
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed asymmetric allylic substitution of racemic/meso substrates
Jun
Li†
a,
Junrong
Huang†
 ab,
Yan
Wang
a,
Yuexin
Liu
a,
Yuxiang
Zhu
*c,
Hengzhi
You
*ab and
Fen-Er
Chen
*abd
ab,
Yan
Wang
a,
Yuexin
Liu
a,
Yuxiang
Zhu
*c,
Hengzhi
You
*ab and
Fen-Er
Chen
*abd
aSchool of Science, Harbin Institute of Technology (Shenzhen), Taoyuan Street, Nanshan District, Shenzhen, 518055, China. E-mail: youhengzhi@hit.edu.cn
bGreen Pharmaceutical Engineering Research Center, Harbin Institute of Technology (Shenzhen), Taoyuan Street, Nanshan District, Shenzhen, 518055, China
cSchool of Pharmaceutical Sciences (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Shenzhen, 518107, China. E-mail: zhuyx86@mail.sysu.edu.cn
dEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai, 200433, China. E-mail: rfchen@fudan.edu.cn
First published on 7th May 2024
Abstract
The synthesis of enantiomerically pure compounds is a pivotal subject in the field of chemistry, with enantioselective catalysis currently standing as the primary approach for delivering specific enantiomers. Among these strategies, Cu-catalyzed asymmetric allylic substitution (AAS) is significant and irreplaceable, especially when it comes to the use of non-stabilized nucleophiles (pKa > 25). Although Cu-catalyzed AAS of prochiral substrates has also been widely developed, methodologies involving racemic/meso substrates are highly desirable, as the substrates undergo dynamic processes to give single enantiomer products. Inspired by the pioneering work of the Alexakis, Feringa and Gennari groups, Cu-catalyzed AAS has been continuously employed in deracemization and desymmetrization processes for the synthesis of enantiomerically enriched products. In this review, we mainly focus on the developments of Cu-catalyzed AAS with racemic/meso substrates over the past two decades, providing an explicit outline of the ligands employed, the scope of nucleophiles, the underlying dynamic processes and their practical applications.
1. Introduction
There is a growing interest in the catalytic and enantioselective synthesis of intricate organic compounds using readily accessible materials.1–6 Among them, AAS has emerged as a reliable approach for the synthesis of enantiomerically pure compounds. Since the pioneering work by the Trost group7–9 on Pd-catalyzed AAS, transition metal-catalyzed AAS has been continuously developed as a powerful tool for the efficient construction of chiral C–C and C–X bonds.10–16 Additionally, AAS has successfully enabled the total synthesis of various natural products and drugs.17–20 This significant transformation was primarily driven by the utilization of second- and third-row transition metals, particularly Pd,21 Rh,22 and Ir,23 which have become dominant catalysts in the total synthesis of numerous natural products. First-row transition metals are used less frequently in AAS reactions despite being more cost-effective and abundant alternatives to second- and third-row metals. Moreover, first-row metals often offer distinct reactivities and mechanistic possibilities compared to their second- and third-row counterparts. Recently, copper has emerged as one of the predominant metal catalysts in metal-catalyzed AAS.24–32 This review aims to elucidate the nomenclature associated with dynamic processes in Cu-catalyzed AAS and provide an overview of their recent advancements and applications in synthesizing enantioenriched molecules. In the first section, fundamental aspects of Cu-catalyzed AAS chemistry, ranging from general mechanisms to dynamic processes, will be discussed.1.1 Cu-catalyzed asymmetric allylic substitution
Compared with Pd/Rh/Ir catalysis, Cu catalysis has its unique features.33,34 Typically, non-stabilized organometallic nucleophiles (pKa > 25) including organozinc, organomagnesium, organolithium, organoaluminum or mild organoboron reagents, are commonly employed in Cu-catalyzed AAS. Recent extensive investigations have proposed a comprehensive mechanism for Cu-catalyzed AAS using hard nucleophiles (Scheme 1a). The first step involves transmetalation between the ligated copper(I) salt A and the organometallic species, resulting in the formation of the organocuprate species B. Upon coordination of A to the allyl substrate, this leads to the subsequent formation of π-complex C. Oxidative addition at the γ-position to the leaving group occurs on copper, yielding [σ + π]-allyl copper(III) species D. The newly formed [σ + π]-allyl complex D can undergo two competitive pathways depending on the nature of X, a non-transferrable substituent at the copper center in D: if X is an electron-withdrawing group such as CN or Cl, reductive elimination becomes favored and forms a branched product along with the regeneration of copper(I) salt A; however, if X is an electron-donating group like an alkyl substituent, isomerization from [σ + π]-allyl complex Dvia π-allyl complex E to [σ + π]-allyl complex F becomes feasible and results in a linear product. It should be noted that the regiochemical outcomes of reactions are dictated not only by the substituent X but also by factors such as steric effects and the presence of chelating groups on ligands, which control site selectivity during bond formation.35–40 Additionally, it is crucial to precisely tune parameters including the structural and electronic properties of the electrophile, leaving group, and nucleophile, as well as reaction conditions such as solvent choice and temperature.11,33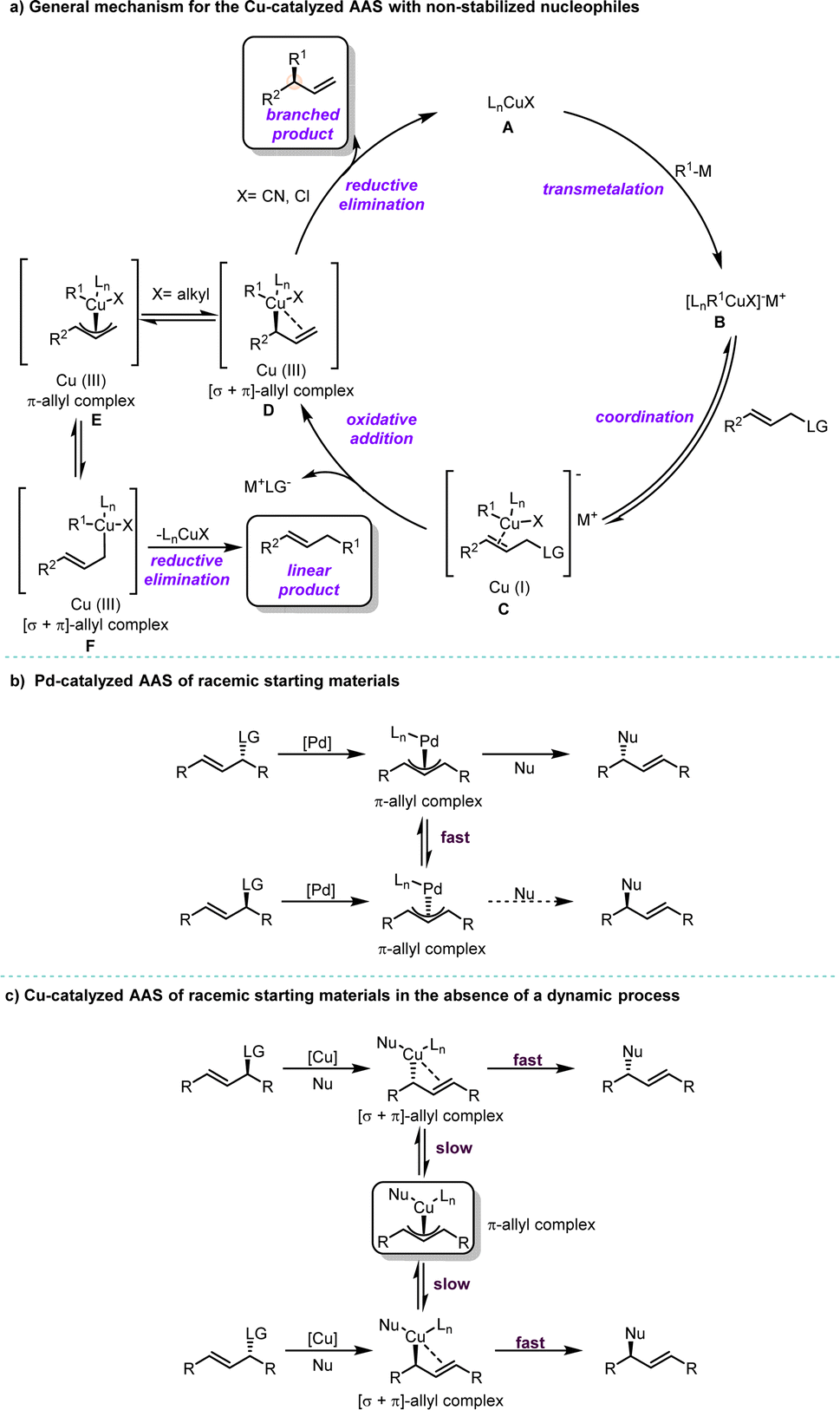 | ||
| Scheme 1 (a) General mechanism for the Cu-catalyzed AAS with non-stabilized nucleophiles; (b) Pd-catalyzed AAS of racemic starting materials; (c) Cu-catalyzed AAS of racemic starting materials in the absence of a dynamic process. | ||
Although the use of stabilized nucleophiles (pKa < 25) in Pd-catalyzed AAS reactions for racemic starting materials is well-established in catalysis and synthesis,10,41 the application of non-stabilized nucleophiles in Cu-catalyzed reactions remains a significant challenge with racemic starting materials. Notably, there are considerable differences between the reaction mechanisms of Pd-catalyzed and Cu-catalyzed AAS reactions. In Pd-catalyzed AAS, both enantiomers of the starting materials are converted to their corresponding π-allyl–Pd intermediates, which undergo rapid interconversion with each other. Subsequently, an enantioselective step takes place, leading to the formation of a new stereogenic center (Scheme 1b). On the other hand, Cu-catalyzed AAS is widely recognized to proceed through regiospecific anti-addition, with SN2′ oxidative addition as the rate-determining step. Unlike in Pd-catalysis, Cu-allyl intermediates lack an efficient pathway for σ to π isomerization (Scheme 1b and c).34 If reductive elimination occurs rapidly, the π-allyl–Cu species will not form. As a result, reactions using racemic starting materials will yield racemic products in the absence of a dynamic process (Scheme 1c).
1.2 Dynamic processes
The development of Cu-catalyzed AAS for prochiral substrates, which has been extensively pursued, mainly relies on the use of organometallic reagents.31,42–48 The recent trend of using unsaturated hydrocarbons as pro-nucleophiles in borylative or reductive couplings with allylic substrates represents a significant development in this field, facilitating the synthesis of products endowed with enhanced functional groups.49–54Methods involving racemic and meso starting materials that undergo dynamic processes to yield single enantiomer products are highly sought after, due to the greater availability of racemic substrates compared to prochiral ones and the more complex chiral induction process in racemic/meso substrates.55 Employing a chiral catalyst allows only one enantiomer of the racemic substrate to selectively engage in the reaction, yielding a product with remarkable enantioselectivity. This typical process is commonly referred to as kinetic resolution (KR).56–60 However, this approach limits the yield of the reaction to 50% in order to ensure optimal enantioselectivity of the final product (Scheme 2a). Unlike the classic KR process, parallel kinetic resolution (PKR) introduces a strategy where both enantiomers of a racemate can be converted into different products.
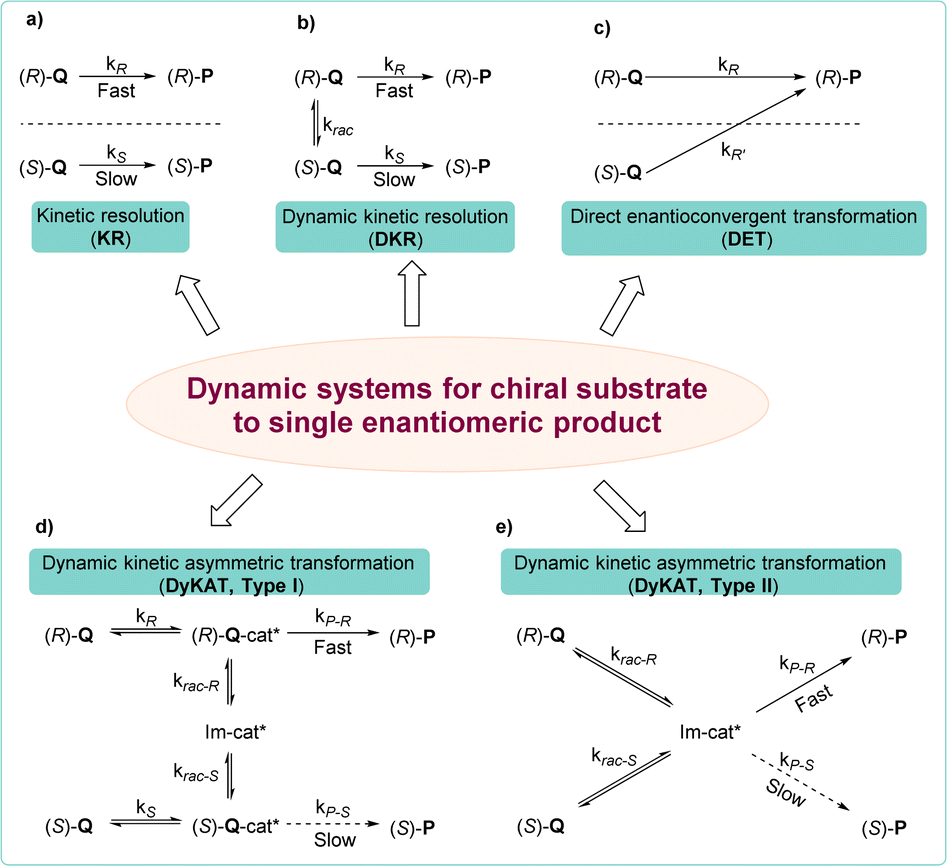 | ||
| Scheme 2 Dynamic processes of a racemate in asymmetric reactions: (a) kinetic resolution; (b) dynamic kinetic resolution; (c) direct enantioconvergent transformation; (d) type I dynamic kinetic asymmetric transformation; (e) type II dynamic kinetic asymmetric transformation. | ||
When it comes to the conversion of both enantiomers of a racemate into a single enantiomer, there are typically three approaches: (1) dynamic kinetic resolution (DKR),61,62 in which the two enantiomers undergo a reversible racemization process before the KR process, which overcomes the 50% maximum yield problem (Scheme 2b); (2) dynamic kinetic asymmetric transformation (DyKAT), in which the equilibration of both enantiomers is facilitated by the presence of a chiral catalyst.63–65 In type I DyKAT (Scheme 2d), both enantiomers bind to the catalyst and undergo equilibration as intermediates. Conversely, in type II DyKAT (Scheme 2e), the racemate interacts with the chiral catalyst to form a prochiral intermediate Im-cat*, resulting in the loss of the stereocenter. These two processes have enabled a diverse range of asymmetric transformations, delivering a fascinating array of chiral building blocks; (3) direct enantioconvergent transformation (DET), in which both enantiomers of the starting material are converted to the same enantiomeric product via two distinct mechanistic pathways (Scheme 2c).66,67 To produce identical enantiomeric products, both pathways must exhibit opposite enantiopreference, thus necessitating an opposite stereochemical course, which means opposite regioselectivity is essential.
The utilization of these strategies has emerged as a flourishing approach over the past decade in asymmetric catalysis, primarily focusing on the bond-forming reactions for the synthesis of enantioenriched compounds. In this review, we focus on providing a detailed and explicit outline of copper catalyst-mediated dynamic asymmetric catalytic processes in AAS.
2. Racemic compounds as starting materials
2.1 Kinetic resolution
Given the value of Cu-catalyzed AAS with racemic substrates, the Feringa group reported pioneering research in 1998 on the catalytic enantioselective addition of simple organozinc reagents to cycloaliphatic vinyloxiranes.68–71 Employing a chiral ligand 2,2′-binaphthyl-based phosphorus amidites L1 and copper(II) triflate Cu(OTf)2, the corresponding allylic alcohol products were obtained via an anti-SN2′-pathway with moderate to high regioselectivity but limited yield (<50%). Using 0.5 equivalents of the nucleophile, the KR of racemic 1.3-cyclohexadiene monoepoxide was achieved, yielding product 2 in 33% yield with 92% enantiomeric excess (e.e.), while recovering the unreacted allylic epoxide 1 with high optical purity. Using ligand L2 and methylidene cycloalkane epoxide 3 as the substrate, the corresponding allylic alcohol 4, derived from a conjugate addition pathway (anti-SN2′), was obtained with excellent regio- and enantioselectivity (88% e.e.) (Scheme 3a). When employing an excess of nucleophiles, these cyclic allylic epoxides underwent the PKR process,72 wherein the two enantiomers within the substrate reacted with nucleophiles via either SN2 or SN2′ pathways, resulting in distinct products (Scheme 3b). Subsequently, a series of KR or PKR processes of racemic epoxides have been developed.73–75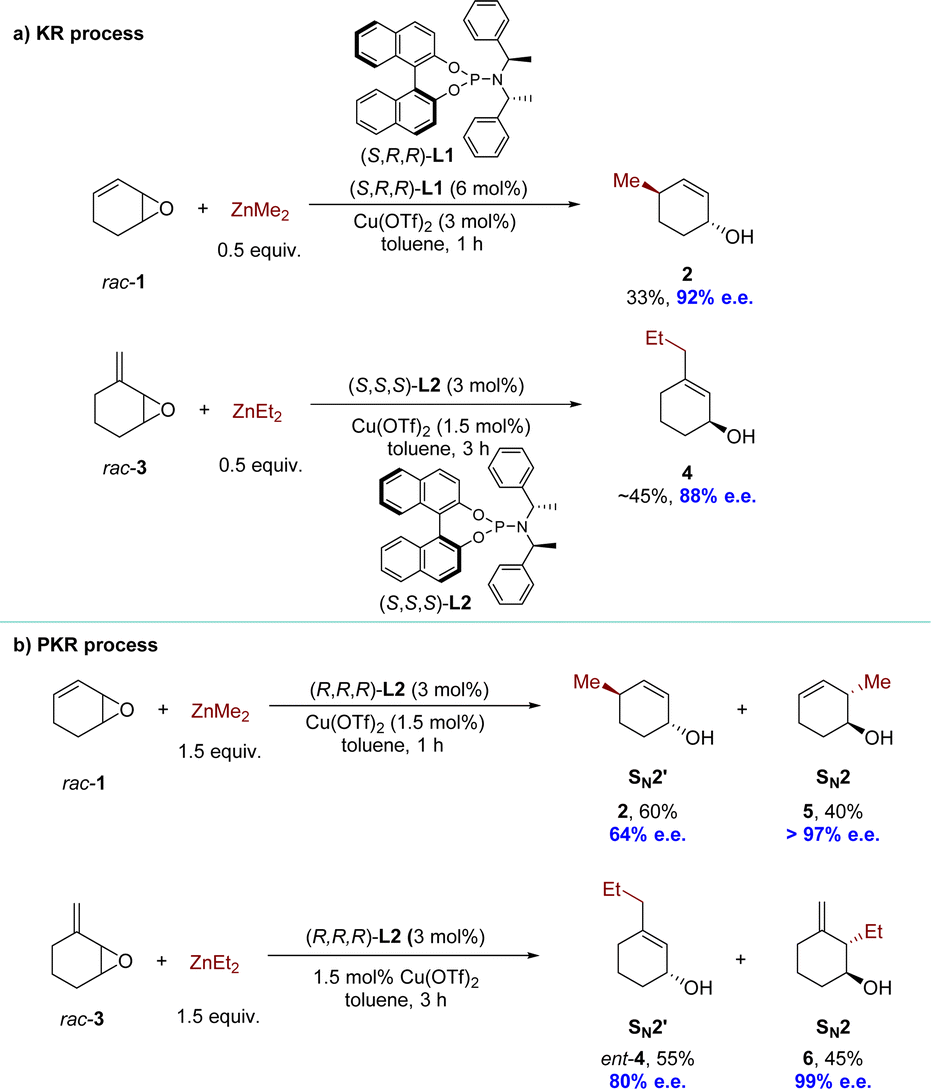 | ||
| Scheme 3 Catalytic enantioselective addition to cycloaliphatic vinyloxiranes: (a) KR process; (b) PKR process. | ||
In 2011, the Alexakis group reported an AAS reaction of acyclic allylic chlorides with Grignard reagents (Scheme 4).76 The conformational flexibility of acyclic substrates, particularly the potential for intramolecular rotation around the carbon–carbon bond, adds an additional layer of complexity to the catalytic processes. This reaction was considered as a stereodivergent parallel kinetic resolution (SKR) process.77 Oxidative addition of the pro-E conformer of (S)-7 was highly favorable, resulting in the formation of the (E)-σ-allyl intermediate. Subsequent reductive elimination, either directly or through the π-allyl complex, would selectively yield the E isomer of alkylation product 8. In stark contrast, an anti-attack on the pro-E conformer of (R)-7 would involve the olefin being approached from its Re face. This possibility would be significantly disfavored; however, the conformational flexibility of an acyclic system allowed rotation around the C3–C4 bond, leading to the formation of the pro-Z conformer of the substrate. The latter could then be attacked on its Si face, producing the (Z)-σ-allyl intermediate. This could be followed by the potential formation of a π-allyl complex and ultimately yielding (Z)-8 after reductive elimination. The facial selectivity could be attributed to steric interactions between the chiral ligand and the vinylic methyl group. Interestingly, the Alexakis group also found a slight difference in the reaction rate between the enantiomers of the substrates. Specifically, (R)-7 exhibits higher reactivity towards nucleophilic reagents. However, it is worth noting that the discrepancy in rates between enantiomers is relatively minimal. Consequently, it is currently a challenge to obtain a single (Z)-8 using this method (up to E/Z = 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :57).
:57).
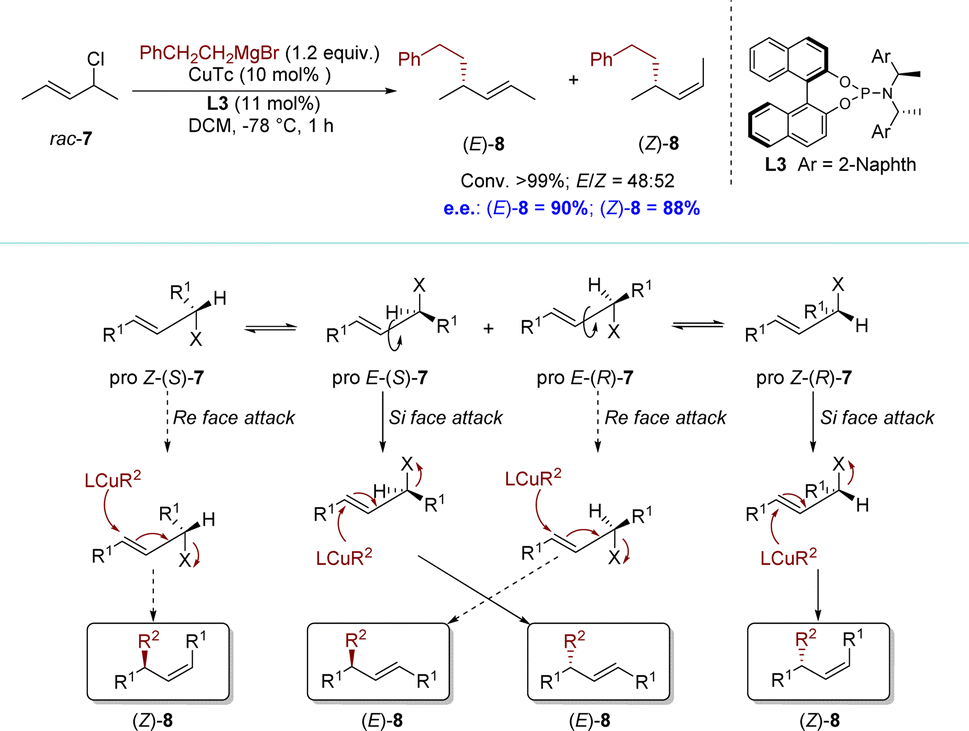 | ||
| Scheme 4 SKR via Cu-catalyzed AAS with acyclic racemic substrates and its plausible reaction mechanism. | ||
Recently, the Fletcher group reported a rare kinetic resolution of allylic chlorides, forming piperidine-based allylic chlorides with high e.e. This AAS process was achieved using Zr-nucleophiles and a copper catalyst.78 The reaction progress of 1-benzyl-3-chloro-1,2,3,6-tetrahydropyridine (rac-9) with 4-phenyl-1-butene was monitored over time (Scheme 5). After 30 minutes at −10 °C, the conversion reached 16%, resulting in the formation of product 10 with a high e.e. of 94%. However, as the less reactive enantiomer of rac-9 was gradually consumed during the reaction, the e.e. of compound 10 decreased by approximately 15%. On the other hand, the e.e. of compound rac-9 increased from 0% to 80% after continuously reacting for about 22 hours. Under optimized conditions, product (R)-9 was obtained in 30% yield with up to 99% e.e. Furthermore, experiments involving D-labelled chloro-tetrahydropyridine and density functional theory (DFT) calculations were conducted to investigate the mechanistic pathways of this process. These studies revealed that the alkylation of (R)-9 was kinetically disfavored compared to (S)-9, primarily via syn-SN2′ pathways.
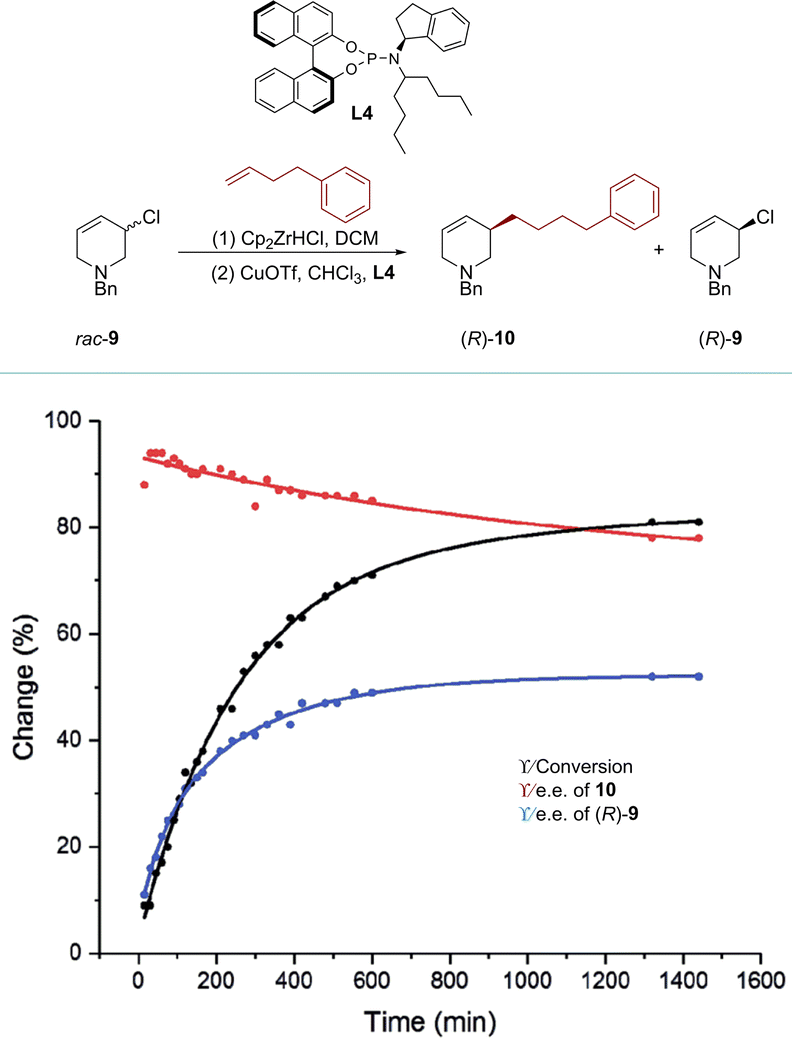 | ||
| Scheme 5 KR of rac-9 using a copper catalyst and an alkyl zirconium reagent (reproduced from ref. 78 with permission from The Royal Society of Chemistry). | ||
More recently, the Ito group developed a novel series of high-performance C2-symmetrical chiral bisphosphine ligands.79 These ligands were modified by incorporating silyl groups into the (R,R)-QuinoxP* ligand backbone. Late on, the group successfully executed a borylative kinetic resolution of acyclic allyl electrophiles using the modified ligand (L5) (Scheme 6). This ligand showed remarkable selectivity and reactivity in the borylative kinetic resolution of rac-11 at low temperatures, achieving s values up to 46.4. The allyl carbonates rac-11 were efficiently converted into the corresponding allyl boronates (S,E)-12 with high selectivity under optimized conditions. Meanwhile, the allyl carbonates (S,Z)-11 were recovered with moderate to high enantioselectivity. The introduction of silyl groups on the ligand proved to be crucial for this process. In contrast, the unmodified ligand (R,R)-QuinoxP* (L6) demonstrated an extremely low conversion of rac-11.
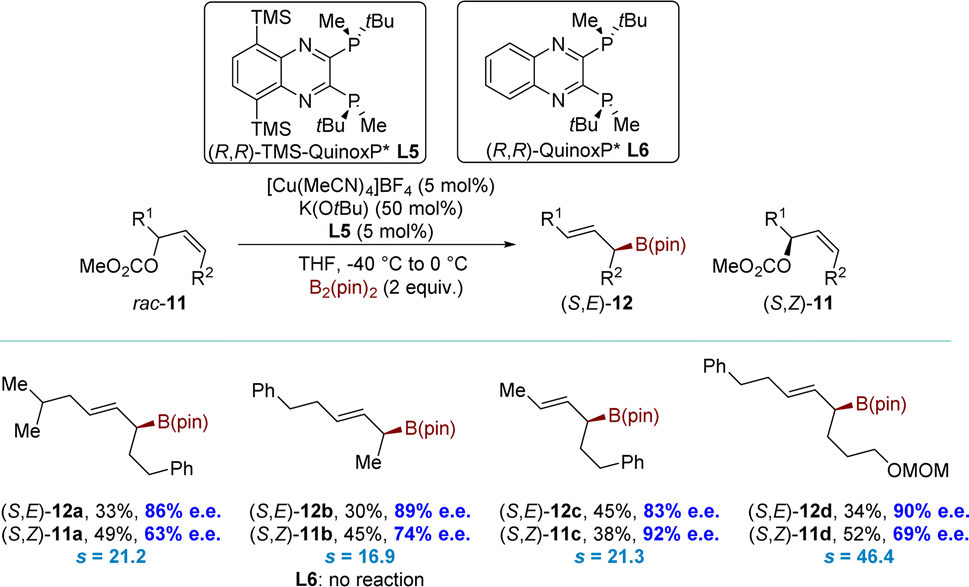 | ||
| Scheme 6 KR of rac-11 using a copper catalyst and bis(pinacolato)diboron [B2(pin)2]. | ||
2.2 Dynamic kinetic resolution
A more universally applicable approach involves the development of dynamic processes, wherein the initial substance undergoes racemization either during the reaction or is recovered post-reaction, racemized, and reintroduced into the reaction conditions. This effectively overcomes the limitation of a maximum yield of 50% in the KR process. In 2018, the Tan group employed functionalized terminal alkynes as pro-nucleophiles to synthesize cyclic 1,4-enynes with 6-, 7- and 8-membered rings, achieving high yields and excellent enantioselectivities under mild reaction conditions (Scheme 7).80 This system was compatible with various terminal alkynes and cyclic allylic bromides. The identification of catalytic intermediates, such as guanidinium cuprate ion pairs and a copper–alkynide complex, was achieved through the utilization of cold-spray ionization mass spectrometry (CSI-MS) and X-ray crystallography (XRD). A direct correlation between the enantiopurity of the catalyst and the reaction product suggests the presence of a copper complex with a singular guanidine ligand during the enantio-determining step.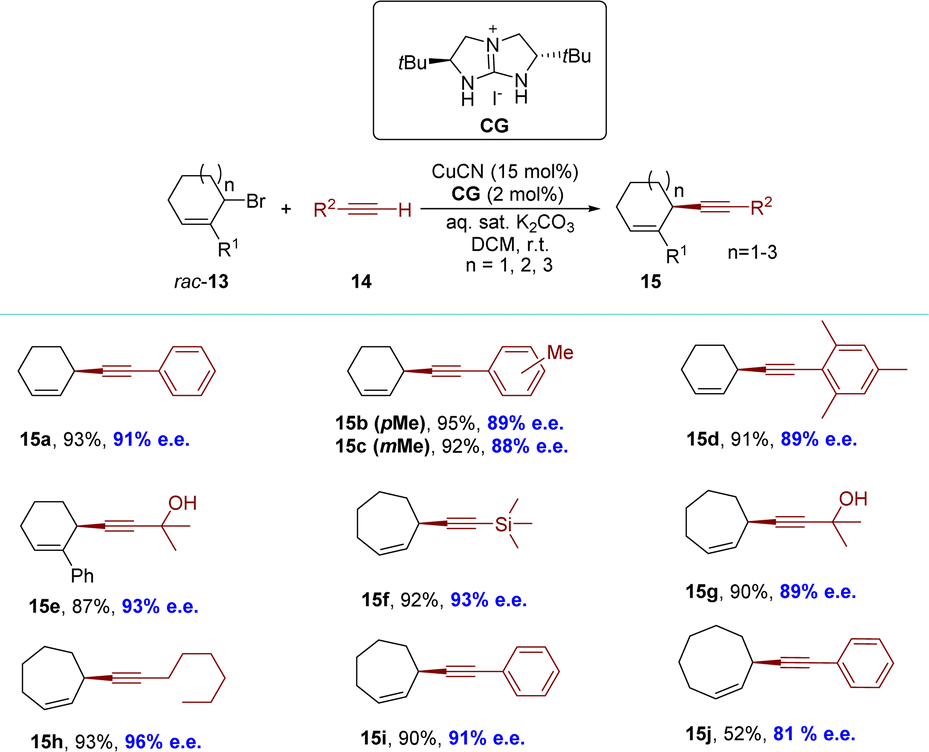 | ||
| Scheme 7 Guanidine–copper catalyzed enantioselective allylic alkynylation of racemic bromides. | ||
To elucidate the stereo-dynamic process of the reaction, the authors conducted a two-dimensional exchange spectroscopy nuclear magnetic resonance (2D EXSY NMR) analysis to investigate the potential ligand-dependent syn-SN2′ reaction between allylic bromide and allylic cyanide. During the initial optimization of the reaction conditions, they introduced the corresponding guanidine (free base) of CG into the reaction mixture (in the absence of iodide) and observed a similar reaction profile, thereby ruling out any involvement of allylic iodide as an intermediate in racemization. Additionally, rapid racemization of allylic bromide occurred when enantioenriched 13 (50% e.e.) was employed with the catalytic system. By employing 2D EXSY NMR spectroscopy, they detected a chemical exchange between Ha(Br) and Hc(Br) protons in 13 in the presence of a guanidine–copper complex (Scheme 8). These findings suggest that the presence of the guanidine–copper complex significantly enhanced racemization rates for allylic bromides, likely through syn-SN2′ isomerization which played a crucial role in successful dynamic kinetic resolution.
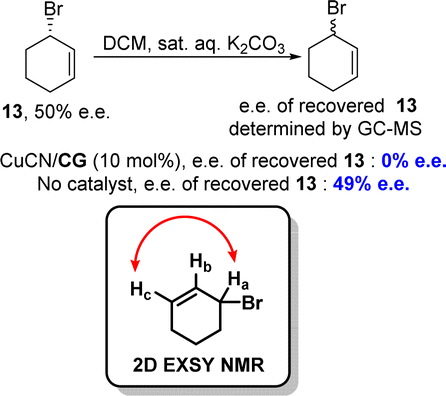 | ||
| Scheme 8 Racemization of enantioenriched allylic bromide 10. | ||
2.3 Dynamic kinetic asymmetric transformation
The concept of dynamic kinetic asymmetric transformation (DyKAT) in allylic substitution was introduced by Trost at the end of the 90s, representing one of the most efficient methodologies for converting a racemic substrate into an enantioenriched product.81,82 Although copper exhibits high catalytic efficiency in promoting the formation of enantioselective allylic C–C bonds using non-stabilized nucleophiles, reports on copper-catalyzed DyKAT were absent until 2010. The Feringa group pioneered the development of a catalytic asymmetric synthesis for chiral enol acetates (Scheme 9).83 The method includes the in situ conversion of α,β-unsaturated aldehydes 16 into α-chloroallylic acetates 17, followed by regio- and enantioselective allylic alkylation of the α-chloroallylic acetates 17 to yield products (Z)-18. This innovative method provides access to highly desirable β-substituted aldehydes 19. The process might operate via a DyKAT process, where one enantiomer of the α-chloroallylic acetates 17 reacts more rapidly than the other with the chiral organocopper key intermediate. The equilibrium between the α-chloroallylic acetates 17 and the α,β-aldehydes 16 likely serves as the racemization mechanism.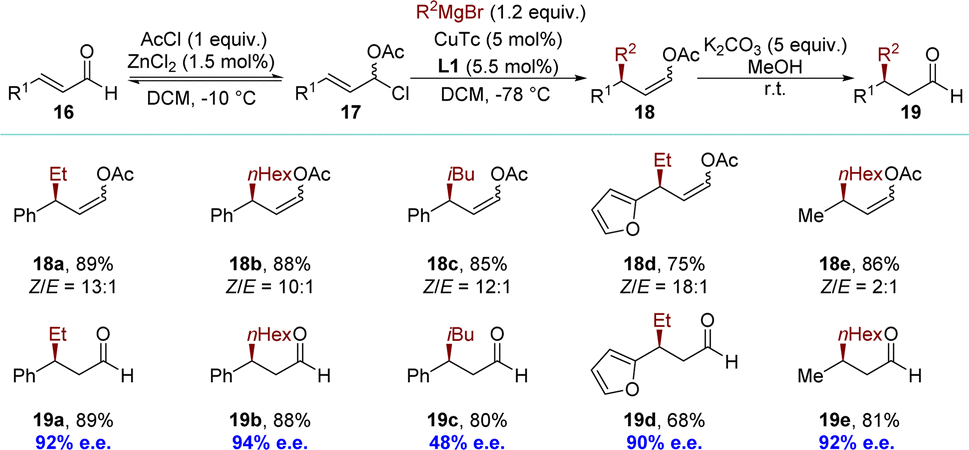 | ||
| Scheme 9 Copper-catalyzed synthesis of chiral enol acetates and aldehydes via allylic alkylation of in situ formed α-chloroallylic acetates. | ||
In 2015, the Fletcher group reported a remarkable copper-catalyzed DyKAT process involving the asymmetric allylic alkylation of racemic cyclic allylic chlorides with alkyl zirconocenes.84–86 This convenient procedure enabled the synthesis of highly enantioenriched products at room temperature (Scheme 10a). The investigation commenced with an examination of the interaction between racemic 3-substituted cyclohexenes and alkyl zirconium species, which were generated in situ from terminal alkenes and Schwartz's reagent (Cp2ZrHCl) (up to 95% e.e.). The study demonstrated the rapid accessibility of crucial structural motifs, as evidenced by the successful asymmetric syntheses of biologically active cyclopentene-containing natural products (hydnocarpic acid, chaulmoogric acid and anthelminthicin C) (Scheme 10b). The scope of nucleophilic reagents was significantly expanded by this research, as it demonstrated compatibility with halogens, alkynes, aromatic rings, ethers, protected alcohols, and the trifluoromethyl group. However, the generation of alkyl zirconium species in this reaction requires the use of terminal alkenes and Schwartz's reagent. The applicability of this method for constructing chiral products containing methyl or secondary alkyl groups is somehow limited by this stipulation. Mechanistic studies suggested that this DyKAT occurred through a rapidly interconverting intermediate, which racemized the substrate by the I anion (Scheme 10c). The racemization of 20, probably through syn-SN2′ displacement,87 and the selective AAS of (S)-20 resulted in products with high e.e. In situ1H-NMR spectroscopy was employed to gather kinetic data, which were then fitted to a model. This data, in conjunction with EXSY and diffusion-ordered spectroscopy (DOSY) experiments, suggested that the catalyst evolves over time to become more efficient and more enantioselective. The initial catalyst is presumed to be dimeric, such as Cu2I2L2. However, as the reaction progressed and the Cl anion was released from the substrate, a more selective catalytic complex containing both I and Cl anions, such as Cu2IClL2, was formed. This evolved catalyst exhibited a threefold increase in reactivity compared to the original catalyst. The structure–activity relationships demonstrated that, in specific cases, remarkable stereochemical control could be achieved by converting racemic mixtures of diastereomers into a single diastereoisomer product with high e.e. After that, the same group successfully applied this method to alkenyl zirconocene reagents, building upon their previous work with well-tuned DYKAT and alkyl zirconocene nucleophiles.88 This elegant demonstration showcases the use of sp2-hybridized nucleophiles in DYKAT (Scheme 10d). Furthermore, this asymmetric allylic alkylation (AAA) system was developed utilizing racemic allylic phosphates and a novel ligand L8.86 This transformation was achieved by utilizing allylic chloride 20, which was formed in situ through the action of TMSCl or from an adventitious Cl anion generated from reaction components (Scheme 10d). The ability to rapidly generate isomerizing allylic halide species in situ from these phosphates 29 is likely to have significant utility in chemistry.
 | ||
| Scheme 10 Addition of alkyl zirconium reagents to racemic cyclic allylic chlorides via DyKAT: (a) dynamic kinetic AAA with alkylzirconocene nucleophiles; (b) concise asymmetric application; (c) proposed mechanism for the DyKAT; (d) extension. | ||
2.4 Direct enantioconvergent transformation
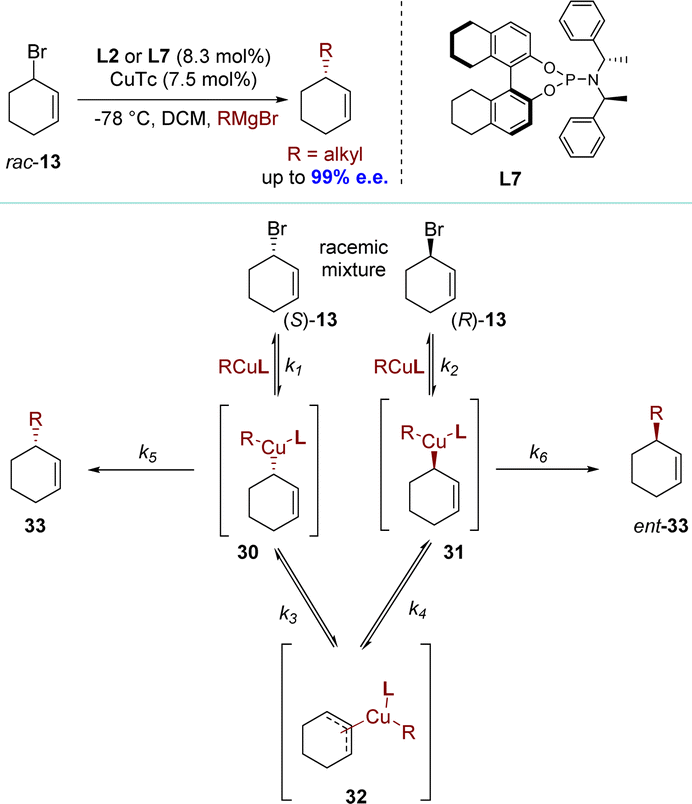
Over the past decade, significant attention has been paid to the synthesis of enantiomerically enriched molecules through DET processes. This process lacks any observable dynamic or reversible characteristics in terms of organic stereogenicity, distinguishing it from conventional DKR and DyKAT processes.In 2009, the Alexakis group reported the alkylation of racemic cyclohex-1-enyl-3-bromide 13. This reaction employed copper(I) thiophene-2-carboxylate (CuTc) with L2 or L7, achieving high efficiency (up to 99% e.e.) with primary alkyl Grignard reagents (Scheme 11).89,90 Based on the experimental results, the authors proposed a plausible reaction mechanism (Scheme 11). The initial step involved ionizing the substrate through metal insertion at the allylic terminus (oxidative addition), while the final step, reductive elimination, led to a mixture of products 33 and ent-33. During these two steps, an equilibrium was established between σ-allyl species 30 and 31, derived from ionization of (S)-13 and (R)-13 respectively, via meso-π-allyl complex 32. Allylic bromides exhibited high reactivity and Cu(III) intermediates presumably underwent equilibration via π-allyl intermediates similar to type 32. Consequently, the rate-determining step shifted towards reductive elimination (k1, k2, K3, K4 ≫ k5, k6). After conducting control experiments to rule out the hypothesis of KR, it was determined that enantio-discrimination resulted from differences in reductive elimination rates, which disrupted intermediate equilibration and favored the formation of products with higher elimination rates.
 | ||
| Scheme 11 Alexakis' work and the proposed mechanism for the DyKAT. | ||
Notably, three years later, the Alexakis group found through DFT calculations that the rate of the oxidative addition step was much lower than the rate of the reductive elimination step (k1, k2 ≪ k5, k6), which was inconsistent with previous assumptions.91 The same group proposed a revised mechanistic route, highlighting the divergent reactivities of both enantiomers of the substrate (Scheme 12a).91 Indeed, (R)-13 underwent an anti-SN2′ oxidative addition, while its antipode reacted in an anti-SN2 manner. This regiodivergent oxidative addition facilitated the formation of a common Cu(III) intermediate, characterized as a cationic σ–π-enyl copper complex, referred to as Int-13. No equilibrations were expected from this key intermediate, and a rapid reductive elimination led to the desired product (R)-33. The absence of any resolving step and the distinct mechanistic pathways for each enantiomer of 13 established this process as a direct enantioconvergent transformation (DET).
 | ||
| Scheme 12 (a) Revised reaction mechanism and the scope of sterically hindered nucleophiles; (b) scope of nucleophiles. | ||
Additionally, the same research group reported further enhancements to the process and expanded the scope of the nucleophiles.91 These advancements included the utilization of both primary alkyl and sterically hindered secondary alkyl Grignard reagents, which effectively demonstrated the versatility of this methodology. However, this catalytic system proved incompatible with Grignard reagents that varied in activity and steric hindrance (Scheme 12b). The efficacy of the allylic bromide substrate improved when employing highly active nucleophiles with less steric hindrance (Me, Et, and nBu); however, its effectiveness decreased with the use of sterically hindered nucleophiles.89,90 On the other hand, the allylic ester substrate outperformed allylic bromide when employing sterically hindered cyclic nucleophiles; however, no product was observed in the presence of a less active methyl Grignard reagent, resulting in only hydrolysis by-products.91
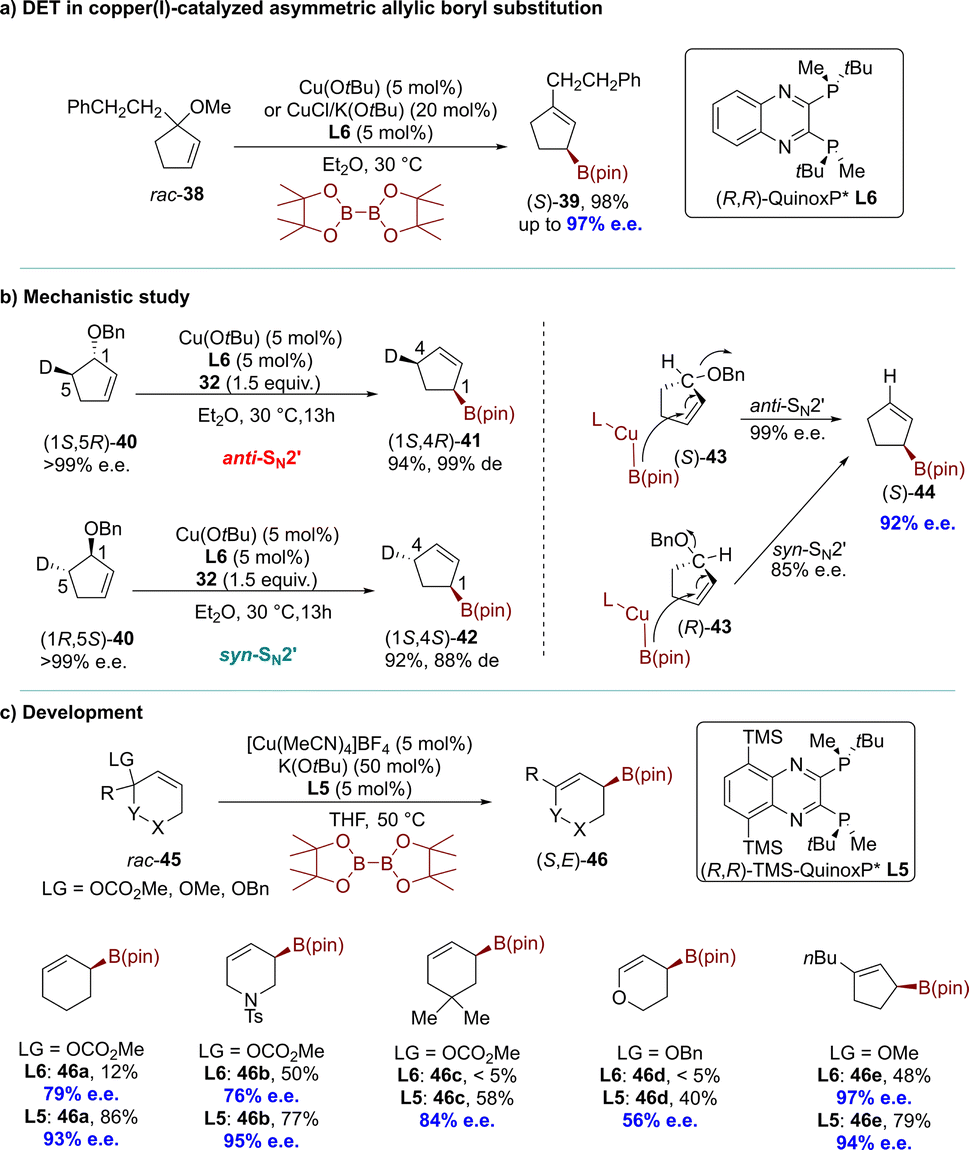
In 2010, the Ito group introduced a DET process promoted by an artificial catalyst.67 The copper(I)-catalyzed AAS of a racemic allylic ether resulted in the formation of a single enantiomer of a chiral α-allylboronate with complete conversion and high enantioselectivity (up to 98% e.e.). One enantiomer of the substrate underwent an anti-SN2′-type reaction, while the other reacted via a syn-SN2′ pathway (Scheme 13a). These products, which could not be synthesized using dynamic procedures, have been utilized for constructing all-carbon quaternary stereocenters. The reaction of rac-38 was initially conducted with B2(pin)2 in the presence of Cu(OtBu) and a chiral phosphine ligand (R,R)-QuinoxP* (L6) in diethyl ether at 30 °C. Complete conversion of rac-38 was achieved within 24 hours in a SN2′ manner, yielding the corresponding allyl boronate (S)-39 in high yield (98%) with excellent enantioselectivity (up to 97% e.e.). Further experiments using deuterated substrates clearly demonstrated that the two enantiomers underwent distinct pathways, namely anti-SN2′ and syn-SN2′, while the chiral centers adjacent to the deuteride remained unchanged. Consequently, this resulted in the formation of two diastereomeric products from each enantiomer. The experimental results could be adequately explained by assuming a direct enantio-convergent reaction. When (R,R)-QuinoxP* (L6) was used as the ligand in the AAS of (S)-43, the nucleophilic attack occurred on the opposite face of the carbon–carbon double bond relative to the leaving group, resulting in the formation of (S)-44 through an anti-SN2′-type pathway with 99% e.e. In contrast, when approaching (R)-43, the same borylcopper(I) intermediate approached from the same side as the leaving group, leading to the formation of (S)-44 through a syn-SN2′-type reaction with 85% e.e (Scheme 13b). A decade later, the Ito group modified (R,R)-QuinoxP* (L6) by introducing the silyl moieties into the ligand's backbone, resulting in (R,R)-5,8-TMS-QuinoxP* (L5).79 The introduction of silyl moieties significantly improved the reactivity and enantioselectivity in the direct enantioconvergent borylation of substrates. The borylation products, including heterocyclic structures, were obtained with higher yield and excellent enantioselectivity, surpassing the results from using (R,R)-QuinoxP*(L6) (Scheme 13c).
 | ||
| Scheme 13 Copper(I)-catalyzed asymmetric allylic boryl substitution via DET and its plausible reaction mechanism: (a) DET in copper(I)-catalyzed asymmetric allylic boryl substitution; (b) mechanistic study; (c) development. | ||
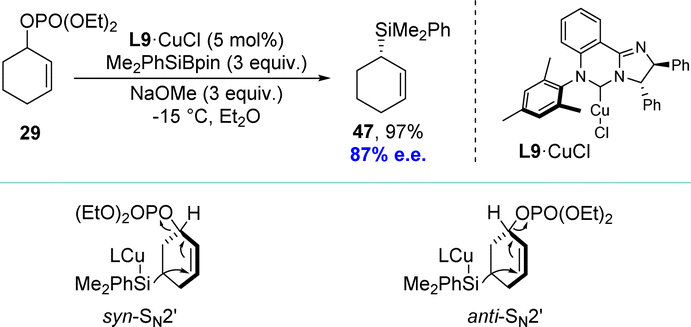
Inspired by the work of the Alexakis and Ito groups, the Oestreich group developed a copper(I)-catalyzed asymmetric allylic silylation of racemic cyclic phosphates using Me2PhSiBpin as the silicon pronucleophile (Scheme 14).92 The group successfully converted racemic cyclic allylic phosphate 29 into highly enantioenriched cyclic allylic silane 47 in near-quantitative yield. The NHC–copper(I) chloride complex L9·CuCl was transformed into the active catalyst L9·CuOMe via ligand metathesis with sodium methoxide, facilitating this reaction. Observations from matched and mismatched substrate/catalyst combinations revealed that both enantiomers undergo SN2′ reactions, each with opposite diastereofacial selectivity. Consequently, the enantiomeric allylic phosphates lead to the identical allylic silane via two separate reaction mechanisms, defining it as a DET process.
 | ||
| Scheme 14 Copper(I)-catalyzed asymmetric allylic silylation via DET and its plausible reaction mechanism. | ||
Given the profitability of the efficient dynamic kinetic copper-catalyzed AAS reaction with a chiral bicyclic guanidine, the Tan group reported a monodentate guanidine–copper(I) complex catalyzed SN2′ borylation process in 2019. This transformation proceeded through a unique DET mechanism. The allyl boronates 48 were obtained with exceptional regioselectivities (up to 99:1 r.r.) and enantioselectivities (up to 99% e.e.). This allowed their subsequent transformation into a wide range of functionalized secondary and tertiary allylic compounds while maintaining their optical purity (Scheme 15).93 The DFT calculations indicated that the reaction follows the DET mechanism, in which the (R)- and (S)-enantiomers of allylic bromide 13 undergo kinetically competitive pathways: (R)-anti oxidative addition and (S)-syn oxidative addition respectively. These pathways led to the formation of a common intermediate intB-R without involving conventional resolving steps. Subsequently, a barrierless B–C bond formation occurred, generating the stereochemically preferred (R)-boronate.
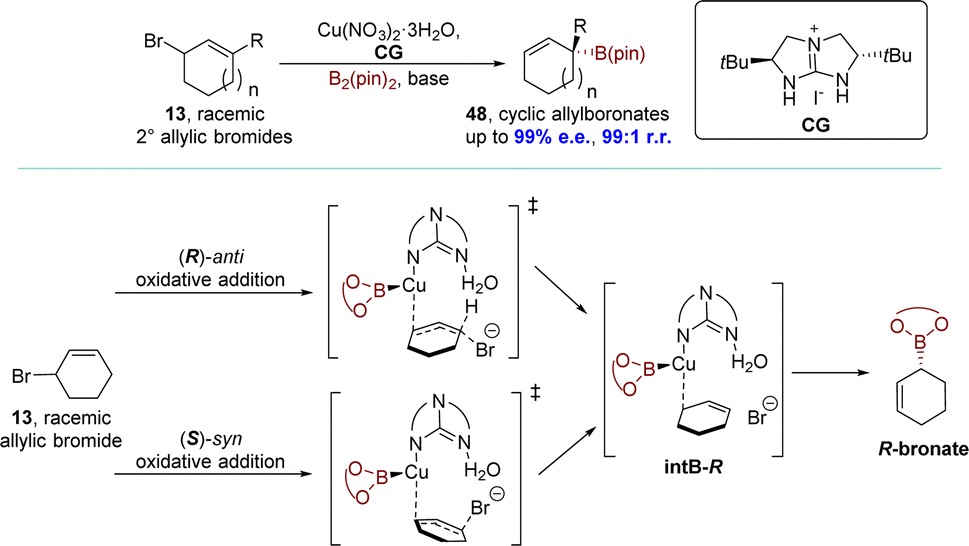 | ||
| Scheme 15 Guanidine–copper complex catalyzed allylic borylation via DET. | ||
In 2022, the Chen group developed a novel Cu-catalyzed AAA process that enabled the enantioconvergent transformation of various racemic cyclic allylic bromides using organolithium reagents, giving the product in high yields with enantioselectivities (Scheme 16).94 Utilizing CuBr·SMe2 and phosphoramidite ligand L7 facilitated convenient access to optically pure cycloalkene derivatives. Furthermore, the catalytic enantioselective allylic alkylation processes have been successfully adapted to continuous flow techniques,95–100 but only with 33% e.e. The difference in the contact mode between batch and flow reactions primarily led to a decrease in enantioselectivities. Typically, in batch reactions, organometallic reagents were introduced dropwise over an extended period into the substrate solution to avert side reactions. However, in flow reactions, substrates and organometallic reagents were mixed simultaneously. As a result, highly active allylic bromides resulted in uncatalyzed reactions or reacted with “ate” and “aggregated” copper species91 which were produced upon the exposure of the chiral catalyst to an excess of organometallic reagents.
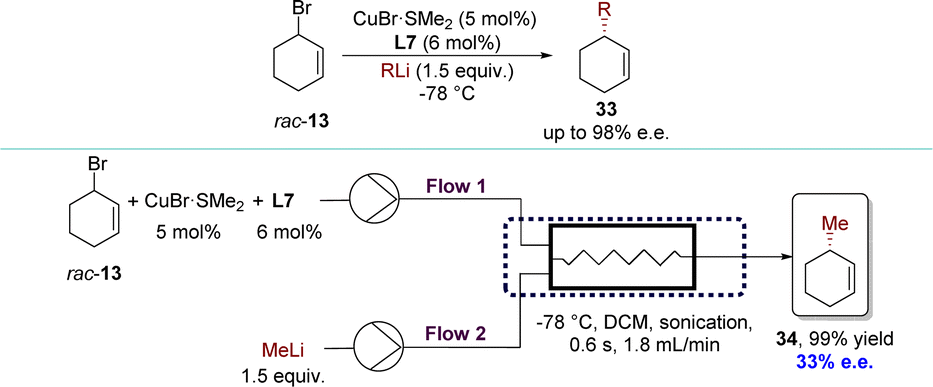 | ||
| Scheme 16 Organolithium reagents in Cu-catalyzed AAA reactions with racemic cyclic substrates via DET. | ||
Recently, the same research group has updated this highly efficient AAA reaction by incorporating racemic inert cyclic allylic ethers in combination with BF3·OEt2 and a Cu complex (Scheme 17).101 This catalytic system demonstrated general effectiveness for challenging, sterically hindered Grignard reagents, accommodating both primary and secondary alkyl groups (Scheme 17a). For mechanistic insight, it is possible that the substrates underwent a similar in situ conversion process as observed by the Fletcher group,86 where racemic 13 was formed from rac-49 in the presence of BF3·OEt2 and the Br anion. Subsequently, the resulting racemic 13 might have undergone the same DET process to form the identical intermediate Int-13 with Grignard reagents as reported by the Alexakis group.91 Finally, Int-13 underwent reductive elimination to yield the desired product 50 with high efficiency and enantioselectivity (Scheme 17b). The method was successfully applied under continuous flow conditions, demonstrating excellent yield and enantioselectivity within a short residence time. Remarkably, benefitting from the in situ conversion strategy, the allylic bromide generated in situ from allylic ethers would react with privileged copper species rapidly, avoiding most of the side reactions under flow conditions. The catalytic system exhibited uninterrupted operation for over 700 minutes without any loss of yield or enantioselectivity, resulting in the production of 1.09 grams of product 50d (84% yield, 91% e.e.) (Scheme 17c).
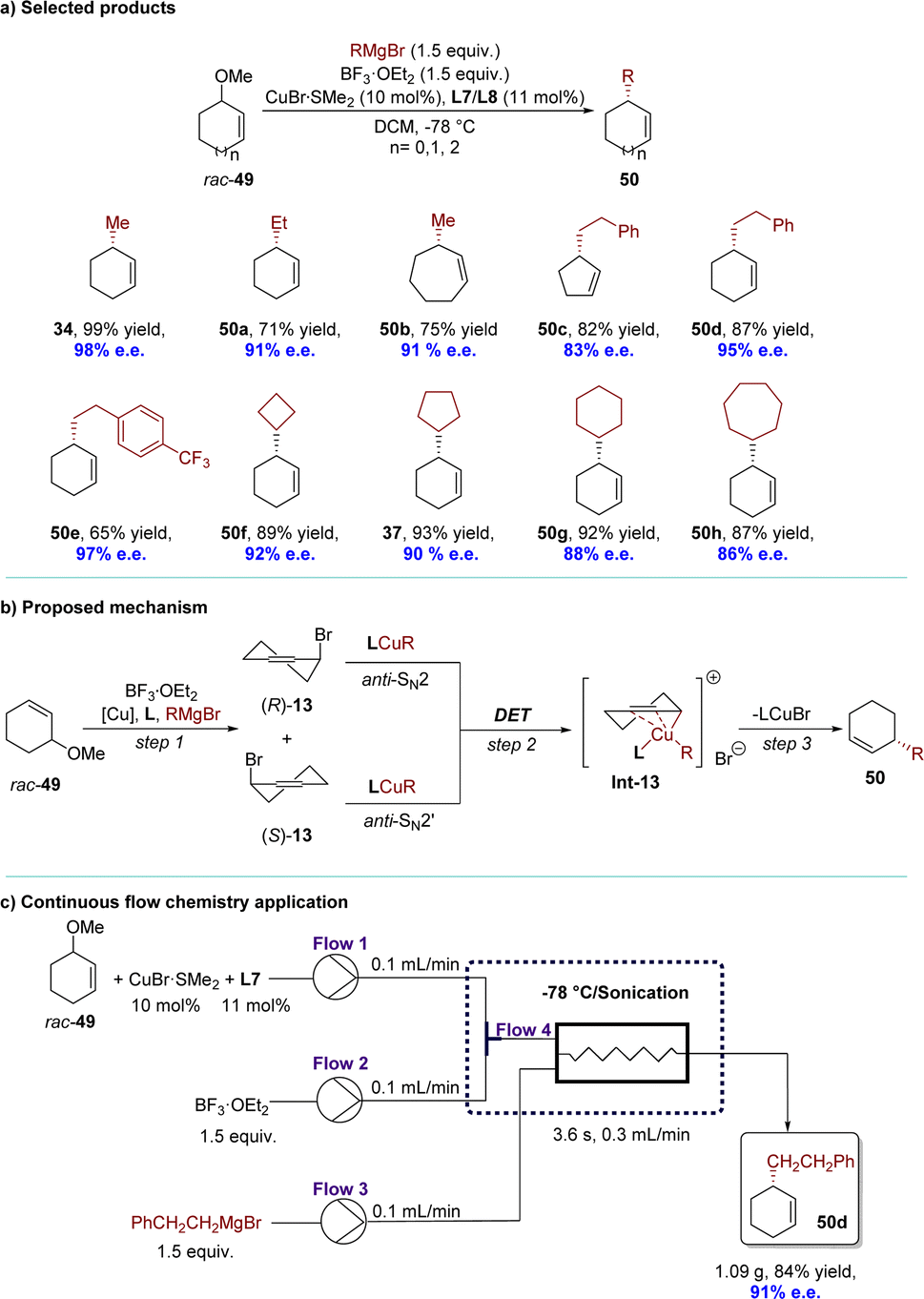 | ||
| Scheme 17 Cu-catalyzed AAA of racemic inert cyclic allylic ethers via DET: (a) selected products; (b) proposed mechanism; (c) continous flow chemistry application. | ||
2.5 Miscellaneous
The kinetic process is not only applied in constructing chiral centers, but also in controlling the Z/E selectivity of reactions. In 2020, the Yun group described the copper-catalyzed stereoconvergent allylation of chiral sp3-hybridized carbon nucleophiles using a racemic mixture of acyclic secondary allylic phosphates (Scheme 18).102 The tandem coupling reaction of vinylarenes, B2(pin)2, and racemic allylic phosphates 52 catalyzed by a copper–ligand L10 complex, led to the formation of chiral alkylboronates possessing an (E)-alkenyl moiety. This transformation proceeded through a direct stereoconvergent allylic coupling, generating a new C(sp3)-stereogenic center. The insertion of a vinyl arene into a chiral ligand-bound copper–boryl complex resulted in the formation of enantiomerically enriched β-boryl benzyl copper species 51. This was followed by direct stereoconvergent C–C bond formation between 51 and the racemic secondary allylic electrophile 52, leading to the desired coupling product 53. Finally, regeneration of the L10*Cu-Bpin species completed the catalytic cycle. This stereoconvergent transformation process, similar to the DET process, involves converting both enantiomers into an (E)-olefin, with the homoallylic chiral center being established during the borylcupration step.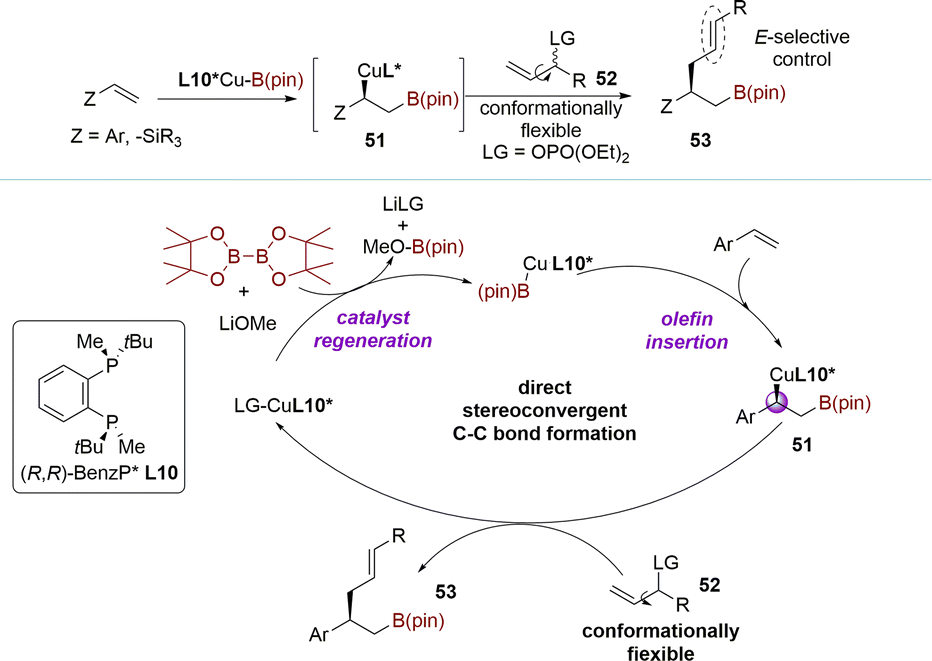 | ||
| Scheme 18 Allylation of chiral alkyl-copper nucleophiles with racemic allylic phosphates via DET. | ||
3. Meso-compounds as starting materials
Next to the presence of racemic substrates, meso compounds constitute a distinct and significant class of substrates containing chiral factors in asymmetric organic reactions.103 Over the past two decades, the desymmetrization strategy for meso compounds has become a crucial, refined, and powerful method in the realm of asymmetric synthesis.104–108 Notably, the Cu-catalyzed AAS has been extensively utilized in desymmetrization processes to synthesize enantiomerically enriched products, often serving as a strategic approach for accessing natural compounds. The typical dynamic process of a Cu-catalyzed AAS reaction for meso substrates can be described as follows: the nucleophile attacks one of the enantiotopic faces of the substrate under the induction of ligands, resulting in an enantiopure product (Scheme 19a). The pioneering work was reported by the Feringa group in 2000 (Scheme 19b).69 Encouraged by preliminary results from the kinetic resolution protocol with methylidene cycloalkane epoxide 3, they explored the possibility of incorporating dialkylzinc reagents into the enantiotopic faces of a symmetrical meso epoxide 54, thereby circumventing the associated inherent limitations of a kinetic resolution process. The first reported catalytic desymmetrization protocol for Cu-catalyzed AAS, using novel symmetrical vinyloxiranes, was disclosed.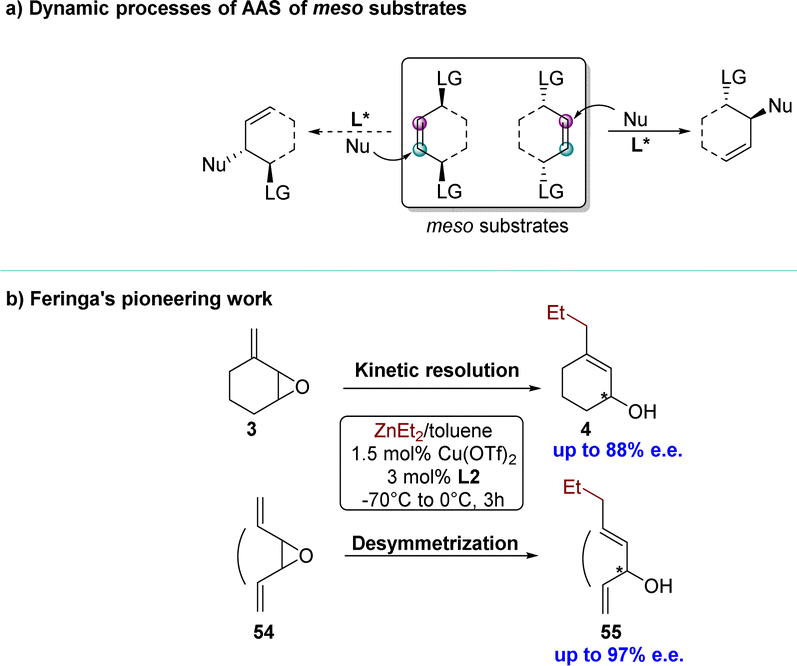 | ||
| Scheme 19 (a) Dynamic processes of AAS of meso substrates (b)_Feringa's pioneering work. | ||
In 2003, the Gennari group introduced a novel highly regio-, diastereo-, and enantioselective desymmetrization of meso, cyclic allylic bisdiethylphosphates. This method employs organozinc reagents and is catalyzed by copper(I) complexes of chiral Schiff base ligands (L11, L12) (Scheme 20).109 For 5-membered ring substrates, the AAS reaction proceeded with high yields and predominantly exhibited anti-SN2′ selectivity, achieving an e.e. of 88%. Unfortunately, the applicability of this reaction to substrates with larger ring sizes proved to be unsuccessful.110 To address this issue, the same group opted to employ phosphoramidite ligands (L1 and L2) and, pleasingly, achieved an e.e. up to 94% for the 6-membered ring product 57b and predominantly one enantiomer (e.e. > 98%) for the 7-membered ring product 57c.
 | ||
| Scheme 20 Desymmetrisation of meso cyclic allylic bis(diethyl phosphates) with organozinc reagents. | ||
The desymmetrization reactions of meso compounds through enantioselective catalysis have emerged as a robust strategy for the synthesis of chiral molecules with multiple stereocenters, which garners significant attention. In 2010, the Ito group reported a remarkable desymmetrization protocol through the enantioselective synthesis of allyl boronates catalyzed by copper(I) (Scheme 21).111 The reaction of the carbonate derivative of meso-diol 58 and B2(pin)2, catalyzed by Cu(OtBu) and a chiral ligand (R,R)-QuinoxP* L6 in toluene at −20 °C, was completed efficiently within 4 hours. Subsequently, benzaldehyde was added at 0 °C to afford compound 60 in high yield (87%) with exceptional diastereo- and enantioselectivities (>99:1 d.r., 97% e.e.). The desymmetrization reaction was employed for the efficient asymmetric synthesis of a precursor to an antiviral medication. This process represented a concise formal synthesis of (−)-abacavir or (−)-carbovir in only five steps (Scheme 21b).112 Coordination of the meso substrate with the chiral borylcopper(I) intermediate yielded complex 64, thereby avoiding the formation of sterically unfavorable 64′. Subsequent addition of borylcopper(I) across the carbon–carbon double bond generates alkylcopper(I) 65, which upon elimination produced the formal anti-SN2′ product allyl boronate 59 and copper(I) carbonate. Regeneration of alkoxycopper(I) was achieved upon the release of carbon dioxide from copper carbonate. Carbonyl addition to 59 proceeded via a 6-membered transition state, positioning the R1 group equatorially, ultimately yielding product 66 after hydrolysis (Scheme 21c).
 | ||
| Scheme 21 Desymmetrization by copper(I)-catalyzed asymmetric borylation of meso-2-alkene-1,4-diol derivatives: (a) new desymmetrization procedure for meso-2-alkene-1,4-diol derivatives; (b) application to established antiviral drugs; (c) proposed mechanism. | ||
An enantioselective SN2′-type allyl–allyl coupling between allyl boronates and (Z)-acyclic/cyclic allylic phosphates was developed by the Sawamura group in 2016 (Scheme 22).113 The reaction was facilitated by the catalysis of a copper(I) complex utilizing a novel phenol/NHC chiral ligand L13, leading to successful application in the desymmetrization of the meso cyclic 1,3-diol diphosphate derivative. The reaction exhibited exceptional regioselectivities and moderate enantioselectivities of the SN2′-type, leading to the formation of chiral 1,5-diene derivatives 68 with a tertiary stereogenic center at the allylic position.
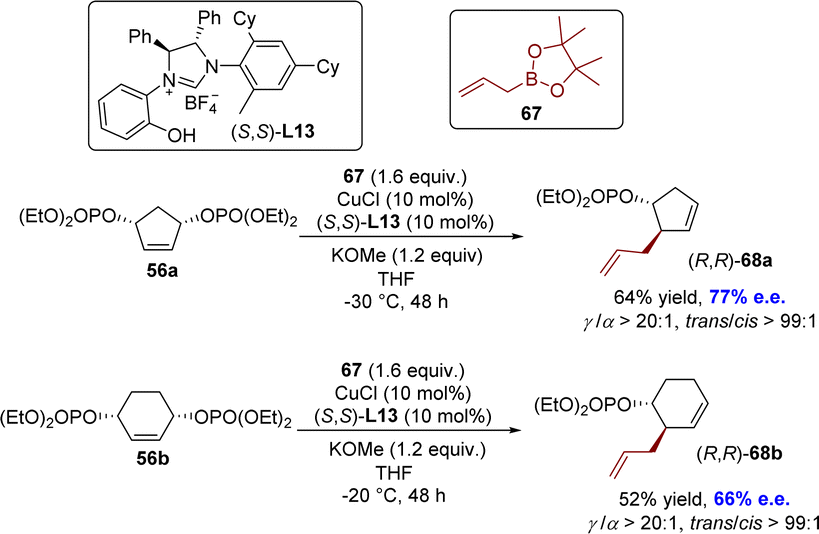 | ||
| Scheme 22 Desymmetrization by copper(I)-catalyzed enantioselective allyl–allyl coupling. | ||
A highly regio- and enantioselective desymmetrization of meso-1,4-dibromocycloalk-2-enes 69 using AAS with organolithium reagents has been successfully achieved by Feringa's group in 2018, resulting in the formation of bromocycloalkenes with excellent diastereoselectivity and enantioselectivity (Scheme 23).114 After careful optimization of reaction conditions, the subsequent utilization of 5- or 6-membered substrates led to the successful incorporation of commercially available alkyllithium reagents, resulting in the formation of AAS products 70 with exceptional enantioselectivities (up to 99:1 e.r). When meso-3,7-dibromo-cycloheptene 71 was employed in the desymmetrization reaction with an alkyllithium reagent, the anticipated products 72 were initially obtained with high diastereoselectivity (>99:1 d.r.) and enantioselectivity (80% to 94% e.e.), as determined by NMR and chiral GC analysis (Scheme 23b). However, during the attempted purification of these 7-membered ring products 72 using flash column chromatography on silica gel, only their corresponding cyclohexene analogs 73 were isolated with high stereospecificity. Experimental and theoretical evidence substantiated a rare type I dyotropic rearrangement involving a [1,2]-alkene shift via transition state TS-74a or a stepwise formal dyotropic rearrangement via an ion-pair intermediate 74b, resulting in regio- and stereospecific ring contraction of bromocycloheptenes.115 Compared to alkyl groups, phenyl substituents are less susceptible to rearrangement reactions. With the employment of L14 as the ligand, 7-membered ring product 75 was successfully isolated in 83% yield with excellent enantioselectivity (90% e.e.).
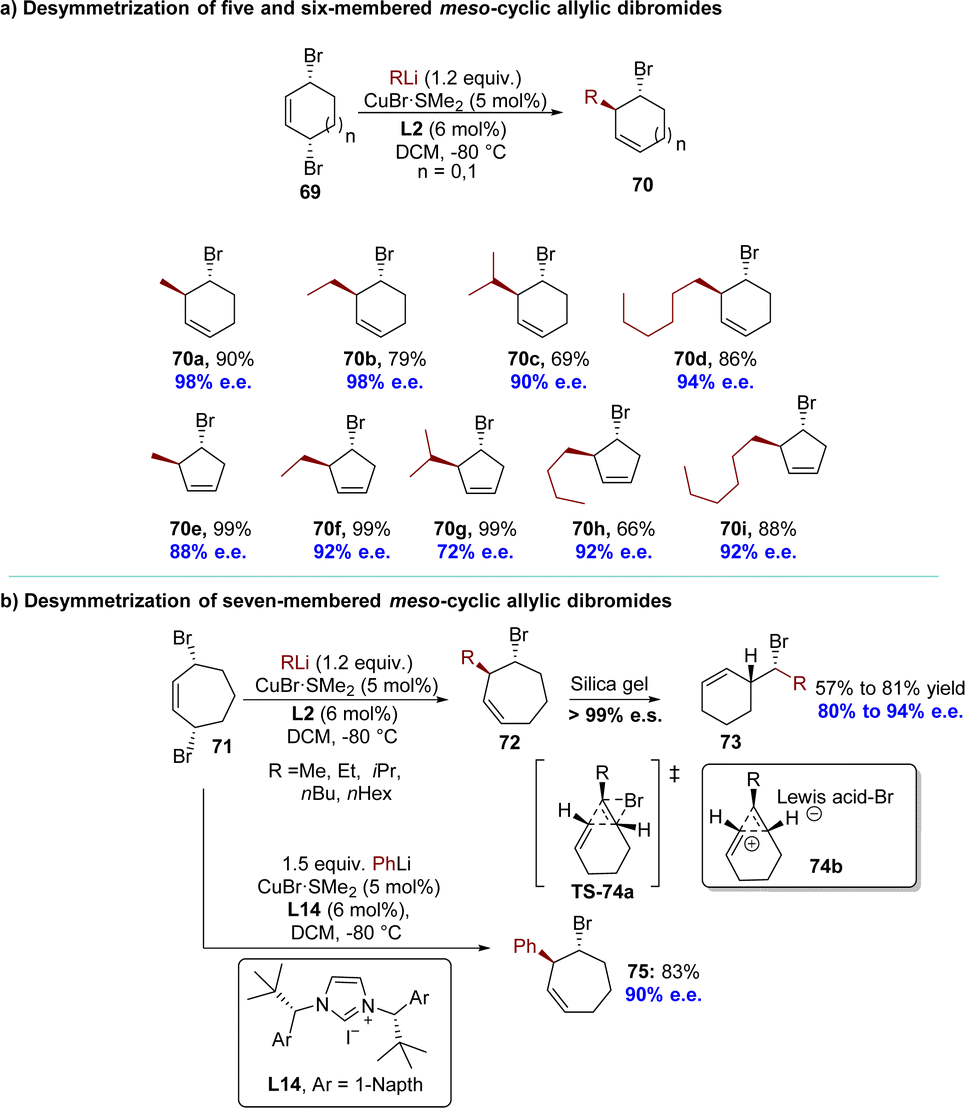 | ||
| Scheme 23 Desymmetrization of meso-dibromocycloalkenes through copper(I)-catalyzed AAS with organolithium reagents: (a) desymmetrization of five and six-membered meso-cyclic allylic dibromides; (b) desymmetrization of seven-membered meso-cyclic allylic dibromides. | ||
Benefiting from accumulated research in organozirconium reagents, the Fletcher group successfully demonstrated a highly enantioselective Cu-catalyzed desymmetrization of diverse meso-cyclic bisphosphates using in situ generated hydrozirconated alkene nucleophiles (Scheme 24).116 To demonstrate the generality of the reaction across various ring sizes, 5-, 6- and 7-membered meso-bisphosphates 56 were subjected to desymmetrization using terminal olefins. The reaction accommodates functional groups that are incompatible with numerous conventional organometallic reagents, thereby enabling access to a wide array of functionalized carbocyclic and heterocyclic structures. The resulting products feature up to three adjacent stereogenic centers, inclusive of quaternary centers and spirocyclic ring systems. Mechanistically, a hybrid species 77 was formed between the Cu–phosphoramidite complex and the hydrozirconated alkene, which coordinated with the bisphosphate to form an alkene donor complex 78. Based on in situ NMR spectroscopic studies of reactant and catalyst mixtures, explicit transmetalation from Zr to Cu was not observed. 78 might undergo a stereo determining anti-SN2′-like process, to generate Cu-(III) σ-allyl species 79, where copper displaced the phosphate. The reductive elimination of intermediate 79 would yield the product 76 and regenerate the Cu–phosphoramidite catalyst (Scheme 24b).
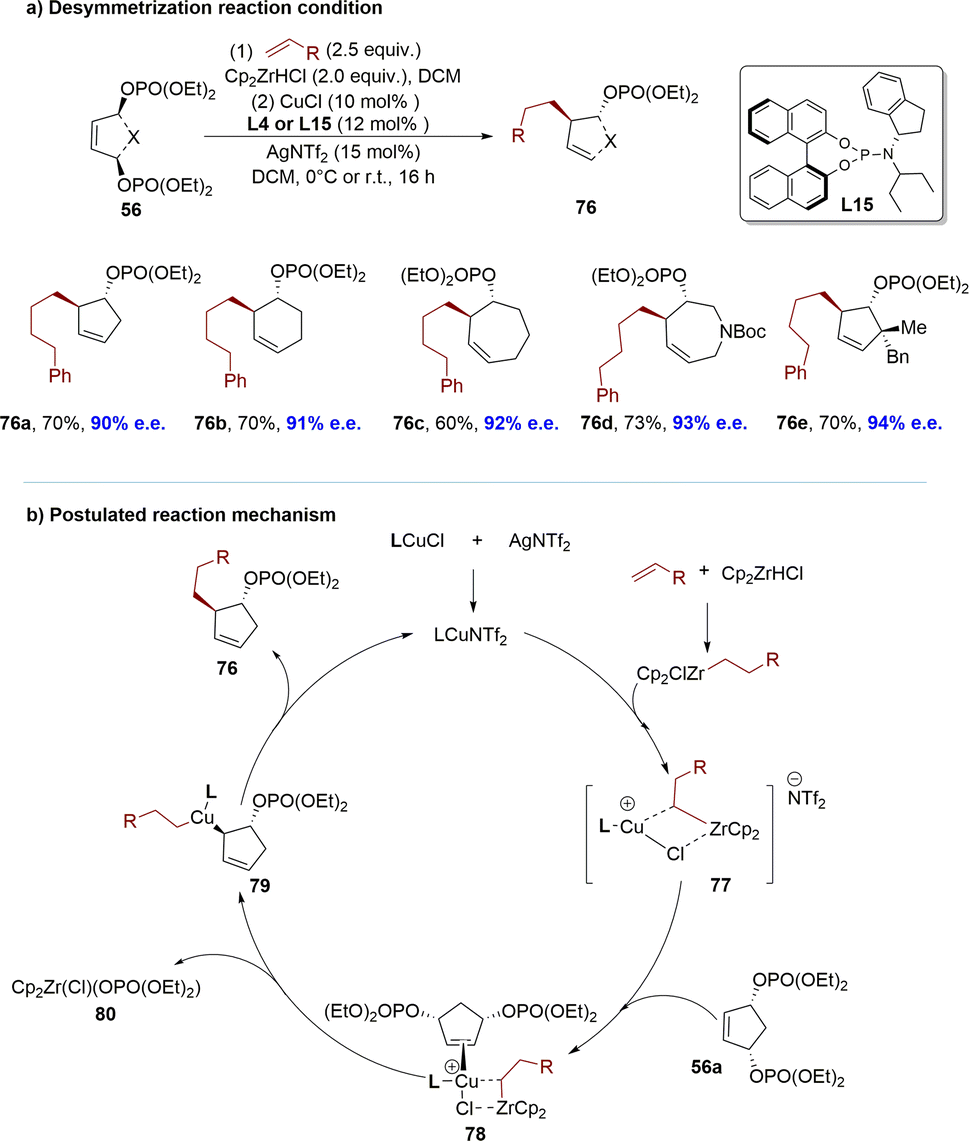 | ||
| Scheme 24 Desymmetrization of meso-bisphosphates using copper catalysis and alkyl zirconocene nucleophiles: (a) desymmetrization reaction condition; (b) postulated reaction mechanism. | ||
Recently, the Fañanás-Mastral group reported a copper-catalyzed coupling between alkynes, B2(pin)2 and meso-dibrocycloalkenes 69 (Scheme 25a).103 This process enabled the desymmetrization of these meso compounds through a net alkyne allylboration reaction using a catalytically generated boron-substituted alkenyl copper intermediate. The method employed a chiral N-heterocyclic carbene (NHC) copper complex for the synthesis of a wide range of chiral cycloalkenes. It is noteworthy that the product contains stereo-defined alkenyl boronate and bromide functionalities, demonstrating excellent regio-, diastereo-, and enantioselectivity. The reaction proceeded excellently with meso-dibromocyclopentene, yielding products with exceptional selectivity when employing phenyl acetylene and methyl 4-ethynylbenzoate as pro-nucleophiles. Interestingly, flash column chromatography afforded 82d and 82e as the products instead of 7-membered compounds 81d and 81e, which could be attributed to a stereospecific ring contraction via dyotropic rearrangement (Scheme 25b). The resulting products underwent an oxidative cyclopropanation reaction by treatment with sodium perborate, yielding valuable building blocks 83 for accessing a diverse range of important chiral structures. This cyclopropanation reaction likely involved the oxidation of the alkenyl boronate to form a boron enolate 84, followed by an intramolecular SN2 substitution, where the carbocycle moiety would be positioned opposite to the sizable pinacolboronate (OBpin) unit (Scheme 25c).
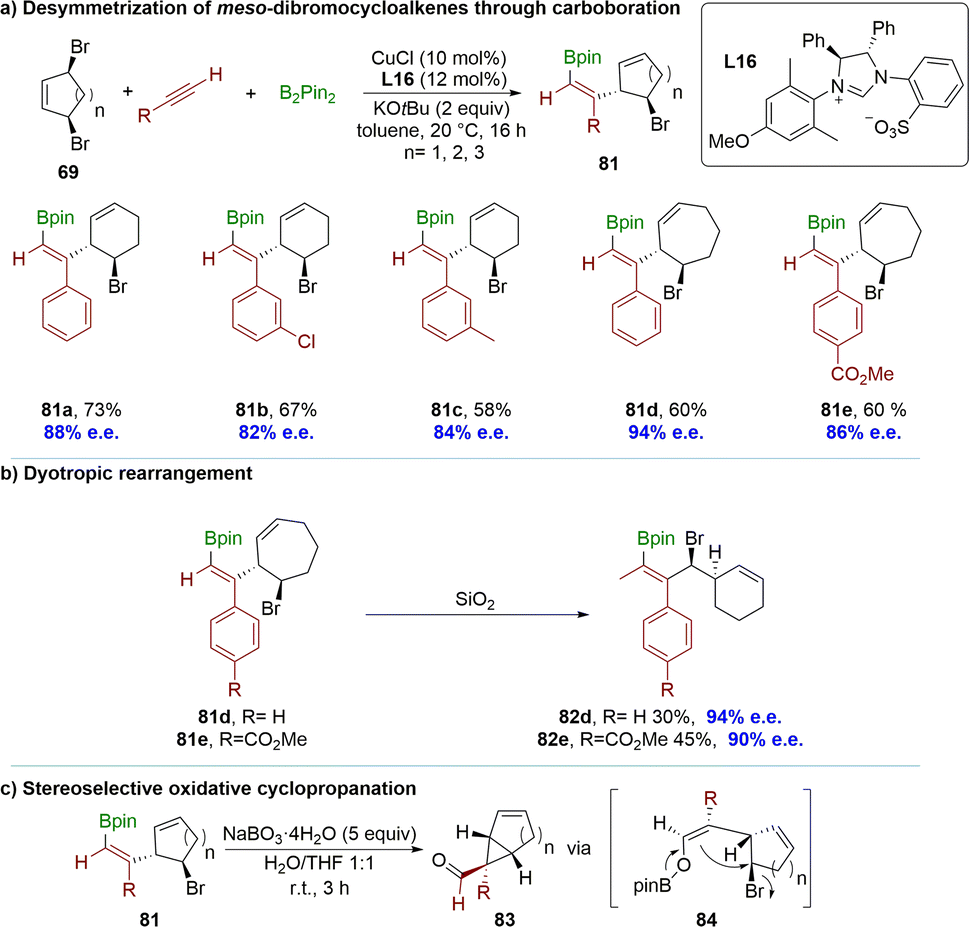 | ||
| Scheme 25 Desymmetrization by copper(I)-catalyzed asymmetric borylative coupling with alkynes: (a) desymmetrization of meso-dibromocycloalkenes through carboboration; (b) dyotropic rearrangement; (c) stereoselective oxidative cyclopropanation. | ||
4. Conclusions
Cu-catalyzed AAS enables the acquisition of enantioenriched building blocks, which are indispensable for the synthesis of bioactive molecules and drugs. Although enantiospecific transformations and additions to prochiral molecules have proven to be valuable tools, the application of racemic or meso starting materials remains underdeveloped despite its immense potential. This is largely attributed to the intricate and nuanced interaction between the substrate and the chiral catalyst. This review provides a comprehensive clarification of the terminology associated with dynamic processes in Cu-catalyzed AAS with racemic and meso substrates, along with an overview of recent advancements.Despite the significant progress in this field, there still exist several challenges: (1) while significant advancements have been made in the development of AAS for cyclic racemic and meso substrates, acyclic substrates have not been as thoroughly investigated. The conformational flexibility of acyclic substrates, especially the potential for intramolecular rotation around the carbon–carbon bond, adds an extra layer of complexity to the catalytic processes. Despite the development of several chiral ligands and additives,76,79 achieving efficient and highly stereoselective allylic substitutions with acyclic racemic substrates remains challenging, with limitations in substrate scope, stereoselectivity and practical applications. (2) Procedures involving unactivated allylic species, such as allylic alcohols, allylic ethers and allylic amines, remain difficult. Notably, their use is advantageous for minimizing the generation of waste by-products and reducing reaction steps, rendering them valuable owing to their inherent stability and prevalence in numerous biologically active compounds;117 (3) despite the numerous reported examples of enantioselective allylic substitution reactions, significant challenges persist in their development, particularly in syntheses involving quaternary center formation; (4) cooperative bimetallic catalysis involving AAS remains unexplored, despite the growing interest in using second- and third-row transition metals like Pd, Rh, and Ir in bimetallic catalysis. While Cu catalysts are primarily employed as nucleophilic activators in AAS,118–123 their application in bimetallic systems remains limited; (5) the potent transformation facilitated by Pd, Rh, and Ir has been successfully employed in the total synthesis of numerous natural products. In contrast, the use of Cu catalysis in producing valuable compounds remains relatively limited. Developing effective catalytic systems with greater tolerance for sterically hindered nucleophilic reagents and a broader range of substrates, including acyclic and heterocyclic types, could enhance the potential of Cu catalysis in total synthesis.
Furthermore, mechanistic investigations play a crucial role in advancing this chemistry by enhancing chemists' comprehension of the demands and challenges associated with the dynamic process of racemic or meso mixtures. These investigations provide an understanding of current limitations and potential opportunities, inspiring novel strategies and targets. Despite the numerous challenges mentioned above, recent developments in Cu-catalyzed AAS have demonstrated their effective solution. These complexities present an exceptional opportunity for synthetic chemists to apply their expertise to this captivating and crucial field of chemistry.
Author contributions
J. L. and J. H. performed the literature search, analyzed the published results, and wrote the manuscript. Y. Z., H. Y. and F. C. structured this review. Y. W. and Y. L. provided key advice and supervised the preparation of the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22302048; no. 82204231) and Guangdong Basic and Applied Basic Research Foundation (No. 2021A1515110366). We are also grateful to Shenzhen Science and Technology Research Fund No. JSGG20201103153807021; GXWD20220811173736002; KCXFZ20230731094904009 and Shenzhen Key Laboratory of Advanced Functional Carbon Materials Research and Comprehensive Application.Notes and references
- G. Zanoni, F. Castronovo, M. Franzini, G. Vidari and E. Giannini, Chem. Soc. Rev., 2003, 32, 115–129 RSC.
- S. Afewerki and A. Córdova, Chem. Rev., 2016, 116, 13512–13570 CrossRef CAS PubMed.
- V. Bhat, E. R. Welin, X. Guo and B. M. Stoltz, Chem. Rev., 2017, 117, 4528–4561 CrossRef CAS.
- A. H. Hoveyda, Y. Zhou, Y. Shi, M. K. Brown, H. Wu and S. Torker, Angew. Chem., Int. Ed., 2020, 59, 21304–21359 CrossRef CAS PubMed.
- M. Diéguez, Chiral Ligands: Evolution of Ligand Libraries for Asymmetric Catalysis, CRC Press, 2021 Search PubMed.
- M. Kamlar, M. Urban and J. Veselý, Chem. Rec., 2023, 23, e202200284 CrossRef CAS PubMed.
- B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292–294 CrossRef CAS.
- B. M. Trost and P. E. Strege, J. Am. Chem. Soc., 1977, 99, 1649–1651 CrossRef CAS.
- B. M. Trost and R. C. Bunt, J. Am. Chem. Soc., 1994, 116, 4089–4090 CrossRef CAS.
- A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS.
- V. Hornillos, J.-B. Gualtierotti and B. L. Feringa, in Progress in Enantioselective Cu(I)-catalyzed Formation of Stereogenic Centers, ed. S. R. Harutyunyan, Springer International Publishing, Cham, 2016, pp. 1–39 Search PubMed.
- L. Süsse and B. M. Stoltz, Chem. Rev., 2021, 121, 4084–4099 CrossRef.
- S. V. Kumar, A. Yen, M. Lautens and P. J. Guiry, Chem. Soc. Rev., 2021, 50, 3013–3093 RSC.
- T. J. Fulton, Y. E. Du and B. M. Stoltz, in Catalytic Asymmetric Synthesis, John Wiley & Sons, Ltd, 2022, pp. 661–704 Search PubMed.
- H. Wu, B. Qu, T. Nguyen, J. C. Lorenz, F. Buono and N. Haddad, Org. Process Res. Dev., 2022, 26, 2281–2310 CrossRef CAS.
- Y. Kobayashi, Catalysts, 2023, 13, 132 CrossRef CAS.
- B. M. Trost and M. L. Crawley, in Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, ed. U. Kazmaier, Springer, Berlin, Heidelberg, 2012, pp. 321–340 Search PubMed.
- Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed.
- B. W. H. Turnbull and P. A. Evans, J. Org. Chem., 2018, 83, 11463–11479 CrossRef CAS PubMed.
- C. Li, S. S. Ragab, G. Liu and W. Tang, Nat. Prod. Rep., 2020, 37, 276–292 RSC.
- O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J.-E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373–4505 CrossRef.
- M. B. Thoke and Q. Kang, Synthesis, 2019, 51, 2585–2631 CrossRef CAS.
- Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed.
- F. Dübner and P. Knochel, Angew. Chem., Int. Ed., 1999, 38, 379–381 CrossRef.
- J. A. Dabrowski, F. Gao and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 4778–4781 CrossRef CAS.
- R. Shintani, K. Takatsu, M. Takeda and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 8656–8659 CrossRef CAS PubMed.
- F. Gao, J. L. Carr and A. H. Hoveyda, Angew. Chem., 2012, 124, 6717–6721 CrossRef.
- M. Fañanás-Mastral, R. Vitale, M. Pérez and B. L. Feringa, Chem.–Eur. J., 2015, 21, 4209–4212 CrossRef PubMed.
- H. Ohmiya, H. Zhang, S. Shibata, A. Harada and M. Sawamura, Angew. Chem., Int. Ed., 2016, 55, 4777–4780 CrossRef CAS PubMed.
- W. Xiong, G. Xu, X. Yu and W. Tang, Organometallics, 2019, 38, 4003–4013 CrossRef CAS.
- C. I. Jette, Z. J. Tong, R. G. Hadt and B. M. Stoltz, Angew. Chem., Int. Ed., 2020, 59, 2033–2038 CrossRef CAS.
- S. Niu, Y. Luo, C. Xu, J. Liu, S. Yang and X. Fang, ACS Catal., 2022, 12, 6840–6850 CrossRef CAS.
- C. A. Falciola and A. Alexakis, Eur. J. Org Chem., 2008, 2008, 3765–3780 CrossRef.
- J.-B. Langlois and A. Alexakis, in Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, ed. U. Kazmaier, Springer, Berlin, Heidelberg, 2012, pp. 235–268 Search PubMed.
- K. Tissot-Croset, D. Polet and A. Alexakis, Angew. Chem., Int. Ed., 2004, 43, 2426–2428 CrossRef CAS.
- K. Tissot-Croset and A. Alexakis, Tetrahedron Lett., 2004, 45, 7375–7378 CrossRef CAS.
- M. Pérez, M. Fañanás-Mastral, P. H. Bos, A. Rudolph, S. R. Harutyunyan and B. L. Feringa, Nat. Chem., 2011, 3, 377–381 CrossRef.
- V. Hornillos, M. Pérez, M. Fañanás-Mastral and B. L. Feringa, J. Am. Chem. Soc., 2013, 135, 2140–2143 CrossRef CAS PubMed.
- M. S. Manna, S. Y. Yoo, M. Sharique, H. Choi, B. Pudasaini, M.-H. Baik and U. K. Tambar, Angew. Chem., Int. Ed., 2023, 62, e202304848 CrossRef CAS PubMed.
- C. Wang, Y. Wang, X. Jia, J. Huang, H. You and F.-E. Chen, J. Catal., 2023, 425, 50–56 CrossRef CAS.
- B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS.
- M. Fañanás-Mastral, M. Pérez, P. H. Bos, A. Rudolph, S. R. Harutyunyan and B. L. Feringa, Angew. Chem., Int. Ed., 2012, 51, 1922–1925 CrossRef.
- C. Jahier-Diallo, M. S. T. Morin, P. Queval, M. Rouen, I. Artur, P. Querard, L. Toupet, C. Crévisy, O. Baslé and M. Mauduit, Chem.–Eur. J., 2015, 21, 993–997 CrossRef CAS.
- J. A. Dabrowski, F. Haeffner and A. H. Hoveyda, Angew. Chem., 2013, 125, 7848–7853 CrossRef.
- L. Qin, M. Sharique and U. K. Tambar, J. Am. Chem. Soc., 2019, 141, 17305–17313 CrossRef CAS PubMed.
- Q. Zhang, S.-W. Zhou, C.-Y. Shi and L. Yin, Angew. Chem., Int. Ed., 2021, 60, 26351–26356 CrossRef CAS.
- S. Guduguntla, J.-B. Gualtierotti, S. S. Goh and B. L. Feringa, ACS Catal., 2016, 6, 6591–6595 CrossRef CAS.
- X. Li, J. Chen and Q. Song, Chem. Commun., 2024, 60, 2462–2471 RSC.
- F. Meng, K. P. McGrath and A. H. Hoveyda, Nature, 2014, 513, 367–374 CrossRef CAS PubMed.
- G. Xu, H. Zhao, B. Fu, A. Cang, G. Zhang, Q. Zhang, T. Xiong and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 13130–13134 CrossRef CAS PubMed.
- G. Xu, B. Fu, H. Zhao, Y. Li, G. Zhang, Y. Wang, T. Xiong and Q. Zhang, Chem. Sci., 2019, 10, 1802–1806 RSC.
- E. Rivera-Chao, M. Mitxelena, J. A. Varela and M. Fañanás-Mastral, Angew. Chem., Int. Ed., 2019, 58, 18230–18234 CrossRef CAS PubMed.
- A. Chaves-Pouso, A. M. Álvarez-Constantino and M. Fañanás-Mastral, Angew. Chem., 2022, 134, e202117696 CrossRef.
- M. Piñeiro-Suárez, A. M. Álvarez-Constantino and M. Fañanás-Mastral, ACS Catal., 2023, 13, 5578–5583 CrossRef PubMed.
- P. Schäfer, M. Sidera, T. Palacin and S. P. Fletcher, Chem. Commun., 2017, 53, 12499–12511 RSC.
- J. Jacques, A. Collet, S. H. Wilen and A. Collet, Enantiomers, Racemates, and Resolutions, Wiley, New York, 1981 Search PubMed.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, 1994 Search PubMed.
- H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef CAS.
- R. M. Kellogg, Acc. Chem. Res., 2017, 50, 905–914 CrossRef CAS PubMed.
- S. M. Morrow, A. J. Bissette and S. P. Fletcher, Nat. Nanotechnol., 2017, 12, 410–419 CrossRef CAS PubMed.
- F. F. Huerta, A. B. E. Minidis and J.-E. Bäckvall, Chem. Soc. Rev., 2001, 30, 321–331 RSC.
- J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5–26 CrossRef CAS.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1999, 121, 3543–3544 CrossRef CAS.
- E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974–4001 CrossRef CAS.
- J. Steinreiber, K. Faber and H. Griengl, Chem.–Eur. J., 2008, 14, 8060–8072 CrossRef CAS PubMed.
- K. Faber, Chem.–Eur. J., 2001, 7, 5004–5010 CrossRef CAS.
- H. Ito, S. Kunii and M. Sawamura, Nat. Chem., 2010, 2, 972–976 CrossRef CAS PubMed.
- F. Badalassi, P. Crotti, F. Macchia, M. Pineschi, A. Arnold and B. L. Feringa, Tetrahedron Lett., 1998, 39, 7795–7798 CrossRef CAS.
- F. Bertozzi, P. Crotti, F. Macchia, M. Pineschi, A. Arnold and B. L. Feringa, Org. Lett., 2000, 2, 933–936 CrossRef CAS.
- F. Bertozzi, P. Crotti, F. Macchia, M. Pineschi and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 930–932 CrossRef CAS.
- M. Pineschi, New J. Chem., 2004, 28, 657–665 RSC.
- M. Pineschi, F. Del Moro, P. Crotti, V. Di Bussolo and F. Macchia, J. Org. Chem., 2004, 69, 2099–2105 CrossRef CAS PubMed.
- R. Millet and A. Alexakis, Synlett, 2007, 2007, 435–438 CrossRef.
- R. Millet and A. Alexakis, Synlett, 2008, 2008, 1797–1800 CrossRef.
- M. Pineschi, V. D. Bussolo and P. Crotti, Chirality, 2011, 23, 703–710 CrossRef CAS PubMed.
- J.-B. Langlois and A. Alexakis, Angew. Chem., Int. Ed., 2011, 50, 1877–1881 CrossRef CAS PubMed.
- L. C. Miller, J. M. Ndungu and R. Sarpong, Angew. Chem., 2009, 121, 2434–2438 CrossRef.
- S. Karabiyikoglu, A. V. Brethomé, T. Palacin, R. S. Paton and S. P. Fletcher, Chem. Sci., 2020, 11, 4125–4130 RSC.
- H. Iwamoto, Y. Ozawa, Y. Takenouchi, T. Imamoto and H. Ito, J. Am. Chem. Soc., 2021, 143, 6413–6422 CrossRef CAS.
- X.-Y. Cui, Y. Ge, S. M. Tan, H. Jiang, D. Tan, Y. Lu, R. Lee and C.-H. Tan, J. Am. Chem. Soc., 2018, 140, 8448–8455 CrossRef CAS PubMed.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1999, 121, 3543–3544 CrossRef CAS.
- B. M. Trost, R. C. Bunt, R. C. Lemoine and T. L. Calkins, J. Am. Chem. Soc., 2000, 122, 5968–5976 CrossRef CAS.
- M. Fañanás-Mastral and B. L. Feringa, J. Am. Chem. Soc., 2010, 132, 13152–13153 CrossRef.
- H. You, E. Rideau, M. Sidera and S. P. Fletcher, Nature, 2015, 517, 351–355 CrossRef CAS.
- E. Rideau and S. P. Fletcher, Beilstein J. Org. Chem., 2015, 11, 2435–2443 CrossRef CAS PubMed.
- E. Rideau, H. You, M. Sidera, T. D. W. Claridge and S. P. Fletcher, J. Am. Chem. Soc., 2017, 139, 5614–5624 CrossRef CAS PubMed.
- A. Streitwieser, E. G. Jayasree, F. Hasanayn and S. S.-H. Leung, J. Org. Chem., 2008, 73, 9426–9434 CrossRef CAS.
- M. Sidera and S. P. Fletcher, Chem. Commun., 2015, 51, 5044–5047 RSC.
- J.-B. Langlois and A. Alexakis, Chem. Commun., 2009, 3868 RSC.
- J.-B. Langlois and A. Alexakis, Adv. Synth. Catal., 2010, 352, 447–457 CrossRef CAS.
- J.-B. Langlois, D. Emery, J. Mareda and A. Alexakis, Chem. Sci., 2012, 3, 1062–1069 RSC.
- L. B. Delvos and M. Oestreich, Synthesis, 2015, 47, 924–933 CrossRef CAS.
- Y. Ge, X. Cui, S. M. Tan, H. Jiang, J. Ren, N. Lee, R. Lee and C. Tan, Angew. Chem., Int. Ed., 2019, 58, 2382–2386 CrossRef CAS PubMed.
- J. Li, X. Song, F. Wu, H. You and F.-E. Chen, Eur. J. Org Chem., 2022, 2022, e202200860 CrossRef CAS.
- J. Liao, S. Zhang, Z. Wang, X. Song, D. Zhang, R. Kumar, J. Jin, P. Ren, H. You and F.-E. Chen, Green Synth. Catal., 2020, 1, 121–133 CrossRef.
- J. Liao, X. Jia, F. Wu, J. Huang, G. Shen, H. You and F.-E. Chen, Org. Chem. Front., 2022, 9, 6640–6645 RSC.
- J. Wang, J. Li, Y. Wang, S. He, H. You and F.-E. Chen, ACS Catal., 2022, 12, 9629–9637 CrossRef CAS.
- L. Wan, G. Kong, M. Liu, M. Jiang, D. Cheng and F. Chen, Green Synth. Catal., 2022, 3, 243–258 CrossRef CAS.
- L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230–4247 RSC.
- X. Song, J. Qing, J. Li, X. Jia, F. Wu, J. Huang, J. Jin and H. You, Chin. J. Org. Chem., 2023, 43, 3174 CrossRef.
- J. Li, X. Song, Y. Wang, J. Huang, H. You and F.-E. Chen, Chem. Sci., 2023, 14, 4351–4356 RSC.
- J. T. Han, S.-T. Kim, M.-H. Baik and J. Yun, Chem.–Eur. J., 2020, 26, 2592–2596 CrossRef CAS PubMed.
- I. Sánchez-Sordo, A. Chaves-Pouso, J. Mateos-Gil, E. Rivera-Chao and M. Fañanás-Mastral, Chem Catal., 2023, 3, 100730 CrossRef.
- T. Suzuki, Tetrahedron Lett., 2017, 58, 4731–4739 CrossRef CAS.
- H. Weißbarth, F. Mühlhaus and A. Gansäuer, Synthesis, 2020, 52, 2940–2947 Search PubMed.
- Y. Xu, T.-Y. Zhai, Z. Xu and L.-W. Ye, Trends Chem., 2022, 4, 191–205 CrossRef CAS.
- P.-J. Yang and Z. Chai, Org. Biomol. Chem., 2023, 21, 465–478 RSC.
- C.-J. Yang, L. Liu, Q.-S. Gu and X.-Y. Liu, CCS Chem. DOI:10.31635/ccschem.024.202403839.
- U. Piarulli, P. Daubos, C. Claverie, M. Roux and C. Gennari, Angew. Chem., 2003, 115, 244–246 CrossRef.
- U. Piarulli, C. Claverie, P. Daubos, C. Gennari, A. J. Minnaard and B. L. Feringa, Org. Lett., 2003, 5, 4493–4496 CrossRef CAS PubMed.
- H. Ito, T. Okura, K. Matsuura and M. Sawamura, Angew. Chem., Int. Ed., 2010, 3, 560–563 CrossRef.
- A. Vázquez-Romero, J. Rodríguez, A. Lledó, X. Verdaguer and A. Riera, Org. Lett., 2008, 10, 4509–4512 CrossRef.
- Y. Yasuda, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2016, 55, 10816–10820 CrossRef CAS.
- S. S. Goh, S. Guduguntla, T. Kikuchi, M. Lutz, E. Otten, M. Fujita and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 7052–7055 CrossRef CAS PubMed.
- S. S. Goh, P. A. Champagne, S. Guduguntla, T. Kikuchi, M. Fujita, K. N. Houk and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 4986–4990 CrossRef CAS PubMed.
- R. Jacques, R. D. C. Pullin and S. P. Fletcher, Nat. Commun., 2019, 10, 21 CrossRef CAS PubMed.
- N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929–7967 RSC.
- J. Mateos, N. Fuentes-Vara, L. Fra, E. Rivera-Chao, N. Vázquez-Galiñanes, A. Chaves-Pouso and M. Fañanás-Mastral, Organometallics, 2020, 39, 740–745 CrossRef CAS.
- R. He, X. Huo, L. Zhao, F. Wang, L. Jiang, J. Liao and W. Zhang, J. Am. Chem. Soc., 2020, 142, 8097–8103 CrossRef CAS PubMed.
- W.-Y. Huang, C.-H. Lu, S. Ghorai, B. Li and C. Li, J. Am. Chem. Soc., 2020, 142, 15276–15281 CrossRef CAS.
- L. Zhao, G. Li, R. He, P. Liu, F. Wang, X. Huo, M. Zhao and W. Zhang, Org. Biomol. Chem., 2021, 19, 1955–1959 RSC.
- L. Xiao, X. Chang, H. Xu, Q. Xiong, Y. Dang and C.-J. Wang, Angew. Chem., Int. Ed., 2022, 61, e202212948 CrossRef CAS.
- B.-K. Zhu, H. Xu, L. Xiao, X. Chang, L. Wei, H. Teng, Y. Dang, X.-Q. Dong and C.-J. Wang, Chem. Sci., 2023, 14, 4134–4142 RSC.
Footnote |
| † These authors contributed equally: Jun Li and Junrong Huang. |
| This journal is © The Royal Society of Chemistry 2024 |