
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrigin of copper as a unique catalyst for C–C coupling in electrocatalytic CO2 reduction†
Jie
Chen
abc,
Benjamin W. J.
Chen
 d,
Jia
Zhang
*d,
Wei
Chen
abe and
Yi-Yang
Sun
*c
d,
Jia
Zhang
*d,
Wei
Chen
abe and
Yi-Yang
Sun
*c
aJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou, 350207, China
bDepartment of Physics, National University of Singapore, 2 Science Drive 3, 117542, Singapore
cState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai, 201899, China. E-mail: yysun@mail.sic.ac.cn
dInstitute of High Performance Computing (IHPC), Agency for Science, Technology and Research (A*STAR), 1 Fusionopolis Way, #16–16 Connexis, 138632, Singapore. E-mail: zhangj@ihpc.a-star.edu.sg
eDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore
First published on 13th May 2024
Abstract
High yields of C2 products through electrocatalytic CO2 reduction (eCO2R) can only be obtained using Cu-based catalysts. Here, we adopt the generalized frontier molecular orbital (MO) theory based on first-principles calculations to identify the origin of this unique property of Cu. We use the grand canonical ensemble (or fixed potential) approach to ensure that the calculated Fermi level, which serves as the frontier orbital of the metal catalyst, accurately represents the applied electrode potentials. We determine that the key intermediate OCCO assumes a U-shape configuration with the two C atoms bonded to the Cu substrate. We identify the frontier MOs that are involved in the C–C coupling. The good alignment of the Fermi level of Cu with these frontier MOs is perceived to account for the excellent catalytic performance of Cu for C–C coupling. It is expected that these new insights could provide useful guidance in tuning Cu-based catalysts as well as designing non-Cu catalysts toward high-efficiency eCO2R.
Electrocatalytic CO2 reduction (eCO2R) is a promising method to convert CO2 to valuable chemicals or fuels.1 Currently, eCO2R to C1 products has achieved high selectivity with faradaic efficiency over 95% and high current density up to hundreds of mA cm−2.2 However, the selectivity and current density for C2 products still cannot meet industrial requirements.3 So far, Cu-based catalysts, which exhibit excellent performance in catalyzing the conversion of CO2 to C1, C2, and even products with longer carbon chains,4 are the only ones capable of achieving a high yield of C2 products from CO2. To achieve higher yield towards C2 products, it is imperative to understand the reaction pathway, especially the rate-determining step.
The rate-determining step towards C2 products is identified to be C–C coupling.5–9 Among various C–C coupling mechanisms, the most established one involves the CO–CO coupling to produce OCCO adsorbed on the catalyst surface.10–12 To ascertain the origin of copper in forming C2 products, it is essential to determine the unique catalytic role of copper in catalyzing the CO–CO coupling. The frontier molecular orbital (MO) theory is a relevant tool for understanding the reaction mechanism at electronic structure level.13 For electrocatalytic reaction, this theory could be generalized by taking the Fermi level of the metal catalyst as a frontier orbital. In such a case, accurately determining the alignment between the Fermi level of the electrode and the MO levels of the adsorbates becomes critically important. The system must be described by the grand canonical ensemble with variable numbers of electrons in the system so that the Fermi level is in accordance with the applied electrode potential. Fixed potential method (FPM),14–18 coupled with the implicit solvent model,19,20 is a state-of-the-art method to simulate electrocatalytic processes in the grand canonical ensemble. In contrast, in the conventional computational hydrogen electrode (CHE) method,21 the total charge of the system is fixed so that the Fermi level of the electrode cannot be directly related to the experimentally applied voltage.22
In this paper, we aim to identify the origin of copper's unique performance in catalyzing the CO–CO coupling. We employed the FPM to ensure the alignment of the Fermi level and the frontier MO levels under the desired applied electrode potentials. We first examined the stability of two debated OCCO configurations under the explicit solvent model to determine the configuration involved in CO–CO coupling. We also established a notation system for the MOs of the OCCO molecule to help track the evolution of the MOs during CO–CO coupling. We then identified the crucial MOs driving the reaction. Last, we conducted a comparative analysis with other metallic catalysts to elucidate why copper uniquely exhibits its remarkable catalytic properties in CO–CO coupling.
Our first-principles study was based on the density functional theory (DFT) as implemented in Vienna Ab initio Simulation Package (VASP).23,24 In the grand canonical ensemble, the electroneutrality of the system was maintained in the presence of countercharges introduced through the implicit solvent model, as implemented in the VASPsol code.19,20 The grand canonical electronic energy (GCEE) can be expressed as,
| F(q) = E(q) − qεF | (1) |
| Ω(q) = F(q) + ZPE + ∫CvdT − TS | (2) |
Regarding the configuration of OCCO on Cu substrate, there are two possibilities. Fig. 1a shows the configuration with one C atom and one O atom bonded to the Cu substrate, which is denoted here as L-shape. The L-shape configuration was first proposed based on DFT calculations without considering the presence of solvent.5,27Fig. 1b shows the configuration with two C atoms bonded to the Cu substrate,28–30 which is denoted as U-shape. The stability of U-shape and L-shape were compared in some calculations without the explicit water solvent.31
 | ||
| Fig. 1 Structures of (a) L-shape OCCO (denoted as *L) and (b) U-shape OCCO (denoted as *U) on Cu(100) surface with explicit water solvent. (c) GCEE of L-shape and U-shape OCCO as a function of added electrons. The value of the neutral L-shape OCCO was taken as the reference. | ||
We calculated the GCEE of the L-shape and U-shape OCCO on Cu substrate with explicitly considering the water solvent. Fig. 1c shows the GCEE curves as a function of added charge (q), which is directly related to the electrode potential, as shown in Fig. S1 in the ESI.† The curves are quadratic functions. As the electrons are injected into the systems, the adsorbed water molecules gradually desorb due to the repulsion between the charged metallic surface and the water molecules. When the desorption happens, discontinuity can be observed on the GCEE curves. The maxima are located at the neutral charge state (q = 0).15,20 The GCEE of the U-shape is consistently lower than that of the L-shape by about 0.4 eV. Our results suggest that the U-shape OCCO should be the actual configuration involved in the CO–CO coupling under experimental conditions.
We investigated the origin of this difference by using the crystal orbital Hamilton population (COHP) analysis,32–35 as shown in Fig. S2 in the ESI.† We found that the Cu–C and Cu–O bonds in the L-shape configuration exhibit more negative values than the Cu–C bonds in the U-shape configuration near the Fermi level, which indicates that the antibonding interactions between the L-shape adsorbate and the Cu surface are stronger than that between the U-shape adsorbate and the Cu surface, resulting in the instability of L-shape OCCO.
We also carried out calculations using the implicit solvent model without explicitly including the water molecules. In this case, the U-shape OCCO adsorbate is unstable. Only when electrons are added to the system, it can be stabilized. In contrast, including the explicit water solvent, the U-shape OCCO adsorbate can be stabilized even when electrons are extracted from the system. These results indicate that the explicit solvent could help the electrons accumulate on the OCCO adsorbate and enhance its stability.
Our study aimed to identify the frontier MOs underlying the CO–CO coupling catalyzed by the Cu surface. For a free CO molecule, it has eight MOs, namely, σs, 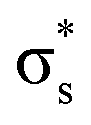 , σp, degenerate πx and πy, degenerate
, σp, degenerate πx and πy, degenerate  and
and  , and
, and  , arranged in increasing order of energy in Fig. 2. For simplicity, we denoted these MOs as s (σs),
, arranged in increasing order of energy in Fig. 2. For simplicity, we denoted these MOs as s (σs), 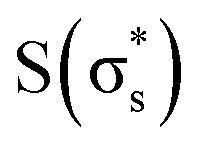 , p (σp), x (πx), y (πy),
, p (σp), x (πx), y (πy), 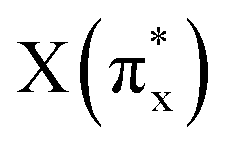 ,
,  and
and 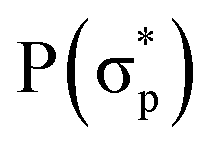 , respectively, where the bonding MOs are labeled with lowercase letters and the antibonding MOs are labeled with capital letters. Because the
, respectively, where the bonding MOs are labeled with lowercase letters and the antibonding MOs are labeled with capital letters. Because the  (or P) MO is too high in energy to play a significant role in the reaction, we did not trace its evolution in this work.
(or P) MO is too high in energy to play a significant role in the reaction, we did not trace its evolution in this work.
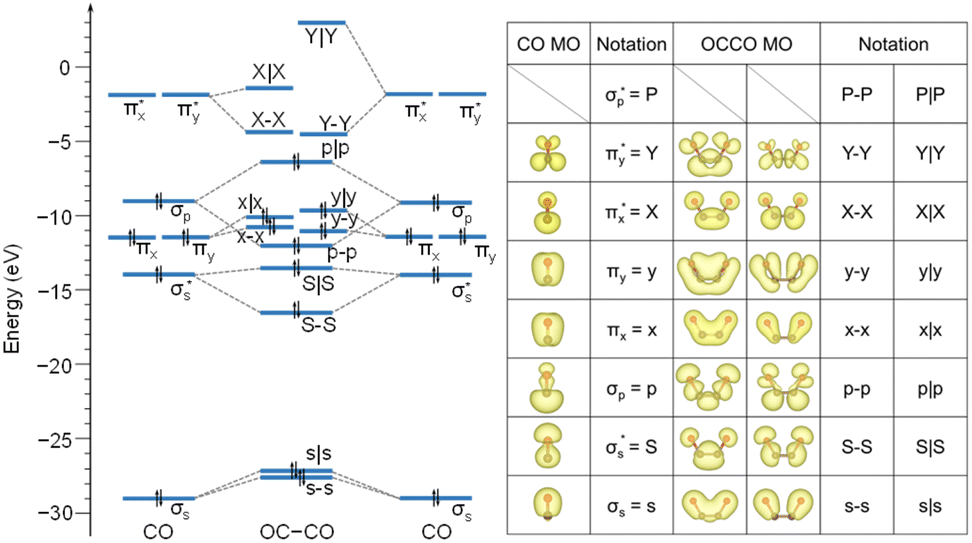 | ||
| Fig. 2 Energy eigenvalues of a free CO and OCCO molecule. Eigenstates have been aligned by referring to vacuum level, and each state has been labeled for ease of reference. Specifically, P, P–P, and P|P MOs are not shown because their eigenvalues are beyond the ranges that we are interested in. The right-hand table provides a visual representation of partial charge density for each MO. | ||
We first analyzed the MOs of a free U-shape OCCO molecule, whose structure is fixed at the one as in Fig. 1b. Coupling the two CO molecules gives rise to a total of 16 bonding and antibonding MOs for the OCCO molecule. We used a hyphen and a vertical bar to represent the bonding and antibonding MOs, respectively. By this notation, the 16 MOs of the free OCCO molecule are denoted by s–s, s|s, S–S, S|S, p–p, p|p, x–x, x|x, y–y, y|y, X–X, X|X, Y–Y, Y|Y, P–P and P|P. From Fig. 2, it is seen that the highest occupied MO (HOMO) for the free OCCO molecule is p|p, while the lowest unoccupied MOs (LUMO) are X–X and Y–Y, which are nearly degenerate.
Next, we investigated the MOs of OCCO adsorbed on the Cu(100) surface by the FPM approach with explicit water solvent. Fig. 3 shows the eigenvalue spectrum at the Γ point of the supercell, where the vertical axis represents the projection of each eigenstate onto the OCCO adsorbate. Clearly, the MOs of OCCO strongly hybridize with the Cu substrate as reflected by the fact that there are more than 16 states having significant projection on the OCCO adsorbate. As shown in Fig. S3 in the ESI,† the Cu 3d orbitals start from about −8 eV below the Fermi level. Within this range, all the MOs of OCCO above the energy level of the p–p orbital hybridize with the Cu 3d orbitals and split into two or more levels. Fig. S4 in the ESI† shows the partial charge densities for the labelled states in Fig. 3, from which the hybridization can be better visualized. The p|p orbital exhibits the largest splitting suggesting that it hybridizes with the Cu 3d orbitals most strongly. Also, it can be seen that at least three MOs carry the p|p character. Based on the pDOS and COHP analysis, the p|p orbitals from −6 to −5 eV, together with the p–p, y–y, x–x and x|x orbitals, contribute to the adsorption of the OCCO molecule, as shown in Fig. S5 in the ESI.†
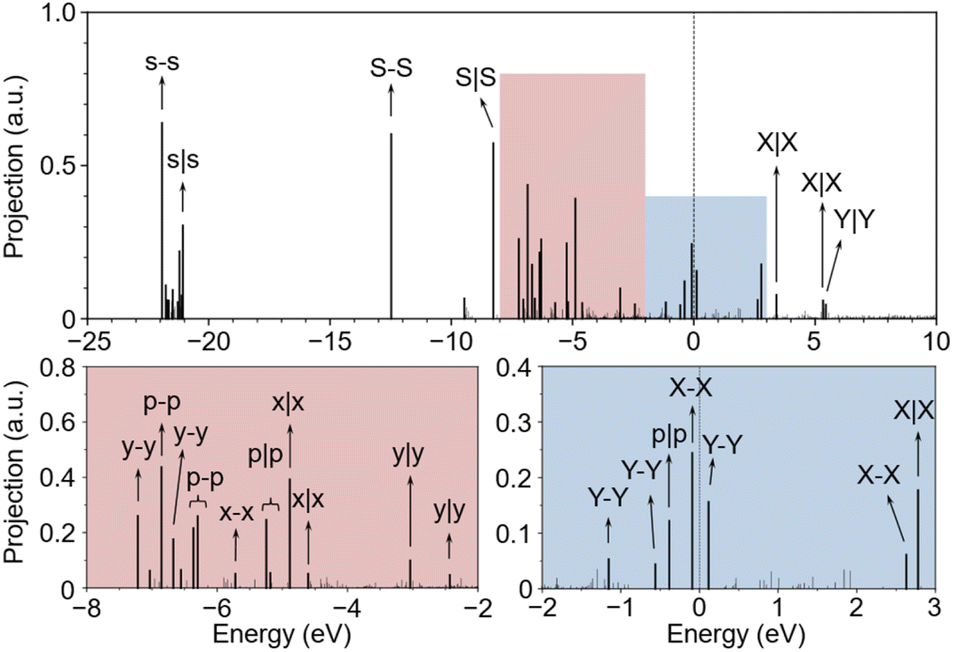 | ||
| Fig. 3 Eigenvalue spectrum of U-shape OCCO on Cu(100) surface at neutral state with explicit water solvent. Upper portion of the figure is full range spectrum, while lower portions are two zoomed-in spectra from −8 eV to −2 eV, and from −2 eV to 3 eV, respectively. The Fermi level, plotted as a dashed line, is set as reference. Notations are labelled near the corresponding MOs. | ||
Comparing Fig. 2 and 3, we can see that the X–X MO is unoccupied in a free OCCO molecule and becomes occupied after adsorbing on the Cu substrate. On the one hand, this result indicates that there is electron transfer from the Cu substrate to OCCO during the adsorption. On the other hand, it suggests that the X–X MO serves as the frontier orbital for the adsorption of OCCO on the Cu substrate. Next, we will show that this X–X MO also plays the role of frontier orbital for the CO–CO coupling.
After understanding the electronic structure of the key intermediate OCCO, we proceeded to study the evolution of the OCCO MOs during the CO–CO coupling. We adopted the dimer method to locate the transition state (TS) between the initial state (IS) and final state (FS).36 The TSs have only one imaginary frequency at all three studied potentials and all the frequency data are listed in Table S1 in the ESI.† Both the forward energy barrier from the IS to the TS and the energy difference between the IS and the FS decrease with the increasing applied potential. However, the backward energy barrier from the FS to the TS increases with the increasing applied potential. As Fig. 4b shows, under −1.0 V (vs. SHE), the forward energy barrier decreases to 0.58 eV and the backward energy barrier increases to 0.34 eV.
 | ||
| Fig. 4 In explicit solvent model, during CO–CO coupling, (a) structures of initial state (IS), transition state (TS) and final state (FS). (b) GCFE profile of CO–CO coupling under different applied potentials. (c) Eigenvalue spectra of IS, TS and FS at −1.0 V (vs. SHE). The X–X MO declining across the Fermi level is colored with orange and the Y–Y MOs emerging upon the transition state are colored with red. We use the notations of OCCO MOs to denote the corresponding MOs in IS to keep the consistency in the following discussion. | ||
Fig. 4c shows the eigenvalue spectra of the IS, TS and FS under −1.0 V (vs. SHE). It is seen that the X–X MO is the LUMO in the IS and TS but becomes the HOMO in the FS, which indicates that the X–X MO serves as the frontier MO in the CO–CO coupling and crosses the Fermi level during the reaction. Interestingly, the Y–Y MO emerges at the TS. To explore the MO evolution along the full reaction path, we also carried out nudged elastic band (NEB) calculations for the CO–CO coupling reaction. However, we are not able to implement the FPM in the NEB calculations yet. Also, convergence can only be achieved by excluding the water solvent. So, these calculations were only used to check the MO evolution along the reaction path. As shown in Fig. S7 in the ESI,† only when the reaction approaches the TS, the Y–Y MO below the X–X and p|p MOs suddenly appears and its energy level shifts down with the structure evolving from the TS to FS, suggesting that the emerged Y–Y state is a bonding state.
More detailed analyses on the MO evolutions are given in Fig. S6–S10 in the ESI.† The results in Fig. S10 in the ESI† show that the position of the y|y MO can be well correlated to the O–C–Cu bond angle. The energy level of the y|y MO is found to increase monotonically from the IS to FS. So, this is an energy-costing mechanism and is responsible for the reaction barrier. In other words, the increasing energy level of the y|y MO explains the bending of the O–C–Cu bond angle, which is necessary for the CO–CO coupling.
The analysis above suggests that the good alignment of the Fermi level of Cu plays a key role in charge transfer and orbital hybridization between the adsorbates (both CO and OCCO) and the substrate. We carried out similar calculations on other common metal catalysts used in electrocatalytic reactions. For a quick examination, no explicit water solvent and charges were added in these calculations. As shown in Fig. S14 in the ESI,† the key MOs X–X and Y–Y are only slightly above the Fermi level on Cu(100) surface. The natural alignment between the Fermi level of the metal catalyst and the energy levels of the frontier MOs plays a key role. As seen in Fig. 2, these orbitals are the C–C bonding MOs. So, the occupation of these MOs is crucial for the C–C coupling reaction. The better aligned these energy levels are, the lower applied potential is required in experiment to initiate the reaction. In comparison, on the (100) surface of Ni, Pt and Pd, the X–X and Y–Y MOs are separated by at least 1 eV from the Fermi level. While it is true that the applied potential can tune these energy levels to align with the Fermi level, for these metal catalysts the required potential may already induce other side reactions such as water splitting, which is consistent with the observation that they are not efficient catalysts for the eCO2R reaction.
Before concluding, we remark that the frontier MOs X–X and Y–Y are not only observed around the Fermi level of Cu substrate in the CO–CO coupling, but also in other possible intermediate candidates discussed in the literatures.12,37 We studied the case of CO–COH coupling by hydrogenating one of the CO monomers. The eigenvalue spectra of IS, TS and FS for the CO–COH coupling are shown in Fig. S15 in the ESI.† It is seen that the MOs X–X and Y–Y emerge around the Fermi level at the TS and remain occupied at the FS. Because of the hydrogenation, the frontier MOs in CO–COH coupling are not as symmetric as in the case of CO–CO coupling. Other than that, the two reaction intermediates are similar in terms of the alignment between the frontier MOs and the Fermi level. These results suggest that the MO analysis given above and the gained insights could be generalized to the C–C coupling reactions involving other possible intermediate species.
In summary, based on grand-canonical first-principles calculations, which can accurately describe the positions of the Fermi level and the molecular orbitals of adsorbates, we study the electrocatalytic CO–CO coupling on Cu(100) surface. Explicit water solvent is included in our calculations. We first show that the key intermediate OCCO adsorbs on the Cu surface in the U-shape configuration under experimental conditions. Regarding the electronic structure, we identify the X–X and Y–Y orbitals to be two frontier molecular orbitals for the CO–CO coupling reaction. As the CO–CO coupling is the rate-determining step for the eCO2R reaction towards the C2 products, the good alignment between the Fermi level of Cu and these frontier MOs is proposed to be the origin of Cu as a unique catalyst for this reaction. Our study provides a new understanding on the mechanism of C–C coupling. It is expected that this understanding could guide the design of new catalysts for C2 production through eCO2R.
Data availability
All data that support the findings of this study are available within the article and the ESI,† or from the corresponding authors upon reasonable request.Author contributions
J. C. conducted all calculations, analysed the data and wrote the manuscript. B. W. J. C. provided guidance for the explicit solvent calculations and revised the manuscript. J. Z. guided the study and revised the manuscript. W. C. supervised the study and revised the manuscript. Y.-Y. S. conceived the idea, analysed the data, guided the study, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. C. acknowledges Professor Lei Wang and Dr Yuke Li for enlightening discussions. The work is supported by the National Research Foundation, Singapore, and A*STAR (Agency for Science, Technology and Research) under its LCER FI program (Award No. U2102d2002) and NUS R&G Postdoc Fellowship Program.References
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
.
- S. Jin, Z. M. Hao, K. Zhang, Z. H. Yan and J. Chen, Advances and Challenges for the Electrochemical Reduction of CO2 to CO: From Fundamentals to Industrialization, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed
- J. Sisler, S. Khan, A. H. Ip, M. W. Schreiber, S. A. Jaffer, E. R. Bobicki, C.-T. Dinh and E. H. Sargent, Ethylene Electrosynthesis: A Comparative Techno-economic Analysis of Alkaline vs. Membrane Electrode Assembly vs. CO2–CO–C2H4 Tandems, ACS Energy Lett., 2021, 6, 997–1002 CrossRef CAS
- S. D. Rihm, M. K. Kovalev, A. A. Lapkin, J. W. Ager and M. Kraft, On the role of C4 and C5 products in electrochemical CO2 reduction via copper-based catalysts, Energy Environ. Sci., 2023, 52, 5013–5050 Search PubMed
- F. Calle-Vallejo and M. T. M. Koper, Theoretical Considerations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS
- H. Xiao, T. Cheng, W. A. Goddard and R. Sundararaman, Mechanistic Explanation of the pH Dependence and Onset Potentials for Hydrocarbon Products from Electrochemical Reduction of CO on Cu (111), J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed
- H. Xiao, T. Cheng and W. A. Goddard, Atomistic Mechanisms Underlying Selectivities in C1 and C2 Products from Electrochemical Reduction of CO on Cu(111), J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed
- Y. Kim, S. Park, S.-J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Time-resolved observation of C–C coupling intermediates on Cu electrodes for selective electrochemical CO2 reduction, Energy Environ. Sci., 2020, 13, 4301–4311 RSC
- H. Peng, M. T. Tang, X. Liu, P. Schlexer Lamoureux, M. Bajdich and F. Abild-Pedersen, The role of atomic carbon in directing electrochemical CO(2) reduction to multicarbon products, Energy Environ. Sci., 2021, 14, 473–482 RSC
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, Identification of Possible Pathways for C–C Bond Formation during Electrochemical Reduction of CO2: New Theoretical Insights from an Improved Electrochemical Model, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed
- T. Cheng, H. Xiao and A. Goddard William, Full atomistic reaction mechanism with kinetics for CO reduction on Cu(100) from ab initio molecular dynamics free-energy calculations at 298 K, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed
- A. J. Garza, A. T. Bell and M. Head-Gordon, Mechanism of CO2 Reduction at Copper Surfaces: Pathways to C-2 Products, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS
- M. Lucking, Y.-Y. Sun, D. West and S. Zhang, A nucleus-coupled electron transfer mechanism for TiO2-catalyzed water splitting, Phys. Chem. Chem. Phys., 2015, 17, 16779–16783 RSC
- C. D. Taylor, S. A. Wasileski, J.-S. Filhol and M. Neurock, First principles reaction modeling of the electrochemical interface: Consideration and calculation of a tunable surface potential from atomic and electronic structure, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 165402 CrossRef
- J.-S. Filhol and M. Neurock, Elucidation of the Electrochemical Activation of Water over Pd by First Principles, Angew. Chem., Int. Ed., 2006, 45, 402–406 CrossRef CAS PubMed
- D. Kim, J. Shi and Y. Liu, Substantial Impact of Charge on Electrochemical Reactions of Two-Dimensional Materials, J. Am. Chem. Soc., 2018, 140, 9127–9131 CrossRef CAS PubMed
- N. Abidi and S. N. Steinmann, How are transition states modeled in heterogeneous electrocatalysis?, Curr. Opin. Electrochem., 2022, 33, 100940 CrossRef CAS
- K. Chan and J. K. Nørskov, Electrochemical Barriers Made Simple, J. Phys. Chem. Lett., 2015, 6, 2663–2668 CrossRef CAS PubMed
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed
- K. Mathew, V. S. C. Kolluru, S. Mula, S. N. Steinmann and R. G. Hennig, Implicit self-consistent electrolyte model in plane-wave density-functional theory, J. Chem. Phys., 2019, 151, 234101 CrossRef PubMed
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
- G. Gao and L.-W. Wang, Substantial potential effects on single-atom catalysts for the oxygen evolution reaction simulated via a fixed-potential method, J. Catal., 2020, 391, 530–538 CrossRef CAS
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- B. W. J. Chen, X. Zhang and J. Zhang, Accelerating explicit solvent models of heterogeneous catalysts with machine learning interatomic potentials, Chem. Sci., 2023, 14, 8338–8354 RSC
- B. W. J. Chen, B. Wang, M. B. Sullivan, A. Borgna and J. Zhang, Unraveling the Synergistic Effect of Re and Cs Promoters on Ethylene Epoxidation over Silver Catalysts with Machine Learning-Accelerated First-Principles Simulations, ACS Catal., 2022, 12, 2540–2551 CrossRef CAS
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, Insights into C-C Coupling in CO2 Electroreduction on Copper Electrodes, ChemCatChem, 2013, 5, 737–742 CrossRef CAS
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, Theoretical Insights into a CO Dimerization Mechanism in CO2 Electroreduction, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed
- H. H. Kristoffersen and K. Chan, Towards constant potential modeling of CO-CO coupling at liquid water-Cu(100) interfaces, J. Catal., 2021, 396, 251–260 CrossRef CAS
- S.-J. Shin, H. Choi, S. Ringe, D. H. Won, H.-S. Oh, D. H. Kim, T. Lee, D.-H. Nam, H. Kim and C. H. Choi, A unifying mechanism for cation effect modulating C1 and C2 productions from CO2 electroreduction, Nat. Commun., 2022, 13, 5482 CrossRef CAS PubMed
- Z. Wei and P. Sautet, Improving the Accuracy of Modelling CO2 Electroreduction on Copper Using Many-Body Perturbation Theory, Angew. Chem., Int. Ed., 2022, 61, e202210060 CrossRef CAS
- R. Dronskowski and P. E. Bloechl, Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids, J. Comput. Chem., 2013, 34, 2557–2567 CrossRef CAS
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Crystal Orbital Hamilton Population (COHP) Analysis As Projected from Plane-Wave Basis Sets, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory, J. Comput. Chem., 2020, 41, 1931–1940 CrossRef CAS PubMed
- G. Henkelman and H. Jónsson, A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS
- E. Pérez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Spectroscopic Observation of a Hydrogenated CO Dimer Intermediate During CO Reduction on Cu(100) Electrodes, Angew. Chem., Int. Ed., 2017, 56, 3621–3624 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02056a |
| This journal is © The Royal Society of Chemistry 2024 |