheng and F. M. Jin, Green Chem., 2021, 23, 430–439 RSC.
- Y. Q. Wang, F. M. Jin, M. Sasaki, Wahyudiono, F. W. Wang, Z. Z. Jing and M. Goto, AIChE J., 2013, 59, 2096–2104 CrossRef CAS.
- Y. Q. Wang, F. M. Jin, X. Zeng, G. D. Yao and Z. Z. Jing, Int. J. Hydrogen Energy, 2013, 38, 760–768 CrossRef CAS.
- L. X. Wu, Y. Yang, J. Cheng, X. G. Wang, Q. Huang and F. M. Jin, React. Chem. Eng., 2022, 7, 839–843 RSC.
- Y. Yang, H. Zhong, R. He, X. Wang, J. Cheng, G. Yao and F. Jin, Green Chem., 2019, 21, 1247–1252 RSC.
- Y. Yang, H. Zhong, G. Yao, R. He, B. Jin and F. Jin, Catal. Today, 2018, 318, 10–14 CrossRef CAS.
- G. Yao, F. Chen, Z. Huo and F. Jin, Int. J. Hydrogen Energy, 2016, 41, 9135–9139 CrossRef CAS.
- G. Yao, J. Duo, B. Jin, H. Zhong, L. Lyu, Z. Ma and F. Jin, J. Energy Chem., 2017, 26, 881–890 CrossRef.
- G. Yao, X. Zeng, Y. Jin, H. Zhong, J. Duo and F. Jin, Int. J. Hydrogen Energy, 2015, 40, 14284–14289 CrossRef CAS.
- X. Zeng, M. Hatakeyama, K. Ogata, J. Liu, Y. Wang, Q. Gao, K. Fujii, M. Fujihira, F. Jin and S. Nakamura, Phys. Chem. Chem. Phys., 2014, 16, 19836–19840 RSC.
- X. Zeng, F. M. Jin, Z. B. Huo, T. Mogi, A. Kishita and H. Enomoto, Energy Fuels, 2011, 25, 2749–2752 CrossRef CAS.
- X. Zeng, F. Jin, G. Yao and Z. Huo, Int. J. Hydrogen Energy, 2016, 41, 9140–9144 CrossRef CAS.
- S. Zhang, Z. B. Huo, D. Z. Ren, J. Luo, J. Fu, L. Li and F. M. Jin, Chin. J. Chem. Eng., 2016, 24, 126–131 CrossRef CAS.
- H. Zhong, Y. Gao, G. D. Yao, X. Zeng, Q. J. Li, Z. B. Huo and F. M. Jin, Chem. Eng. J., 2015, 280, 215–221 CrossRef CAS.
- H. Zhong, L. Ma, Y. Y. Zhu, B. B. Jin, T. F. Wang, Y. G. Wang and F. M. Jin, J. Supercrit. Fluids, 2020, 157, 104717 CrossRef CAS.
- H. Zhong, H. S. Yao, J. Duo, G. D. Yao and F. M. Jin, Catal. Today, 2016, 274, 28–34 CrossRef CAS.
- Y. J. Zhu, Y. Yang, X. G. Wang, H. Zhong and F. M. Jin, Energy Sci. Eng., 2019, 7, 881–889 CrossRef CAS.
- J. Duo, F. Jin, Y. Wang, H. Zhong, L. Lyu, G. Yao and Z. Huo, Chem. Commun., 2016, 52, 3316–3319 RSC.
- C. He, G. Tian, Z. Liu and S. Feng, Org. Lett., 2010, 12, 649–651 CrossRef CAS PubMed.
- F. M. Jin, Y. Gao, Y. J. Jin, Y. L. Zhang, J. L. Cao, Z. Wei and R. L. Smith, Energy Environ. Sci., 2011, 4, 881–884 RSC.
- B. Wu, Y. Gao, F. M. Jin, J. L. Cao, Y. X. Du and Y. L. Zhang, Catal. Today, 2009, 148, 405–410 CrossRef CAS.
- A. Steinfeld, Int. J. Hydrogen Energy, 2002, 27, 611–619 CrossRef CAS.
- N. J. Pester, K. Ding and W. E. Seyfried, Geology, 2014, 42, 255–258 CrossRef CAS.
- X. Liu, Y. Guo, D. H. Xu and Q. Q. Guan, J. Cleaner Prod., 2022, 366, 132978 CrossRef CAS.
- X. Liu, Y. Guo, A. Dasgupta, H. He, D. Xu and Q. Guan, Renewable Energy, 2022, 183, 627–650 CrossRef CAS.
- A. A. Peterson, F. Vogel, R. P. Lachance, M. Fröling, M. J. Antal and J. W. Tester, Energy Environ. Sci., 2008, 1, 32–65 RSC.
- M. Akizuki, T. Fujii, R. Hayashi and Y. Oshima, J. Biosci. Bioeng., 2014, 117, 10–18 CrossRef CAS PubMed.
- S. S. Toor, L. Rosendahl and A. Rudolf, Energy, 2011, 36, 2328–2342 CrossRef CAS.
- V. Lehr, M. Sarlea, L. Ott and H. Vogel, Catal. Today, 2007, 121, 121–129 CrossRef CAS.
- N. H. Sleep, Origins Life Evol. Biospheres, 1986, 16, 179–180 CrossRef.
- B. Herschy, A. Whicher, E. Camprubi, C. Watson, L. Dartnell, J. Ward, J. R. Evans and N. Lane, J. Mol. Evol., 2014, 79, 213–227 CrossRef CAS PubMed.
- V. Sojo, B. Herschy, A. Whicher, E. Camprubi and N. Lane, Astrobiology, 2016, 16, 181–197 CrossRef CAS PubMed.
- J. Fiebig, A. B. Woodland, J. Spangenberg and W. Oschmann, Geochim. Cosmochim. Acta, 2007, 71, 3028–3039 CrossRef CAS.
- A. D. Aubrey, H. J. Cleaves and J. L. Bada, Origins Life Evol. Biospheres, 2009, 39, 91–108 CrossRef CAS PubMed.
- Y. Q. Chen, Z. Z. Jing, J. J. Miao, Y. Zhang and J. J. Fan, Int. J. Hydrogen Energy, 2016, 41, 9123–9127 CrossRef CAS.
- H. Takahashi, T. Kori, T. Onoki, K. Tohji and N. Yamasaki, J. Mater. Sci., 2007, 43, 2487–2491 CrossRef.
- G. Tian, C. He, Y. Chen, H. M. Yuan, Z. W. Liu, Z. Shi and S. H. Feng, ChemSusChem, 2010, 3, 323–324 CrossRef CAS PubMed.
- G. Tian, H. Yuan, Y. Mu, C. He and S. Feng, Org. Lett., 2007, 9, 2019–2021 CrossRef CAS PubMed.
- X. Zeng, G. D. Yin, Y. Y. Zhou and J. F. Zhao, Molecules, 2022, 27, 7371 CrossRef CAS PubMed.
- B. Jin, L. Luo and L. Xie, ACS Omega, 2021, 6, 11280–11285 CrossRef CAS PubMed.
- H. W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Organometallics, 2012, 31, 8225–8236 CrossRef CAS.
- F. Jin, X. Zeng, J. Liu, Y. Jin, L. Wang, H. Zhong, G. Yao and Z. Huo, Sci. Rep., 2014, 4, 4503 CrossRef PubMed.
- Y. Le, H. Zhong, Y. Yang, R. T. He, G. D. Yao and F. M. Jin, J. Energy Chem., 2017, 26, 936–941 CrossRef.
- L. Y. Wang, G. D. Yao, Z. Z. Jing and F. M. Jin, Adv. Mater. Res., 2014, 1073–1076, 39–42 Search PubMed.
- H. Zhong, L. Wang, Y. Yang, R. He, Z. Jing and F. Jin, ACS Appl. Mater. Interfaces, 2019, 11, 42149–42155 CrossRef CAS PubMed.
- J. Liu, X. Zeng, M. Cheng, J. Yun, Q. Li, Z. Jing and F. Jin, Bioresour. Technol., 2012, 114, 658–662 CrossRef CAS PubMed.
- Z. Shen, Y. L. Zhang and F. M. Jin, Green Chem., 2011, 13, 820–823 RSC.
- Y. Q. Wang, F. W. Wang, C. J. Li and F. M. Jin, Int. J. Hydrogen Energy, 2016, 41, 9128–9134 CrossRef CAS.
- A. Costine, J. S. C. Loh, F. Busetti, C. A. Joll and A. Heitz, Ind. Eng. Chem. Res., 2013, 52, 5572–5581 CrossRef CAS.
- C. A. Ramírez-López, J. R. Ochoa-Gómez, S. Gil-Río, O. Gómez-Jiménez-Aberasturi and J. Torrecilla-Soria, J. Chem. Technol. Biotechnol., 2011, 86, 867–874 CrossRef.
- H. Kishida, F. Jin, X. Yan, T. Moriya and H. Enomoto, Carbohydr. Res., 2006, 341, 2619–2623 CrossRef CAS PubMed.
- Z. Shen, M. Y. Gu, M. Zhang, W. J. Sang, X. F. Zhou, Y. L. Zhang and F. M. Jin, RSC Adv., 2014, 4, 15256–15263 RSC.
- W. B. Yan, B. B. Jin, J. Cheng, X. Y. Shi, H. Zhong and F. M. Jin, ACS Sustainable Chem. Eng., 2022, 10, 5374–5383 CrossRef CAS.
- J. Su, L. Yang, X. Yang, M. Lu, B. Luo and H. Lin, ACS Sustainable Chem. Eng., 2014, 3, 195–203 CrossRef.
- J. Park, A. H. Valekar, K. R. Oh, A. Awad, I. H. Song, C. Yoo, J. J. An and Y. K. Hwang, Chem. Eng. J., 2023, 463, 142410 CrossRef CAS.
- Y. Yang, H. Zhong, J. Cheng, Y. H. Hu, R. L. Smith and F. Jin, Next Energy, 2023, 1, 100037 CrossRef.
- M. Andérez-Fernández, E. Pérez, A. Martín and M. D. Bermejo, J. Supercrit. Fluids, 2018, 133, 658–664 CrossRef.
- M. Andérez-Fernández, S. Ferrero, J. P. S. Queiroz, E. Pérez, C. M. Álvarez, Á. Martín and M. D. Bermejo, J. Taiwan Inst. Chem. Eng., 2022, 139, 104504 CrossRef.
- M. Andérez-Fernández, E. Pérez, S. Ferrero, C. M. Álvarez, J. Gumiel, Á. Martín and M. D. Bermejo, Chem. Eng. J., 2023, 453, 139741 CrossRef.
- K. R. Oh, A. H. Valekar, G. Y. Cha, M. Lee, C. Yoo and Y. K. Hwang, J. CO2 Util., 2022, 60, 101981 CrossRef CAS.
- G. Ding, J. Su, C. Zhang, K. Tang, L. Yang and H. Lin, ChemSusChem, 2018, 11, 2029–2034 CrossRef CAS PubMed.
- J. C. Li, P. D. Zhu, H. Zhong, Y. Yang, J. Cheng, Y. G. Wang and F. M. Jin, ACS Sustainable Chem. Eng., 2021, 9, 4791–4800 CrossRef CAS.
- E. Smidt and K. Meissl, Waste Management, 2007, 27, 268–276 CrossRef CAS PubMed.
- M. Anderez-Fernandez, E. Perez, A. Martin, J. McGregor and M. D. Bermejo, ACS Sustainable Chem. Eng., 2022, 10, 16948–16957 CrossRef CAS PubMed.
- K. Ragaert, L. Delva and K. Van Geem, Waste Management, 2017, 69, 24–58 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS.
- Y. Li, M. Wang, X. Liu, C. Hu, D. Xiao and D. Ma, Angew Chem. Int. Ed. Engl., 2022, 61, e202117205 CrossRef CAS PubMed.
- X. Liang, M. Wang and D. Ma, J. Am. Chem. Soc., 2024, 146, 2711–2717 CrossRef CAS PubMed.
- W. J. Chen, Y. C. Jiao, Y. Liu, M. Wang, F. Zhang and D. Ma, CCS Chem., 2023, 1–8 Search PubMed.
- L. H. Lu, H. Zhong, T. F. Wang, J. N. Wu, F. M. Jin and T. Yoshioka, Green Chem., 2020, 22, 352–358 RSC.
- Y. Zhuge, G. Fan, Y. Lin, L. Yang and F. Li, Dalton Trans., 2019, 48, 9161–9172 RSC.
- L. Lin, X. He, X. G. Zhang, W. Ma, B. Zhang, D. Wei, S. Xie, Q. Zhang, X. Yi and Y. Wang, Angew. Chem., 2022, 135, e202214959 CrossRef.
- Z. Liu, E. Huang, I. Orozco, W. Liao, R. M. Palomino, N. Rui, T. Duchon, S. Nemsak, D. C. Grinter, M. Mahapatra, P. Liu, J. A. Rodriguez and S. D. Senanayake, Science, 2020, 368, 513–517 CrossRef CAS PubMed.
- Y. Zhu, Y. Yang, X. Wang, H. Zhong and F. Jin, Energy Sci. Eng., 2019, 7, 881–889 CrossRef CAS.
- S. Saedy, M. A. Newton, M. Zabilskiy, J. H. Lee, F. Krumeich, M. Ranocchiari and J. A. van Bokhoven, Catal. Sci. Technol., 2022, 12, 2703–2716 RSC.
- L. Y. Lyu, F. M. Jin, H. Zhong, H. J. Chen and G. D. Yao, RSC Adv., 2015, 5, 31450–31453 RSC.
- Y. Le, G. D. Yao, H. Zhong, B. B. Jin, R. T. He and F. M. Jin, Catal. Today, 2017, 298, 124–129 CrossRef CAS.
- J. Q. Zhao, Q. Yang, R. Shi, G. I. N. Waterhouse, X. Zhang, L. Z. Wu, C. H. Tung and T. R. Zhang, NPG Asia Mater., 2020, 12, 5 CrossRef CAS.
- L. Wang, Y. Dong, T. Yan, Z. Hu, F. M. Ali, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- S. B. Ning, H. Xu, Y. H. Qi, L. Z. Song, Q. Q. Zhang, S. X. Ouyang and J. H. Ye, ACS Catal., 2020, 10, 4726–4736 CrossRef CAS.
- D. Mateo, J. Albero and H. García, Appl. Catal., B, 2018, 224, 563–571 CrossRef CAS.
- Y. H. Pei, W. Gu, S. Cheng, S. H. Xiao, C. L. Wang, Y. Yang, H. Zhong and F. M. Jin, ACS Catal., 2023, 13, 12082–12091 CrossRef CAS.
- J. Su, L. Yang, M. Lu and H. Lin, ChemSusChem, 2015, 8, 813–816 CrossRef CAS PubMed.
- Q. Y. Bi, J. D. Lin, Y. M. Liu, X. L. Du, J. Q. Wang, H. Y. He and Y. Cao, Angew Chem. Int. Ed. Engl., 2014, 53, 13583–13587 CrossRef CAS PubMed.
- L. G. Lundsted, J. Am. Chem. Soc., 1949, 71, 323 CrossRef CAS PubMed.
- G. Bredig and S. R. Carter, Ber. Dtsch. Chem. Ges., 2006, 47, 541–545 CrossRef.
- T. Mandal, A. Kumar, J. Panda, T. Kumar Dutta and J. Choudhury, Angew Chem. Int. Ed. Engl., 2023, 62, e202314451 CrossRef CAS PubMed.
- S. Wang, S. Hou, C. Wu, Y. Zhao and X. Ma, Chin. Chem. Lett., 2019, 30, 398–402 CrossRef CAS.
- Y. Shen, Q. Zheng, Z. N. Chen, D. Wen, J. H. Clark, X. Xu and T. Tu, Angew Chem. Int. Ed. Engl., 2021, 60, 4125–4132 CrossRef CAS PubMed.
- S. Mukhopadhyay, M. S. Naeem, G. Shiva Shanker, A. Ghatak, A. R. Kottaichamy, R. Shimoni, L. Avram, I. Liberman, R. Balilty, R. Ifraemov, I. Rozenberg, M. Shalom, N. Lopez and I. Hod, Nat. Commun., 2024, 15, 3397 CrossRef CAS PubMed.
- Z. Wang, H. Li, T. Dong, Y. Geng, X. Tian, R. Chang, J. Lai, S. Feng and L. Wang, Chem. Eng. J., 2024, 489, 151238 CrossRef CAS.
- N. Thonemann and A. Schulte, Environ. Sci. Technol., 2019, 53, 12320–12329 CrossRef CAS PubMed.
- L. Ai, S. F. Ng and W. J. Ong, ChemSusChem, 2022, 15, e202200857 CrossRef CAS PubMed.
- R. Aldaco, I. Butnar, M. Margallo, J. Laso, M. Rumayor, A. Dominguez-Ramos, A. Irabien and P. E. Dodds, Sci. Total Environ., 2019, 663, 738–753 CrossRef CAS PubMed.
- S. Kibria Nabil, S. McCoy and M. G. Kibria, Green Chem., 2021, 23, 867–880 RSC.
- H. H. Khoo, I. Halim and A. D. Handoko, J. CO2 Util., 2020, 41, 101229 CrossRef CAS.
- J. Q. Wang, X. J. Liu, J. J. Ma, S. Zhang, H. R. Liu, Y. L. Dong and Q. Y. Yang, Green Chem. Eng., 2023 DOI:10.1016/j.gce.2023.10.003.
- G. Z. S. Ling, J. J. Foo, X. Q. Tan and W. J. Ong, ACS Sustainable Chem. Eng., 2023, 11, 5547–5558 CrossRef CAS.
- G. Zang, P. Sun, A. Elgowainy and M. Wang, Environ. Sci. Technol., 2021, 55, 5248–5257 CrossRef CAS PubMed.
- Y. Khojasteh-Salkuyeh, O. Ashrafi, E. Mostafavi and P. Navarri, J. CO2 Util., 2021, 50, 101608 CrossRef CAS.
- J. Xu, J. Cheng, R. T. He, J. Q. Lu, C. L. Wang, H. Zhong and F. M. Jin, Front. Environ. Sci. Eng., 2023, 17, 127 CrossRef CAS.
- K. Lee, H. Yan, Q. M. Sun, Z. H. Zhang and N. Yan, Acc. Mater. Res., 2023, 4, 746–757 CrossRef CAS.
- H. Luo, J. Barrio, N. Sunny, A. Li, L. Steier, N. I. Shah, I. E. L. Stephens and M. M. Titirici, Adv. Energy Mater., 2021, 11, 2101180 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |