
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel fungal diphenyl ether biosynthetic gene clusters encode a promiscuous oxidase for elevated antibacterial activities†
Qingpei
Liu‡
 a,
Shuaibiao
Gao‡
a,
Jin
Fang‡
a,
Yifu
Gong
a,
Yiling
Zheng
a,
Yao
Xu
a,
Dan
Zhang
a,
Jiayuan
Wei
a,
Liangxiu
Liao
a,
Ming
Yao
a,
Wenjing
Wang
a,
Xiaole
Han
c,
Fusheng
Chen
de,
István
Molnár
*b and
Xiaolong
Yang
*a
a,
Shuaibiao
Gao‡
a,
Jin
Fang‡
a,
Yifu
Gong
a,
Yiling
Zheng
a,
Yao
Xu
a,
Dan
Zhang
a,
Jiayuan
Wei
a,
Liangxiu
Liao
a,
Ming
Yao
a,
Wenjing
Wang
a,
Xiaole
Han
c,
Fusheng
Chen
de,
István
Molnár
*b and
Xiaolong
Yang
*a
aSchool of Pharmaceutical Sciences, South-Central Minzu University, Wuhan 430074, P.R. China. E-mail: 2019001@mail.scuec.edu.cn
bVTT Technical Research Centre of Finland, FI-02044 VTT, Espoo, Finland. E-mail: istvan.molnar@vtt.fi
cSchool of Chemistry and Materials Science, South-Central Minzu University, Wuhan 430074, P.R. China
dSchool of Life Sciences, Guizhou Normal University, Guiyang 550025, P.R. China
eCollege of Food Science and Technology, Huazhong Agricultural University, Wuhan 430070, P.R. China
First published on 29th July 2024
Abstract
Diphenyl ethers (DPEs) are produced by filamentous fungi using polyketide synthases (PKSs) directly, or via Cu oxidase-catalyzed oxidative rearrangements of benzophenone intermediates. Here, we use heterologous expression to reveal a third route towards DPEs in Preussia isomera that relies on an oxidative multienzyme cascade to convert a PKS-generated, ester-linked didepside to depsidones and further to DPEs, and apply comparative genomics to identify conserved biosynthetic gene clusters for this pathway in multiple fungi. The distribution of DPE products is modulated by the expression chassis upon pathway reconstitution. Among the post-PKS enzymes, the DpeH tyrosinase shows considerable substrate promiscuity towards synthetic DPE analogues. By creating hybrid enzymes with a DpeH orthologue from Aspergillus nidulans, we identify the C-terminal region of DpeH to alter substrate recognition. Our work highlights an evolutionarily conserved way to produce DPEs, and provides enzymatic tools to generate DPE analogues with broad spectrum antibiotic activity against multidrug-resistant human pathogens.
Naturally occurring didepsides, encompassing two hydroxybenzoic acid moieties connected by an ester linkage, are common lichen metabolites and have also been isolated from a limited number of fungi. Didepsides display antitumor, antimalarial, antibacterial, antifouling, and various human enzyme-inhibitory activities.1 The ester linkage may form by oxidative coupling or rearrangements after the release of orsellinic acid or its derivatives from polyketide synthase enzymes (PKSs).1,2 However, PKSs directly affording didepsides have also been reported recently, among which DrcA from Aspergillus duricaulis,3 MollE from Ovatospora sp.,4 and DepH from A. sp. SCSIO SX7S7
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 condense acyl carrier protein-bound intermediates to form ester-linked didepsides (Fig. 1A). The enzymes encoded by gene clusters containing such PKSs may further elaborate the didepsides. Thus, cytochrome P450 monooxygenases (P450s) such as MollD and DepG install an ether bond to form depsidones (DEPs),4,5 while nonribosomal peptide synthetases (such as DrcB) or prenyltransferases (e.g., MollF) form composite natural products (Fig. 1A).3,4
5 condense acyl carrier protein-bound intermediates to form ester-linked didepsides (Fig. 1A). The enzymes encoded by gene clusters containing such PKSs may further elaborate the didepsides. Thus, cytochrome P450 monooxygenases (P450s) such as MollD and DepG install an ether bond to form depsidones (DEPs),4,5 while nonribosomal peptide synthetases (such as DrcB) or prenyltransferases (e.g., MollF) form composite natural products (Fig. 1A).3,4
 | ||
| Fig. 1 Biosynthetic gene clusters in fungi and models for the biosynthesis of (A) didepsides; and (B) DPEs. | ||
Previously, we identified a novel PKS, Preu6 (renamed DpeA here) from Preussia isomera XL1326, which utilizes collaborating starter acyl transferase (SAT) and thioesterase (TE) domains to form didepside 1 (Fig. 2D).6,7 In addition to dpeA, the corresponding cluster contains eight genes, dpeB–dpeI (GenBank accession PP925597, Fig. 2A). We reconstituted this cluster and delineated the reaction order of the encoded enzymes using stepwise heterologous expression in Saccharomyces cerevisiae BJ5464-NpgA8,9 (Table S1 and Fig. S1†). The results revealed that DpeB (P450, 49% identity to DepG of A. sp. SCSIO SX7S75) and DpeD (decarboxylase, 42% identity to DepF) transform didepside 1 to two DEPs (2 and 3), similar to the reactions catalyzed by their DepGF orthologues during the formation of unguinol (Fig. 1A).5 Next, DpeC (predicted α/β-hydrolase), DpeE (putative methyltransferase with a Methyltransf_23 conserved domain, pfam13489), DpeF (deduced methyltransferase with a Methyltransf_2 conserved domain, pfam00891), and DpeH (putative tyrosinase) convert DEP 3 to a series of DPEs, 4–8, among which DPE 8 is a new compound (Fig. 2B and D).
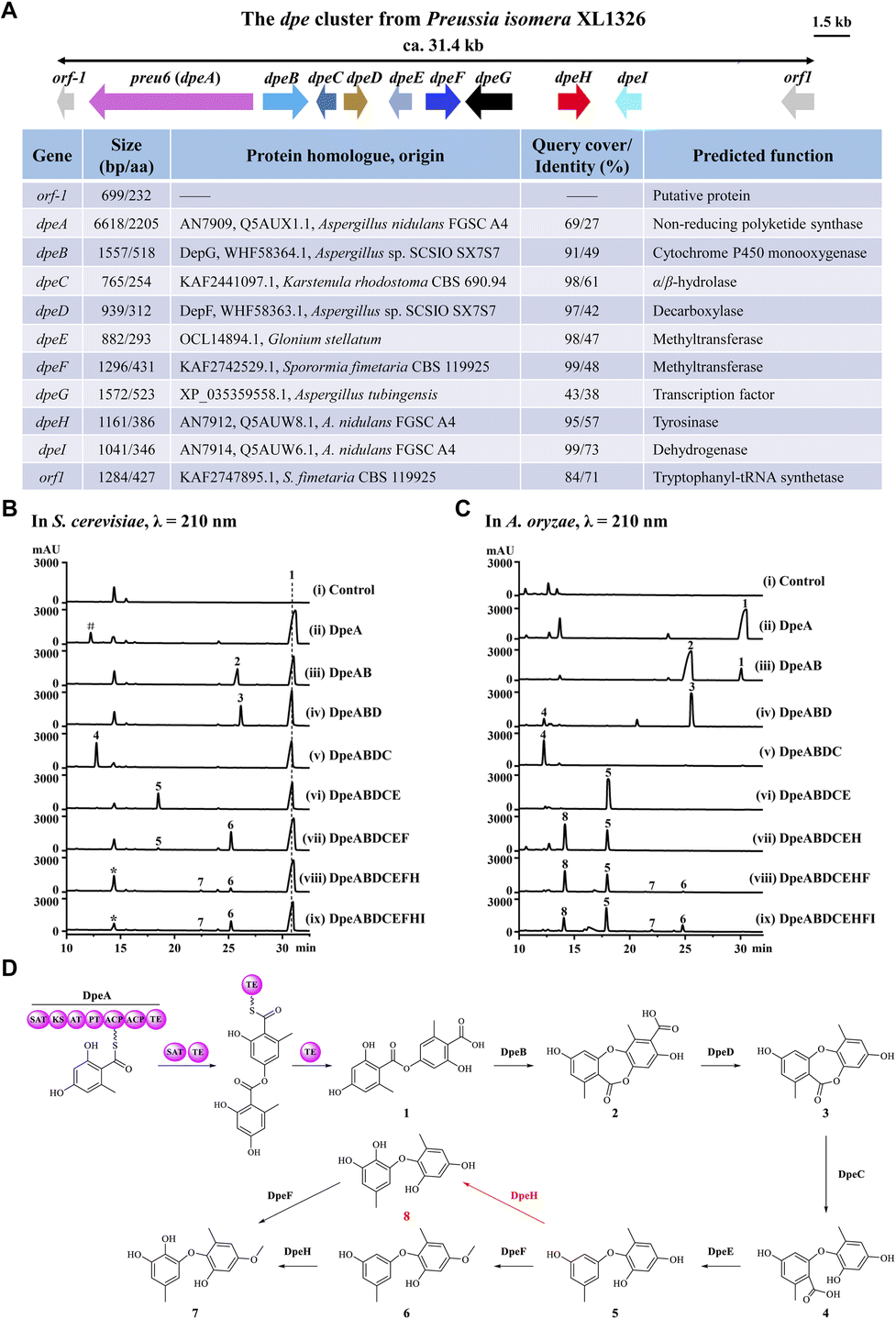 | ||
| Fig. 2 Characterization of the dpe gene cluster in Preussia isomera. (A) Schematic representation and annotation of the dpe cluster. (B and C) Product profiles of S. cerevisiae BJ5464-NpgA or A. oryzae NSAR1 transformants expressing the indicated gene combinations, respectively. Control, the corresponding empty vectors. *, a mixture of an endogenous yeast metabolite with trace amounts of 8 (detected by MS); #, orsellinic acid. (D) Biosynthetic model for DPEs 7 and 8. | ||
Since the production of 7 was very inefficient in the yeast system, we could not detect any further transformations with DpeI, a putative dehydrogenase (73% identity to AN7914 of A. nidulans with an unknown function10,11). Thus, we introduced the dpe biosynthetic gene combinations defined in BJ5464-NpgA into A. oryzae NSAR1.12 The biosynthetic steps catalyzed by DpeABDCE to yield compounds 1–5 were identical in the two systems, although small amounts of 3 were also converted to 4 by endogenous enzymes in A. oryzae (Fig. 2C and S2†). DpeE, a predicted SAM-dependent methyltransferase (Fig. S3A†), acted as a decarboxylase in both chassis to convert 4 to 5, as precedented by other enzymes with an apparent methyltransferase fold.13 This decarboxylase activity was not dependent on the presence of SAM as verified by in vitro reconstitution of the recombinant DpeE enzyme (Fig. S3B and C†). Remarkably, A. oryzae NSAR1 preferred to transform 5 to 8 using DpeH (Fig. S4†), in contrast to the yeast BJ5464-NpgA that utilized DpeF to convert 5 to 6 (Fig. 2B). Despite the higher productivity of the A. oryzae chassis, DpeI remained apparently nonfunctional, nor did purified, recombinant DpeI catalyze any conversions of 1–8in vitro (Fig. S5†). Thus, the function of DpeI, if any, requires further investigation. Meanwhile, 8 was shown to be transformed to 7 using in vivo biocatalytic conversions with DpeF-producing S. cerevisiae or A. oryzae strains (Fig. S6†).
Taken together, the dpe cluster relies on the transformation of a PKS-generated, ester-linked didepside to DEPs, and further to various DPEs in a multistep, multi-enzyme, oxidative reaction cascade. This is different from the ors cluster of A. nidulans FGSC A4 where PKS AN7909 alone affords a DPE;10,14 or from the pta cluster of Pestalotiopsis fici where the Cu oxidase PtaE catalyzes an oxidative rearrangement to generate the DPE product (Fig. 1B).15
To date, approximately 170 DPEs have been isolated from 46 fungal species (Table S2†). Upon alignment of the 31 available genome sequences of DPE producers (Table S2†) to the known clusters ors, pta, and dpe, we identified putative DPE clusters in 20 species (Table S3†), although the existence of divergent DPE clusters cannot be excluded in the remaining species either. The identified clusters could be classified into three categories. Twelve DPE clusters belong to Type I (ors-like clusters; Fig. 3A), featuring PKSs with high similarity to AN7909. These are predicted to produce diorcinolic acid-like DPEs utilizing these PKSs alone.10,14 Correspondingly, the DPE compounds isolated from these 12 species are overwhelmingly diorcinol derivatives (Table S4†). Three DPE clusters belong to Type II (pta-like clusters; DPE products similar to pestheic acid; Fig. 3B and Table S5†), in which PtaE-like Cu oxidases are predicted to form the ether linkage.15 Finally, five DPE clusters belong to Type III (dpe-like clusters; Fig. 3C), featuring conserved PKSs, P450s, and DpeD-like decarboxylases that presumably generate DEPs en route to DPEs (Table S6†). Three of the five Type III clusters (those from Corynespora, Boeremia, and Aspergillus spp.) lack DpeC orthologues that would hydrolyze the ester bond of DEPs to form DPEs. We hypothesize that endogenous hydrolases encoded elsewhere in the genomes of these species perform this function, just as seen in A. oryzae NSAR1 (Fig. 2C and S2†). Interestingly, all three cluster types are present in genus Aspergillus, potentially enriching the variety of DPEs in these fungi.
 | ||
| Fig. 3 Distribution of the proposed DPE biosynthetic gene clusters in fungi. (A) ors-like gene clusters (Type I), in which the PKSs alone may produce DPEs. (B) pta-like gene clusters (Type II), in which conserved PKSs and PtaE-like Cu oxidases are predicted to be involved in the formation of DPEs. (C) dpe-like gene clusters (Type III), in which conserved PKSs and P450s may produce DEPs that are further converted to DPEs. Arrows with identical colors indicate orthologous genes. The numbers above the genes indicate the percent identities of the encoded proteins to their orthologues in the corresponding model cluster. | ||
Next, we tested the activities of compounds 1–8 against eight antibiotic-resistant bacteria. DEP 3 was active against Gram-positive bacteria, while DPEs 7 and 8 with the C-2′ hydroxy functionality installed by DpeH displayed broad-spectrum antibacterial activities. Remarkably, the novel DPE 8 (yield: 259.8 ± 15.9 mg L−1 in A. oryzae; Fig. S7†) exhibited potent activities against multidrug-resistant Staphylococcus epidermidis, methicillin-resistant Sta. aureus (MIC = 6.25 μg mL−1), and carbapenems-resistant Acinetobacter baumannii and Klebsiella pneumoniae (MIC = 12.5 μg mL−1; Table S7†).
Then, we turned to AN7912, an orthologue (57% identity) of DpeH in the ors cluster. AN7912 hydroxylates diorcinol (Fig. 1B)11 and accepts the simplified substrate 9 (Fig. 4C), but this enzyme could not convert 5 to 8 (Fig. 4B), or 6 to 7 (Fig. S8†). In contrast, DpeH converts 9, 5, and 6 to their hydroxylated derivatives (9a/b, 8, and 7, respectively; Fig. 4B and S8†). The AlphaFold2-predicted16 structures of AN7912 and DpeH differed both at their N-termini (M1–K59 in DpeH; M1–R58 in AN7912) and C-termini (L346–Q386 in DpeH; L344–P369 in AN7912; Fig. 4A and S9†). Replacing either or both terminal regions of AN7912 with those of DpeH showed that hybrid M2 (AN7912[M1–L343] + DpeH[L346–Q386]) could transform compounds 5 to 8, and 6 to 7 while chimera M1 (DpeH[M1–K59] + AN7912[E59–P369]) could not, revealing that the C-terminal region modulates substrate recognition. Correspondingly, hybrid M4 (DpeH[M1–L345] + AN7912[L344–P369]), and truncated enzyme M5 (DpeH[M1–L345]) lost the ability to convert 5 to 8, or 6 to 7 (Fig. 4B and S8†). Importantly, all tested chimeras (M1–M4) and truncated enzymes (M5–M6) retained their activities towards simplified substrate 9.
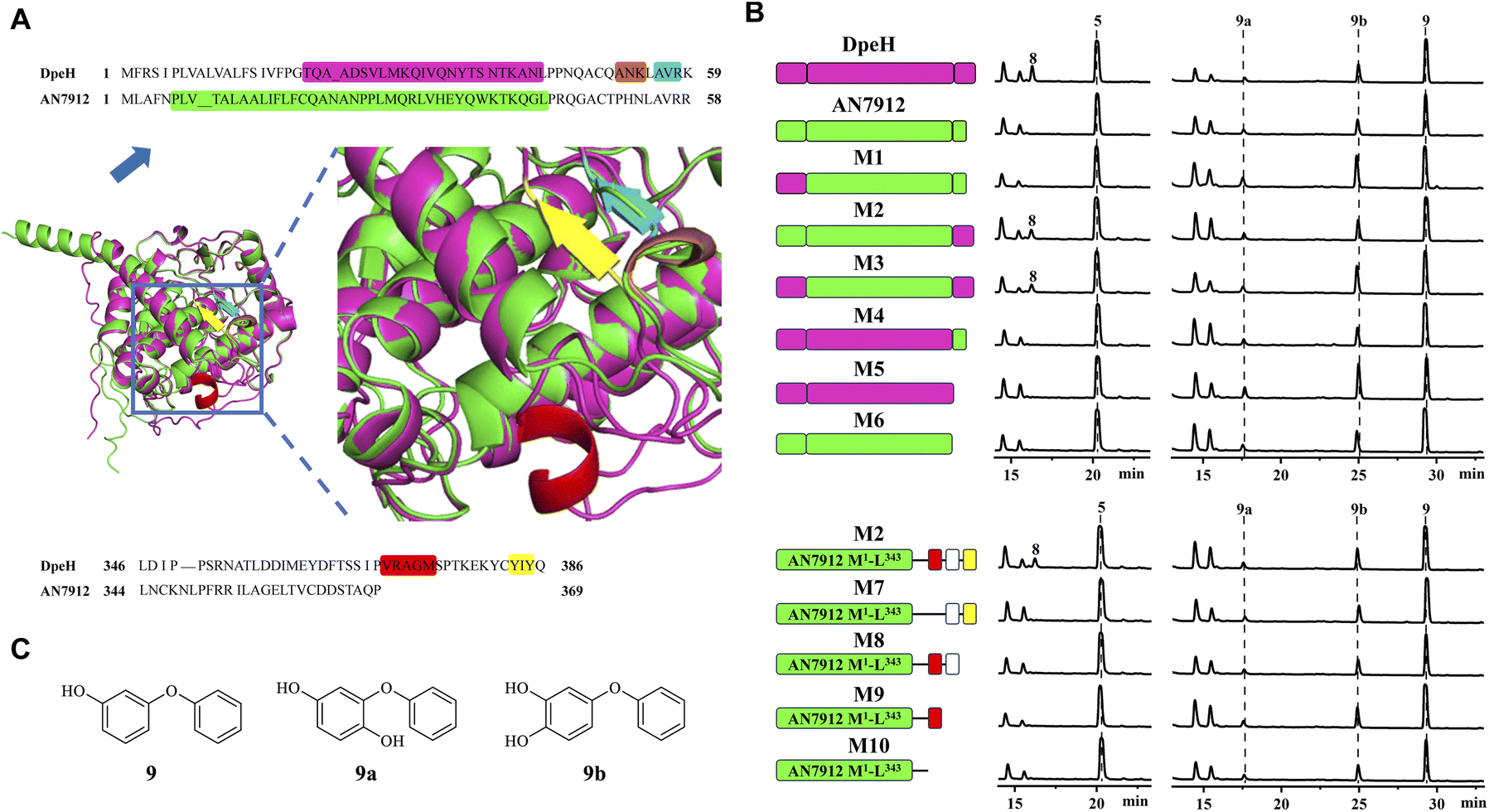 | ||
| Fig. 4 Structural elements of DpeH that modulate substrate recognition. (A) Protein structural differences between DpeH (magenta) and AN7912 (green) predicted by AlphaFold2. Brown or cyan highlights, α-helix or β-sheet, respectively, in the N-terminal region of DpeH (top); red or yellow highlights, α-helix or β-sheet, respectively, in the C-terminal region of DpeH (bottom). (B) Product profiles (210 nm) of the A. oryzae NSAR1 strains expressing the indicated enzymes and challenged with substrates 5 or 9, respectively. In mutant enzymes M1–M10, AN7912-derived regions are shown in green. In mutants M1–M5, segments from DpeH are in magenta. In mutants M7–M10 (derived from M2), missing boxes indicate deletions; red boxes show the α-helix, white boxes represent the linker, and yellow boxes indicate the β-sheet of DpeH. (C) Structures of compounds 9, 9a and 9b. | ||
The C-terminal region of DpeH possesses a short α-helix (V370–M374), a linker (S375–C382), and a short β-sheet (Y383–Y385) that are all missing from AN7912 (Fig. 4A). Deleting the α-helix from chimera M2 (i.e., mutant M7: M2-Δ[V368–M372]) or progressively truncating the M2 hybrid enzyme (i.e., mutants M8–M10) revealed that all these structural elements are necessary to produce 8 from 5, and 7 from 6, but their presence or absence does not affect the hydroxylation of substrate 9 (Fig. 4B and S8†).
Finally, we tested the substrate promiscuities of DpeH and AN7912 with various synthetic DPE analogues using biotransformation in the A. oryzae chassis. DPE 11 was partially converted to 11c by an unknown endogenous enzyme of A. oryzae (Fig. 5 and S10†). AN7912 showed a narrow substrate spectrum, affording only minute amounts of 17a from 17, but rejecting all other substrates (Fig. 5B). In contrast, DpeH turned out to be a promiscuous enzyme accepting all tested DPEs to generate novel “unnatural” products (Fig. 5A and D). The regiospecificity of the reaction was strict, with hydroxylation at C-2′ in all cases. With DPE 17, double hydroxylation at C-3 and C-2′ was also observed, as may have been expected considering the symmetrical structure of this substrate (Fig. 5A). Importantly, mutant M2 (AN7912[M1–L343] + DpeH[L346–Q386]) was just as promiscuous as DpeH, and afforded the same biotransformation products (Fig. S11†), confirming the essential role of the C-terminal region of DpeH in determining substrate specificity.
 | ||
| Fig. 5 Substrate promiscuity of DpeH and AN7912. (A and B) Product profiles (210 nm) of the A. oryzae NSAR1 strains expressing DpeH or AN7912, and fed with substrates 10–17, respectively. ※, 17a was detectable only with MS. (C and D) Structures of substrates 10–17, DpeH products 10a–17a and 17b, and host-derived product 11c. *, New compounds. | ||
Conclusions
In summary, we characterized the dpe biosynthetic gene cluster of Preussia isomera via heterologous expression in S. cerevisiae and A. oryzae chassis. Most predicted DPE biosynthetic clusters in fungi seem to rely on a conserved PKS to directly form DPE analogues from orsellinic acid-type nascent, PKS-bound monomers. Rarer DPE clusters afford their products by enlisting a PtaE-like Cu oxidase to catalyze the oxidative rearrangement of polyketide-derived benzophenones. In contrast, dpe-type clusters utilize a conserved PKS to produce analogues of the ester-linked didepside lecanoric acid 1. These are then converted to DPEs by a multienzyme cascade. With the dpe cluster, oxidative conversion of 1 to DEP 2 is catalyzed by DpeB, followed by decarboxylation to 3 by DpeD, oxidative ring opening to 4 by DpeC, and another decarboxylation to 5 by DpeE. DPE 5 may then be converted to the novel broad-spectrum antibiotic 8 by DpeH, or methylated by DpeF to yield 6 which is then hydroxylated by DpeH to yield another broad-spectrum antibiotic, DPE 7. Interestingly, the preference to produce 8 or 6/7 seems to be determined by the chassis, with DPE 8 dominating in Aspergillus, while DPEs 6 and 7 preferred in Saccharomyces. DpeH is a promiscuous enzyme that recognizes a variety of DPE analogues, and faithfully transforms them to their C-2′ hydroxylated derivatives. AN7912, the DpeH orthologue encoded in the ors cluster of A. nidulans is a much less promiscuous catalyst than DpeH. However, transplanting a short C-terminal region of DpeH (41 amino acids) broadens the substrate specificity of AN7912, and allows the resulting chimera to accept DPEs 5 and 10–17 as its substrates. Our work not only clarifies a novel pathway towards DPEs in filamentous fungi, but also provides a useful tool for synthetic biology to produce novel “unnatural” DPE analogues and highlights protein engineering strategies to broaden the substrate specificity of DpeH orthologues. Meanwhile, DPEs 7 and 8 may further be evaluated as potent broad-spectrum antibiotics against multidrug-resistant Gram-positive and Gram-negative pathogens on the WHO global priority list.Data availability
Materials and methods, additional tables and figures, and spectroscopic data are available in the ESI.† The dpe gene cluster of P. isomera XL1326 has been deposited in GenBank under accession code PP925597. The genome sequences used for comparative genomics were obtained from the NCBI GenBank or the Joint Genome Institute (JGI) MycoCosm databases (Table S2†).Author contributions
Conceptualization, project administration, supervision, and resources, Q. L., I. M., and X. Y.; methodology, investigation, and data curation, Q. L., S. G., J. F., Y. G., Y. Z., Y. X., D. Z., and J. W.; formal analysis, Q. L., S. G., J. F., Y. G., L. L., M. Y., W. W., X. H., and F. C.; writing – original draft, Q. L., S. G., and J. F.; writing – review & editing, Q. L., I. M., and X. Y.; funding acquisition, Q. L., X. Y., W. W., and I. M.Conflicts of interest
I. M. has disclosed financial interests in TEVA Pharmaceuticals Hungary which are unrelated to the subject of the research presented here. All other authors declare no competing financial interests.Acknowledgements
This research was supported by the National Natural Science Foundation of China (82073714 to X. Y., 22007102 to Q. L., 82373757 to W. W.); the Knowledge Innovation Program of Wuhan-Shuguang Project (2022020801020406 to Q. L.); the National Key Research and Development Program of China (2023YFF1104001 to X. Y.); the Key project at central government level: the ability establishment of sustainable use for valuable Chinese medicine resources (2060302) (2060302-2101-20 to Q. L.); the Fundamental Research Funds for the Central Universities, South-Central Minzu University (CZQ23045 to Q. L.); and VTT Technical Research Centre of Finland (to I. M.). We express special thanks to Prof. Ikuro Abe (The University of Tokyo) for providing the Aspergillus host strain and expression vectors, and to the members of the laboratories of Prof. Lixia Pan (Guangxi Academy of Sciences) for their useful discussions about the protein structures. We also acknowledge support from the Instrumental Analysis Center of School of Pharmaceutical Sciences, South-Central Minzu University. This article is dedicated to Prof. A. A. Leslie Gunatilaka (SW Center for Natural Products Research, The University of Arizona) upon his retirement.References
- S. R. M. Ibrahim, A. Sirwi, B. G. Eid, S. G. A. Mohamed and G. A. Mohamed, Fungal depsides-naturally inspiring molecules: biosynthesis, structural characterization, and biological activities, Metabolites, 2021, 11, 683 CrossRef CAS PubMed
.
- E. Stocker-Wörgötter, Metabolic diversity of lichen-forming ascomycetous fungi: culturing, polyketide and shikimate metabolite production, and PKS genes, Nat. Prod. Rep., 2008, 25, 188 RSC
- L. Chen, X. Wei and Y. Matsuda, Depside bond formation by the starter-unit acyltransferase domain of a fungal polyketide synthase, J. Am. Chem. Soc., 2022, 144, 19225 CrossRef CAS PubMed
- X. Zhao, Y. Chen, T. Long, Z. Liu, Q. Zhang, H. Zhang, Y. Yan, C. Zhang and Y. Zhu, Genome mining and biosynthetic reconstitution of fungal depsidone mollicellins reveal a dual functional cytochrome P450 for ether formation, J. Nat. Prod., 2023, 86, 2046 CrossRef CAS PubMed
- J. Yang, Z. Zhou, Y. Chen, Y. Song and J. Ju, Characterization of the depsidone gene cluster reveals etherification, decarboxylation and multiple halogenations as tailoring steps in depsidone assembly, Acta Pharm. Sin. B, 2023, 13, 3919 CrossRef PubMed
- Q. Liu, D. Zhang, S. Gao, X. Cai, M. Yao, Y. Xu, Y. Gong, K. Zheng, Y. Mao, L. Yang, D. Yang, I. Molnár and X. Yang, Didepside formation by the nonreducing polyketide synthase Preu6 of Preussia isomera requires interaction of starter acyl transferase and thioesterase domains, Angew. Chem., Int. Ed., 2023, 62, e202214379 CrossRef CAS PubMed
- Q. Liu, D. Zhang, Y. Xu, S. Gao, Y. Gong, X. Cai, M. Yao and X. Yang, Cloning and functional characterization of the polyketide synthases based on genome mining of Preussia isomera XL-1326, Front. Microbiol., 2022, 13, 819086 CrossRef PubMed
- C. Wang, X. Wang, L. Zhang, Q. Yue, Q. Liu, Y. M. Xu, A. A. L. Gunatilaka, X. Wei, Y. Xu and I. Molnár, Intrinsic and extrinsic programming of product chain length and release mode in fungal collaborating iterative polyketide synthases, J. Am. Chem. Soc., 2020, 142, 17093 CrossRef CAS PubMed
- X. Wang, C. Wang, L. Duan, L. Zhang, H. Liu, Y. Xu, Q. Liu, T. Mao, W. Zhang, M. Chen, M. Lin, A. A. L. Gunatilaka, Y. Xu and I. Molnár, Rational reprogramming of O-methylation regioselectivity for combinatorial biosynthetic tailoring of benzenediol lactone scaffolds, J. Am. Chem. Soc., 2019, 141, 4355 CrossRef CAS PubMed
- C. Feng, Q. Wei, C. Hu and Y. Zou, Biosynthesis of diphenyl ethers in fungi, Org. Lett., 2019, 21, 3114 CrossRef CAS PubMed
- A. Ninomiya, S. I. Urayama and D. Hagiwara, Antibacterial diphenyl ether production induced by co-culture of Aspergillus nidulans and Aspergillus fumigatus, Appl. Microbiol. Biotechnol., 2022, 106, 4169 CrossRef CAS PubMed
- Z. Cao, S. Li, J. Lv, H. Gao, G. Chen, T. Awakawa, I. Abe, X. Yao and D. Hu, Biosynthesis of clinically used antibiotic fusidic acid and identification of two short-chain dehydrogenase/reductases with converse stereoselectivity, Acta Pharm. Sin. B, 2019, 9, 433 CrossRef PubMed
- D. Yan and Y. Matsuda, Biosynthetic elucidation and structural revision of brevione E: Characterization of the key dioxygenase for pathway branching from setosusin biosynthesis, Angew. Chem., Int. Ed., 2022, 61, e202210938 CrossRef CAS PubMed
- Q. Wei, Z. P. Wang, X. Zhang and Y. Zou, Diaryl ether formation by a versatile thioesterase domain, J. Am. Chem. Soc., 2022, 144, 9554 CrossRef CAS PubMed
- X. Xu, L. Liu, F. Zhang, W. Wang, J. Li, L. Guo, Y. Che and G. Liu, Identification of the first diphenyl ether gene cluster for pestheic acid biosynthesis in plant endophyte Pestalotiopsis fici, Chembiochem, 2014, 15, 284 CrossRef CAS PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 96, 583 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Material and methods, additional tables and figures, and NMR data. See DOI: https://doi.org/10.1039/d4sc01435a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |