
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increased CO2/N2 selectivity by stepwise fluorination in isoreticular ultramicroporous metal–organic frameworks†
Tuo
Di
a,
Yukihiro
Yoshida
 *a,
Ken-ichi
Otake
b,
Susumu
Kitagawa
b and
Hiroshi
Kitagawa
*a
*a,
Ken-ichi
Otake
b,
Susumu
Kitagawa
b and
Hiroshi
Kitagawa
*a
aDivision of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-Oiwakecho, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: yoshiday@ssc.kuchem.kyoto-u.ac.jp; kitagawa@kuchem.kyoto-u.ac.jp
bInstitute for Integrated Cell-Material Sciences (iCeMS), Kyoto University Institute for Advanced Study, Kyoto University, Yoshida Ushinomiya-cho, Sakyo-ku, Kyoto 606-8501, Japan
First published on 16th May 2024
Abstract
Exploration of porous adsorbents with high CO2/N2 selectivity is of great significance for reducing CO2 content in the atmosphere. In this study, a series of isoreticular ultramicroporous fluorinated metal–organic frameworks (MOFs) were prepared to explore the benefits of fluorinated ultramicropores in improving CO2/N2 selectivity. Gas adsorption measurements revealed that the increase in the number of fluorine atoms in a ligand molecule leads to the increased CO2 uptakes and CO2/N2 selectivity. Theoretical calculations indicate that the interaction between the fluorine atoms and adsorbed CO2 molecules enhances the CO2-philicity, offering useful insight into the improvement of CO2/N2 selectivity in isoreticular frameworks.
Introduction
Excessive emissions of greenhouse gases, primarily carbon dioxide (CO2), can lead to the intensification of the greenhouse effect, resulting in global temperature imbalance.1–4 It is imperative to develop appropriate and effective methods to address the CO2 emitted from production and daily activities. Until now, carbon capture and sequestration (CCS) and direct air capture (DAC) technologies, which can efficiently capture CO2 from emission sources, are acknowledged as effective approaches for CO2 treatment.5–8 Exploration of efficient CO2 capture materials that meet the needs of above-mentioned scenarios is of utmost importance in reducing the impact of greenhouse effect. Although traditional inorganic adsorbents such as activated carbon and zeolite have shown prominent gas separation selectivities,9 their low structural designability has limited their use as CO2 capture adsorbents at low concentration in ambient air and indoor confined spaces.Metal–organic frameworks (MOFs) have emerged as a new family of highly promising porous materials for CO2 capture because of their structural and chemical tunability.10–12 Given the self-assembling construction of metal ions/clusters and organic linkers via strong coordination bonds, MOFs have provided new avenues to accurately regulate both their surface and interior by judiciously selecting metal knots and organic ligands to regulate their CO2-philicity.13,14 It has been found that the interpenetration of microporous frameworks to produce ultramicropores (pore size: ca. 0.5 nm) leads to higher CO2 affinity, thus higher CO2/N2 selectivity, than those of the parent microporous MOF.15 In addition, chemical modifications, such as fluorination of component ligands, can also lead to increased CO2-philicity, because the favourable interactions between the quadrupole of CO2 and fluorine atoms can greatly enhance CO2 affinity.16–19 Thus, it is possible that the combination of fluorination with the ultramicroporous structure offers an optimal platform to systematically investigate the effect of fluorination on CO2/N2 selectivity. To this end, it is necessary to choose a series of isostructural MOFs with different degrees of fluorination, because uncertainties caused by different structural characteristics inevitably reduce the rationality of the obtained results. It should be mentioned that the fluorination of ligands greatly affects the framework structure in many cases,20–22 which will hamper our research to examine the fluorination effect on the gas sorption properties.
Chun and co-workers reported a family of pillar-layered MOFs; ZnII-paddlewheel dimers coordinate with bis-bidentate terephthalate (BDC2−) linkers to construct two-dimensional (2D) square grids, which are connected by bis-monodentate 1,4-diazabicyclo[2.2.2]octane (DABCO) linkers.23 Of particular importance is that the three-dimensional (3D) framework structure renders the ultramicropores (pore size ∼0.6 nm) and even remains unchanged by replacing hydrogen atoms on BDC ligands with other functional groups such as methyl and halogen groups,23–26 which is inherently suitable for the present research.
In this study, we selected the ultramicroporous Zn2(BDC)(tmBDC)(DABCO) (tmBDC2−: tetramethylterephthalate) as a parent MOF because of its high stability against water vapour compared with its analogues.27 Crystallographic, optical, and sorption studies confirmed the successful substitution of BDC ligands with 2-fluoro-BDC (1FBDC2−) or 2,5-difluoro-BDC (2FBDC2−) ligands while maintaining the framework structure. Theoretical calculations based on sorption isotherms demonstrated that increasing the fluorine content leads to a pronounced increase in CO2/N2 selectivity under low CO2 concentration conditions, primarily driven by intermolecular interaction between the fluorine atoms and adsorbed CO2 molecules. The present work would provide deeper insights into the rational design of ultramicroporous MOFs for controlling CO2/N2 selectivity.
Methods
Materials and chemicals
Zinc(II) nitrate hexahydrate (Zn(NO3)2·6H2O; 99%), hydrochloric acid (HCl; 35–37 wt%), potassium permanganate (KMnO4; 99%), and N,N-dimethylformamide (DMF; super dehydrated) were used as purchased from Fujifilm Wako Pure Chemical Industries, Ltd. Terephthalic acid (H2BDC; 99%) and 1,4-diazabicyclo[2.2.2]octane (DABCO; 98%) were obtained from Tokyo Chemical Industry Co., Ltd. 2,3,5,6-Tetramethylterephthalic acid (H2tmBDC, 95%) and 3-fluoro-4-methylbenzoic acid (98%) were acquired from Combi-Blocks Inc. 2,5-Difluoroterephthalic acid (H22FBDC, 95%) was obtained from Enamine Ltd. All these chemicals were used without purification. 2-Fluororoterephthalic acid (H21FBDC) was synthesized according to the reported literature28 with modest modifications (ESI and Fig. S1†).Syntheses
Characterization
Powder X-ray diffraction (PXRD) measurements were performed with a Rigaku MiniFlex600 with 2θ ranging from 5–50° using Cu Kα radiation (λ = 1.5418 Å). Fourier transform infrared (FT-IR) spectra were measured using a PerkinElmer Spectrum 100 spectrometer in attenuated total reflection (ATR) mode. Thermal stability was characterized by thermogravimetric analysis (TGA) on a Bruker TG-DTA2000SA at a heating rate of 5 K min−1 under N2 atmosphere. The N2 (77 K and 273 K) and CO2 (273 K and 298 K) gas adsorption and desorption measurements were performed using a BELSORP-max volumetric adsorption system. Before each measurement, the activated powder sample was activated at 343 K for 12 h. Ideal Adsorbed Solution Theory (IAST) selectivity simulation was performed based on a graphical user interface software, GraphIAST.29 Single-crystal X-ray diffraction (SCXRD) measurements were performed on a Rigaku XtaLAB PRO diffractometer equipped with a HyPix-6000HE detector with a graphite monochromatized Mo Kα radiation (λ = 0.71069 Å) at 100 K. A single crystal was cooled by a stream of cooled nitrogen gas. Breakthrough experiments were conducted on a BELCAT II-Cryo-10 apparatus. A mixture of CO2/N2 (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 v/v) was passed through a column packed with powdered samples (ca. 0.3 g) at 273 K. The samples were activated at 70 °C for 20–30 min prior to the experiments. Geometry optimizations of all CO2-adsorbed MOFs were carried out based on grand canonical Monte Carlo (GCMC) simulations using Materials Studio software (Accelrys) based on the universal force field (UFF).30
:85 v/v) was passed through a column packed with powdered samples (ca. 0.3 g) at 273 K. The samples were activated at 70 °C for 20–30 min prior to the experiments. Geometry optimizations of all CO2-adsorbed MOFs were carried out based on grand canonical Monte Carlo (GCMC) simulations using Materials Studio software (Accelrys) based on the universal force field (UFF).30
Results and discussion
Crystal structures of the three MOFs were determined by SCXRD measurements, in which as-synthesized crystals were utilized for DMOF-0F and DMOF-1F, whereas heat-treated crystals (see below) were employed for DMOF-2F. Because the guest solvent molecules in the pore space could not be refined due to their disordered nature, the SQUEEZE function in PLATON software31 was utilized to remove the residual electron density. The PLATON/SQUEEZE procedure estimated a residual electron count of 175 e−, corresponding to 1.1 DMF molecules per Zn2(1FBDC)(tmBDC)(dabco) unit. Here, we elaborate only on the refined structures of two fluorinated MOFs, DMOF-1F and DMOF-2F, since the structure of the non-fluorinated MOF (i.e., DMOF-0F) has already been reported in the literature.23 In fact, the fluorinated MOFs are isomorphous with the reported DMOF-0F. As shown in Fig. 1a–c, 2D (two-dimensional) square grids were constructed by bridging Zn dimers with bis-bidentate BDC-type ligands in a square-planar geometry in the ab plane. The paddlewheel units are connected by nitrogen atoms of DABCO molecules along the c axis, thereby expanding the planar network into a 3D structure. However, their space groups are not identical; namely, DMOF-1F belongs to the I4/mcm space group, whereas DMOF-2F crystallizes in the P4/mmm space group as in DMOF-0F.23 In the crystals, xFBDC (x = 1 or 2) and tmBDC ligands coexist in a disordered manner, where they have different orientations with a dihedral angle of 73.70° for x = 1, and 50.67° for x = 2 (Fig. 1d). The equivalent occupancy between xFBDC and tmBDC of 0.5 is consistent with the elemental analysis data. In addition, considering the fact that the xFBDC ligands can coordinate to Zn ions in a random manner (Fig. S2 and S3†), fluorine atoms in xFBDC ligands are disordered over four sites with an equivalent occupancy of 0.25 for DMOF-1F (Fig. 1b) and over two orientations, i.e., 2,5- and 3,6-positions, with an equivalent occupancy of 0.5 for DMOF-2F (Fig. 1c). All three MOFs possess large 3D inter-connected voids with one-dimensional channels running along the c axis with windows through the a and b axes. Although the fluorine atoms slightly protrude into the channels, there are very little difference in the sizes of pores and windows, with an average diameter of ca 0.6 nm in all three MOFs. The procedure to estimate the pore sizes is shown in Fig. S4.† It is noteworthy that these pores are apparently larger than the kinetic diameter of common gas molecules (e.g., N2 (0.364 nm) and CO2 (0.330 nm)), which allows their practical application in gas transportation and storage. In addition, it is expected that the surface density of fluorine atoms in the pores has a significant effect on the affinity for various gas molecules,32,33 which would be manifested in the adsorption isotherms, as discussed below. | ||
| Fig. 1 Expanded cubic-like structure of (a) DMOF-0F, (b) DMOF-1F, and (c) DMOF-2F, in which only one orientation of the disordered xFBDC ligand is shown. (d) Disorder of 1FBDC and tmBDC in DMOF-1F. Hydrogen atoms are omitted for clarity. Colour code: C: grey; F: orange; N: blue; O: red; Zn: green. | ||
PXRD measurement was utilized to confirm the phase purity of the polycrystalline samples. As shown in Fig. 2a and S5,† the PXRD patterns of the three as-synthesized MOFs match well with those simulated from the SCXRD data without any trace impurity peaks, indicating the high purity of the polycrystalline samples. FT-IR spectroscopy was applied to investigate the chemical structure of the as-synthesized MOFs. In Fig. 2b, the peak at 749 cm−1 can be assigned to the stretching vibration of Zn–O bonds originating from the dinuclear paddlewheel units.34 The peaks at 1619 is ascribed to the stretching vibration of carboxylate groups,35 whereas the peak at 1057 cm−1 is due to the asymmetric stretching vibration of N–C3 bonds in pillar DABCO molecules.34 In addition, the C–F stretching band was observed at 1227 cm−1 (DMOF-1F) and 1184 cm−1 (DMOF-2F) in fluorinated MOFs. The observation of conspicuous characteristic peaks of DMF, which was used as the reaction solvent, at 1664 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1256 cm−1 (C–N stretching), and 660 cm−1 (OC–N bending) indicates the inclusion of DMF in the as-synthesized MOFs.36 Thermal stability of the as-synthesized MOFs was evaluated using TGA in the temperature range from room temperature to 600 °C under N2 atmosphere. As shown in Fig. 2c, all the TG profiles exhibit similar thermal behaviour, showing a huge weight loss below 200 °C. Combining the results from FT-IR, we conclude that the decrease in weight is primarily caused by the removal of solvent DMF molecules adsorbed in the channel and on the surface of particles. Subsequently, there is a plateau between 200 and 300 °C followed by a weight loss possibly due to the removal of terephthalate or DABCO molecules. Based on the TG profiles, it can be concluded that the three MOFs are thermally stable up to approximately 300 °C under N2 atmosphere, which is comparable to those of some well-known MOFs; e.g., HKUST-1 (Cu3(BTC)2; H3BTC: 1,3,5-benzenetricarboxylic acid; up to ca. 240 °C)37 and MOF-801 (Zr6O4(OH)4(fumarate)6; up to ca. 260 °C).38
O stretching), 1256 cm−1 (C–N stretching), and 660 cm−1 (OC–N bending) indicates the inclusion of DMF in the as-synthesized MOFs.36 Thermal stability of the as-synthesized MOFs was evaluated using TGA in the temperature range from room temperature to 600 °C under N2 atmosphere. As shown in Fig. 2c, all the TG profiles exhibit similar thermal behaviour, showing a huge weight loss below 200 °C. Combining the results from FT-IR, we conclude that the decrease in weight is primarily caused by the removal of solvent DMF molecules adsorbed in the channel and on the surface of particles. Subsequently, there is a plateau between 200 and 300 °C followed by a weight loss possibly due to the removal of terephthalate or DABCO molecules. Based on the TG profiles, it can be concluded that the three MOFs are thermally stable up to approximately 300 °C under N2 atmosphere, which is comparable to those of some well-known MOFs; e.g., HKUST-1 (Cu3(BTC)2; H3BTC: 1,3,5-benzenetricarboxylic acid; up to ca. 240 °C)37 and MOF-801 (Zr6O4(OH)4(fumarate)6; up to ca. 260 °C).38
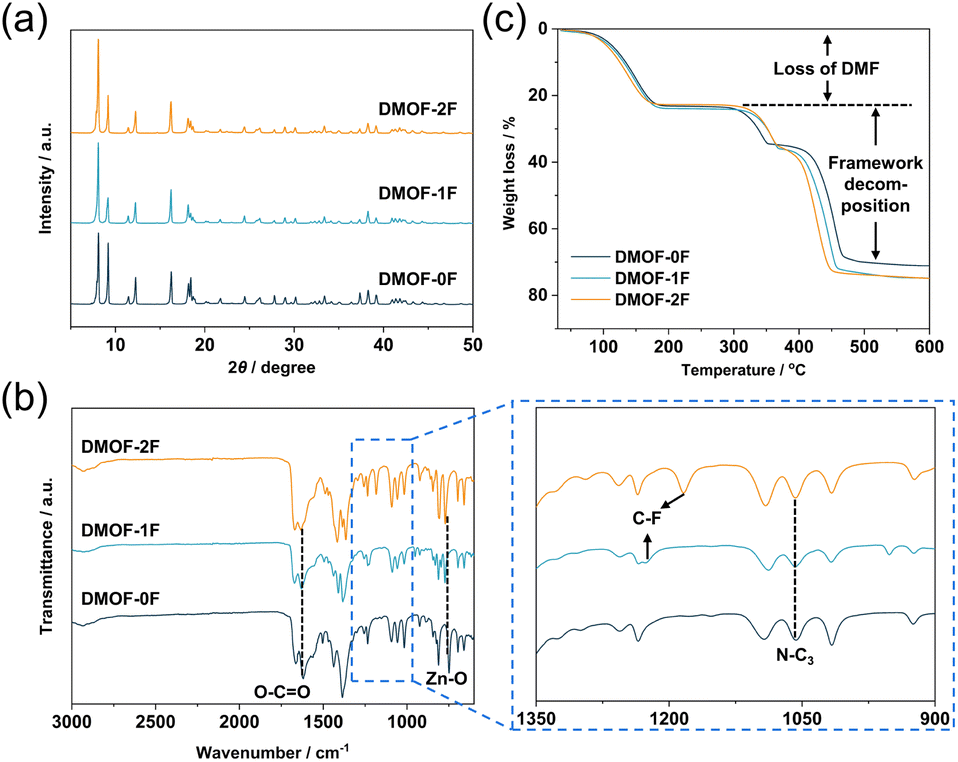 | ||
| Fig. 2 (a) PXRD patterns, (b) FT-IR spectra, and (c) TGA profiles of as-synthesized DMOF-0F (dark green), DMOF-1F (pale blue), and DMOF-2F (orange). In (b), enlarged spectra in the range of 900–1350 cm–1 are shown in the panel to the right. | ||
Because the DMF molecules occupy the pore space throughout the frameworks, appropriate methods should be applied to completely remove the solvent species in the pores before subsequent adsorption measurements. After plenty of attempts, we finally determined that the dynamic vacuum at elevated temperature is the optimal method to achieve the complete removal of DMF in the pores while keeping the framework structure. All the activated MOFs after thermal treatment at 120 °C for DMOF-0F and DMOF-1F and 105 °C for DMOF-2F for 10–12 h exhibit the PXRD patterns similar to those of as-synthesized ones with respect to the peak positions and widths (Fig. S6†). Given that the vibrational bands of DMF molecules (1664, 1256, and 660 cm−1) and weight loss due to DMF evaporation disappear in the FT-IR spectra (Fig. S7†) and TG profiles (Fig. S8†), respectively, it is obvious that DMF molecules are completely removed through the dynamic vacuum, and therefore, the activated MOFs are available for gas adsorption measurements.
Porous properties of the three MOFs were evaluated by N2 adsorption/desorption measurements at 77 K. As shown in Fig. 3a, the three MOFs exhibit a steep increase in the low-pressure region (P/P0 < 0.01), suggesting the presence of abundant permanent micropores throughout the frameworks. Subsequently, they exhibit negligible N2 uptakes in the relative pressure range 0.1–0.8 with a typical type-I isotherm in the IUPAC classification.39 In the high-pressure region (P/P0 > 0.9), all three isotherms show a slight increase, indicating the presence of macropores arising from the inter-particulate voids due to the loose packing of particles. The uptake of N2 adsorption at P/P0 = 0.97 in DMOF-0F (263 cm3 g−1) is lower than those of DMOF-1F (308 cm3 g−1) and DMOF-2F (313 cm3 g−1). The Brunauer–Emmett–Teller (BET) surface areas of DMOF-0F, 1F, and 2F are estimated to be 949, 1123, and 1225 m2 g−1 (Table 1), respectively, which are close to the reported value for DMOF-0F (1100 m2 g−1).23 The total pore volumes (Vtotal) calculated at a P/P0 of 0.97 are 0.41, 0.48, and 0.48 cm3 g−1, respectively. Pore size distribution calculated using the micropore analysis (MP) method shows that the three MOFs possess prominent ultramicropores with a size of ca. 0.6 nm (Fig. 3b), which is consistent with that expected from the crystallographic data (ca. 0.6 nm).
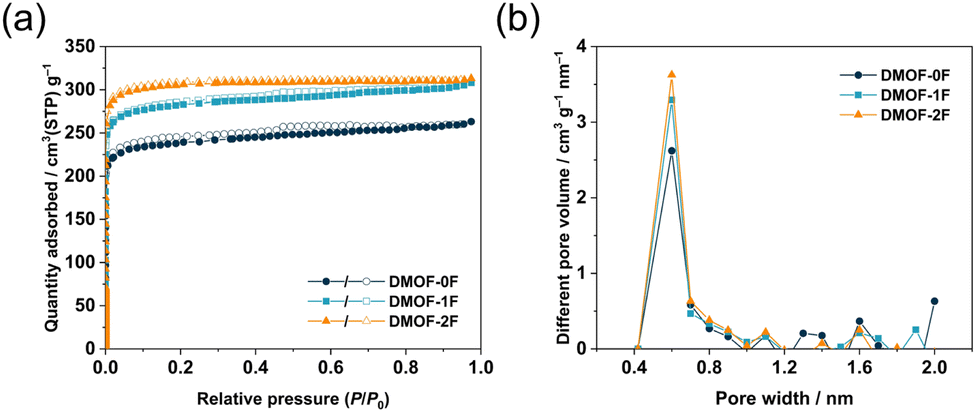 | ||
| Fig. 3 (a) N2 adsorption (closed symbols) and desorption (open symbols) isotherms at 77 K and (b) pore size distribution of DMOF-0F (dark green circles), DMOF-1F (pale blue squares), and DMOF-2F (orange triangles). | ||
Considering the relatively high BET surface areas (SBET), the present MOFs with comparable ultramicropore sizes with different fluorination environments allow us to investigate the effect of fluorination on CO2-philicity, because several fluorinated MOFs have been reported to exhibit Coulomb interactions between the negatively-charged fluorine atoms and positively-charged carbon atom in CO2 molecules.33Fig. 4a and b display the CO2 sorption isotherms of the three MOFs collected at 298 and 273 K, respectively. At both temperatures, all the MOFs show a gradual increase in CO2 uptake over the whole pressure region with a minimal hysteresis loop, indicating the physisorption-dominant process. They exhibit a relatively high CO2 uptake, possibly as a consequence of their high SBET and Vtotal. The CO2 uptake at 273 K follows the order DMOF-0F (102.1 cm3 g−1, 4.55 mmol g−1, 20.0 wt%) < DMOF-1F (106.1 cm3 g−1, 4.73 mmol g−1, 20.8 wt%) < DMOF-2F (107.4 cm3 g−1, 4.79 mmol g−1, 21.1 wt%). Although the lower Vtotal value of DMOF-0F estimated from the N2 sorption isotherm at 77 K may lead to the lower CO2 uptake, it is apparent that the higher CO2 uptake in DMOF-2F than that of DMOF-1F with comparable Vtotal is attributed to the higher fluorine content. These values are comparable or superior to some well-known MOFs, such as SNU-4 (Zn2(abtc)(DMF)2; H4abtc: 3,3′,5,5′-azobenzenetetracarboxylic acid, 20.6 wt%, surface area 1460 m2 g−1),40 SNU-5′ (Cu2(abtc)(DMF)2; 19.2 wt%, surface area 1260 m2 g−1),40 Zn2(BTTB)(DMF)2 (H4BTTB: 1,2,4,5-tetrakis(4-carboxyphenyl)benzene; 19.7 wt%, surface area 1370 m2 g−1),18 and SNU-21 (Cu2(TCM)(H2O)2, H4TCM: tetrakis[4-(carboxyphenyl)oxamethyl]methane; 18.4 wt%, surface area 905 m2 g−1).41 The reversible isotherm suggests that the MOFs under study can release the adsorbed CO2 molecules without the need of any additional thermal energy, which is advantageous for the real-world CO2 capture and regeneration of porous materials.42 The PXRD patterns after the CO2 adsorption/desorption measurement (Fig. S9†) confirmed the retention of the structure during the sorption process.
 | ||
| Fig. 4 CO2 adsorption (closed symbols) and desorption (open symbols) isotherms of DMOF-0F (dark green circles), DMOF-1F (pale blue squares), and DMOF-2F (orange triangles) at (a) 298 K and (b) 273 K. (c) Isosteric heats of adsorption (Qst) for CO2 calculated from the data at 298 and 273 K. | ||
To examine the thermodynamics of the CO2 adsorption, isosteric heat of adsorption (Qst) toward CO2 were calculated from the Clausius–Clapeyron equation:43 ln(P2/P1) = Qst × (T2 − T1)/(RT1T2), where Pn and Tn (n = 1 or 2) denote the pressure and temperature values, respectively, for the nth isotherm, and R is the gas constant expressed in the appropriate unit (8.314 J K−1 mol−1), using the adsorption isotherms at 298 and 273 K. As shown in Fig. 4c, the Qst values are estimated to be 17.2–25.7 kJ mol−1 for DMOF-0F, 19.1–25.5 kJ mol−1 for DMOF-1F, and 20.2–26.7 kJ mol−1 for DMOF-2F, depending on the degree of CO2 loading, which lie in the typical range for CO2 adsorption of MOFs.9,44 It is noteworthy that the Qst values, which are lower than the energy of the chemical bonds, is again indicative of the physisorption process (<40 kJ mol−1),45 which facilitates the controlled release of CO2 gas as mentioned above.42
It is noteworthy that for the removal of CO2 from flue gas in coal power plants (also known as post-combustion CO2 capture), the CO2 concentration must be reduced to a certain level with respect to N2. Thus, it is necessary to explore adsorbents that exhibit a high CO2/N2 selectivity to enable efficient capture of CO2 under such conditions. The CO2 and N2 adsorption isotherms of the MOFs under study were compared to investigate the adsorption selectivity of CO2 over N2. As shown in Fig. 5a–c, the adsorption amount of CO2 is remarkably higher than that of N2 at 273 K and the CO2/N2 selectivity is estimated to be 8.4, 11.3, and 14.8 for DMOF-0F, DMOF-1F, and DMOF-2F, respectively, based on the initial slope method (Fig. 5d–f).42 Although the selectivity values are less than those of a certain number of MOFs reported so far,1 the results deliver the message that fluorination has a favourable effect on CO2/N2 selectivity. Dynamic breakthrough measurements using a gas mixture of CO2/N2 (15:85) at 273 K demonstrate the effective separation via a one-step experiment, although no significant differences were found among the three MOFs (Fig. S10†). The difference in CO2/N2 selectivity depending on the number of fluorine atoms (8.4–14.8) is more pronounced compared with that of ultramicroporous fluorinated MOF, Ni(tpt)(xF-OPA)(H2O) (tpt: 2,4,6-tri(4-pyridinyl)-1,3,5-triazine; H2OPA: o-phthalic acid; 13.8–15.8), with 1D channels of ca. 0.62 nm in diameter, in which 4-fluoro-OPA (x = 1; TKL-105), 3,6-difluoro-OPA (x = 2; TKL-106), and 3,4,5,6-tetrafluoro-OPA (x = 4; TKL-107) were utilized as a bridging ligand.46 Although the factors that determine the selectivity remain unclear in these systems, the CH⋯F hydrogen bonds between the pyridine moieties of and fluorine atoms of xF-OPA within the pore walls in TKL-106 and 107 would suppress the intermolecular CF⋯C(CO2) interactions between the xF-OPA ligands and CO2 molecules. Although DMOF-1F and DMOF-2F also involve CH⋯F hydrogen bonds, the C–H groups are in the fast-rotating DABCO ligands47 which may allow the effective intermolecular CF⋯C(CO2) interactions between xFBDC ligands and CO2 molecules. Park and co-workers reported the increased CO2/N2 selectivity by the partial substitution of 2-amino-BDC (NH2BDC2−) with 2,3,5,6-tetrafluoro-BDC (4FBDC2−) in amino-functionalized MIL-101(Cr) with a chemical composition of Cr3O(OH)(H2O)2(NH2BDC)3;48 the CO2/N2 selectivity in the 4FBDC-introduced MOFs (92–108) is significantly higher than that of the pristine MOF (51). However, the authors concluded that the increased CO2 adsorption in 4FBDC-introduced MOFs arises mainly from the decreased pore sizes (1.7 to 0.9 cm3 g−1) rather than the increased fluorine content. On the other hand, we succeeded in improving the CO2/N2 selectivity mainly by controlling the number of fluorine atoms in a BDC ligand while properly keeping the porous properties (0.41 to 0.48 cm3 g−1). Thus, the present result is the first demonstration of fluorinated ultramicroporous MOFs to control the CO2/N2 selectivity at a significant level by controlling the fluorine content (Table S1†).
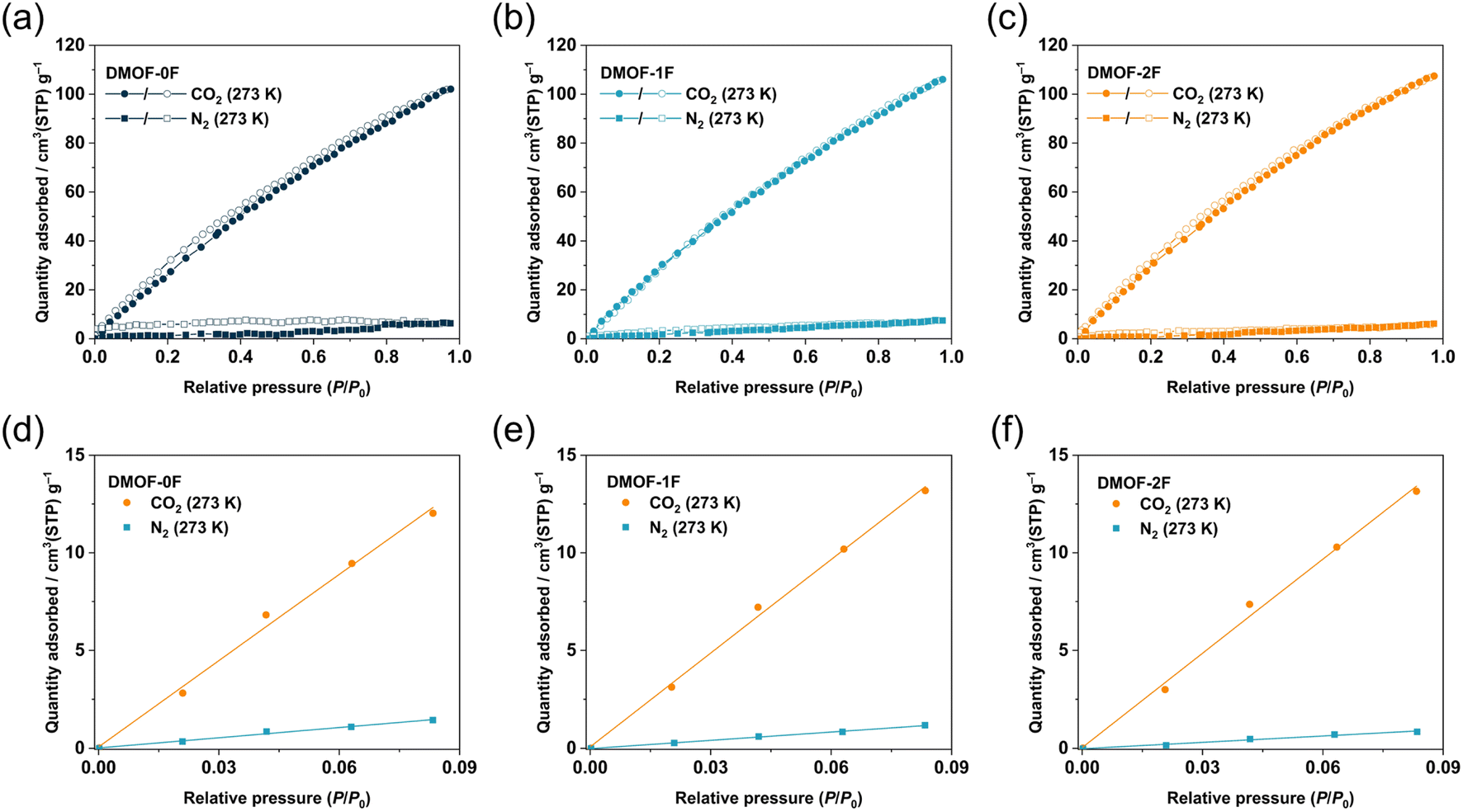 | ||
| Fig. 5 CO2 and N2 sorption isotherms of at 273 K for (a) DMOF-0F, (b) DMOF-1F, and (c) DMOF-2F. Initial slope fitting for CO2 and N2 adsorption isotherms at 273 K for (d) DMOF-0F, (e) DMOF-1F, and (f) DMOF-2F. | ||
To demonstrate the ability of the three MOFs to capture trace CO2, their CO2/N2 selectivity at atmospheric concentration (500 ppm for CO2) was calculated using IAST.29 As illustrated in Fig. 6a, the CO2/N2 selectivity varies depending on the fluorination and increases with increasing pressure. The selectivity in the low-pressure region (<0.3 bar) is in the order DMOF-0F (12.4) < DMOF-1F (14.5) < DMOF-2F (21.9), which is consistent with the results obtained from the initial slope method (Fig. 6b). The difference between DMOF-0F and DMOF-1F (2.1) is less pronounced than that between DMOF-1F and DMOF-2F (7.4), which is consistent with the calculated Qst values (Table 1).
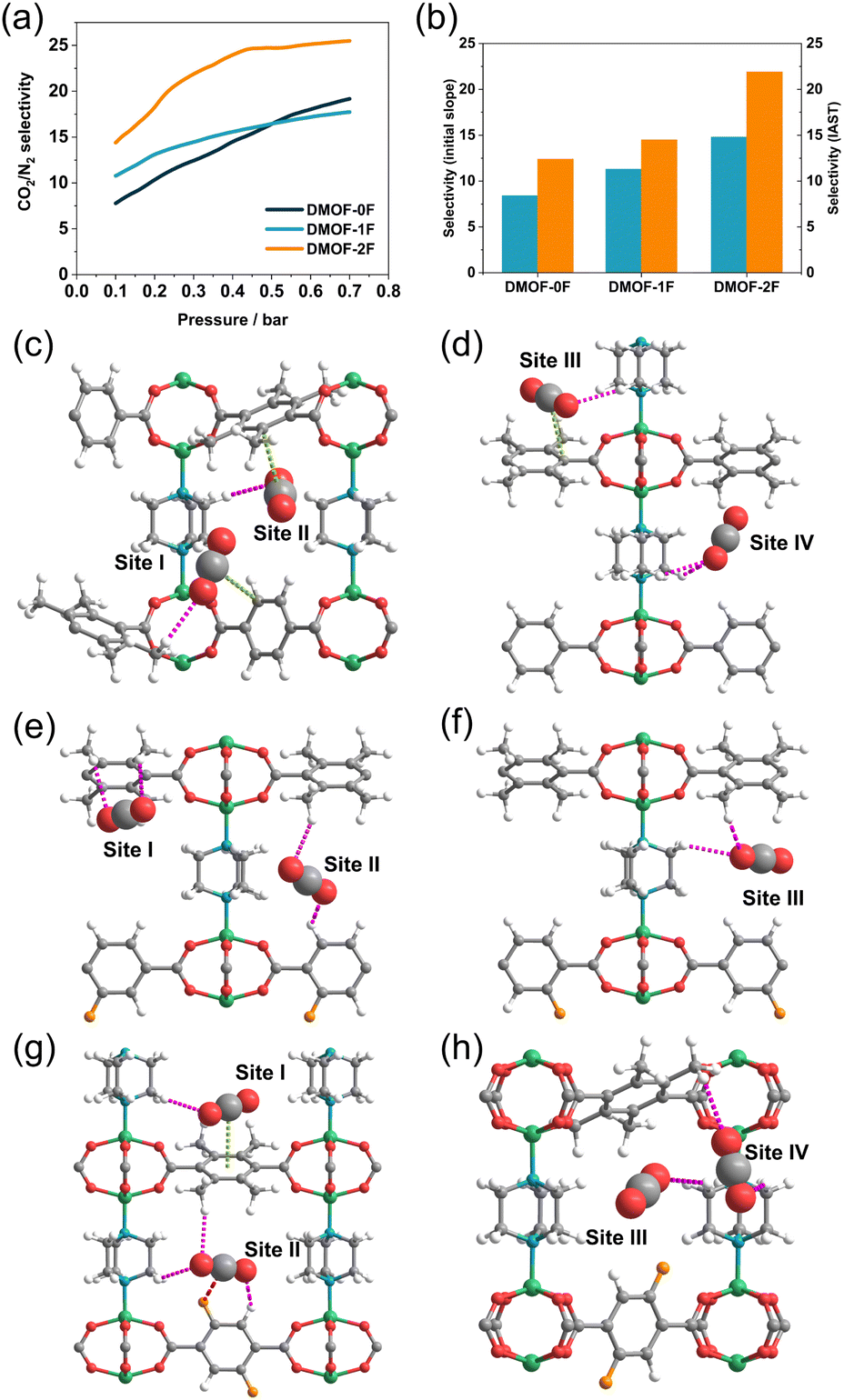 | ||
| Fig. 6 (a) Simulated IAST selectivity of CO2/N2 for DMOF-0F (dark green line), DMOF-1F (pale blue line), and DMOF-2F (orange line) under atmospheric CO2 concentration (i.e., 500 ppm of CO2 to N2). (b) CO2/N2 selectivity of three MOFs based on initial slope method (pale blue) and IAST method (orange). Simulated interactions between the framework and adsorbed CO2 molecules in (c and d) DMOF-0F, (e and f) DMOF-1F, (g and h) DMOF-2F, where CO2 molecules with no effective interactions are emitted. CH⋯O(CO2), CF⋯C(CO2), and π⋯C(CO2) interactions are shown in pink, red, and light green dotted lines, respectively. | ||
GCMC simulations were conducted to elucidate the origin of the CO2 capture behaviour at the microscopic level. The geometric structures of the MOFs were optimized with the assumption that there are four CO2 molecules in each 1D channel in the unit cell at P/P0 = 0.3 at 273 K. In DMOF-0F, there are several CH⋯O(CO2) hydrogen bonding interactions between the tmBDC or DABCO ligands and CO2 molecules with an H⋯O distance of 2.88 Å (Site I), 2.89 Å (Site II), 2.94 Å (Site III), and 2.97 and 3.01 Å (Site IV) (vs. the sum of the van der Waals radii: 2.72 Å49) as shown in Fig. 6c, d and Table S2.† Such interactions were found in CO2-incorporated DMOF-1F framework, with a distance of 3.02 and 3.04 Å (Site I), 2.98 and 2.98 Å (Site II), and 2.90 and 3.00 Å (Site III) (Fig. 6e and f). In addition, DMOF-0F shows several π⋯C(CO2) interactions19,50 between CO2 molecules and benzene rings of BDC-based ligands, with the nearest C⋯C distance of 3.45 Å (Site I), 3.60 Å (Site II), and 3.53 Å (Site III). However, there is no significant interaction between the fluorine atoms and CO2 molecules in the pore of DMOF-1F, although a Coulomb attractive force is expected between the negatively-charged fluorine atoms and positively-charged carbon atom in CO2 molecules.33 We note that the increase in the number of CO2 molecules in a channel in the unit cell from 4 (corresponding to P/P0 ∼ 0.3) to 6 (corresponding to P/P0 ∼ 0.4) results in the formation of short CF⋯C(CO2) contacts with a distance of 3.30 Å (vs. the sum of the van der Waals radii: 3.17 Å;49 Fig. S11†). In DMOF-2F (Fig. 6g and h), CO2 molecules in the pores exhibit a significant interaction with fluorine atoms with an F⋯C distance of 3.33 Å (Site II in Fig. 6g). Such a favourable CF⋯C(CO2) interaction has also been found in fluorinated Ce-MIL-140 (CeO(4FBDC)),51 in which an CF⋯C(CO2) interaction with a distance of 3.3 Å results in a higher affinity for CO2 and boosts the CO2 selectivity. Therefore, it is apparent that the CF⋯C(CO2) interaction plays a pivotal role in the higher CO2/N2 selectivity in DMOF-2F than those of DMOF-0F and DMOF-1F.
Conclusions
In this study, a series of isoreticular MOFs, namely, DMOF-0F, DMOF-1F, and DMOF-2F, possessing ultramicropores with varying degrees of fluorination were synthesized through the rational design of component species and framework structure. Both experimental and theoretical studies demonstrate that the CO2/N2 selectivity in the MOFs under study is increased by incorporating a higher number of fluorine atoms into the ultramicropore walls. It appears that the strong CF⋯C(CO2) interaction between 2FBDC ligands and CO2 molecules in DMOF-2F, predicted based on the Monte Carlo simulation, makes it the best adsorbent for CO2 among the three MOFs. Given that the study of the relationship between the fluorine content and CO2/N2 selectivity in ultramicroporous MOFs is still in its infancy, this work provides deeper insight into the design and synthesis of MOFs with controllable CO2/N2 selectivity.Data availability
Additional details regarding experimental and calculation data are given in the ESI.†Author contributions
T. D., Y. Y., and H. K. conceived the research and designed the experiments. T. D. performed all the experiments, measurements, and calculations. Dynamic breakthrough measurements were performed by T. D. with the support of K.-i. O. and S. K. T. D., Y. Y., and H. K. cowrote the manuscript. All the authors discussed and commented on the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Tokutaro Komatsu (Nihon University) for his useful advice on the GCMC simulations. This work was supported by the ACCEL program (JPMJAC1501) of the Japan Science and Technology Agency (JST), JSPS KAKENHI (Grant No. 20H02708 and 20H05623). T. D. was supported by JST SPRING (Grant No. JPMJSP2110).References
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS PubMed.
- G. Cui, J. Wang and S. Zhang, Chem. Soc. Rev., 2016, 45, 4307–4339 RSC.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chem and K. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- X. Zhu, W. Xie, J. Wu, Y. Miao, C. Xiang, C. Chen, B. Ge, Z. Gan, F. Yang, M. Zhang, D. O'Hare, J. Li, T. Ge and R. Wang, Chem. Soc. Rev., 2022, 51, 6574–6651 RSC.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- S. Chowdhury, Y. Kumar, S. Shrivastava, S. K. Patel and J. S. Sangwai, Energy Fuels, 2023, 37, 10733–10757 CrossRef CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- L. Jiang, W. Liu, R. Q. Wang, A. Gonzalez-Diaz, M. F. Rojas-Michaga, S. Michailos, M. Pourkashanian, X. J. Zhang and C. Font-Palma, Prog. Energy Combust. Sci., 2023, 95, 101069 CrossRef.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- D. M. Venturi, M. S. Notari, R. Bondi, E. Mosconi, W. Kaiser, G. Mercuri, G. Giambastiani, A. Rossin, M. Taddei and F. Costantino, ACS Appl. Mater. Interfaces, 2022, 14, 40801–40811 CrossRef CAS PubMed.
- A. S. Palakkal and R. S. Pillai, J. Phys. Chem. C, 2020, 124, 16975–16989 CrossRef CAS.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- J. R. Fried and N. Hu, Polymer, 2003, 44, 4363–4372 CrossRef CAS.
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS PubMed.
- Y.-S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 15, 2131–2134 RSC.
- A. Torrisi, C. Mellot-Draznieks and R. G. Bell, J. Chem. Phys., 2009, 130, 194703 CrossRef PubMed.
- D. Smets and U. Ruschewitz, Z. Anorg. Allg. Chem., 2020, 646, 1157–1167 CrossRef CAS.
- Y. Wang, Z. Liu, Y. Li, Z. Bai, W. Liu, Y. Wang, X. Xu, C. Xiao, D. Sheng, J. Diwu, J. Su, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, J. Am. Chem. Soc., 2015, 137, 6144–6147 CrossRef CAS PubMed.
- R. Christoffels, C. Breitenbach, J. Weber, L. Körtgen, C. Tobeck, M. Wilhelm, S. Mathur, J. Neudörfl, M. Sadegh, Z. Farid, M. Maslo, E. Strub and U. Ruschewitz, Cryst. Growth Des., 2022, 22, 681–692 CrossRef CAS.
- H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem. – Eur. J., 2005, 11, 3521–3529 CrossRef CAS PubMed.
- L. K. Cadman, J. K. Bristow, N. E. Stubbs, D. Tiana, M. F. Mahon, A. Walsh and A. D. Burrows, Dalton Trans., 2016, 45, 4316–4326 RSC.
- K. Uemura, Y. Yamasaki, F. Onishi, H. Kita and M. Ebihara, Inorg. Chem., 2010, 49, 10133–10143 CrossRef CAS PubMed.
- N. C. Burtch, H. Jasuja, D. Dubbeldam and K. S. Walton, J. Am. Chem. Soc., 2013, 135, 7172–7180 CrossRef CAS PubMed.
- H. Jasuja, N. C. Burtch, Y.-G. Huang, Y. Cai and K. S. Walton, Langmuir, 2013, 29, 633–642 CrossRef CAS.
- S. T. Meek, J. J. Perry, S. L. Teich-McGoldrick, J. A. Greathouse and M. D. Allendorf, Cryst. Growth Des., 2011, 11, 4309–4312 CrossRef CAS.
- E. Dautzenberg, S. van Hurne, M. M. J. Smulders and L. C. P. M. de Smet, Comput. Phys. Commun., 2022, 280, 108494 CrossRef CAS.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- D. Balestri, I. Bassanetti, S. Canossa, C. Gazzurelli, A. Bacchi, S. Bracco, A. Comotti and P. Pelagatti, Cryst. Growth Des., 2018, 18, 6824–6832 CrossRef CAS.
- S. Kumar, B. Mohan, C. Fu, V. Gupta and P. Ren, Coord. Chem. Rev., 2023, 476, 214876 CrossRef CAS.
- L. Peng, S. Wu, X. Yang, J. Hu, X. Fu, Q. Huo and J. Guan, RSC Adv., 2016, 6, 72433–72438 RSC.
- M. Nara, H. Torii and M. Tasumi, J. Phys. Chem., 1996, 100, 19812–19817 CrossRef CAS.
- G. Durgaprasad, D. N. Sathyanarayana and C. C. Patel, Bull. Chem. Soc. Jpn., 1971, 44, 316–322 CrossRef CAS.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- G. Wißmann, A. Schaate, S. Lilienthal, I. Bremer, A. M. Schneider and P. Behrens, Microporous Mesoporous Mater., 2012, 152, 64–70 CrossRef.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- Y.-G. Lee, H. R. Moon, Y. E. Cheon and M. P. Suh, Angew. Chem., Int. Ed., 2008, 47, 7741–7745 CrossRef CAS PubMed.
- T. K. Kim and M. P. Suh, Chem. Commun., 2011, 47, 4258–4260 RSC.
- H. Gao, L. Ding, H. Bai, A. Liu, S. Li and L. Li, J. Mater. Chem. A, 2016, 4, 16490–16498 RSC.
- H. Pan, J. A. Ritter and P. B. Balbuena, Langmuir, 1998, 14, 6323–6327 CrossRef CAS.
- J. M. Simmons, H. Wu, W. Zhou and T. Yildirim, Energy Environ. Sci., 2011, 4, 2177–2185 RSC.
- B. von Oepen, W. Kördel and W. Klein, Chemosphere, 1991, 22, 285–304 CrossRef CAS.
- D.-S. Zhang, Z. Chang, Y.-F. Li, Z.-Y. Jiang, Z.-H. Xuan, Y.-H. Zhang, J.-R. Li, Q. Chen, T.-L. Hu and X.-H. Bu, Sci. Rep., 2013, 3, 3312 CrossRef.
- N. C. Burtch, A. Torres-Knoop, G. S. Foo, J. Leisen, C. Sievers, B. Ensing, D. Dubbeldam and K. S. Walton, J. Phys. Chem. Lett., 2015, 6, 812–816 CrossRef CAS PubMed.
- J. M. Park, G.-Y. Cha, D. Jo, K. H. Cho, J. W. Yoon, Y. K. Hwang, S.-K. Lee and U.-H. Lee, Chem. Eng. J., 2022, 444, 136476 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- A. Torrisi, C. Mellot-Draznieks and R. G. Bell, J. Chem. Phys., 2010, 132, 044705 CrossRef PubMed.
- Z. Zhang, S. B. Peh, R. Krishna, C. Kang, K. Chai, Y. Wang, D. Shi and D. Zhao, Angew. Chem., Int. Ed., 2021, 60, 17198–17204 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of H21FBDC and additional experimental results such as 1H NMR spectra, PXRD patterns, FT-IR spectra, TG profiles, and breakthrough curves. CCDC 2336743 (DMOF-1F) and 2336744 (DMOF-2F). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01525h |
| This journal is © The Royal Society of Chemistry 2024 |