
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly chemoselective oxidative dimerization of indolosesquiterpene alkaloids: a biomimetic approach to dixiamycin†‡
Mintu
Munda§
 a,
Ayan
Mondal§
b,
Nanda Kishore
Roy
b,
Ranjit
Murmu
b,
Sovan
Niyogi
b and
Alakesh
Bisai
*ab
a,
Ayan
Mondal§
b,
Nanda Kishore
Roy
b,
Ranjit
Murmu
b,
Sovan
Niyogi
b and
Alakesh
Bisai
*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal Bypass Road, Bhopal 462 066, Madhya Pradesh, India. E-mail: alakeshb@gmail.com
bDepartment of Chemistry, Indian Institute of Science Education and Research Kolkata, Mohanpur Campus, Kalyani, Nadia 741 246, West Bengal, India. E-mail: alakesh@iiserkol.ac.in
First published on 1st May 2024
Abstract
Dimeric indolosesquiterpene alkaloids, typically N–N- and C–N-linked xiamycin dimers, feature a pentacyclic framework with four contiguous stereogenic centers at the periphery of a trans-decalin scaffold to which a carbazole unit is attached. In comparison with actual biosynthetic dixiamycin derivatives, we designed C–C-linked xiamycin dimers, aiming to use them as a powerful tool to create unique scaffolds as drug candidates. In this work, we disclose the first synthetic route to access a C–C dimeric indolosesquiterpene skeleton, featuring a hypervalent iodine (PIFA)-catalyzed oxidative dimerization reaction in a single-step operation with overwhelming control over the chemoselectivity and regioselectivity. This strategy has been successfully applied to the synthesis of a C–C dimer of xiamycin A (3) and xiamycin A methyl ester (15) that demonstrates a new synthetic pathway for dimeric indolosesquiterpene alkaloids.
Introduction
Chemoselective C–C bond formation between two identical (homo-coupling) or different organic entities (cross-coupling) remains a significant challenge in the field of organic chemistry.1,2 As a pivotal structural moiety, dimeric heterocycles, specifically dimeric indolosesquiterpenoids are widely present in various natural products, biologically active molecules, and functional materials.3 In this regard, naturally occurring dimeric indolosesquiterpenoids, such as dixiamycin A (1a), dixiamycin B (1b), or dixiamycin C (2), are worth mentioning (Fig. 1).4 These alkaloids have been isolated from marine-derived actinomycetes.4a Consequently, the direct and efficient synthesis of dimeric indolosesquiterpenoid frameworks has garnered considerable interest within the synthetic chemistry community. Structurally, an indolosesquiterpenoid-based complex natural product (such as xiamycin A)5 contains a carbazole fused to a trans-decalin core with four contiguous stereocenters, two of which are all-carbon quaternary centers. These indolosesquiterpenoids have demonstrated noteworthy biological properties.4a Given their intricate structural attributes and significant bioactivity, numerous research groups have directed their efforts towards the efficient total synthesis of these alkaloids.6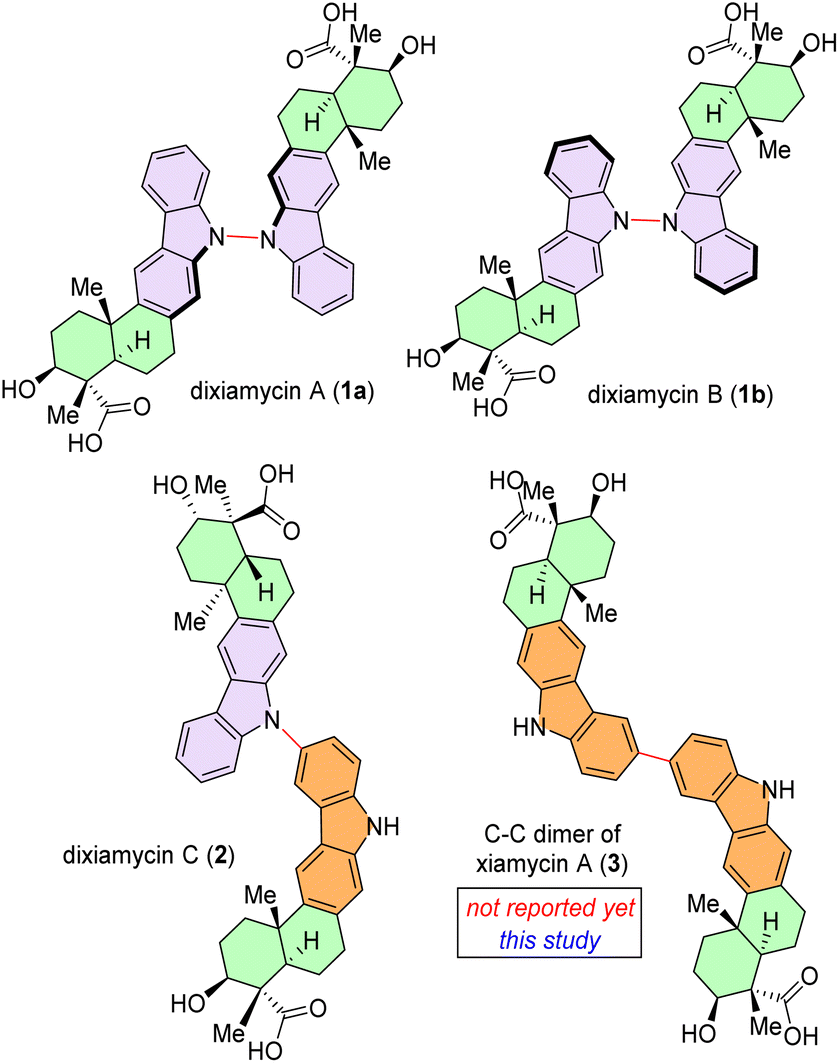 | ||
| Fig. 1 Naturally occurring dimeric indolosesquiterpenoids, dixiamycin A (1a), dixiamycin B (1b), and dixiamycin C (2) and an unnatural C–C dimer of xiamycin A (3). | ||
In 2014, Baran et al. reported the first total synthesis of dixiamycin B (1b) via the electrochemical oxidative N–N dimerisation of xiamycin A (5). However, this effort could not afford the total synthesis of dixiamycin A (an atropisomer of dixiamycin B) (1a), and the authors observed a range of unidentified compounds, possibly C–C and C–N dimers, without offering any characterization for these species (Fig. 2A).6a In 2015, Ang Li et al. reported the first total synthesis of dixiamycin C6bvia crucial late-stage C–N bond formation between TMSE-protected xiamycin A (6) and its bromo derivative (7) using Buchwald's protocol7 (Fig. 2B). Recently, our group reported the first asymmetric total synthesis of dixiamycin A (1a) in addition to its atropisomer, dixiamycin B (1b), via Cu(I)-catalyzed aerobic oxidation to construct an N–N bond.8b
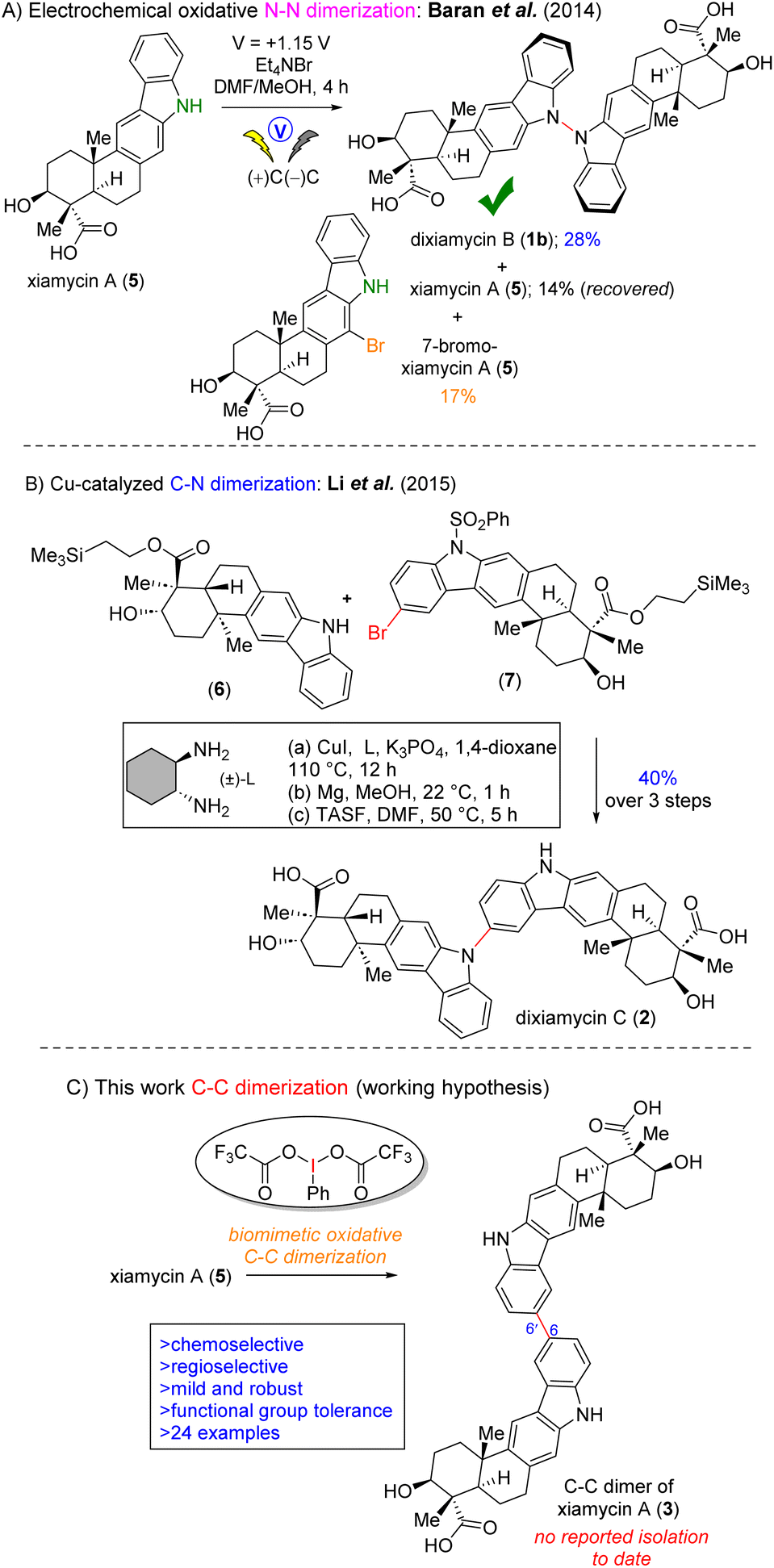 | ||
| Fig. 2 Dimerization of xiamycins and our strategy. (A) Total synthesis of dixiamycin B (1b) via N–N bond formation. (B) Total synthesis of dixiamycin C (2) via C–N bond formation. (C) Our hypothesis of biomimetic oxidative C–C dimerization of xiamycin A (5). | ||
Biosynthetically, all N–N and C–N coupled xiamycin dimers are initiated by XiaH4c mediated N-cation radical formation via single electron transfer (SET) and followed by deprotonation, resulting in xiamycin N-radical Int-1. This xiamycin N-radical (Int-1) could show mesomeric structures where spin density resides on different carbon atoms, among them C6 (Int-3) and C21 (Int-2). Eventually, all N–N and C–N coupled dimers were formed by homocoupling reactions of these radicals at different sites (Scheme 1).4b,c,8b
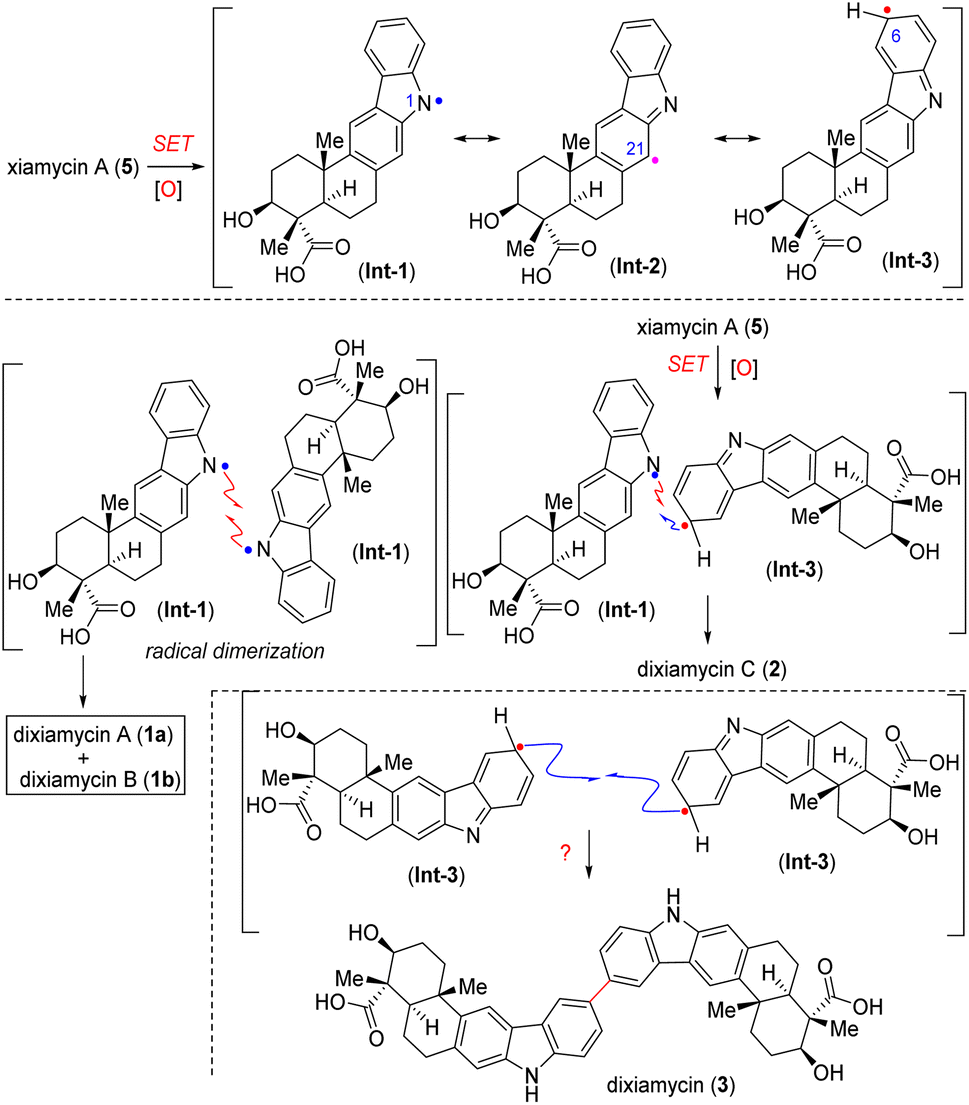 | ||
| Scheme 1 Biosynthetic hypothesis for the oxidative dimerization of xiamycin A (5). | ||
Herein, we report the discovery and full characterization of a C–C dimer of xiamycin A (3), which had been overlooked in the biogenesis (Scheme 1). Therefore, developing a protocol for the regioselective C–C dimerisation of xiamycin A (5) through a biomimetic pathway is still a fascinating goal and may open up a new molecular space for the study of the structure–activity relationship. The aryl–aryl coupling of pentacyclic indolosesquiterpenoids with various functional groups, such as ester, acid, primary alcohol, silyl ether, benzylic ketone, benzylic alcohol and carbazoline N-substituents took place under extremely mild conditions. The reaction exhibits high levels of chemoselectivity (functional group tolerance) and regioselectivity (coupling occurs exclusively at C-6) (Fig. 2C).
Initially, we hypothesized that an appropriate oxidant (hypervalent iodine(III), PIFA) could oxidize carbazole (Cz) to a carbazole radical (Ċz) via two-electron oxidation and perhaps a homocoupling could be achieved between the two species. However, there were still numerous potential challenges to this proposed transformation, including: (1) securing an oxidant that could chemoselectively make a C–C bond, (2) controlling the regioselectivity (coupling occurs exclusively at C-6), (3) inhibiting overoxidation of the carbazole scaffold and secondary hydroxyl group. Hence, we developed a radical-mediated protocol for the C6–C6′ dimerization of xiamycin A (5) by exploiting the hypervalent iodine(III) reagent,9 phenyliodine bis(trifluoroacetate) (PIFA) as an oxidant and BF3·Et2O as an additive (Fig. 2C), which leads to efficient and mild conditions for generating chemoselective and regioselective C–C bond formation.
It is important to mention that Orita and Adachi reported FeCl3-mediated oxidative C–C dimerization on carbazole to obtain 3,3′-bicarbazolyl.10 Later, Venkatakrishnan reported DDQ as a metal-free organic oxidant mediated oxidative C–C coupling on a carbazole scaffold.11 Although these coupling reactions are elegant, demonstration of their synthetic utility in complex natural products and broad functional group tolerance has never been achieved. Therefore, there is a strong need for a robust strategy that can address these limitations and fulfil the desire for a versatile approach.
Results and discussion
To evaluate the feasibility of our hypothesis, we started optimization with deoxy-xiamycin A methyl ester (8a) as a model substrate. Initially, various oxidants were screened based on their known or predicted ability to facilitate the direct dimerization reaction (Table 1). At the outset, compound 8a was treated with 1.0 equiv. of hypervalent iodine catalyst (PIFA) in CH2Cl2 under an inert atmosphere but did not provide the desired dimerization product (9a) (Table 1, entry 1).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Oxidant | Additive | Solvent | Temp | Time | Yield (9a)a,b |
| a Yield of the isolated product. b All reactions were performed on 0.15 mmol scale; oxidant was 1.0 equiv.; and additive was 1.0 equiv. c Multitude of spots and no desired product formation. d Decomposition of the rest of the mass balance. e 50 mol% PIFA was used. [CAN: ceric ammonium nitrate; MsOH: methanesulfonic acid; TTBP: 2,4,6-tri-tert-butyl-4-methylpyrimidine; and TMSI: iodotrimethylsilane]. | ||||||
| 01 | PhI(OCOCF3)2 | None | CH2Cl2 | 25 °C | 12 h | —c |
| 02 | FeCl3 | None | CHCl3 | 25 °C | 6 h | <8% |
| 03 | DDQ | MsOH | CHCl3 | 25 °C | 6 h | <15d |
| 04 | CAN | None | H2O | 25 °C | 12 h | —c |
| 05 | AgSbF6 | TTBP | (CH2Cl)2 | 25 °C | 2 h | —c |
| 06 | PhI(OCOCF3)2 | Et3N | (CH2Cl)2 | 0 °C | 6 h | —c |
| 07 | PhI(OCOCF3)2 | BF3·OEt2 | CH2Cl2 | 0 °C | 2 h | 27% |
| 08 | PhI(OCOCF3)2 | BF3·OEt2 | CH2Cl2 | −40 °C | 2 h | 43% |
| 09 | PhI(OCOCF 3 ) 2 | BF 3 ·OEt 2 | CH 2 Cl 2 | −78 °C | 2 h | 77% |
| 10 | PhI(OCOCF3)2 | HFIP | CH2Cl2 | −78 °C | 2 h | 64% |
| 11 | PhI(OCOCF3)2 | BBr3 | CH2Cl2 | −78 °C | 2 h | 36% |
| 12 | PhI(OCOCF3)2 | TMSI | CH2Cl2 | −78 °C | 2 h | 55% |
| 13 | PhI(OCOCF3)2e | BF3·OEt2 | CH2Cl2 | −78 °C | 2 h | 31% |
| 14 | PhI(OCOCH3)2 | BF3·OEt2 | CH2Cl2 | −78 °C | 2 h | 62% |
| 15 | PhIO | BF3·OEt2 | CH2Cl2 | −78 °C | 2 h | <5% |
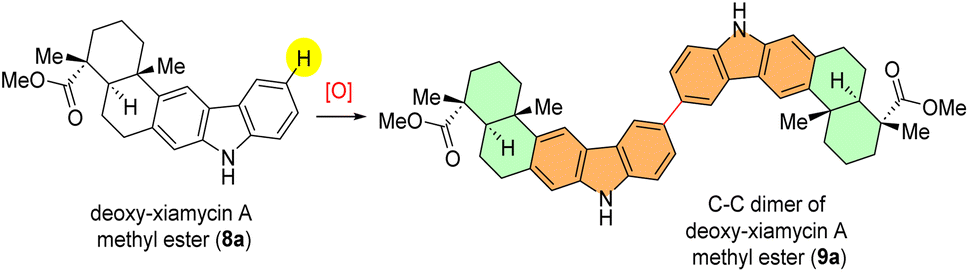
Interestingly, upon treating deoxy-xiamycin A methyl ester (8a) with FeCl3 (ref. 10) as the oxidant, a small yield (8%) of the desired dimerized product (9a) was obtained, displaying a sole regioisomer (Table 1, entry 2). Due to initial positive outcomes, a more thorough examination and refinement of the direct dimerization procedure were undertaken. Next, keeping in mind literature reports, compound 8a was charged with organic oxidant 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)11 in the presence of methanesulphonic acid (MsOH) as an additive. Unfortunately, this approach did not give desired results, with only a modest 15% yield (Table 1, entry 3). Despite various attempts using various common oxidants, such as ceric ammonium nitrate (CAN) and silver hexafluoroantimonate (AgSbF6),12 none of them were successful in producing the desired dimerized product (Table 1, entries 4 and 5). Next, considering the role of additives in activating a hypervalent iodine (PIFA) catalyst, a mixture of substrate and triethyl amine as a base additive was treated with PIFA as an oxidant, resulting in complete recovery of the starting material (8a) (Table 1, entry 6). Following that, the possibility of improving the reaction by incorporating Lewis acids as acid additives was explored. Encouragingly, it was found that the dimerized product (9a) was obtained in 27% yield as a single regioisomer at a temperature of 0 °C by employing BF3–Et2O as an additive (Table 1, entry 7). To investigate the reaction further, temperature screening was performed (Table 1, entries 8 and 9).
Remarkably, at a temperature of −78 °C, the reaction displayed high efficiency with 77% yield of the dimerized product (9a). To further showcase the influence of the solvent, the model reaction was performed in hexafluoroisopropanol (HFIP) solvent, but no enhancement was observed in product formation (Table 1, entry 10). Next, to study the effect of additives on the model reaction, different additives (Table 1, entries 11 and 12) were used, which decreased the efficiency of the reaction substantially. Furthermore, when the content of PIFA was decreased from 1.0 equiv. to 0.5 equiv., a decrease in the formation of 9a to 31% was observed (Table 1, entry 13). Later, the efficacy of optimized conditions with PIDA as an oxidant was tested, which resulted in a satisfactory (62%) yield for dimerization (Table 1, entry 14). However, when iodosobenzene (PhIO) was used as an oxidant, the desired product (9a) was obtained in a trace amount (Table 1, entry 15). Importantly, we have not observed the formation of N–N or C–N dimers of indolosesquiterpene derivatives. This suggests that N–N or C–N dimers represent the product that forms rapidly due to kinetic factors (the kinetically controlled product, KCP), whereas the more stable C–C dimer is the product favored by thermodynamic considerations (the thermodynamically controlled product, TCP). It is believed that C–C dimerization goes through the initial formation of an N–N bond, which then quickly forms the most stable C–C dimer.
With established optimal conditions in hand, the applicability of the PIFA-mediated oxidative dimerization reaction was tested with a wide range of the indolosesquiterpene derivatives. Scheme 2 summarizes various N-substituents from carbazole that were examined during investigation of this reaction. Carbazole derivatives bearing electron-donating groups (e.g., methyl, ethyl, benzyl, carbazoline ethyl acetate, prenyl, and propargyl) showed good reactivity and provided dimerised products in good yields (9b–9g).
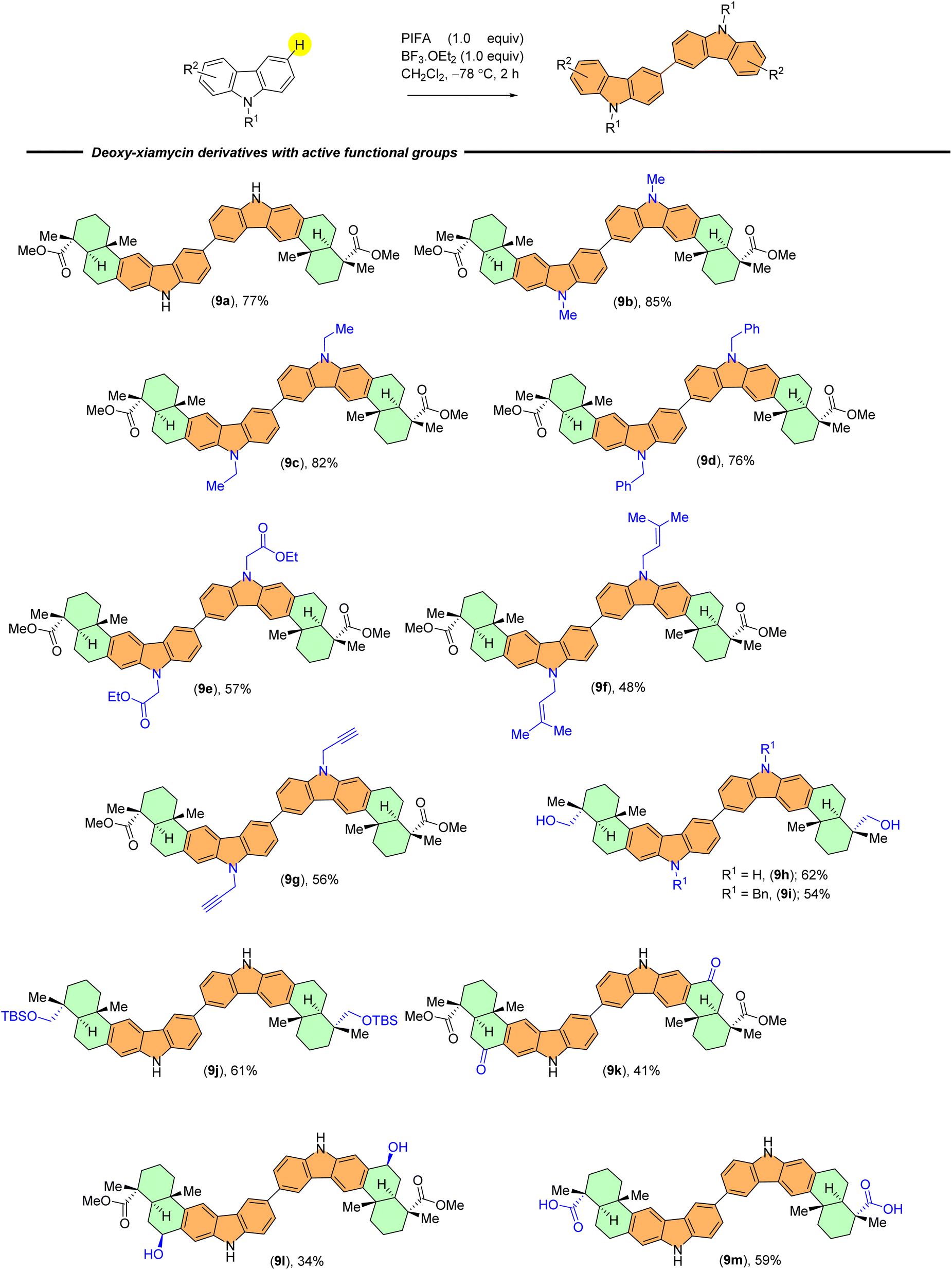 | ||
| Scheme 2 C–C dimers from indolosesquiterpenoids scaffold library. | ||
Moreover, indolosesquiterpenes containing diverse reactive functional groups, such as primary alcohol (9h and 9i), silyl ether (9j), benzylic ketone (9k), benzylic alcohol (9l), and carboxylic acid (9m) groups exhibited good compatibility and yielded expected products in moderate yields. Notably, yields obtained with benzylic ketone and benzylic alcohol were comparatively lower, possibly due to potential overoxidation (Scheme 2).
The scope of substrates was further broadened in our study to include diastereomeric and regioisomeric scaffolds (Scheme 3). We were able to efficiently produce 10a–c, which are the C-16 epimers of 9a–b, 9d, along with their N-substituted counterparts (10d–10e), yielding satisfactory results. Additionally, we found that our established protocol is capable of accommodating regioisomeric derivatives of 9a–b, 9d and 9h, providing yields ranging from 55% to 78% (11a–d) (Scheme 3). We observed successful reactions in all instances, leading to the formation of desired dimers with yields ranging from 34% to 85%.
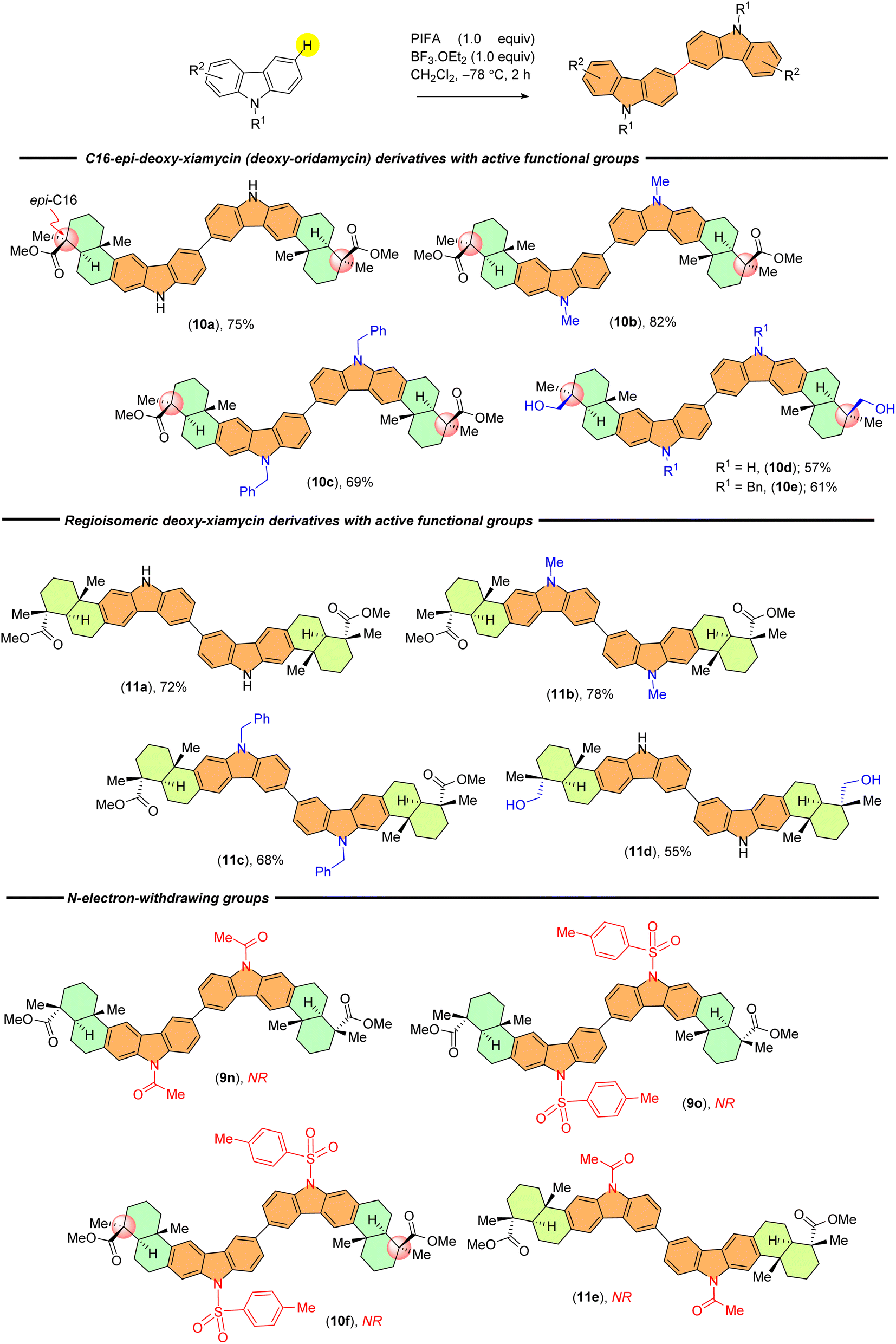 | ||
| Scheme 3 C–C dimers from indolosesquiterpenoids scaffold library. | ||
Mechanistically, the reaction commences with the generation of cation radical intermediate Int-A from carbazoles via a single-electron oxidation (SET) mediated by PIFA-BF3.OEt2 (pathway A, Scheme 4).13 The formation of a cation radical intermediate is further supported by the introduction of electron-withdrawing groups (acetyl 9n, 11e and p-toluenesulfonyl 9o, 10f) on the carbazole ring within the indolosesquiterpene motif, resulting in the inhibition of dimerization (Scheme 3). This is likely to be attributed to the destabilization of the cation radical intermediate formed during the reaction (pathway A, Scheme 4). Subsequently, Int-A engages with compound Cz, leading to the formation of cation radical intermediate Int-B, which then undergoes another single-electron oxidation (SET), resulting in the formation of a dication intermediate (Int-C). This intermediate (Int-C) promptly undergoes deprotonation, yielding the desired C–C dimerized product BCz (pathway A, Scheme 4).
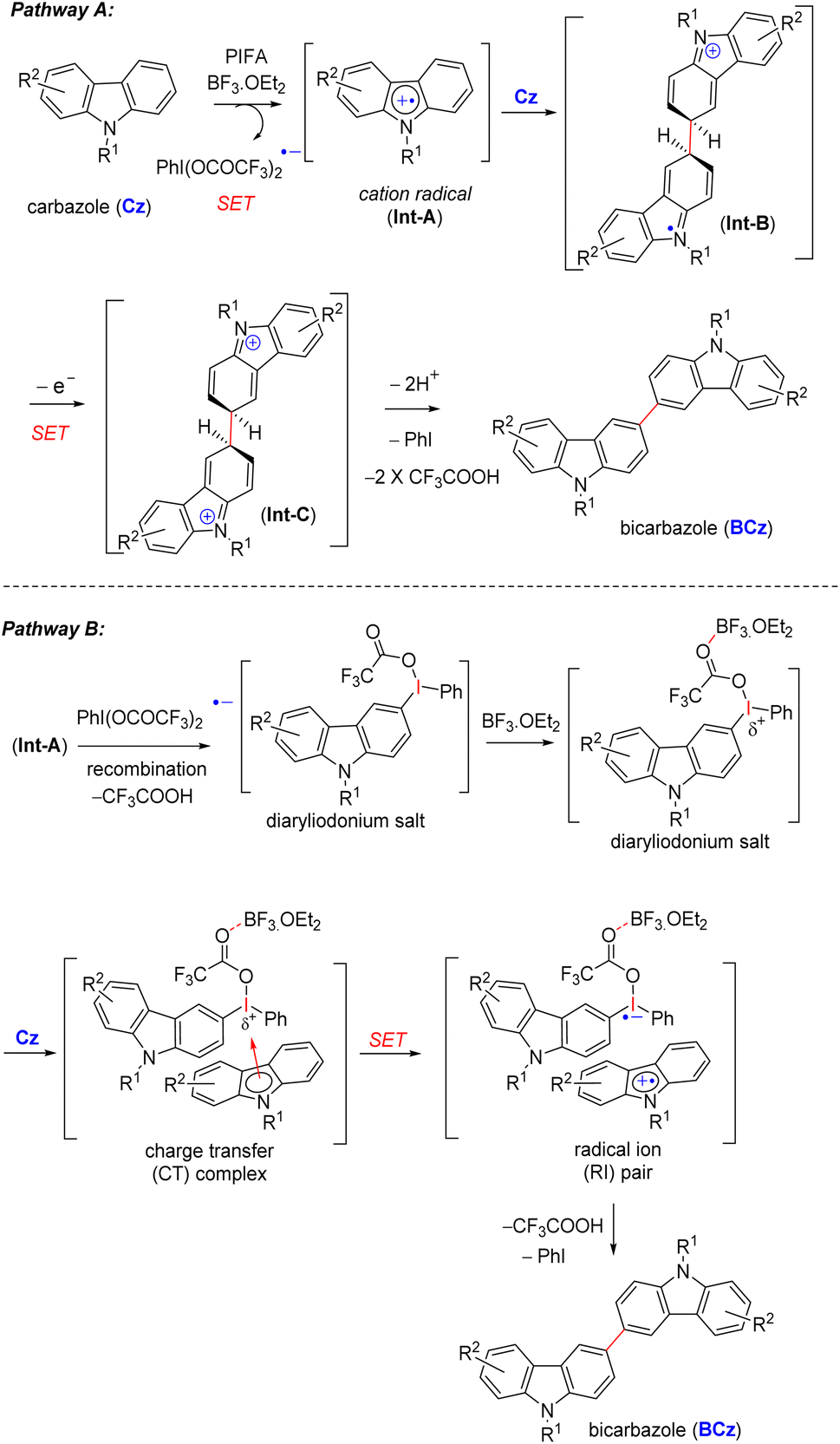 | ||
| Scheme 4 Proposed mechanism for PIFA-mediated oxidative dimerization. | ||
In an alternative pathway, in accordance with the proposal by Kita et al.,14 a cation radical intermediate (Int-A) competes with the PIFA-derived radical anion, leading to the formation of a diaryliodonium salt (pathway B, Scheme 4).15 BF3·OEt2 appears to play a crucial role by facilitating the initial interaction between the diaryliodonium salt and carbazole compound (Cz). This interaction results in the formation of a charge transfer (CT) complex, where BF3·OEt2 enhances the electrophilicity of the iodine atom in the diaryliodonium salt by coordinating with its oxygen atom.16 Consequently, BF3·OEt2 assists in the smooth generation of cation radicals [a radical ion (RI) pair] from the carbazole compound (Cz), enabling the transfer of the electron-rich heteroaryl ring (pathway B, Scheme 4).17
Therefore, a few control experiments were conducted to shed light on the probable mechanism of the PIFA-mediated oxidative dimerization (Scheme 5). Encouraged by the study of Boger et al.15b on the intermolecular coupling reaction of vindoline with a β-keto ester, we undertook similar studies using a carbazole scaffold such as 8a. Towards this end, a reaction of carbazole 8a with 1.5 equivalents of PIFA and BF3·OEt2 for the in situ preparation of diaryliodonium salt17 followed by a reaction with β-keto ester did not afford the intermolecular coupling product (Scheme 5). Instead, we were able to isolate 19% of C–C coupled carbazole dimer 9a from this reaction mixture. In another sequence, a reaction of carbazole 8a with 1.5 equivalents of PIFA and BF3·OEt2 to generate the radical cation followed by a reaction with well-known radical inhibitor TEMPO led to the formation of a TEMPO-adduct that was detected via the HRMS analysis of the crude reaction mixture. This reaction was also associated with the formation of 27% yield of C–C coupled carbazole dimer 9a (Scheme 5). Together with the literature reports, our results suggest that direct dimerization of a radical cation might be operative for the oxidative dimerization process (pathway A, Scheme 4).
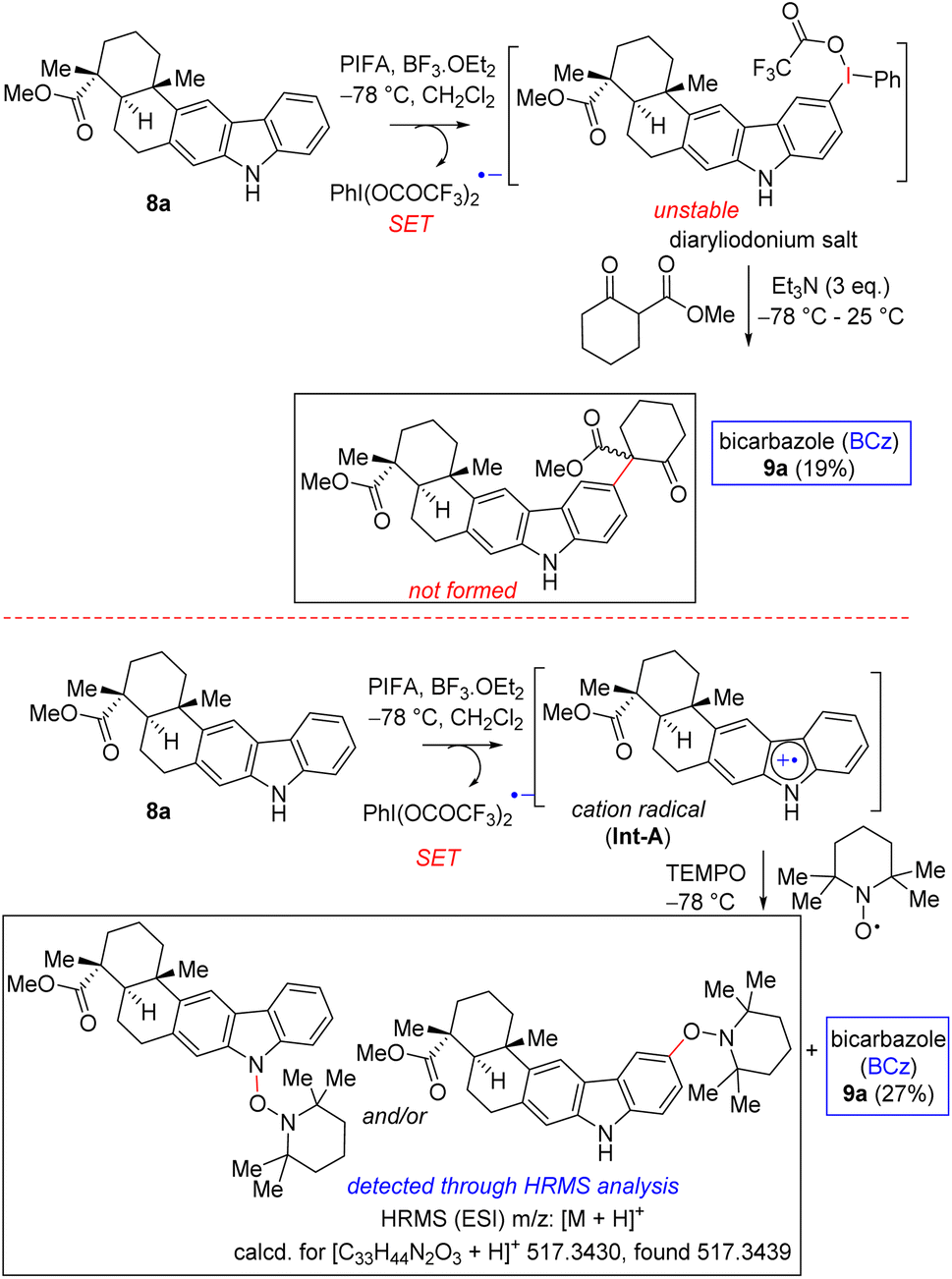 | ||
| Scheme 5 Control experiments using carbazole 8a. | ||
The utilization of this methodology opens up promising avenues for the synthesis of complex alkaloid structures and expands our understanding of their chemical diversity. By applying well-established oxidative dimerization conditions, we successfully synthesized two different sets of unnatural dimeric indolosesquiterpene alkaloids in a convergent manner (Scheme 7). Using our previously published procedure,8a we embarked on a synthesis journey starting from abietic acid (12), ultimately yielding xiamycin D (13) as a single diastereomer (Scheme 6). This intricate process involved 15 sequential steps and resulted in an overall 12% yield. After obtaining xiamycin D, our focus shifted towards the selective dehydroxylation reaction aimed at synthesizing xiamycin A methyl ester (14). To achieve this, we used a BF3·OEt2/Et3SiH18 mediated procedure, resulting in the successful synthesis of xiamycin A methyl ester (14) in 65% yield (Scheme 6). Subsequent hydrolysis of compound 14 with lithium hydroxide19 gave us 82% yield of xiamycin A (5), which was spectroscopically identical to the natural product (Scheme 6).
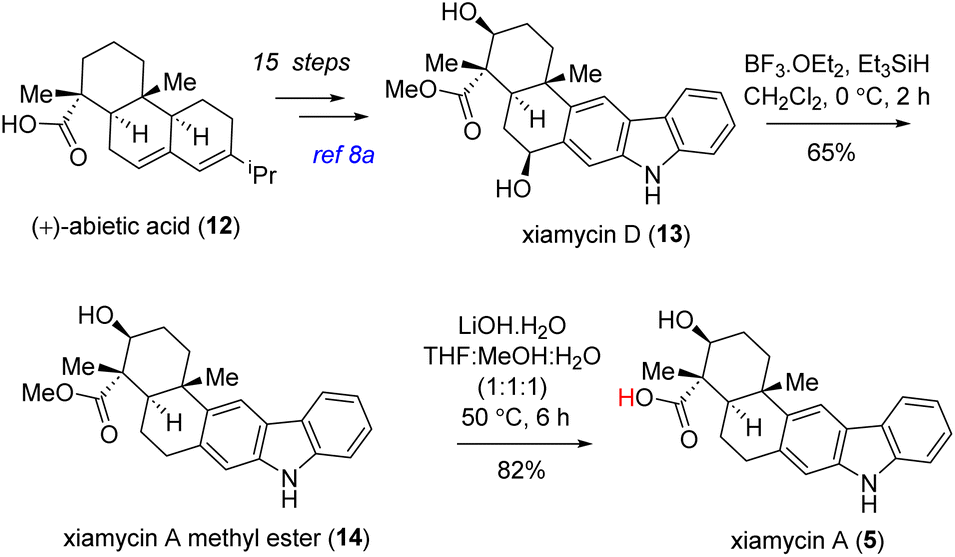 | ||
| Scheme 6 Formal syntheses of naturally occurring antiviral indolosesquiterpene alkaloids, xiamycin A methyl ester (14) and xiamycin A (5). | ||
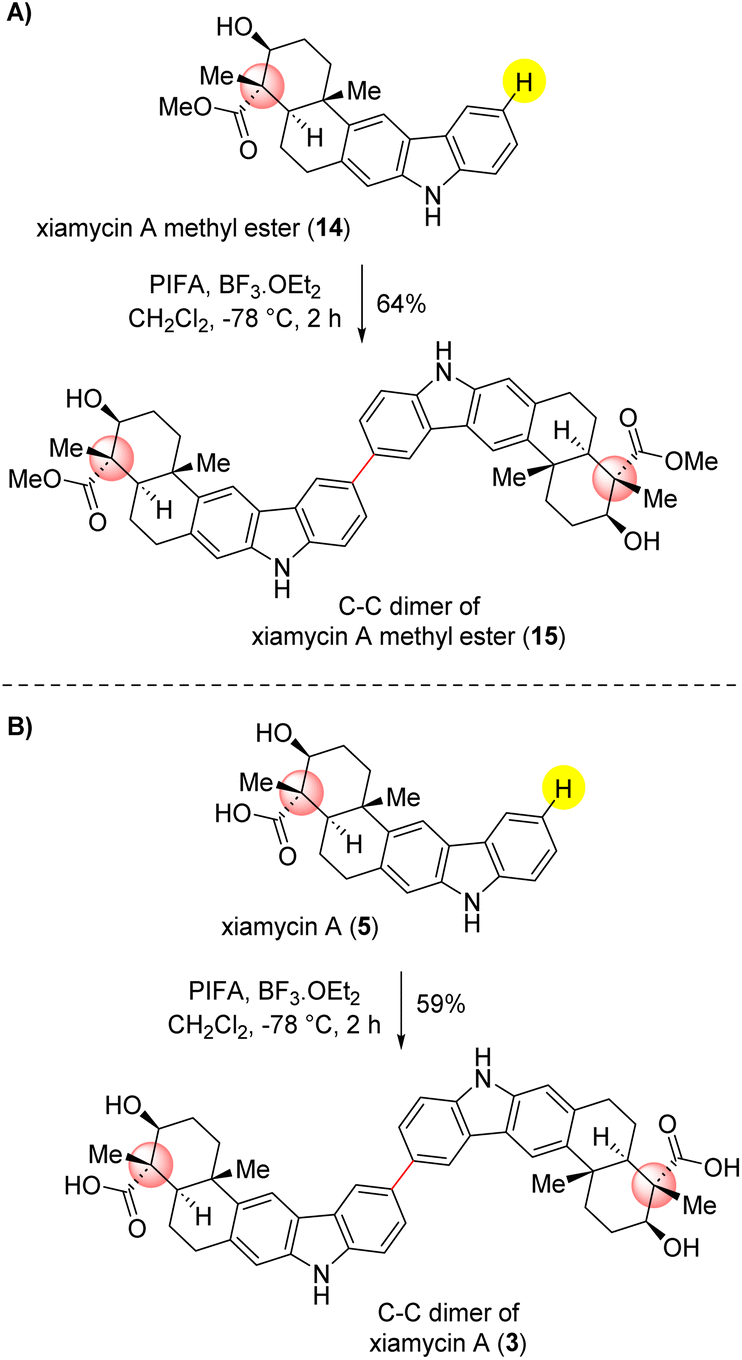 | ||
| Scheme 7 Application to dimerization of naturally occurring indolosesquiterpenoids. (A) dimerization of xiamycin A methyl ester (14). (B) dimerization of xiamycin A (5). | ||
Compound 3 can also be synthesized by treating compound 15 with lithium hydroxide through a saponification reaction (see ESI‡ for details). This significant achievement demonstrates the effectiveness of our optimized methodology in producing these structurally important compounds with noteworthy potential in various applications. As shown in Scheme 7, the PIFA-mediated dimerization of xiamycin A methylester (14) delightfully afforded 64% yield of C–C dimer 15 under the optimized conditions. Along similar lines, the dimerization of xiamycin A (5) furnished a C–C dimer of xiamycin A (3) in 59% yield, clearly demonstrating the potential of this approach. Compounds 15 and 3, currently unidentified, hold the possibility of being concealed within the realms of nature, awaiting discovery. However, there is an expectation that they will be revealed in the near future.
Conclusions
In summary, we have developed an innovative approach to directly combine carbazole compounds, resulting in the formation of a new C–C bond. This breakthrough method opens up exciting possibilities for synthesizing diverse compounds and expanding the scope of carbazole chemistry. This reaction demonstrates a wide substrate scope through the dimerization of substituted carbazoles with esters, acids, ketones, alcohols (1°, 2°, and benzylic 2°), and silyl ethers. To highlight the practicality of our method in the context of complex natural products, we successfully accomplished the formal total synthesis of xiamycin A (5) and xiamycin A methyl ester (14). By employing our methodology, we have achieved the successful synthesis of an unnatural C–C dimer of xiamycin A (3) and xiamycin A methyl ester (15). This achievement highlights the effectiveness of our methodology and its potential significance in further studies and applications.Data availability
Experimental details and spectral analysis are available free of charge from the ESI‡ available with this article.Author contributions
A. B. conceived and supervised this project. M. M. and A. M. investigated the key oxidative dimerization reactions leading to C–C dimer of xiamycins. N. K. R., R. M. and S. N. synthesized the starting materials for the dimerization process. A. B. and M. M. wrote the original draft of the manuscript which was edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the SERB [CRG/2023/000782], [SCP/2022/000486] and STARS-MoE (2023/0753) is gratefully acknowledged. MM, AM, NKR, RM, and SN, thank the CSIR for research fellowships (SRFs). AB is a SERB-STAR Fellow and sincerely acknowledges the SERB [STR/2020/000061] for their generous support.Notes and references
- (a) Review: K. Nagaraju and D. Ma, Chem. Soc. Rev., 2018, 47, 8018–8029 RSC; (b) P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2004, 126, 7450–7451 CrossRef CAS PubMed; (c) L. Bai, Y. Ma and X. Jiang, J. Am. Chem. Soc., 2021, 143, 20609–20615 CrossRef CAS PubMed.
- H. Ueda, S. Sato, K. Noda, H. Hakamata, E. Kwon, N. Kobayashi and H. Tokuyama, Angew. Chem., Int. Ed., 2023, 62, e202302404 CrossRef CAS PubMed.
- Review: I. S. Marcos, R. F. Moro, I. Costales, P. Basabe and D. Díez, Nat. Prod. Rep., 2013, 30, 1509–1526 RSC.
- (a) Isolation: Q. Zhang, A. Mandi, S. Li, Y. Chen, W. Zhang, X. Tian, H. Zhang, H. Li, W. Zhang, S. Zhang, J. Ju, T. Kurtan and C. Zhang, Eur. J. Org Chem., 2012, 5256–5262 CrossRef CAS; (b) Z. Xu, M. Baunach, L. Ding and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 10293–10297 CrossRef CAS PubMed; (c) M. Baunach, L. Ding, T. Bruhn, G. Bringmann and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 9040–9043 CrossRef CAS PubMed; (d) Q. Zhang, H. Li, Y. Sun, L. Yu, Y. Zhu, H. Zhu, L. Zhang, S. Li, Y. Shen, C. Tian, A. Li, H. Liu and C. Zhang, Chem. Sci., 2017, 8, 5067–5077 RSC.
- Isolation of xiamycin A, see: (a) L. Ding, J. Münch, H. Goerls, A. Maier, H.-H. Fiebig, W.-H. Lin and C. Hertweck, Bioorg. Med. Chem. Lett., 2010, 20, 6685–6687 CrossRef CAS PubMed; (b) L. Ding, A. Maier, H. H. Fiebig, W. H. Lin and C. Hertweck, Org. Biomol. Chem., 2011, 9, 4029–4031 RSC; (c) H. Li, Q. Zhang, S. Li, Y. Zhu, G. Zhang, H. Zhang, X. Tian, S. Zhang, J. Ju and C. Zhang, J. Am. Chem. Soc., 2012, 134, 8996–9005 CrossRef CAS PubMed; (d) Isolation of xiamycins C-E, see; S.-H. Kim, T.-K.-Q. Ha, W. K. Oh, J. Shin and D.-C. Oh, J. Nat. Prod., 2016, 79, 51–58 CrossRef CAS PubMed.
- (a) B. R. Rosen, E. W. Werner, A. G. O'Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571–5574 CrossRef CAS PubMed; (b) Z. Meng, H. Yu, L. Li, W. Tao, H. Chen, M. Wan, P. Yang, D.-J. Edmonds, J. Zhong and A. Li, Nat. Commun., 2015, 6, 6096–7003 CrossRef CAS PubMed; (c) A. H. Trotta, Org. Lett., 2015, 17, 3358–3361 CrossRef CAS PubMed; (d) J. Feng, F. Noack and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 12364–12367 CrossRef CAS PubMed; (e) A. H. Trotta, J. Org. Chem., 2017, 82, 13500–13516 CrossRef CAS PubMed; (f) M. Pfaffenbach, I. Bakanas, N. R. O'Connor, J. L. Herrick and R. Sarpong, Angew. Chem., Int. Ed., 2019, 58, 15304–15308 CrossRef CAS PubMed; (g) D. H. Dethe and M. Shukla, Chem. Commun., 2021, 57, 10644–10646 RSC.
- A. Klapars, J. C. Antilla, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7727–7729 CrossRef CAS PubMed.
- (a) M. Munda, R. Nandi, V. R. Gavit, S. Kundu, S. Niyogi and A. Bisai, Chem. Sci., 2022, 13, 11666–11671 RSC; (b) R. Nandi, S. Niyogi, S. Kundu, V. R. Gavit, M. Munda, R. Murmu and A. Bisai, Chem. Sci., 2023, 14, 8047–8053 RSC.
- (a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed; (b) L. F. Silva Jr. and B. Olofsson, Nat. Prod. Rep., 2011, 28, 1722 RSC.
- B. Li, H. Nomura, H. Miyazaki, Q. Zhang, K. Yoshida, Y. Suzuma, A. Orita, J. Otera and C. Adachi, Chem. Lett., 2014, 43, 319–321 CrossRef CAS.
- S. Mallick, S. Maddala, K. Kollimalayan and P. Venkatakrishnan, J. Org. Chem., 2019, 84, 73–93 CrossRef CAS PubMed.
- K. A. D'angelo, C. K. Schissel, B. L. Pentelute and M. Movassaghi, Science, 2022, 375, 894–899 CrossRef PubMed.
- T. Dohi, K. Morimoto, A. Maruyama and Y. Kita, Org. Lett., 2006, 8, 2007–2010 CrossRef CAS PubMed.
- For biaryl synthesis via an oxidative process involving diaryliodonium-mediated cross-over pathways, see; (a) T. Dohi, M. Ito, N. Yamaoka, K. Morimoto, H. Fujioka and Y. Kita, Angew. Chem., Int. Ed., 2010, 49, 3334–3337 CrossRef CAS PubMed; (b) N. Yamaoka, K. Sumida, I. Itani, H. Kubo, Y. Ohnishi, S. Sekiguchi, T. Dohi and Y. Kita, Chem.–Eur. J., 2013, 19, 15004–15011 CrossRef CAS PubMed.
- (a) T. Dohi, N. Yamaoka and Y. Kita, Tetrahedron, 2010, 66, 5775–5785 CrossRef CAS; (b) T. C. Turner, K. Shibayama and D. L. Boger, Org. Lett., 2013, 15, 1100–1103 CrossRef CAS PubMed.
- We sincerely thank the reviewers for their suggestions on the mechanistic consideration for PIFA-mediated oxidative dimerization process following a SET-mechanism..
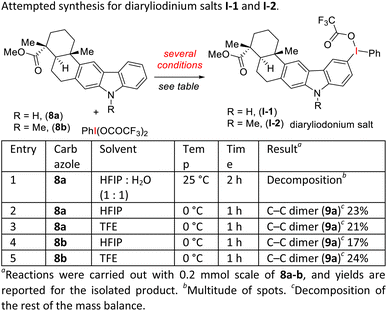
- Despite rigorous attempts to isolate the diaryliodonium salt using carbazoles 8a-b following the established literature procedures,14,15 we were not successful. If fact in most of these cases, significant amount of the C–C dimer (bicarbazole) was obtained (see, below scheme). This is probably due to its highly reactive nature of carbazole motifs under the oxidation mediated by hypervalent iodine(III) reagent, PIFA
 .
. - A. Maity, A. Roy, M. K. Das, S. De, M. Naskar and A. Bisai, Org. Biomol. Chem., 2020, 18, 1679–1684 RSC.
- Y. Sun, P. Chen, D. Zhang, M. Baunach, C. Hertweck and A. Li, Angew. Chem., Int. Ed., 2014, 53, 9012–9016 CrossRef CAS PubMed.
Footnotes |
| † This work is dedicated respectfully to Professor Vinod K. Singh, IIT Kanpur, on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01396d |
| § MM and AM contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |