
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoconfinement of polyoxometalates in cyclodextrin: computational inspections of the binding affinity and experimental demonstrations of reactivity modulation†
Mireia
Segado-Centellas‡
 *a,
Clément
Falaise‡
*b,
Nathalie
Leclerc
b,
Gabrielle
Mpacko Priso
b,
Mohamed
Haouas
b,
Emmanuel
Cadot
b and
Carles
Bo
a
*a,
Clément
Falaise‡
*b,
Nathalie
Leclerc
b,
Gabrielle
Mpacko Priso
b,
Mohamed
Haouas
b,
Emmanuel
Cadot
b and
Carles
Bo
a
aInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science & Technology (BIST), Av. Països Catalans 16, 43007 Tarragona, Spain
bInstitut Lavoisier de Versailles, CNRS, UVSQ, Université Paris-Saclay, 45 Avenue des Etats-Unis, 78035, Versailles, France. E-mail: clement.falaise@uvsq.fr
First published on 5th September 2024
Abstract
Chaotropic polyoxometalates (POMs) form robust host–guest complexes with γ-cyclodextrin (γ-CD), offering promising applications in catalysis, electrochemical energy storage, and nanotechnology. In this article, we provide the first computational insights on the supramolecular binding mechanisms using density-functional theory and classical molecular dynamics simulations. Focusing on the encapsulation of archetypal Keggin-type POMs (PW12O403−, SiW12O404− and BW12O405−), our findings reveal that the lowest-charged POM, namely PW12O403− spontaneously confines within the wider rim of γ-CD, but BW12O405− does not exhibit this behaviour. This striking affinity for the hydrophobic pocket of γ-CD originates from the structural characteristics of water molecules surrounding PW12O403−. Moreover, through validation using 31P NMR spectroscopy, we demonstrate that this nanoconfinement regulates drastically the POM reactivity, including its capability to undergo electron transfer and intermolecular metalate Mo/W exchanges. Finally, we exploit this nanoconfinement strategy to isolate the elusive mixed addenda POM PW11MoO403−.
Introduction
Polyoxometalates (POMs) represent an extensive class of anionic metal–oxo clusters built from early transition metals in their highest oxidation states.1,2 Although many POM structures have been reported,1,3,4 only a few are used in practical applications. Among them, the Keggin-type phosphometalates formulated PM12O403− (M = Mo6+ or W6+) are widely studied in fundamental and applied research and have found real-life applications in catalysis, analysis, or histology.5 These Keggin-type POMs are very popular and have been manipulated by several generations of chemists, however, their ability to interact with non-ionic organic matter in aqueous solution was largely underestimated until the publication of two articles in 2015.6,7 One reported that the PMo12O403− anion forms robust host–guest complexes with organic macrocycles in water6 and the other one shows that the POM PW12O403− anion tends to be adsorbed at the surface of micelles.7 The origins of these self-assembly processes were debated.8 Today, the community agrees on the fact that the strong affinity between organic substances and POMs having a low charge density, such as the Keggin-type phosphometalates, is mainly due to their striking hydration shell, classifying POMs as (super)chaotropic ions.9–18It has become increasingly evident that water transcends its role as merely a “solvent” and can be seen as a matrix that actively connects and interacts closely with ions. The structure and dynamics of the hydration shell around ions appear to influence the behaviour of both the ions and the water molecules, creating an interplay between the two entities. Recent insights show conceptual similarity between large ions and weakly solvated ions in the Hofmeister series, categorized as chaotropic.16 The name comes from the idea that these ions disrupt the structure of water, a notion currently subject to controversy.19,20 POMs exhibiting behaviour akin to chaotropic ions but with heightened intensity, are labelled as (super)chaotropic.19,21 Since the first mention of the chaotropicity of POMs by Thouvenot in 1978,22 the terms “high energy water” or “disordered water” are usually used to describe the water surrounding POMs. This description remains quite vague and unsatisfying however probing experimentally the water structure at solute/liquid interfaces is extremely challenging.23–26 In context, using theoretical tools appears as an efficient strategy to investigate the origin of the chaotropic effect. In recent literature, the study of the chaotropic system's effects has emerged as a topic of growing interest. This is underscored by the recent publication of two computational studies—one classifying nano-ions within the Hofmeister series and the other investigating the water's ordering in salt solutions.19,27 In both cases, very simple spherical models were used to describe the nano-ions. The chaotropicity of POMs and the related phenomena are unexplored, indeed previous simulations about the solution behaviour of POMs were mostly focused on ion-pairing effects.28–34 Actually, the only molecular dynamics studies of POMs performed under the prism of the chaotropic effect were conducted to link the potential affinity of POMs with hen egg-white lysozyme, although these studies were not supported by experimental data.35,36
The complexation of Keggin-type POMs with γ-cyclodextrin (notated γ-CD), a natural oligosaccharide made up of eight glucopyranose units, represents certainly one of the most significant chaotropically-driven systems reported so far since it leads to potential applications in catalysis, energy storage, iodine capture, or nanotechnologies.37–42 The adequate size matching between γ-CD cavity and the Keggin-type POM leads to a wide variety of host–guest complexes.6,15,43–47 As observed, the global charge density of the Keggin-type POMs dramatically influences both the binding constant and the nature of the aggregates.15,43 For instance, solution investigations using NMR evidenced that both primary and secondary faces contribute to the POM's complexation.15 Calorimetric studies revealed that this process is orthogonal to the classical hydrophobic effect since it is enthalpically driven accompanied by an entropic penalty.15 It is worth noting that this embedment within the hydrophobic cavity can be viewed as a nanoconfinement similar to those observed in Mo-storage proteins, in which the protein binding pocket acts not only as a template and nucleation site for polynuclear Mo-based entities but also offers a protecting barrier again hydrolytic attacks.48,49 In biological systems, nanoconfinement represents a widely used strategy to ensure vital functions such as oxygen transport in the blood or enzyme catalysis. In context, the supramolecular association between Keggin-type POMs and γ-CD represents appealing models for studying the chemical reactivity of POMs entrapped in a hydrophobic pocket.
In this article, we first report the computational inspection of the supramolecular binding process of three archetypal Keggin-type POM (PW12O403−, SiW12O404− and BW12O405−) with γ-CD using Density-Functional Theory (DFT) and classical Molecular Dynamics (MD) simulations. This theoretical study shows that the super-chaotropic character of PW12O403− is responsible for its strong affinity with γ-CD host. Then, we demonstrate experimentally using 31P NMR studies that the nanoconfinement of PW12O403− provides a way to control its chemical reactivity, allowing tuning of the intermolecular electron-transfer rates and the exchange of metalate ions between Keggin-type POMs. Finally, we employ this strategy to prepare the elusive mixed-metal Keggin anion PW11MoO403− with high purity.
Results and discussion
Theoretical studies of the encapsulation process
To gain insight into the forces that govern the association process between Keggin-type POM and the γ-CD, we first performed theoretical investigations using DFT and MD simulations. Geometries of three POMs (PW12O403−, SiW12O404−, and BW12O405−), γ-CD, and their 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 supramolecular adducts were fully optimized using DFT and an implicit solvent model. Various types of assemblies that have been previously observed in crystal structures were found, including the host–guest associations with the primary, secondary face, and external interaction.15,45 Additionally, two other aggregates, named side-primary and side-secondary assemblies (see Fig. 1), were identified. To compare the optimized structures with X-ray structures, we focused on the distances between γ-CD and POMs. All these distances fall within the range of O–H distances observed in X-ray crystal structures.13,15,45 (for more details see Section 2.a of ESI†).
:1 supramolecular adducts were fully optimized using DFT and an implicit solvent model. Various types of assemblies that have been previously observed in crystal structures were found, including the host–guest associations with the primary, secondary face, and external interaction.15,45 Additionally, two other aggregates, named side-primary and side-secondary assemblies (see Fig. 1), were identified. To compare the optimized structures with X-ray structures, we focused on the distances between γ-CD and POMs. All these distances fall within the range of O–H distances observed in X-ray crystal structures.13,15,45 (for more details see Section 2.a of ESI†).
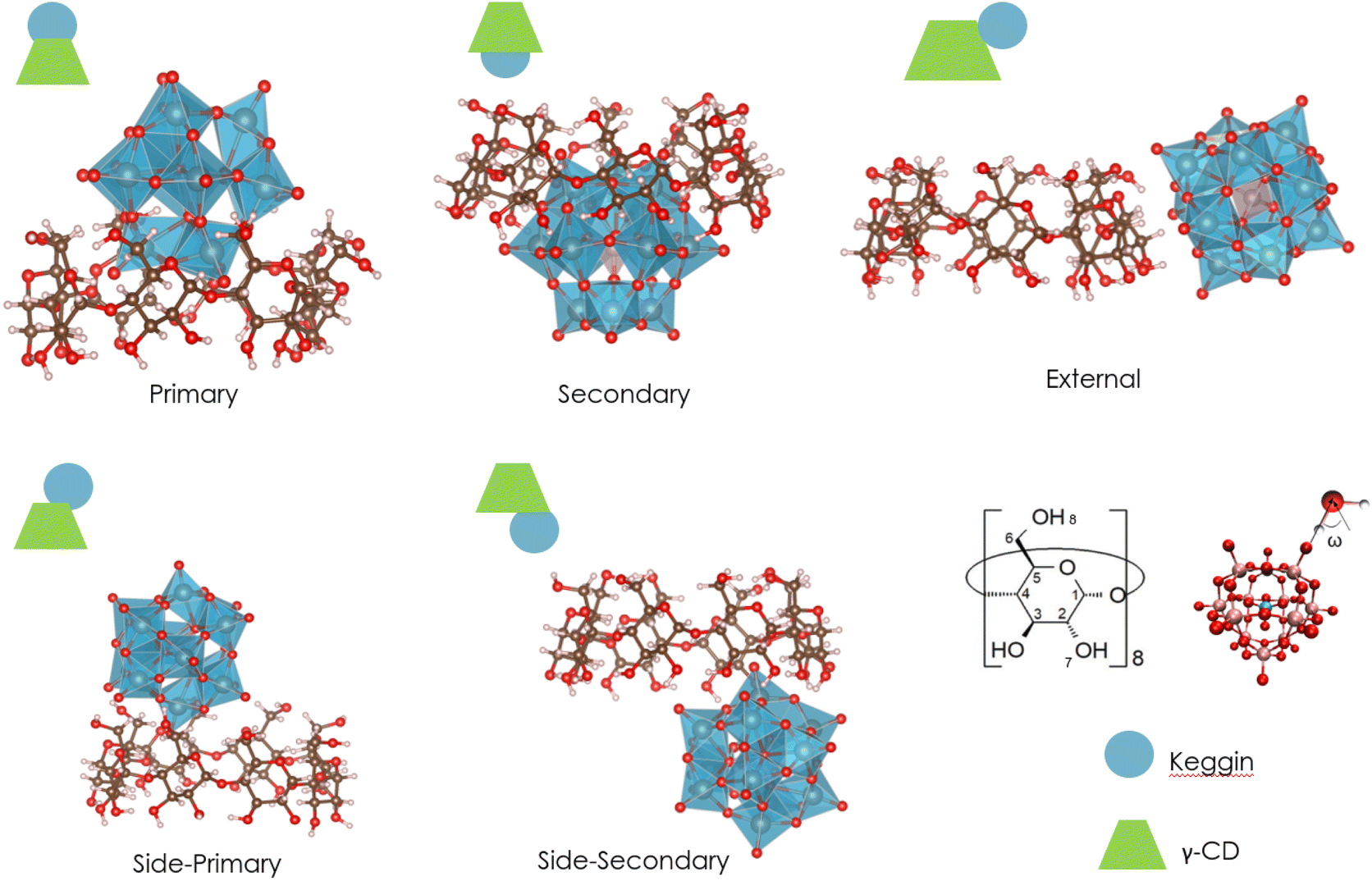 | ||
| Fig. 1 Illustration of the different types of 1:1 supramolecular adducts resulting from the association of Keggin-type POM and γ-CD that have been optimized using DFT and the implicit solvent model. | ||
To computationally determine preferential configurations along the series, two factors were investigated using DFT methods: the POM size and host–guest interaction energies. The computed molecular surface area for PW12O403−, SiW12O404−, and BW12O405− is 1687.53, 1679.82 and 1662.71 Å2, respectively. Considering the insignificant differences in their molecular area, we confirm that size is not the determining factor for diverse preferential configurations observed experimentally. To determine the host–guest binding energies between POM and γ-CD in water solution, we dissected the binding process using a standard thermodynamic cycle: ΔGbindaq = ΔGbindgas + ΔGsolv whereas ΔGsolv = POM-CDΔGsolv − POMΔGsolv − γ-CDΔGsolv (for more details see Section 1.a of ESI†). The DFT Gibbs solvation and binding energies are reported in Table 1. This indicates the POM-CD binding is an enthalpic driven process with and small entropic penalty. Importantly, ΔG solvation energies play an important role in conformers relative energies. For the most charged system [BW12O40]5− the desolvation energy effect is more significant than POM-CD intermolecular interactions, resulting in the most stable conformer being the one with least contact, referred to as external. For medium charged system [SiW12O40]4− in gas phase and when solvent is included implicitly, secondary configuration is the most preferred. For the most chaotropic system [PW12O40]3−, the penalty of ΔG solvation energies for secondary configuration, the most penetrating one, is half that of [BW12O40]5− highlighting the importance of enthalpy as a driving binding force for chaotropic systems. Hence, what is crucial here is the effect of the anion on the surrounding water molecules, as expected for the chaotropically-driven self-assemblies.50 Thus, the dynamic solvation sphere of the POMs is likely to play a significant role in the formation of the adducts. To incorporate the effects of hydration and the structural and dynamic properties of water, we employed explicit classical MD simulations.
| Primary | Secondary | External | |
|---|---|---|---|
| PW 12 O 40 3− | |||
| ΔH(gas) | −123.6 | −159.8 | −65.4 |
| −TΔS(gas) | 16.8 | 18.7 | 22.5 |
| ΔGsolv | 95.2 | 93.6 | 51.5 |
| ΔGbindaq | −11.6 | −47.5 | 8.6 |
|
|||
| SiW 12 O 40 4− | |||
| ΔH(gas) | −165.9 | −210.5 | −107.9 |
| −TΔS(gas) | 16.3 | 17.6 | 21.8 |
| ΔGsolv | 137.3 | 143.8 | 90.4 |
| ΔGbindaq | −12.3 | −49.1 | 4.3 |
|
|||
| BW 12 O 40 5− | |||
| ΔH(gas) | −221.9 | −277.6 | −134.2 |
| −TΔS(gas) | 16.6 | 19.3 | 20.9 |
| ΔGsolv | 190.7 | 205.5 | 53.9 |
| ΔGbindaq | −14.6 | −52.8 | −59.4 |
We computed the Radial Distribution Function (RDF) between the center of the Keggin-type POMs and the γ-CD center to analyse the average number of supramolecular adducts formed over time (see Fig. 2). Firstly, our goal was to determine the stability of various adducts. To achieve this, we conducted MD simulations using two initial box systems: one with all POMs complexed by the primary face and another with all POMs bounded to the secondary faces. Secondly, we aimed to identify the predominant adduct formed in aqueous solution. For this purpose, we initialized a periodic simulation box with POMs and γ-CD randomly distributed as free molecules, not complexed (see Fig. S1†). It's worth noting that each supramolecular assembly had a specific distance between the POM and center of γ-CD. Consequently, each peak observed in the RDF corresponds to one specific supramolecular adduct.
 | ||
| Fig. 2 Radial distribution function between the center of mass of Keggin-type anions and γ-CD in explicit water. | ||
Regarding the low-charged PW12O403−, when starting from the secondary configuration (green curve in Fig. 2), the secondary adducts remain stable from beginning to end of the simulation, exhibiting the highest peak intensity compared to other starting systems confirming the highest stability of the secondary configuration. There is also an appearance over time of a lower intensity peak around 800–1000 pm, which corresponds to the newly proposed side-secondary adduct. On the other hand, when starting from the primary configuration (pink curve), the primary adducts show stability over time. However, when starting from an initial free configuration (black curve), various adducts are formed during the simulation, but the secondary and external adducts exhibit the highest intensities in contrast with side adducts. For the free initial state simulation of PW12O403−, note that for longer simulation times, the peak corresponding to the external site decrease intensity, thus demonstrating that although the system did not reach equilibrium, the position and intensity of the peak corresponding to the secondary structure persists. This demonstrates that the POM and CD assemble spontaneously in explicit water solution, and that the preferred binding mode derived from the simulations precisely coincides with that observed in experiments.
In the medium-charged SiW12O404−, when starting from the secondary and primary configurations, some initial supramolecular assemblies persist over all the simulations. However, when starting from a free configuration, the predominant adducts formed in solution are the side-primary and external adducts, with minor contributions from the side-secondary adducts observed in the spatial distribution functions within the range of 800–1000 pm. Overall, the intensity of the peaks is lower compared to PW12O403−. One intriguing aspect of this result is the appearance of the newly proposed side-primary configuration as one of the two most prevalent adducts.
Moving on to the high-charged Keggin-type POMs, namely BW12O405−, when starting from the secondary and primary configurations, both do not remain stable over time. In all initial cases, only the external adducts are formed during the dynamics. The RDF exhibits peaks of extra low intensity in all cases.
We compared our predictions with experimental findings obtained from 1H NMR data (titration experiment) that provide valuable insights into the formation and stability of supramolecular association of Keggin-type POMs and γ-CD in water.15 Specifically, the chemical shifts of H3, H5, and H6 exhibit distinct alterations after the addition of various equivalents of Keggin-type POMs (see Table 2). These observed changes in chemical shifts indicate different interactions and recognition processes. The most exposed hydrogens of γ-CD to POM are H3 and H5, located inside the cavity, as well as H6, located on the methoxy group outside the ring. In addition, other hydrogen atoms able to form hydrogen bonds with the Keggin anions are the in-ring and out-ring hydroxo groups, labelled H7 and H8, respectively (see Fig. 1).
| POM | Δδ H3 | Δδ H5 | Δδ H6 |
|---|---|---|---|
| PW12O403− | 0.53 | 0.36 | 0.28 |
| SiW12O404− | 0.03 | 0.16 | 0.30 |
| BW12O405− | 0.03 | 0.18 | 0.33 |
For PW12O403−, the experimental chemical shifts exhibit the highest shift for H3, followed by smaller shifts for H5 and H6.15 Our MD simulations indicate that main configurations, secondary, external, and to a lesser extent, side-secondary and side-primary adducts, assemble over time. The structural DFT parameters calculated for complexation by the secondary face reveal close contacts between the POM H3 and H5. Additionally, our proposed new solution structure, the side-primary adduct, exhibits close contact with H6. The observed chemical shifts can be explained utilizing MD by the presence of a combination of secondary and to a lesser extent side-primary, and external adduct configurations. The experimental 1H NMR study revealed that the addition of SiW12O404− to γ-CD induces a strong shift of the H6 signal and a smaller one for H5.15 Our MD simulations reveal that silicotungstate anions spontaneously assemble mostly external and side-primary adducts. Optimized DFT structures further confirm the presence of close contacts between the side-primary configuration and H6, while side-secondary configurations predominantly interact with the proton of hydroxo H7. The observed chemical shifts align well with the co-existence of side-primary and external adduct configurations. The dynamic behaviour of side- and external adduct systems is particularly intriguing, as the analysis of the MD trajectories for external configurations reveals a region of possible near-side primary configurations where the POM exhibits some motion over time. Some of these configurations involve potential interactions between the POM and H5. Similarly, in the case of BW12O405−, the highest chemical shift is observed for H6, followed by a smaller shift for H5. From our MD simulations, it is evident that only external adducts are spontaneously formed, which correlates with the observed chemical shift in H6. These findings demonstrate a good agreement between the experimentally observed chemical shifts and the adduct configurations formed during molecular dynamics simulations. Also, similar chemical shifts can originate from different adducts, highlighting the complexity of solution assemblies. The combination of NMR spectroscopy and MD simulations is a useful methodology for extracting valuable information about the assembly behaviour in solution and provides information on the structural characteristics of the adducts.
Our results reveal distinct patterns of adduct formation based on the charge density of the Keggin-type POMs. For the POM exhibiting the lowest charge density, the secondary and external adducts dominate in solution. In the case of Keggin with the global anionic charge of −4, there is less aggregation, and both side-primary and external adducts are observed with higher abundance. However, for the highest charged Keggin, the formation of adducts is quite limited since only external adducts are observed, implying an interaction without losing its hydration shell. Oppositely, the deep penetration of PW12O403− within the hydrophobic cavity of the CD allows its full dehydration.
Globally, the MD results are consistent with the experimental results and explain the tendencies of the Keggin-type anions within the Hofmeister series.
Nanoconfinement effect on the intermolecular electron transfer
According to previously published experimental observations15 that are supported by the theoretical study presented in this article, the compound PW12O403− can be easily embedded within the cavity of the macrocyclic host. This encapsulation process can be viewed as a nanoconfinement in which the γ-CD acts as a protecting barrier. This process is known to improve the hydrolytic stabilities of the POM guest.44 In this section, we explore experimentally the effect of this nanoconfinement on the rate of electron transfer. Understanding and tuning the electron transfer rate involving reduced POMs is crucial because this class of compounds is currently widely employed as a redox mediator to shuttle electrons in many catalytic or electrochemical processes.51–53To highlight the nanoconfinement effect of PW12O403− embedded within γ-CD, we revisited the classical Baker's experiment54,55 in which he studied the electron transfer between one-electron reduced POM and its non-reduced form using 31P NMR measurements. It must be noted, that all of our experiments were carried out at room temperature and involved equimolar aqueous solution of one-electron reduced and non-reduced form ([PW12O403−] = [PW12O403−] = 25 mM in 0.1 M HCl), and various equivalents of γ-CD (from 0 to 6 equivalents).
In the absence of γ-CD, the 31P NMR signal exhibits only one narrow signal (Δν1/2 = 3 Hz) situated at −12.7 ppm (Fig. 3). The presence of this signal, located exactly at the 31P NMR chemical shift corresponding to the mean value of the chemical shifts of oxidized (−14.8 ppm) and one-electron reduced (−10.7 ppm) POMs, indicates that electron exchange within the donor–acceptor tandem occurs in a fast-exchange regime in the NMR time scale. This result is in good agreement with the observations of Baker.54,55 Interestingly, adding γ-CD (from 0 to 3 eq.) to the redox mixture PW12O403−:PW12O404− has a dramatic effect on the 31P NMR spectra as evidenced first by the gradual broadening and then the splitting of the single resonances into two signals (see Fig. 3). Modifications of the 31P NMR spectra highlight the gradual transition from a fast to a slow electron exchange regime featured by the presence of two 31P resonances at −10.7 and −14.8 ppm corresponding to the 31P NMR chemical shifts of PW12O404− and PW12O403−, respectively (see Fig. 3). We note that these two signals become narrower as the γ-CD content increases up to three. This study shows that the presence of γ-CDs in the vicinity of POMs prevents intermolecular electron transfer by insulating the POMs into a protective barrier. This nanoconfinement effect reduces the probability of direct POM–POM contacts, which are responsible for intermolecular electron transfer. Actually, such an observation should be related to the high binding constants previously reported for the oxidized Keggin PW12O403− ion which forms the host–guest complex POM@(γ-CD)2 as predominant species under these conditions (K1:1 = 90000 M−1 and K1:2 = 1500 M−1).15
 | ||
| Fig. 3 Evolution of the 31P NMR spectrum of an equimolar mixture of non-reduced and one-electron-reduced Keggin-type POM ([PW12O40]3− = [PW12O40]4− = 25 mM) in the presence of γ-CD (from 0 to 3 eq. of γ-CD per POM). T = 294 K. The reduced PW12O404− was prepared in situ by adding sodium dithionite to the solutions. | ||
To go a step further, quantitative determination of the electron rate transfer can be performed. For intermediate/slow exchange regime, the electron rate constant Ket for the equimolar mixture PW12O403−/PW12O404− can be calculated from eqn (1) and (2):
| ket = π(Δvmixox − Δv0ox)/[PW12O404−] | (1) |
| ket = π(Δvmixred − Δv0red)/[PW12O403−] | (2) |
For the intermediate/fast exchange regime, the Ket values can be determined using eqn (3).
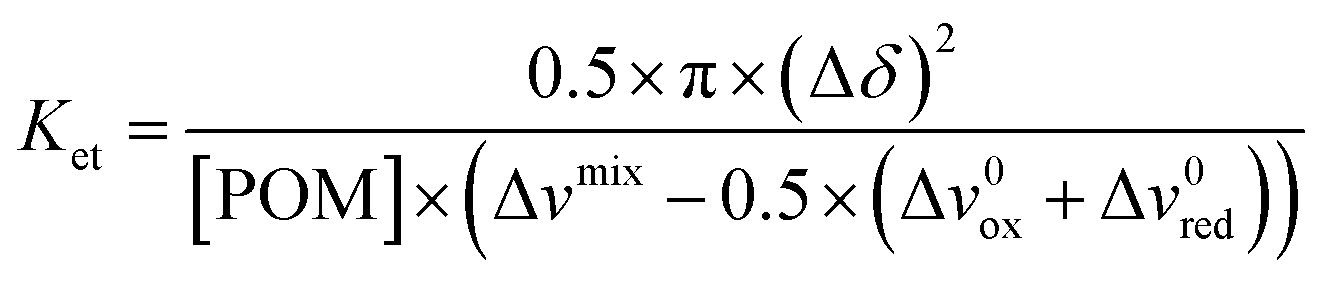 | (3) |
The logKet values determined for the different γ-CD/POM ratios are showed in Fig. 4 except for the ratio 1.5 where the 31P NMR linewidths are too large to be determined properly. In the absence of γ-CD, the Ket determined for the equimolar mixture PW12O403−/PW12O404− is about 6.3 × 106 L mol−1 s−1. Remarkably, the Ket dramatically decreases by several orders of magnitude through the adding of only a few equivalents of γ-CD (see Table S1†), and reach a value of about 330 L mol−1 s−1 when 3 equivalents of γ-CD are introduced in the solution. Although it is known that host–guest supramolecular interactions decreases the PW12O403− reducibility (shift of the first redox event by 150 mV toward more negative potential),15 the dramatic decrease of the electron-rate transfer observed herein should be the consequence of the insulating nature of the γ-CD that shun or at least limit the intermolecular electron transfer. We showed herein that the encapsulation of Keggin-type POM in a hydrophobic cavity lead to a huge variation of POM reactivity toward the electron transfer in homogeneous systems. This observation, supported by previous results showing γ-CD acts as a protecting barrier toward hydrolytic attacks,44 we anticipated the nanoconfinement effect produced by γ-CD cavity offer also a route to control metalate ions exchange between POM species.
 | ||
| Fig. 4 Plot of logKet for electron self-exchange between PW12O403− and PW12O404− as a function of γ-CD concentration. | ||
Nanoconfinement effect on the intermolecular metalate Mo/W exchanges
Although POMs are described as robust and stable species, these species are always involved in dynamic equilibria in aqueous solution.2,56 Such chemical equilibria result from partial decondensation/condensation sequences that give rise to slow chemical exchange of oxometalate ions between POM species. Such a process is strongly favoured by the amphoteric nature of water while it becomes fully cancelled in non-protic solvent. Furthermore, such a phenomenon becomes remarquable from mixtures of polyoxomolybdate and polyoxotungstate ions, whose chemical exchange leads to a large distribution of mixed-metal POMs.57 For instance, van Bekkum and co-workers showed that the Mo/W interconversion processes between PW12O403− and PMo12O403− are highly efficient in water while no exchange occurs in organic media.58 As γ-CD is able to create a hydrophobic environment in the POM vicinity, this could cancel intermolecular metalate exchanges in aqueous solution. Then, we monitored by 31P NMR spectroscopy, aqueous mixtures containing H3[PW12O40] (25 mM) and H3[PMo12O40] (25 mM) and various amounts of CD (from 0 to 100 mM). These experiments were carried out at room temperature and solution equilibration was monitored by 31P NMR.In the absence of γ-CD, the spectrum recorded after 24 hours consists of a statistical distribution of 31P NMR resonances centered at about −8.5 ppm (see Fig. 5). The chemical shift of the central phosphorous of Keggin-type POMs is known to be sensitive to the ratio of molybdenum to tungsten and the 31P NMR spectrum is consistent with a complete distribution including all the possible mixed-metal POMs PW12−xMoxO403− in solution. It must be noted the spectra are very complex due to the existence of positional isomers for all POMs with x varying from 2 to 10. For instance, the theoretical number of positional isomers for PW6Mo6O403− is 48.59 We also note that the 31P NMR signals of PW12O403− and PMo12O403− completely disappeared.
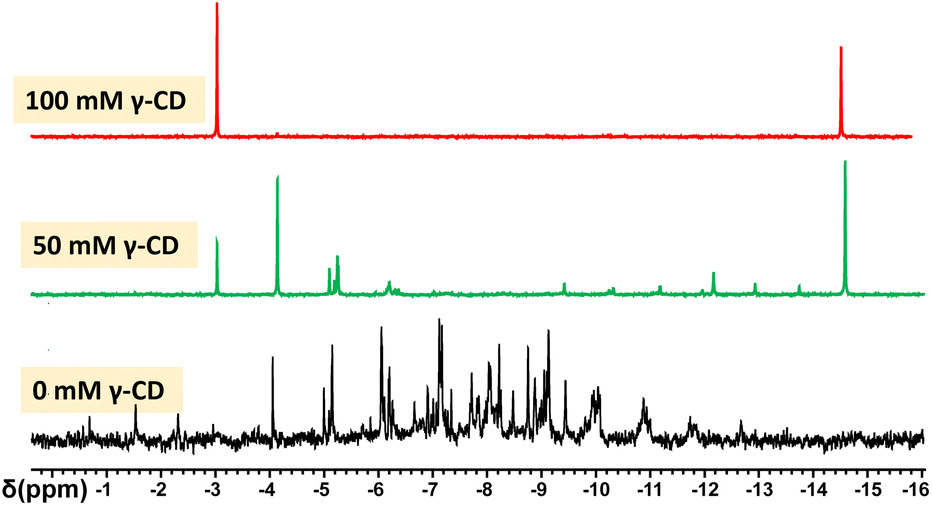 | ||
| Fig. 5 31P NMR spectrum of 24 h aged aqueous solutions in which PW12O403− (25 mM), PMo12O403− (25 mM), and various amounts of γ-CD (from 0 to 100 mM) have been introduced. | ||
The experiments performed in the presence of variable quantities of γ-CD (50 and 100 mM) show that the intermolecular metalate Mo/W exchanges are dramatically slowed down by the presence of the γ-CD (see Fig. 5). In the presence of 100 mM of γ-CD, the spectrum measured after 24 hours exhibits only two peaks observed at −3 and −14.8 ppm, corresponding to the homometallic Keggin anions PMo12O403− and PW12O403−, respectively.
When 50 mM in γ-CD (CD/POM = 1) is introduced, the situation is quite complex since a multimodal distribution of 31P resonances is observed after 24 hours. In this condition, the exchange process is strongly slowed down and it becomes possible to study the intermediate stages of the metalate exchanges (31P NMR spectra and the species distribution are shown in Fig S2 and S3†). This evolution revealed a different reactivity between the POM rich in Mo centers and those rich in W centers. Similarly, quantitative analysis evidences that the quantities of mixed metal POM rich in W centers (x = 1 or 2) remain very low during all processes while the POM entities rich in Mo centers (x = 10 or 11) reach highest concentration at the beginning of the process. This reveals that W exchange process involving PW11MoO403− and PW10Mo2O403− species proceeds faster than PW12O403− and that the kinetic stability of Mo-rich Keggin globally increases by introducing W centres in the metal oxo frameworks. All these observations are fully consistent with the well-known POM behaviour suggesting that the kinetic stability of POM is higher for W-rich POM than for Mo-rich POMs.2,14 Nevertheless, it appears the complexation of the Keggin-type anion by γ-CD provides a way to control to a large extent the metalate exchange processes.
To quantify the impact of the γ-CD concentration on the intermolecular metalate exchanges, we determined the decrease of homometallic POMs concentration can be roughly described as first-order kinetics (Fig. S4†). The rate constant (k) and half-life of a first-order reaction (t1/2) for the decomposition of PMo12O403− and PW12O403− are reported in Table S2.† These results evidenced that the embedment POMs within the macrocyclic host produced a dramatic increase of the half-life of POMs that is multiplied by more than three orders of magnitude when only two equivalents of γ-CD are introduced in the solution (Table S2†).
Toward the stereoselective synthesis of elusive POM
In the previous parts, we demonstrated that the chaotropically-driven complexation of Keggin-type POMs by γ-CD host inhibits intermolecular exchanges. Such a behaviour can be employed as a tool to improve the stereoselectivity of POM synthesis. As a proof of concept, we report in this section the preparation of the molybdenum monosubstituted Keggin-type phosphotungstate PW11MoO403−.The classical way to prepare monosubstituted Keggin-type POMs consists of a condensation process of a metalate species to a monovacant polyoxotungstate XW11O39n− (X = PV or SiIV). Although this process is highly selective for the formation of the SiW11MoO404− ion, that of the phosphato analogue PW11MoO403− is hindered by formation of numerous side-products mainly driven by the M/W metalate exchanges. Indeed, addition of the cationic MoO22+ species to a solution containing preformed PW11O397− as Li+ salt leads to in a few minutes the fast precipitation of an insoluble pale-yellow solid while the supernatant analysis by 31P NMR revealed that the desired anion PW11MoO403− correspond to only 18% of the total compounds. The other side-products are mainly mixed-metal POMs PW12−xMoxO403− (with x from 0 to 3) (see Fig. 6). The situation is dramatically different when six equivalents of γ-CD per PW11O397− precursor were initially introduced in the synthesis solution. In such conditions, no precipitation was observed and the 31P NMR analysis of the mixture contains PW11MoO403− with high purity (>97%) as revealed by 31P NMR analysis (see Fig. 6). This result can be easily understood if we consider that the chaotropic PW11MoO403− ions is protected until its formation by γ-CD. The resulting host–guest aggregate creates a hydrophobic shell that stabilizes chaotropic POMs by cancelling out metalate exchange.
 | ||
| Fig. 6 31P NMR spectra of the supernatant solutions resulting from the addition of cationic MoO22+ species (1.2 eq. per monovacant POM) into a solution of PW11O397− containing no γ-CD or six equivalents of γ-CD. The final pH of both solutions was 0.6. | ||
Conclusion
The supramolecular assemblies resulting from the host–guest association between a Keggin-type POM and γ-CD, have been inspected for the first time using computational methods. Our results indicate that the strength and the binding modes are controlled by the global charge of the POM. This conclusion is in excellent agreement with published experimental observations15,43 in which the chaotropic effect was considered as the main driving force of the encapsulation process.This article does not only decipher the forces behind the encapsulation process of POMs by macrocyclic hosts but also demonstrates experimentally that the striking affinity of super-chaotropic POMs with γ-CD allows it to modulate to a large extent its physical-chemical behaviour such as efficiency of electron transfers or metalates exchange. Furthermore, the chatropically-induced nanoconfinement can be exploited to mimic hydrophobic conditions in aqueous solution.
All these results, both theoretical and experimental, confirm that the solvent effect, often neglected, is ubiquitous in polyoxometalate chemistry, and provide a better understanding of aggregation processes with hydrophobic organic substances and, in fine, alterations to their intrinsic physical-chemical properties.
Data availability
All computational and experimental supporting data and procedures are available in the ESI.†Author contributions
MSC carried out the computational studies under the mentorship of CB who was also responsible for funding acquisition. NL and GMP performed the study about the intermolecular electron transfer under the supervision of CF and EC. MH and CF carried out the study concerning the intermolecular metalate exchanges. NL and CF carried out the experimental part dedicated to the stereoselective synthesis. All authors contributed to the conceptualization of the project, which originated from an initial idea by MSC and CF. MSC and CF created the original draft, which was subsequently edited and reviewed by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the ICIQ-CERCA Program/Generalitat de Catalunya and MICINN (PID2020-112806RB-I00, CEX2019-000925-S), the CNRS (IRN-“smart molecular oxides”), the UVSQ, the Paris Ile-de-France Region-DIM “Respore”, the French National Research Agency (ANR-11-LABX-0039-grant).References
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- N. I. Gumerova and A. Rompel, Chem. Soc. Rev., 2020, 49, 7568–7601 RSC.
- E. Al-Sayed and A. Rompel, ACS Nanosci. Au, 2022, 2, 179–197 CrossRef CAS PubMed.
- P. Yang and U. Kortz, Acc. Chem. Res., 2018, 51, 1599–1608 CrossRef CAS PubMed.
- D. E. Katsoulis, Chem. Rev., 1998, 98, 359–388 CrossRef CAS PubMed.
- Y. Wu, R. Shi, Y.-L. Wu, J. M. Holcroft, Z. Liu, M. Frasconi, M. R. Wasielewski, H. Li and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 4111–4118 CrossRef CAS PubMed.
- B. Naskar, O. Diat, V. Nardello-Rataj and P. Bauduin, J. Phys. Chem. C, 2015, 119, 20985–20992 CrossRef CAS.
- C. Drummond, L. Pérez-Fuentes and D. Bastos-González, J. Phys. Chem. C, 2019, 123, 28744–28752 CrossRef CAS.
- S. Bhattacharya, A. Barba-Bon, T. A. Zewdie, A. B. Müller, T. Nisar, A. Chmielnicka, I. A. Rutkowska, C. J. Schürmann, V. Wagner, N. Kuhnert, P. J. Kulesza, W. M. Nau and U. Kortz, Angew. Chem., Int. Ed., 2022, 61, e202203114 CrossRef CAS PubMed.
- T. Buchecker, P. Schmid, S. Renaudineau, O. Diat, A. Proust, A. Pfitzner and P. Bauduin, Chem. Commun., 2018, 54, 1833–1836 RSC.
- M. Hohenschutz, I. Grillo, O. Diat and P. Bauduin, Angew. Chem., Int. Ed., 2020, 59, 8084–8088 CrossRef CAS PubMed.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596–612 CrossRef CAS PubMed.
- S. Khlifi, J. Marrot, M. Haouas, W. E. Shepard, C. Falaise and E. Cadot, J. Am. Chem. Soc., 2022, 144, 4469–4477 CrossRef CAS PubMed.
- C. Falaise, M. A. Moussawi, S. Floquet, P. A. Abramov, M. N. Sokolov, M. Haouas and E. Cadot, J. Am. Chem. Soc., 2018, 140, 11198–11201 CrossRef CAS PubMed.
- S. Yao, C. Falaise, A. A. Ivanov, N. Leclerc, M. Hohenschutz, M. Haouas, D. Landy, M. A. Shestopalov, P. Bauduin and E. Cadot, Inorg. Chem. Front., 2021, 8, 12–25 RSC.
- K. I. Assaf and W. M. Nau, Org. Biomol. Chem., 2023, 21, 6636–6651 RSC.
- A. Barba-Bon, N. I. Gumerova, E. Tanuhadi, M. Ashjari, Y. Chen, A. Rompel and W. M. Nau, Adv. Mater., 2024, 36, 2309219 CrossRef CAS PubMed.
- A. Kondinski, Polyoxometalates, 2024, 3, 9140058 CrossRef.
- P. Dullinger and D. Horinek, J. Am. Chem. Soc., 2023, 145, 24922–24930 CAS.
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- C. Falaise, S. Khlifi, P. Bauduin, P. Schmid, W. Shepard, A. A. Ivanov, M. N. Sokolov, M. A. Shestopalov, P. A. Abramov, S. Cordier, J. Marrot, M. Haouas and E. Cadot, Angew. Chem., Int. Ed., 2021, 60, 14146–14153 CrossRef CAS PubMed.
- R. Thouvenot, Etude par spectrométrie de vibration de polyanions du W(VI) et du Mo(VI), PhD thesis, Université Pierre et Marie Curie, 1978.
- J. D. Smith, C. D. Cappa, B. M. Messer, W. S. Drisdell, R. C. Cohen and R. J. Saykally, J. Phys. Chem. B, 2006, 110, 20038–20045 CrossRef CAS PubMed.
- Ph. Wernet, D. Nordlund, U. Bergmann, M. Cavalleri, M. Odelius, H. Ogasawara, L. Å. Näslund, T. K. Hirsch, L. Ojamäe, P. Glatzel, L. G. M. Pettersson and A. Nilsson, Science, 2004, 304, 995–999 CrossRef CAS PubMed.
- Y. Zubavicus and M. Grunze, Science, 2004, 304, 974–976 CrossRef CAS PubMed.
- H. J. Bakker and J. L. Skinner, Chem. Rev., 2010, 110, 1498–1517 CrossRef CAS PubMed.
- R. Shi, A. J. Cooper and H. Tanaka, Nat. Commun., 2023, 14, 4616 CrossRef CAS PubMed.
- A. Chaumont and G. Wipff, Phys. Chem. Chem. Phys., 2008, 10, 6940–6953 RSC.
- A. Chaumont and G. Wipff, C. R. Chim., 2012, 15, 107–117 CrossRef CAS.
- X. López, C. Nieto-Draghi, C. Bo, J. B. Avalos and J. M. Poblet, J. Phys. Chem. A, 2005, 109, 1216–1222 CrossRef PubMed.
- T. Rahman, E. Petrus, M. Segado, N. P. Martin, L. N. Palys, M. A. Rambaran, C. A. Ohlin, C. Bo and M. Nyman, Angew. Chem., Int. Ed., 2022, 61, e202117839 CrossRef CAS PubMed.
- D. J. Sures, S. A. Serapian, K. Kozma, P. I. Molina, C. Bo and M. Nyman, Phys. Chem. Chem. Phys., 2017, 19, 8715–8725 RSC.
- F. Leroy, P. Miró, J. M. Poblet, C. Bo and J. Bonet Ávalos, J. Phys. Chem. B, 2008, 112, 8591–8599 CrossRef CAS PubMed.
- M. Segado, M. Nyman and C. Bo, J. Phys. Chem. B, 2019, 123, 10505–10513 CrossRef CAS PubMed.
- A. Solé-Daura, J. M. Poblet and J. J. Carbó, Chem.–Eur. J., 2020, 26, 5799–5809 CrossRef PubMed.
- A. Solé-Daura, V. Goovaerts, K. Stroobants, G. Absillis, P. Jiménez-Lozano, J. M. Poblet, J. D. Hirst, T. N. Parac-Vogt and J. J. Carbó, Chem.–Eur. J., 2016, 22, 15280–15289 CrossRef PubMed.
- P. Schmid, G. Jost, X. Graß, D. Touraud, O. Diat, A. Pfitzner and P. Bauduin, Green Chem., 2022, 24, 2516–2526 RSC.
- P. Yang, W. Zhao, A. Shkurenko, Y. Belmabkhout, M. Eddaoudi, X. Dong, H. N. Alshareef and N. M. Khashab, J. Am. Chem. Soc., 2019, 141, 1847–1851 CrossRef CAS PubMed.
- L. Ni, H. Li, H. Xu, C. Shen, R. Liu, J. Xie, F. Zhang, C. Chen, H. Zhao, T. Zuo and G. Diao, ACS Appl. Mater. Interfaces, 2019, 11, 38708–38718 CrossRef CAS PubMed.
- F. Yang, M. Moors, D. A. Hoang, S. Schmitz, M. Rohdenburg, H. Knorke, A. Charvat, X.-B. Wang, K. Yu. Monakhov and J. Warneke, ACS Appl. Nano Mater., 2022, 5, 14216–14220 CrossRef CAS.
- L. Ni, J. Gu, X. Jiang, H. Xu, Z. Wu, Y. Wu, Y. Liu, J. Xie, Y. Wei and G. Diao, Angew. Chem., Int. Ed., 2023, 135, e202306528 CrossRef.
- H. Guo, L. Li, X. Xu, M. Zeng, S. Chai, L. Wu and H. Li, Angew. Chem., Int. Ed., 2022, 61, e202210695 CrossRef CAS PubMed.
- S. Yao, C. Falaise, S. Khlifi, N. Leclerc, M. Haouas, D. Landy and E. Cadot, Inorg. Chem., 2021, 60, 7433–7441 CrossRef CAS PubMed.
- S. Yao, C. Falaise, N. Leclerc, C. Roch-Marchal, M. Haouas and E. Cadot, Inorg. Chem., 2022, 61, 4193–4203 CrossRef CAS PubMed.
- Z.-G. Jiang, W.-T. Mao, D.-P. Huang, Y. Wang, X.-J. Wang and C.-H. Zhan, Nanoscale, 2020, 12, 10166–10171 RSC.
- W. Guan, G. Wang, B. Li and L. Wu, Coord. Chem. Rev., 2023, 481, 215039 CrossRef CAS.
- B. Zhang, W. Guan, F. Yin, J. Wang, B. Li and L. Wu, Dalton Trans., 2018, 47, 1388–1392 RSC.
- B. Kowalewski, J. Poppe, U. Demmer, E. Warkentin, T. Dierks, U. Ermler and K. Schneider, J. Am. Chem. Soc., 2012, 134, 9768–9774 CrossRef CAS PubMed.
- J. Poppe, E. Warkentin, U. Demmer, B. Kowalewski, T. Dierks, K. Schneider and U. Ermler, J. Inorg. Biochem., 2014, 138, 122–128 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Angew. Chem., Int. Ed., 2018, 57, 13968–13981 CrossRef CAS PubMed.
- A. D. Stergiou and M. D. Symes, Catal. Today, 2022, 384–386, 146–155 CrossRef CAS.
- B. Rausch, M. D. Symes, G. Chisholm and L. Cronin, Science, 2014, 345, 1326–1330 CrossRef CAS PubMed.
- O. Tomita, H. Naito, A. Nakada, M. Higashi and R. Abe, Sustainable Energy Fuels, 2022, 6, 664–673 RSC.
- M. Kozik and L. C. W. Baker, J. Am. Chem. Soc., 1990, 112, 7604–7611 CrossRef CAS.
- M. Kozik, C. F. Hammer and L. C. W. Baker, J. Am. Chem. Soc., 1986, 108, 7627–7630 CrossRef CAS PubMed.
- M. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, Berlin Heidelberg, 1983 Search PubMed.
- M. A. Leparulo-Loftus and M. T. Pope, Inorg. Chem., 1987, 26, 2112–2120 CrossRef CAS.
- M. A. Schwegler, J. A. Peters and H. van Bekkum, J. Mol. Catal., 1990, 63, 343–351 CrossRef CAS.
- M. T. Pope and T. F. Scully, Inorg. Chem., 1975, 14, 953–954 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01949k |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |