
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent catalytic behaviors of assembled organogold(I) clusters derived from enyne cyclization†
Qian
Liu
,
Xiao-Yi
Zhai
,
Rui-Jun
Jian
and
Liang
Zhao
 *
*
Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: zhaolchem@mail.tsinghua.edu.cn
First published on 24th June 2024
Abstract
Homogeneous gold catalysis has attracted much recent attention due to diverse activation modes of gold(I) towards unsaturated organic groups. Because of attractive aurophilic interaction, structural transformations of metalated species into high nuclear clusters are often proposed in gold catalysis, while to date little is known about their assembly behaviors and catalytic activity. In this work, based on stoichiometric Au(I)-mediated enyne cyclization reactions, we achieve a discrete vicinal dicarbanion-centered Au4 intermediate and three assembled Au11, Au28, and Au14 clusters held together by several aryl dicarbanions. Spectral monitoring, kinetic and theoretical investigations confirm that these discrete and assembled intermediates display four different pathways upon catalyzing the cyclization reaction of the same 1,5-enyne substrate. The discrete Au4 cluster undergoes a full protodeauration process to generate active [Au(PPh3)]+ species for catalytic use. In contrast, the net-like Au11 cluster experiences a substrate-induced dissociation to generate a semi-stable Au10 unit and an active [alkyne-Au(PPh3)]+ fragment for further transformation. The dumbbell-like Au28 cluster is prone to cleavage of the central Au–Au linkage and each Au14 moiety exposes a coordination unsaturated site to activate a substrate molecule. However, the synthetic closed-Au14 cluster with full ligand protection is no longer catalytically active.
Introduction
Structural evolution of catalytically active species widely exists in transition metal-catalyzed reactions.1–3 Therein, mononuclear coordination metal catalysts experience the dissociation of labile ligands to recombine as polynuclear active species or undergo reduction by substrates, ligands, solvents, or additives to in situ generate metal clusters or even nanoclusters.4–10 Not only can they directly activate substrates by exposing coordination unsaturated metal sites,11,12 but they can also act as a reservoir to dissociate into smaller clusters as real active species for catalytic use.13–15 Such metal clusters and nanoclusters show high diversity in nuclearity numbers, charges, configurations and peripheral ligands, promisingly allowing screening of highly active species to achieve excellent efficiency and selectivity.16,17 Therefore, the exploration of catalytically active cluster species is of great significance to reveal detailed mechanisms and discover novel organic transformations.In general, active metal cluster species are in situ generated during the induction period of metal-catalyzed reactions, which are highly reactive and temporarily stabilized by reactants or additional ligands along with coordinative solvents as identified by mass spectroscopy.5,18,19 However, metalated products formed in the late stage of catalytic reactions have been rarely explored regarding their particular role as catalytically active species. In this context, a vast number of Au(I)-catalyzed reactions bear much attention20–22 because attractive aurophilic interaction between closed-shell d10 Au(I) atoms facilitates the formation of polyaurated reaction intermediates.23–30 As gold(I) complexes act as a superior Lewis acid to activate unsaturated π-systems,31 coordination of a substrate to Au(I) followed by the attack of a nucleophile often generates isolable organogold intermediates. They are generally believed to undergo a subsequent protodeauration process to output desired organic products and regenerate gold(I) catalytic species.32–34 However, some isolated organogold intermediates featuring gem-diaurated motifs have been found resistant to the protodeauration-driven recycling process.35–37 Therefore, structural evolution and catalytic activity investigations of metalated organic compounds based on stoichiometric gold-mediated organic transformations may provide new insights into the mechanism of Au(I) catalysis.
Herein, we report the isolation and structural characterization of a series of differently assembled organogold clusters based on Au(I)-mediated enyne cyclization reactions (Scheme 1). By using different σ-aurated enyne substrates, we obtain a discrete vicinal dicarbanion-bonded tetragold complex [Au4(PPh3)4(LOMe)][NTf2]2 (2a, LOMe = 1,3-dimethoxyphenanthrene diide, and NTf2 = bis-(trifluoromethanesulfonyl)imidate) and two assembled clusters [Au11(PPh3)6(LPh)4][NTf2]3 (2b, LPh = 1-phenylnaphthalene diide) and [Au28(PPh3)8(LMe)12][NTf2]4 (2c, LMe = 1-methylnaphthalene diide). Moreover, 2b can undergo further transformation to form a tetradecanuclear cluster [Au14(PPh3)5(LPh)6][NTf2]2 (2d). These discrete and assembled intermediates display four different pathways upon catalyzing the cyclization reaction of a 1,5-enyne substrate. 2a undergoes a full protodeauration to generate unsaturated [Au(PPh3)]+ species for catalytic use. As for 2b, a substrate-induced dissociation of the net-like Au11 core leads to a semi-stable Au10 unit and an active [alkyne-Au(PPh3)]+ fragment for further transformation (pathway I in Scheme 1). The Au10 cluster recombines with the recycling [Au(PPh3)]+ unit to recover the Au11 core. In contrast, 2c initiates the catalytic reaction by exposing an unsaturated site through the cleavage of a Au–Au linkage in the dumbbell-like Au28 core (pathway II in Scheme 1). Lastly, the closed tetradecanuclear cluster 2d with full ligand protection has no catalytic capacity.
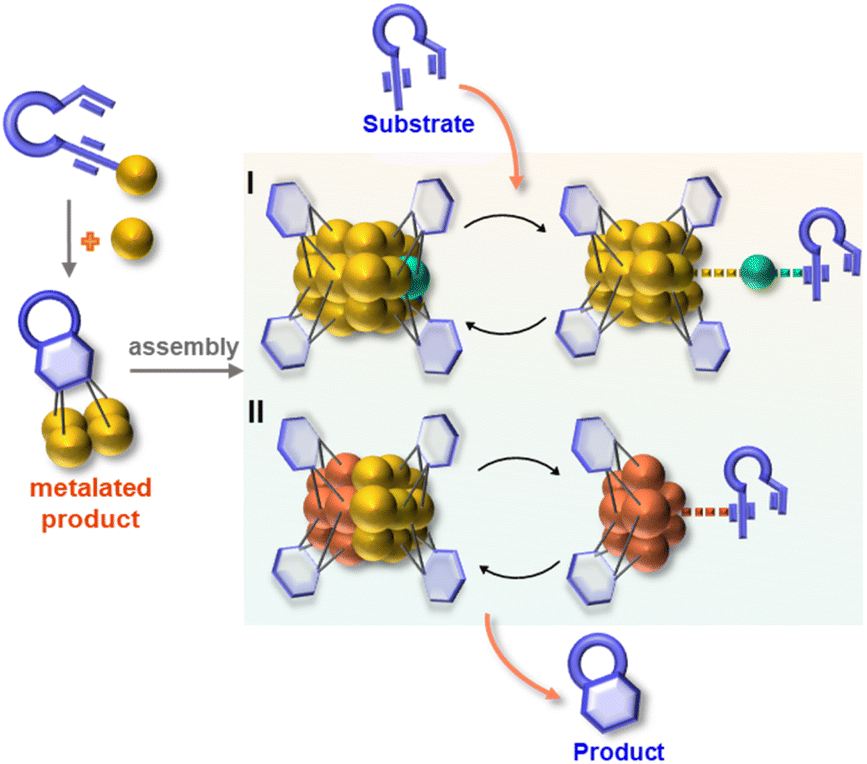 | ||
| Scheme 1 Assembly and catalytic behaviors of organometallic clusters. | ||
Results and discussion
Synthesis and assembly of organogold(I) clusters
In view of the reported reaction pathway for enyne cyclization reactions,38 we herein select σ-aurated alkyne compounds 1a, 1b and 1c as substrates. As 1a was treated with a stoichiometric amount of [PPh3Au](NTf2) in 1,2-dichloroethane (DCE), the solution colour gradually changed from light yellow to yellow brown. Electrospray ionization mass spectroscopy (ESI-MS) of the reaction mixture revealed four peaks corresponding to polymetalated species [Au4(PPh3)4(LOMe)]2+, [Au3(PPh3)3(LOMe)]+, [Au3(PPh3)2(LOMe)]+, and [Au2(PPh3)2(LOMeH)]+ (LOMe = 1,3-dimethoxyphenanthrene diide) (Fig. S1†). Further screening suggested that the optimized 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio of 1a and [PPh3Au](NTf2) gave a satisfactory isolated yield (35%) for the yellow crystalline compound 2a. The formula of 2a was determined as [(AuPPh3)4(LOMe)][NTf2]2 by single-crystal X-ray diffraction analysis (Fig. 1a). 2a has a phenanthrene dianionic center in a μ4-C,C-η2,η2 mode to bind with a butterfly-shaped tetranuclear ring, which is further strengthened by a close Au–O interaction (2.7570(3) Å). The two carbanionic centers each attach to a gem-digold unit with the Au–C bond lengths in the range of 2.108(14)–2.214(12) Å, a bit longer than that of the reported CAu2 species.27,28,39–41 The Au–Au distances of the Au4 unit can be classified into two shorter edges (2.7825(8) and 2.7859(8) Å) and two longer ones (2.9791(7) and 3.0472(8) Å). The crystal structure of 2a is in good agreement with the detailed proton and phosphorus atom assignments as shown in the 1H and 31P NMR spectra (Fig. S4†). To understand the detailed electronic structure of 2a, we performed adaptive natural density partitioning (AdNDP) analysis.42 The AdNDP analysis explores that the [C2Au4] unit of 2a is composed of two σ-type 3c–2e and one pseudo-π-type 6c–2e bonds. These findings imply that the cluster formation is promoted by aurophilic interaction and multi-centered bonding between the 6s group orbitals of gold atoms and the 2p orbitals of carbon atoms (Fig. S6†).
:3 ratio of 1a and [PPh3Au](NTf2) gave a satisfactory isolated yield (35%) for the yellow crystalline compound 2a. The formula of 2a was determined as [(AuPPh3)4(LOMe)][NTf2]2 by single-crystal X-ray diffraction analysis (Fig. 1a). 2a has a phenanthrene dianionic center in a μ4-C,C-η2,η2 mode to bind with a butterfly-shaped tetranuclear ring, which is further strengthened by a close Au–O interaction (2.7570(3) Å). The two carbanionic centers each attach to a gem-digold unit with the Au–C bond lengths in the range of 2.108(14)–2.214(12) Å, a bit longer than that of the reported CAu2 species.27,28,39–41 The Au–Au distances of the Au4 unit can be classified into two shorter edges (2.7825(8) and 2.7859(8) Å) and two longer ones (2.9791(7) and 3.0472(8) Å). The crystal structure of 2a is in good agreement with the detailed proton and phosphorus atom assignments as shown in the 1H and 31P NMR spectra (Fig. S4†). To understand the detailed electronic structure of 2a, we performed adaptive natural density partitioning (AdNDP) analysis.42 The AdNDP analysis explores that the [C2Au4] unit of 2a is composed of two σ-type 3c–2e and one pseudo-π-type 6c–2e bonds. These findings imply that the cluster formation is promoted by aurophilic interaction and multi-centered bonding between the 6s group orbitals of gold atoms and the 2p orbitals of carbon atoms (Fig. S6†).
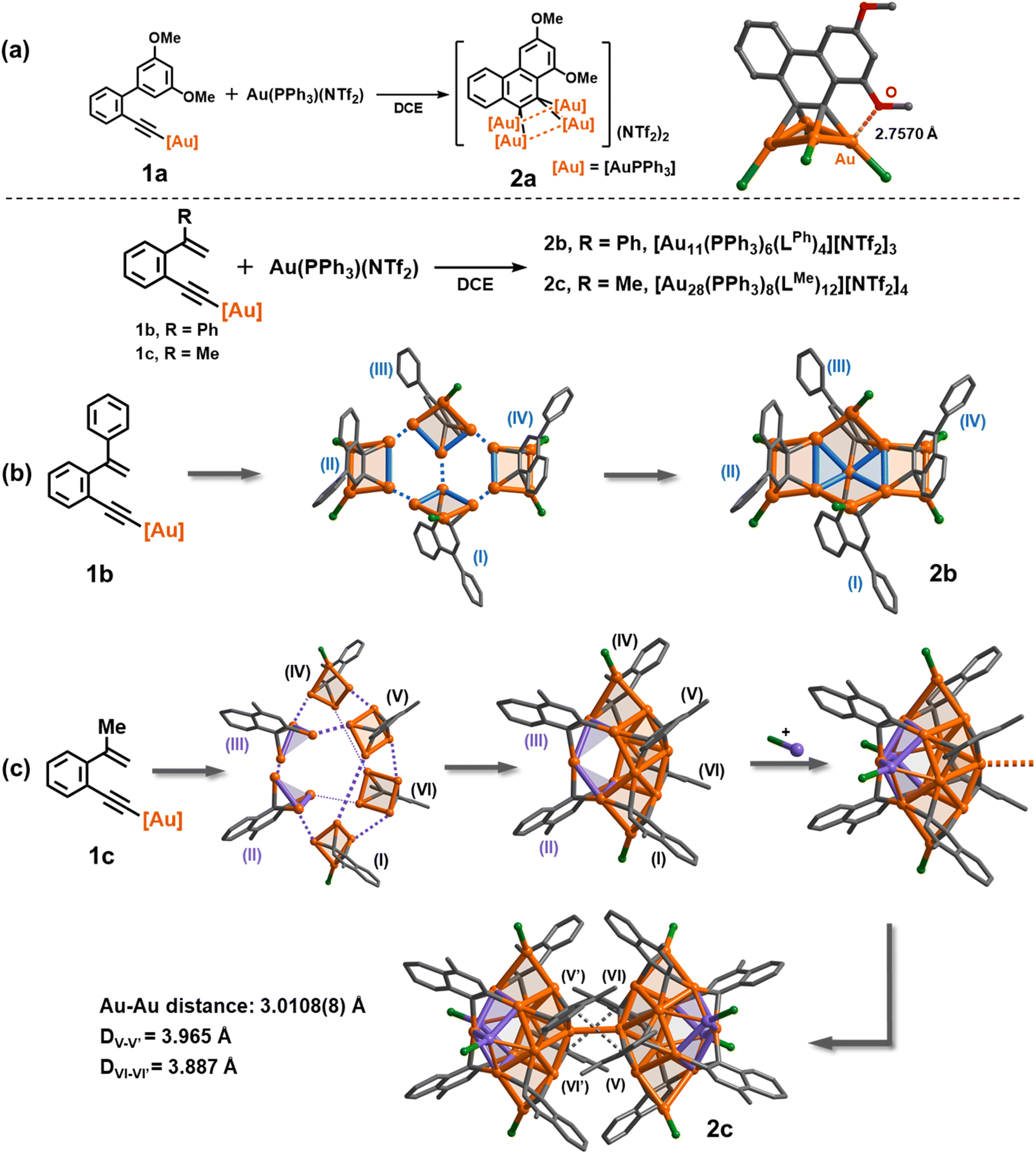 | ||
| Fig. 1 Synthesis, crystal structures and assembled patterns of (a) discrete vicinal dicarbanion-bonded tetragold cluster 2a, (b) net-like cluster 2b, and (c) dumbbell-like cluster 2c. Hydrogen atoms, phenyl groups of PPh3 ligands and peripheral NTf2− counter anions are omitted for clarity. Color coding: Au, orange and purple; C, gray; O, red; P, green. | ||
Next, we investigated the transformation of 1b as a representative alkenyl substrate. Under the same synthetic conditions as those for 1a, the solution of 1b turned brownish. The in situ ESI-MS analysis showed three distinct peaks, namely [Au2(PPh3)2(LPhH)]+, [Au3(PPh3)2(LPh)]+, and [Au3(PPh3)3(LPh)]+ (LPh = 1-phenylnaphthalene diide). However, no peaks corresponding to a similar tetranuclear cluster structure as observed in 2a were detected (Fig. S7†). Diffusion of diethyl ether into the brown solution deposited orange crystals of 2b, whose molecular formula was identified as [Au11(PPh3)6(LPh)4][NTf2]3 by single-crystal X-ray diffraction analysis. As shown in Fig. 1b and S8,†2b shows a net-like Au11 structure that comprises four vicinal dicarbanion-bonded Au4 quadrilaterals (I–VI) fused together by sharing five gold atoms. The remaining six unshared gold atoms are coordinated by a PPh3 ligand each. Owing to steric hindrance, the four 1-phenylnaphthalene groups are alternatively located above (II and IV) and beneath (I and III) the extended Au11 plane. Therein, the Au–C bonds are in the range of 2.125(5)–2.193(6) Å (average 2.157 Å), comparable to the average bond length of 2a. Notably, in comparison with the Au–Au distances of 2a, 2b has a shorter Au–Au edge in a CAu2 moiety (average 2.714 Å), suggesting the formation of more robust gem-digold units. In addition, besides the low abundance peak of [Au11(PPh3)6(LPh)4]3+ in the ESI-MS spectrum of 2b, a few prominent peaks were observed in the spectrum, indicating the presence of missing PPh3 or [Au(PPh3)]+ species (Fig. S9†). This observation suggests that 2b readily undergoes dissociation into smaller fragments under the conditions of mass spectroscopy. In contrast, the 1H NMR spectrum of 2b reveals a series of fine splitting peaks ranging from 5.8 to 8.9 ppm, and the 31P NMR spectrum shows two peaks at 32.23 and 36.88 ppm corresponding to two inequivalent PPh3 ligands as shown in the crystal structure of 2b (Fig. S10†). This evidence demonstrates that the structure of 2b in solution is consistent with its crystal structure.
To further understand the influence of ligands on cluster assembly, a new alkenyl substrate 1c was introduced. In this case, the alkenyl α-position is substituted with a less bulky methyl group. This modification allows for a comparison of the ligand effect on the cluster assembly, providing insights into how different ligands impact the formation and structure of the clusters. The in situ ESI-MS analysis of the reaction mixture revealed several major peaks that can be assigned to bi- or tri-nuclear species such as [Au2(PPh3)2(LMeH)]+ and [Au3(PPh3)3(LMe)]+ (Fig. S12†). Surprisingly, we also observed a minor peak corresponding to a high-nuclear assembled species [Au14(PPh3)4(LMe)6]2+. X-ray crystallographic analysis of the acquired complex 2c revealed its molecular formula as [Au28(PPh3)8(LMe)12][NTf2]4. 2c consists of two identical [Au14(PPh3)4(LMe)6]2+ (MeAu14) fragments linked by a Au–Au linkage (3.0108(8) Å) and strengthened by two-fold π–π stacking between methylnaphthalenes (DV–V′ = 3.965 Å and DVI–VI′ = 3.867 Å, Fig. 1c and S13†). Theoretical studies show that these interactions provide a stabilization energy of 22.7 kcal mol−1 (Fig. S14†). In view of the fact that the common Au–Au bond dissociation energy is about 5–15 kcal mol−1,24 we conjecture that the π–π stacking of methylnaphthalene is an essential factor for stabilizing the dimeric form of 2c (Fig. S15†).43 Each closed-MeAu14 fragment is formed through the assembly of four vicinal dicarbanion-bonded Au4 quadrilaterals (I, IV, V and VI) and two complementary Au3 triangles (II and III) by sharing gold atoms. Additionally, two PPh3-bonded gold atoms are attached to the fragment. This arrangement of shared gold atoms and bonding interactions contributes to the overall structure and stability of the MeAu14 fragment. In contrast to the assembled structure of 2b, 2c has slightly long Au–Au edges (average 2.730 and 3.084 Å for CAu2 units and cross-unit edges, respectively) as well as long Au–C bonds (average 2.163 Å). The ESI-MS spectrum of 2c displayed two dicationic cluster ion peaks at m/z = 2323.6321 and 2454.6816 that can be assigned to [Au14(PPh3)4(LMe)6]2+ and [Au14(PPh3)5(LMe)6]2+ (MeAu14-PPh3), respectively (Fig. S16†). The obtained result indicates that the central Au–Au linkage in 2c is susceptible to cleavage and thus yields two MeAu14 fragments.
Based on the discrete cluster structure in 2a and assembled structures in 2b and 2c, we speculate that the driving force for assembly may originate from electronic properties of different in situ generated vicinal dicarbanions. It is expected that the moderate electron-donating ability of LPh and LMe relative to LOMe makes it insufficient to stabilize four positive [AuPPh3]+ units and causes further assembly of discrete aryl dianion-centered polygold units by sharing gold atoms. This assumption is also supported by the calculated atomic dipole moment corrected Hirshfeld population (ADCH) charges of two central carbons in the basic [C2Au4] units of 2a, 2b and 2c. The sum of charges for 2a, 2b and 2c is −0.556, −0.422 and −0.174, respectively, suggesting that the electronic nature of organic skeletons indeed has an important influence on the assembly of [C2Au4] units. In addition, previous studies have illustrated that a gem-diaurated species decomposes into [Au(PPh3)2]+ in the solution state.35,44 Combined with in situ ESI-MS spectra, we propose that 1b and 1c first generate discrete [C2Au3] units, and free [Au(PPh3)]+ seizes the PPh3 ligand on [C2Au3] to lead to [Au(PPh3)2]+ and expose a Au site. The latter coordination unsaturation facilitates the occurrence of assembly.
In view of the open structure of 2b relative to the closed architecture of 2c, we speculate that the gold atoms in 2b may also be accessible as a Lewis acid.45–49 We then employed 2b to activate the σ-aurated substrate 1b. The 1H NMR monitoring showed gradual disappearance of the alkenyl proton doublets of 1b at 5.73 and 5.39 ppm (Fig. S21†). This process is accompanied by colour change from orange to deep red. Subsequently, diffusion of diethyl ether into the deep red solution led to red crystals of 2d with the formula [Au14(PPh3)5(LPh)6][NTf2]2 (Fig. 2a). The closed-Au14 cluster core in 2d is very similar to the MeAu14 of 2c. Of note, all dianionic centers adopt a μ3-C,C-η1,η2 mode to form several [C2Au3] units. Thus, the structure of 2d can be envisioned as six [C2Au3] units fused together by sharing nine gold atoms. To complete the structure, vacant vertices of the fused cluster are filled with three equatorial and two axial [AuPPh3]+ units (Fig. S22†). In comparison with 2a–2c, short Au–C distances (average 2.121 Å) in 2d indicate strong interaction between organic skeletons and the central gold cluster core. Due to the existence of a tight layer of ligand protection, 2d no longer has the ability to activate 1b towards further structural transformation.
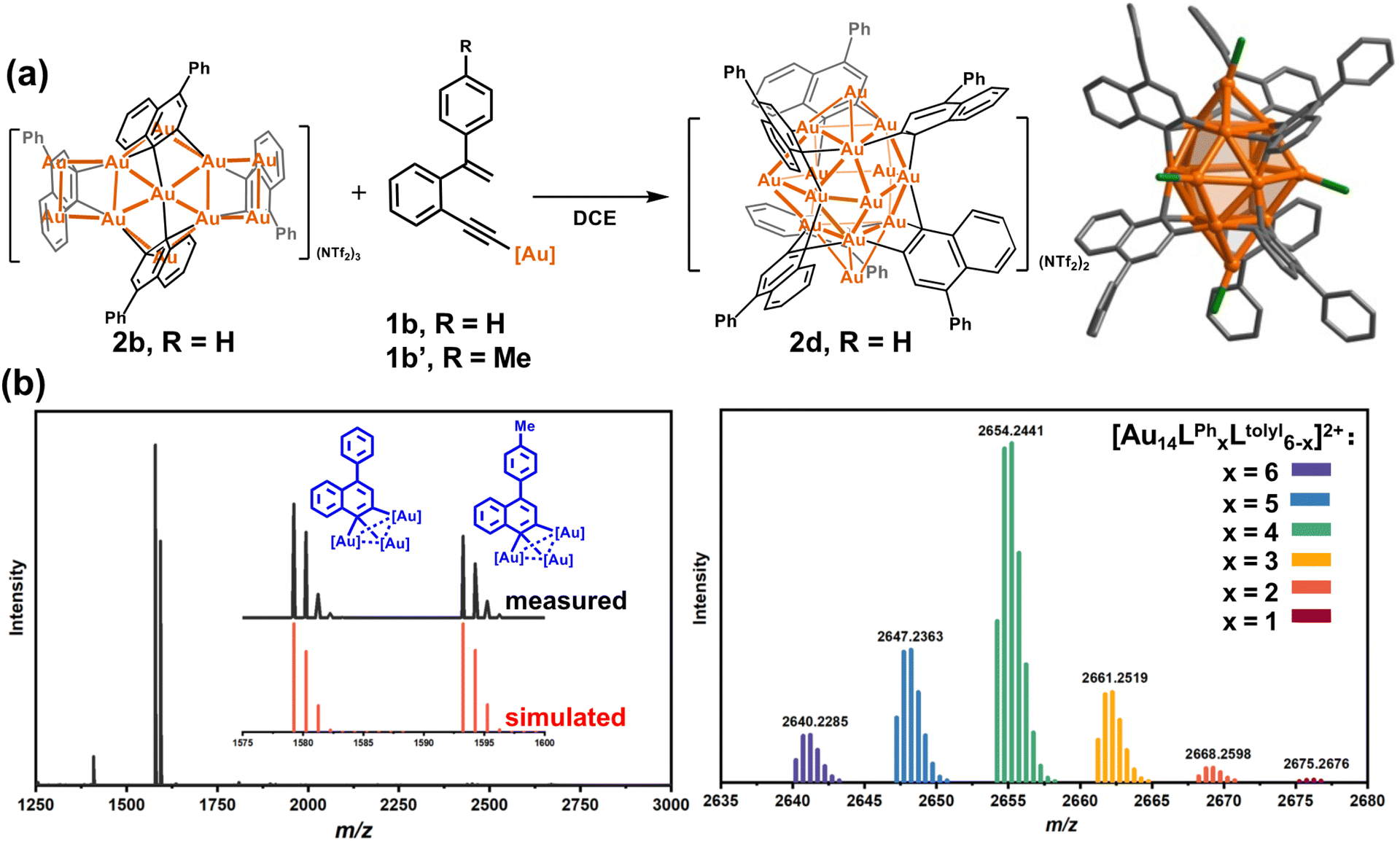 | ||
| Fig. 2 (a) The 2b-to-2d transformation and crystal structure of 2d. Phenyl groups of PPh3 ligands, hydrogen atoms, and peripheral NTf2− counter anions are omitted for clarity. Color coding: Au, yellow; C, gray; P, green. (b) (Left) The in situ ESI mass spectra of the crossover experiment between 2b and 1b′, and (right) ESI-MS of deposited microcrystals. | ||
To further investigate the detailed construction process of 2d, we performed a crossover experiment by using 2b to activate the tolyl-substituted substrate 1b′. The in situ ESI-MS spectrum discloses that 1b′ undergoes a similar cyclization to yield a tolyl-[C2Au3] species. Meanwhile, 2b disrupts its assembled structure to regenerate many Ph-substituted [C2Au3] fragment species (Fig. 2b). Furthermore, diffusion of diethyl ether into the resulting maroon solution deposited crimson microcrystals. ESI-MS analysis on the microcrystal sample showed a normal distribution of Au14 nanoclusters with a random combination of Ph- and tolyl-substituted [C2Au3] from 6:0 to 1:5 (Fig. 2b). We thus conceive that 2b actually serves as a metastable species to dissociate and activate the transformation of substrates. Then, the newly formed Ph-substituted [C2Au3] units plus the dissociated species of 2b promote the formation of the closed-cluster product 2d.
Catalytic behaviors of organogold clusters
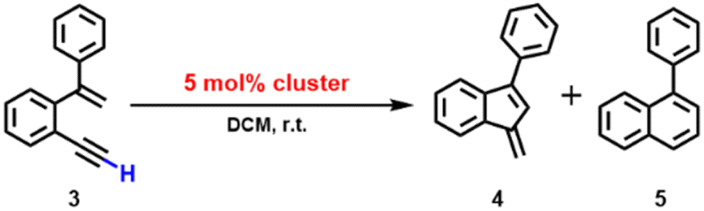
Inspired by the above 2b-mediated cyclization of 1b and 1b′, we then tried to clarify the catalytic possibility of 2a–2d in the cyclization reaction of 1,5-enynes. 2a–2d show a typical butterfly-shaped, net-like, dumbbell-like and closed cluster structure, respectively. Upon the use of 3 mol% 2a as catalyst, the cyclization of 1-ethynyl-2-(1-phenylvinyl)benzene (3) took place smoothly, which finally shows a high conversion ratio (>99%) and yields a mixture of two products 4 and 5 in a ratio of 4.5:1. Such selectivity is similar to the catalytic result (a 4:1 mixture) based on a mononuclear gold(I) catalyst.38 In the absence of 3, 2a kept its structure intact and stable as shown in the 1H NMR monitoring (Fig. S24†). Nevertheless, the monitoring further showed that along with the catalytic reaction the characteristic peaks of 2a gradually disappeared and the protodeaurated product 1,3-dimethoxyphenanthrene was simultaneously detected (Fig. S25†). Previous studies have revealed that the gem-diaurated species often experience the dissociation of a gold unit followed by protonolysis of the remaining Au–C bond to release organic products and recycle the mononuclear gold(I) catalytic species.36 We thus believe that 2a undergoes a similar protodeauration process to generate [Au(PPh3)]+ units as a true catalytic species.
The 2b-catalyzed cyclization of 3 also resulted in a mixture of 4 and 5 in a 4:1 ratio. However, in contrast to the full protodeauration process of 2a, chemical shifts of 2b in 1H and 31P NMR spectra showed negligible change (Fig. 3a and S28†). After the reaction, 2b could be recovered as a crystalline solid in a yield of 32%. Surprisingly, the catalyst loading can be lowered to 0.2 mol% such that the reaction proceeds with a turnover number (TON) of 403 (Fig. S29†). Subsequent kinetic studies based on 1H NMR monitoring indicated that the decline of the alkyne proton signal at 2.97 ppm obeys pseudo-first-order kinetics. Along with the increase in catalyst loading from 1% to 5%, it also follows a first-order kinetic relationship dependent on the concentration of 2b (Fig. 3b and c). Thus, the reaction orders of both substrate and catalyst are determined as one. We then employed ESI-MS to trace the entire reaction to make clear the true catalytic species. In order to avoid the interference of the organic skeleton of 2b with the catalytic product of 3, a tolyl-substituted substrate 3′ was used for a crossover experiment. The detection of a typical gem-diaurated species (m/z values of 1135.2021) as shown in many mononuclear gold-based catalysis demonstrates that [Au(PPh3)]+ may be the true catalytically active species (Fig. S30†). Accordingly, we conceive that a substrate molecule first attaches onto the Au11 kernel and then induces its dissociation to generate a [alkyne-Au(PPh3)]+ fragment and a Au10 unit. The former adduct further undergoes a 6-endo-dig or 5-exo-dig cyclization to give organic products. After the catalytic transformation, the Au10 intermediate combines with the post reaction free [Au(PPh3)]+ unit by aurophilic interaction to rebuild the Au11 compound 2b (Fig. 3d and S31†).
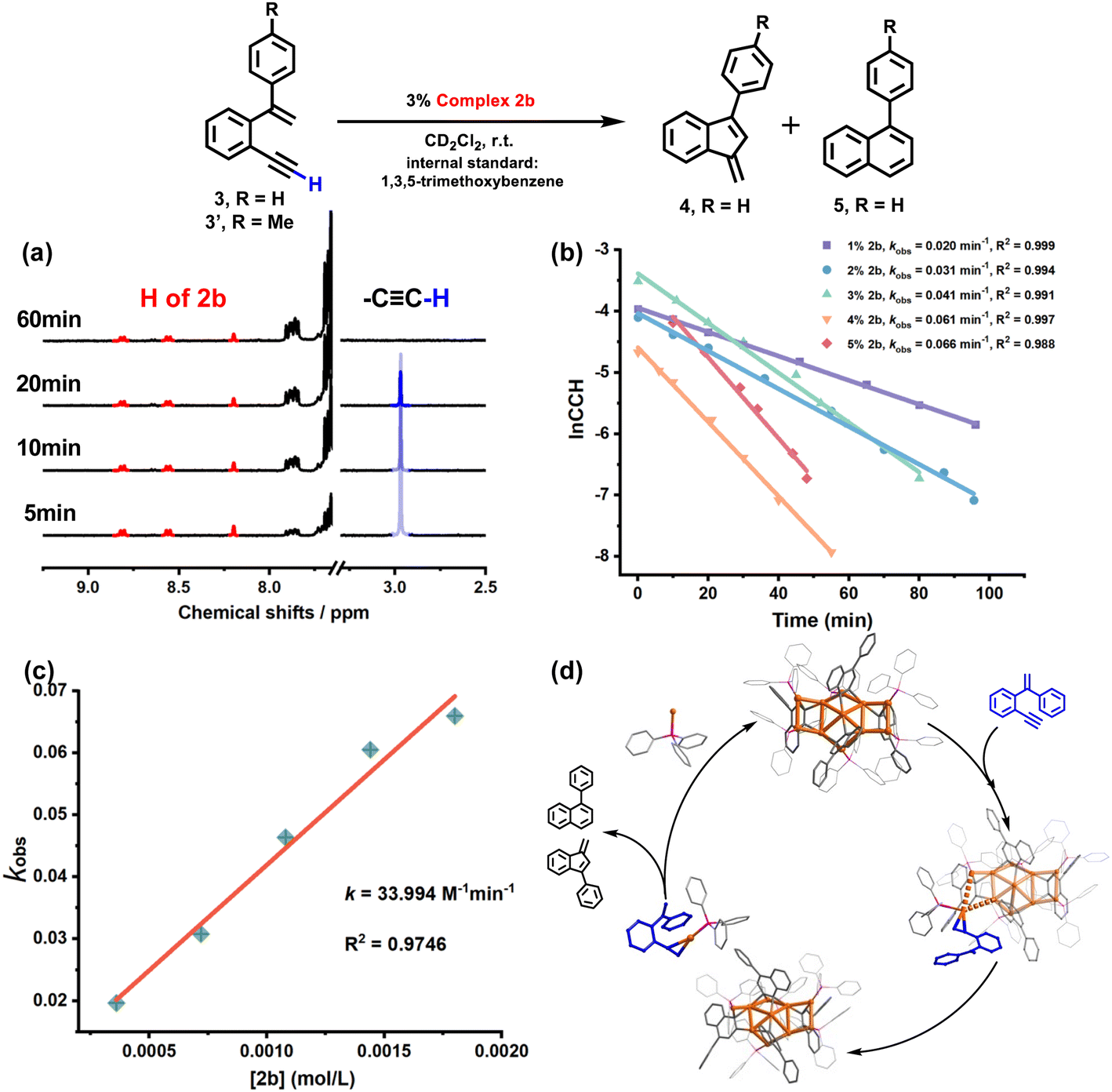 | ||
| Fig. 3 (a) 1H NMR monitoring of the 2b-catalyzed cyclization reaction. Red and blue lines represent the proton NMR peaks of 2b and the alkyne substrate, respectively. (b) Pseudo-first-order kinetic curves dependent on the concentration of 2b. (c) Kinetic curves of 2b dependent on the catalyst concentration. (d) Proposed mechanism for the cyclization reaction catalyzed by 2b. | ||
As for the 2c-catalyzed cyclization reaction (3 mol%), it displayed a very high conversion close to 100% and a 2:1 mixture of 4 and 5. The unchanged chemical shifts and 17% crystal recovery of 2c imply a catalytic process similar to that the 2b-based one (Fig. S32†). And with 0.2 mol% loading of the catalyst, the TON of this reaction is 341 (Fig. S33†). However, 1H NMR kinetic studies revealed that the 2c-catalyzed reaction is a good fit with a pseudo-first-order of the substrate but a half-order with the catalyst concentration (Fig. S34†). As shown in the ESI-MS spectrum of 2c (Fig. S16†), 2c is prone to dissociating into two MeAu14 and thereby exposes an unsaturated coordination site. During the catalytic reaction, the methyl proton signal of 2c at 2.67 ppm changed from broad to narrow (Fig. S32†), which is probably ascribed to the conformational adjustment along with the cleavage of 2c to MeAu14. In addition, ESI-MS monitoring of the reaction mixture clearly shows characteristic signals of the dissociated species MeAu14 (m/z 2323.6966) and MeAu14-PPh3 ([Au14(PPh3)5(LMe)6]2+, m/z 2454.7042) and no peaks corresponding to any gem-diaurated species. The latter implies that no mononuclear gold species dissociate from 2c (Fig. S35†). Consequently, we assume that the catalytic cycle of the dumbbell-like cluster 2c is initiated by the cleavage of the central Au–Au linkage to produce two MeAu14 units, each of which exposes a coordination unsaturated site to activate the alkyne group and complete the cyclization reaction (Fig. 4 and S36†).
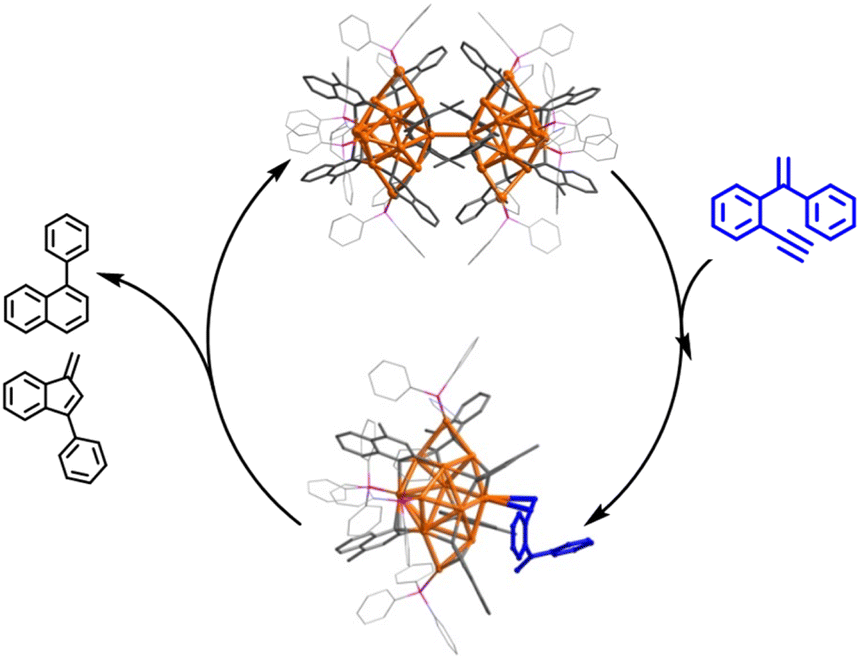 | ||
| Fig. 4 Proposed mechanism for the activation of 3 by 2c. | ||
In contrast, despite having a similar tetradecanuclear kernel as MeAu14, the closed-cluster 2d has no catalytic activity for 3 and other 1,5-enyne substrates (Fig. S37†). In view of the composition difference between MeAu14 and 2d, we believe that such catalytic inertness of 2d should be ascribed to the loss of unsaturated Au site due to strong coordination of a PPh3 ligand. Combining the above experiments, the catalytic performances of 2a–2d are summarized in Table 1.
|
|
||||
|---|---|---|---|---|
| Entry | Cluster | Conversion (%) | Selectivity (4/5) | TON |
| 1 | 2a | >99 | 4.5:1 |
|
| 2 | 2b | >99 | 4:1 |
403 |
| 3 | 2c | >99 | 2:1 |
341 |
| 4 | 2d | 0 | 0 | |
DFT calculations and mechanistic insight
To further rationalize our proposed mechanisms for the catalytic reactions of 2a–2c, we then performed density functional theory (DFT) calculations to complement experimental kinetic studies. The geometry optimizations and single point energy calculations for the three catalytically active clusters and their intermediates in the respective key steps were conducted. First, we investigated the dissociation of a [Au(PPh3)]+ or PPh3 moiety from 2a–2c. In addition, the pathway for the dissociation of 2c into two MeAu14 units was considered as well (Fig. S38†). The calculated results suggest that the PPh3 ligands strongly attach onto each metal cluster core with a very high dissociation energy (77.8–88.6 kcal mol−1). Although the dissociation energy of a [Au(PPh3)]+ species out from the cluster cores of 2a–2c is relatively small (29.6–47.6 kcal mol−1), such a process is still energetically unfavored.In accordance with the proposed mechanisms vide supra, we then designed new possible intermediates of the 2a and 2b-catalyzed reactions by dissociating a [alkyne-Au(PPh3)]+ species (type I in 2a-I and 2b-I) or replacing a PPh3 on the cluster core by an alkyne substrate molecule (type II in 2a-II and 2b-II), respectively (Fig. 5a and S39†). Also, there are two chemically non-equivalent Au sites in each cluster structure that need to be considered. They are labelled as L(left)/R(right) in 2a and L(left)/M(middle) in 2b. As shown in Fig. 5b, the process to obtain the intermediate 2b-I by forming a [alkyne-Au(PPh3)]+ species is exothermic, while the formation of the PPh3-replaced intermediate 2b-II needs a high energy of around 30 kcal mol−1. Such a 2b-I-based reaction pathway and cyclization reaction selectivity (Fig. S40 and S41†) are in agreement with the above experimental studies. Similar calculation studies on 2a show the same trend in terms of energy requirements. Moreover, the formation of the favored 2a-I possibly causes a subsequent protodeauration process to sequentially cleave the remaining carbon-polymetallic bonding and generate [AuPPh3]+ species for catalytic use. As for the 2c-based transformation, the binding of an alkyne molecule with MeAu14 largely compensates the energy requirement for the Au28-to-MeAu14 dissociation in 2c by breaking the central Au–Au linkage, finally reducing the single point energy change to −46.4 kcal mol−1 (Fig. 5b). Hence, these cluster intermediates with different architectures experience distinct bond breaking modes (Au–C and Au–Au for 2a and 2b; Au–Au for 2c) in the same catalytic system, finally resulting in distinct catalytic pathways.
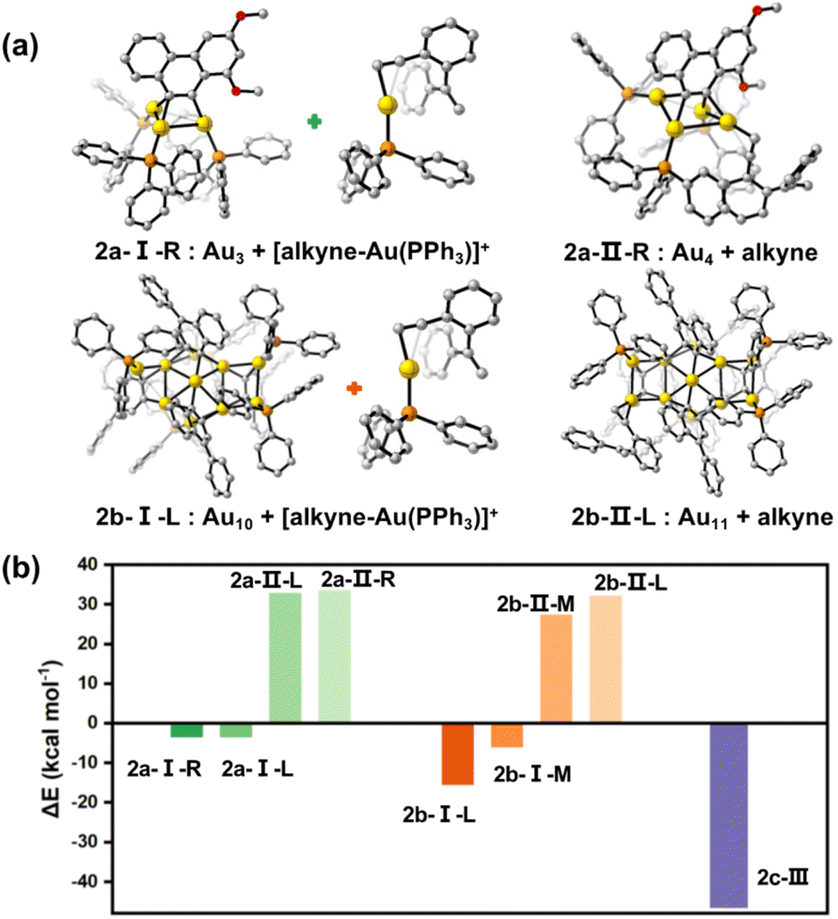 | ||
| Fig. 5 DFT results for energies (kcal mol−1) of intermediates of 2a–2c according to the proposed mechanisms. (a) The calculated intermediate structures of 2a and 2b in the catalytic cyclization of enyne substrates. Only one dissociating site is shown here, and the other sites are shown in Fig. S39.† (b) Comparison of the energy differences for different activation types. | ||
Conclusions
In summary, with the isolation of four vicinal dicarbanion-bonded organometallic clusters 2a–2d from the cyclization of aurated enynes, we demonstrate that organogold clusters generated from the reaction transformation can be further assembled into nano-sized polynuclear clusters to function as catalytically active species. With the combination of detailed NMR kinetic monitoring, ESI-MS spectra, and DFT calculations, we illustrate that multiple aurophilic interactions and carbon–gold multi-center bonding in assemblies provide significant support for the stabilization of active intermediates. The differently assembled structures experience distinct pathways in the catalytic cyclization of enyne substrates. The dumbbell-like cluster 2c tends to break the central Au–Au linkage to expose a catalytic site, while the net-like cluster 2b drives the reaction by dissociating an [alkyne-Au(PPh3)]+ adduct from its Au11 cluster core. The poor catalytic activity of the closed cluster 2d may provide a rationale for catalyst deactivation in transition metal catalysis. Compared with the well-established synthesis of gold nanoclusters by reduction, this work presents a novel assembly pathway based on stoichiometric gold(I)-mediated reactions. The assembly behavior observed in this context should also be taken into consideration when studying other systems with metallophilic interactions. Not only the field of gold catalysis, but other transition metal catalytic systems also require the scrutiny of such catalytically active organometal species.Experimental
Materials and methods
2′-Iodoacetophenone, o-iodobenzophenone, 1-bromo-2-iodobenzene, 2-iodobenzoyl chloride and AgNTf2 were purchased from Energy Chemical. Anhydrous tetrahydrofuran and Au(PPh3)Cl were purchased from J&K Scientific. All other reagents and solvents were used without further purification. 1H, 13C and 31P NMR were carried out on a JEOL ECX-400 MHz instrument. High resolution electrospray ionization mass spectrometry (ESI-MS) spectra were obtained on a Thermo Scientific Exactive Orbitrap instrument. The synthesis of σ-aurated substrates 1a–1c and 1,5-enyne substrates 3–3′ is included in the ESI.†Computational details
Structures of all these clusters for calculation were built on the basis of single-crystal structures. Due to the huge system and the limitation of computing resources, the free energy calculation was replaced by single point energy. Geometry optimizations and single point energy calculations for all compounds were performed using the ORCA 5.0.2 program.48,49 Geometry optimizations for the compounds considered here were optimized with the hybrid functional B3LYP50,51 with Grimme GD3(BJ) dispersion correction52,53 and the def2-SVP basis set.54,55 Single point energy was calculated based on the optimized structures with the hybrid functional B3LYP with Grimme GD3(BJ) dispersion correction and a def2-SVP basis set for C, H, P and def2-TZVP basis set54,55 for Au. To speed up the calculations, density fitting together with the chain of spheres approximations as implemented in ORCA (RIJCOSX)56 was used, with the auxiliary basis def2/J.57 The geometry of models was visualized using CYLview20 software.58Molecular thermochemistry properties of mononuclear species were evaluated using the Gaussian 09 program.59 Geometry optimizations as well as frequency calculations for all mononuclear species considered here were optimized with the hybrid functional B3LYP50,51 with Grimme GD3(BJ) dispersion correction52,53 and the def2-SVP basis set.54,55 Geometric optimizations were performed without restriction in the experimental solvent dichloromethane. The integral equation formalism polarizable continuum (IEFPCM) solvation model with SMD radii60 was used for solvent effect corrections. The optimized structures were confirmed to have no imaginary vibrational mode for intermediates and only one imaginary vibrational mode for each transition state. Transition states were further confirmed by connecting proper stationary points through intrinsic reaction coordinate (IRC) calculations. Thermal corrections were obtained by frequency calculations using the same method on optimized structures within the harmonic potential approximation under 298.15 K and 1 atm pressure. Single point energy was calculated based on the optimized structures with the hybrid functional B3LYP with Grimme GD3(BJ) dispersion correction and a def2-TZVP basis set.54,55 AdNDP analysis42 was performed using NBO 6.061 and Multiwfn 3.8 software.62 Hirshfeld,63 Mulliken, and ADCH64 atomic charges were calculated using Multiwfn 3.8 software. The molecular orbitals and AdNDP orbitals were visualized using Multiwfn 3.8 and VMD 1.9.3.65
Data availability
The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC-2286449 (2a), CCDC-2286452 (2b), CCDC-2286453 (2c) and CCDC-2286454 (2d). For full characterization data including NMR and ESI-MS spectra and experimental details, see the ESI.† Any further relevant data are available from the authors upon reasonable request.Author contributions
L. Z. conceived and supervised the project. The synthetic experiments, structural characterization, and catalytic studies were carried out by Q. L. DFT calculations were performed by X. Z. R. J. provided assistance in ESI-MS measurements. Q. L., X. Z., and L. Z. co-wrote the manuscript. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (22025105, 22350002 and 21821001) is gratefully acknowledged. The authors thank the Tsinghua Xuetang Talents Program for providing computational resources.Notes and references
- A. S. Kashin and V. P. Ananikov, J. Org. Chem., 2013, 78, 11117–11125 CrossRef CAS PubMed.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef CAS.
- L.-C. Liu and A. Corma, Trends Chem., 2020, 2, 383–400 CrossRef CAS.
- J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Domínguez, A. Leyva-Pérez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS PubMed.
- A. Leyva-Perez, J. Oliver-Meseguer, P. Rubio-Marques and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 11554–11559 CrossRef CAS PubMed.
- J. Oliver-Meseguer, A. Leyva-Pérez and A. Corma, ChemCatChem, 2013, 5, 3509–3515 CrossRef CAS.
- J. Oliver-Meseguer, L. Liu, S. Garcia-Garcia, C. Canos-Gimenez, I. Dominguez, R. Gavara, A. Domenech-Carbo, P. Concepcion, A. Leyva-Perez and A. Corma, J. Am. Chem. Soc., 2015, 137, 3894–3900 CrossRef CAS PubMed.
- M. Neumeier, U. Chakraborty, D. Schaarschmidt, V. de la Pena O'Shea, R. Perez-Ruiz and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2020, 59, 13473–13478 CrossRef CAS.
- C.-H. Li, S. Song, Y.-L. Li, C. Xu, Q.-Q. Luo, Y.-L. Guo and X.-M. Wang, Nat. Commun., 2021, 12, 3813 CrossRef CAS PubMed.
- E. Bayram, J. C. Linehan, J. L. Fulton, J. A. Roberts, N. K. Szymczak, T. D. Smurthwaite, S. Ozkar, M. Balasubramanian and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 18889–18902 CrossRef CAS.
- J. Cordon, G. Jimenez-Oses, J. M. Lopez-de-Luzuriaga and M. Monge, Nat. Commun., 2017, 8, 1657 CrossRef PubMed.
- T. N. Gieshoff, U. Chakraborty, M. Villa and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2017, 56, 3585–3589 CrossRef CAS PubMed.
- Y. S. Panova, A. S. Kashin, M. G. Vorobev, E. S. Degtyareva and V. P. Ananikov, ACS Catal., 2016, 6, 3637–3643 CrossRef CAS.
- B. D. Briggs, N. M. Bedford, S. Seifert, H. Koerner, H. Ramezani-Dakhel, H. Heinz, R. R. Naik, A. I. Frenkel and M. R. Knecht, Chem. Sci., 2015, 6, 6413–6419 RSC.
- D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Muller-Bunz, A. M. Trzeciak and M. Albrecht, Chem.–Eur. J., 2012, 18, 6055–6062 CrossRef CAS.
- S. Sandl, F. Schwarzhuber, S. Pollath, J. Zweck and A. Jacobi von Wangelin, Chem.–Eur. J., 2018, 24, 3403–3407 CrossRef CAS.
- M. A. Rivero-Crespo, A. Leyva-Perez and A. Corma, Chem.–Eur. J., 2017, 23, 1702–1708 CrossRef CAS PubMed.
- X.-F. Jiang, H. Huang, Y.-F. Chai, T. L. Lohr, S.-Y. Yu, W. Lai, Y.-J. Pan, M. Delferro and T. J. Marks, Nat. Chem., 2016, 9, 188–193 CrossRef PubMed.
- B. L. Tran, J. L. Fulton, J. C. Linehan, J. A. Lercher and R. M. Bullock, ACS Catal., 2018, 8, 8441–8449 CrossRef CAS.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed.
- L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953–965 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- (a) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC; (b) N. Mirzadeh, S. H. Priver, A. J. Blake, H. Schmidbaur and S. K. Bhargava, Chem. Rev., 2020, 120, 7551–7591 CrossRef CAS PubMed.
- Z.-N. Wu, Y.-H. Du, J.-L. Liu, Q.-F. Yao, T.-K. Chen, Y.-T. Cao, H. Zhang and J.-P. Xie, Angew. Chem., Int. Ed., 2019, 58, 8139–8144 CrossRef CAS PubMed.
- Q.-S. Zheng, S. Borsley, G. S. Nichol, F. Duarte and S. L. Cockroft, Angew. Chem., Int. Ed., 2019, 58, 12617–12623 CrossRef CAS PubMed.
- T. A. Nguyen, E. Daiann Sosa Carrizo, H. Cattey, P. Fleurat-Lessard, J. Roger and J. C. Hierso, Chem.–Eur. J., 2022, 28, e202200769 CrossRef CAS PubMed.
- E. S. Smirnova and A. M. Echavarren, Angew. Chem., Int. Ed., 2013, 52, 9023–9026 CrossRef CAS PubMed.
- J. Yuan, T.-T. Sun, X. He, K. An, J. Zhu and L. Zhao, Nat. Commun., 2016, 7, 11489 CrossRef CAS.
- K. Xiao, Y. Xue, B. Yang and L. Zhao, CCS Chem., 2021, 3, 555–565 CrossRef CAS.
- D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed.
- E. Soriano and J. Marco-Contelles, Acc. Chem. Res., 2009, 42, 1026–1036 CrossRef CAS PubMed.
- E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed.
- A. Collado, D. J. Nelson and S. P. Nolan, Chem. Rev., 2021, 121, 8559–8612 CrossRef CAS PubMed.
- G. Seidel, C. W. Lehmann and A. Furstner, Angew. Chem., Int. Ed., 2010, 49, 8466–8470 CrossRef CAS PubMed.
- A. S. Hashmi, I. Braun, P. Nosel, J. Schadlich, M. Wieteck, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 4456–4460 CrossRef CAS PubMed.
- D. Weber, M. A. Tarselli and M. R. Gagne, Angew. Chem., Int. Ed., 2009, 48, 5733–5736 CrossRef CAS PubMed.
- T. Shibata, Y. Ueno and K. Kanda, Synlett, 2006, 2006, 0411–0414 CrossRef.
- K. Xiao, Y. Zhao, J. Zhu and L. Zhao, Nat. Commun., 2019, 10, 5639 CrossRef CAS.
- Q.-H. Wei, L.-Y. Zhang, G.-Q. Yin, L.-X. Shi and Z.-N. Chen, J. Am. Chem. Soc., 2004, 126, 9940–9941 CrossRef CAS.
- W.-D. Si, C.-K. Zhang, M. Zhou, Z. Wang, L. Feng, C.-H. Tung and D. Sun, Sci. Adv., 2024, 10, eadm6928 CrossRef CAS PubMed.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- Q.-F. Yao, L.-M. Liu, S. Malola, M. Ge, H.-Y. Xu, Z.-N. Wu, T.-K. Chen, Y. T. Cao, M. F. Matus, A. Pihlajamäki, Y. Han, H. Häkkinen and J.-P. Xie, Nat. Chem., 2023, 15, 230–239 CrossRef CAS PubMed.
- J. E. Heckler, M. Zeller, A. D. Hunter and T. G. Gray, Angew. Chem., Int. Ed., 2012, 51, 5924–5928 CrossRef CAS PubMed.
- G. Li, H. Abroshan, Y.-X. Chen, R.-C. Jin and H. J. Kim, J. Am. Chem. Soc., 2015, 137, 14295–14303 CrossRef CAS PubMed.
- R. R. Nasaruddin, Q.-F. Yao, T.-K. Chen, M. J. Hülsey, N. Yan and J.-P. Xie, Nanoscale, 2018, 10, 23113–23121 RSC.
- J.-H. Yu, Z.-R. Yuan, J. Xu, J.-G. Wang, T.-D. Li, Y.-Z. Li and D. Sun, Chem. Sci., 2023, 14, 6564–6571 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- C. Y. Legault, CYLview20, Université de Sherbrooke, 2020, https://www.cylview.org Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- T. Lu and F. Chen, J. Theor. Comput. Chem., 2012, 11, 163–183 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2286449 and 2286452–2286454. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01618a |
| This journal is © The Royal Society of Chemistry 2024 |