
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePuCl3{CoCp[OP(OEt)2]3}: transuranic elements entering the field of heterometallic molecular chemistry†
Thomas E.
Shaw‡
a,
Zachary R.
Jones‡
a,
Sara L.
Adelman
 *a,
Nickolas H.
Anderson
a,
Eric G.
Bowes
a,
Eric D.
Bauer
*a,
David
Dan
a,
Jan
Klouda
a,
Karah E.
Knope
*b,
Stosh A.
Kozimor
*a,
Molly M.
MacInnes
a,
Veronika
Mocko
a,
Francisca R.
Rocha
a,
Harrison D.
Root
a,
Benjamin W.
Stein
a,
Joe D.
Thompson
a and
Jennifer N.
Wacker
ab
*a,
Nickolas H.
Anderson
a,
Eric G.
Bowes
a,
Eric D.
Bauer
*a,
David
Dan
a,
Jan
Klouda
a,
Karah E.
Knope
*b,
Stosh A.
Kozimor
*a,
Molly M.
MacInnes
a,
Veronika
Mocko
a,
Francisca R.
Rocha
a,
Harrison D.
Root
a,
Benjamin W.
Stein
a,
Joe D.
Thompson
a and
Jennifer N.
Wacker
ab
aLos Alamos National Laboratory (LANL), P. O. Box 1663, Los Alamos, New Mexico 87545, USA. E-mail: sadelman@lanl.gov; stosh@lanl.gov
bDepartment of Chemistry, Georgetown University, 37th and O Streets NW, Washington, D.C. 20057, USA. E-mail: karah.knope@georgetown.edu
First published on 15th July 2024
Abstract
Recent advances enabled the discovery of heterometallic molecules for many metals: main group, d-block, lanthanides, and some actinides (U, Th). These complexes have at least two different metals joined by bridging ligands or by direct metal–metal bonding interactions. They are attractive because they can enable chemical cooperativity between metals from different parts of the periodic table. Some heterometallics provide access to unique reactivity and others exhibit physical properties that cannot be accessed by homometallic species. We envisioned that transuranic heterometallics might similarly enable new transuranic chemistry, though synthetic routes to such compounds have yet to be developed. Reported here is the first synthesis of a molecular transuranic complex that contains plutonium (Pu) and cobalt (Co). Our analyses of PuCl3{CoCp[OP(OEt)2]3} showed Pu(IV) and Co(III) were present and suggested that the Pu(IV) oxidation state was stabilized by the electron donating phosphite ligands. This synthetic method – and the demonstration that Pu(IV) can be stabilized in a heterobimetallic molecular setting – provides a foundation for further exploration of transuranic multimetallic chemistry.
Introduction
Recent synthetic advances have enabled discovery of heterometallic molecules for many elements including those from the main, d-block, lanthanide, and even some actinide (uranium and thorium) elements.1–26 These heterometallic compounds are defined as having two – or more – different metals within one discrete molecule, where the metals are fixed in proximity through bridging ligands or direct metal–metal bonds. Combining properties from elements on opposite sides of the periodic table within one molecule unlocks potential for new cooperative chemistry that would otherwise be inaccessible to homometallic species. The application space for heterometallics is now quite broad and ranges from catalysis to conductive materials, photophysical applications, and molecular magnets.1–3,12–14,27–31 Based on those successes, we posited that preparation of plutonium (Pu) heterometallic molecules could be a gateway for new and interesting chemistry, provided appropriate synthetic pathways can be developed. This notion was fueled by the isolation of heterometallic extended solids reported from the transuranic solid-state and intermetallic communities.32–35 Perhaps the most notable result from those synthetic efforts was the discovery of PuCoGa5. This intermetallic enabled plutonium superconductivity to be observed for the first time and advanced fundamental understanding of plutonium's electronic structure and physical properties.32Herein, we report the isolation of the first discrete heterobimetallic molecule for plutonium, namely plutonium(IV) trichloride (cyclopentadienyl)tris(diethylphosphito)cobalt(III), PuCl3{CoCp[OP(OEt)2]3}. This compound was prepared by reacting an acidic Pu4+(aq) residue with Na{CoCp[OP(OEt)2]3}, affectionately referred to in the literature as the Kläui ligand.36–46 Our synthetic methodology was attractive because it could be carried out without exclusion of air and moisture. It also enabled large amounts (>300 mg) of PuCl3{CoCp[OP(OEt)2]3} to be isolated in modest crystalline yield (70%) and in high purity. Although many plutonium molecules have been characterized using a wide variety of techniques, limited access to plutonium and the substantial infrastructure needed to safety handle large quantities of plutonium often limits the extent to which a given plutonium compound can be studied. Access to 300 mg of PuCl3{CoCp[OP(OEt)2]3} facilitated rigorous characterization by single crystal X-ray diffraction, nuclear magnetic resonance (NMR) spectroscopy, variable temperature magnetometry, cyclic voltammetry, and optical spectroscopy. The results indicated that PuCl3{CoCp[OP(OEt)2]3} contained discrete Pu(IV) and Co(III) metal cations within the same molecule. These data also showcased that Pu(IV) was stabilized by the electron donating properties of the bridging phosphite ligands. It is our hope that the discovery of PuCl3{CoCp[OP(OEt)2]3} will enable synthetic access to higher order multi-metallic transuranic molecules (e.g., tri-, tetra-, penta-metallic, and beyond) and potentially inspire others to begin studying heterometallic transuranic molecular chemistry.
Results and discussion
Synthesis
The plutonium used in this study was recovered from residues generated by previous research campaigns using a published Pu-recycling procedure.47 This process generated an oxidation state pure and chemically pure stock-solution of Pu4+(aq) in HCl(aq) (6 M). Heating an aliquot from that Pu4+(aq) stock-solution on a hot block under a stream of filtered air afforded an acidic Pu4+(aq) residue used to prepare PuCl3{CoCp[OP(OEt)2]3}. The acidic nature of the Pu4+(aq) residue raised concern for further reactivity, e.g. would residual HCl(aq) decompose the CoCp[OP(OEt)2]31− fragment. To alleviate these types of concerns, numerous scoping studies were conducted that targeted previously reported MCl3{CoCp[OP(OEt)2]3} (M = Ce, U) complexes using (NH4)2Ce(NO3)6 and UCl4 surrogate starting materials dissolved in HCl(aq).48–50 These studies suggested that the Na{CoCp[OP(OEt)2]3} reagent was tolerant to the acidic conditions associated with the Pu4+(aq) acidic residue and enabled us to optimize experimental conditions for crystallization of the heterometallic product. Moving forward, we established a robust protocol that reproducibly provided single crystals of PuCl3{CoCp[OP(OEt)2]3} on a small scale (3 mg scale of Pu). Subsequent efforts demonstrated that the synthesis could be scaled-up 100-fold, generating >300 mg of PuCl3{CoCp[OP(OEt)2]3} in 70% crystalline yield.The PuCl3{CoCp[OP(OEt)2]3} heterobimetallic complex was prepared by a salt metathesis reaction that occurred when the acidic Pu4+(aq) residue was reacted with crystalline Na{CoCp[OP(OEt)2]3} in THF (Scheme 1). In this reaction, a chloride ligand associated with the Pu4+(aq) residue was displaced by the CoCp[OP(OEt)2]31− monoanion, which consequently outcompeted neutral THF and H2O solvents for complexation of Pu(IV). During the synthesis the Pu4+(aq) residue did not completely dissolve in THF. It formed a suspension instead. Only after combining that orange slurry with the THF solution of Na{CoCp[OP(OEt)2]3} did homogeneity occur. Stirring the resulting solution overnight generated a brown solution and a white precipitate. That solid – presumedly NaCl – was removed in two steps. First, the mixture was filtered through a Celite column and the solvent was removed. Second, dichloromethane was added to the resulting residue and that mixture was filtered through another Celite column. The title complex was then isolated as a dark green powder following the removal of the solvent.
 | ||
| Scheme 1 Reaction that generated PuCl3{CoCp[OP(OEt)2]3}. | ||
Structure
Green, rectangular prisms of PuCl3{CoCp[OP(OEt)2]3} suitable for X-ray diffraction were reproducibly obtained from dichloromethane solutions layered with a toluene counter solvent in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Data were collected at room temperature to avoid icing imposed from the tertiary containment method required for safe handling of radioactive single crystal samples.47 Despite having significantly disordered chloride and ethoxy functional groups, the room temperature single crystal X-ray diffraction data from PuCl3{CoCp[OP(OEt)2]3} were modeled in the monoclinic P21/n space group (see Fig. 1 and ESI†). Within this heterobimetallic compound were the duo of Pu(IV) and Co(III) cations bridged by three phosphite ligands. The inner coordination sphere around Pu(IV) was comprised of three chloride ligands and three oxygen atoms from the bridging phosphites. These chlorine and oxygen atoms were arranged in a distorted octahedron with approximate C3v symmetry around Pu(IV). With respect to Co(III), the structure was best described using the “three-legged piano stool” analogy often invoked to describe molecules with one cyclopentadienide and three monodentate ligands.
:1 ratio. Data were collected at room temperature to avoid icing imposed from the tertiary containment method required for safe handling of radioactive single crystal samples.47 Despite having significantly disordered chloride and ethoxy functional groups, the room temperature single crystal X-ray diffraction data from PuCl3{CoCp[OP(OEt)2]3} were modeled in the monoclinic P21/n space group (see Fig. 1 and ESI†). Within this heterobimetallic compound were the duo of Pu(IV) and Co(III) cations bridged by three phosphite ligands. The inner coordination sphere around Pu(IV) was comprised of three chloride ligands and three oxygen atoms from the bridging phosphites. These chlorine and oxygen atoms were arranged in a distorted octahedron with approximate C3v symmetry around Pu(IV). With respect to Co(III), the structure was best described using the “three-legged piano stool” analogy often invoked to describe molecules with one cyclopentadienide and three monodentate ligands.
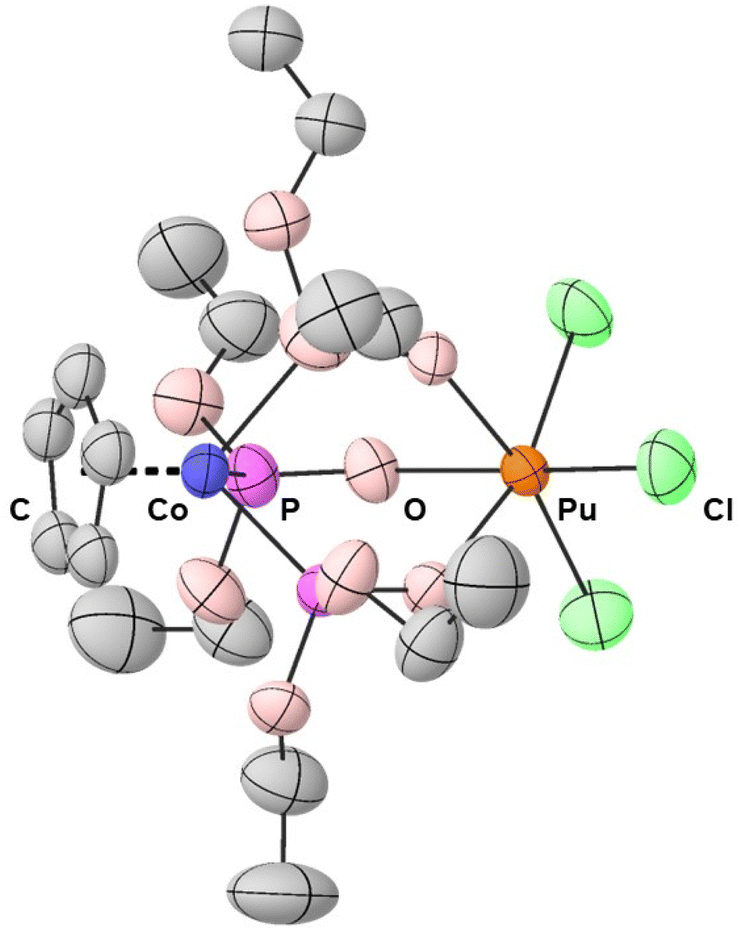 | ||
| Fig. 1 Thermal ellipsoid plot (30% probability) generated from the single crystal X-ray diffraction data from PuCl3{CoCp[OP(OEt)2]3}. Hydrogen atoms have been omitted. | ||
Defining metrics from the PuCl3{CoCp[OP(OEt)2]3} structure were the Co⋯Pu, Pu–Cl, Pu–Ophosphite, Co–Pphosphite, and Co–CCp bond distances. Comparing these bond distances with those reported previously from MCl3{CoCp[OP(OEt)2]3} (M = Ce, Ti, Tc)48,51,52 showed regularity and predictability when the six-coordinate metal ionic radii of the M(IV) cations were accounted for (Table 1). For example, a Co⋯M distance of 3.3 Å was obtained for each complex when the six-coordinate metal ionic radii were subtracted from the Co⋯M distances. This value of 3.3 Å was also similar to the ionic radius corrected Co⋯U distance in the structurally-related UCl3(THF){CoCp[OP(OEt)2]3} compound. Comparison with the later compound, UCl3(THF){CoCp[OP(OEt)2]3}, further indicated that increasing the M(IV) coordination number by adding a neutral ligand had no impact on the Co⋯M distance.49 All of these Co⋯M distances were far too long than what would be anticipated for Co–M bonds. As testament, the Co⋯Pu distance in PuCl3{CoCp[OP(OEt)2]3} was over 1 Å longer than the Co–U bond distance observed by Bart, Thomas, and coworkers in their related UX[CoI(Ph2PNiPr)]3 (X = Ph2PNiPr, I) structures.53 Other structural metrics worth noting were the three equivalent Pu–Ophosphite bond distances that ranged from 2.216(5) to 2.231(4) Å and the three equivalent Pu–Cl bond distances that ranged from 2.567(7) to 2.576(5) Å. These Pu–Ophosphite and Pu–Cl distances averaged 2.222 ± 0.008 Å and 2.572 ± 0.005 Å, respectively. Uncertainty for all average bond distances have been reported as the standard deviation from the mean at 1σ and denoted by the “±” symbol.
| Bond | Pu(IV) this work (Å) | PuCl62− (Å) | Ce(IV) (Å) | U(IV) (Å) | Ti(IV) (Å) | Tc(IV) (Å) |
|---|---|---|---|---|---|---|
| a The uncertainty for average distances were reported as the standard deviation of the mean at 1σ. b Uncertainty for these uranium distances were difficult to extract from the reported primary literature. | ||||||
| M–O(1) | 2.218(4) | — | 2.226(3) | 2.2358b | 1.956(6) | 2.035(2) |
| 2.013(2) | ||||||
| M–O(2) | 2.216(5) | — | 2.233(3) | 2.2607b | 1.962(7) | 2.027(2) |
| 2.028(2) | ||||||
| M–O(3) | 2.231(4) | — | 2.244(3) | 2.2607b | 1.979(6) | 2.026(2) |
| 2.016(2) | ||||||
| M⋯Co | 4.1958(9) | 4.2044(7) | 4.289(5) | 3.973(2) | 3.9269(5) | |
| 3.9518(5) | ||||||
| M–Omeana | 2.222(8) | — | 2.234(9) | 2.25(1) | 1.97(1) | 2.029(4) |
| 2.019(8) | ||||||
| M–Omeana corrected | 1.362(8) | — | 1.365(9) | 1.36(1) | 1.36(1) | 1.378(4) |
| 1.374(8) | ||||||
| M–Cl(1) | 2.567(7) | 2.5757 | 2.5749(10) | 2.6480b | 2.305(3) | 2.3036(7) |
| 2.568(4) | 2.3171(7) | |||||
| M–Cl(2) | 2.575(5) | 2.5935 | 2.5903(11) | 2.6679b | 2.304(3) | 2.3098(7) |
| 2.572(7) | 2.3105(7) | |||||
| M–Cl(3) | 2.569(6) | 2.5943 | 2.5840(12) | 2.6679b | 2.322(3) | 2.3136(7) |
| 2.568(5) | 2.3290(7) | |||||
| M–Clmeana | 2.571(4) | 2.59(1) | 2.583(8) | 2.66(1) | 2.31(1) | 2.309(5) |
| 2.570(2) | 2.319(9) | |||||
| M–Clmeana corrected | 1.711(4) | 1.734(7) | 1.713(8) | 1.77(1) | 1.71(1) | 1.664(5) |
| 1.710(2) | 1.674(9) | |||||
The three Co–Pphosphite distances ranged from 2.160(2) to 2.166(18) Å, averaged 2.162 ± 0.003 Å, and were equivalent within the measurement uncertainty. These distances were slightly shorter than the Co–Pphosphite distances in the Na{CoCp[OP(OEt)2]3} precursor: 2.187(2) and 2.190(2) Å.54 They were also equivalent to the average Co–Pphosphite distances previously reported in the other MCl3{CoCp[OP(OEt)2]3} complexes (Table 1). The five Co–CCp distances in PuCl3{CoCp[OP(OEt)2]3} ranged from 2.064(12) to 2.093(12) Å and had a Co–Ccentroid distance equal to 1.699 Å. These distances also matched the Co–CCp distances reported previously in the MCl3{CoCp[OP(OEt)2]3} complexes shown in Table 1. Regularities in the Pu–ligand and Co–ligand bond distances within PuCl3{CoCp[OP(OEt)2]3} and the other reported MCl3{CoCp[OP(OEt)2]3} complexes highlighted ubiquitous steric saturation and stability within this heterobimetallic framework for Co(III), Pu(IV), and the other Ce(IV), Ti(IV), and Tc(IV) cations.
Nuclear magnetic resonance spectroscopy (NMR)
Solution-phase 1H and 31P NMR data from PuCl3{CoCp[OP(OEt)2]3} and Na{CoCp[OP(OEt)2]3} were provided in Fig. 2 and in the ESI.† These data indicated that PuCl3{CoCp[OP(OEt)2]3} was stable when dissolved in deuterated chloroform. In contrast to the diamagnetic Na{CoCp[OP(OEt)2]3} precursor, spectra from PuCl3{CoCp[OP(OEt)2]3} exhibited a paramagnetic contact shift associated with the Pu(IV) cation (Fig. 2). For instance, the 1H NMR spectrum from Na{CoCp[OP(OEt)2]3} contained three resonances: a singlet from the Cp1− ligand at 5.18 ppm, a broad multiplet from the methylene protons on the phosphite functional group at 4.09 ppm, and a triplet corresponding to the methyl protons at 1.27 ppm. In the paramagnetic product, the Cp1− singlet shifted downfield by 1.53 ppm to 6.71 ppm. It also shifted the methyl triplet upfield to 0.73 ppm and the methylene resonance downfield to 3.03 ppm. Another defining feature for the PuCl3{CoCp[OP(OEt)2]3} spectrum was that the methylene resonance was split into a complicated multiplet, which we described as a doublet of pentets (J = 92.0, 7.8 Hz). These methylene protons were diasteriotopic and experienced geminal proton–proton coupling, vicinal proton–proton coupling, and coupling to adjacent phosphorous atoms. Consistent with this interpretation were the relative peak intensities (5 for Cp1−, 12 for the methylene, and 18 for the methyl) and similarity between chemical shift values from PuCl3{CoCp[OP(OEt)2]3} and the previously reported uranium analogue, UCl3{CoCp[OP(OEt)2]3}. The spectrum from UCl3{CoCp[OP(OEt)2]3} had Cp1−, methylene, and methyl resonances at 6.45, 3.04, and 0.50 ppm, respectively.50 | ||
| Fig. 2 (Left) 31P NMR spectra from PuCl3{CoCp[OP(OEt)2]3} (top) and Na{CoCp[OP(OEt)2]3} (bottom) dissolved in CDCl3. (Right) 1H NMR spectra from PuCl3{CoCp[OP(OEt)2]3} (top) and Na{CoCp[OP(OEt)2]3} (bottom) dissolved in CDCl3. The molecular fragments illustrate our interpretation of the NMR data (methyl – blue; methylene – green; cyclopentadienyl – orange; phosphorus – pink). | ||
The 31P NMR spectrum from PuCl3{CoCp[OP(OEt)2]3} was impacted by the paramagnetic Pu(IV) cation, as well. It displayed a single phosphite resonance at 24.5 ppm. This value was substantially shifted upfield from the 114.61 ppm resonance associated with the diamagnetic Na{CoCp[OP(OEt)2]3} precursor. The 31P phosphite chemical shift from PuCl3{CoCp[OP(Oet)2]3} was quite different than that observed for the Ce(IV) (diamagnetic) and U(IV) (paramagnetic) analogues, which displayed phosphite resonances at 124.2 and 191.4 ppm, respectively.48,50 The 31P NMR data also suggested that there was only one phosphorous containing compound in solution, namely PuCl3{CoCp[OP(OEt)2]3}. Evaluating this data alongside the 1H NMR data provided no evidence of CoCp[OP(OEt)2]31− decomposition or ligand redistribution and highlighted the solution-phase stability of PuCl3{CoCp[OP(OEt)2]3}.
Magnetometry
Variable-temperature magnetic susceptibility measurements were made on PuCl3{CoCp[OP(OEt)2]3} to determine to the 5f- and 3d-electron occupancies for the Pu and Co metals. Data collected from the paramagnetic PuCl3{CoCp[OP(OEt)2]3} complex was compared with data from a known Pu(IV) standard, (Me4N)2PuCl6 (Fig. 3).55 It is often difficult to collect reproducible magnetic data on radioactive samples because magnetic measurements require extreme precision regarding sample mass. Unfortunately, many actinide synthetic campaigns are carried out on the 1 to 10 mg scale and it is hard to accurately transfer those small quantities into holders that provide radiological containment for magnetic measurements. We reduced the impact from this variable by developing synthetic methods and sample handling procedures that were compatible with large (hundreds of milligrams) crystalline sample sizes. As such, the masses for the magnetometry samples used for both compounds shown in Fig. 3 were determined with confidence and concerns regarding errors from mass dependence of the measurements were low.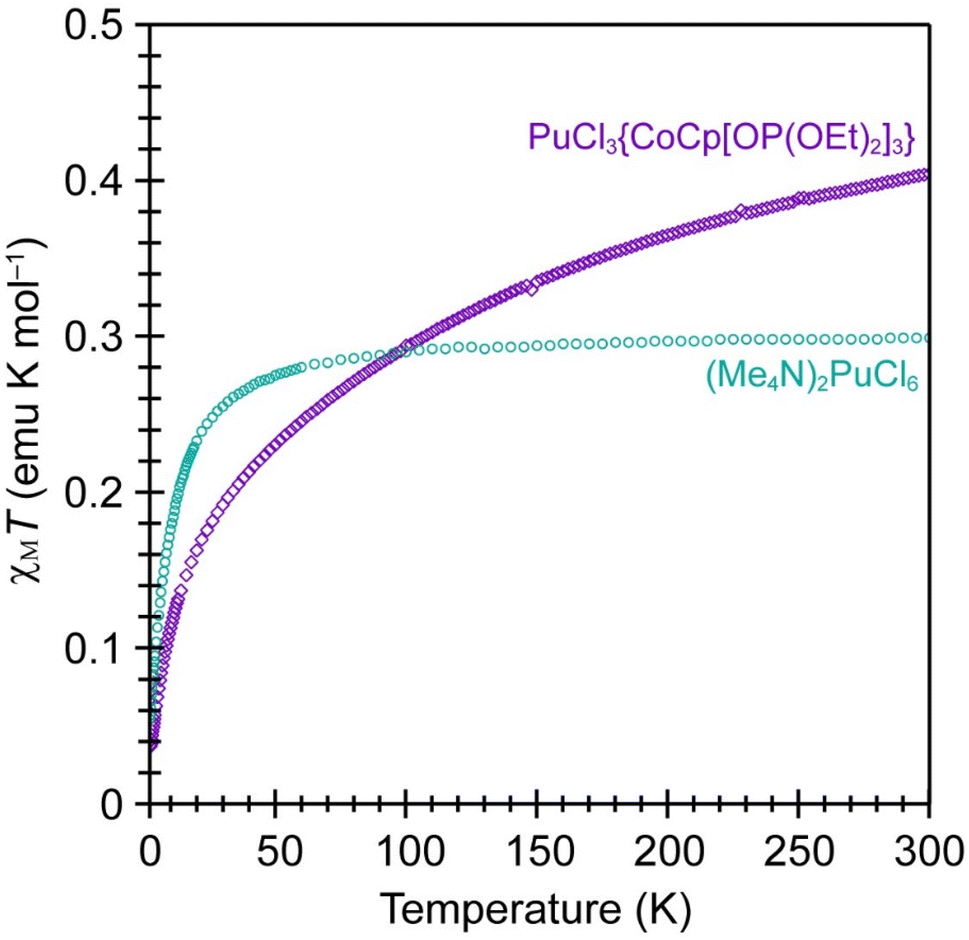 | ||
| Fig. 3 Variable temperature magnetic susceptibility in a 5 T field from PuCl3{CoCp[OP(OEt)2]3} (purple trace) and (Me4N)2PuCl6 (teal trace). | ||
Data from PuCl3{CoCp[OP(OEt)2]3} were quite similar to those from PuCl62−. Both compounds exhibited a drop in χMT as the temperature was lowered, decreasing to approximately 0.04 emu K mol−1 at low temperature (near 5 K). These data suggested that the number of unpaired 5f-electrons in PuCl3{CoCp[OP(OEt)2]3} was identical to that in (Me4N)2PuCl6, both having the 5f4 electron configuration associated with Pu(IV). Note, that χMT approached an approximate value of 0.4 emu K mol−1 at high temperature for PuCl3{CoCp[OP(OEt)2]3}. This value was similar to the high-temperature χMT data obtained from (Me4N)2PuCl6, which saturated at 0.3 emu K mol−1. These values were substantially suppressed from what would be expected for a free Pu(IV) cation (as expected), where χMT would saturate at 0.9 emu K mol−1 (the Currie constant) and the effective magnetic moment (μeff) would equal 2.68 μB. Like (Me4N)2PuCl6,55 the reduced Currie constant (saturated χMT) in PuCl3{CoCp[OP(OEt)2]3} was most naturally explained by ligand field splitting of the 5f4 multiplet. Overall, these comparisons suggested that PuCl3{CoCp[OP(OEt)2]3} contained diamagnetic Co(III) and paramagnetic Pu(IV).
Optical spectroscopy (UV-vis-NIR spectroscopy)
The single crystal UV-vis-NIR absorption spectrum from PuCl3{CoCp[OP(OEt)2]3} validated conclusions based on the magnetometry data and indicated that the compound contained Pu(IV) and Co(III) (Fig. 4). Confidence in the oxidation state assignments came through comparisons with UV-vis data we collected on the Na{CoCp[OP(OEt)2]3} precursor (see ESI†) and the aforementioned Pu(IV) standard, (Me4N)2PuCl6 (Fig. 4). The spectrum from PuCl3{CoCp[OP(OEt)2]3} was similar to PuCl62− in that both data sets contained absorbance features characteristic of Pu(IV), most notably between 10000 and 16000 cm−1 (600 to 1000 nm).56 Oxidation state purity for the PuCl3{CoCp[OP(OEt)2]3} sample was clear, as the UV-vis spectrum displayed no evidence of lower [Pu(III)] or higher (PuO2n+; n = 1, 2) oxidation state plutonium species.56,57 We inferred that PuCl3{CoCp[OP(OEt)2]3} must have Co(III) to maintain charge neutrality, which was also consistent with the magnetometry analyses described above.
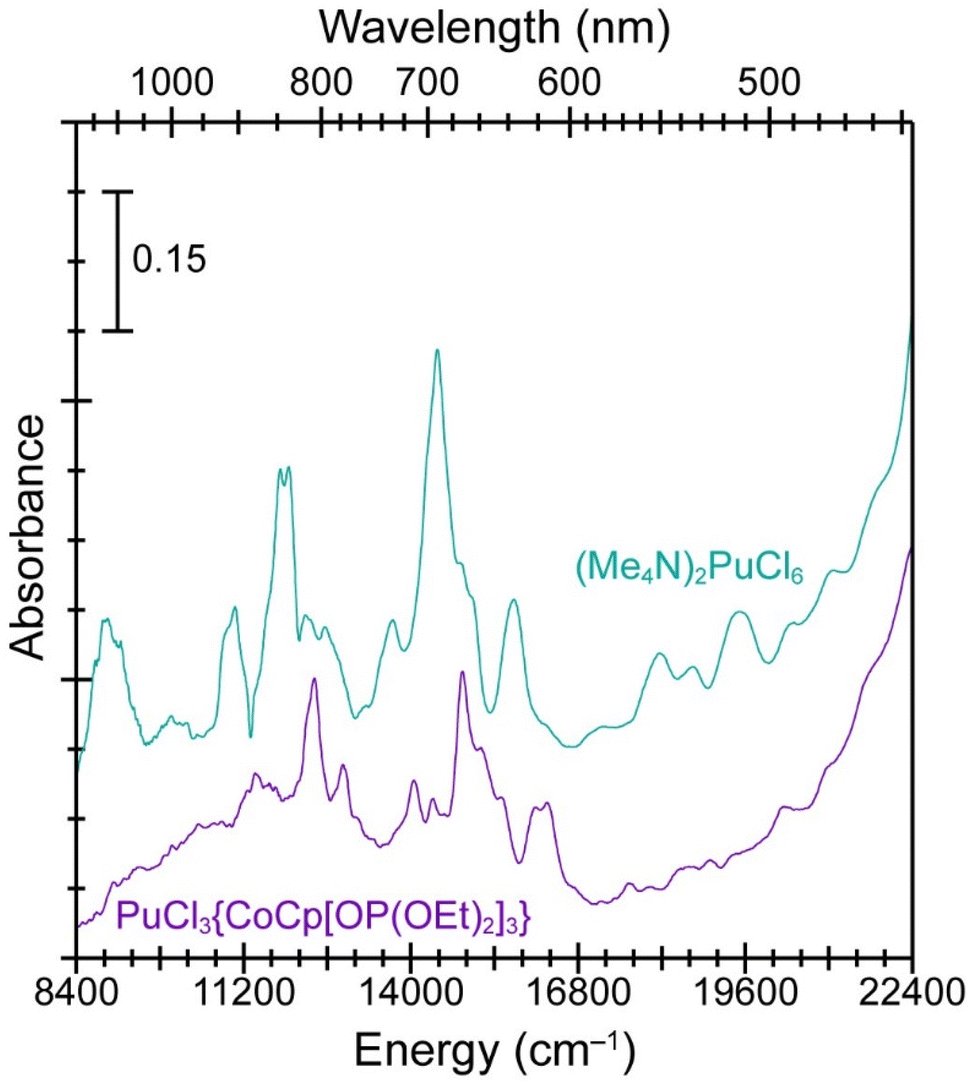 | ||
| Fig. 4 The solid-state UV-vis-NIR absorption spectra from PuCl3{CoCp[OP(OEt)2]3} (purple trace, bottom) vs. (Me4N)2PuCl6 (teal trace, top). | ||
Comparing single crystal UV-vis data from (Me4N)2PuCl6 with PuCl3{CoCp[OP(OEt)2]3} provided an experimental glimpse into how the Pu(IV) ligand field was impacted by changing from Pu–Cl to Pu–Ophosphite bonding interactions. For example, moving from a homoleptic and octahedral chloride ligand environment in PuCl62− to the heteroleptic and pseudo-octahedral environment in PuCl3{CoCp[OP(OEt)2]3} had a substantial impact on the Pu(IV) absorbance peak energies. We observed a hypsochromic energy shift for the PuCl3{CoCp[OP(OEt)2]3} absorbance peaks between 10000 and 16000 cm−1 (600 to 1000 nm) by approximately 430 cm−1vs. PuCl62−. A possible explanation was that substituting three Pu–Cl bonds in PuCl62− for three Pu–Ophosphite bonds in PuCl3{CoCp[OP(OEt)2]3} increased the valence orbital energy splitting. That change could increase the Pu(IV) 5f → 5f and 5f → 6d absorption transition energies between 10000 and 16000 cm−1 (600 to 1000 nm) and likely resulted from increased Pu ← Ophosphitevs. Pu ← Cl electron donation.
Electrochemistry
Further evidence that the Pu ← Ophosphite electron donation was larger than Pu ← Cl came from electrochemical analyses of PuCl3{CoCp[OP(OEt)2]3} vs. (Me4N)2PuCl6 (Fig. 5). These data were collected in acetonitrile solutions, with a [Bu4N][PF6] supporting electrolyte and using a standard three electrode cell (glassy carbon working, platinum counter, and platinum pseudo reference electrodes). Data were referenced to the FeCp21+/FeCp2 internal standard redox couple. Cyclic voltammograms from PuCl3{CoCp[OP(OEt)2]3} showed a single quasi-reversible wave attributable to the Pu(IV)/Pu(III) redox event with a half-potential (E½) of −0.33 V and an anodic to cathodic peak separation (ΔEp) of 0.163 V (scan rate = 0.2 V s−1). The Pu(IV)/Pu(III) redox event from PuCl3{CoCp[OP(OEt)2]3} was substantially shifted negatively (>0.3 V) in comparison to the Pu(IV)/Pu(III) redox event observed from PuCl62−: E½ was near zero (−0.04 V) and ΔEp equaled 0.095 V (scan rate = 0.2 V s−1). These data highlighted that the Pu–Ophosphite bond stabilized electron deficient and high-oxidation state Pu(IV) over electron rich and low-oxidation state Pu(III). Identifying ligand environments that stabilize Pu(IV) in organic solvents is special. Although there exist examples where Pu(IV) remains when complexed in organic media,58–63 many ligand scaffolds induce auto-reduction of Pu(IV) to Pu(III). Examples include – but are not limited to – dithiophosphinate,64 oximes,65–68 hydroxamic acids,69–72 hydroxylamines,73,74 and many others. In contrast, the data reported here showcased that bridging phosphite ligands in CoCp[OP(OEt)2]31− formed stable bonds with Pu(IV) in organic solvents, more so than the homoleptic Cl1− ligand environment within PuCl62−. At this time, it remains difficult to predict ligand sets that provide Pu(IV) stability vs. those that incite reduction of Pu(IV). We wonder if the Co(III) is contributing inductively in some way toward that Pu(IV) stability and are currently investigating ways to enhance electronic and magnetic communication pathways between the two metals in this system.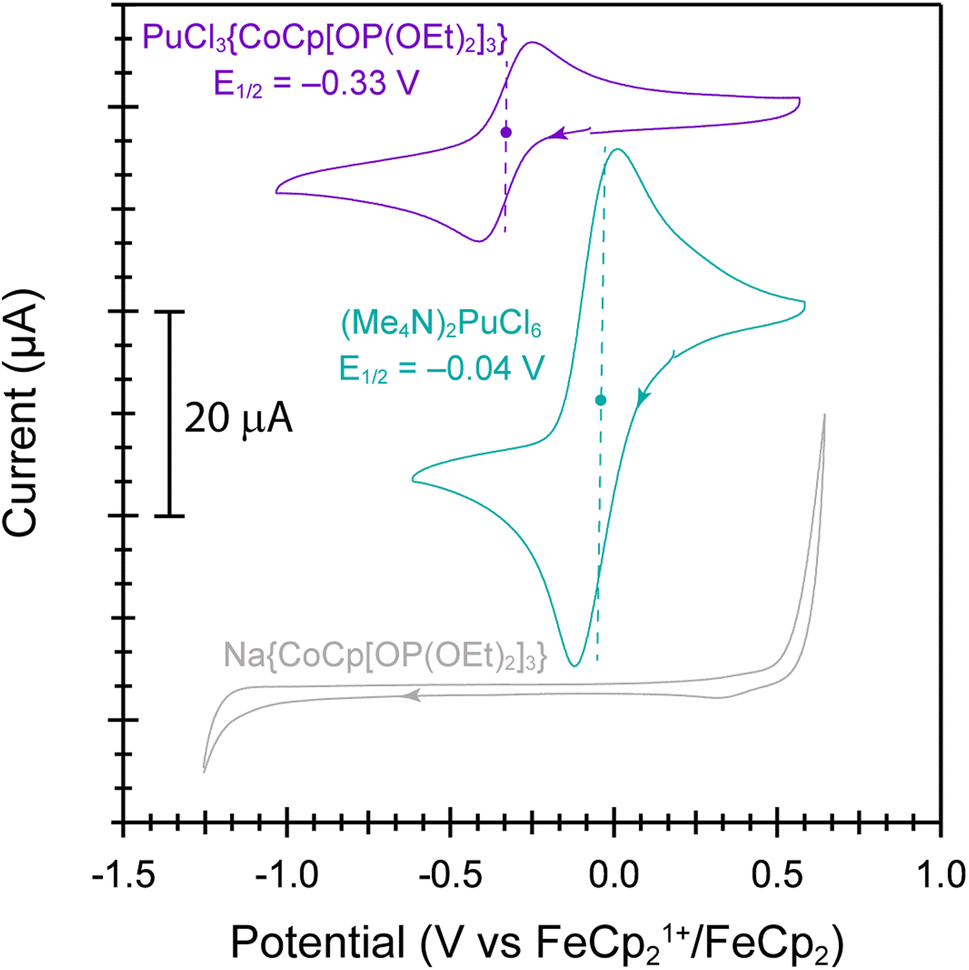 | ||
| Fig. 5 Cyclic voltammograms from acetonitrile solutions that contained PuCl3{CoCp[OP(OEt)2]3} (purple trace), (Me4N)2PuCl6 (teal trace), or Na{CoCp[OP(OEt)2]3} (gray trace) dissolved alongside [Bu4N][PF6] (0.1 M) in MeCN (2 mL). The dashed vertical lines and solid circles indicate the position of the reduction potential (E½) for the Pu4+/Pu3+ redox events. Arrows show the scanning direction. The working electrode was a glassy carbon disk electrode, the counter electrode was a platinum wire, and a separate platinum wire was used as the pseudo-reference electrode. The potentials were referenced internally to the FeCp21+/FeCp2 redox couple. Both voltammograms were collected at a scan rate of 0.200 V s−1. | ||
Outlook
We report the synthesis and characterization of the first heterobimetallic transuranic molecule. Large quantities of PuCl3{CoCp[OP(OEt)2]3} were prepared in modest, crystalline yield (70%) via the salt metathesis reaction between Na{CoCp[OP(OEt)2]3} and an acidic Pu4+(aq) residue suspended in THF. The synthetic procedure could be carried out under ambient conditions (without the exclusion of air and moisture) and on a large scale (hundreds of milligrams). As a result, appreciable amounts of PuCl3{CoCp[OP(OEt)2]3} were accessible for thorough characterization by X-ray diffraction, NMR, UV-vis-NIR spectroscopy, cyclic voltammetry, and magnetic susceptibility measurements. These data indicated that PuCl3{CoCp[OP(OEt)2]3} possessed two separate metals – Pu(IV) and Co(III) – and that the Pu(IV) oxidation state was stabilized in organic media. We assumed this stability was related to the highly electron donating phosphite ligands, but could not disregard the possibility a stabilizing inductive effect imparted by the Co(III) cation. Future efforts are focused on better understanding the electronic structure of PuCl3{CoCp[OP(OEt)2]3} and identifying ways to enhance Pu(IV) and Co(III) electronic and magnetic communication pathways. This PuCl3{CoCp[OP(OEt)2]3} heterobimetallic complex has potential to serve as a precursor for higher order multi-metallic species, which is currently being pursued. More broadly speaking, developing synthetic routes to plutonium heterometallic molecules will establish a novel platform for scientific exploration within the photophysical, electron transfer, and magnetic experimental spaces and holds potential for improving understanding of actinide electronic structure and bonding. Such developments could lead to new chemistries and unveil unique properties for plutonium that would otherwise be difficult to observe and characterize.Experimental
General consideration
All manipulations were carried out without exclusion of air and moisture. Optima grade aqueous solutions of nitric acid [HNO3(aq)] and hydrochloric acid [HCl(aq)] were obtained commercially (Fisher Scientific). Water was deionized and passed through a Barnstead water purification system until a resistivity of 18.2 MΩ cm−1 was achieved. Tetrahydrofuran (THF; Sigma Aldrich, anhydrous), Celite 545 (Fischer), dichloromethane (CH2Cl2; Sigma Aldrich), toluene (Sigma Aldrich), acetonitrile (MeCN; Sigma Aldrich), tetrabutylammonium hexafluorophosphate ([Bu4N][PF6], Sigma Aldrich), deuterated chloroform (CDCl3, Cambridge Isotopes), and heptane (Sigma Aldrich) were obtained commercially and used as received. The sodium (cyclopentadienyl)tris(diethylphosphito)cobalt(III) salt, Na(Cp)Co[OP(OEt)2]3, was prepared from bis(cyclopentadienyl)cobalt(II) and diethyl phosphite, as described previously.46 The tetramethyl ammonium plutonium(IV) hexachloride [(Me4N)2PuCl6]55 was prepared as previously described.The plutonium used in this study contained a blend of 238Pu [half-life (t½) = 87.7(1)y], 239Pu [t½ = 24110(30)y], 240Pu [t½ = 6561(7)y], 241Pu [t½ = 14.329(29)y], and 242Pu [t½ = 3.75(33) × 105y].75 Relative isotopic ratios by mass were approximately 0.015% for 238Pu, 95.65% for 239Pu, 6.1% for 240Pu, 0.224 for 241Pu, and 0.025% for 242Pu. Caution! These radionuclides and their progeny isotopes present serious health threats owing to their radioactive properties and α-, β-, and γ-emissions. Hence, all studies that involved manipulation of these isotopes were conducted in a radiation laboratory equipped with HEPA filtered hoods, continuous air monitors, negative pressure gloveboxes, and monitoring equipment appropriate for α-, β-, and γ-particle detection. Free-flowing solids were handled within negative pressure gloveboxes equipped with HEPA filters. These boxes were used to protect the worker from loose contamination and did not contain inert atmospheres. Entrance to the laboratory space was controlled with a hand and foot monitoring instrument for α-, β-, and γ-emitting isotopes and a full-body personnel contamination monitoring station.
Spectroscopic measurements: UV-vis-NIR, NMR
Single crystal absorption ultraviolet-visible-near infrared (UV-vis-NIR) measurements were made on single crystals dispersed on a quartz slide under oil using a Craic Technologies microspectrophotometer. Data were collected from 9091 to 40000 cm−1 (1100 nm to 250 nm). Solution-phase absorption UV-vis-NIR measurements were recorded on a Varian Cary 6000i spectrophotometer in screw cap quartz cuvettes. Nuclear magnetic resonance (NMR) experiments were conducted on a Bruker Avance III 400 MHz NMR spectrometer, with samples that had been doubly contained in a Teflon liner (as previously described).76 Spectra were processed using MestReNova v10.
Single crystal X-ray diffraction measurements
Single crystal X-ray diffraction studies were performed at room temperature (T = 288 K) on a Bruker Quest D8 diffractometer equipped with an IμS 3.0 microfocus source™ (Mo Kα radiation, λ = 0.71073 Å) and a CPAD Photon II™ area detector. Green, rectangular prisms of PuCl3{CoCp[OP(OEt)2]3} suitable for X-ray diffraction were reproducibly obtained from dichloromethane solutions layered with a toluene counter solvent in a 1:1 ratio. Single crystals of PuCl3{CoCp[OP(OEt)2]3} were mounted on MiTeGen Micromounts™ in mineral oil. Three layers of containment were employed to encapsulate the plutonium crystals, as has been previously described.47 Data were collected at room temperature to avoid icing imposed from the tertiary containment method required for safe handling of highly radioactive single crystal samples. Data was collected with the Apex3 software package using omega and phi scans.77 Frame integration, including Lorentz-polarization corrections, and final cell parameter calculations were carried out using SAINT software.77 A multi-scan absorption correction was applied to the data using the SADABS program.77 The structure was solved using Intrinsic Phasing methods. The structural refinement was performed using SHELXL78 within the ShelXle79 software interface. See Tables S1–S5† for crystallographic parameters. Figures were produced using VESTA and Olex2.80,81
All non-hydrogen atoms were located in the Fourier difference map and refined anisotropically, resulting in a structural model that consisted of a Pu(IV) bound to three chlorides and one tridentate CoCp[OP(OEt)2]31− anion per asymmetric unit. The chlorides, cyclopentadienyl ring, and all but one ethoxide group were disordered over two positions and their respective site occupancies allowed to refine freely. The disordered groups were refined with both positional and displacement restraints; the distances between pairs and groups of atoms were restrained to be similar, the atomic displacement parameters of disordered atoms near one another were refined to be similar, and several atoms were restrained to be approximately isotropic. Finally, the displacement parameters of bonded atoms within the entire structure were restrained to be alike. Hydrogen atoms were placed in calculated positions and their Ueq values were assigned values 1.2 times that of their parent atom. CCDC deposit number: 2323718.
Electrochemistry
A CH Instruments 620C potentiostat was used for all voltametric measurements. In a HEPA-filtered fume hood, a three-electrode cell was used, consisting of a glassy carbon disk working electrode (0.3 cm diameter), a platinum wire pseudo reference electrode (0.1 cm diameter), and a platinum-wire counter electrode (0.1 cm diameter). All electrodes were purchased from CH Instruments and the working electrode was cleaned and polished with a felt polishing pad and a mixture of alumina powder (0.05 μm) suspended in water. The cell consisted of a glass cylindrical body and a PTFE cap with holes to hold the electrodes in place. A small section of PTFE tubing (22 gauge) was inserted through a hole in the PTFE cap and used to flow argon into the cell. An electrolyte stock solution of [Bu4N][PF6] (0.1 M) was prepared in MeCN. For the voltametric measurements, an aliquot from the electrolyte solution (2 mL) was added to the cell and the solution was de-aerated with argon (purge 5 min). Argon was continuously flowed through the headspace of the cell during all subsequently described voltametric measurements. After characterizing the potential window for the electrolyte solution and confirming that no electron transfer processes occurred within that window, the electrolyte solution was replaced with either a green solution containing a few crystals of PuCl3{CoCp[OP(OEt)2]3} and (Bu4N)(PF6) (77 mg, 0.2 mmol) dissolved in MeCN (2 mL) or an orange solution containing a few crystals of (Me4)2PuCl6 and [Bu4N][PF6] (77 mg, 0.2 mmol) dissolved in MeCN (2 mL). Homogeneity was achieved by stirring and sparging the solution with argon (5 min) prior to collection of the cyclic voltammograms. The potentials from all data were referenced to the ferrocenium/ferrocene (FeCp21+/FeCp2) redox couple by adding ferrocene to the PuCl3{CoCp[OP(OEt)2]3} electrolyte solution after measurements were taken.Sample preparation for magnetometry
The magnetic susceptibility measurements were conducted on PuCl3{CoCp[OP(OEt)2]3} (120.9 mg, 0.150 mmol). The sample was weighed in a negative pressure glovebox used for radiological controls. Then, crystallites of the sample were loaded into a plastic soda straw and sandwiched between two plugs of quartz wool (99.6 mg). The top of the straw was equipped with a Delrin plug and the bottom of the straw was equipped with a Delrin plug that had a porous titanium frit (2 μm). Both plugs were sealed with Stycast (2850FT) epoxy. The holder provided a single layer of containment for the radioactive material and the frit enabled the helium gas (in the magnetometer) to contact the sample, which facilitated temperature equilibration during the magnetic susceptibility measurements. Magnetic susceptibility measurements were performed from 2.5 K to 300 K in a magnetic field of H = 5 T in a Quantum Design Magnetic Properties Measurement System-3. Data included a dominant contribution from the sample as well as a weak diamagnetic contribution from the quartz wool. The latter of which was well defined – as it had been measured independently – and was subtracted from the total susceptibility. The molar susceptibility in units of emu/mol is given by: χM = NA(gμB)2J(J + 1)/3kBT, where NA is Avagadro's number, g is the Lande g-factor, J is total electronic momentum, and kB is Boltzmann's constant. In c.g.s. units (which gives χM in units of emu mol−1), 3kB/NAμB2 = 8, so χMT = p2/8 = C, where p is the effective magnetic moment p = g[J(J + 1)]1/2 and C is the Curie constant. The Lande factor g = 3/2 + ½[S(S + 1) − L(L + 1)]/[J(J + 1)], where S is the spin quantum number and L is the angular momentum quantum number. By Hund's rules, the Curie constant is, for f4: S = 2, L = 6 and J = 4, then g = 0.6 and p = 2.68; χMT = (p2/8) = 0.9 emu K mol−1.Preparation of the Pu4+(aq) stock solution
Plutonium was obtained as a residue used previously in other research campaigns. This residue was dissolved in a small volume of HNO3(aq) (5 to 10 mL; 16 M), and subsequently heated on a hot plate under a stream of filtered flowing air until a soft dryness was reached. The resulting residue was dissolved again in HNO3(aq) (5 to 10 mL; 16 M) and the process was repeated two more times. This plutonium nitrate residue was purified as described previously.47,55 At a high-level, this two-step purification involved (1st) precipitating the Pu3+(aq) using HF(aq) (28 M) and (2nd) purification by anion exchange chromatography. On occasion we have observed small amounts of plutonium reduction on the anion exchange resin that generates a small amount of Pu3+(aq). When this occurred, the blue Pu3+(aq) contaminant was separated easily from the reddish-orange Pu4+(aq) product because Pu3+(aq) passes through the anion exchange column at a faster rate than Pu4+(aq) during the elution process. For example, passing slightly acidic HCl(aq) (50 mL of H2O to five drops of HCl(aq); 12 M) through the column pushed the blue Pu3+(aq) fraction off the column first. Then, the reddish-orange Pu4+(aq) fraction was independently isolated. The eluate HCl(aq) concentration was then adjusted to approximately 6 M by doubling the Pu4+(aq) eluate solution volume and adding HCl(aq) (12 M). For example, 7 mL of HCl(aq) (12 M) was added if the eluted Pu4+(aq) fraction was 7 mL in volume. An aliquot (100 μL) of the final stock solution was added to a quartz cuvette charged with HClO4(aq) (1 M, 1 mL), and assayed by UV-vis-NIR absorption spectroscopy. The absorption peak at 21277 cm−1 (470 nm; ε470nm = 56.5 L mol−1 cm−1) was used to quantify the concentration of the Pu4+(aq) stock solution, which was found to be 0.127 M.
Plutonium(IV) trichloride (cyclopentadienyl)tris(diethylphosphito)cobalt(III), PuCl3{CoCp[OP(OEt)2]3}
Under ambient conditions and with no efforts to exclude air or moisture, a polyethylene falcon cone (50 mL) was charged with an aliquot of the aforementioned Pu4+(aq) stock solution (0.127 M in Pu4+, 151.2 mg, 0.633 mmol) in HCl(aq) (5 mL, 6 M). The solution was heated on a hot plate under a stream of filtered flowing air until a soft dryness was achieved. The resulting residue was suspended in THF (7.5 mL). Most of the Pu4+(aq) residue dissolved. The resulting red slurry was transferred into a Pyrex falcon cone (50 mL) that contained an orange solution of Na{CoCp[OP(OEt)2]3} (340.0 mg, 0.610 mmol) dissolved in THF (7.5 mL). Combining the plutonium mixture and the Na{CoCp[OP(OEt)2]3} solutions resulted in a homogenous brown solution. A white solid, presumably sodium chloride (NaCl), precipitated as the solution stirred overnight. Insoluble byproducts were removed by passing the solution through a filter stick charged with Celite. The eluent was exposed to a stream of filtered air until a soft dryness was achieved. The majority of the resulting residue dissolved in CH2Cl2 (5 mL) however, a stubborn white solid (likely NaCl) lingered. Removal of this persistent precipitate was achieved only after the mixture was filtered through two additional Celite columns. The resulting homogenous dark green solution was subsequently evaporated to soft dryness at room temperature under a stream of filtered air, which left a green solid. This solid was dissolved in a small amount of CH2Cl2 (5 mL) and layered with toluene (5 mL). Dark green and rectangular single crystals of PuCl3{CoCp[OP(OEt)2]3} formed after this solution sat at room temperature for 48 h. After the mother liquor was decanted, the crystals were washed with heptane, and dried under air. This enabled the title compound, PuCl3{CoCp[OP(OEt)2]3}, to be isolated (0.3735 g, 0.4619 mmol) in 70% crystalline yield; based on Na{CoCp[OP(OEt)2]3}.1H NMR, (CDCl3, 20 °C, 400.13 MHz): δ 6.71 ppm (s, 5H); δ 3.03 (dp, J = 91, 7.8 Hz, 12H); δ 0.73 ppm (t, J = 6.9 Hz, 18H). 31P (CDCl3, 20 °C, 202.46 MHz): δ 24.5 ppm (s, 1P).
UV-vis-NIR (cm−1): 12406.95, 12866.60; 14064.70 sh; 14388.49 sh; 14880.95 sh; 163313.21 sh; 16077.17 sh; 22123.89 CT.
Data availability
Crystallographic data for PuCl3{CoCp[OP(OEt)2]3} has been deposited at the CCDC (deposit number: 2323718). The rest of the data supporting this article have been included as part of the main text or within the ESI.†Author contributions
All of the authors contributed to writing, reviewing, and editing the document. Thomas E. Shaw conducted electrochemical studies and formal analyses. Zachary R. Jones conducted the initial synthetic explorations and project design. Sara L. Adelman conducted experiment design. Nickolas H. Anderson contributed to synthesis and experimental design. Eric G. Bowes helped conduct the plutonium synthesis and sample preparation. Eric D. Bauer led the magnetic measurements. David Dan conducted magnetic measurements. Jan Klouda conducted electrochemical studies. Karah E. Knope was the contributed to structural analysis and experimental design. Stosh A. Kozimor provided experimental design, conceptualization, formal analyses, plutonium reprocessing. Molly M. MacInnes conducted electrochemical experiments. Veronika Mocko conducted electrochemical measurements. Francisca R. Rocha conducted electrochemical measurements. Harrison D. Root conducted synthetic experimental studies and NMR studies. Benjamin W. Stein oversaw the spectroscopic measurements. Joe D. Thompson conducted magnetic measurements. Jennifer N. Wacker conducted crystallographic measurements and solved the crystal structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry program (2020LANLE372) for funding the majority of this work. Additional support came from LANL's LDRD projects for electrochemical measurements (20220054DR and 20190154ER) and magnetic measurements (20200045ER). We thank the Glenn T. Seaborg Institute (Jones), the Natural Sciences and Engineering Research Council of Canada Postdoctoral Fellowship (Bowes), and the DOE, Office Of Science, Office of Workforce Development for Teachers and Scientists, Office of Science Graduate Student Research (SCGSR) program, which is administered by the Oak Ridge Institute for Science and Education (ORISE) and managed by ORAU for the DOE (contract number DE-SC0014664) (Wacker) for graduate student funding. KEK was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Early Career Research Program under Award DE-SC0019190. LANL is an affirmative action/equal opportunity employer managed by Triad National Security, LLC, for the National Nuclear Security Administration of the U.S. DOE.References
- J. Campos, Nat. Rev. Chem., 2020, 4, 696 CrossRef CAS PubMed.
- F.-F. Chen, Z.-Q. Chen, Z.-Q. Bian and C.-H. Huang, Coord. Chem. Rev., 2010, 254, 991 CrossRef CAS.
- T. Lathion, A. Fürstenberg, C. Besnard, A. Hauser, A. Bousseksou and C. Piguet, Inorg. Chem., 2020, 59, 1091 CrossRef CAS PubMed.
- P. Sharma, D. R. Pahls, B. L. Ramirez, C. C. Lu and L. Gagliardi, Inorg. Chem., 2019, 58, 10139 CrossRef CAS PubMed.
- J. Shen, T. Rajeshkumar, G. Feng, Y. Zhao, S. Wang, L. Maron and C. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202303379 CrossRef CAS PubMed.
- G. Feng, M. Zhang, P. Wang, S. Wang, L. Maron and C. Zhu, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 17645 Search PubMed.
- J. A. Hlina, J. R. Pankhurst, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2016, 138, 3333 CrossRef CAS.
- R. S. Sternal, C. P. Brock and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 8270 CrossRef CAS.
- A. J. Ayres, A. J. Wooles, M. Zegke, F. Tuna and S. T. Liddle, Inorg. Chem., 2019, 58, 13077 CrossRef CAS PubMed.
- A. L. Ward, W. W. Lukens, C. C. Lu and J. Arnold, J. Am. Chem. Soc., 2014, 136, 3647 CrossRef CAS PubMed.
- C. Z. Ye, I. D. Rosal, M. A. Boreen, E. T. Ouellette, D. R. Russo, L. Maron, J. Arnold and C. Camp, Chem. Sci., 2022, 14, 861 RSC.
- A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 8826 CrossRef CAS.
- M. Carmona, R. Pérez, J. Ferrer, R. Rodríguez, V. Passarelli, F. J. Lahoz, P. García-Orduña and D. Carmona, Inorg. Chem., 2022, 61, 13149 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002 CrossRef CAS.
- I. Čorić and P. L. Holland, J. Am. Chem. Soc., 2016, 138, 7200 CrossRef.
- B. H. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041 CAS.
- T. Le Borgne, E. Rivière, J. Marrot, J.-J. Girerd and M. Ephritikhine, Angew. Chem., Int. Ed., 2000, 39, 1647 CrossRef CAS.
- D. Patel, F. Moro, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 10388 CrossRef CAS PubMed.
- C. Camp, D. Toniolo, J. Andrez, J. Pécaut and M. Mazzanti, Dalton Trans., 2017, 46, 11145 RSC.
- B. M. Gardner, J. McMaster, F. Moro, W. Lewis, A. J Blake and S. T. Liddle, Chem.–Eur. J., 2011, 17, 6909 CrossRef CAS.
- B. M. Gardner, D. Patel, A. D. Cornish, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Chem.–Eur. J., 2011, 17, 11266 CrossRef CAS PubMed.
- V. Mougel, L. Chatelain, J. Pécaut, R. Caciuffo, E. Colineau, J.-C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011 CrossRef CAS.
- L. Cahtelain, J. P. S. Walsh, J. Pécaut, F. Tuna and M. Mazzanti, Angew. Chem., Int. Ed., 2014, 53, 13434 CrossRef PubMed.
- P. Yang, E. Zhou, G. Hou, G. Zi, W. Ding and M. D. Walter, Chem.–Eur. J., 2016, 22, 13845 CrossRef CAS.
- E. Lu, A. J. Wooles, M. Gregson, P. J. Cobb and S. T. Liddle, Angew. Chem., Int. Ed., 2018, 57, 6587 CrossRef CAS.
- M. A. Boreen, T. D. Lohrey, G. Rao., D. Britt, L. Maron and J. Arnold, J. Am. Chem. Soc., 2019, 141, 5144 CrossRef CAS.
- O. Kahn, Adv. Inorg. Chem., 1995, 43, 179 CrossRef CAS.
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev., 2012, 54, 1 CrossRef CAS.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28 CrossRef CAS.
- S. Sinhababu, Y. Lakliang and N. P. Mankad, Dalton Trans., 2022, 51, 6129 RSC.
- A. Lachguar, A. V. Pichugov, T. Neumann, Z. Dubrawski and C. Camp, Dalton Trans., 2024, 53, 1393 CAS.
- N. J. Curro, T. Caldwell, E. D. Bauer, L. A. Morales, M. J. Graf, Y. Bang, A. V. Balatsky, J. D. Thompson and J. L. Sarrao, Nature, 2005, 434, 622 CAS.
- N. Budantseva, G. Andreev, M. Sokolova and A. Fedoseev, Inorg. Chem., 2021, 60, 18395 CrossRef CAS PubMed.
- C. Tamain, B. A. Chapelet, M. Rivenet, F. Abraham, R. Caraballo and S. Grandjean, Inorg. Chem., 2013, 52, 4941 CrossRef CAS.
- J. N. Cross, P. M. Duncan, E. M. Villa, E. V. Alekseev, C. H. Booth and T. E. Albrecht-Schmitt, J. Am. Chem. Soc., 2013, 135, 2769 CrossRef CAS.
- W. Kläui, H. O. Asbahr, G. Schramm and U. Englert, Chem. Ber./Recl, 1997, 130, 1223 CrossRef.
- I. Goldberg, H. Shinar, G. Navon and W. Kläui, J. Incl. Phenom., 1987, 5, 181 CrossRef CAS.
- H. Shinar, G. Navon and W. Kläui, J. Am. Chem. Soc., 1986, 108, 5005 CrossRef CAS.
- H. Werner, H. Neukomm and W. Kläui, Helv. Chim. Acta, 1977, 60, 326 CrossRef CAS.
- M. Glaum, W. Kläui, B. W. Skelton and A. H. White, Aust. J. Chem., 1997, 50, 1047 CrossRef CAS.
- W. Kläui and T. Hardt, J. Organomet. Chem., 1998, 553, 241 CrossRef.
- W. Kläui, M. Glaum, E. Hahn and T. Lugger, Eur. J. Inorg. Chem., 2000, 21 CrossRef.
- W. Kläui, W. Peters, N. Liedtke, S. Trofimenko, A. L. Rheingold and R. D. Sommer, Eur. J. Inorg. Chem., 2001, 693 CrossRef.
- W. Kläui, N. Mocigemba, A. Weber-Schuster, R. Bell, W. Frank, D. Mootz, W. Poll and H. Wunderlich, Chem.–Eur. J., 2002, 8, 2335 CrossRef.
- W. Kläui and P. C. Kunz, Inorg. Synth., 2010, 35, 125 Search PubMed.
- W. Kläui, Z. Naturforsch., 1979, 34B, 1403 CrossRef.
- S. K. Cary, K. S. Bowland, J. N. Cross, S. A. Kozimor and B. L. Scott, Polyhedron, 2017, 126, 220 CrossRef CAS.
- G.-C. Wang, Y.-M. So, K.-L. Wong, K.-C. Au-Yeung, H. H.-Y. Sung, I. D. Williams and W.-H. Leung, Chem.–Eur. J., 2015, 21, 16126 CrossRef CAS PubMed.
- M. Wedler, J. W. Gilje, M. Noltemeyer and F. T. Edelmann, J. Organomet. Chem., 1991, 411, 271 CrossRef CAS.
- D. Baudry, M. Ephritikhine, W. Kläui, M. Lance, M. Nierlich and J. Vigner, Inorg. Chem., 1991, 30, 2333 CrossRef CAS.
- T. C. H. Lam, E. Y. Y. Chan, W.-L. Mak, S. M. F. Lo, I. D. Williams, W.-T. Wong and W.-H. Leung, Inorg. Chem., 2003, 42, 1842 CrossRef CAS PubMed.
- A. C. Grunwald, C. Scholtysik, A. Hagenbach and U. Abram, Inorg. Chem., 2020, 59, 9396 CrossRef CAS PubMed.
- J. W. Napoline, S. J. Kraft, E. M. Matson, P. E. Fanwick, S. C. Bart and C. M. Thomas, Inorg. Chem., 2013, 52, 12170 CrossRef CAS.
- W. Kläui, D. Matt, F. Balegroune and D. Grandjean, Acta. Cryst., 1991, C47, 1614 Search PubMed.
- A. M. Mounce, H. Yasuoka, G. Koutroulakis, J. A. Lee, H. Cho, F. Gendron, E. Zurek, B. L. Scott, J. A. Trujillo, A. K. Slemmons, J. N. Cross, J. D. Thompson, S. A. Kozimor, E. D. Bauer, J. Autschback and D. L. Clark, Inorg. Chem., 2016, 55, 8371 CrossRef CAS PubMed.
- R. E. Wilson, Y.-J. Hu and H. Nitsche, Radiochim. Acta, 2005, 93, 203 CrossRef CAS.
- M. R. Antonio, C. W. Williams, J. A. Sullivan, S. Skanthakumar, Y.-J. Hu and L. Soderholm, Inorg. Chem., 2012, 51, 5274 CAS.
- C. J. Windorff, C. A. P. Goodwin, J. M. Sperling, T. E. Albrecht-Schönzart, Z. Bai, W. J. Evans, Z. K. Huffman, R. Jeannin, B. N. Long, D. P. Mills, T. N. Poe and J. W. Ziller, Inorg. Chem., 2023, 62, 18136 CAS.
- J. Su, T. Cheisson, A. McSkimming, C. A. P. Goodwin, I. M. DiMucci, T. Albrecht-Schönzart, B. L. Scott, E. R. Batista, A. J. Gaunt, S. A. Kozimor, P. Yang and E. J. Schelter, Chem. Sci., 2021, 12, 13343 RSC.
- C. Berger, C. Marie, D. Guillaumont, C. Tamain, T. Dumas, T. Dirks, N. Boubals, E. Acher, M. Laszczyk and L. Berthon, Inorg. Chem., 2020, 59, 1823 CrossRef CAS.
- D. Lemire, T. Dumas, C. Marie, F. Giusti, G. Arrachart, S. Dourdain and S. Pellet-Rostaing, Eur. J. Inorg. Chem., 2023, 26, e202300461 CrossRef CAS.
- L. Yao, W. Junli, L. Yuhang, W. Hui, W. Wentao, L. Baole and Y. Taihong, RSC Adv., 2024, 14, 560 RSC.
- C. Berger, E. Moreau, C. Marie, D. Guillaumont, A. Beillard and L. Berthon, Solvent Extr. Ion Exch., 2021, 40, 290 CrossRef.
- J. A. Macor, J. L. Brown, J. N. Cross, S. R. Daly, A. J. Gaunt, G. S. Girolami, M. T. Janicke, S. A. Kozimor, M. P. Neu, A. C. Olson, S. D. Reilly and B. L. Scott, Dalton Trans., 2015, 44, 18923 RSC.
- X.-B. Li, Q.-Y. Wu, C.-Z. Wang, J.-H. Lan, M. Zhang, Z.-F. Chai and W.-Q. Shi, J. Phys. Chem. A, 2023, 127, 7479 CrossRef CAS.
- V. S. Koltunov, R. J. Taylor, S. M. Baranov, E. A. Mezhov, V. G. Pastushchak and I. May, Radiochim. Acta, 2000, 88, 65 CrossRef CAS.
- V. S. Koltunov, V. I. Marchenko, G. I. Zhuravleva and O. A. Savilova, Radiochemistry, 2001, 43, 334 CrossRef CAS.
- V. S. Koltunov, S. M. Baranov and V. G. Pastushchak, Radiochemistry, 2001, 43, 346 CrossRef CAS.
- P. Tkac, M. Precek and A. Pulenova, Proc. Global, 2009, 6 Search PubMed.
- P. Tkac, M. Precek and A. Paulenova, Inorg. Chem., 2009, 48, 11935 CrossRef CAS PubMed.
- R. J. Taylor and I. May, Czech J. Phys., 1999, 49, 617 CrossRef CAS.
- R. J. Taylor, C. Mason, R. Cooke and C. Boxall, J. Nucl. Sci. Technol., 2002, 39, 278 CrossRef.
- V. I. Marchenko, K. N. Dvoeglazov, O. A. Savilova and V. I. Volk, Radiochemistry, 2012, 54, 459 CrossRef CAS.
- S. L. Yarbro, S. B. Schreiber, E. M. Ortiz and R. L. Ames, J. Radioanal. Nucl. Chem., 1998, 235, 21 CrossRef CAS.
- National Nuclear Data Center and Brookhaven National Laboratory, 2021, accessed April 21, 2021. https://www.nndc.bnl.gov/nudat2/.
- B. W. Stein, S. A. Kozimor and V. Mocko, The Plutonium Handbook, ed. D. L Clark, D. A. Geeson and R. J. Hanrahan Jr, American Nuclear Society, La Grange Park, Illinois, 2nd Edition, 2019, p. 2951 Search PubMed.
- APEX3, SADABS, SAINT, SHELXTL, XCIF, and XPREP, Bruker AXS, Inc., Madison, WI, 2016.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- O. Dolomanov, L. Bourhis, R. Gildea, J. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Structural metrics and additional 1H NMR spectra. CCDC 2323718. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01767f |
| ‡ Contributed equally and are joint first authors. |
| This journal is © The Royal Society of Chemistry 2024 |