
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDenitrogenative dismantling of heteroaromatics by nucleophilic substitution reactions with diazomethyl compounds†
Soumen
Biswas
a,
Claire
Empel
b,
Luis Mario
Sanchez-Palestino
ac,
Hadi
Arman
a,
Rene M.
Koenigs
 *b and
Michael P.
Doyle
*a
*b and
Michael P.
Doyle
*a
aDepartment of Chemistry, The University of Texas at San Antonio One UTSA Circle, San Antonio, TX 78249, USA. E-mail: michael.doyle@utsa.edu
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, D-52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de
cEscuela Superior de Medicina, Instituto Politécnico Nacional, Mexico City 11340, Mexico
First published on 17th June 2024
Abstract
Nucleophiles from deprotonation of diazomethyl compounds having diverse electron withdrawing groups react with 4-carboxylato-1,2,3-triazines at the 6-position to extrude dinitrogen and produce diazovinylketoesters compounds with five or six linear contiguous sp2-hybridized carbons, whereas these same nucleophiles react with 4-carboxylato-1,2,3-triazine 1-oxides, also at the 6-position, to form pyrazolines with the expulsion of nitrous oxide and cyanocarboxylate. This disparity is due to the significant difference in reactivity of the nucleophilic addition products.
Introduction
Heterocyclic compounds containing three nitrogen atoms in an unsaturated six-membered ring exist as three isomers depending on the relative placement of the nitrogen atoms. The least well known triazines are those with three contiguous nitrogen atoms. 1,2,3-Triazines are known to be the source of a variety of N-heterocycles1 and important pharmaceutical targets.2 Despite their relevance across different areas, their reactions and reactivities have only been explored to a very limited extent. For example, the very fundamental reaction of triazines with nucleophiles that generally results in the extrusion of dinitrogen poses intrinsic challenges for site-specific, chemoselective substitution reactions, yet only a few investigations with basic amidine and enolate nucleophiles,3 hydrides, alcoholates, or thiolates have explored this reactivity.4A further important aspect when studying the chemistry of heterocycles, lies in the formation of their N-oxides, which can fundamentally alter the reactivity of the parent heterocycle.2b Although the oxidation of pyridine to pyridine N-oxide is straightforward, oxidation of the 1,2,3-triazine core is not selective.5 We have recently discovered specific access to 4-carboxylato-1,2,3-triazine 1-oxides through reactions of vinyldiazoacetates with tert-butyl nitrite,6–8 and from them by deoxygenation to identically substituted 1,2,3-triazine-4-carboxylates.7 In reactions with carbon nucleophiles, both triazine systems were observed to undergo reactions with carbon nucleophiles exclusively at the 6-position,4 making possible a rich array of pyridines via formal inverse electron demand Diels–Alder reactions (Scheme 1a).8 Despite these advances, a concise study that explores and directly compares similarities and differences in the reactivity of 1,2,3-triazines and their N-oxides remains an important task and would provide key insights into the chemical properties and applications of both 1,2,3-triazine heterocycles.
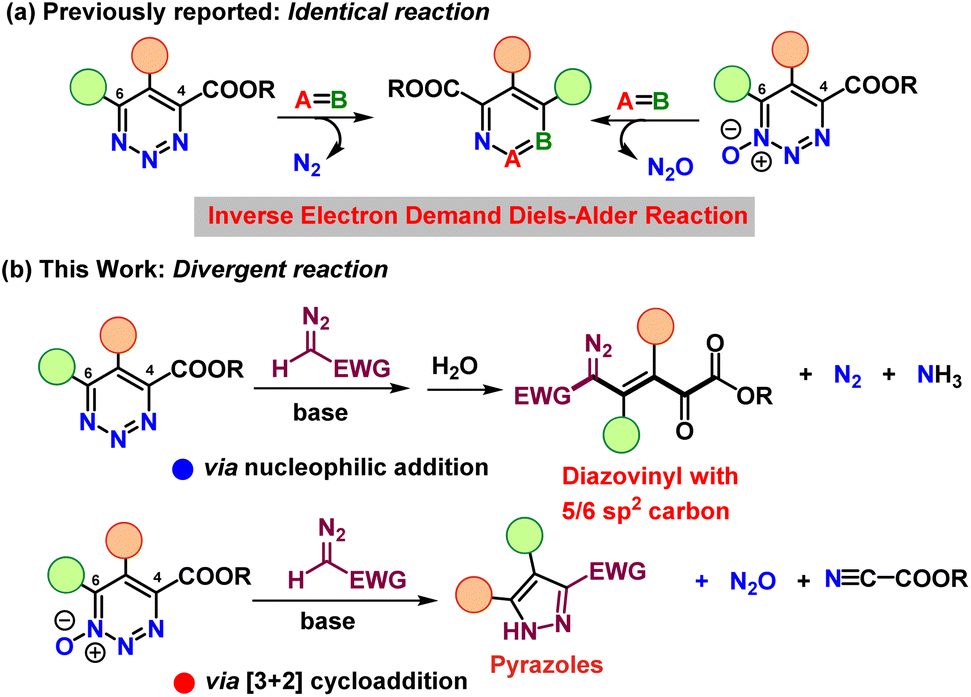 | ||
| Scheme 1 Identical (a) and divergent (b) reactions of 1,2,3-triazines and their 1-oxides. | ||
Bearing in mind the reactivity of the 1,2,3-triazine system towards nucleophiles, we considered that these heterocycles could be formidable substrates for the nucleophilic introduction of a diazo functional group and thereby provide new classes of diazo compounds. Specifically, diazomethyl compounds attached to strong electron-withdrawing groups are relatively strong acids capable of proton removal from the diazo carbon.9 We envisioned that these nucleophiles would be capable of addition to the triazine core of 1,2,3-triazines and 1,2,3-triazine 1-oxides, but the outcome of each transformation was uncertain. Thus, we were surprised to discover that 1,2,3-triazine-4-carboxylates underwent exclusive nucleophilic addition at the 6-position to give unexpected diazovinylketoesters with five or six linear contiguous sp2-hybridized carbons, while nucleophilic attack on 4-carboxylato-1,2,3-triazine 1-oxides by the same nucleophiles also occurred exclusively at the 6-position but formed pyrazole derivatives via an uncommon electroreversion (Scheme 1b).
Results and discussion
We commenced our investigation with the reaction of ethyl 5-phenyl-1,2,3-triazine-4-carboxylate 1a and diethyl diazomethylphosphonate 2a (the Seyferth–Gilbert reagent)10 in an acetonitrile solution containing cesium carbonate. Upon quenching the reaction with water when 1a was fully consumed, a single product was produced whose structural connectivity was determined spectroscopically to be phosphonyl diazovinylketoester 3a (Scheme 2). Isolated in 82% yield, this compound possesses five contiguous sp2-hybridized carbons linking diazo, alkene, ketone carbonyl, and ester functional groups to a phosphonate group. The only uncertainty was the stereochemistry about the carbon–carbon double bond, and this was inferred from rhodium acetate catalyzed dinitrogen extrusion that produced indene derivative 4a. Organic bases DBU and DABCO were found to be inefficient for the condensation reaction giving 38% and 25% yield, respectively, of 3a (see Table S1 in ESI†).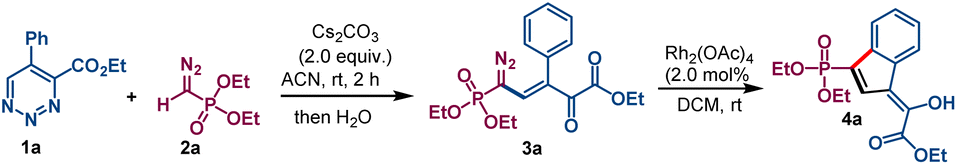 | ||
| Scheme 2 Cesium carbonate promoted condensation of ethyl 5-phenyl-1,2,3-triazine-4-carboxylate (1a) with diethyl diazomethylphosphonate (2a) and subsequent dirhodium acetate catalyzed aromatic C–H insertion. Condensation reaction was performed by addition of Cs2CO3 (0.2 mmol) to 1.0 mL ACN solution of compound 1a (0.1 mmol) and 2a (0.12 mmol). C–H insertion reactions was performed by dropwise addition of 3a (0.1 mmol) in 1.0 mL of DCM (30 min) to Rh2(OAc)4 in 1.0 mL DCM. | ||
This result stimulated us to examine the reaction scope for the formation of similar compounds with diazomethyl substituents having different electron-withdrawing groups. Widely used ethyl diazoacetate 2b gave the similar vinyldiazo compound 3b that contains six contiguous sp2-hybridized carbons in moderate yield. This process proved to be general as other diazomethyl compounds with different electron withdrawing groups, including amide (2c), ketone (2d), sulfone (2e) and trifluoromethyl (2f), provided the desired substituted diazovinylketoester compounds in good yields (Scheme 3). However, diazoacetonitrile failed to trigger the reaction, remaining intact in the reaction mixture under similar reaction conditions, probably due to its lower proton acidity.11 Variation of the substituent on the aryl group at the 5-position of the 1,2,3-triazine revealed that election withdrawing groups (F and CF3) furnished higher yields of diazovinylketoester products as compared to those from 1a or the reactant with an electron donating group (OMe). In addition, naphthyl substituted diazovinylketoester 3j was obtained in good yield. Aliphatic functional groups such as cyclopropyl, tert-(butyldimethylsilyl)oxyethyl, ethyl, n-propyl, n-pentyl, phenethyl and homoallyl were also well tolerated, giving vinyldiazo compounds 3k–q with moderate to good yields. The 1,2,3-triazine 1-oxide bearing a benzyl ester instead of an ethyl ester reacted in similar fashion to generate diazovinyl compound 3r in high yield. 5-Bromo-1,2,3-triazine 1n, which is representative of symmetrical 1,2,3-triazines that are formed by alternative synthetic methodologies,12 was treated with the Seyferth–Gilbert reagent 2a under the same conditions. Without a substituent at either the 4- or 6-positions, a comparable ring opening reaction to that in Scheme 3A was expected to form a terminal aldehyde. Indeed, bromo-substituted vinyldiazo-phosphonate 5a having a terminal aldehyde functional group was isolated in good yield (Scheme 3B). Moreover, ethyl diazoacetate furnished the corresponding bromo substituted diazo compound 5b in moderate yield. The multiple functionalities of these diazovinylketo-esters/-aldehydes and their ease of formation offers new polyfunctional platforms for a broad spectrum of chemical transformations. As diazo compounds they provide structural diversity that is limited in current methodologies.13 As β,γ-unsaturated-α-ketoesters, these condensation products offer new opportunities for conjugate addition reactions.14
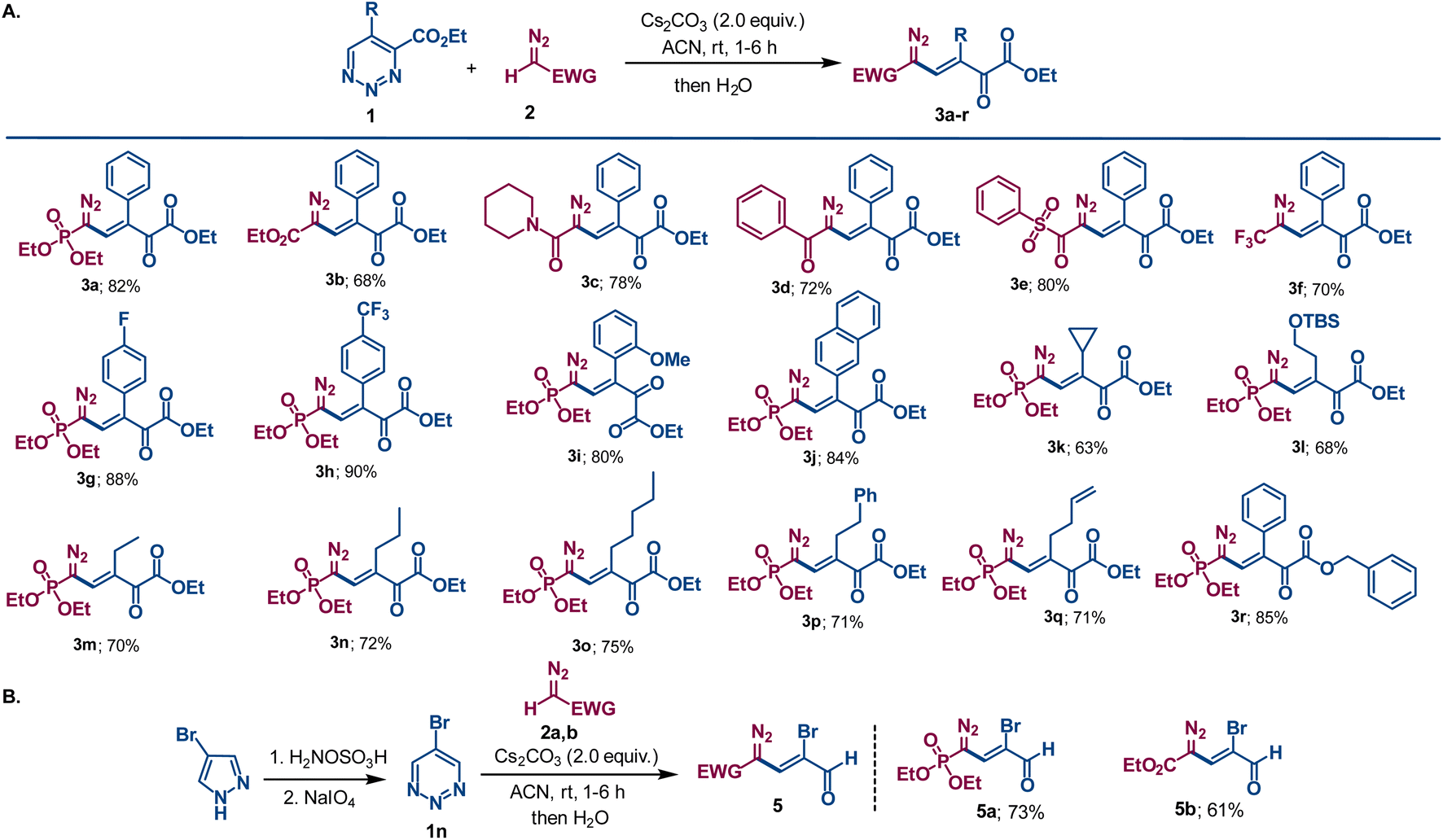 | ||
| Scheme 3 (A) Cesium carbonate promoted condensation of 1,2,3-triazine-4-carboxylates (1) with various diazomethyl derivatives (2). Reactions were performed a 0.1 mmol scale by addition of Cs2CO3 (0.2 mmol) to 1.0 mL ACN solution of compound 1 and 2. A 20% molar excess of the dioazomethyl compound 2 was employed. (B) Alternative synthetic route to 1,2,3-triazine and its condensation reaction with diazomethyl reactants. | ||
As an example of their potential in catalytic metal carbene reactions, the diazovinylketoesters formed from 1a were treated with a catalytic amount of Rh2(OAc)4 in DCM at room temperature, forming 1,3-difunctionalized indene derivatives 4b–4f in excellent yields (Scheme 4). Since diazo compounds are also known to undergo aromatic C–H insertion in the presence of blue LED light,15 when compound 3a was exposed to blue LED light (450 nm), instead of using Rh2(OAc)4, the identical indene derivative 4a was isolated in good yield. These formal C–H insertion reactions occur by addition to the proximal ortho position of the aromatic ring followed by hydrogen transfer.16 Apparent from the data in Scheme 4, the electron withdrawing group has no obvious impact on product yield. The crystal structure of compound 4f (see ESI†) confirmed the atomic connectivity and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double geometry of the diazovinylketoesters. Notable is the enol structure that connects the phosphonate group through the indene ring and imparts aromatic character to the five-membered ring. The functionalities provided by this transformation offer intriguing templates for complex indene scaffolds.17 Attempted aliphatic C–H insertion with diazovinylketoester 3l having the pendant (tert-butyldimethylsilyl)oxy)ethyl group gave cyclopentene derivative 4i in 81% isolated yield; however, attempts to achieve enantiocontrol with traditional chiral dirhodium(II) carboxylate and copper(I)-Box catalysts had limited success (see SI). Compound 4i was obtained with only moderate ee (48%) with the optimum chiral dirhodium catalyst Rh2(s-TCPTTL)4.18 Diazovinyl derivative 3q having a pendant homoallyl group underwent exclusive C–H insertion at the allylic position, instead of cyclopropanation, to form 4j in 72% isolated yield with Rh2(OAc)4 catalysis, but low enantioselectivity (30% ee) was achieved with catalysis by Rh2(s-TCPTTL)4.
C double geometry of the diazovinylketoesters. Notable is the enol structure that connects the phosphonate group through the indene ring and imparts aromatic character to the five-membered ring. The functionalities provided by this transformation offer intriguing templates for complex indene scaffolds.17 Attempted aliphatic C–H insertion with diazovinylketoester 3l having the pendant (tert-butyldimethylsilyl)oxy)ethyl group gave cyclopentene derivative 4i in 81% isolated yield; however, attempts to achieve enantiocontrol with traditional chiral dirhodium(II) carboxylate and copper(I)-Box catalysts had limited success (see SI). Compound 4i was obtained with only moderate ee (48%) with the optimum chiral dirhodium catalyst Rh2(s-TCPTTL)4.18 Diazovinyl derivative 3q having a pendant homoallyl group underwent exclusive C–H insertion at the allylic position, instead of cyclopropanation, to form 4j in 72% isolated yield with Rh2(OAc)4 catalysis, but low enantioselectivity (30% ee) was achieved with catalysis by Rh2(s-TCPTTL)4.
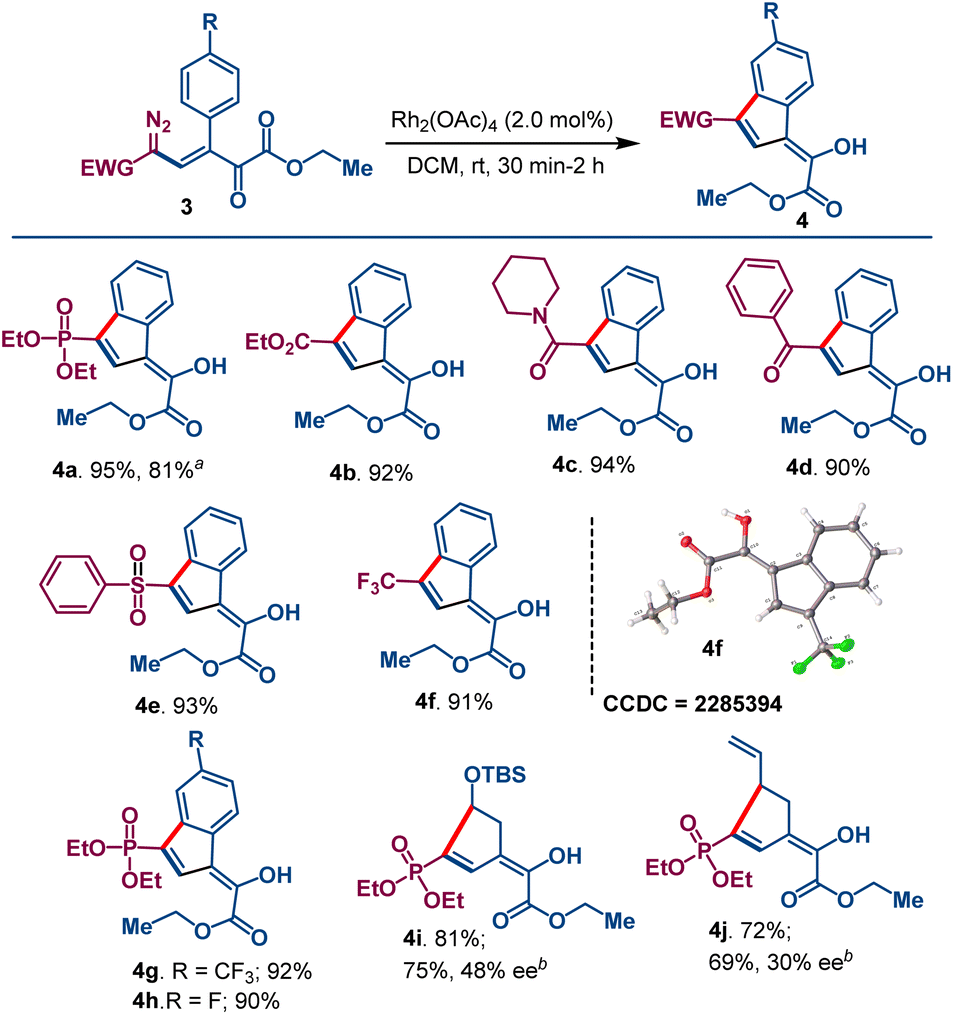 | ||
| Scheme 4 Rh(II)-catalyzed insertion reactions of diazovinylketoeaster compounds. Reactions were performed a 0.1 mmol scale by dropwise addition of 3 in 1.0 mL of DCM (30 min) to Rh2(OAc)4 in 1.0 mL DCM. aReaction was performed a 0.1 mmol scale of 3 in 1.0 mL of DCM (2 h) using blue LED 450 nm. bUsing catalyst Rh2(s-TCPTTL)4. | ||
The presence of multiple electrophilic centers in diazovinylketoester compounds encouraged us to examine the fate of 3a,b in reactions with different nucleophiles. Surprisingly, the reactions with borohydride, the phenyl Grignard reagent, and the nitromethane anion formed by triethylamine (Henry reaction)19 all occurred exclusively at the keto group in high yields (Scheme 5). In contrast, the reaction of nitromethane with strong organic base DBU led to pyrazole derivatives 9a,b having the nitromethyl group. However, when compounds 3a,b were treated with sodium cyanide, conjugate addition of cyanide took place to form pyrazole derivatives 10a,b in 81–83% isolated yields – a process that required the elimination of COCOOEt. Moreover, addition of the diazomethylsulfonyl anion to the carbonyl group of diazovinyl compound 3e furnished derivative 11 with good yield.
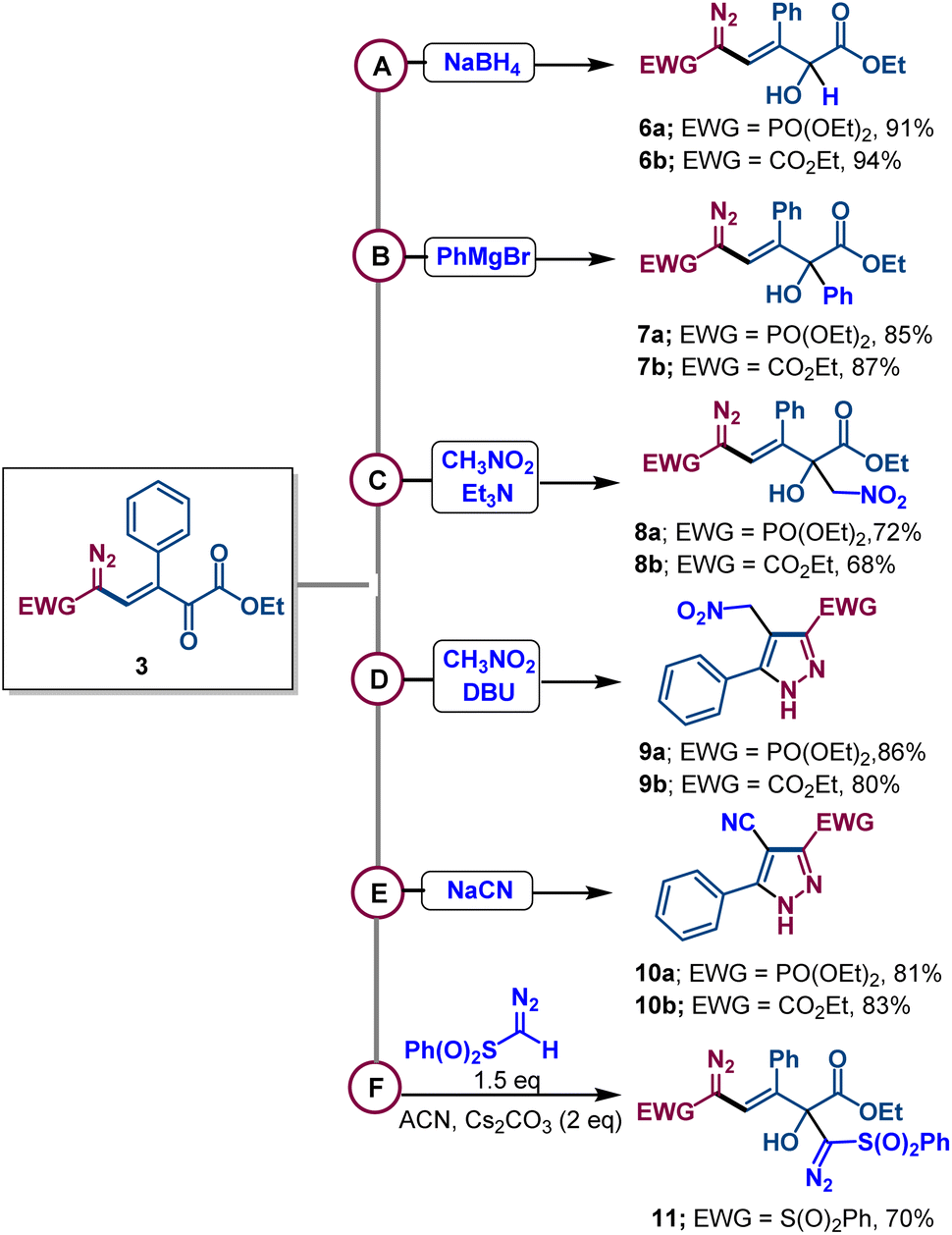 | ||
| Scheme 5 Addition of different nucleophiles to diazovinylketoeaster compound 3. All the reactions were performed on a 0.1 mmol scale of 3. (A) NaBH4 (1.0 eq.), EtOH, rt, 30 min; (B) PhMgBr (1.5 eq.), THF 0 °C to rt, 30 min; (C) CH3NO2, Et3N (2.0 eq.), rt, 12 h; (D) CH3NO2, DBU (2.0 eq.), rt, 6 h; (E) NaCN (2.0 eq.), EtOH rt, 12 h; (F) sulfonyldiazo (1.5 eq.), Cs2CO3 (2.0 eq.), ACN, rt, 2 h. | ||
Nucleophilic reactions of 1,2,3-triazine 1-oxides often have different outcomes compared with their 1,2,3-triazine analogues.4 Consequently, we were interested to know the outcome of reactions between diazomethyl-derived carbanions and the N-oxides of 1,2,3-triazines. Since the 6-position is the most electrophilic site for carbanion addition,4,7 the anion from the diazomethyl compound was expected to react at the 6-position, as did the 1,2,3-triazine, and by electroreversion and the expulsion of nitrous oxide we anticipated the same products as were obtained from the 1,2,3-triazines. However, when the same reaction was performed between 5-phenyl-1,2,3-triazine-4-carboxylate 1-oxide 12a and the Seyferth–Gilbert reagent 2a in the presence of Cs2CO3, pyrazole derivative 13a was isolated in good yield (eqn (1)). This surprising result requires the net loss of the ester functionality, as well as nitrous oxide and CN. The missing units, ethyl cyanocarboxylate (NC–COOEt)20 and N2O21 were identified (NCCOOEt by NMR and MS and nitrous oxide by IR).
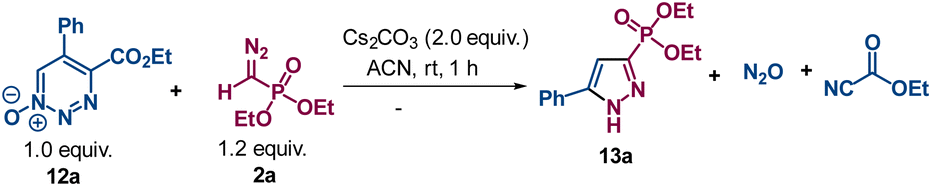 | (1) |
The pyrazole ring is widely found as the core structure in a large variety of compounds that possess important agrochemical and pharmaceutical activities, and many synthetic methodologies have been developed for their synthesis.22 Access to them from diazo compounds is particularly noteworthy23 because the diazo compound supplies one carbon and the two adjacent nitrogens of their core structure. Intense recent interest has been directed to the synthesis of fluorinated24 and phosphorolated25 pyrazoles via dipolar cycloaddition reactions of activated alkynes and allenes with fluorinated and phosphorolated diazo compounds. Two reaction pathways are dominant in these reactions. One is the classic dipolar [3 + 2]-cycloaddition that occurs in neutral media or with a Lewis acid catalyst,26 and is completed by hydrogen migration, and the second is base removal of the acidic proton on the diazo carbon followed by nucleophilic addition, cyclization, and reprotonation,27 but neither of them account for pyrazole formation from 1,2,3-triazine 1-oxides.
Reaction conditions were optimized using different bases. Organic bases were less effective. Of the inorganic bases surveyed, cesium carbonate was optimum, and a higher product yield of 13a was obtained from the reaction of 12a with diazomethyl-phosphonate 2a performed in acetonitrile than in THF (see Table S2 in ESI†). Using the optimum conditions with cesium carbonate, diazomethyl compounds with ester (2b), amide (2c), ketone (2d), sulfone (2e), trifluoromethyl (2f) and cyano (2g) electron withdrawing groups were treated with ethyl 5-phenyl-1,2,3-triazine-4-carboxylate-1-oxide 12a, and pyrazole derivatives 13b–13g were formed in good yields (Scheme 6). The diazo compounds with phosphonate, amide, and sulfone electron withdrawing groups reacted faster than did those with ester, ketone, or cyano groups.
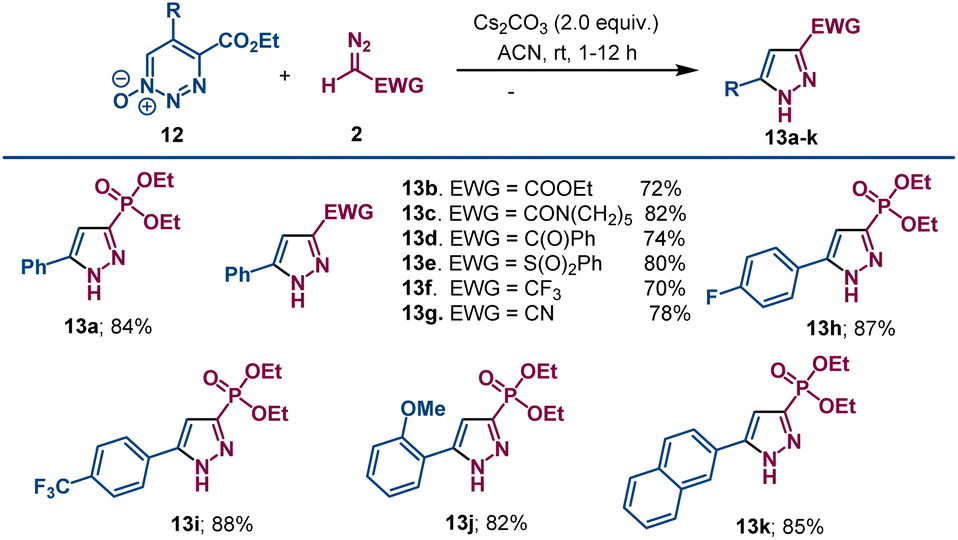 | ||
| Scheme 6 Pyrazole formation from 1,2,3-triazine 1-oxides and diazomethyl compounds. Reactions were performed a 0.1 mmol scale by addition of Cs2CO3 (0.2 mmol) to 1.0 mL ACN solution of compound 12 and 2. A 20% molar excess of the dioazomethyl compound 2 was employed. | ||
The reaction of 1,2,3-triazine 1-oxide 12a with α-diazoacetophenone 2d produced pyrazole 13d and a reaction intermediate 13d′ with a pyrazole-fused ring whose structure was confirmed by X-ray crystallography. Pyrazole 13d was isolated as the sole product from 13d′ upon further treatment with cesium carbonate (Scheme 7).
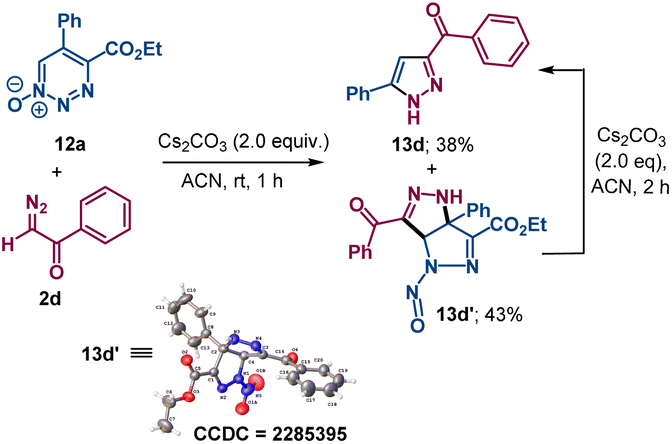 | ||
| Scheme 7 Pyrazole formation from reaction of 12a with the anion from 2dvia intermediate 13d′. Reaction was performed on a 0.1 mmol scale by addition of Cs2CO3 (0.2 mmol) to 1.0 mL ACN solution of compound 12a and 2d (0.12 mmol). | ||
DFT calculations on the reaction mechanism suggest an energetically favored deprotonation of the diazoalkane to form the corresponding diazomethyl anion (Fig. 1). This deprotonation is favored for a broad range of different diazoalkanes, which is in line with the observed tolerance of different electron-withdrawing groups (for details please see Schemes 3 and S1 in the ESI†). In a first reaction step, the diazomethyl anion undergoes nucleophilic addition to the 6-position of 1,2,3-triazine or 1,2,3-triazine-1-oxide to generate INT1A or its oxidised analogue INT1B, respectively (Fig. 1a). The reactivity of INT1A and INT1B, however, differs significantly and rationalizes the divergent reaction outcome.
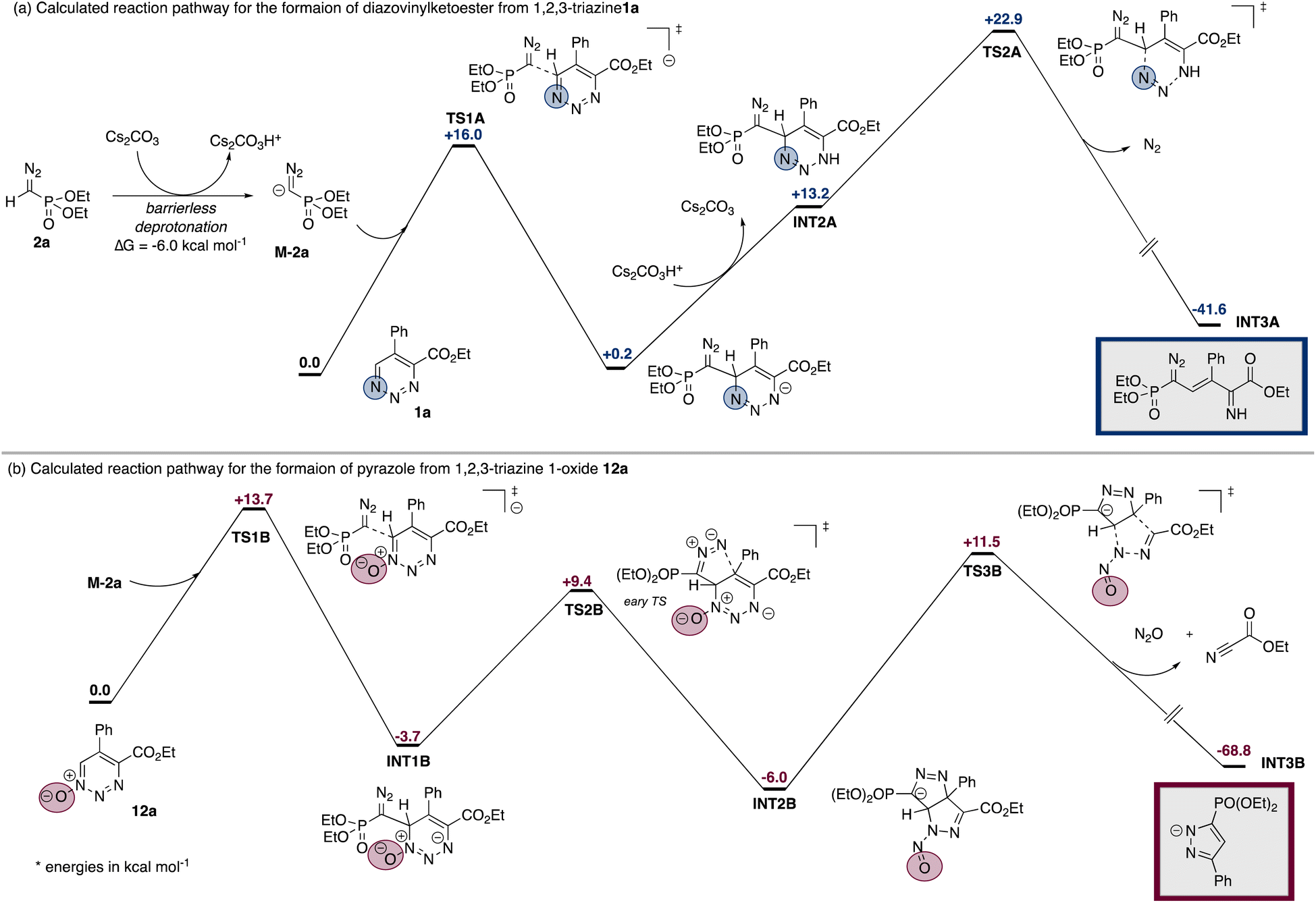 | ||
| Fig. 1 DFT assessment of the reaction pathways to diazovinylketoesters (from 1,2,3-triazines) and pyrazoles (from 1,2,3-triazine 1-oxides). The pathways diverge following nucleophilic addition at the 6-position. | ||
A reprotonation step initiates the reaction pathway for rupture of the triazine ring and formation of the open-chain product 3a. In the case of INT1a such re-protonation is followed by the cleavage of nitrogen gas to form INT3AviaTS2A (ΔG = +22.9 kcal mol−1). In the presence of water INT3A will provide the desired product 3a. For the reaction of the triazine-1-oxide-based intermediate INT1B, such re-protonation would give INTB4 and is significantly higher in energy (see Scheme S3 in ESI†). Moreover, in this case the cleavage of N2O viaTS4B requires a total activation energy of +53.6 kcal mol−1 and is therefore not feasible.†
The surprising formation of the pyrazole product is a result of a cyclization reaction starting from the oxidized intermediate INT1B (Fig. 1b). This cyclization gives a product of a formal [3 + 2] cycloaddition in which the N-atom of the diazo functional group reacts at the 5-position of the former 1,2,3-trizine-1-oxide with an activation energy of +13.1 kcal mol−1 (TS2B). Interestingly, TS2B is an early transition state that directly leads to a complex rearrangement of the molecular framework and the formation of INT2B, which makes the back reaction unfavorable. In a last step, the cleavage of N2O and ethyl cyanocarboxylate takes place to furnish pyrazole derivative INT3B (ΔG = +17.5 kcal mol−1) (eqn (1)). Notably, the reaction pathway viaINT2B is confirmed for the reaction with α-diazoacetophenone 2d (Scheme 7).
In the case of triazine, the related [3 + 2] cycloaddition from INT1A would proceed viaTS3A (ΔG = +18.4 kcal mol−1) to form INT4A, which is higher in energy by +7.6 kcal mol−1 compared to INT1A (see Scheme S2 in ESI†). In this case, no subsequent skeletal rearrangement occurs and therefore, the equilibrium of this reaction clearly lies on the side of INT1A and therefore disfavors the formation of the pyrazole product. In the pyrazole forming pathway, the main difference thus lies in an equilibrium of a stepwise [3 + 2] cycloaddition reaction and a subsequent skeletal rearrangement, which is favored for triazine-1-oxide.
In conclusion, we describe divergent reactions of 1,2,3-triazines and 1,2,3-triazine 1-oxides with diazomethyl compounds in the presence of cesium carbonate. Initial nucleophilic attack occurs at the 6-position of both the triazine and triazine 1-oxide, but the triazine produces diazovinylketoesters with five to six contiguous sp2 carbons, and the triazine 1-oxide gives rise to pyrazole derivatives through extrusion of ethyl cyanocarboxylate and N2O. Nucleophiles from deprotonation of diazomethyl compounds having phosphonate, ester, amide, sulfone, trifluoromethyl and cyano functional groups have been reacted with both triazines and triazine 1-oxides to identify the breadth of this protocol. Moreover, a Rh(II)-catalyzed transformation of aryl-substituted vinyldiazoketoesters provides substituted indene scaffolds with excellent yields. Reactions with the phosphonate-vinyldiazoketoester having pendant functionalized alkyl substituents, instead of aryl, undergo C–H insertion, but enantioselectivity is limited with the optimum catalyst that has been used. Nucleophilic addition reactions on this polyfunctionalized template occur at the ketone carbonyl group, except in the reaction with cyanide in which conjugate addition is exclusive. A detailed mechanistic study has been described based on reaction intermediates, reaction byproducts, and DFT energy calculations.
Data availability
The data supporting the findings of this study are available within the article and its ESI.†Author contributions
All authors contributed to the manuscript, and all authors have given their approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RMK acknowledges Deutsche Forschungsgemeinschaft for their financial support. MPD acknowledges the Welch Foundation (AX-1871) for their financial support.Notes and references
- F.-G. Zhang, Z. Chen, X. Tang and J.-A. Ma, Chem. Rev., 2021, 121, 14555–14593 CrossRef CAS PubMed.
- (a) A. M. Mfuh and O. V. Larionov, Curr. Med. Chem., 2015, 22, 2819–2857 CrossRef CAS PubMed; (b) O. V. Larionov, Heterocyclic N-Oxides, Springer International Publishing, Cham, Switzerland, 2018 Search PubMed; (c) S. Cascioferro, B. Parrino, V. Spano, A. Carbone, A. Montalbano, P. Barraja and P. Diana, Eur. J. Med. Chem., 2017, 142, 74–86 CrossRef CAS PubMed.
- (a) Y. Zhang, H. Luo, Q. Lu, Q. An, Y. Li, S. Li and Z. Tang, Chin. Chem. Lett., 2021, 32, 393–396 CrossRef CAS; (b) Z. C. Wu, K. N. Houk, D. L. Boger and D. Svatunek, J. Am. Chem. Soc., 2022, 144, 10921–10928 CrossRef CAS PubMed; (c) Y.-F. Yang, P. Yu and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 18213–18221 CrossRef CAS PubMed; (d) H. Luo, Y. Li, Y. Zhang, Q. An, M. Xu, S. Li, J. Li and B. Li, J. Org. Chem., 2022, 87, 2590–2600 CrossRef CAS PubMed.
- L. De Angelis, G. C. Haug, G. Rivera, S. Biswas, A. Al-Sayyed, H. Arman, O. Larionov and M. P. Doyle, J. Am. Chem. Soc., 2023, 145, 13059–13068 CrossRef CAS PubMed.
- (a) A. Ohsawa, H. H. Arai, H. OhnishiI and H. Igeta, J. Chem. Soc., Chem. Commun., 1980, 1182–1183 RSC; (b) A. Ohsawa, H. Arai, H. Ohnishi, T. Kaihoh, K. Yamaguchi, H. Igeta and Y. litaka, Chem. Pharm. Bull., 1986, 34, 109 CrossRef CAS; (c) S. I. Nagai, T. Ueda and J. Het, Chem, 2000, 37, 1663–1664 CAS.
- L. De Angelis, H. Zheng, M. T. Perz, H. Arman and M. P. Doyle, Org. Lett., 2021, 23, 6542–6546 CrossRef CAS PubMed.
- G. Rivera, L. De Angelis, A. Al-Sayyed, S. Biswas, H. Arman and M. P. Doyle, Org. Lett., 2022, 24, 6543–6547 CrossRef CAS PubMed.
- S. Biswas, L. De Angelis, G. Rivera, H. Arman and M. P. Doyle, Org. Lett., 2023, 25, 1104–1108 CrossRef CAS PubMed.
- Y. Zhang and J. Wang, Chem. Commun., 2009, 5350–5361 RSC.
- M. Marinozzi, F. Pertusati and M. Serpi, Chem. Rev., 2016, 116, 13991–14055 CrossRef CAS PubMed.
- P. K. Mykhailiuk and R. M. Koenigs, Chem.–Eur. J., 2020, 26, 89–101 CrossRef CAS PubMed.
- (a) E. D. Anderson and D. L Boger, J. Am. Chem. Soc., 2011, 133, 12285–12292 CrossRef CAS PubMed; (b) H. Sugimura, R. Takeuchi, S. Ichikawa, E. Nagayama and I. Sasaki, Org. Lett., 2018, 20, 3434–3437 CrossRef CAS PubMed; (c) H. Neunhoeffer, R. Bopp and W. Diehl, Liebigs Ann. Chem., 1993, 4, 367–373 CrossRef.
- (a) M. Regitz and G. Maas, Diazo Compounds. Properties and Synthesis, Academic Press, Orlando, FL, 1986 Search PubMed; (b) J. Wang , and D. Qiu, Recent developments of diazo compounds in organic synthesis, World Scientific, London, 2020 Search PubMed.
- Q. Tang, K. Fu, P. Ruan, S. Dong, Z. Su, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 11846–11851 CrossRef CAS PubMed.
- J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS.
- M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, John Wiley & Sons, Inc., New York, NY, 1998, ch. 6 Search PubMed.
- (a) N. Ahmed, Stud. Nat. Prod. Chem., 2016, 51, 383–434 CAS; (b) P. Prasher and M. Sharma, ChemistrySelect, 2021, 6, 2658–2677 CrossRef CAS; (c) H. Gao, J. A. Katzenellenbogen, R. Garg and C. Hansch, Chem. Rev., 1999, 99, 723–744 CrossRef CAS PubMed.
- M. Yamawaki, H. Tsutsui, S. Kitagaki, M. Anada and S. Hashimoto, Tetrahedron Lett., 2002, 43, 9561–9564 CrossRef CAS.
- C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org Chem., 2007, 16, 2561–2574 CrossRef.
- F. M. Betzler, T. M. Klapötke and S. M. Sproll, Eur. J. Org Chem., 2013, 509–514 CrossRef CAS.
- Nitrous oxide, NISTWeb, National Institute of Standards and Technology, https://webbook.nist.gov/cgi/cbook.cgi?ID=C10024972%26Type=IR-SPEC%26Index=1, accessed Search PubMed.
- (a) R. Singh, R. Kaur, P. Ahlawat, P. Kaushik and K. Singh, Org. Prep. Proced. Int., 2021, 53, 317–351 CrossRef CAS; (b) G. Küçükgüzel and S. Şenkardeş, Eur. J. Med. Chem., 2015, 97, 786–815 CrossRef PubMed; (c) S. S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed.
- S. P. Chandrasekharan, A. Dhami, S. Kumara and K. Mohanan, Org. Biomol. Chem., 2022, 20, 8787–8817 RSC.
- (a) P. K. Mykhailiuk, Chem. Rev., 2021, 121, 1670–1715 CrossRef CAS PubMed; (b) P. Yamanushkin, K. Kaya, M. A. M. Feliciano and B. Gold, J. Org. Chem., 2022, 87, 3868–3873 CrossRef CAS PubMed; (c) J. S. S. Neto and G. Zeni, Chem. - Eur. J., 2020, 26, 8175–8189 CrossRef CAS PubMed.
- (a) M. Dhameja and J. Pandey, Asian J. Org. Chem., 2018, 7, 1502–1523 CrossRef CAS; (b) Y.-T. Tian, F.-G. Zhang and J.-A. Ma, J. Org. Chem., 2021, 86, 3574–3582 CrossRef CAS PubMed; (c) L. Devi, G. Sharma, R. Kant, S. Shukla and N. Rastogi, Org. Biomol. Chem., 2021, 19, 4123–4126 RSC; (d) K. Ruffell, F. R. S. Smith, M. T. Green, S. M. Nicolle, M. Inman, W. Lewis, C. J. Hayes and C. J. Moody, Eur. J. Chem., 2021, 27, 13703–13708 CrossRef CAS PubMed.
- M. P. Sammes and A. R. Katritzky, Adv. Heterocycl. Chem., 1983, 34, 1–52 CrossRef CAS.
- (a) S. Muruganantham, S. M. Mobin and I. N. N. Namboothiri, Org. Lett., 2007, 9, 1125–1128 CrossRef PubMed; (b) R. Kumar, D. Verma, S. M. Mobin and I. N. N. Namboothiri, Org. Lett., 2012, 14, 4070–4073 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2285394 and 2285395. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01578a |
| This journal is © The Royal Society of Chemistry 2024 |