
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent carbene–copper(I)–amide polymers for efficient host-free solution-processed OLEDs†
Yao
Tan
,
Ao
Ying
,
Jianlong
Xie
,
Guohua
Xie
and
Shaolong
Gong
 *
*
College of Chemistry and Molecular Sciences, Hubei Key Laboratory on Organic and Polymeric Optoelectronic Materials, Wuhan University, Wuhan 430072, China. E-mail: slgong@whu.edu.cn
First published on 17th June 2024
Abstract
Luminescent metallopolymers have attracted broad interest in the fields of healthcare and organic electronics. However, polymeric emitters based on earth-abundant metal complexes are scarce. Here, two series of Cu(I) polymers, PMAC-x and PCAAC-x (x = 1–3) have been developed using two kinds of Cu(I)-based carbene–metal–amide (CMA) complexes as side-chain emitter units to combine with a nonconjugated polystyrene backbone. These Cu(I) polymers emit via distinct thermally activated delayed fluorescence or dominant phosphorescence, inherited from the grafted Cu(I)-based CMA units. Particularly, the PMAC-x polymers exhibit high photoluminescence quantum efficiencies of up to 0.78, short emission lifetimes of down to 0.66 μs, and fast radiative rates of up to 106 s−1 in neat films. Thanks to the good encapsulation effect of the polystyrene backbone, these Cu(I) polymers not only demonstrate favorable moisture stability but also show significant aggregation-induced emission. The resultant host-free solution-processed organic light-emitting diodes (OLEDs) achieve outstanding electroluminescence performance with a record external quantum efficiency of 13.8% at a practical luminance of ∼100 nits, representing state-of-the-art device efficiency for metallopolymer-based OLEDs. This work not only presents the first example of CMA polymers but also provides the future direction of polymeric emitters from earth-abundant metal complexes for the OLED application.
Introduction
Since the first metal-containing polymer was reported as early as 1955, metallopolymers have attracted paramount attention in the chemistry and materials science fields, due to their broad-range applications in catalysis, healthcare, and semiconductors.1–7 Amongst them, luminescent noble-metal polymers usually have the combined advantages of phosphorescent metal emitters and polymeric materials in delivering efficient phosphorescence properties, good solubility, and excellent processability simultaneously.8–11 Therefore, luminescent noble-metal polymers have been widely used as emitters for solution-processed organic light-emitting diodes (OLEDs). The first example of a phosphorescent iridium(III) polymer was reported by Kim et al. in 2003 (Fig. 1A), which incorporated side-chain carbazole and iridium(III) complex units into a polyethylene backbone.12 The resultant OLED emitted a green light with a maximum external quantum efficiency (EQE) of 4.4%. Afterwards, various phosphorescent metallopolymers containing Ir(III) or Pt(II) emissive cores were developed.13–16 In 2013, Lee and co-workers developed a series of polynorbornene polymers with side-chain Ir(III) emitters and host moieties.17 The respective non-doped solution-processed OLEDs exhibited high EQEs of up to 13.3% (Fig. 1A). Similarly, a handful of metallopolymer emitters based on rare-earth metals such as Eu have been developed as well, yet their electroluminescence (EL) efficiencies are unsatisfactory due to their low photoluminescence quantum yields (ΦPL).18–21 Despite the promising results, the introduction of noble or rare-earth metals inevitably brings out resource and environmental issues, impeding the potential commercialization of luminescent metallopolymers. In this sense, first-row transition metal complexes such as copper(I) and zinc(II) complexes are optional earth-abundant candidates for the construction of luminescent metallopolymers. Nonetheless, this kind of metallopolymer based on earth-abundant metals is rarely exploited and normally exhibit poor EL efficiencies (Fig. 1A).22–24 In this context, the search for new classes of efficient polymeric emitters based on earth-abundant metals is of vital importance for the further development of metallopolymer-based OLEDs. | ||
| Fig. 1 (A) Selected examples of luminescent metallopolymers in the literature. (B) Two series of Cu(I) polymers in this work. | ||
Luminescent Cu(I) complexes have long been appealing candidates in replacing noble-metal complexes, due to high earth abundance and the absence of low-lying metal-centered states.25–27 The long-term focus has been on four- or three-coordinate Cu(I) complexes. Most of these complexes, however, undergo obvious structure distortion in excited states, limiting their emission efficiencies.28,29 The recent discovery of two-coordinate carbene–metal–amide (CMA) complexes has inspired the rapid development of two-coordinate Cu(I) complexes.30–32 This new kind of Cu(I)–CMA complex characterizes linear structures to suppress conformation deformation upon excitation (Jahn–Taller effect), giving rise to excellent photoluminescence efficiency. Meanwhile, most of the Cu(I)–CMA complexes emit via thermally activated delayed fluorescence (TADF) with short emission lifetimes, originating from the low-lying excited states with dominant ligand–ligand charge transfer (LLCT) nature and minor metal–ligand charge transfer character. 33,34 With these unique emission features, two-coordinate Cu(I)–CMA complexes become the mainstream focus towards the OLED application.35–37 Despite the promising development, Cu(I)–CMA polymers, that show great potential in solution-processed OLEDs, have not been reported to date.
For the present study, we conceive that the integration of Cu(I)–CMA complexes into polymers is a promising approach for constructing high-efficiency and cost-effective metallopolymer emitters. To prove this concept, we have successfully constructed two panels of nonconjugated Cu(I) polymers, PMAC-x and PCAAC-x (x = 1–3), using a side-chain engineering strategy on a polystyrene backbone. Pendant Cu(I)–CMA complexes and 9-phenyl-9H-carbazole (PhCz) moieties were employed as guest and host units, respectively (Fig. 1B). These Cu(I) polymers exhibited sky-blue phosphorescence or yellow TADF emission, originating from different Cu(I)–CMA guest units. In particular, the PMAC-x polymers exhibited a high ΦPL of up to 0.78 and large radiative rate constants (kr) of up to 106 s−1 in neat films. Intriguingly, in contrast to the prototypical Cu(I)–CMA complexes, these Cu(I) polymers afforded superb moisture stability and significant aggregation-induced emission (AIE) properties, mainly due to the good encapsulation effect from the polystyrene backbone. Host-free solution-processed OLEDs based on these Cu(I) polymers delivered promising EQEs of up to 13.8% at a luminance of ∼100 nits, which is the champion value for OLEDs based on metallopolymers. Moreover, the optimal OLED maintained a high EQE of 10.0% together with a slow efficiency roll-off value of 27.5% at a practical luminance of ∼1000 nits.
Results and discussion
Two series of nonconjugated Cu(I) polymers, PMAC-x and PCAAC-x (x = 1–3), were prepared by radical copolymerization employing styrene-substituted carbazole as the host unit and two classes of styrene-substituted Cu(I)–CMA monomers (MAC–Cu–PhCz and CAAC–Cu–PhCz) as guest units at different host/guest molar feed ratios of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 90:10, and 85:15 (Scheme S3, ESI†), respectively. The synthetic routes of all the styrene-substituted monomers can be found in the ESI (Schemes S1 and S2, ESI†). The structures of the key Cu(I)–CMA monomers and the target Cu(I) polymers were confirmed by 1H NMR and 13C NMR spectra as well as elemental analysis (Fig. S27–S44, ESI†). These Cu(I) polymers exhibited relatively high weight-average molecular weights (Mw) of 29.0–56.4 kDa with relatively narrow polydispersity indices of 2.07–2.19, indicating that different types and concentrations of Cu(I)–CMA monomers had a small influence on radical polymerization (Table S1, ESI†). Atomic emission spectroscopy measurements demonstrated that the actual Cu element contents of these Cu(I) polymers were very similar to the calculated values from the molar feed ratios, verifying that Cu(I)–CMA monomers were effectively copolymerized at different feed ratios (Table S1, ESI†). In addition, high molecular weights endowed all the Cu(I) polymers with high thermal decomposition temperatures (Td) of >368 °C (Fig. S2 and Table S1, ESI†). Moreover, all the Cu(I) polymers exhibited distinct glass transition temperatures (Tg) ranging from 211 to 227 °C. Furthermore, these Cu(I) polymers exhibited favourable solubility in common organic solvents, such as dichloromethane, tetrahydrofuran, and chlorobenzene. Therefore, these Cu(I) polymers are feasible candidates as neat-film emitters for solution-processed OLEDs.
:5, 90:10, and 85:15 (Scheme S3, ESI†), respectively. The synthetic routes of all the styrene-substituted monomers can be found in the ESI (Schemes S1 and S2, ESI†). The structures of the key Cu(I)–CMA monomers and the target Cu(I) polymers were confirmed by 1H NMR and 13C NMR spectra as well as elemental analysis (Fig. S27–S44, ESI†). These Cu(I) polymers exhibited relatively high weight-average molecular weights (Mw) of 29.0–56.4 kDa with relatively narrow polydispersity indices of 2.07–2.19, indicating that different types and concentrations of Cu(I)–CMA monomers had a small influence on radical polymerization (Table S1, ESI†). Atomic emission spectroscopy measurements demonstrated that the actual Cu element contents of these Cu(I) polymers were very similar to the calculated values from the molar feed ratios, verifying that Cu(I)–CMA monomers were effectively copolymerized at different feed ratios (Table S1, ESI†). In addition, high molecular weights endowed all the Cu(I) polymers with high thermal decomposition temperatures (Td) of >368 °C (Fig. S2 and Table S1, ESI†). Moreover, all the Cu(I) polymers exhibited distinct glass transition temperatures (Tg) ranging from 211 to 227 °C. Furthermore, these Cu(I) polymers exhibited favourable solubility in common organic solvents, such as dichloromethane, tetrahydrofuran, and chlorobenzene. Therefore, these Cu(I) polymers are feasible candidates as neat-film emitters for solution-processed OLEDs.
To evaluate the electronic structures and the related optical properties of these Cu(I) polymers, the density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were conducted on the corresponding oligomers (PMAC and PCAAC) containing one guest and three host units. The highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) are predominantly located on the amide and carbene ligands of the Cu(I)–CMA moieties of the oligomers (Fig. S4, ESI†), respectively. This result is in accordance with those of the reported prototypical Cu(I)–CMA complexes.29,30 The same amide ligands supported these two oligomers having similar HOMO values of about −4.30 eV. Due to the weaker electrophilic ability of the CAAC ligand, the LUMO value of the PCAAC oligomer (−1.61 eV) was slightly shallower than that of the PMAC oligomer (−1.88 eV). Consequently, the S1/T1 energy levels (2.49/2.29 eV) of the PCAAC oligomer were slightly higher than those of the PMAC oligomer (2.21/2.05 eV) (Fig. S5, ESI†). It is noteworthy that in the optimized S1 configurations, both the Cu(I) oligomers took a perpendicular orientation between the carbene and amide ligands of the Cu(I)–CMA units, different from the coplanar orientation obtained based on the optimized S0 structures. Larger spatial separations of frontier molecular orbital (FMO) distributions (Fig. S5, ESI†) were observed and thus the ΔEST values decreased to 0.02 eV for both Cu(I) oligomers in the perpendicular configuration. In this regard, the theoretical ΔEST values are in the range of 0.02–0.16 and 0.02–0.20 eV for the PMAC and PCAAC oligomers, respectively. Additionally, according to the natural transition orbital analysis of S1/T1 excited states for the two oligomers, electrons and holes were mainly located on the carbene and amide ligands of the Cu(I)–CMA moieties, respectively, illustrating the obvious LLCT nature of their excited states (Fig. S6, ESI†). Moreover, cyclic voltammetry measurements were performed to study the HOMO and LUMO energy levels of the Cu(I) polymers. As shown in Fig. S7 (ESI†), the LUMO energy levels of PMAC-x were calculated to be about −2.75 eV, which were significantly deeper than those of PCAAC-x (ca. −2.58 eV). This measured LUMO trend is in line with the theoretically calculated results.
The UV/visible absorption spectra of the Cu(I) polymers in dilute toluene solution (1 × 10−4 M) and neat films are shown in Fig. 2A and S9 (ESI†). All the Cu(I) polymers demonstrated intense, vibronically structured absorption bands (<350 nm) in toluene solution, which were assigned to π–π* transitions of the host moieties according to the absorption spectra of the homopolymer (PPhCz) based on PhCz (Fig. S8B, ESI†). The additional broad and weak absorption bands or tails at approximately 425 nm for PMAC-x and 380 nm for PCAAC-x were ascribed to the LLCT transition from the amide to carbene ligands in the corresponding Cu(I)–CMA moieties (see insets of Fig. 2A). This is further supported by the intensified absorption with the increment of the Cu(I)–CMA content (Table 1). Moreover, all the Cu(I) polymers exhibited strong absorption bands within 350 nm in neat films (Fig. S9, ESI†), originating from the host moieties. Additionally, the intensities of LLCT absorptions were gradually enhanced with the increment of Cu(I)–CMA contents in neat films, consistent with the tendency observed in the solution state.
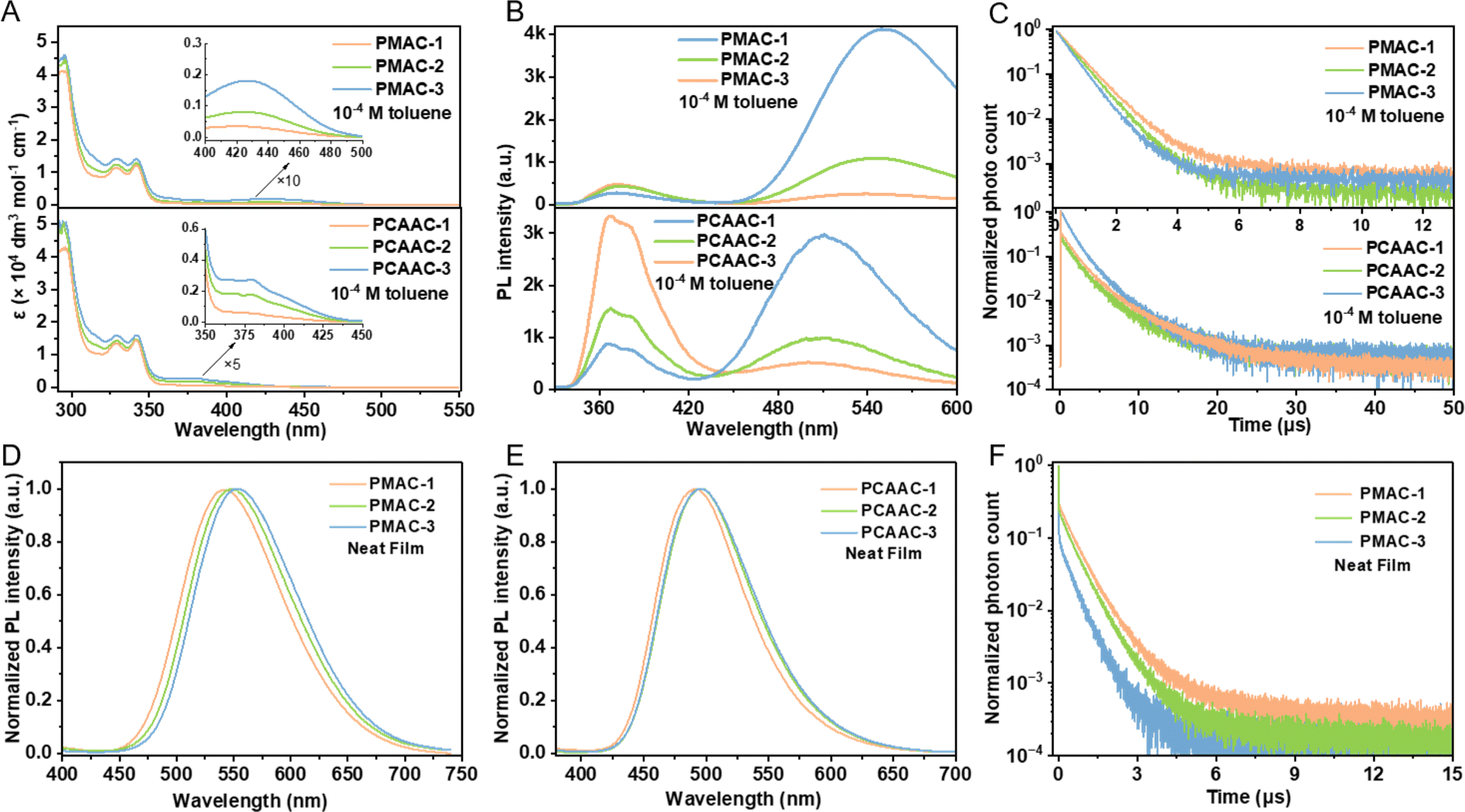 | ||
| Fig. 2 (A) UV/visible absorption spectra of PMAC-x (top) and PCAAC-x (bottom) in toluene solutions (1 × 10−4 M, 300 K). (B) PL spectra of PMAC-x (top) and PCAAC-x (bottom) under 320 nm excitation in toluene solutions (1 × 10−4 M, 300 K). (C) Normalized transient PL decays of PMAC-x (top) and PCAAC-x (bottom) under 375 nm excitation in degassed toluene solutions (1 × 10−4 M, 300 K). Normalized PL spectra of (D) PMAC-x under 380 nm excitation and (E) PCAAC-x under 360 nm excitation in neat films. (F) Normalized transient PL decays of PMAC-x under 375 nm excitation in neat films. | ||
| Polymers | λ abs [nm] (ε [×104 dm3 mol−1 cm−1]) | λ PL [nm] | Φ PL | τ d [μs] | λ PL [nm] | τ d [μs] | Φ PL | k r [105 s−1] | k nr [105 s−1] |
|---|---|---|---|---|---|---|---|---|---|
| In toluene | Neat film | ||||||||
| a The lifetime given is the weighted average. b k r = ΦPL/τd. c k nr = kr/ΦPL − kr. | |||||||||
| PMAC-1 | 295 (4.11)/329 (1.12)/341 (1.20)/425 (0.03) | 369/540 | 0.34 | 0.78 | 545 | 0.82 | 0.78 | 9.5 | 2.7 |
| PMAC-2 | 295 (4.38)/329 (1.24)/341 (1.28)/425 (0.08) | 369/544 | 0.38 | 0.67 | 548 | 0.73 | 0.71 | 9.7 | 4.0 |
| PMAC-3 | 295 (4.59)/329 (1.44)/341 (1.40)/425 (0.18) | 369/549 | 0.45 | 0.58 | 555 | 0.66 | 0.66 | 10.0 | 5.2 |
| PCAAC-1 | 295 (4.23)/329 (1.32)/341 (1.41)/380 (0.06) | 367/510 | 0.21 | 3.1 | 493 | 93.3 | 0.50 | 0.05 | 0.05 |
| PCAAC-2 | 295 (5.07)/329 (1.40)/341 (1.41)/380 (0.17) | 367/510 | 0.24 | 2.8 | 495 | 81.5 | 0.44 | 0.05 | 0.06 |
| PCAAC-3 | 295 (5.07)/329 (1.57)/341 (1.61)/380 (0.27) | 367/510 | 0.31 | 2.6 | 496 | 54.9 | 0.38 | 0.07 | 1.1 |
Significantly, the photoluminescence (PL) spectra of Cu(I) polymers in the solution and neat films were also recorded and are summarized in Table 1. As shown in Fig. 2B, the PL spectra of PMAC-x and PCAAC-x in toluene solutions displayed two main emission bands in the high and low energy regions. All the polymers exhibited similar high-energy emissions peaking at ∼367 nm, which is consistent with the PL spectra of the homopolymer PPhCz in toluene (Fig. S8C, ESI†). Therefore, the high-energy emission regions are ascribed to the fluorescence emission from the host moieties. The low-energy emission regions (λ > 450 nm) belong to the LLCT emission of the respective Cu(I)–CMA units. Similar to the absorption spectra, PMAC-x displayed significant PL red-shifts with respect to PCAAC-x, in good agreement with the theoretical results. Notably, the LLCT emissions were gradually intensified with the increased content of Cu(I)–CMA moieties in the polymers. This indicates an energy transfer process from the pendant host units to the Cu(I)–CMA units within the copolymers. By contrast, the PL spectra of PMAC-x and PCAAC-x in neat films featured single-peak profiles at ∼548 and 495 nm from the corresponding Cu(I)–CMA monomers, respectively (Fig. 2D and E). This result pinpoints that the energy transfer from the host unit to the Cu(I)–CMA units occurs more completely in thin films.
To carry out further investigation into exciton dynamics, transient PL spectra of the Cu(I) polymers in solutions and neat films were measured (Fig. 2C, F and Table 1). All the polymers delivered short emission lifetimes (0.58–0.78 μs for PMAC-x and 2.6–3.1 μs for PCAAC-x) in degassed toluene. Moreover, the decay lifetimes of these polymers decreased as the content of Cu(I)–CMA monomers increased, which is ascribed to a more complete energy transfer from the host to Cu(I)–CMA monomers. Similarly, the emission lifetimes in neat films followed the same order of 0.82 μs (PMAC-1) > 0.73 μs (PMAC-2) > 0.66 μs (PMAC-3). Impressively, the neat films of the PMAC-x polymers manifested a high ΦPL of 0.66–0.78. Combined with the sub-μs-scale lifetimes, the PMAC-x polymers achieved fast radiative rate constants (kr) of up to 1.0 × 106 s−1 (Table 1), comparable to high-efficiency noble-metal phosphors. Differently, the neat films of PCAAC-x polymers showed longer emission lifetimes of 54.9–93.3 μs, implying the presence of the long-lived locally excited triplet state (3LE) as discussed below.
To further picture the emission mechanism of these Cu(I) polymers, we compared the emission spectra of Cu(I) polymers in neat films under argon, air, and oxygen conditions (Fig. S11, ESI†). Due to the triplet-quenching effect of oxygen, the emission intensities of all the Cu(I) polymers obviously declined in the air and oxygen atmosphere. Notably, the PL intensities of PCAAC-x were more O2-sensitive, implying that the longer-lived 3LE states may be involved in the emission processes of these polymers. Subsequently, we selected PMAC-3 and PCAAC-3 as examples to record temperature-dependent PL spectra and lifetimes. As depicted in Fig. 3A, PMAC-3 displayed broad and CT-featured profiles along with gradually blue-shifted emissions from 77 to 300 K, coupled with the sharply decreased emission lifetimes. Furthermore, the temperature-dependent exciton lifetimes of PMAC-3 can be well-fitted to the Boltzmann-type equation (eqn (S1)†), confirming its distinct TADF character. According to the Arrhenius plot (eqn (S2), ESI†), the ΔEST value of PMAC-3 was found to be 0.05 eV (Fig. S14A, ESI†), falling in the range of the theoretical ΔEST value (0.02–0.16 eV) for the PMAC oligomer. Differently, a vibronic-structured and long-lived emission (τ = 12.45 ms) was observed for PCAAC-3 at 77 K. As the temperature increased from 77 to 200 K, the emission of PCAAC-3 exhibited broad and featureless profiles along with the decreased average lifetimes of down to 2.1 ms, indicating that 3LE emissions are gradually weakened. Upon increasing the temperature from 200 to 300 K, PCAAC-3 displayed CT-featured emission profiles along with sharply reduced average lifetimes, indicative of a thermally activated process. Nevertheless, a large proportion of the phosphorescence component of PCAAC-3 was observed at low or even room temperatures (Table S3, ESI†), establishing the phosphorescence-dominated emission origin of PCAAC-3. As expected, the kr of PCAAC-3 was very small (in the 103 s−1 order, Table 1). Furthermore, we noted that the phosphorescence spectrum of 3-phenyl-9H-carbazole (3-PhCz) is consistent with the emission profile of PCAAC-3 below 150 K, suggesting that the long-lived emission component of PCAAC-3 stems from the 3LE phosphorescence of the amide unit (Fig. S13C, ESI†). Compared with the unsubstituted carbazole ligand, the additional phenylene unit enlarges the π-conjugation and thus lowers the 3LE energy level to 2.70 eV. Furthermore, to roughly get the 3CT energy level of PCAAC-3, the μs-scale lifetime (τ1) components from 200 to 300 K, attributed to the TADF process, were well fitted by using the Arrhenius plot, affording an ΔEST value of 0.10 eV between 1CT and 3CT. In this context, PCAAC-3 has very similar energy levels of 3LE and 3CT excited states and thus the phosphorescence from the 3LE state could easily become dominant via a fast conversion process between 3CT and 3LE (Fig. 3F). Comparatively, the stronger LLCT nature of the Cu(I)–CMA monomer in PMAC-3 afforded lower S1/T1 energy levels, making TADF the dominant origin through the effective up-conversion from 3CT to 1CT (Fig. 3E).
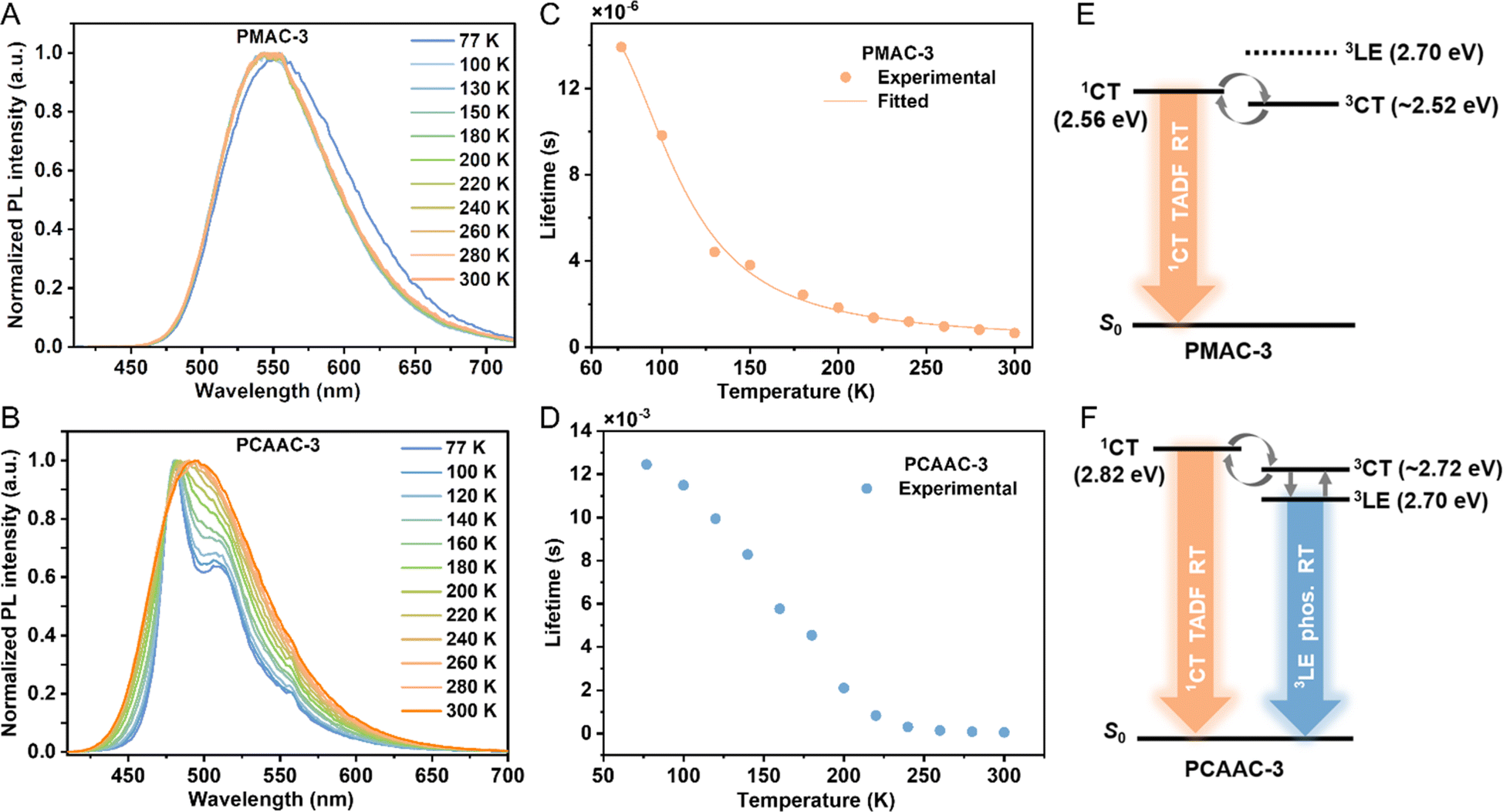 | ||
| Fig. 3 Normalized PL spectra of (A) PMAC-3 and (B) PCAAC-3 under 375 nm excitation at different temperatures from 77 to 300 K. (C) Fitting line of the temperature-dependent lifetime data from 77 to 300 K (symbols) according to eqn (S1)† (line) of PMAC-3. (D) Temperature-dependent lifetime data from 77 to 300 K of PCAAC-3. Schematic energy diagrams of the relevant states for (E) PMAC-3 and (F) PCAAC-3. | ||
Intriguingly, these Cu(I) polymers exhibited superior moisture stability compared with the reference CMA complexes. After adding D2O to the polymers, the 1H NMR spectra of PMAC-3 and PCAAC-3 retained the same proton signals (Fig. S15 and S17, ESI†) compared with the initial spectra. Even after being stored for one week, the 1H NMR spectra of the Cu(I) polymers remained the same. For comparison, we also measured the 1H NMR spectra of the reference Cu(I)–CMA complexes. After adding D2O to the reference complexes for a few minutes, the complexes significantly decomposed along with the presence of new 1H NMR signals (Fig. S16 and S18, ESI†). Impressively, all the Cu(I) polymers afforded AIE properties in THF/H2O mixed solutions (Fig. 4, S19 and S20, ESI†). Subsequently, PMAC-3 and PCAAC-3 were selected as examples to further discuss the AIE feature in detail. As shown in Fig. 4A–D, PMAC-3 and PCAAC-3 gave weak PL intensity peaking at ∼575 and 520 nm, respectively, in dilute THF solution (10−4 M). When the water proportions (fw) increased from 0% to 10%, the PL intensities were decreased together with blue-shifted PL spectra, which could be assigned to the enhanced LLCT effect caused by the increased solvent polarity. Significantly, as the water fraction increased from 0% to 90%, the emission intensity of both Cu(I) polymers dramatically increased, suggesting the apparent AIE properties of PMAC-3 and PCAAC-3. This phenomenon could be associated with restricted ligand–ligand rotation of the Cu(I)–CMA monomers under aggregation conditions. Previous studies showed that limiting the ligand–ligand rotation of CMA complexes can lead to elongated lifetimes.38 Thus, the transient PL spectra for PMAC-3 and PCAAC-3 in THF/H2O mixed solutions were also measured. The decay lifetimes of both Cu(I) polymers were gradually increased with an increase in the water fraction, indicative of aggregation-induced ligand–ligand rotation limitation of Cu(I)–CMA units (Fig. 4E and F). Furthermore, at the 90% water fraction, the decay lifetimes of PMAC-3 and PCAAC-3 reached 508 ns and 2.7 μs, respectively, which are basically consistent with those in degassed toluene solution (Table S4†). This result further established that the nonconjugated polystyrene backbone has an excellent encapsulation effect on the pendent Cu(I)–CMA units.
 | ||
| Fig. 4 (A) PMAC-3 and (B) PCAAC-3 in THF/water mixtures with different water fractions (fw) under 380 nm and 370 nm excitation. Emission intensity vs. fw of (C) PMAC-3 and (D) PCAAC-3. Inset: photos in THF/water mixtures (fw = 0 and 90%) under 365 nm excitation. Lifetimes vs. fw of (E) PMAC-3 and (F) PCAAC-3. | ||
Inspired by the high ΦPL and AIE properties, we evaluated the EL performance of these Cu(I) polymers in solution-processed OLEDs. Before the device characterization, we used atomic force microscopy to study the film-forming ability of the Cu(I) polymers. The spin-coated neat films of the Cu(I) polymers afforded a smooth surface with a root-mean-square roughness of <1 nm (Fig. S22, ESI†), feasible for the fabrication of solution-processed OLEDs. The host-free OLEDs consisted of a common configuration of indium tin oxide (ITO)/poly(3,4-ethylene dioxythiophene)-doped poly(styrene sulfonate) (PEDOT:PSS) (50 nm)/emitting layers (EMLs)/(oxybis(2,1-phenylene))bis(diphenylphosphine oxide) (DPEPO) (10 nm)/1,3,5-tri(m-pyrid-3-yl-phenyl)benzene (TmPyPB) (50 nm)/lithium 8-quinolate (Liq) (1 nm)/Al (100 nm) (Fig. 5A and S23, ESI†). The neat films of PMAC-x and PCAAC-x are used as EMLs for devices A1–A3 and B1–B3, respectively. As shown in Fig. 5B and E, devices A1–A3 and B1–B3 delivered yellow and sky-blue EL emissions peaking at 540–548 nm and 484–486 nm, respectively, without residual emission from the host unit. These results indicated that these EL profiles were only emitted from the Cu(I)–CMA units by complete energy transfer from the host moieties to the emitter units. Moreover, the EL spectra of all the Cu(I) polymers exhibited blue shifts of 5–10 nm compared to their respective PL spectra, most likely due to the optical cavity effects. As listed in Table 2, the turn-on voltages of these devices were gradually decreased with the increased contents of Cu(I)–CMA monomers (Fig. 5C and F), demonstrating that the electrical properties of the devices can be improved by increasing the molar ratio of Cu(I)–CMA units in the polymers. Meanwhile, as the content of Cu(I)–CMA moieties increased, the EML thickness based on PMAC-x and PCAAC-x exhibited decreased tendencies (Fig. S26, ESI†). However, with the exception of device A3, all the other devices still demonstrated relatively higher turn-on voltages exceeding 5.5 V compared with the most reported solution-processed OLEDs (<4 V), which could stem from the thicker EMLs (>50 nm) constructed by the Cu(I) polymers (Fig. S26, ESI†). Inspiringly, by virtue of high ΦPL and fast kr, PMAC-x supported devices exhibited promising device efficiencies. Particularly, the device A3 based on PMAC-3 delivered an outstanding EQE of 13.8% at a luminance of ∼100 nits, which is the best device performance for solution-processed OLEDs based on metallopolymers (Fig. 5F). Additionally, due to the shortest emission lifetime of PMAC-3 among these Cu(I) polymers, device A3 exhibited a slow efficiency roll-off of 7% at 300 cd m−2 and 27.5% at 1000 cd m−2. Comparatively, the devices B1–B3 based on PCAAC-x only achieved EQEs of 2.6–6.3%, mainly due to the lower ΦPL.
 | ||
| Fig. 5 (A) Device structure and energy level diagram of devices A1–A3. EQE–current density curves for devices (B) A1–A3 and (E) B1–B3. Inset: EL spectra of the corresponding devices. Luminance–voltage (left) and current density–voltage (right) curves of devices (C) A1–A3 and (F) B1–B3. (D) Summary of efficient Ir(III) polymers previously reported in the literature.17,39–50 | ||
| Device | Polymer | V on [V] | Maximum value/300 cd m−2/1000 cd m−2 | ELpeake [nm] | L max [cd m−2] | CIE (x, y) | ||
|---|---|---|---|---|---|---|---|---|
| EQEb [%] | CEc [cd A−1] | PEd [lm W−1] | ||||||
| a The turn-on voltage recorded at a luminance of 2 nits. b Maximum external quantum efficiency. c Current efficiency. d Power efficiency and the values at 300 and 1000 nits. e The EL spectra recorded at 13 V for devices A1–A3 and 15 V for devices B1–B3. f Maximum luminance. | ||||||||
| A1 | PMAC-1 | 7.0 | 9.1/7.1/3.1 | 28.4/22.1/13.5 | 9.4/6.6/3.1 | 540 | 981 | 0.35, 0.54 |
| A2 | PMAC-2 | 5.5 | 12.7/10.8/6.4 | 40.0/33.9/20.0 | 17.9/13.3/6.3 | 542 | 2194 | 0.37, 0.54 |
| A3 | PMAC-3 | 4.5 | 13.8/12.8/10.0 | 43.2/40.0/31.3 | 20.9/20.9/13.1 | 548 | 5357 | 0.39, 0.55 |
| B1 | PCAAC-1 | 7.0 | 5.6/1.4/— | 12.3/3.1/— | 4.3/0.8/— | 484 | 502 | 0.22, 0.35 |
| B2 | PCAAC-2 | 6.5 | 6.3/3.1/— | 14.7/7.2/— | 5.5/2.4/— | 486 | 667 | 0.22, 0.37 |
| B3 | PCAAC-3 | 6.5 | 2.6/1.7/— | 6.0/3.9/— | 2.2/1.2/— | 486 | 618 | 0.23, 0.37 |
Conclusion
In summary, a new class of Cu(I) polymers has been successfully constructed by incorporating Cu(I)–CMA complexes and PhCz units as pendant chromophores and host units into a non-conjugated polystyrene backbone. To the best of our knowledge, this is the first example of CMA polymers. Impressively, these Cu(I) polymers display significant TADF or phosphorescence nature, profiting from the unique photophysical properties of Cu(I)–CMA emitter units. Among them, PMAC-x demonstrates a large kr of up to 106 s−1, short exciton lifetime of down to 0.66 μs, and outstanding ΦPL of up to 0.78 in neat films. Intriguingly, in contrast with prototypical Cu(I)–CMA complexes, the presence of the nonconjugated backbone enhances moisture and air stability of Cu(I) polymers and then results in evident AIE properties. Based on the above characterization, host-free solution-processed OLEDs based on these Cu(I) polymers deliver promising device performance. The device based on PMAC-3 exhibits a high EQE of 13.8% at ∼100 nits, representing a record efficiency for metallopolymer-based OLEDs. This work sheds light on the potential of Cu(I)–CMA polymers for constructing high-efficiency solution-processed OLEDs.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Author contributions
S. G. conceived and designed the project. Y. T. synthesized the polymers. Y. T. and A. Y. measured photophysical properties. Y. T., J. X., and G. X. fabricated and characterized the devices. Y. T. performed quantum chemical calculations. S. G. supervised this research. Y. T., A. Y., and S. G. co-wrote the manuscript. All the authors discussed the results and contributed to the manuscript.Conflicts of interest
S. G. and Y. T. are inventors on a patent application related to this work (CN patent application no. 2024102080555) filed by Wuhan University. The other authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52022071). The authors wish to acknowledge the supercomputing system in the Supercomputing Center of Wuhan University for numerical calculations. We are grateful to the Core Facility of Wuhan University for inductively coupled plasma-atomic emission spectroscopy and atom force microscopy.References
- F. S. Arimoto and A. C. Haven Jr, J. Am. Chem. Soc., 1955, 77, 6295–6297 CrossRef CAS.
- G. R. Whittell and I. Manners, Adv. Mater., 2007, 19, 3439–3468 CrossRef CAS.
- Y. Yan, J. Zhang, L. Ren and C. Tang, Chem. Soc. Rev., 2016, 45, 5232–5263 RSC.
- M. Y. Wong, J. Electron. Mater., 2017, 46, 6246–6281 CrossRef CAS.
- A. Liang, L. Ying and F. Huang, J. Inorg. Organomet. Polym. Mater., 2014, 24, 905–926 CrossRef CAS.
- A. Verma, P. Chaudhary, R. K. Tripathi, A. Singh and B. C. Yadav, J. Inorg. Organomet. Polym. Mater., 2022, 32, 2807–2826 CrossRef CAS.
- C.-L. Ho and W.-Y. Wong, Top. Curr. Chem., 2016, 374, 64 CrossRef PubMed.
- B. Liu, F. Dang, Z. Tian, Z. Feng, D. Jin, W. Dang, X. Yang, G. Zhou and Z. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 16360–16374 CrossRef CAS PubMed.
- S. Gong, C. Yang and J. Qin, Chem. Soc. Rev., 2012, 41, 4797–4807 RSC.
- X. Yang, G. Zhou and W.-Y. Wong, J. Mater. Chem. C, 2014, 2, 1760–1778 RSC.
- F. Xu, H. U. Kim, J.-H. Kim, B. J. Jung, A. C. Grimsdale and D.-H. Hwang, Prog. Polym. Sci., 2015, 47, 92–121 CrossRef CAS.
- C.-L. Lee, N.-G. Kang, Y.-S. Cho, J.-S. Lee and J.-J. Kim, Opt. Mater., 2003, 21, 119–123 CrossRef CAS.
- J. Jiang, W. Yang and Y. Cao, J. Inorg. Organomet. Polym. Mater., 2007, 17, 37–55 CrossRef CAS.
- P. T. Furuta, L. Deng, S. Garon, M. E. Thompson and J. M. J. Fréchet, J. Am. Chem. Soc., 2004, 126, 15388–15389 CrossRef CAS PubMed.
- K. Feng, C. Zuniga, Y.-D. Zhang, D. Kim, S. Barlow, S. R. Marder, J. L. Brédas and M. Weck, Macromolecules, 2009, 42, 6855–6864 CrossRef CAS.
- S. Jhulki, M. W. Cooper, S. Barlow and S. R. Marder, Mater. Chem. Front., 2019, 3, 1699–1721 RSC.
- J. H. Park, T.-W. Koh, J. Chung, S. H. Park, M. Eo, Y. Do, S. Yoo and M. H. Lee, Macromolecules, 2013, 46, 674–682 CrossRef CAS.
- Z. Zhang, Y.-N. He, L. Liu, X.-Q. Lü, X.-J. Zhu, W.-K. Wong, M. Pan and C.-Y. Su, Chem. Commun., 2016, 52, 3713–3716 RSC.
- Y. Hirai, T. Nakanishi and Y. Hasegawa, J. Lumin., 2016, 170, 801–807 CrossRef CAS.
- X.-Y. Chen, X. Yang and B. J. Holliday, J. Am. Chem. Soc., 2008, 130, 1546–1547 CrossRef CAS PubMed.
- Y. Hirai, T. Nakanishi, K. Miyata, K. Fushimi and Y. Hasegawa, Mater. Lett., 2014, 130, 91–93 CrossRef CAS.
- (a) A. I. Il’icheva, Y. P. Barinova, L. N. Bochkarev and V. A. Il’ichev, Russ. J. Appl. Chem., 2012, 85, 1711–1717 CrossRef; (b) A. I. Ilicheva, L. N. Bochkarev, V. A. Ilichev and Y. D. Semchikov, Russ. J. Gen. Chem., 2015, 85, 1140–1145 CrossRef CAS.
- (a) J. Ohshita, T. Kai, Y. Adachi, K. Yamaji, M. Nakamura, S. Watase, S. Mori and N. Matsuyama, Appl. Organomet. Chem., 2019, 34, e5306 CrossRef; (b) J. Zhao, B. Liu, Z. Feng, D. Jin, W. Dang, X. Yang, G. Zhou, Z. Wu and W.-Y. Wong, Polym. Chem., 2017, 8, 6368–6377 RSC.
- S. Mondal, D. C. Santra, S. Roy, Y. S. L. V. Narayana, T. Yoshida, Y. Ninomiya and M. Higuchi, ACS Appl. Mater. Interfaces, 2023, 15, 42912–42919 CrossRef CAS PubMed.
- (a) Y. Liu, S.-C. Yiu, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2018, 375, 514–557 CrossRef CAS; (b) C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS; (c) H. Yersin, R. Czerwieniec, M. Z. Shafikov and A. F. Suleymanova, ChemPhysChem, 2017, 18, 3508–3535 CrossRef CAS PubMed.
- (a) R. Czerwieniec, J. Yu and H. Yersin, Inorg. Chem., 2011, 50, 8293–8301 CrossRef CAS PubMed; (b) M. Osawa, M. Hoshino, M. Hashimoto, I. Kawata, S. Igawa and M. Yashima, Dalton Trans., 2015, 44, 8369–8378 RSC; (c) D. Volz, Y. Chen, M. Wallesch, R. Liu, C. Fléchon, D. M. Zink, J. Friedrichs, H. Flügge, R. Steininger, J. Göttlicher, C. Heske, L. Weinhardt, S. Bräse, F. So and T. Baumann, Adv. Mater., 2015, 27, 2538–2543 CrossRef CAS PubMed.
- N. Zhang, Y. Li, S. Han, Y. Wei, H. Hu, R. Huo, C. Duan, J. Zhang, C. Han, G. Xie and H. Xu, Angew. Chem., Int. Ed., 2023, 62, e202305018 CrossRef CAS PubMed.
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS.
- C. Sandoval-Pauker, M. Santander-Nelli and P. Dreyse, RSC Adv., 2022, 12, 10653–10674 RSC.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed.
- (a) S. Shi, M. C. Jung, C. Coburn, A. Tadle, D. Sylvinson, M. R, P. I. Djurovich, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 3576–3588 CrossRef CAS PubMed; (b) J. Li, L. Wang, Z. Zhao, X. Li, X. Yu, P. Huo, Q. Jin, Z. Liu, Z. Bian and C. Huang, Angew. Chem., Int. Ed., 2020, 59, 8210–8217 CrossRef CAS PubMed.
- (a) A. S. Romanov, S. T. E. Jones, Q. Gu, P. J. Conaghan, B. H. Drummond, J. Feng, F. Chotard, L. Buizza, M. Foley, M. Linnolahti, D. Credgington and M. Bochmann, Chem. Sci., 2020, 11, 435–446 RSC; (b) T.-Y. Li, J. Schaab, P. I. Djurovich and M. E. Thompson, J. Mater. Chem. C, 2022, 10, 4674 RSC.
- (a) C. N. Muniz, J. Schaab, A. Razgoniaev, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2022, 144, 17916–17928 CrossRef CAS PubMed; (b) M. Gernert, L. Balles-Wolf, F. Kerner, U. Müller, A. Schmiedel, M. Holzapfel, C. M. Marian, J. Pflaum, C. Lambert and A. Steffen, J. Am. Chem. Soc., 2020, 142, 8897–8909 CrossRef CAS PubMed.
- (a) R. Tang, S. Xu, T. L. Lam, G. Cheng, L. Du, Q. Wan, J. Yang, F. F. Hung, K. H. Low, D. L. Phillips and C. M. Che, Angew. Chem., Int. Ed., 2022, 61, e202203982 CrossRef CAS PubMed; (b) H. J. Wang, Y. Liu, B. Yu, S. Q. Song, Y. X. Zheng, K. Liu, P. Chen, H. Wang, J. Jiang and T.-Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202217195 CrossRef CAS PubMed.
- (a) L. Zhan, A. Ying, Y. Qi, K. Wu, Y. Tang, Y. Tan, Y. Zou, G. Xie, S. Gong and C. Yang, Adv. Funct. Mater., 2021, 31, 2106345 CrossRef CAS; (b) Q. Gu, F. Chotard, J. Eng, A.-P. M. Reponen, I. J. Vitorica-Yrezabal, A. W. Woodward, T. J. Penfold, D. Credgington, M. Bochmann and A. S. Romanov, Chem. Mater., 2022, 34, 7526–7542 CrossRef CAS PubMed; (c) A. Ying, Y. Tan and S. Gong, Adv. Opt. Mater., 2024, 12, 2303333 CrossRef CAS; (d) A. Ying, L. Zhan, Y. Tan, X. Cao, C. Yang and S. Gong, Sci. China: Chem., 2023, 66, 2274–2282 CrossRef CAS.
- (a) A. Ying and S. Gong, Chem.–Eur. J., 2023, 29, e2023018 Search PubMed; (b) T.-Y. Li, S.-J. Zheng, P. I. Djurovich and M. E. Thompson, Chem. Rev., 2024, 124, 4332–4392 CrossRef CAS PubMed.
- A. Ying, Y. Ai, C. Yang and S. Gong, Angew. Chem., Int. Ed., 2022, 61, e202210490 CrossRef CAS PubMed.
- Z. Ma, L. Chen, J. Ding, L. Wang, X. Jing and F. Wang, Adv. Mater., 2011, 23, 3726–3729 CrossRef CAS PubMed.
- H. Zhen, C. Jiang, W. Yang, J. Jiang, F. Huang and Y. Cao, Chem.–Eur. J., 2005, 11, 5007–5016 CrossRef CAS PubMed.
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2012, 134, 15189–15192 CrossRef CAS PubMed.
- H. Zhen, J. Luo, W. Yang, Q. Chen, L. Ying, Z. Jianhua, H. Wu and Y. Cao, J. Mater. Chem., 2007, 17, 2824–2831 RSC.
- L. Ying, J. Zou, A. Zhang, B. Chen, W. Yang and Y. Cao, J. Organomet. Chem., 2009, 694, 2727–2734 CrossRef CAS.
- W.-Y. Lai, M. N. Balfour, J. W. Levell, A. K. Bansal, P. L. Burn, S.-C. Lo and I. D. W. Samuel, Macromolecules, 2012, 45, 2963–2971 CrossRef CAS.
- W.-Y. Lai, J. W. Levell, A. C. Jackson, S.-C. Lo, P. V. Bernhardt, I. D. W. Samuel and P. L. Burn, Macromolecules, 2010, 43, 6986–6994 CrossRef CAS.
- F. Xu, J.-H. Kim, H. U. Kim, J.-H. Jang, K. S. Yook, J. Y. Lee and D.-H. Hwang, Macromolecules, 2014, 47, 7397–7406 CrossRef CAS.
- H. Zhen, C. Luo, W. Yang, W. Song, B. Du, J. Jiang, C. Jiang, Y. Zhang and Y. Cao, Macromolecules, 2006, 39, 1693–1700 CrossRef CAS.
- S. Tokito, M. Suzuki, F. Sato, M. Kamachi and K. Shirane, Org. Electron., 2003, 4, 105–111 CrossRef CAS.
- L. Ying, Y. Xu, W. Yang, L. Wang, H. Wu and Y. Cao, Org. Electron., 2009, 10, 42–47 CrossRef CAS.
- S. Tokito, M. Suzuki and F. Sato, Thin Solid Films, 2003, 445, 353–357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental and synthetic procedures, photophysical and OLED characterization, and computational details. See DOI: https://doi.org/10.1039/d4sc01865f |
| This journal is © The Royal Society of Chemistry 2024 |