
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural response of G protein binding to the cyclodepsipeptide inhibitor FR900359 probed by NMR spectroscopy†
Christian
Bonifer
 a,
Wiebke
Hanke
b,
Jonas
Mühle
c,
Frank
Löhr
a,
Johanna
Becker-Baldus
a,
Jessica
Nagel
d,
Gebhard F. X.
Schertler
c,
Christa E.
Müller
d,
Gabriele M.
König
b,
Daniel
Hilger
*e and
Clemens
Glaubitz
*a
a,
Wiebke
Hanke
b,
Jonas
Mühle
c,
Frank
Löhr
a,
Johanna
Becker-Baldus
a,
Jessica
Nagel
d,
Gebhard F. X.
Schertler
c,
Christa E.
Müller
d,
Gabriele M.
König
b,
Daniel
Hilger
*e and
Clemens
Glaubitz
*a
aInstitute of Biophysical Chemistry, Centre of Biomolecular Magnetic Resonance, Goethe University Frankfurt, Max-von-Laue Str. 9, 60438 Frankfurt, Germany. E-mail: glaubitz@em.uni-frankfurt.de
bInstitute for Pharmaceutical Biology, University of Bonn, Nussallee 6, 53115 Bonn, Germany
cDivision of Biology and Chemistry, Laboratory of Biomolecular Research, Paul Scherrer Institute, Forschungsstr. 111, 5232 Villigen PSI, Switzerland
dDepartment of Pharmaceutical & Medicinal Chemistry, Pharmaceutical Institute, University of Bonn, An der Immenburg 4, 53121 Bonn, Germany
eDepartment of Pharmaceutical Chemistry, University of Marburg, 35037 Marburg, Germany. E-mail: daniel.hilger@pharmazie.uni-marburg.de
First published on 4th July 2024
Abstract
The cyclodepsipeptide FR900359 (FR) and its analogs are able to selectively inhibit the class of Gq proteins by blocking GDP/GTP exchange. The inhibitor binding site of Gq has been characterized by X-ray crystallography, and various binding and functional studies have determined binding kinetics and mode of inhibition. Here we investigate isotope-labeled FR bound to the membrane-anchored G protein heterotrimer by solid-state nuclear magnetic resonance (ssNMR) and in solution by liquid-state NMR. The resulting data allowed us to identify regions of the inhibitor which show especially pronounced effects upon binding and revealed a generally rigid binding mode in the cis conformation under native-like conditions. The inclusion of the membrane environment allowed us to show a deep penetration of FR into the lipid bilayer illustrating a possible access mode of FR into the cell. Dynamic nuclear polarization (DNP)-enhanced ssNMR was used to observe the structural response of specific segments of the Gα subunit to inhibitor binding. This revealed rigidification of the switch I binding site and an allosteric response in the α5 helix as well as suppression of structural changes induced by nucleotide exchange due to inhibition by FR. Our NMR studies of the FR-G protein complex conducted directly within a native membrane environment provide important insights into the inhibitors access via the lipid membrane, binding mode, and structural allosteric effects.
Introduction
The protein superfamily of G protein-coupled receptors (GPCRs) is involved in a plethora of cellular signaling pathways across different organisms. Those integral membrane proteins are activated by extracellular ligands, which induce a conformational change in the receptor upon binding, leading to the transduction of this extracellular signal to the intracellular side, where the eponymous G protein is located.1 G proteins are membrane-anchored heterotrimers of the Gα, Gβ and Gγ subunit, of which the Gα subunit forms the main contact site with the GPCR. Gα consists of an alpha-helical domain and a GTP-binding domain that acts as a GTPase by binding and hydrolyzing the nucleotide guanosine triphosphate (GTP).2 In the inactive state, Gα is bound to guanosine diphosphate (GDP) and Gβγ. Upon GPCR activation, the conformational changes of the receptor lead to a release of GDP from Gα and binding of GTP, which is readily available inside the cell in a concentration surplus over GDP of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3 The GTP-bound active state of Gα dissociates from the Gβγ domains and the GPCR to interact with downstream effectors of the activated signaling pathway, leading to further cellular responses to the external ligand (Fig. 1, top). Once the bound GTP is hydrolyzed, the G protein reverts back into its inactive form and the heterotrimer can bind the GPCR again.4 This is the general model of signal transduction via GPCRs and a large number of those receptors have evolved to accommodate a likewise large number of different ligands and activators. But this wide range of receptors is coupled to only a relatively small number of G protein families, highlighting their relevance as a central element in this signaling cascade. The different G protein families couple to different GPCRs and also initiate distinct downstream signaling pathways. Gs proteins stimulate adenylate cyclase, while Gi proteins inhibit this enzyme. This influence on the intracellular cyclic adenosine monophosphate (cAMP) levels regulates further downstream effects. Gq/11 proteins signal via phospholipase C (PLC), which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2), leading to the release of the second messengers inositol trisphosphate (IP3) and diacyl glycerol (DAG), while G12/13 proteins activate Rho guanine nucleotide exchange factors (GEFs) leading to remodeling of the cytoskeleton.5,6
:1.3 The GTP-bound active state of Gα dissociates from the Gβγ domains and the GPCR to interact with downstream effectors of the activated signaling pathway, leading to further cellular responses to the external ligand (Fig. 1, top). Once the bound GTP is hydrolyzed, the G protein reverts back into its inactive form and the heterotrimer can bind the GPCR again.4 This is the general model of signal transduction via GPCRs and a large number of those receptors have evolved to accommodate a likewise large number of different ligands and activators. But this wide range of receptors is coupled to only a relatively small number of G protein families, highlighting their relevance as a central element in this signaling cascade. The different G protein families couple to different GPCRs and also initiate distinct downstream signaling pathways. Gs proteins stimulate adenylate cyclase, while Gi proteins inhibit this enzyme. This influence on the intracellular cyclic adenosine monophosphate (cAMP) levels regulates further downstream effects. Gq/11 proteins signal via phospholipase C (PLC), which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2), leading to the release of the second messengers inositol trisphosphate (IP3) and diacyl glycerol (DAG), while G12/13 proteins activate Rho guanine nucleotide exchange factors (GEFs) leading to remodeling of the cytoskeleton.5,6
 | ||
| Fig. 1 General model of Gq protein function and inhibition by FR. Extracellular ligands bind to membrane-embedded GPCRs, which conduct the signal to the heterotrimeric G protein. The latter is lipid-anchored to the membrane and the Gα subunit is GDP bound in its inactive state. Upon ligand-induced conformational changes of the GPCR, the G protein undergoes nucleotide exchange of GDP for GTP and the subunits dissociate. Gα and Gβγ are now able to conduct the signal to other downstream effectors in the signaling pathway (top). Once GTP is hydrolyzed by the Gα subunit, the system reverts back to its inactive state (not shown). In the case of FR-inhibition, the selective binding of the cyclodepsipeptide to Gαq proteins blocks the nucleotide exchange and thus keeps the G protein in its inactive heterotrimeric state, prohibiting signal conduction (bottom). | ||
Further investigation of GPCR signaling pathways is also supported by the ability to selectively regulate sections of those pathways.7,8 Selective inhibition of specific G protein families, among other approaches, helps to elucidate the often complex and interconnected cellular signaling events linked to GPCR activation.9 One such selective inhibitor is the cyclic depsipeptide FR900359 (FR), which was isolated from the plant Ardisia crenata10 and has been found to be produced by the plant's endosymbiont Candidatus Burkholderia crenata.11,12 FR acts as a guanine nucleotide dissociation inhibitor (GDI) by blocking the exchange of GDP for GTP upon GPCR-mediated G protein activation.13 It binds selectively to Gαq proteins at the hinge region between the alpha-helical and the GTP-binding domain. FR blocks the conformational changes that accompany nucleotide exchange, thereby keeping the protein in the GDP-bound inactive state and inhibiting further downstream signaling (Fig. 1, bottom). FR is highly selective for the Gq/11 family and shows a longer residence time than the structurally similar Gq inhibitor YM-254890 (YM).14–17 Additionally, FR biosynthesis has been established in the cultivable bacterium Chromobacterium vaccinii MWU205,18 making this molecule a suitable tool to study Gq-related signaling pathways, including the effects of PLC signaling or second messenger effects of IP3 and DAG. Previous studies have shown that FR-mediated Gq inhibition could be therapeutically relevant, for example in asthma treatment15,19 or uveal melanoma,20,21 as the inhibition also affects the GTPase-deficient Gq mutant Q209L, which is a hallmark of this type of cancer.22
An X-ray crystal structure of YM bound to a chimeric Gi/q protein with the N-terminal helix of Gαi1 provided structural information about the binding mode of the cyclic depsipeptide inhibitor between linker 1 and switch I.23 Further analysis by nuclear magnetic resonance (NMR) spectroscopy of FR and YM in solution highlighted the solvent-dependent presence of cis/trans isomers with different proposed binding affinities.24,25 The development of FR- and YM-radioligands14 enabled kinetic studies, pointing to a binding mode of the inhibitor to Gαqvia conformational selection, and highlighting the role of hydrophobic interactions with the isopropyl anchor 2 in FR, leading to a longer residence time compared to YM.16,17 Supported by molecular dynamics (MD) simulations13,25 the current model for FR-mediated locking of the protein in the GDP-bound form has been proposed, involving rigidification of various parts of the Gα domain upon inhibitor binding, which is yet to be experimentally verified. It has also been shown that FR-insensitive G proteins of different families have similar binding sites as Gq, which can be turned into FR-binding sites by certain mutations that stabilize the hydrophobic interaction network of the inhibitor.21,26,27 This greatly enhances the ability of FR to act as a tool to study G protein signaling more broadly.
In this study we investigate 13C15N isotope-labeled FR in solution by liquid-state NMR (L-NMR) spectroscopy and provide an extensive resonance assignment in aqueous solution. We compare this solubilized state with the Gq heterotrimer-bound state and observe the dynamics of the bound inhibitor in the native-like environment of the lipid membrane, which plays a vital part in G protein function28 by solid-state NMR (ssNMR) spectroscopy. In addition, we investigate membrane penetration of FR to understand the access route to the cytoplasmic site of the plasma membrane. Finally, we utilize dynamic nuclear polarization (DNP)-enhanced ssNMR and a unique pair labeling approach to study site-specific effects of inhibitor and nucleotide binding in a FR-sensitive mutant of Gαi.
Experimental
Preparation of isotope-labeled FR
For large-scale production of isotope-labeled FR, Chromobacterium vaccinii MWU205 (German Collection of Microorganisms and Cell Cultures GmbH) was grown in M9 minimal medium (33.7 mM Na2HPO4, 22 mM KH2PO4, 8.55 mM NaCl, 9.35 mM 15NH4Cl, 21.49 mM U-13C-glucose, 1 mM MgSO4, 0.3 mM CaCl2, 134 μM EDTA, 31 μM FeCl3, 6.2 μM ZnCl2, 0.76 μM CuCl2, 0.42 μM CoCl2, 1.62 μM H3BO3, and 0.081 μM MnCl2) supplemented with 50 μg mL−1 carbenicillin. For the optimized M9 culture medium for FR production, 5 mM of U-13C-propionic acid (pH 7) were additionally added. Cultivation was performed at 25 °C for 48 h. Extraction was performed with n-butanol at a 1:1 ratio over night at 180 rpm. After centrifugation at 4000 rpm for 10–15 min, the upper phase of the extract was collected and the solvent was removed by evaporation. The extract was weighed. If not mentioned otherwise, the samples were diluted with LC/MS grade methanol to 1 mg mL−1 for measurement with LC-MS/MS.
LC-MS/MS data were recorded on a micrOTOF-QII mass spectrometer (Bruker) with ESI-source coupled with a HPLC Dionex Ultimate 3000 (Thermo Scientific) using an EC10/2 Nucleoshell C18 2.7 μm column (Macherey-Nagel). The column temperature was 25 °C. MS data were acquired over a range from 100 to 3000 m/z in positive mode. Auto MS/MS fragmentation was achieved with rising collision energy (35–50 keV over a gradient from m/z 500–2000) with a frequency of 4 Hz for all ions over a threshold of 100. High-performance liquid chromatography (HPLC) begins with 90% H2O containing 0.1% AcOH. The gradient starts after 1 min to 100% acetonitrile (0.1% AcOH) in 20 min. A 5 μL amount of a 1 mg mL−1 sample solution (MeOH) was injected to a flow of 0.3 mL min−1. Data analysis was performed using Bruker Compass DataAnalysis Version 4.2 (Build: 383.1).
For isolation of uniformly 13C15N-labeled FR, the crude butanolic extract was fractionated on a Grace Reveleris X2 flash chromatography system with integrated evaporative light scattering (ELSD) and UV-Vis detection via a Reveleris C18 flash column (220 g, 40 μm). A stepwise gradient solvent system of increasing polarity and a flow rate of 65 mL min−1 was used starting with 50/50 H2O/MeOH for 13 min, then changing to 30/70 H2O/MeOH within 1 min and held again for 13 min. The gradient was then changed within 1 min to 25/75 H2O/MeOH and held for 22 min, then within 1 min to 20/80 H2O/MeOH, held for 13 min, then within 1 min to 15/85 H2O/MeOH and held for 15 min. Finally, the gradient was changed within 1 min to 100% MeOH and held for an additional 10 min. According to the measured ELSD and UV signals, a FR containing fraction was collected at 70 min. Final purification was done by HPLC with a semi-preparative Macherey-Nagel Nucleodur C18 column (250 × 8 mm, 5 μm) using an isocratic elution with 20/80 H2O/MeOH (flow 2.0 mL min−1). Uniformly 13C15N-labeled FR was isolated as a white powder (tR: 20 min, 40 mg).
For the small-scale cultures to examine the impact of propionic acid on the FR/FR-2 production ratio, C. vaccinii was grown in unlabeled M9 minimal medium (composition as mentioned above) supplemented with 50 μg mL−1 carbenicillin to avoid contamination. Three cultures with 50 mL M9 medium were inoculated with C. vaccinii and supplemented with 5 mM propionic acid (pH 7), three cultures were inoculated with C. vaccinii but not supplemented, and three cultures were not inoculated. Cultivation was performed at 25 °C for 48 h. Extraction was performed 1:1 with n-butanol over night at 180 rpm. Afterwards, the upper phase was collected using centrifugation at 4000 rpm for 10–15 min followed by evaporation. The extract was weighed. If not mentioned otherwise, the concentration of samples diluted with LC/MS grade methanol was 1 mg mL−1 for measurement with LC/MS. The average AUC for m/z 988.5 and m/z 1002.5 of the controls were deducted from the AUC of each inoculated sample.
High-performance liquid chromatography mass spectrometry (HPLC/MS) data was recorded on a Waters 2695 separation module, which was coupled to a Waters 996 photodiode array detector, and a Waters QDa detector with electrospray ionization source. For separation a gradient elution with mobile phases A (acetonitrile/water 5/95 with 5 mM ammonium acetate and 40 μL acetic acid per Liter) and B (acetonitrile/water 95/5 with 5 mM ammonium acetate and 40 μL acetic acid per liter) on a Waters X Bridge Shield RP18 column (100 × 2.1 mm; 3.5 μm) at 25 °C were used (flow of 0.3 mL min−1, 80/20 A/B to 0/100 A/B within 20 min and hold for 10 min). MS data was collected in positive and negative mode in the range between m/z 140–1250 and additionally in the positive single ion mode for the mass trace of FR (m/z 1002.5; M + H+). HPLC was carried out either using a Waters HPLC system, controlled by Waters Millennium software, consisting of a 600E pump, a 996 PDA detector, and a 717 plus autosampler or on a Waters Breeze HPLC system equipped with a 1525μ dual pump, a 2998 photodiode array detector, and a Rheodyne 7725i injection system.
Insect cell expression of Gq
For the expression of the Gq heterotrimer, three baculoviruses generated by the BestBac method (Expression Systems) were coinfected encoding the wild-type human Gαq subunit, the wild-type human Gβ1γ2 subunits, and the rat guanine nucleotide exchange factor Ric-8A. To facilitate purification of the Gq heterotrimer, a His6-tag with an HRV-3C protease cleavage site was attached at the amino terminus of the Gβ1 subunit. Trichoplusia ni cells (Expression Systems) were infected with the baculoviruses at a cell density of 3 mill per mL followed by incubation of 48 hours at 27 °C. After harvest by centrifugation, cells were lysed in a hypotonic buffer composed of 10 mM Tris, pH 7.5, 100 μM MgCl2, 5 mM β-ME, 10 μM GDP and protease inhibitors (20 μg mL−1 leupeptin, 160 μg mL−1 benzamidine). The membrane fraction was collected by centrifugation and solubilized with 20 mM HEPES, pH 7.5, 100 mM NaCl, 1% Na-cholate, 0.05% DDM, 5 mM MgCl2, 5 mM β-ME, 5 mM imidazole, 10 μM GDP and protease inhibitors. After homogenization with a Dounce homogenizer, the solubilization reaction was incubated for 45 min at 4 °C. After centrifugation, the soluble fraction was loaded onto Ni2+-chelating Sepharose (Cytiva, Munich, Germany) followed by a gradual detergent exchange into 0.1% DDM. The protein was eluted in buffer supplemented with 200 mM imidazole and dialyzed overnight against 20 mM HEPES, pH 7.5, 100 mM NaCl, 0.1% DDM, 1 mM MgCl2, 5 mM β-ME and 10 μM GDP together with HRV-3C protease to cleave off the amino-terminal His6-tag. Cleaved His6-tag, uncleaved fractions and HRV-3C protease were removed by Ni2+-chelating Sepharose. The cleaved G protein was dephosphorylated by lambda protein phosphatase (NEB, Frankfurt, Germany), calf intestinal phosphatase (NEB, Frankfurt, Germany), and antarctic phosphatase (NEB, Frankfurt, Germany) in the presence of 1 mM MnCl2. Lipidated Gq heterotrimer was isolated using a MonoQ 5/50 GL column (Cytiva, Munich, Germany). The protein was bound to the column in buffer A (20 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM MgCl2, 0.1% DDM, 100 μM TCEP, 10 μM GDP) and washed in buffer A. The Gq heterotrimer was eluted with a linear gradient of 0 to 50% buffer B (buffer A + 1 M NaCl). The main peak containing isoprenylated Gq heterotrimer was collected and dialyzed against 20 mM HEPES, pH 7.5, 100 mM NaCl, 0.1% DDM, 100 μM TCEP and 10 μM GDP. The protein was concentrated to 26 mg mL−1, 20% glycerol was added, and the protein was flash frozen in liquid nitrogen and stored at −80 °C until use.Gq protein reconstitution
Liposomes for the reconstitution of the Gq heterotrimer were prepared from POPC, POPE, and cholesterol (Avanti Polar Lipids). The lipid powder was weighed in a 5:4:1 molar ratio and dissolved in chloroform:methanol 1:1 (v/v) to ensure proper mixing. The dissolved lipids were dried overnight on a rotary evaporator at 80 mbar, followed by two hours at 10 mbar to remove the solvents and form a lipid film. This film was resuspended in lipid buffer (20 mM HEPES pH 7.5, 100 mM NaCl) for 1 hour at 42 °C. The lipid suspension was subjected to 5 freeze–thaw cycles and subsequent 11 extrusion cycles through a 0.4 μm membrane to form evenly sized liposomes. The liposomes were used right away for reconstitution or stored at −20 °C. Freshly thawed liposomes were re-extruded before usage.
For reconstitution, the Gq protein was diluted with Gq buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 50 μM GDP, 100 μM TCEP, 1 mM MgCl2, 0.1% DDM) to a concentration of 1 mg mL−1. The protein solution was mixed with the extruded liposome solution at a molar lipid-to-protein ratio (LPR) of 250:1 and stirred for 30 minutes at room temperature. After reconstitution, the detergent was removed by rotating incubation of the sample with bio-beads (Bio-Rad) over night at 4 °C. Once the proteoliposome solution turned turbid again, the bio-beads were removed and the sample was pelleted by ultracentrifugation (30.000×g, 30 minutes). The sample was resuspended in 1 mL lipid buffer and for paramagnetic NMR-doping, 10 μL Gd3+-DOTA [100 mM] was added. After rotating incubation for 30 minutes at 4 °C, the sample was ultracentrifuged again (222.600×g, 30 minutes) to create a dense NMR pellet that was centrifuged into a 3.2 mm or 4 mm NMR-rotor for ssNMR experiments.
E. coli expression of Gαi/q
The expression plasmid for Gαi/q was generated by subcloning the sequence of the mutated GNAI1 gene from an insect cell vector into the pET16b vector for E. coli expression. The gene carried the mutations V50I, K54R, Y69F, V72L, K180P, V185I, T187Y, and H188P necessary for sensitivity of the protein towards FR. Subcloning was performed by amplifying the target gene via PCR and digesting the insert as well as the target plasmid with the restriction enzymes NdeI and XhoI (Thermo Fisher Scientific). Subsequent ligation and transformation into DH5α cells (New England Biolabs GmbH) allowed the confirmation of the subcloned sequence by plasmid isolation and sequencing. In the E. coli expression system, Gαi/q was N-terminally tagged by a 10x His-Tag connected to the protein by a 10 aa linker and put under the expression control of the lac operon. Additionally, the bla gene, leading to ampicillin resistance, on the pET16b vector was used for successful transformant selection.Protein expression was conducted by transforming the expression plasmid into chemically competent E. coli BL21 cells (Thermo Fisher Scientific) via heat shock. After plating on ampicillin-containing agar plates and incubation over night at 37 °C, a single colony was picked and added to 100 mL LB-medium with 0.1 mg mL−1 ampicillin as preculture. The preculture was grown overnight at 37 °C at an agitation of 180 rpm. The main culture consisted of 1 L TB-medium with 0.1 mg mL−1 ampicillin and was inoculated with 10 mL of preculture. During incubation at 37 °C and agitation at 220 rpm the optical density at 600 nm (OD600) was measured to monitor bacterial growth. At an OD600 of 0.6 the cultures were harvested by centrifugation (6400×g, 10 minutes) and remains of the TB-medium were washed away by resuspending the cells in minimal M9-medium and repeated centrifugation. The washed cell pellet was resuspended in and added to 600 mL expression medium of defined M9 medium with different supplements, depending on the chosen NMR-labeling scheme for the sample. For unlabeled protein expression, defined M9-medium was used, supplemented with amino acids, glucose and NH4Cl. For specifically labeled samples, the used amino acids were exchanged for their labeled counterparts. 13C-valine (230 mg L−1) and 15N-proline (100 mg L−1) were used for the VP-labeled sample, 13C15N-arginine (400 mg L−1) and 15N-methionine (250 mg L−1) were used for the RM-labeled sample and 13C-valine (230 mg L−1) and 15N-phenylalanine (130 mg L−1) were used for the VF-labeled sample. For the latter, 4-Hydroxyphenylpyruvic acid (800 mg L−1), a precursor for tyrosine, was also added to prevent metabolic scrambling.29 After adaptation to the expression medium for 1 hour at 37 °C at an agitation of 220 rpm, the expression was induced by the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) to the medium. After induction, the culture was kept at 20 °C for 17 hours at an agitation of 250 rpm. The cells were harvested by centrifugation (6400×g, 10 minutes) and stored at −80 °C or used for protein purification.
For lysis, the cell pellet was dissolved in buffer A (25 mM Tris pH 7.5, 0.5 M NaCl, 5 mM TCEP, 50 mM imidazole, 10% (v/v) glycerol) and passed four times through a cell disruptor at 1.8 bar. The lysed cells were ultracentrifuged (250.000×g, 1 hour) to remove debris and cellular membranes and the supernatant was incubated two hours at 4 °C with buffer A pre-equilibrated TALON cobalt-containing metal affinity resin (Takara Bio) for batch binding and immobilized metal affinity chromatography. After binding, the resin was added to a gravity flow column and washed with 20 column volumes (CV) buffer A to remove unspecifically bound protein from the column. For elution the column was incubated 2 times with 3 CV buffer B (25 mM Tris pH 7.5, 0.5 M NaCl, 5 mM TCEP, 500 mM imidazole, 10% (v/v) glycerol). The eluate was pooled and concentrated with a 10.000 MWCO concentrator (Merck) for buffer exchange to buffer C (25 mM HEPES pH 7.5, 0.15 M NaCl, 2 mM DTT).
Biochemical characterization
Protein concentration was determined via absorption measurements at 280 nm conducted at a nanodrop spectrophotometer with an expected protein mass of 44.01 kDa and an extinction coefficient of 35.870 M−1 cm−1 for Gαi/q. Further analysis and purification by size exclusion chromatography was performed on a Bio-Rad NGC chromatography system with a Superdex 200 Increase 10/300 GL column (GE Healthcare). Protein-containing fractions were identified by their absorption at 280 nm and collected for further experiments. For SDS-PAGE analysis the protein samples were diluted with 4x SDS-PAGE sample buffer (62.5 mM Tris pH 6.8, 0.1 g L−1 LDS, 100 g L−1 glycerol, 255 mM β-mercaptoethanol, 50 mg L−1 bromophenol blue) and 20 μL were loaded into a SurePAGE 4–12% gel (as well as 5 μL of protein marker (PageRuler Prestained Protein Ladder, Thermo Scientific). The gel was run in SDS running buffer (50 mM MES, 50 mM Tris, 1 g L−1 SDS, 1 mM EDTA) for 50 minutes at 150 V for protein band separation. For the detection of the Gγ subunit a 4–20% Mini-PROTEAN gel (Bio-Rad) was run in Rotiphorese buffer (Roth). The gel was stained overnight in staining solution (20% ethanol, 10% glacial acetic acid, 2.5 g L−1 Coomassie R-250) and destained for two hours in destaining solution (20% ethanol, 10% glacial acetic acid).Radioligand binding studies
The purified chimeric Gαi/q protein was preincubated with TALON immobilized metal affinity chromatography (IMAC) resin beads (Takara Bio) for 1–2 h at 4 °C with gentle shaking (0.025 μL beads per 0.1 mL of protein solution in 50 mM Tris–HCl, pH 7.4; final protein amount per sample: 5 μg). Each sample contained 70 μL of assay buffer (50 mM Tris–HCl, pH 7.4), 5 μL of dimethyl sulfoxide (DMSO) or the test compound (dissolved in DMSO), 50 μL of [3H]PSB-15900 (FR-derived radioligand; 28 Ci/mmol; Pharmaron, Cardiff, UK)14 in assay buffer (final concentration: 5 nM) and 125 μL of protein solution. Total binding of [3H]PSB-15900 was determined using 5 μL of DMSO, and non-specific binding was determined using 5 μL of unlabeled FR900359 (final concentration: 5 μM). For competition binding experiments, dilutions from 30 μM to 0.003 μM of unlabeled FR900359, dissolved in DMSO, were added. Incubation was performed for 2 h at 4 °C with gentle shaking. Incubation was terminated by rapid filtration through Whatman GF/C glass fiber filters (GE Healthcare Life Sciences Whatman™, USA) and washing with 4 × 3 mL of ice-cold buffer containing 50 mM Tris–HCl (pH 7.4), 0.1% of bovine serum albumin (BSA), and 0.1% of Tween 20. After punching out the filters, 2.5 mL of scintillation cocktail (ProSafe FC+, Meridian Biotechnologies Ltd, Surrey, UK) were added and the samples were directly measured using a liquid scintillation counter. Data analysis was carried out with GraphPad Prism version 8.0. For calculating KD- and Bmax-values, the one-site homologous equation was used. Data represent means ± standard deviation of three independent experiments, performed in duplicates.Liquid-state NMR experiments
Assignment experiments were recorded on 13C15N-labeled FR [11.65 mM] dissolved in Gq buffer with the addition of 5% D2O and 0.15 mM 2,2-dimethyl-2-silapentan-5-sulfonat (DSS) for referencing. The sample was recorded at an 800 MHz Avance III HD spectrometer with a 5 mm 1H{13C/15N} TCI CryoProbe (Bruker) at a temperature of 298 K. 2D 15N-1H-BEST-TROSY, 2D 15N-1H-TROSY-H(N)CACB, 2D 15N-1H-TROSY-H(N)COC, 2D 15N-1H-TROSY-H(NCA)CO, 2D 13C-1H-HSQC, 2D 13C-detected CON, 2D 13C-detected (HCA)CON, 3D HCN and 3D (H)CCH-TOCSY were recorded for FR resonance assignment. Solvent effects were studied on a sample of the 13C15N-labeled FR dissolved in either Gq buffer with the addition of 5% D2O and 0.15 mM DSS for referencing or CDCl3 referenced to the solvent. The sample in aqueous Gq buffer was recorded at an 800 MHz spectrometer, the CDCl3-dissolved sample at a 1.2 GHz Avance Neo spectrometer with a 3 mm 1H{13C/15N} TCI CryoProbe (Bruker). For both samples 2D 15N-1H-BEST-TROSY spectra were recorded. 1H-1H-NOESY spectra of 13C15N-FR in Gq buffer without detergent to avoid detection of FR-detergent NOE signals were recorded at 1.2 GHz and 298 K with different mixing times (50/100/200/400/800 ms) with referencing to DSS. A sample of the same composition was used to study dynamics of FR in solution at 298 K. At a 600 MHz spectrometer 15N T1 and T2 relaxation times were determined by gradient-selected, sensitivity enhanced HSQC-based experiments and picosecond dynamics were investigated by recording a TROSY version of a {1H}15N-hetNOE experiment. The contribution of chemical exchange was determined by recording 15N-CPMG relaxation dispersion experiments at both 600 and 950 MHz 1H frequencies. The data was analyzed with Dynamics Center 2.8.5 (Bruker) and the Carver Richards Model was utilized to fit the data.30Solid-state NMR experiments
In order to study binding of FR to liposome membranes and the Gq protein, two samples were prepared. The first sample consisted of extruded POPC:POPE:Cholesterol (5:4:1) liposomes, while the second sample contained 836 μg Gq protein reconstituted into the same liposome composition at a molar LPR of 250:1. The samples were pelleted into a 3.2 mm magic-angle spinning (MAS) rotor and in order to study FR binding, 13C15N-labeled FR from a 30 mM stock solution in d6-DMSO was added directly into the rotor at a 2:1 molar surplus over the reconstituted protein. The experiments were conducted at a nominal temperature of 290 K for liposomes in the liquid-crystalline phase and at a nominal temperature of 260 K for the gel phase. Since the sample temperature inside the rotor under MAS conditions can be higher, a more precise sample temperature during the experiment was calculated based on the chemical shift of the H2O signal.31 Based on this calculation the nominal temperatures of 260 K and 290 K corresponded to 275 K and 305 K, respectively. A 600 MHz Avance Neo spectrometer (Bruker) with a 3.2 mm MAS HCN efree probe (Bruker) was used for the experiment and a MAS rate of 14 kHz was chosen. 13C-CP with 83.33 kHz proton decoupling and 13C-INEPT experiments were recorded.
To assess the membrane penetration depth, 13C15N-labeled FR was also added to POPC:POPE:Cholesterol liposomes and 1H-1H-NOESY spectra with different mixing times (50/100/200/400/800 ms) were recorded before and after the addition of FR. A MAS rate of 14 kHz at a 600 MHz spectrometer and nominal temperatures of 290 K for the liquid-crystalline phase and 260 K for the gel phase were chosen. Spin-Lattice relaxation times T1 in the NOESY analysis were determined by an exponential decay fit of the diagonal aromatic signal for the different mixing times.
Two samples of the same makeup as the samples to study FR binding to membranes with or without Gq protein were used to study GTP hydrolysis and hydrolysis inhibition by FR at a nominal temperature of 310 K, corresponding to a calculated sample temperature of 326 K, in the liquid-crystalline phase. Those experiments were conducted with a 4 mm rotor at a 600 MHz spectrometer with a 4 mm MAS HX probe (Bruker) and a MAS rate of 8 kHz was chosen. GTP-hydrolysis was observed by 31P-detection with 100 kHz proton decoupling over a 48 hour spanning series of experiments after the addition of 5 μL of a 17 mM solution of GTP:MgCl2 (1:1 molar ratio). Quantification of β-GTP peaks was done by fitting of the measured β-GTP signal in Origin 2017 with the non-linear Lorentz peak function. The fit error for the peak area was given as error in the respective figure.
Assignment experiments were recorded on a sample of 5 mg reconstituted Gq protein incubated with 13C15N-labeled FR from a 30 mM stock solution in d6-DMSO (2:1 molar surplus) and subsequently washed two times with lipid buffer. The experiments were conducted at a nominal temperature of 260 K for liposomes in the gel phase. For the 13C-assignment, a set of 2D 13C13C-PDSD experiments was recorded with 20/50/200 ms mixing time and for the 15N-assigment, 2D HNCO (0.8/3 ms 15N-CP contact time) and 2D HNCA (0.8/3 ms 15N-CP contact time) experiments were conducted. All assignment experiments were recorded using a 3.2 mm rotor at a 600 MHz spectrometer with a 3.2 mm MAS HCN efree probe, at a MAS rate of 14 kHz and with 83.33 kHz proton decoupling except for the 13C13C-PDSD (200 ms mixing time), which was recorded at an 850 MHz Avance III spectrometer (Bruker) with a 3.2 mm MAS HCN efree probe at a MAS rate of 14 kHz and with 83.33 kHz proton decoupling. NMR-spectra were processed using the software Topspin 4.0.9, resonance assignment was supported by CCPN Version 3.32 The same sample was also used for 2D 15N-SLF (0.8 ms 15N-CP contact time) experiments at identical measurement conditions to determine NH dipolar couplings of Gq-bound 13C15N-labeled FR. Due to the chosen MAS frequency of 14 kHz the R1852 recoupling sequence was applied at 63 kHz. The total echo time was set to 5 ms and 6.6 ms acquisition time was chosen for the indirect dimension. The dipolar coupling was obtained from the peak splitting using the theoretical scaling factor for the applied R1825 recoupling sequence of 0.3038.33 The dipolar order parameter was calculated from the ratio between the measured dipolar coupling and the theoretical NH dipolar coupling value of 11648 Hz, corresponding to an effective bond length of 1.015 Å.34,35
For all samples the phase of the lipid bilayer was determined from the observed 1H line width of the lipid signals. The 1H and 13C referencing in all MAS-NMR experiments was done indirectly to DSS via13C-CP on 1–13C-alanine at 179.85 ppm. 31P referencing was done indirectly to 10% trimethyl phosphate using the 31P signal of triethylphosphine sulfide (TEPS) at 55.58 ppm.
DNP experiments
The protein response to inhibitor binding was studied by HNCO(CX) experiments with 20 ms mixing time recorded on DNP samples of frozen Gαi/q protein solution. The polarizing agent AsymPolPOK36 was dried from a chloroform:methanol (1:1) stock solution to form a film that was resuspended with 1 mg of protein solution that was concentrated to a volume of 15 μL, subsequently 15 μL of d8-glycerol was added. The final concentration of radical in the mixture with the protein sample in the 3.2 mm NMR rotor was 10 mM. The samples were measured at a DNP setup consisting of a 400 MHz Avance Neo spectrometer (Bruker) with a 3.2 mm Cryo MAS HCN probe (Bruker) and a 263 GHz Gyrotron as microwave source. All experiments were done at a nominal temperature of 105 K. A MAS rate of 8 kHz and proton decoupling of 100 kHz were chosen. To investigate the effects of inhibitor binding, the protein solution was incubated with unlabeled FR or YM from a 10 mM DMSO stock solution at an inhibitor to protein molar ratio of 1.5:1 at 4 °C overnight. To study the effects of nucleotide binding, 5 μL of GDP was added to the samples from a 17 mM solution of GDP:MgCl2 (3:1 molar ratio) directly into the rotor. The GTP-bound state was generated by subsequent addition of 2 μL of GTP [0.1 M] and 2 μL AlF4 salt solution (150 mM MgCl2, 75 mM NaF, 15 mM AlCl3). DNP enhancement was measured by comparison of 13C-CP spectra with and without microwave irradiation. Referencing was done indirectly using the right peak of d8-glycerol at 64.78 ppm.
Results and discussion
Preparation of isotope-labeled FR
To conduct a broader range of more complex NMR experiments on FR, isotope labeling of this compound was required. FR production by Chromobacterium vaccinii MWU205 in a chemically defined minimal medium (M9) was established.37 Subsequent exchange of the nitrogen source NH4Cl for 15NH4Cl and the carbon source glucose for the uniformly 13C-labeled glucose enabled the production of uniformly 13C15N-labeled FR. A first cultivation experiment followed by n-butanol extraction, fractionation via flash chromatography, and investigation via liquid chromatography-mass spectrometry (LC-MS/MS) (Fig. S1†) verified the successful isotope incorporation and generation of uniformly 13C15N-labeled FR. However, a large-scale 10 L cultivation experiment only yielded 3 mg 13C15N-FR, at the same time a considerable amount of 55 mg 13C15N-FR-2 was purified. This FR-derivative features N-acetyl-β-hydroxyleucine instead of N-propionyl-β-hydroxyleucine as side chain (Fig. S1 B†). FR/FR-2 production titers showed similar responses when comparing C. vaccinii in complex lysogeny broth (LB) medium and chemically defined minimal M9 medium. In both media, the production peaked after 36 h of cultivation but afterwards the amount of FR and FR-2 decreased in LB medium, while its production in M9 medium remained stable over the course of 7.5 days. While the production titer curve shape was similar for both compounds, the produced amount, indicated by the area under the curve (AUC), was higher for FR-2 in M9 medium compared to FR (Fig. 2A and B).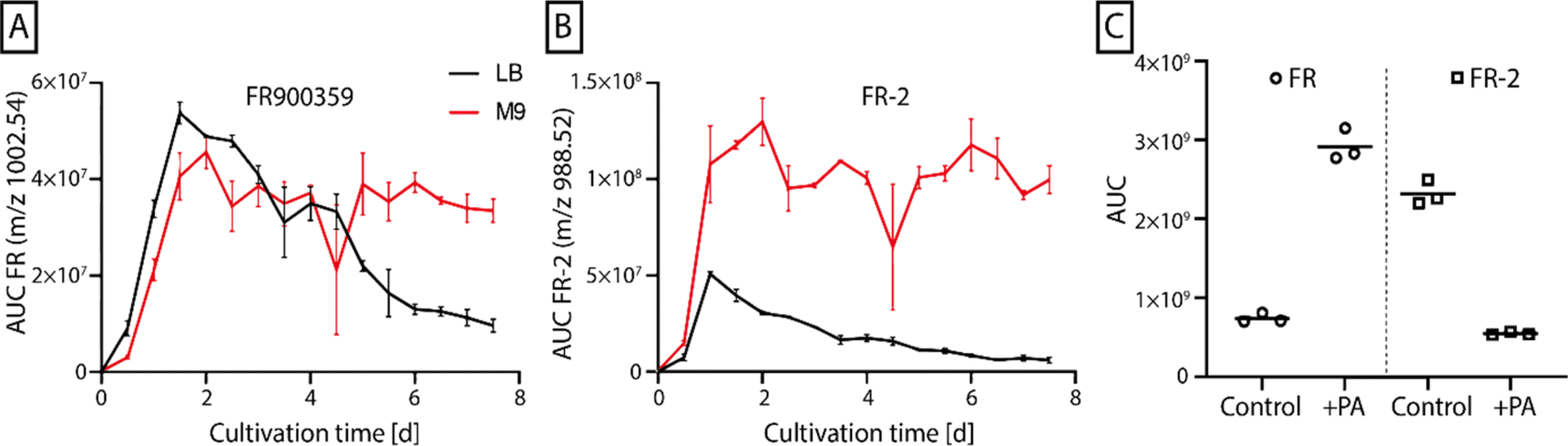 | ||
| Fig. 2 Production of FR900359 measured via LC/MS as AUC for the m/z 1002.54 (A) and FR-2 measured as AUC for the m/z 988.52 (B) by Chromobacterium vaccinii MWU205 cultivated in LB medium (black) and M9 medium (red) over 7.5 d. Error bars show the standard deviation of two repeats. (C) Comparison of FR/FR-2 production cultures in M9 medium without (control) and with addition of 5 mM propionic acid (+PA). Experiments were performed three times. | ||
Since the only difference between the compounds was the incorporation of acetic acid in FR-2 instead of propionic acid (PA) in FR, the observed deviating production levels could be due to a different abundance of PA in the applied media. This was explored in a small-scale experiment with C. vaccinii grown in M9 medium with added PA. Analysis of n-butanol extracts by LC/MS depicted an increase of FR production due to the addition of PA, while FR-2 production levels did decrease (Fig. 2C). This suggested a lower production of PA by C. vaccinii in M9 medium, causing the difference in FR/FR-2 production levels. After optimization by addition of 13C-labeled PA to 4.5 L isotope-labeled culture medium, the production of uniformly 13C15N-labeled FR was enhanced and 40 mg of the labeled compound were purified.
FR resonance assignment in aqueous solution
Uniformly 13C15N-labeled FR was analyzed by L-NMR to confirm successful isotope labeling and assign 1H, 13C and 15N resonances. This analysis was conducted in aqueous buffer, which corresponds to the Gq protein buffer to allow for comparison between the chemical shifts in solvent and Gq-bound FR. The obtained 1H chemical shift were overall comparable with the chemical shifts in H2O/D2O determined in a previous study24 showing only a small uniform perturbation due to the change in buffer. Differences were larger for 13C chemical shifts as they had been determined in organic solvent only. Due to the isotope labeling employed here, also 15N chemical shifts of FR could be determined. Also, 1H-15N correlation spectra of FR in various solvents (aqueous buffer, CDCl3) showed similar sensitivity of the chemical shifts towards the chosen solvent, leading to strong solvent dependent signal shifts (Fig. S2†), highlighting the necessity to choose the appropriate solvent to allow for chemical shift perturbation (CSP) analysis due to protein binding. The four protonated nitrogen atoms were visible in [15N, 1H]-BEST-TROSY spectra (Fig. S3†) together with weaker signals from isotope labeled co-purified impurities. A combination of HCN- and CON-experiments allowed for the detection of three methylated nitrogen residues. In total, all 1H and 15N and 47 of 49 13C resonances were assigned in aqueous buffer (Table S1†).Expression and reconstitution of the Gq heterotrimer
The wild-type human G protein Gq was expressed in Trichoplusia ni (Tni) insect cells using a baculovirus expression system to purify lipid-modified, heterotrimeric G protein capable of associating with the plasma membrane. Gq possesses three lipid moieties: two S-palmitoylations at the N-terminal cysteine residues C9 and C10 of the Gαq subunit, and geranylgeranylation at the C-terminal cysteine C68 of the Gγ2 subunit. These post-translational modifications have been shown to be important for membrane targeting of the G protein heterotrimer.38,39 To increase the yield of functional Gq heterotrimer, it was co-expressed with the rat glutathione S-transferase (GST)-tagged GEF resistance to inhibitors of cholinesterase 8A (Ric-8A), known to fold nascent Gα subunits (Gαi/o, Gα12/13 and Gαq/11) prior to G protein heterotrimer formation.40,41 The Gq heterotrimer was subsequently solubilized from the membrane of the Tni cells using detergents sodium cholate and n-dodecyl-β-D-maltoside (DDM) and purified in DDM to homogeneity using Ni2+-affinity and ion exchange chromatography. The purified protein was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and gel filtration (Fig. S4 A and B†).1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC): 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE):Cholesterol (5:4:1 mol%) liposomes were chosen as suitable membrane environment for the heterotrimeric G protein to conduct ssNMR measurements, reflecting the high amount of PE in the inner leaflet of the plasma membrane, where Gq is located in vivo.42 Cholesterol was added as it is an important constituent of mammalian membranes and effects lipid dynamics and phase transition behavior. In order to assess successful reconstitution of Gq heterotrimer into these liposomes, several samples were taken during the proteoliposome preparation and analyzed by SDS-PAGE for their protein content (Fig. S4 C†). The two bands at ∼40 kDa correspond to Gαq (42.1 kDa) and Gβ1 (37.7 kDa) showing unreconstituted G protein in the supernatant and reconstituted protein in the proteoliposome pellet. The latter gives rise to smeared bands due to the uneven migratory behavior of liposomes compared to lipid-free samples in the gel. No signal was observed in the supernatant of the washing step, thus excluding the presence of loosely liposome-associated protein. The weaker signal for Gγ2 (7.9 kDa) could only be detected on higher percentage SDS-PAGE.
Analysis of Gq-bound FR in liposomes
Gq proteoliposomes were incubated with 13C15N-FR and subsequently MAS ssNMR experiments were recorded. FR was added in surplus and differentiation between bound and free FR was achieved by using cross-polarization (CP) based and insensitive nuclei enhanced by polarization transfer (INEPT) based experiments, respectively.43,44 The experiments were conducted in the gel phase without freezing the sample as those conditions allow for effective CP transfer without freeze-immobilization of soluble compounds. Comparison of the 13C-CP spectra of the G protein proteoliposomes before and after the addition of 13C15N-FR showed strong additional signals in the FR-treated sample from immobilized FR (Fig. 3A). In addition, the J-coupling based INEPT spectrum, which selects for signals from mobile components, showed the free FR in both samples that was not immobilized by binding to the protein. In a control experiment with protein-free liposomes, no FR signals were observed in the 13C-CP spectra, whereas signals were still present in the INEPT experiment, revealing the absence of unspecific binding of FR to the lipids (Fig. 3B). Interestingly, when the control experiments were repeated at a higher temperature, resulting in the more mobile liquid-crystalline membrane phase of the liposomes, FR gave rise to signals in the 13C-CP spectrum, indicating some interaction with the protein-free liposomes (Fig. 3C). A possible explanation for this observation could be that the increased fluidity of the membrane resulted in a more efficient penetration and interaction of FR with the bilayer than in the more densely packed gel phase. As membranes in vivo are in the liquid-crystalline phase, this could provide one mode of access for the inhibitor to cross the plasma membrane and reach its G protein target anchored at the inner leaflet of the lipid bilayer.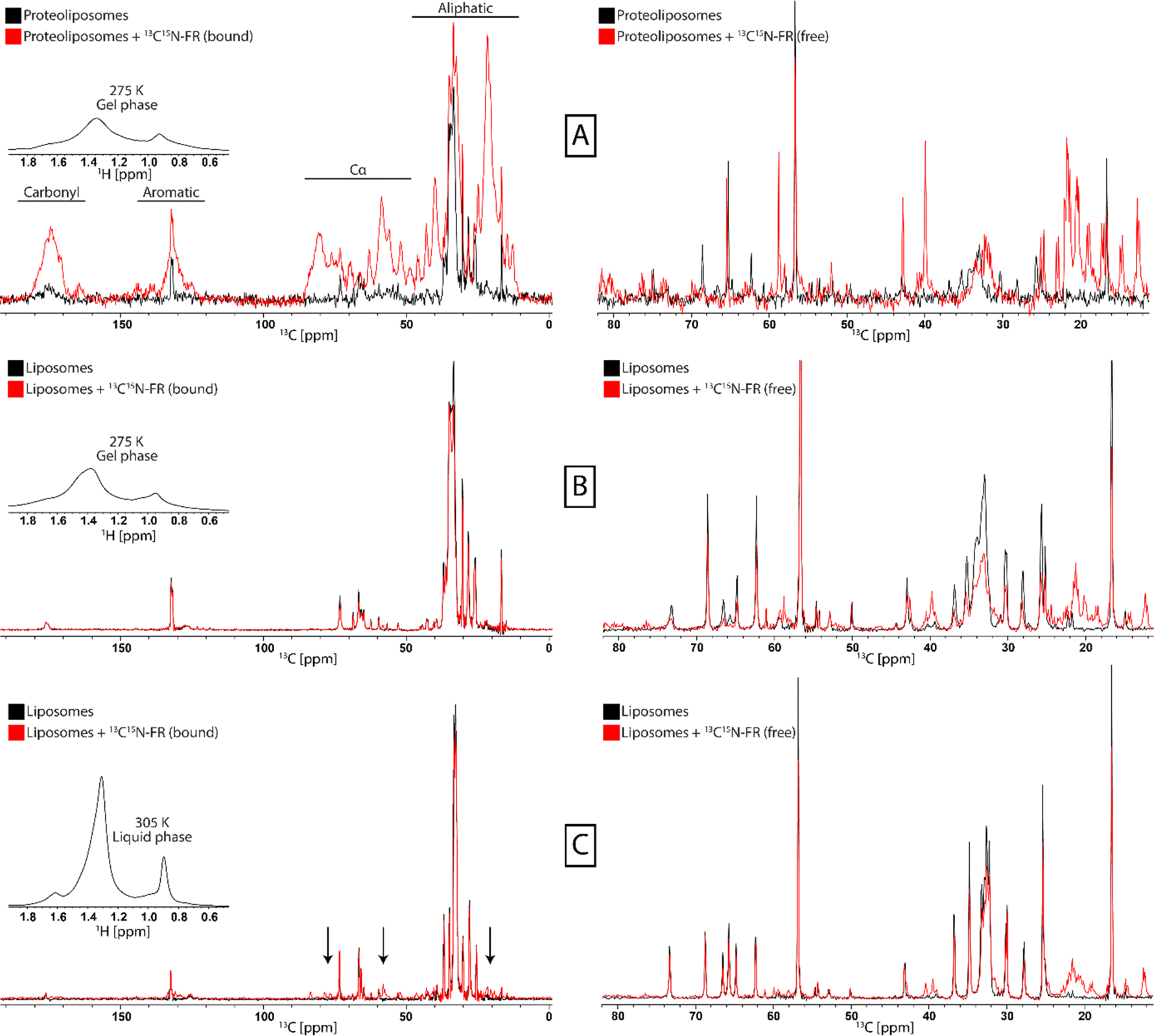 | ||
| Fig. 3 Characterization of interactions between FR and Gq proteoliposomes and liposomes by ssNMR. 13C-CP spectra for the detection of immobile components (left) and 13C-INEPT spectra for the detection of mobile components (rights) were recorded for Gq proteoliposomes in the gel phase (A), POPC:POPE:Cholesterol (5:4:1) liposomes in the gel phase (B), and POPC:POPE:Cholesterol (5:4:1) liposomes in the liquid-crystalline phase (C). In the presence of 13C15N-labeled FR (red) additional strong signals of immobilized inhibitor molecules appear only in the Gq proteoliposome sample, indicating binding to the protein. In addition, for the liposomes in the liquid-crystalline phase some weak signals also appear, pointing to membrane-embedded FR (arrows). The respective membrane phase is monitored by 1H-spectra (insert). The respective CH2 resonances are broad in the gel phase and narrow in the liquid crystalline phase. | ||
FR-membrane interactions
To further investigate the interactions between FR and the lipid bilayer in the liquid-crystalline phase, the membrane penetration was assessed by 1H-1H-NOESY spectra, in which cross-peaks arise from spatially close protons. Here we focused on the cross-peaks arising from the aromatic region (7–8 ppm) of the FR molecule as they do not overlap with any signal from the lipid bilayer (Fig. S5†). Strong cross-peaks were observed in ssNMR 1H-1H-NOESY experiments between these aromatic protons and the lipid molecules (Fig. 4A), namely the CH2 acyl chains (1.3 ppm) and the choline head group (3.24 ppm). Interestingly, also a cross-peak was observed at 0.89 ppm, which could either arise from a contact to the terminal CH3 group of the lipid molecules or from intramolecular correlations within the FR molecule from the N-acetylhydroxyleucine moiety. This intramolecular correlation was investigated by conducting the same experiments as L-NMR 1H-1H-NOESY on the FR molecule alone in solution (Fig. S6†). It was determined that this contact was rather long-ranged, corresponding to a slow nuclear Overhauser effect (NOE)-build-up. In contrast, the build-up of the cross-peak at 0.89 ppm in the NOESY spectra recorded on the liposome samples was much faster (Fig. 4B), despite similar T1 times in both samples (Fig. S7†). This NOE-build-up resembled the observed curve for the short-ranged NOE between protons in the aromatic ring and was therefore determined to correspond to a short-ranged contact to the lipid CH3 group. No contacts between the aromatic region of FR and the acyl chain region of the membrane lipids were detected when the experiments were repeated in the gel phase (Fig. S8†). This shows that FR can penetrate deeply into the lipid bilayer only in the liquid-crystalline phase, opening up a way of access to enter the cell and reach its G protein target. | ||
| Fig. 4 NOE build-up for selected intramolecular FR–FR and intermolecular FR-lipid contacts. (A) The aromatic region of the ssNMR 1H-1H-NOESY (400 ms mixing time) shows cross-peaks that represent intermolecular contacts of the aromatic ring of the phenyllactic acid moiety of FR to various parts of POPC lipids. (B) The normalized cross-peak intensities of ssNMR and L-NMR NOESY spectra were plotted against the experimental mixing times (50/100/200/400/800 ms). The putative FR-lipid contact (black) shows a fast build-up similar to the short-range NOE curve for contacts inside of the aromatic ring of FR (red) in contrast to the intramolecular FR contact between the aromatic ring and the isopropyl group of the N-acetylhydroxyleucine moiety (blue). | ||
FR activity in Gq proteoliposomes
To validate the functionality of the reconstituted G protein, GTP turnover experiments were performed on Gq proteoliposomes using time-resolved 31P-NMR, which can be used to directly follow changes in GTP, GDP, and free phosphate concentrations over time. Incubation of the proteoliposomes with GTP resulted in a complete consumption of the nucleotide within 20 hours, whereas in the control sample without G protein it was stable over this time range, confirming protein-driven GTP hydrolysis. No GDP build-up could be detected during this period due to the instability of GDP under the experimental conditions, leading to auto-hydrolysis, resulting in GMP-specific resonances. When this experiment was repeated with proteoliposomes that were preincubated with FR, a strong inhibition of GTPase activity could be detected, as FR blocks the exchange of GDP for GTP (Fig. 5).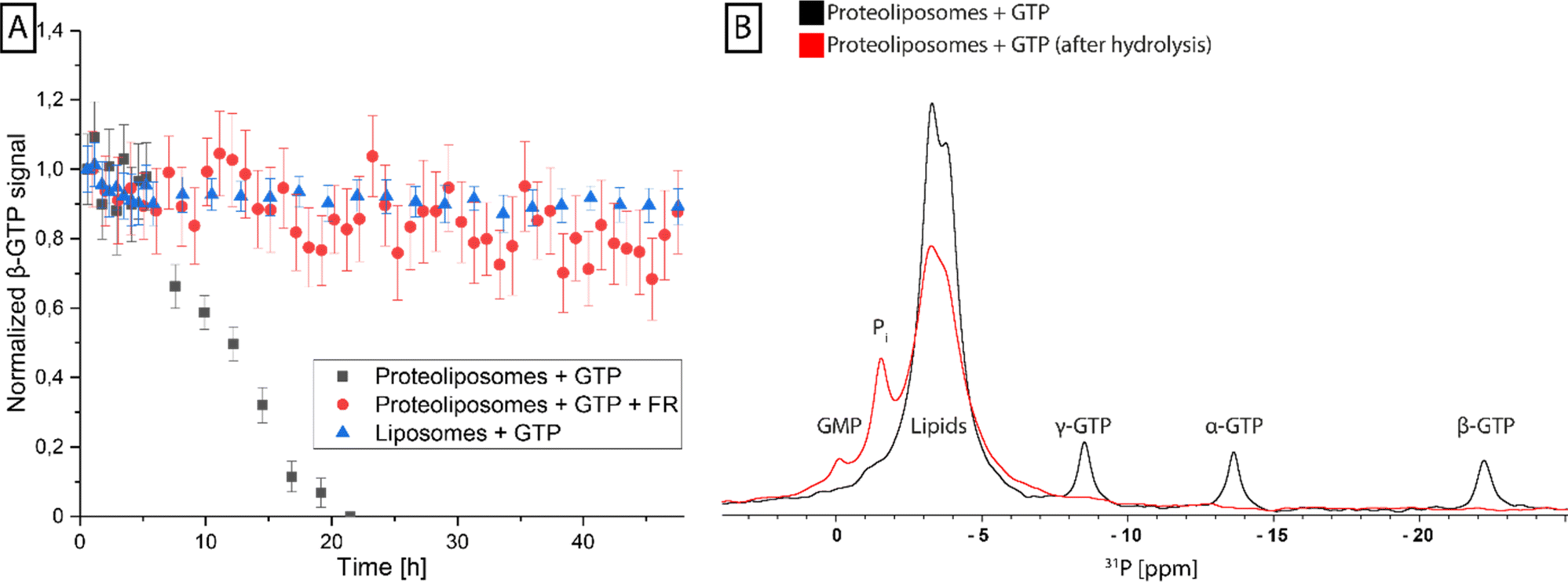 | ||
| Fig. 5 Analysis of GTP hydrolysis of reconstituted Gq protein by ssNMR. (A) Several 31P-spectra were recorded in a real-time analysis to observe the β-GTP signal in the presence of reconstituted Gq protein (gray), FR-inhibited reconstituted Gq protein (red) and POPC:POPE:Cholesterol liposomes (blue). The β-GTP signal was fitted using a Lorentzian peak function and the normalized peak integrals were plotted against the experimental time. Error bars correspond to area error of the peak fit. (B) 31P-spectra of proteoliposomes after the addition of GTP show three distinct signals corresponding to the α-, β- and γ-phosphates of GTP in addition to the phosphate signals of the lipids (black). After the hydrolysis reaction, only the lipid signal remains and two new resonances of inorganic phosphate (Pi) and GMP appear (red). | ||
Chemical shift changes in Gq-bound FR
To compare the signals of G protein-bound FR with free FR, 13C-13C correlation, NCA and NCO spectra were recorded. Protonated and methylated nitrogen atoms could be differentiated in these experiments using different 15N-CP contact times (Fig. S9 + S10†). The acquired spectra were compared to the L-NMR data in aqueous buffer and the assignment was transferred by assigning ssNMR signals with similar chemical shifts as detected by L-NMR. This was validated by the 13C-13C-correlation network to neighboring carbons (Fig. S11–S14†) and nitrogen–carbon correlations found in the NCA and NCO spectra. In total 40 of 49 carbons were detected of which 36 could be assigned unambiguously in the bound state as well as all of the nitrogen resonances (Table S1†). Based on these data, chemical shift differences between the free and bound state were calculated and regions of larger perturbations (>1 ppm) were identified (Fig. 6 + Table S1†).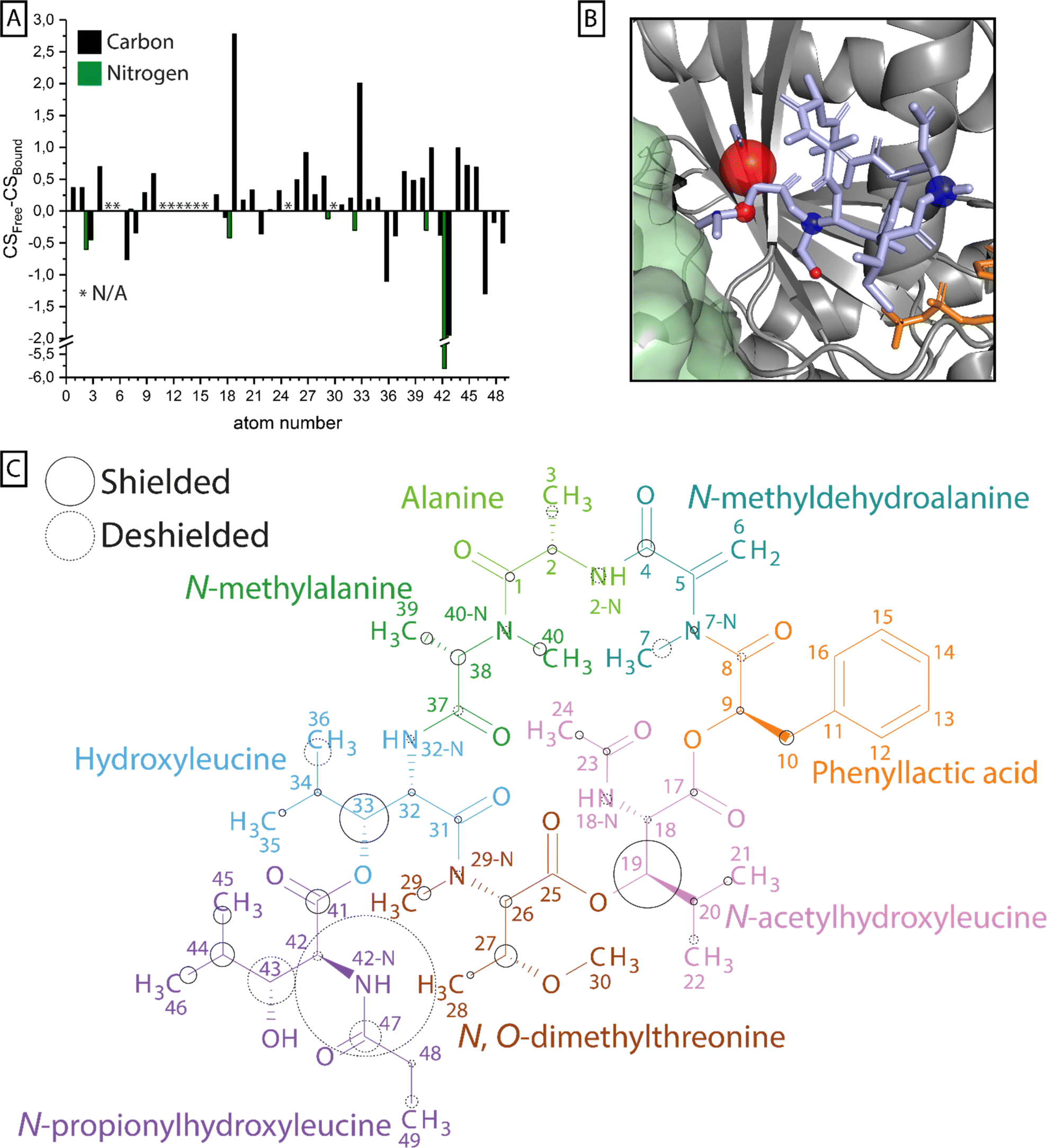 | ||
| Fig. 6 Chemical shift perturbation upon FR binding to Gq. (A) For all carbon (black) and nitrogen (green) atoms in 13C15N-labeled FR that could be assigned in solution by L-NMR and in the protein-bound state by ssNMR, the difference in chemical shift is plotted in a bar chart. (B) Nuclei with chemical shift perturbations ≥ 1 ppm are highlighted by spheres and mapped onto the X-ray structure of YM (light blue) in its binding pocket in the Gα subunit (gray) (PDB 3AH8).23 The size of the spheres corresponds to the magnitude of the shift, while the color indicates shielding (blue) and deshielding (red). In this structure the Gβ subunit (green) and a bound GDP molecule (orange) are also depicted. (C) The calculated chemical shift perturbation was normalized and mapped onto the FR molecular structure. The size of the circles indicates the magnitude of the shift, while the stroke shows shielding (solid) or deshielding (dashed) of the nucleus upon binding. The coloring scheme of FR was adapted from ref. 45. | ||
Previous docking studies have shown that except for the N-methylalanine and the alanine moieties, modifications in the cyclodepsipeptide inhibitors are not well tolerated in terms of Gq binding and inhibitory activity.24 CSP analysis can highlight parts of the molecule that undergo changes in their chemical environment upon Gq binding, potentially indicating their relevance to the inhibitor's functionality. Those changes can be a result of structural rearrangements, forming or dissolving of interactions like H-bonds, or different solvent accessibility. While resonance assignment of FR in solution was less ambiguous due to strong and narrow signals, ssNMR assignment of the Gq-bound form in the presence of the membrane environment was more complex. The absence of fast isotropic tumbling of the observed system leads to partial line broadening caused by slow conformational exchange or structural heterogeneity, making the assignment more challenging. Therefore, especially for chemically similar groups like the methyl groups in the isopropyl moieties or the aromatic ring in FR, a reliable analysis of the chemical shift perturbation was not possible. Multiple regions of the inhibitor molecule displayed changes in the chemical shift upon binding, among those most prominently the NH group of the extracyclic N-propionylhydroxyleucine moiety (ΔCS = 5.81 ppm). Noticeable chemical shift perturbations (ΔCS ≥ 1 ppm) were also found in other parts of the N-propionylhydroxyleucine moiety as well as in the isopropyl groups of N-acetylhydroxyleucine and hydroxyleucine. The large cluster of chemical shift perturbation in the N-propionylhydroxyleucine moiety of FR fits the current binding model in which interactions are formed between the cyclodepsipeptide inhibitor and residues of switch I in Gq like hydrogen bonds between E191 and the free hydroxyl/amide group and I189, which is part of the hydrophobic interaction network that coordinates the isopropyl group in this anchor 1 region of the inhibitor molecule.23 Formation of a hydrogen bond also explains the large CSP observed for 42-NH, since it has been shown that an increase in N–H bond length leads to deshielding of the nitrogen nucleus.46 The methyl groups of the three isopropyl moieties in the inhibitor molecules displayed different chemical shifts in solution, indicating no completely free and fast rotation of those groups even in the unbound state (Table S1†). Once bound to the G protein, only the methyl groups of hydroxyleucine could be assigned unambiguously due to their large difference in chemical shift, which even increased upon binding. The latter observation fits the proposed hydrophobic interactions of this group with I190 and T187 and also explains the large CSP observed for 33-C.23 The last notable CSP in N-acetylhydroxyleucine fits the previously proposed relevance of this FR-specific lipophilic anchor 2 region, leading to a longer residence time compared to YM.17 The N-methylalanine and the alanine moieties have not been identified as part of the pharmacophore of the inhibitor molecule and accordingly no large CSPs were found for those residues.24
Earlier studies have found that cyclodepsipeptide inhibitors occur in different isomeric forms, with the ratio depending on the solvent and varying between FR and YM.25,47 The difference between the isomers lies in the conformation of the various amide groups in the inhibitor molecule. While in the X-ray structure (PDB: 3AH8)23 the G protein-bound YM displays a trans amide bond between N-methyldehydroalanine and phenyllactic acid (trans YM), the predominant conformation of both FR and YM in water features a cis amide bond at this position (cis FR/YM). By comparing published chemical shifts of the isomers, we identified four carbon resonances that displayed a large sensitivity towards the cis/trans conformation (CSP ∼ 3–10 ppm).48 Of those resonances, two (26, 29) are close to the amide bond between N, O-dimethylthreonine and hydroxyleucine and one (40) is close to the amide bond between N-methylalanine and alanine. Both amide bonds are in different conformations in cis/trans YM, respectively. We find that for all three resonances, which show a high sensitivity towards the isomer conformation and are close to amide bonds, our detected chemical shifts correspond to the cis isomer. This indicates that FR is not only mainly in the cis conformation in solution but also when it is bound to its G protein target. This observation aligns with published molecular dynamics simulations.25 These simulations confirmed that the cis YM structure, the major isomer in aqueous solution, also fits the electron density map of the bound YM in the X-ray structure and displays a higher affinity for the binding site compared to the trans YM.25
Dynamics of bound FR
Dynamics of the inhibitor bound to the protein were assessed with a separated local field (SLF) experiment to determine dipolar coupling parameters of the protonated nitrogens, which are well dispersed in the molecule. 1D Slices were taken from the 2D spectra according to the previously determined expected chemical shift of the amides. The signal splitting revealed similar order parameters for amides 2-NH and 18-NH of 0.74 and 0.76 respectively. For amide 32-NH, a slightly higher order parameter of 0.82 was measured. Additionally, at the chemical shift of 42-NH, a slightly more mobile population was detected with an order parameter of 0.68. Analysis of these values must take into account the additional signals in the SLF-experiment that do not correspond to the expected chemical shift regions, due to Gq-binding impurities in the sample. Because of this, no detailed analysis can be conducted, however, it can be concluded that FR is bound to Gq in a relatively rigid binding mode. All regions of the cyclodepsipeptide exhibit similar dynamics. The observed lower order parameter of 42-NH in the N-propionylhydroxyleucine moiety fits the previously observed large CSP for this nucleus. Since the N–H bond length increases upon formation of an hydrogen bond, the NH dipolar coupling decreases, leading to reduced splitting in the SLF spectrum.49 Moreover, the 32-NH amide in the hydroxyleucine moiety has been identified as being the most rigid part of the structure (Fig. 7). In comparison, the measurement of μs/ms-dynamics relaxation parameters of free tumbling FR in solution showed reduced T2 values for 2-NH and 32-NH, hinting at reduced mobilty of the amides that are part of the cyclic backbone of FR compared to the side chain amides 18-NH and 42-NH. Additionally, relaxation dispersion experiments detected higher rates of chemical exchange for 2-NH and to a lesser degree 32-NH, showing that those parts of the inhibitor sample different chemical environments. Measurement of the heteronuclear NOE displayed high mobility for all NH groups in the range of ps-dynamics (Table S2†).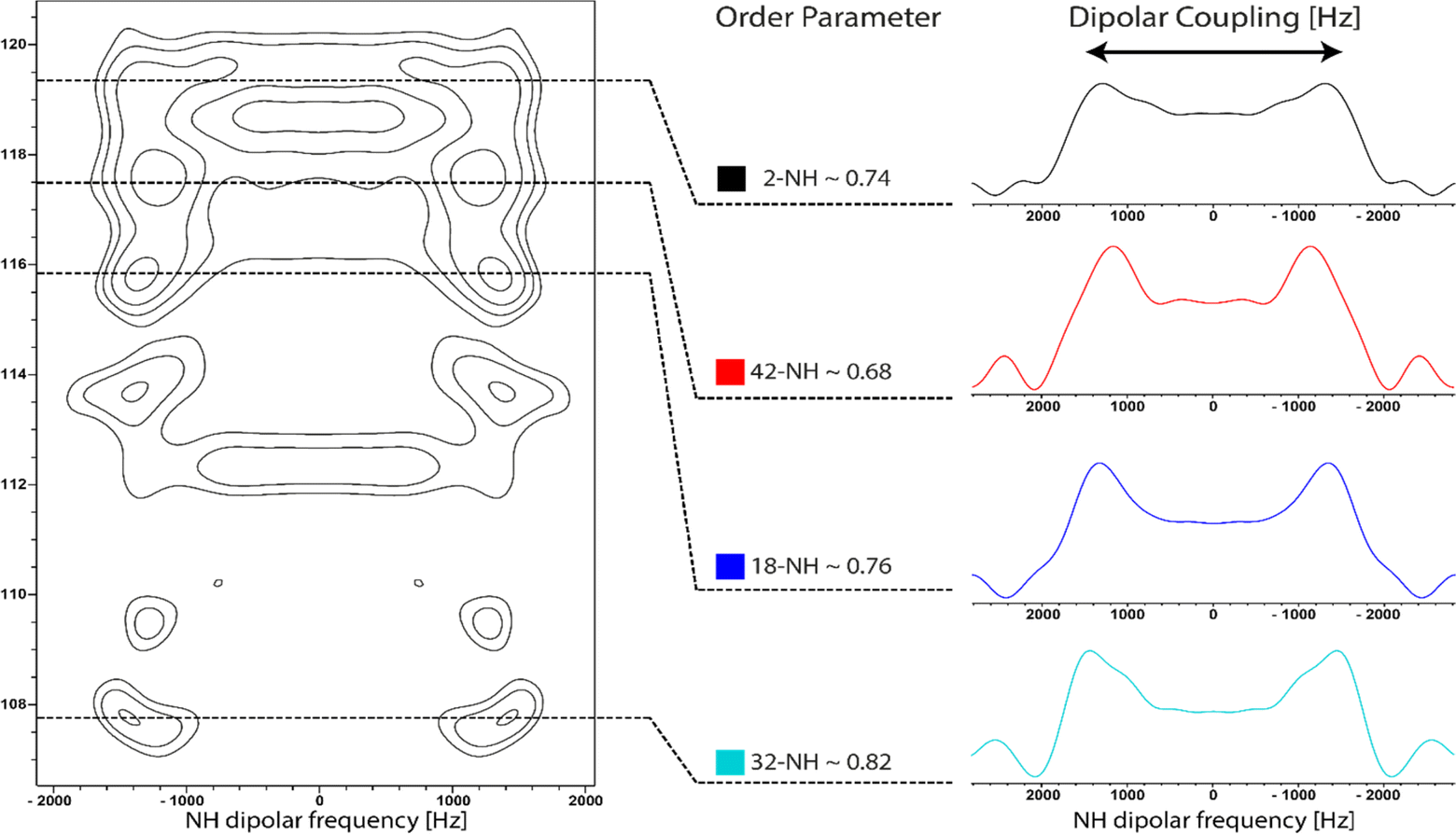 | ||
| Fig. 7 Analysis of inhibitor NH dynamics by ssNMR. 2D 15N-SLF spectrum of 13C15N-FR bound to reconstituted Gq protein. Slices corresponding to the chemical shift of the four amide nitrogens in FR were isolated and dipolar order parameters were calculated. Based on the accuracy of the determination of the dipolar splitting an error of ± 0.02 is estimated for the calculated order parameters. | ||
Spatially resolved conformational response of the G protein
Multiple regions of the protein are either directly involved in binding of cyclodepsipeptide inhibitors, as shown by structural data on bound YM,23 or show an indirect response to binding of the inhibitor, as proposed by MD simulations.13,25 To investigate the conformational response of the G protein to inhibitor binding, structural alterations of the G protein itself must be detected. This requires isotope labeling, which is challenging for the Gq protein due to the necessity for expression in insect cells. Instead, the FR-sensitive Gαi protein mutant Gαi/q, which can be expressed and isotope labeled in E. coli, was chosen as a model.21 The gene for Gαi/q was subcloned from an insect cell expression plasmid (kindly provided by the Schertler group, PSI Villigen) into the pET16b vector for E. coli expression. After purification, biochemical analysis confirmed that the protein was homogeneous and pure, with a yield suitable for NMR experiments (Fig. S15†). Notably, the yield did not decrease upon isotope labeling. The chimeric Gαi/q protein was further characterized by radioligand binding studies using the FR-derived [3H]PSB-15900 as a radioligand.14 The soluble recombinant protein bearing a 10x histidine tag was attached to metal affinity chromatography beads, which allowed to perform filtration assays to separate bound from unbound radioligand. Competition binding studies with increasing concentrations of FR were performed resulting in displacement of the radioligand (Fig. S16†). A KD value of 0.271 ± 0.069 μM was estimated for FR and a Bmax value of 36.3 ± 8.8 pmol mg−1 of protein was calculated. These results clearly show that the employed chimeric Gαi/q protein is properly folded and binds FR with high affinity.In order to collect information about conformational changes in a spatially resolved manner, a unique pair labeling approach was chosen,50 resulting in signals in the NMR spectra from a unique pair of neighboring amino acids in NCO type experiments. We selected the V179-P180 pair in switch I, the R242-M243 pair in switch III, and the V335-F336 pair in the α5 helix, all of which contain amino acids not affected by metabolic scrambling in E. coli. The VP pair is directly involved in FR binding, whereas the RM and VF pair are more distant but predicted by MD simulations to respond to inhibitor binding (Fig. 8).13,25 To obtain sufficient signal intensity in the experiments, spectra were recorded using DNP-enhanced ssNMR. This hybrid electron paramagnetic resonance (EPR)-ssNMR method can enhance sensitivity by orders of magnitude. It requires sample doping with polarizing agents and data acquisition under cryogenic conditions. Under those conditions, the full sampled conformational space is conserved and reflected in NMR line shapes, which can then be analyzed. All measured samples displayed a signal enhancement in the range of 60–80-fold. By comparing the NCO peak position and line shape, local changes can be observed between the free and inhibitor-bound G protein. The experiment showed a clear narrowing of the 13C valine signals in the NCOCX spectrum of the VP-pair sample upon binding of both FR and YM. The VF-pair in the α5 helix of Gαi/q also showed a response to FR-binding, evidenced by the decrease of the two peaks flanking the main signal. For the RM-pair in switch III, no big changes were detectable (Fig. 9 + S17†). Consequently, the switch I region, which is directly involved in inhibitor binding, seems to show a pronounced response to the addition of cyclodepsipeptide inhibitors. Additionally, the more distant region of the α5 helix, despite having no direct contact with the inhibitor binding site, shows a structural response to FR binding, as suggested by the previously published MD simulation data. However, the proposed effect on the switch III region could not be confirmed.
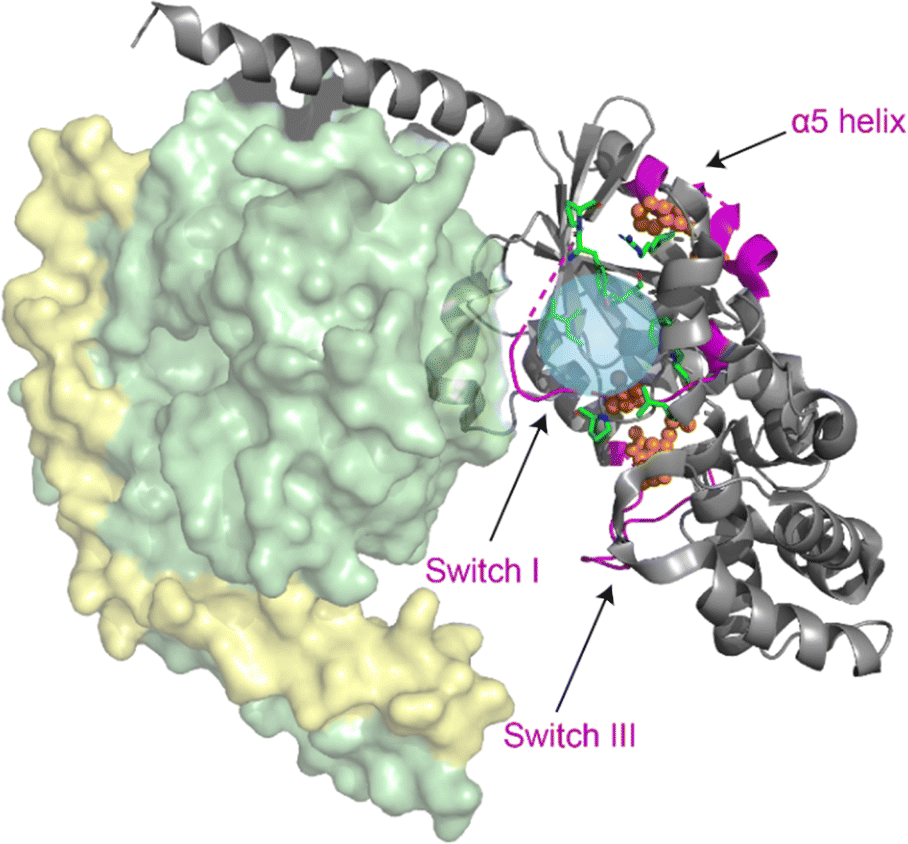 | ||
| Fig. 8 Structural model of the Gi/q heterotrimer (modified from PDB: 1GP2). Eight mutations (green) were introduced into the Gαi protein to generate FR-binding Gαi/q. Probed structural motifs (purple) and the detected unique pairs (orange spheres) are highlighted. The blue sphere marks the inhibitor binding site. DNP-enhanced ssNMR experiments were conducted on the isolated Gαi/q subunit. | ||
 | ||
| Fig. 9 Spatially resolved protein response to FR binding by DNP-enhanced ssNMR. 13C-NCOCX spectrum of frozen Gαi/q solution (black). Labeling schemes were chosen to detect one unique pair signal in switch I (A), switch III (B) or in the α5 helix (C). Addition of FR to the samples leads to narrowing of the signal in switch I, no effects in switch III and changes in the intensity of the flanking signals in the α5 helix (red). | ||
The same set of samples was also used to study the impact of nucleotide binding on the conformation of the protein and to assess how this is affected by prior inhibition with FR. The switch I and II regions in guanine nucleotide-binding proteins generally and additionally switch III in Gα proteins are sensitive towards the bound nucleotide and undergo structural changes upon the release of the γ-phosphate during GTP-hydrolysis.51,52 It has been shown previously that the structural dynamics of the Gα subunit is dependent on the presence and type of nucleotide bound to the Ras domain.53,54 While the apo and GDP-bound protein are more flexible, the GTP-bound G protein adopts a more rigid conformation. For each sample spectra were recorded under the following conditions: Gαi/q, Gαi/q + GDP, Gαi/q + GDP + GTP/AlF4, Gαi/q + FR, Gαi/q + FR + GDP, Gαi/q + FR + GDP + GTP/AlF4. Addition of AlF4 occupies the binding site of the third phosphate group of GTP in the G protein and thereby mimics GTP binding.55 Line shape analysis of the NCO signal revealed structural changes between the initial state after purification, which should be at least partially nucleotide-free, GDP-bound, and GTP-bound state in the switch I and III regions but only minor changes for the α5 helix. This was expected since the switch regions in G proteins are the main conformational responders to nucleotide binding.56 Comparison of the observed changes with samples that were preincubated with FR showed a strong suppression of those effects due to the presence of the inhibitor. This confirms its mode of action, which involves keeping the protein locked in the inactive GDP-bound state (Fig. 10).
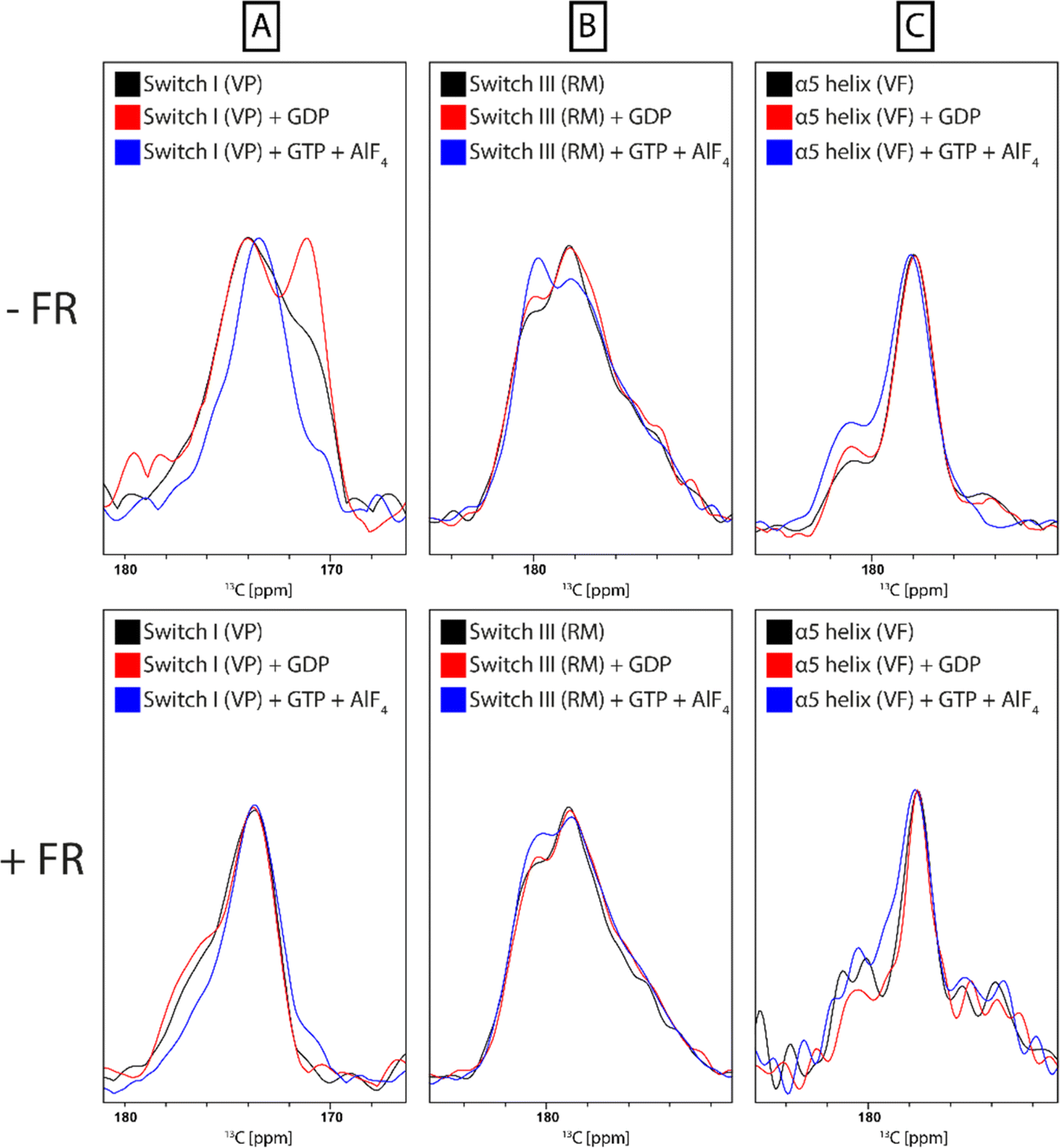 | ||
| Fig. 10 Spatially resolved protein response to nucleotide binding by DNP-enhanced ssNMR. 13C-NCOCX spectrum of frozen Gαi/q solution (black). Labeling schemes were chosen to detect one unique pair signal in switch I (A), switch III (B) or in the α5 helix (C). Signal line shapes are changing for switch I and switch III upon addition of GDP (red) or trapping of the protein in the GTP-bound state by addition of GTP and AlF4 (blue). Only minor changes are observed in the α5 helix. Line shape deviations from the state without nucleotide addition (black) that are visible without prior FR incubation (top) are attenuated upon prior protein incubation with FR (bottom). | ||
Conclusions
The class of cyclodepsipeptide inhibitors provides a powerful tool for studying GPCR-G protein signaling due to their mode of selective inhibition of nucleotide exchange in Gαq. This inhibitory mechanism can be transferred by mutagenesis to other G proteins, making this instrument even more versatile.21,26,27 Purification of the inhibitor FR900359 in a 13C15N isotope-labeled form enabled the characterization of this cyclodepsipeptide in a membrane environment and also directly bound to its G protein target. Here, we present an extensive resonance assignment of FR in aqueous solution and bound to the membrane-anchored Gq protein heterotrimer. Chemical shift perturbation analysis between those two states, which both mimic native-like states, revealed parts of the inhibitor molecule that are particularly affected by protein binding. This is in line with previous docking studies, kinetic and structural data.17,23,24 Our findings show that FR is in the cis conformation in both the solubilized as well as in the Gq-bound state. This supports previous MD simulations proposing a higher binding affinity for the cis compared to the trans isomer found in the G protein-bound X-ray structure of YM.25 In a recently solved X-ray structure of FR in complex with G11 the cis isomer was also observed and a contact between Gβ and the N-propionylhydroxyleucine moiety of the inhibitor was identified.57 The large CSP observed for 42-NH indicates a FR-Gq hydrogen bond which is compatible with the heterotrimer-stabilizing hydrogen bond network between FR and Gq as derived from the before mentioned X-ray structure. Although FR does not interact with our liposomes in the gel phase, bound FR was detected by cross-polarization experiments in the physiologically relevant liquid-crystalline phase. Further investigation of intramolecular and intermolecular FR-lipid contacts by 1H-1H-NOESY experiments revealed full penetration of membranes by FR in the more fluid liquid-crystalline phase, providing a mode of access for the small molecule into the cell. The dynamics of the FR molecule in the bound state were studied by SLF-experiments on the well dispersed nitrogen atoms in the inhibitor molecule, showing an overall rigid FR structure in the bound state. The weaker dipolar coupling for the N–H bond in the N-propionylhydroxyleucine moiety again points to the formation of a hydrogen bond network in this part of the inhibitor binding pocket, possibly involving the Gβ subunit.57 On the protein side of the system, we observed the hydrolysis activity of the membrane-anchored Gq protein in real-time by 31P-NMR, as well as the inhibitory effects of FR on the hydrolysis reaction due to the diminished nucleotide exchange. For a spatially-resolved analysis of structural changes in the G protein upon inhibitor binding, we utilized the aforementioned sensitization of otherwise FR-insensitive G protein families. Gαi/q, a FR-sensitive mutant of Gαi, displayed high affinity binding of FR in a radioligand binding assay and the system was analyzed by DNP-enhanced ssNMR. Based on the chosen unique pair labeling scheme, the line shape analysis of the signals revealed spatially resolved responses of the protein. We could demonstrate an allosteric response in the α5-helix in addition to the expected effect in the direct binding site (switch I). This finding supports previous MD simulations13,25 but a predicted response within the switch III region was not observed. Additionally, the effects of nucleotide binding on the switch regions were observed as changes of the conformational space of those structural motifs upon switching between the GDP- or GTP-bound state. In line with the cyclodepsipeptide's mode of action as a GDI, these observed changes could be suppressed by prior incubation of the protein with FR. In summary, L-NMR, ssNMR, as well as DNP-enhanced experiments allowed for the acquisition of a broad range of diverse information about the FR-G protein system. In conjunction with other structural and biochemical data, these methods help assess the current model of the FR binding mode. Especially, the native-like sample conditions, like inclusion of the membrane and an aqueous environment, that can be combined with NMR, make this method a valuable lens through which one can study such systems.Data availability
NMR raw data can be obtained via the Goethe University data repository (https://gude.uni-frankfurt.de/).Author contributions
CB prepared samples, carried out solid-state NMR experiments, performed data analysis, interpreted data, prepared figures and wrote the paper. WH and GK produced labeled FR, analyzed samples and contributed to figures and paper writing. JM and GS provided plasmids and initial protocols and contributed to discussions and experimental design. FL carried out solution-state NMR experiments and performed data analysis. JB helped in performing solid-state NMR experiments, helped with data interpretation and discussion and contributed to the project design. JN and CM provided the radioligand binding studies and contributed to paper writing. DH provided Gq, helped with data interpretation and contributed to paper writing. CG designed the project, contributed to experimental design, data interpretation, discussion and paper writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Evi Kostenis, University of Bonn for helpful and inspiring discussion. This work was funded by DFG FOR 2372 “G-protein signaling cascades: using new molecular probes and active substances to develop new pharmacological concepts”, project number 273251628. WH acknowledges support by a DBU PhD scholarship 20018/568. DH thanks Betsy White, Stanford University for help with the purification of Gq. Jan H. Voss, University of Bonn is acknowledged for providing material for initial experiments. Stefan Kehraus, University of Bonn is acknowledged for help with FR quality control. Christian Czupalla, Institute of Biophysics, Goethe University is acknowledged for support with phase transition controls. Ingrid Weber is acknowledged for technical support.References
- W. M. Oldham and H. E. Hamm, Heterotrimeric G protein activation by G-protein-coupled receptors, Nat. Rev. Mol. Cell Biol., 2008, 9, 60–71 CrossRef CAS PubMed.
- S. R. Sprang, Invited review: activation of G proteins by GTP and the mechanism of Galpha-catalyzed GTP hydrolysis, Biopolymers, 2016, 105, 449–462 CrossRef CAS.
- J. L. Bos, H. Rehmann and A. Wittinghofer, GEFs and GAPs: critical elements in the control of small G proteins, Cell, 2007, 129, 865–877 CrossRef CAS.
- J. P. Mahoney and R. K. Sunahara, Mechanistic insights into GPCR-G protein interactions, Curr. Opin. Struct. Biol., 2016, 41, 247–254 CrossRef CAS PubMed.
- G. Milligan and E. Kostenis, Heterotrimeric G-proteins: a short history, Br. J. Pharmacol., 2006, 147(Suppl 1), S46–S55 CAS.
- M. I. Simon, M. P. Strathmann and N. Gautam, Diversity of G proteins in signal transduction, Science, 1991, 252, 802–808 CrossRef CAS.
- D. M. Gill and R. Meren, ADP-ribosylation of membrane proteins catalyzed by cholera toxin: basis of the activation of adenylate cyclase, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3050–3054 CAS.
- R. E. West Jr., J. Moss, M. Vaughan, T. Liu and T. Y. Liu, Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site, J. Biol. Chem., 1985, 260, 14428–14430 CrossRef CAS.
- A. P. Campbell and A. V. Smrcka, Targeting G protein-coupled receptor signalling by blocking G proteins, Nat. Rev. Drug Discovery, 2018, 17, 789–803 CrossRef CAS.
- M. Fujioka, S. Koda, Y. Morimoto and K. Biemann, Structure of Fr900359, a Cyclic Depsipeptide from Ardisia-Crenata Sims, J. Org. Chem., 1988, 53, 2820–2825 CrossRef CAS.
- A. Carlier, L. Fehr, M. Pinto-Carbo, T. Schaberle, R. Reher, S. Dessein, G. Konig and L. Eberl, The genome analysis of Candidatus Burkholderia crenata reveals that secondary metabolism may be a key function of the Ardisia crenata leaf nodule symbiosis, Environ. Microbiol., 2016, 18, 2507–2522 CrossRef CAS.
- M. Crusemann, R. Reher, I. Schamari, A. O. Brachmann, T. Ohbayashi, M. Kuschak, D. Malfacini, A. Seidinger, M. Pinto-Carbo, R. Richarz, T. Reuter, S. Kehraus, A. Hallab, M. Attwood, H. B. Schioth, P. Mergaert, Y. Kikuchi, T. F. Schaberle, E. Kostenis, D. Wenzel, C. E. Muller, J. Piel, A. Carlier, L. Eberl and G. M. Konig, Heterologous Expression, Biosynthetic Studies, and Ecological Function of the Selective Gq-Signaling Inhibitor FR900359, Angew Chem. Int. Ed. Engl., 2018, 57, 836–840 CrossRef.
- R. Schrage, A. L. Schmitz, E. Gaffal, S. Annala, S. Kehraus, D. Wenzel, K. M. Bullesbach, T. Bald, A. Inoue, Y. Shinjo, S. Galandrin, N. Shridhar, M. Hesse, M. Grundmann, N. Merten, T. H. Charpentier, M. Martz, A. J. Butcher, T. Slodczyk, S. Armando, M. Effern, Y. Namkung, L. Jenkins, V. Horn, A. Stossel, H. Dargatz, D. Tietze, D. Imhof, C. Gales, C. Drewke, C. E. Muller, M. Holzel, G. Milligan, A. B. Tobin, J. Gomeza, H. G. Dohlman, J. Sondek, T. K. Harden, M. Bouvier, S. A. Laporte, J. Aoki, B. K. Fleischmann, K. Mohr, G. M. Konig, T. Tuting and E. Kostenis, The experimental power of FR900359 to study Gq-regulated biological processes, Nat. Commun., 2015, 6, 10156 CrossRef CAS PubMed.
- M. Kuschak, V. Namasivayam, M. Rafehi, J. H. Voss, J. Garg, J. G. Schlegel, A. Abdelrahman, S. Kehraus, R. Reher, J. Kuppers, K. Sylvester, S. Hinz, M. Matthey, D. Wenzel, B. K. Fleischmann, A. Pfeifer, A. Inoue, M. Gutschow, G. M. Konig and C. E. Muller, Cell-permeable high-affinity tracers for Gq proteins provide structural insights, reveal distinct binding kinetics and identify small molecule inhibitors, Br. J. Pharmacol., 2020, 177, 1898–1916 CrossRef CAS.
- J. G. Schlegel, M. Tahoun, A. Seidinger, J. H. Voss, M. Kuschak, S. Kehraus, M. Schneider, M. Matthey, B. K. Fleischmann, G. M. Konig, D. Wenzel and C. E. Muller, Macrocyclic Gq Protein Inhibitors FR900359 and/or YM-254890-Fit for Translation?, ACS Pharmacol. Transl. Sci., 2021, 4, 888–897 CrossRef CAS PubMed.
- J. H. Voss, J. Nagel, M. Rafehi, R. Guixa-Gonzalez, D. Malfacini, J. Patt, S. Kehraus, A. Inoue, G. M. Konig, E. Kostenis, X. Deupi, V. Namasivayam and C. E. Muller, Unraveling binding mechanism and kinetics of macrocyclic Galpha(q) protein inhibitors, Pharmacol. Res., 2021, 173, 105880 CrossRef CAS PubMed.
- J. H. Voss, M. Crusemann, C. R. O. Bartling, S. Kehraus, A. Inoue, G. M. Konig, K. Stromgaard and C. E. Muller, Structure-affinity and structure-residence time relationships of macrocyclic G alpha(q) protein inhibitors, Iscience, 2023, 26(4), 106492 CrossRef CAS PubMed.
- C. Hermes, R. Richarz, D. A. Wirtz, J. Patt, W. Hanke, S. Kehraus, J. H. Voss, J. Kuppers, T. Ohbayashi, V. Namasivayam, J. Alenfelder, A. Inoue, P. Mergaert, M. Gutschow, C. E. Muller, E. Kostenis, G. M. Konig and M. Crusemann, Thioesterase-mediated side chain transesterification generates potent Gq signaling inhibitor FR900359, Nat. Commun., 2021, 12, 144 CrossRef CAS.
- M. Matthey, R. Roberts, A. Seidinger, A. Simon, R. Schroder, M. Kuschak, S. Annala, G. M. Konig, C. E. Muller, I. P. Hall, E. Kostenis, B. K. Fleischmann and D. Wenzel, Targeted inhibition of Gq signaling induces airway relaxation in mouse models of asthma, Sci. Transl. Med., 2017, 9(407), eaag2288 CrossRef.
- D. Lapadula, E. Farias, C. E. Randolph, T. J. Purwin, D. McGrath, T. H. Charpentier, L. Zhang, S. Wu, M. Terai, T. Sato, G. G. Tall, N. Zhou, P. B. Wedegaertner, A. E. Aplin, J. Aguirre-Ghiso and J. L. Benovic, Effects of Oncogenic Galpha(q) and Galpha(11) Inhibition by FR900359 in Uveal Melanoma, Mol. Cancer Res., 2019, 17, 963–973 CrossRef CAS PubMed.
- M. D. Onken, C. M. Makepeace, K. M. Kaltenbronn, S. M. Kanai, T. D. Todd, S. Wang, T. J. Broekelmann, P. K. Rao, J. A. Cooper and K. J. Blumer, Targeting nucleotide exchange to inhibit constitutively active G protein alpha subunits in cancer cells, Sci. Signaling, 2018, 11(546), eaao6852 CrossRef.
- S. Annala, X. Feng, N. Shridhar, F. Eryilmaz, J. Patt, J. Yang, E. M. Pfeil, R. D. Cervantes-Villagrana, A. Inoue, F. Haberlein, T. Slodczyk, R. Reher, S. Kehraus, S. Monteleone, R. Schrage, N. Heycke, U. Rick, S. Engel, A. Pfeifer, P. Kolb, G. Konig, M. Bunemann, T. Tuting, J. Vazquez-Prado, J. S. Gutkind, E. Gaffal and E. Kostenis, Direct targeting of Galphaq and Galpha11 oncoproteins in cancer cells, Sci. Signaling, 2019, 12(573), eaau5948 CrossRef PubMed.
- A. Nishimura, K. Kitano, J. Takasaki, M. Taniguchi, N. Mizuno, K. Tago, T. Hakoshima and H. Itoh, Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13666–13671 CrossRef CAS PubMed.
- R. Reher, T. Kuhl, S. Annala, T. Benkel, D. Kaufmann, B. Nubbemeyer, J. P. Odhiambo, P. Heimer, C. A. Bauml, S. Kehraus, M. Crusemann, E. Kostenis, D. Tietze, G. M. Konig and D. Imhof, Deciphering Specificity Determinants for FR900359-Derived Gq alpha Inhibitors Based on Computational and Structure-Activity Studies, ChemMedChem, 2018, 13, 1634–1643 CrossRef CAS.
- D. Tietze, D. Kaufmann, A. A. Tietze, A. Voll, R. Reher, G. Konig and F. Hausch, Structural and Dynamical Basis of G Protein Inhibition by YM-254890 and FR900359: An Inhibitor in Action, J. Chem. Inf. Model., 2019, 59, 4361–4373 CrossRef CAS PubMed.
- D. Malfacini, J. Patt, S. Annala, K. Harpsoe, F. Eryilmaz, R. Reher, M. Crusemann, W. Hanke, H. Zhang, D. Tietze, D. E. Gloriam, H. Brauner-Osborne, K. Stromgaard, G. M. Konig, A. Inoue, J. Gomeza and E. Kostenis, Rational design of a heterotrimeric G protein alpha subunit with artificial inhibitor sensitivity, J. Biol. Chem., 2019, 294, 5747–5758 CrossRef CAS PubMed.
- M. W. Boesgaard, K. Harpsoe, M. Malmberg, C. R. Underwood, A. Inoue, J. M. Mathiesen, G. M. Konig, E. Kostenis, D. E. Gloriam and H. Brauner-Osborne, Delineation of molecular determinants for FR900359 inhibition of Gq/11 unlocks inhibition of Galphas, J. Biol. Chem., 2020, 295, 13850–13861 CrossRef CAS PubMed.
- O. Vogler, J. M. Barcelo, C. Ribas and P. V. Escriba, Membrane interactions of G proteins and other related proteins, Biochim. Biophys. Acta, 2008, 1778, 1640–1652 CrossRef.
- R. M. Rasia, B. Brutscher and M. J. Plevin, Selective Isotopic Unlabeling of Proteins Using Metabolic Precursors: Application to NMR Assignment of Intrinsically Disordered Proteins, Chembiochem, 2012, 13, 732–739 CrossRef CAS PubMed.
- J. P. Carver and R. E. Richards, General 2-Site Solution for Chemical Exchange Produced Dependence of T2 Upon Carr-Purcell Pulse Separation, J. Magn. Reson., 1972, 6, 89 CAS.
- J. P. Demers, P. Fricke, C. Shi, V. Chevelkov and A. Lange, Structure determination of supra-molecular assemblies by solid-state NMR: practical considerations, Prog. Nucl. Magn. Reson. Spectrosc., 2018, 109, 51–78 CrossRef CAS.
- S. P. Skinner, R. H. Fogh, W. Boucher, T. J. Ragan, L. G. Mureddu and G. W. Vuister, CcpNmr AnalysisAssign: a flexible platform for integrated NMR analysis, J. Biomol. NMR, 2016, 66, 111–124 CrossRef CAS PubMed.
- G. Hou, I. J. Byeon, J. Ahn, A. M. Gronenborn and T. Polenova, 1H-13C/1H-15N heteronuclear dipolar recoupling by R-symmetry sequences under fast magic angle spinning for dynamics analysis of biological and organic solids, J. Am. Chem. Soc., 2011, 133, 18646–18655 CrossRef CAS PubMed.
- L. Yao, B. Vogeli, J. Ying and A. Bax, NMR determination of amide N-H equilibrium bond length from concerted dipolar coupling measurements, J. Am. Chem. Soc., 2008, 130, 16518–16520 CrossRef CAS.
- V. Chevelkov, U. Fink and B. Reif, Accurate determination of order parameters from 1H,15N dipolar couplings in MAS solid-state NMR experiments, J. Am. Chem. Soc., 2009, 131, 14018–14022 CrossRef CAS PubMed.
- F. Mentink-Vigier, I. Marin-Montesinos, A. P. Jagtap, T. Halbritter, J. van Tol, S. Hediger, D. Lee, S. T. Sigurdsson and G. De Paepe, Computationally Assisted Design of Polarizing Agents for Dynamic Nuclear Polarization Enhanced NMR: The AsymPol Family, J. Am. Chem. Soc., 2018, 140, 11013–11019 CrossRef CAS PubMed.
- W. Hanke, J. Patt, J. Alenfelder, J. H. Voss, M. M. Zdouc, S. Kehraus, J. B. Kim, G. V. Grujicic, V. Namasivayam, R. Reher, C. E. Muller, E. Kostenis, M. Crusemann and G. M. Konig, Feature-Based Molecular Networking for the Targeted Identification of Gq-Inhibiting FR900359 Derivatives, J. Nat. Prod., 2021, 84(7), 1941–1953 CrossRef CAS.
- P. B. Wedegaertner, D. H. Chu, P. T. Wilson, M. J. Levis and H. R. Bourne, Palmitoylation Is Required for Signaling Functions and Membrane Attachment of G(Q)Alpha and G(S)Alpha, J. Biol. Chem., 1993, 268, 25001–25008 CrossRef CAS.
- J. B. Higgins and P. J. Casey, The role of prenylation in G-protein assembly and function, Cell. Signalling, 1996, 8, 433–437 CrossRef CAS.
- P. Chan, M. Gabay, F. A. Wright, W. Kan, S. S. Oner, S. M. Lanier, A. V. Smrcka, J. B. Blumer and G. G. Tall, Purification of heterotrimeric G protein alpha subunits by GST-Ric-8 association: primary characterization of purified G alpha(olf), J. Biol. Chem., 2011, 286, 2625–2635 CrossRef CAS PubMed.
- M. Gabay, M. E. Pinter, F. A. Wright, P. Chan, A. J. Murphy, D. M. Valenzuela, G. D. Yancopoulos and G. G. Tall, Ric-8 Proteins Are Molecular Chaperones That Direct Nascent G Protein α Subunit Membrane Association, Sci. Signaling, 2011, 4(200), ra79 CrossRef PubMed.
- O. Vogler, J. Casas, D. Capo, T. Nagy, G. Borchert, G. Martorell and P. V. Escriba, The Gbetagamma dimer drives the interaction of heterotrimeric Gi proteins with nonlamellar membrane structures, J. Biol. Chem., 2004, 279, 36540–36545 CrossRef.
- A. Pines, J. S. Waugh and M. G. Gibby, Proton-Enhanced Nuclear Induction Spectroscopy - Method for High-Resolution Nmr of Dilute Spins in Solids, J. Chem. Phys., 1972, 56, 1776 CrossRef CAS.
- G. A. Morris and R. Freeman, Enhancement of Nuclear Magnetic-Resonance Signals by Polarization Transfer, J. Am. Chem. Soc., 1979, 101, 760–762 CrossRef CAS.
- R. Reher, M. Kuschak, N. Heycke, S. Annala, S. Kehraus, H. F. Dai, C. E. Muller, E. Kostenis, G. M. Konig and M. Crusemann, Applying Molecular Networking for the Detection of Natural Sources and Analogues of the Selective Gq Protein Inhibitor FR900359, J. Nat. Prod., 2018, 81, 1628–1635 CrossRef CAS PubMed.
- S. Kuroki, S. Ando, I. Ando, A. Shoji, T. Ozaki and G. A. Webb, Hydrogen-Bonding Effect on N-15 Nmr Chemical-Shifts of the Glycine Residue of Oligopeptides in the Solid-State as Studied by High-Resolution Solid-State Nmr-Spectroscopy, J. Mol. Struct., 1990, 240, 19–29 CrossRef CAS.
- M. Taniguchi, K. Suzumura, K. Nagai, T. Kawasaki, J. Takasaki, M. Sekiguchi, Y. Moritani, T. Saito, K. Hayashi, S. Fujita, S. Tsukamoto and K. Suzuki, YM-254890 analogues, novel cyclic depsipeptides with Galpha(q/11) inhibitory activity from Chromobacterium sp. QS3666, Bioorg. Med. Chem., 2004, 12, 3125–3133 CAS.
- H. Zhang, A. L. Nielsen, M. W. Boesgaard, K. Harpsoe, N. L. Daly, X. F. Xiong, C. R. Underwood, L. M. Haugaard-Kedstrom, H. Brauner-Osborne, D. E. Gloriam and K. Stromgaard, Structure-activity relationship and conformational studies of the natural product cyclic depsipeptides YM-254890 and FR900359, Eur. J. Med. Chem., 2018, 156, 847–860 CrossRef CAS PubMed.
- X. Zhao, J. L. Sudmeier, W. W. Bachovchin and M. H. Levitt, Measurement of NH bond lengths by fast magic-angle spinning solid-state NMR spectroscopy: a new method for the quantification of hydrogen bonds, J. Am. Chem. Soc., 2001, 123, 11097–11098 CrossRef CAS PubMed.
- M. Kainosho and T. Tsuji, Assignment of the three methionyl carbonyl carbon resonances in Streptomyces subtilisin inhibitor by a carbon-13 and nitrogen-15 double-labeling technique. A new strategy for structural studies of proteins in solution, Biochemistry, 1982, 21, 6273–6279 CAS.
- M. V. Milburn, L. Tong, A. M. deVos, A. Brunger, Z. Yamaizumi, S. Nishimura and S. H. Kim, Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins, Science, 1990, 247, 939–945 CrossRef CAS PubMed.
- I. R. Vetter and A. Wittinghofer, The guanine nucleotide-binding switch in three dimensions, Science, 2001, 294, 1299–1304 CAS.
- D. Goricanec, R. Stehle, P. Egloff, S. Grigoriu, A. Pluckthun, G. Wagner and F. Hagn, Conformational dynamics of a G-protein alpha subunit is tightly regulated by nucleotide binding, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3629–E3638 CrossRef CAS.
- Y. Toyama, H. Kano, Y. Mase, M. Yokogawa, M. Osawa and I. Shimada, Dynamic regulation of GDP binding to G proteins revealed by magnetic field-dependent NMR relaxation analyses, Nat. Commun., 2017, 8, 14523 CrossRef CAS.
- J. Bigay, P. Deterre, C. Pfister and M. Chabre, Fluoroaluminates activate transducin-GDP by mimicking the gamma-phosphate of GTP in its binding site, FEBS Lett., 1985, 191, 181–185 CAS.
- D. G. Lambright, J. P. Noel, H. E. Hamm and P. B. Sigler, Structural determinants for activation of the alpha-subunit of a heterotrimeric G protein, Nature, 1994, 369, 621–628 CrossRef CAS PubMed.
- J. Mühle, M. J. Rodrigues, J. Alenfelder, L. Jürgenliemke, R. Guixà-González, A. Bacchin, F. Andres, M. Hennig, H. Schihada, M. Crusemann, G. Konig, E. Kostenis, G. Schertler and X. Deupi, Cyclic peptide inhibitors stabilize Gq/11 heterotrimers, bioRxiv, 2023, preprint, DOI:10.1101/2023.10.24.563737.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01950d |
| This journal is © The Royal Society of Chemistry 2024 |