
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProBID-Net: a deep learning model for protein–protein binding interface design†
Zhihang
Chen
,
Menglin
Ji
,
Jie
Qian
,
Zhe
Zhang
,
Xiangying
Zhang
 ,
Haotian
Gao
,
Haojie
Wang
,
Renxiao
Wang
* and
Yifei
Qi
*
,
Haotian
Gao
,
Haojie
Wang
,
Renxiao
Wang
* and
Yifei
Qi
*
Department of Medicinal Chemistry, School of Pharmacy, Fudan University, 826 Zhangheng Road, Shanghai 201203, People's Republic of China. E-mail: wangrx@fudan.edu.cn; yfqi@fudan.edu.cn
First published on 30th October 2024
Abstract
Protein–protein interactions are pivotal in numerous biological processes. The computational design of these interactions facilitates the creation of novel binding proteins, crucial for advancing biopharmaceutical products. With the evolution of artificial intelligence (AI), protein design tools have swiftly transitioned from scoring-function-based to AI-based models. However, many AI models for protein design are constrained by assuming complete unfamiliarity with the amino acid sequence of the input protein, a feature most suited for de novo design but posing challenges in designing protein–protein interactions when the receptor sequence is known. To bridge this gap in computational protein design, we introduce ProBID-Net. Trained using natural protein–protein complex structures and protein domain–domain interface structures, ProBID-Net can discern features from known target protein structures to design specific binding proteins based on their binding sites. In independent tests, ProBID-Net achieved interface sequence recovery rates of 52.7%, 43.9%, and 37.6%, surpassing or being on par with ProteinMPNN in binding protein design. Validated using AlphaFold-Multimer, the sequences designed by ProBID-Net demonstrated a close correspondence between the design target and the predicted structure. Moreover, the model's output can predict changes in binding affinity upon mutations in protein complexes, even in scenarios where no data on such mutations were provided during training (zero-shot prediction). In summary, the ProBID-Net model is poised to significantly advance the design of protein–protein interactions.
Introduction
Natural proteins, despite their vital roles as carriers of various life activities, have limitations due to their specific working environment and finite lifespan. To address these limitations, the ability to create entirely novel proteins from scratch using computational algorithms becomes essential. This process is known as computational protein design (CPD). The primary objective of CPD is to identify specific combinations of amino acid residues on a native scaffold that can fold into desired protein structures with precise functions. Additionally, CPD can be utilized to optimize existing native proteins or complexes to enhance their stability or modify their functions to serve specific purposes. This powerful approach allows researchers to engineer proteins tailored to specific needs, going beyond what nature has provided and offering great potential for various applications in biomedicine and beyond.1Designing proteins is very challenging because of the vast search space of sequences and structures. Before the recent surge of AI algorithms, the most advanced methods still relied on hand-crafted energy functions and heuristic sampling algorithms, which frequently produce suboptimal solutions and are computationally intensive and time-consuming. Classical CPD methods, such as Rosetta Design,2 typically demand predefined protein secondary structures or specific folding modes, then select appropriate amino acids or short peptides using energy functions, perform sequence structure optimization iterations, and finally generate output sequences by ranking energy function scoring results.2–6 In recent years, there have been numerous impressive achievements7–14 in protein design through classical CPD methods, including self-assembly,10 immune signaling,12,13 enzyme,15 targeted therapeutics,14,16 and protein switches,11,17 demonstrating the tremendous potential of designed proteins.1–6
With the advent of deep learning, CPD research has rapidly transformed from knowledge-based to data-driven methods in recent years.18 Artificial deep neural networks are capable of extracting protein features from existing data, generating integrated statistical motifs, and storing them in millions of parameters for inference in different protein design applications. Several deep network architectures have been widely used in protein research and have made a significant impact. Early AI-protein-design models, including SPIN19 and SPIN2,20 have achieved a sequence recovery rate of about 34%. Later on, SPROF,21 ProDCoNN22 and our prior research, referred to as DenseCPD,23 have employed a 3D-CNN as the model architecture. These approaches have notably enhanced sequence recovery. More recently, Graph Neural Network (GNN) is employed to predict residue interactions and residue types in proteins. In this context, proteins are treated as graphs, with nodes representing residues, and the prediction becomes a graph classification problem. Among them, graph based models, such as ProtTrans,24 GVP,25 StructGNN,26 AlphaDesign,27 ESM-IF, ProteinMPNN,28 PiFold,29 SPIN-CGNN and VFN,30 have exhibited notable successes in the realm of protein design and have improved sequence recovery to 50–55%. Notably, LM-DESIGN,31 ABACUS-R32 and ProDESIGN-LE,33 ESM-IF, LM-DESIGN,31 and CarbonDesign34 have used various AI models including large language Model (LLM) and AlphaFold2 architectures to achieve high accuracy in sequence design or generation. The designs of some of these models have been verified by wet experiments and exhibited impressive success rate.4,5,32,35–37
Protein–protein interactions play a vital role in numerous biological processes as they form the foundation of many molecular machines responsible for multiple functions.38,39 Understanding these interactions in detail can provide crucial insights into the functions of protein complexes and has significant implications for medical and drug research. Classical CPD methods often leverage information extracted from native complex structures. This involves the strategic placement of naturally occurring protein scaffolds guided by hotspot residues, followed by the generation of binders through methodologies such as library selection14 or antibody modification.40–42 Subsequently, computational saturation mutagenesis is employed to optimize the affinity and specificity of the protein binder.43–45
Although the deep learning models mentioned above have demonstrated impressive results in designing individual protein units by predicting the joint probability of residues under given backbone constraints or generating direct sequences, there is a notable lack of models specifically tailored for protein binder design. Thus, developing such binder design models is an important area of research that holds great potential for advancing our understanding of protein–protein interactions and would be valuable in identifying suitable binding proteins for a given target protein structure. Among the mentioned models, those of both Rosetta2 and ProteinMPNN35 can be employed for the specific task of designing protein binder interfaces.
In this study, we aimed to develop a specialized model for the design and optimization of protein–protein interface residues. We used DenseNet46 to recognize three-dimensional structural data of protein interface residues. The resulting network, named Protein Binding Interface Design Network (ProBID-Net), was trained to learn the correlations between target residues and their surrounding interface environment, based on the distribution of residue backbone atoms found in known receptor protein chains.
As a result, ProBID-Net has effectively acquired knowledge regarding protein–protein interaction from interfaces and achieved an impressive sequence recovery rate of 52.7% on an independent test set and 43.9% on an external test set. It exhibited low perplexity in interface residue prediction and high conservation of hydrophobic positions. We predicted the complex structure of the designs with AlphaFold-Multimer, and found that the predicted structure was in good agreement with the design target, which further verified the foldability and binding specificity of the model design sequence. In addition, the predicted probability of each amino acid on the protein interface residues can be used as a zero-shot prediction of binding affinity change caused by mutations, providing a reference for binding affinity modification.
Results and discussion
Sequence recovery and perplexity
The ProBID-Net architecture comprises DenseNet models featuring three Dense Blocks and were trained on the training set of QSalign47 labelled heterodimers and domain–domain interfaces. Subsequently, three distinct non-redundant test sets, namely the TS920, de novo set, and Folddock set, were employed for evaluation. Each protein–protein complex of these test sets exhibited a sequence identity with those in the training set of less than 40%.Model performance was evaluated using perplexity and average recovery rate of residues located at the interfaces of ligand protein. We defined residues on the unknown chain with CA atoms within a distance of <8 Å from any atom on the known receptor chain as the target interface residues. Meanwhile, interface sequence recovery is measured by reading structures in test sets and then calculating the percent identity between them by iterating over all residues on ligand protein interfaces. Perplexity is a measure used in information theory to quantify how well a probability distribution or probability model predicts a sample. As shown in Table 1, the model achieved an average interface sequence recovery of 37.7% on TS920, 37.6% on the de novo set and 32.8% on the Folddock set.
| Average interface recovery (%) ↑ | ||||||
|---|---|---|---|---|---|---|
| Model no. | 1 | 2 | 3 | 4 | 5 | Average |
| a Trained with growth rate = 70. b The domain–domain interfaces are not included in the training dataset, the training set solely comprises chain–chain interface structures extracted from heterodimers. c The format for the numbers is “Average Interface Recovery ± Standard Deviation” in percentage (%). | ||||||
| TS920 | 37.9 ± 12.8 | 38.0 ± 12.9 | 35.9 ± 12.2 | 40.2 ± 13.4 | 36.7 ± 12.5 | 37.7 |
| De novo | 39.4 ± 13.9 | 35.9 ± 13.0 | 35.0 ± 13.4 | 40.8 ± 14.3 | 37.0 ± 13.8 | 37.6 |
| Folddock | 32.6 ± 11.4 | 32.7 ± 11.4 | 31.5 ± 11.1 | 35.1 ± 12.1 | 32.1 ± 11.4 | 32.8 |
In order to increase the amount of protein–protein interface structural data, we hypothesize that the evolutionary conservation of protein domains aligns closely with that observed at the fold level, potentially leading to an augmentation of protein interface datasets. We assembled an additional dataset focused on domain–domain interfaces through the segmentation of domains in multi-domain protein chains according to CATH.48Table 2 provides a comprehensive evaluation of the average interface residue sequence recovery and perplexity for two ProBID-Net models that were respectively trained on datasets from pure chain–chain interface data and on the set with the addition of domain–domain interface structure data. For comparison, 1000 sequences were generated using ProteinMPNN and Rosetta Design (using the ref 2015 energy function) for each complex in the three test sets and the sequence recovery and predictive perplexity were compared.
| Model | Average interface recovery (%) ↑ | Perplexity↓ |
|---|---|---|
| a The format for the table is “Average Interface Recovery ± Standard Deviation” in percentage (%). | ||
| TS920 | ||
| ProBID-Net-CC | 40.2 ± 13.4 | 3.91 |
| ProBID-Net | 52.7 ± 16.5 | 3.02 |
| ProteinMPNN | 36.7 ± 18.6 | 6.06 |
| Rosetta fast design | 43.2 ± 14.6 | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| De novo set | ||
| ProBID-Net-CC | 40.8 ± 14.3 | 3.87 |
| ProBID-Net | 43.9 ± 10.4 | 3.67 |
| ProteinMPNN | 43.0 ± 14.5 | 4.12 |
| Rosetta fast design | 42.6 ± 13.8 | — |
|
||
| Folddockset | ||
| ProBID-Net-CC | 35.1 ± 12.1 | 4.63 |
| ProBID-Net | 37.6 ± 11.7 | 4.28 |
| ProteinMPNN | 39.3 ± 18.4 | 8.11 |
| Rosetta fast design | 40.5 ± 16.2 | — |
The enhancement observed in ProBID-Net trained on both chain–chain interface and domain–domain interface sets (ProBID-Net) relative to the version trained on the chain–chain interface (ProBID-Net-CC) suggests an increased confidence in accurate protein interface sequence design when incorporating domain–domain interface structure data into the training set.
ProBID-Net achieved a remarkable sequence recovery rate of 52.7% on the independent heterodimer test set (TS920), surpassing the performance of ProteinMPNN (36.7%) and Rosetta (43.2%). Moreover, on the de novo protein–protein complex test set (de novo set) and the Folddock test set (Folddock set), ProBID-Net achieved sequence recovery rates of 43.9% and 37.6%, respectively. Notably, ProBID-Net demonstrated better performance in recovery scores on both the TS920 and de novo set compared to both ProteinMPNN and Rosetta Design. However, ProBID-Net does not achieve the highest performance on the Folddock set. We attribute this outcome to the removal of all structures from the Folddock dataset that exhibited high similarity to those in the ProBID-Net training set, reducing the number of complexes from 2734 to 1106. The remaining complexes in the Folddock set display significant differences from the training set, which likely contributed to the observed reduction in performance. In contrast, the CATH4.2 dataset, used for training ProteinMPNN, was not subjected to a similar structural dereplication process relative to the Folddock set. This lack of filtering enabled ProteinMPNN to more easily predict the correct interface residues.
Regarding the perplexity of interface residues, ProBID-Net consistently outperformed the other two models on all test sets. This robust performance underscores the efficacy of ProBID-Net in designing protein–protein interaction interfaces. In Fig. 1, we plotted the distribution of sequence recovery rates for both models on TS920, de novo sets, and Folddock set. Additionally, metrics such as residue type precision, recall and F1_score of ProBID-Net and ProteinMPNN are present in Fig. S1.† The flexibility of certain positions in protein structural interfaces, allowing the replacement of amino acids without compromising the stability of the structure and potentially enhancing binding strength, highlights the dynamic nature of these regions. Our objective was to conduct a thorough assessment of our model for interface residues, deviating from established norms to develop a nuanced understanding of the variations under natural conditions. To accomplish this, we utilized the BLOSUM score, a comprehensive metric that combines BLOSUM62 (ref. 49) values and probabilities predicted by ProBID-Net. The calculation of this score follows a similar approach to the evaluation methodology used in SPIN-CGNN.50,51 This score serves as an enlightening metric, effectively capturing the intricacies associated with both perplexity and the amino acid substitution.
 | ||
| Fig. 1 The distribution of sequence recovery rates for both ProBID-Net and ProteinMPNN on TS920 (A), de novo set (B), and Folddock set (C). The violin plots represent the interface residue sequence recovery from 920 heterodimers in TS920, 62 heterodimers in the de novo set, and 1106 heterodimers in the Folddock set. | ||
As presented in Table 3, ProBID-Net demonstrated better performance compared to ProteinMPNN across all three test sets, as indicated by the relative BLOSUM. These findings underscore the heightened efficacy of ProBID-Net in capturing evolutionary information pertaining to interface residues, positioning it as a robust model with great capabilities compared to other deep learning counterparts.
| Model | Average hydrophobic conservation (%) ↑ | AA substitutions (relative BLOSUM score) ↑ |
|---|---|---|
| a The format for the table is “Average Interface Recovery ± Standard Deviation” in percentage (%). | ||
| TS920 | ||
| ProBID-Net-CC | 74.43 ± 8.6 | 0.343 |
| ProBID-Net | 77.78 ± 9.5 | 0.467 |
| ProteinMPNN | 76.79 ± 16.3 | 0.218 |
| Rosetta fast design | 73.46 ± 10.3 | — |
|
||
| De novo set | ||
| ProBID-Net-CC | 77.97 ± 8.3 | 0.351 |
| ProBID-Net | 76.09 ± 7.9 | 0.360 |
| ProteinMPNN | 73.34 ± 11.8 | 0.283 |
| Rosetta fast design | 70.21 ± 11.4 | — |
|
||
| Folddockset | ||
| ProBID-Net-CC | 70.68 ± 10.5 | 0.280 |
| ProBID-Net | 71.56 ± 9.8 | 0.306 |
| ProteinMPNN | 69.02 ± 16.7 | 0.246 |
| Rosetta fast design | 67.39 ± 13.6 | — |
Amino acid substitutions in fixed backbone protein design methods were acquired by calculating the position-wise confusion matrix, elucidating the concordance between predicted and native amino acids at interface positions. Such a confusion matrix can be compared to the BLOSUM62 matrix to examine the likelihood of amino acid replacements in protein interfaces. As shown in Fig. 2, the confusion matrix of ProBID-Net presented a similar pattern to the BLOSUM62 matrix. Notably, most positive substitutions in the BLOSUM62 matrix align with positive values in the confusion matrix of ProBID-Net. This alignment signifies a congruence between the model's predictions and amino acid substitution probabilities in protein interfaces.
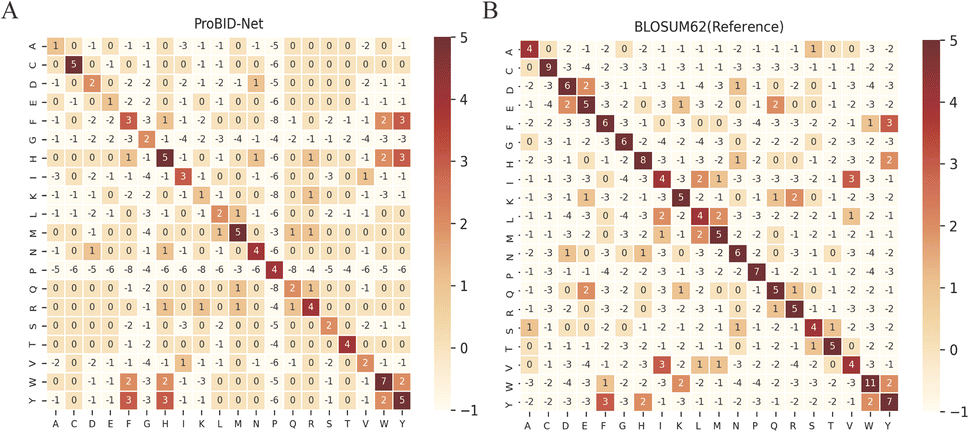 | ||
| Fig. 2 Confusion matrix of ProBID-Net. (A) In comparison to the reference matrix BLOSUM62. (B) On all three test sets. Positive values (colored red) indicate preferred substitutions between amino acids. | ||
To evaluate the sensitivity of recovery to the initial placement of interacting partners, we re-docked the protein–protein complexes from the test set using HDOCKlite v1.1.52 This procedure involved splitting all complexes from the three test sets and performing local protein–protein docking to generate slightly altered conformations compared to the wild-type complexes. The interfaces were then redesigned based on these modified conformations. The docked ligand proteins were categorized according to their root mean square deviation (RMSD) from the crystal structures: 0–1 Å, 1–5 Å, and >5 Å (Table S1†). The recovery rate for each group was calculated and compared to the recovery rate for the crystal structures (Fig. S2†). The results demonstrate a clear trend: as RMSD increases, the accuracy of the model in predicting interfacial residues decreases. These results suggest that the ProBID-Net model is relatively sensitive to the initial structural placement of the input protein–protein interaction interface, underscoring the importance of a high-quality initial structure for accurate predictions.
Hydrophobicity conservation
In soluble proteins, hydrophobic residues such as leucine and alanine are typically buried within the protein core, while hydrophilic residues dominate the surface, facilitating solvent accessibility. In contrast, the protein–protein interface requires a high proportion of hydrophobic residues to foster hydrophobic interactions and ensure complex binding affinity and stability.53 This phenomenon underscores the critical role of conserving hydrophobic residues in protein binders, a key factor in maintaining protein–protein interactions throughout evolution and a pivotal metric for assessing the efficacy of designed protein binders. While protein sequence recovery is an important metric in evaluating protein design models, the true test of a designed protein sequence lies in its ability to fold into the desired 3D structure and perform its intended function. Recently, it has been shown that adaptations of AlphaFold2 (ref. 54) (AF) for protein complexes (FoldDock55) can rival high-throughput yeast-two-hybrid and mass spectrometry screens in identifying PPIs. To assess the performance of ProBID-Net, we conducted tests on a set of protein–protein complexes from the de novo set.To gauge the proficiency of ProBID-Net in predicting analogous amino acids, we further scrutinized the conservation of hydrophobic and hydrophilic sequence positions in designed sequences by categorizing residues into hydrophobic (Ile, Leu, Met, Phe, Cys, Trp, Pro, Val, Ala, and Gly) and hydrophilic (Ser, Thr, Asn, Gln, Asp, Glu, His, Arg, Lys, and Tyr) based on the native sequence. Table 4 illustrates that ProBID-Net displays a high conservation in hydrophobicity protein interface positions.
| ProBID-Net-CC | ProBID-Net | ProteinMPNN | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Top1 | Top3 | 0.3 | 0.5 | Top1 | Top3 | 0.3 | 0.5 | Top1 | Top3 | 0.3 | 0.5 | |
| a Top1 & Top3 signifies the preservation of hydrophobicity in the top 1 and 3 amino acids possessing the highest predicted probabilities. b 0.3 & 0.5 indicate the preservation of hydrophobicity within the cumulative probability of 0.3 & 0.5. | ||||||||||||
| TS920 | 74.43 | 95.31 | 81.78 | 89.43 | 77.78 | 96.17 | 85.52 | 92.43 | 76.79 | 93.95 | 84.88 | 90.80 |
| De novo set | 77.97 | 93.78 | 83.15 | 90.15 | 76.09 | 93.68 | 83.90 | 91.01 | 73.34 | 91.23 | 80.12 | 88.41 |
| Foldock set | 70.68 | 92.36 | 78.59 | 87.24 | 71.56 | 93.46 | 82.03 | 90.16 | 69.02 | 90.68 | 78.34 | 86.59 |
AlphaFold2 folding validation
Specifically, we replaced the interface residues of the ligand protein with the amino acid predicted with the highest probability by ProBID-Net. The AlphaFold2 v2.3.1 multimer module was employed to assess the foldability of the designed sequences. This evaluation involved predicting the 3D structure of protein complexes formed by the designed sequences and comparing them to the crystal structures from the Protein Data Bank (PDB). Key metrics, including Predict Aligned Error (PAE), TM-score and RMSD, were utilized to examine the structural disparities between the predicted and actual structures. ProBID-Net achieved a similar TM-score with ProteinMPNN for many complexes but also showed more complexes with better TM-score (Fig. 3). As depicted in Fig. 4, a majority of protein complex-binding proteins within the de novo set, redesigned by both ProBID-Net, adopted a binding pattern reminiscent of the crystal structure. | ||
| Fig. 3 Comparison of global TM-score between predicted structures of designed sequences from ProBID-Net and ProteinMPNN. Structure prediction was performed using AlphaFold-Multimer. | ||
 | ||
| Fig. 4 Predicted complex structures of ProBID-Net designs using AlphaFold-Multimer. The receptor and ligand protein in the X-ray structures are colored in green and cyan. The predicted structure of the receptor and ligand protein are colors in yellow and orange. The chain id in each figure refers to the chain of the ligand protein. | ||
Fig. 5A–D depict various protein–protein complexes (PDB ID 6DM9, 6F0F, 7A48, 7XFR) as examples. In these instances, ProBID-Net demonstrated better recovery of the complex structure compared to ProteinMPNN. Visualization of these complex interfaces revealed that the binders designed by ProBID-Net displayed closer fit to the native structures and also smaller PAE. Notably, the inter-chain PAE of the model predicted by ProBID-Net appeared lower than that of the model assembled by ProteinMPNN, suggesting a higher accuracy in the inter-chain orientation of the ProBID-Net model compared to ProteinMPNN.
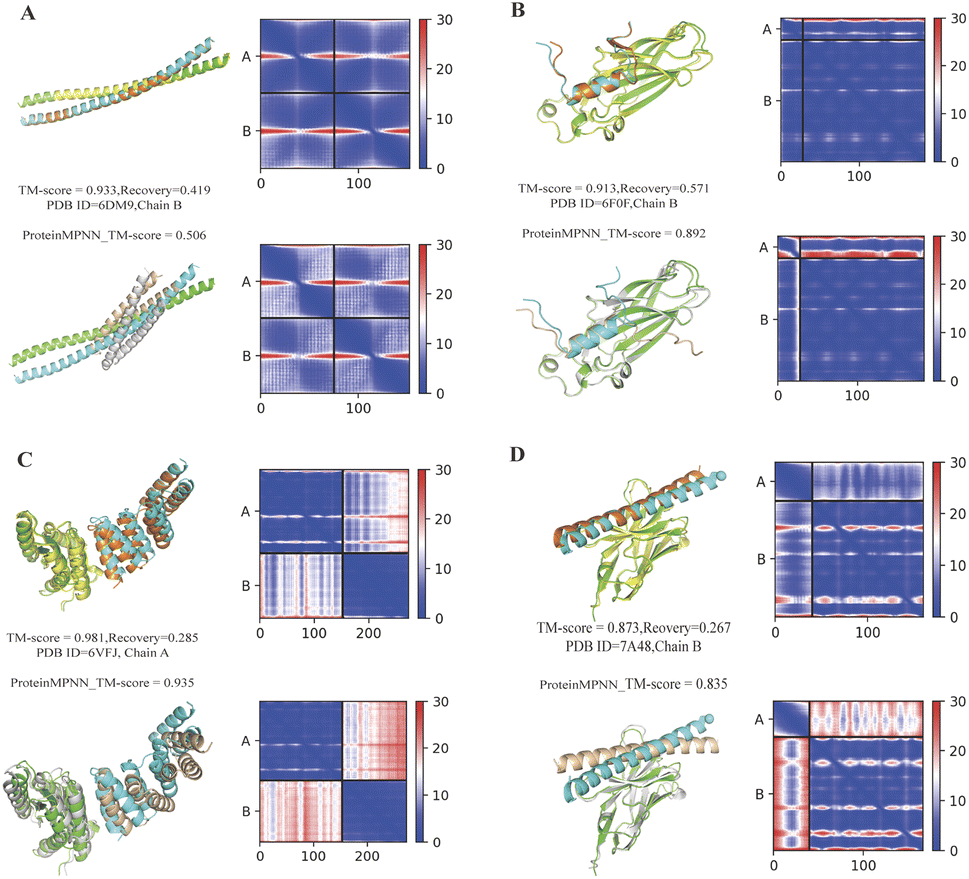 | ||
| Fig. 5 Comparison of structures of binder sequences designed by ProBID-Net and ProteinMPNN, predicted with AlphaFold-Multimer, and aligned to native complex structures (colored by chain). The AlphaFold-Multimer PAE heatmap is presented, depicting the PAE between all pairs of residues in Angstroms. In each subfigure, the upper row shows results from ProBID-Net and the bottom row from ProteinMPNN. | ||
Correlation between ProBID-Net prediction and protein–protein binding affinity
The predictive capabilities of ProBID-Net regarding the influence of mutations on the binding affinity of protein complexes were assessed in a zero-shot setting. To this end, the binding affinity caused by these mutations from the SKEMPI v2.0 database56 was used as test data. The predictions from mutations on protein–protein complexes and the changes of ProBID-Net were used to classify whether a single mutation increased or decreased the binding of the complex. The classification used the logarithm of the ratio (Pmutant/Pwildtype) of the difference in amino acid probabilities yielding a ROCAUC value of 0.66 (Fig. 6). In the context of the study, an elevated ROCAUC serves as an indicator of the model's heightened ability to discriminate between mutations classified as advantageous and deleterious. | ||
| Fig. 6 The ROC of ProBID-Net zero-shot prediction of affinity change caused by mutation in the SKEMPI v2.0 dataset. The ROC line in the graph serves as a reference, representing the ROC of an imaginary model making random predictions pertaining to the advantageous and deleterious effects associated with binding energy in residue mutations. | ||
The zero-shot evaluation focusing on protein–protein binding affinity assesses a model's potential to extrapolate its knowledge and forecast mutation effects on binding affinity without direct training on the precise mutations within the evaluation dataset. This result suggests that ProBID-Net demonstrates some ability to predict the impact of mutations on the stability of protein complexes, although further improvements may be needed for more accurate predictions. To further evaluate the performance of ProBID-Net in zero-shot binding affinity prediction, we utilized the large-scale data from MaveDB,57 which is a publicly accessible database (https://www.mavedb.org) that collects datasets derived from various analyses of variant effects, such as those obtained from deep mutation scanning (DMS) or Massively parallel Reporting Analysis (MPRA) experiments. For our investigation, we curated five protein–protein complex systems from MaveDB, specifically focusing on three datasets with a substantial sample size exceeding 300 data points. In order to explore whether ProBID-Net could discover beneficial mutations within the same protein, potentially optimizing the binding affinity of the complexes, we employed both ProBID-Net and ProteinMPNN to redesign the interface residues of the binding regions in the C. thermocellum cohesin fragment (PDB ID: 2VN5),58 IgG and IGg-FC (PDB ID: 1FCC)59 complexes, and SARS-CoV-2 Receptor Binding Domain and ACE2 (ref. 60) complex (Fig. 7). The analysis outcomes revealed that ProBID-Net (with a ROCAUC of 0.73, 0.67, and 0.57) surpassed ProteinMPNN (with a ROCAUC of 0.62, 0.67, and 0.38) in the correlation test of protein–protein complex binding energy. By redesigning the interfacial sequence, ProBID-Net demonstrated its efficacy in diminishing the binding free energy, thereby augmenting the stability of protein–protein complexes. These findings suggest that ProBID-Net holds promise in guiding and refining protein–protein complex interactions by facilitating beneficial mutations in interface residues. This, in turn, culminates in an enhancement of the binding affinity and overall stability of the protein complexes.
 | ||
| Fig. 7 Comparison of ROC Curves for ProBID-Net (Left) and ProteinMPNN (Right) predictions against binding free energy changes from MaveDB. (A) C. thermocellum cohesin fragment (PDB ID: 2VN5); (B) an IgG and IgG–Fc complex (PDB ID: 1FCC); (C) SARS-CoV-2 receptor binding domain and ACE2 complex (PDB ID: 6M0J). | ||
To explore the relationship between protein–protein binding affinity and recovery rate, we compared protein–protein complexes from the three test sets with data from the PDBbind61 dataset. This process identified 117 complexes with binding affinities reported as Kd, Ki, or IC50, which were then converted to ΔΔG values. We plotted the recovery rates of ProBID-Net for each complex against their binding affinities (Fig. S3†). The analysis shows that the overall recovery rate is independent of the binding affinity.
To evaluate the performance of ProBID-Net in recovering hotspot and non-hotspot residues, we utilized the MIX hotspot structural dataset from a recent study.62 Hotspot residues were defined as interfacial residues with a ΔΔG > 2 kcal mol−1 upon mutation to alanine, which resulted in 440 hotspot residues and 1902 non-hotspot residues. ProBID-Net was used to redesign these residues and the average recovery rates for hotspots and non-hotspots were 0.334 and 0.472. The detailed recovery rate for each protein–protein complex is illustrated in Fig. S4.† These results indicate that ProBID-Net exhibits lower prediction accuracy for hotspot residues compared to non-hotspot residues. This discrepancy may be due to the highly dynamic nature of hotspot residues, which can participate in multiple binding conformations depending on the interacting partner, making accurate prediction more challenging.
Conclusions
ProBID-Net has successfully learned the characteristics of protein–protein interactions from the interfaces of protein heterodimer complexes. This enables the model to furnish predictions regarding the amino acid probability on each residue within a given protein backbone structure, leveraging insights garnered from the protein receptor binding sites. We broadened the training set by incorporating domain–domain interface structure data into the training set, thus allowing for a more comprehensive representation of diverse interaction patterns. ProBID-Net outperforms ProteinMPNN with a higher protein sequence recovery rate and lower perplexity on all the independent heterodimer test sets. Furthermore, the model exhibits pronounced conservation in hydrophobicity positions, underscoring its robust capacity to capture evolutionary information pertaining to protein interface residues.The primary objective behind developing ProBID-Net was to enhance its focus on the structural training set specific to protein–protein interaction interfaces. In addition, domain–domain interfaces were also considered as a subset of protein–protein interfaces and incorporated into the supplementary training set. This approach significantly expanded the size of the training dataset. Our test results demonstrate that this strategy effectively improves the model's performance, particularly in recovering interface residue sequences across various test sets. The inclusion of domain–domain interfaces as part of the training data contributed to this enhancement by providing a broader and more diverse dataset for the model to learn from, thus strengthening its ability to accurately predict interface residues.
The reliability of redesigned protein complex sequences was validated through AlphaFold-Multimer verification experiments, which demonstrated the foldability and specificity of the binding sites. Notably, the lower PAE observed in sequences designed by ProBID-Net implies a heightened accuracy in inter-chain orientation compared to counterparts designed by ProteinMPNN.
In the context of zero-shot prediction, ProBID-Net exhibited a notable correlation between mutations occurring in residues at the protein–protein complex interface and a consequential decrease in binding free energy in complexes sourced from the SKEMPI and MaveDB database.
In conclusion, ProBID-Net emerges as a promising tool with substantial potential. Its utility extends to enhancing the binding stability of protein–protein complexes and facilitating the redesign of highly specific, compatible binding proteins. This is achieved through a nuanced understanding of the structural nuances within protein receptor binding sites, making ProBID-Net a valuable asset in the realm of computational protein design.
Materials and methods
Datasets
The overall data collection and processing workflow are shown in Fig. 8. We employed the PDB structures labeled as heterodimers by QSalignHET,47 which comprised 3821 structures with the confidence category of “Very High”, “High” and “Medium”. By treating one of the chains as the design target in turn, we derived a dataset of 7642 (3821 × 2) complex structures. | ||
| Fig. 8 The overview of dataset collection, clustering and partitioning processes and the number of protein–protein interface structures contained in each dataset, where the green arrow indicates the integration of similar structures from the test set into the training set. | ||
Given the limited availability of protein–protein complexes within PDB, particularly within the realm of heterodimer chain–chain interfaces, it becomes imperative to explore alternative approaches. Recognizing the similarity between domain–domain and chain–chain interfaces, we consider the former as a unit of evolutionary conservation and leverage domain–domain interface structures extracted from the PDB using Sen's protein interface library. This library, comprising 27885 clusters with structural similarity, serves as a valuable resource for training our model. To enhance the diversity of our dataset, we meticulously curate multi-domain chains from heterodimers mentioned above, resulting in a comprehensive dataset of 19481 domain–domain interface residue structures. This enriched dataset, integrated into the training set, empowers our model to better comprehend the intricacies of protein interactions, ultimately enhancing its predictive capabilities.
For de novo designed protein–protein complexes, we conducted an exhaustive search for recent heterodimeric complex structures12,13,63–75 published from 2018 to 2023 in the PDB. We used specific keywords “de novo design” and applied additional filters, such as ensuring that each complex had a total number of polymer instances (chains) equal to 2 and a number of distinct protein entities greater than or equal to 2. These structures were then clustered using MMseqs2 (ref. 76) with a 30% sequence identity cutoff, resulting in a set of 89 unique structures.
Additionally, we collected six heterodimeric complexes from the CASP15 competition and compiled a total of 2728 protein complexes with known pairwise interfaces from recent studies,55,77 forming the “Folddock complex set”. These complexes were carefully chosen to meet certain criteria, including a resolution between 1 and 5 Å, sequence length between 30 and 1200, and a sequence identity of less than 30% within the Folddock complex set. To remove the redundancy between the QSalignHET set and the other two test sets, all the complexes in the four datasets were clustered with a sequence identity of 40% and a coverage of 0.8 using MMseqs2: mmseqs easy-cluster Heterodimers_chain Heterodimer_chain_clu tmp--cov-mode 0-c 0.8--min-seq-id 0.4--threads 8.
This procedure resulted in the formation of 2996 distinct clusters, each containing representative and component chains.
After clustering, for the de novo set and Folddock set, structures with identical PDB IDs or those sharing the same cluster as the heterodimers identified by QSalignHET were systematically excluded. As a result, 17 pairs of complexes in the initial de novo set, and 1622 pairs in the initial Folddock set were moved to the training set. This process resulted in the generation of 62 structures in the de novo set and 1106 structures in the Folddock set. Additionally, 10% of the clusters in the chain–chain interface set were chosen to constitute an independent test set, distinct from the other 90%. The remaining 90%, in conjunction with domain–domain interface data, constituted the training dataset. The PDB IDs for all test sets, along with the corresponding ligand chain IDs, are presented in Tables S2–S4.†
Network architecture
The distribution of constituent atoms of residues on both the target chain and the neighboring chain is taken into consideration by evaluating them within a three-dimensional grid box. This process involves quantifying the densities of these atoms and subsequently storing this information in the characteristic atomic channels corresponding to the 20 natural amino acids. This method of representing structural data ensures the preservation of the natural features of the protein structure and prevents the risk of information loss that could arise from arbitrary extraction. Its efficacy and efficiency have been demonstrated through its successful application in DenseCPD.23After preprocessing the PDB structures, certain components such as nucleic acids, small molecule ligands, and short peptides with less than 20 amino acids were removed to focus specifically on the protein–protein interactions. Each target residue and its neighbouring residues on another chain were then positioned in a standardized orientation, with the N atom situated on the y = 0 plane and x < 0. The Cα atom of the target residue was set at the origin (0, 0, 0), and the Cβ atom was placed on the positive Z axis.
To represent the atomic coordinates, a density distribution was created on a 20 Å × 20 Å × 20 Å grid box with a grid size of 1 Å. The grid box was centered at (0, 0, 2) Å to encompass more neighbouring residues in contact with the target residue. The distribution of atom densities was achieved using the Gaussian function  , where d is the distance between the atom and the center, and r is the atom's radius. The radii of N, C, Cα, Cβ, and O atoms were assigned as 0.755, 0.817, 0.817, 0.821, and 0.695 Å, respectively. These radii were determined based on the van der Waals radius from the CHARMM36 force field, ensuring that each atom's density would be 0.05 at a distance of its van der Waals radius.
, where d is the distance between the atom and the center, and r is the atom's radius. The radii of N, C, Cα, Cβ, and O atoms were assigned as 0.755, 0.817, 0.817, 0.821, and 0.695 Å, respectively. These radii were determined based on the van der Waals radius from the CHARMM36 force field, ensuring that each atom's density would be 0.05 at a distance of its van der Waals radius.
The encoding of the 20 natural amino acids, each possessing distinct characteristic atoms, necessitated consideration of the specific atom count associated with each amino acid type. For instance, glycine (Gly) comprises only 4 atoms (CA, C, N, and O), while phenylalanine (Phe) has 11 atoms (CA, CB, C, CD1, CD2, CE1, CE2, CG, CZ, N, and O). Crucially, the target amino acid is characterized by five essential atoms: CA, CB, C, N, and O. Consequently, the encoding process involved representing the 20 amino acids using 173 atomic types, and the densities of these atomic types were meticulously stored in dedicated grid boxes. This resulted in a data size for a target residue interface structure of 20 × 20 × 20 × 173, reflecting the grid arrangement for each interface structure.
The structures of the target residue and its neighbors were analysed using DenseNet, which is composed of several dense blocks connected by transition blocks. Each dense block contains multiple convolution blocks with a bottleneck operation, followed by batch normalization, ReLU activation, and a (3 × 3 × 3) convolution. All the outputs from the convolution blocks are connected to each other within the same dense block.
To enhance computational efficiency and reduce the input feature map size, the bottleneck operation was used in the transition blocks and involved batch normalization, ReLU activation, and a (1 × 1 × 1) convolution. These transition blocks perform compression and pooling operations with a compression rate of 0.5 between the dense blocks.
The depth of the DenseNet was adjustable, with variations in the number of dense blocks and convolution blocks. The growth rate, representing the size of the feature map in the output of a convolution block, determined the number of feature maps. In this study, three dense blocks, each comprising six convolution blocks and a growth rate of 70, were employed.
Finally, the output of ProBID-Net is processed through a softmax layer, generating 20 values that sum to 1. These values represented the probabilities of the 20 natural amino acids for the target residue.
Training
To ensure robustness and assess the model's performance, the complex structures in the training set were divided into five equal groups based on their clustering. This facilitated 5-fold cross-validation, where the model was trained and evaluated on different combinations of the data groups to ensure a thorough and reliable assessment of its predictive capabilities.The training was carried out for 35 epochs, and early stopping was implemented with a patience of 5 epochs to avoid unnecessary iterations. The categorical cross-entropy was utilized as the loss function, and the Adam optimization method with a learning rate of 0.001 and a batch size of 16 were employed for training. To prevent overfitting during training, we applied a weight decay of 10−4 to the convolution layers.
ProBID-Net was implemented using TensorFlow with the Keras library (http://keras.io). During the training process, a categorical cross-entropy loss function was employed, and optimization was performed using the Adam algorithm for 35 epochs. After that, the training was extended by adding the domain–domain interface data and continuing for another 35 epochs. For optimization with stochastic gradient descent (SGD), early stopping was implemented, and the learning rate and batch size remained the same as during the Adam optimization. The output of the neural network is the probabilities of the 20 amino acids for the target residue.
Performance measure
 | (1) |
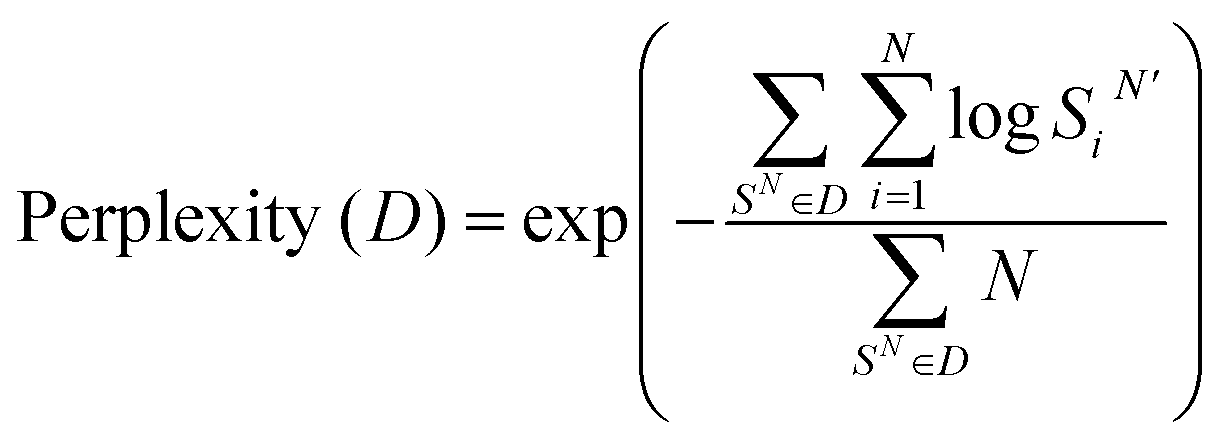 | (2) |
Hydrophobicity conservation
We examined the conservation of hydrophobic and hydrophilic sequence positions of design sequences by defining hydrophobic (Ile, Leu, Met, Phe, Cys, Trp, Pro, Val, Ala and Gly) and hydrophilic (Ser, Thr, Asn, Gln, Asp, Glu, His, Arg, Lys and Tyr) residue positions according to the native sequence.BLOSUM62 substitution matrix
The substitution scores between native sequences and designed sequences were calculated using: | (3) |
BLOSUM62 substitution matrix
The BLOSUM score is calculated as the weighted sum of BLOSUM62 (ref. 78) values based on predicted probabilities: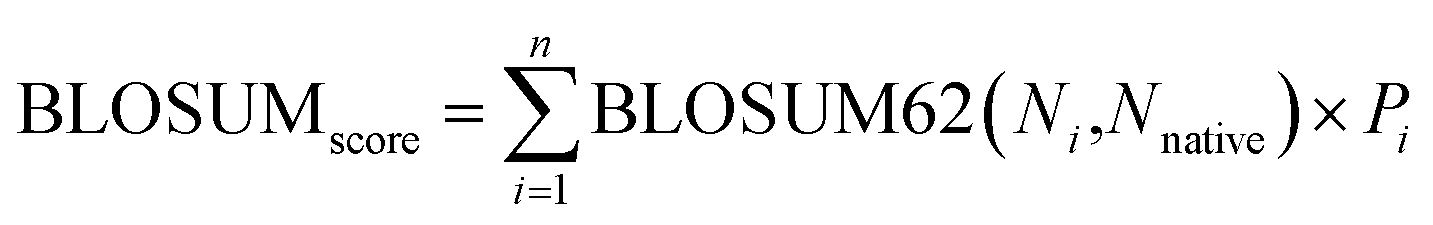 | (4) |
The normalization is done by dividing the BLOSUM score of the methods by the BLOSUM score of the native sequence:
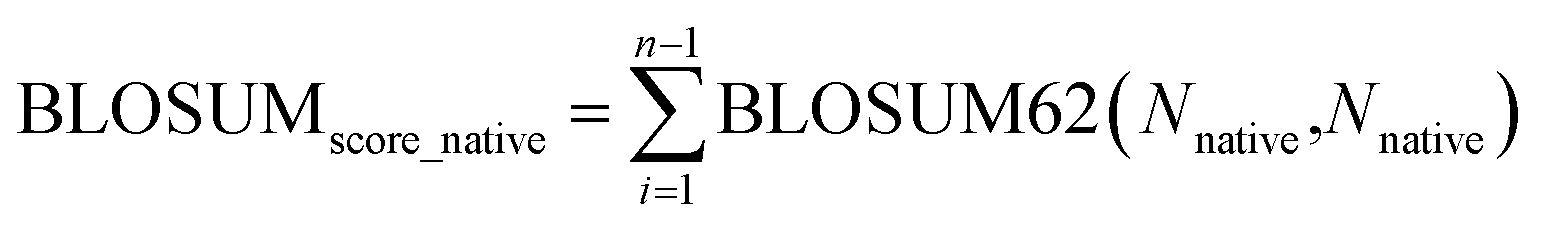 | (5) |
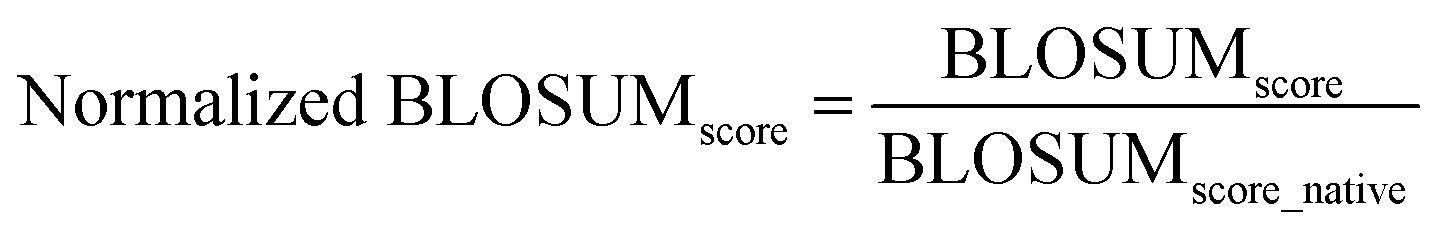 | (6) |
The sums are over all positions in the sequence. This notation reflects the mathematical representation of the BLOSUM score calculation and its normalization.
Zero-shot evaluation on protein–protein binding affinity
Zero-shot evaluation on the protein–protein binding affinity refers to assessing the performance of ProBID-Net predicting the impact of mutations on the binding affinity of protein complexes without any specific training on the exact mutations in question. | (7) |
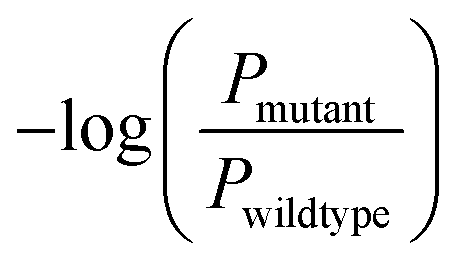 is the negative logarithm of the ratio of amino acid type probabilities after and before mutation, and ROCAUC is the Receiver Operating Characteristic Area Under the Curve.
is the negative logarithm of the ratio of amino acid type probabilities after and before mutation, and ROCAUC is the Receiver Operating Characteristic Area Under the Curve.
The process involves using a dataset containing mutations within protein–protein complexes and the corresponding changes in binding affinity. The model is then tasked with binary classification, determining whether a given mutation leads to an increase in the stability of the complex. The evaluation is conducted without explicit training on these specific mutations, relying on the model's ability to extrapolate from its training data to make accurate predictions for novel mutations.
Data availability
The PDB IDs of all test sets are available in the ESI.† ProBID-Net code is available at https://github.com/ComputArtCMCG/ProBID-NET. The model checkpoint trained on both chain–chain interface and domain–domain interface is available at https://figshare.com/s/ebbd5184c0a46fb2b179.Author contributions
Z. C. conducted data collection and the experiments, performed data analysis, and wrote the manuscript. M. J. and J. Q. contributed to experimental design and data analysis. Z. Z., X. Z., H. G., and H. W. assisted in model training and protein structure encoding. R. W. and Y. Q. conceived the study, supervised the research, and provided critical revisions to the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Grant No. 22033001, 81725022, 82173739, 81430083, 21661162003, 21472227), the Ministry of Science and Technology of China (National Key Research Program, Grant No. 2016YFA0502302), and Science and Technology Commission of Shanghai Municipality (Grant No. 20S11900500). The computations in this research were performed using the CFFF platform of Fudan University.Notes and references
- P.-S. Huang, S. E. Boyken and D. Baker, The coming of age of de novo protein design, Nature, 2016, 537, 320–327, DOI:10.1038/nature19946.
- A. Leaver-Fay, M. Tyka, S. M. Lewis, O. F. Lange, J. Thompson, R. Jacak, K. Kaufman, P. D. Renfrew, C. A. Smith, W. Sheffler, I. W. Davis, S. Cooper, A. Treuille, D. J. Mandell, F. Richter, Y. E. Ban, S. J. Fleishman, J. E. Corn, D. E. Kim, S. Lyskov, M. Berrondo, S. Mentzer, Z. Popović, J. J. Havranek, J. Karanicolas, R. Das, J. Meiler, T. Kortemme, J. J. Gray, B. Kuhlman, D. Baker and P. Bradley, ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules, Methods Enzymol., 2011, 487, 545–574, DOI:10.1016/b978-0-12-381270-4.00019-6.
- P. S. Huang, Y. E. Ban, F. Richter, I. Andre, R. Vernon, W. R. Schief and D. Baker, RosettaRemodel: a generalized framework for flexible backbone protein design, PLoS One, 2011, 6, e24109, DOI:10.1371/journal.pone.0024109.
- P. Xiong, M. Wang, X. Zhou, T. Zhang, J. Zhang, Q. Chen and H. Liu, Protein design with a comprehensive statistical energy function and boosted by experimental selection for foldability, Nat. Commun., 2014, 5, 5330, DOI:10.1038/ncomms6330.
- P. Xiong, X. Hu, B. Huang, J. Zhang, Q. Chen and H. Liu, Increasing the efficiency and accuracy of the ABACUS protein sequence design method, Bioinformatics, 2020, 36, 136–144, DOI:10.1093/bioinformatics/btz515.
- S. Liang, Z. Li, J. Zhan and Y. Zhou, De novo protein design by an energy function based on series expansion in distance and orientation dependence, Bioinformatics, 2021, 38, 86–93, DOI:10.1093/bioinformatics/btab598.
- O. Khersonsky, R. Lipsh, Z. Avizemer, Y. Ashani, M. Goldsmith, H. Leader, O. Dym, S. Rogotner, D. L. Trudeau, J. Prilusky, P. Amengual-Rigo, V. Guallar, D. S. Tawfik and S. J. Fleishman, Automated Design of Efficient and Functionally Diverse Enzyme Repertoires, Mol. Cell, 2018, 72, 178–186, DOI:10.1016/j.molcel.2018.08.033.
- A. A. Glasgow, Y.-M. Huang, D. J. Mandell, M. Thompson, R. Ritterson, A. L. Loshbaugh, J. Pellegrino, C. Krivacic, R. A. Pache, K. A. Barlow, N. Ollikainen, D. Jeon, M. J. S. Kelly, J. S. Fraser and T. Kortemme, Computational design of a modular protein sense-response system, Science, 2019, 366, 1024–1028, DOI:10.1126/science.aax8780.
- A. Glasgow, J. Glasgow, D. Limonta, P. Solomon, I. Lui, Y. Zhang, M. A. Nix, N. J. Rettko, S. Zha, R. Yamin, K. Kao, O. S. Rosenberg, J. V. Ravetch, A. P. Wiita, K. K. Leung, S. A. Lim, X. X. Zhou, T. C. Hobman, T. Kortemme and J. A. Wells, Engineered ACE2 receptor traps potently neutralize SARS-CoV-2, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 28046–28055, DOI:10.1073/pnas.2016093117.
- H. Shen, J. A. Fallas, E. Lynch, W. Sheffler, B. Parry, N. Jannetty, J. Decarreau, M. Wagenbach, J. J. Vicente, J. Chen, L. Wang, Q. Dowling, G. Oberdorfer, L. Stewart, L. Wordeman, J. De Yoreo, C. Jacobs-Wagner, J. Kollman and D. Baker, De novo design of self-assembling helical protein filaments, Science, 2018, 362, 705–709, DOI:10.1126/science.aau3775.
- R. A. Langan, S. E. Boyken, A. H. Ng, J. A. Samson, G. Dods, A. M. Westbrook, T. H. Nguyen, M. J. Lajoie, Z. Chen, S. Berger, V. K. Mulligan, J. E. Dueber, W. R. P. Novak, H. El-Samad and D. Baker, De novo design of bioactive protein switches, Nature, 2019, 572, 205–210, DOI:10.1038/s41586-019-1432-8.
- K. Mohan, G. Ueda, A. R. Kim, K. M. Jude, J. A. Fallas, Y. Guo, M. Hafer, Y. Miao, R. A. Saxton, J. Piehler, V. G. Sankaran, D. Baker and K. C. Garcia, Topological control of cytokine receptor signaling induces differential effects in hematopoiesis, Science, 2019, 364, eaav7532, DOI:10.1126/science.aav7532.
- D.-A. Silva, S. Yu, U. Y. Ulge, J. B. Spangler, K. M. Jude, C. Labão-Almeida, L. R. Ali, A. Quijano-Rubio, M. Ruterbusch, I. Leung, T. Biary, S. J. Crowley, E. Marcos, C. D. Walkey, B. D. Weitzner, F. Pardo-Avila, J. Castellanos, L. Carter, L. Stewart, S. R. Riddell, M. Pepper, G. J. L. Bernardes, M. Dougan, K. C. Garcia and D. Baker, De novo design of potent and selective mimics of IL-2 and IL-15, Nature, 2019, 565, 186–191, DOI:10.1038/s41586-018-0830-7.
- L. Cao, I. Goreshnik, B. Coventry, J. B. Case, L. Miller, L. Kozodoy, R. E. Chen, L. Carter, A. C. Walls, Y. J. Park, E. M. Strauch, L. Stewart, M. S. Diamond, D. Veesler and D. Baker, De novo design of picomolar SARS-CoV-2 miniprotein inhibitors, Science, 2020, 370, 426–431, DOI:10.1126/science.abd9909.
- J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction, Science, 2010, 329, 309–313, DOI:10.1126/science.1190239.
- A. Chevalier, D. A. Silva, G. J. Rocklin, D. R. Hicks, R. Vergara, P. Murapa, S. M. Bernard, L. Zhang, K. H. Lam, G. Yao, C. D. Bahl, S. I. Miyashita, I. Goreshnik, J. T. Fuller, M. T. Koday, C. M. Jenkins, T. Colvin, L. Carter, A. Bohn, C. M. Bryan, D. A. Fernández-Velasco, L. Stewart, M. Dong, X. Huang, R. Jin, I. A. Wilson, D. H. Fuller and D. Baker, Massively parallel de novo protein design for targeted therapeutics, Nature, 2017, 550, 74–79, DOI:10.1038/nature23912.
- W. M. Dawson, E. J. M. Lang, G. G. Rhys, K. L. Shelley, C. Williams, R. L. Brady, M. P. Crump, A. J. Mulholland and D. N. Woolfson, Structural resolution of switchable states of a de novo peptide assembly, Nat. Commun., 2021, 12, 1530, DOI:10.1038/s41467-021-21851-8.
- W. Ding, K. Nakai and H. Gong, Protein design via deep learning, Briefings Bioinf., 2022, 23, bbac102, DOI:10.1093/bib/bbac102.
- Z. Li, Y. Yang, E. Faraggi, J. Zhan and Y. Zhou, Direct prediction of profiles of sequences compatible with a protein structure by neural networks with fragment-based local and energy-based nonlocal profiles, Proteins, 2014, 82, 2565–2573, DOI:10.1002/prot.24620.
- J. O'Connell, Z. Li, J. Hanson, R. Heffernan, J. Lyons, K. Paliwal, A. Dehzangi, Y. Yang and Y. Zhou, SPIN2: predicting sequence profiles from protein structures using deep neural networks, Proteins, 2018, 86, 629–633, DOI:10.1002/prot.25489.
- S. Chen, Z. Sun, L. Lin, Z. Liu, X. Liu, Y. Chong, Y. Lu, H. Zhao and Y. Yang, To Improve Protein Sequence Profile Prediction through Image Captioning on Pairwise Residue Distance Map, J. Chem. Inf. Model., 2020, 60, 391–399, DOI:10.1021/acs.jcim.9b00438.
- Y. Zhang, Y. Chen, C. Wang, C. C. Lo, X. Liu, W. Wu and J. Zhang, ProDCoNN: protein design using a convolutional neural network, Proteins, 2020, 88, 819–829, DOI:10.1002/prot.25868.
- Y. Qi and J. Z. H. Zhang, DenseCPD: Improving the Accuracy of Neural-Network-Based Computational Protein Sequence Design with DenseNet, J. Chem. Inf. Model., 2020, 60, 1245–1252, DOI:10.1021/acs.jcim.0c00043.
- A. Elnaggar, M. Heinzinger, C. Dallago, G. Rehawi, Y. Wang, L. Jones, T. Gibbs, T. Feher, C. Angerer, M. Steinegger, D. Bhowmik and B. Rost, ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning, IEEE Trans. Pattern Anal. Mach. Intell., 2022, 44, 7112–7127, DOI:10.1109/tpami.2021.3095381.
- B. Jing, S. Eismann, P. Suriana, R. J. L. Townshend and R. O. J. A. Dror, Learning from Protein Structure with Geometric Vector Perceptrons, arXiv, 2021, preprint, 10.48550/arXiv.2009.01411, DOI:10.48550/arXiv.2009.01411.
- J. Ingraham, V. K. Garg, R. Barzilay and T. Jaakkola, Generative Models for Graph-Based Protein Design, NeurIPS, 2019, 15820–15831 Search PubMed.
- Z. Gao, C. Tan and S. Z. J. A. Li, AlphaDesign: a graph protein design method and benchmark on AlphaFoldDB, arXiv, 2022, preprint, 10.48550/arXiv.2202.01079, DOI:10.48550/arXiv.2202.01079.
- C. Hsu, R. Verkuil, J. Liu, Z. Lin, B. Hie, T. Sercu, A. Lerer and A. Rives, Learning inverse folding from millions of predicted structures, Proceedings of the 39th International Conference on Machine Learning, 2022, 162, 8946–8970 Search PubMed.
- Z. Gao, C. Tan and S. Z. Li, PiFold: Toward effective and efficient protein inverse folding, Eleventh International Conference on Learning Representations, 2023 Search PubMed.
- W. Mao, M. Zhu, Z. Sun, S. Shen, L. Y. Wu, H. Chen and C. J. A. Shen, De novo protein design using geometric vector field networks, arXiv, 2023, preprint, arXiv:2310.11802, DOI:10.48550/arXiv.2310.11802.
- Z. Zheng, Y. Deng, D. Xue, Y. Zhou, Y. Fei and Q. J. b. Gu, Structure-informed Language Models Are Protein Designers, arXiv, 2023, preprint, arXiv:2302.01649, DOI:10.48550/arXiv.2302.01649.
- Y. Liu, L. Zhang, W. Wang, M. Zhu, C. Wang, F. Li, J. Zhang, H. Li, Q. Chen and H. Liu, Rotamer-free protein sequence design based on deep learning and self-consistency, Nat. Comput. Sci., 2022, 2, 451–462, DOI:10.1038/s43588-022-00273-6.
- B. Huang, T. Fan, K. Wang, H. Zhang, C. Yu, S. Nie, Y. Qi, W.-M. Zheng, J. Han, Z. Fan, S. Sun, S. Ye, H. Yang and D. Bu, Accurate and efficient protein sequence design through learning concise local environment of residues, Bioinformatics, 2023, 39, btad122, DOI:10.1093/bioinformatics/btad122.
- M. Ren, C. Yu, D. Bu and H. Zhang, Accurate and robust protein sequence design with CarbonDesign, Nat. Mach. Intell., 2024, 6, 536–547, DOI:10.1038/s42256-024-00838-2.
- J. Dauparas, I. Anishchenko, N. Bennett, H. Bai, R. J. Ragotte, L. F. Milles, B. I. M. Wicky, A. Courbet, R. J. de Haas, N. Bethel, P. J. Y. Leung, T. F. Huddy, S. Pellock, D. Tischer, F. Chan, B. Koepnick, H. Nguyen, A. Kang, B. Sankaran, A. K. Bera, N. P. King and D. Baker, Robust deep learning–based protein sequence design using ProteinMPNN, Science, 2022, 378, 49–56, DOI:10.1126/science.add2187.
- I. Anishchenko, S. J. Pellock, T. M. Chidyausiku, T. A. Ramelot, S. Ovchinnikov, J. Hao, K. Bafna, C. Norn, A. Kang, A. K. Bera, F. DiMaio, L. Carter, C. M. Chow, G. T. Montelione and D. Baker, De novo protein design by deep network hallucination, Nature, 2021, 600, 547–552, DOI:10.1038/s41586-021-04184-w.
- J. Wang, S. Lisanza, D. Juergens, D. Tischer, J. L. Watson, K. M. Castro, R. Ragotte, A. Saragovi, L. F. Milles, M. Baek, I. Anishchenko, W. Yang, D. R. Hicks, M. Expòsit, T. Schlichthaerle, J. H. Chun, J. Dauparas, N. Bennett, B. I. M. Wicky, A. Muenks, F. DiMaio, B. Correia, S. Ovchinnikov and D. Baker, Scaffolding protein functional sites using deep learning, Science, 2022, 377, 387–394, DOI:10.1126/science.abn2100.
- I. R. Humphreys, J. Pei, M. Baek, A. Krishnakumar, I. Anishchenko, S. Ovchinnikov, J. Zhang, T. J. Ness, S. Banjade, S. R. Bagde, V. G. Stancheva, X.-H. Li, K. Liu, Z. Zheng, D. J. Barrero, U. Roy, J. Kuper, I. S. Fernández, B. Szakal, D. Branzei, J. Rizo, C. Kisker, E. C. Greene, S. Biggins, S. Keeney, E. A. Miller, J. C. Fromme, T. L. Hendrickson, Q. Cong and D. Baker, Computed structures of core eukaryotic protein complexes, Science, 2021, 374, eabm4805, DOI:10.1126/science.abm4805.
- W. Dai, A. Wu, L. Ma, Y.-X. Li, T. Jiang and Y.-Y. Li, A novel index of protein-protein interface propensity improves interface residue recognition, BMC Syst. Biol., 2016, 10, 112, DOI:10.1186/s12918-016-0351-7.
- Y. Chen, Y. N. Zhang, R. Yan, G. Wang, Y. Zhang, Z. R. Zhang, Y. Li, J. Ou, W. Chu, Z. Liang, Y. Wang, Y. L. Chen, G. Chen, Q. Wang, Q. Zhou, B. Zhang and C. Wang, ACE2-targeting monoclonal antibody as potent and broad-spectrum coronavirus blocker, Signal Transduct. Targeted Ther., 2021, 6, 315, DOI:10.1038/s41392-021-00740-y.
- J. Adolf-Bryfogle, O. Kalyuzhniy, M. Kubitz, B. D. Weitzner, X. Hu, Y. Adachi, W. R. Schief and R. L. Dunbrack, Jr., RosettaAntibodyDesign (RAbD): a general framework for computational antibody design, PLoS Comput. Biol., 2018, 14, e1006112, DOI:10.1371/journal.pcbi.1006112.
- D. Baran, M. G. Pszolla, G. D. Lapidoth, C. Norn, O. Dym, T. Unger, S. Albeck, M. D. Tyka and S. J. Fleishman, Principles for computational design of binding antibodies, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10900–10905, DOI:10.1073/pnas.1707171114.
- A. Glasgow, J. Glasgow, D. Limonta, P. Solomon, I. Lui, Y. Zhang, M. A. Nix, N. J. Rettko, S. Zha, R. Yamin, K. Kao, O. S. Rosenberg, J. V. Ravetch, A. P. Wiita, K. K. Leung, S. A. Lim, X. X. Zhou, T. C. Hobman, T. Kortemme and J. A. Wells, Engineered ACE2 receptor traps potently neutralize SARS-CoV-2, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28046–28055, DOI:10.1073/pnas.2016093117.
- J. Shirian, V. Arkadash, I. Cohen, T. Sapir, E. S. Radisky, N. Papo and J. M. Shifman, Converting a broad matrix metalloproteinase family inhibitor into a specific inhibitor of MMP-9 and MMP-14, FEBS Lett., 2018, 592, 1122–1134, DOI:10.1002/1873-3468.13016.
- V. Arkadash, G. Yosef, J. Shirian, I. Cohen, Y. Horev, M. Grossman, I. Sagi, E. S. Radisky, J. M. Shifman and N. Papo, Development of High Affinity and High Specificity Inhibitors of Matrix Metalloproteinase 14 through Computational Design and Directed Evolution, J. Biol. Chem., 2017, 292, 3481–3495, DOI:10.1074/jbc.M116.756718.
- G. Huang, Z. Liu, L. van der Maaten and K. Q. Weinberger, Densely Connected Convolutional Networks, Proc. IEEE Int. Conf. Comput. Vis., 2016, 4700–4708 Search PubMed.
- S. Dey, D. W. Ritchie and E. D. Levy, PDB-wide identification of biological assemblies from conserved quaternary structure geometry, Nat. Methods, 2018, 15, 67–72, DOI:10.1038/nmeth.4510.
- I. Sillitoe, N. Bordin, N. Dawson, V. P. Waman, P. Ashford, H. M. Scholes, C. S. M. Pang, L. Woodridge, C. Rauer, N. Sen, M. Abbasian, S. Le Cornu, S. D. Lam, K. Berka, I. H. Varekova, R. Svobodova, J. Lees and C. A. Orengo, CATH: increased structural coverage of functional space, Nucleic Acids Res., 2021, 49, D266–d273, DOI:10.1093/nar/gkaa1079.
- S. R. Eddy, Where did the BLOSUM62 alignment score matrix come from?, Nat. Biotechnol., 2004, 22, 1035–1036, DOI:10.1038/nbt0804-1035.
- D. Song, J. Chen, G. Chen, N. Li, J. Li, J. Fan, D. Bu and S. C. Li, Parameterized BLOSUM Matrices for Protein Alignment, IEEE/ACM Trans. Comput. Biol. Bioinform., 2015, 12, 686–694, DOI:10.1109/tcbb.2014.2366126.
- X. Zhang, H. Yin, F. Ling, J. Zhan and Y. Zhou, SPIN-CGNN: improved fixed backbone protein design with contact map-based graph construction and contact graph neural network, PLoS Comput. Biol., 2023, 19, e1011330, DOI:10.1371/journal.pcbi.1011330.
- Y. Yan, H. Tao, J. He and S. Y. Huang, The HDOCK server for integrated protein-protein docking, Nat. Protoc., 2020, 15, 1829–1852, DOI:10.1038/s41596-020-0312-x.
- C. Yan, F. Wu, R. L. Jernigan, D. Dobbs and V. Honavar, Characterization of protein-protein interfaces, Protein J., 2008, 27, 59–70, DOI:10.1007/s10930-007-9108-x.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589, DOI:10.1038/s41586-021-03819-2.
- P. Bryant, G. Pozzati and A. Elofsson, Improved prediction of protein-protein interactions using AlphaFold2, Nat. Commun., 2022, 13, 1265, DOI:10.1038/s41467-022-28865-w.
- J. Jankauskaite, B. Jiménez-García, J. Dapkunas, J. Fernández-Recio and I. H. Moal, SKEMPI 2.0: an updated benchmark of changes in protein-protein binding energy, kinetics and thermodynamics upon mutation, Bioinformatics, 2019, 35, 462–469, DOI:10.1093/bioinformatics/bty635.
- D. Esposito, J. Weile, J. Shendure, L. M. Starita, A. T. Papenfuss, F. P. Roth, D. M. Fowler and A. F. Rubin, MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect, Genome Biol., 2019, 20, 223, DOI:10.1186/s13059-019-1845-6.
- C. A. Kowalsky and T. A. Whitehead, Determination of binding affinity upon mutation for type I dockerin-cohesin complexes from Clostridium thermocellum and Clostridium cellulolyticum using deep sequencing, Proteins, 2016, 84, 1914–1928, DOI:10.1002/prot.25175.
- C. A. Olson, N. C. Wu and R. Sun, A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain, Current Biol., 2014, 24, 2643–2651, DOI:10.1016/j.cub.2014.09.072.
- T. N. Starr, A. J. Greaney, S. K. Hilton, D. Ellis, K. H. D. Crawford, A. S. Dingens, M. J. Navarro, J. E. Bowen, M. A. Tortorici, A. C. Walls, N. P. King, D. Veesler and J. D. Bloom, Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding, Cell, 2020, 182, 1295–1310, DOI:10.1016/j.cell.2020.08.012.
- Z. Liu, M. Su, L. Han, J. Liu, Q. Yang, Y. Li and R. Wang, Forging the Basis for Developing Protein-Ligand Interaction Scoring Functions, Acc. Chem. Res., 2017, 50, 302–309, DOI:10.1021/acs.accounts.6b00491.
- Y. Zhang, S. Yao and P. Chen, Prediction of hot spots towards drug discovery by protein sequence embedding with 1D convolutional neural network, PLoS One, 2023, 18, e0290899, DOI:10.1371/journal.pone.0290899.
- D. D. Sahtoe, A. Coscia, N. Mustafaoglu, L. M. Miller, D. Olal, I. Vulovic, T. Y. Yu, I. Goreshnik, Y. R. Lin, L. Clark, F. Busch, L. Stewart, V. H. Wysocki, D. E. Ingber, J. Abraham and D. Baker, Transferrin receptor targeting by de novo sheet extension, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2021569118, DOI:10.1073/pnas.2021569118.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millán, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Accurate prediction of protein structures and interactions using a three-track neural network, Science, 2021, 373, 871–876, DOI:10.1126/science.abj8754.
- L. T. Dang, Y. Miao, A. Ha, K. Yuki, K. Park, C. Y. Janda, K. M. Jude, K. Mohan, N. Ha, M. Vallon, J. Yuan, J. G. Vilches-Moure, C. J. Kuo, K. C. Garcia and D. Baker, Receptor subtype discrimination using extensive shape complementary designed interfaces, Nat. Struct. Mol. Biol., 2019, 26, 407–414, DOI:10.1038/s41594-019-0224-z.
- P. Hosseinzadeh, P. R. Watson, T. W. Craven, X. Li, S. Rettie, F. Pardo-Avila, A. K. Bera, V. K. Mulligan, P. Lu, A. S. Ford, B. D. Weitzner, L. J. Stewart, A. P. Moyer, M. Di Piazza, J. G. Whalen, P. Greisen Jr, D. W. Christianson and D. Baker, Anchor extension: a structure-guided approach to design cyclic peptides targeting enzyme active sites, Nat. Commun., 2021, 12, 3384, DOI:10.1038/s41467-021-23609-8.
- Y. K. Lau, V. Baytshtok, T. A. Howard, B. M. Fiala, J. M. Johnson, L. P. Carter, D. Baker, C. D. Lima and C. D. Bahl, Discovery and engineering of enhanced SUMO protease enzymes, J. Biol. Chem., 2018, 293, 13224–13233, DOI:10.1074/jbc.RA118.004146.
- V. K. Mulligan, S. Workman, T. Sun, S. Rettie, X. Li, L. J. Worrall, T. W. Craven, D. T. King, P. Hosseinzadeh, A. M. Watkins, P. D. Renfrew, S. Guffy, J. W. Labonte, R. Moretti, R. Bonneau, N. C. J. Strynadka and D. Baker, Computationally designed peptide macrocycle inhibitors of New Delhi metallo-β-lactamase 1, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2012800118, DOI:10.1073/pnas.2012800118.
- S. J. Caldwell, I. C. Haydon, N. Piperidou, P. S. Huang, M. J. Bick, H. S. Sjöström, D. Hilvert, D. Baker and C. Zeymer, Tight and specific lanthanide binding in a de novo TIM barrel with a large internal cavity designed by symmetric domain fusion, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30362–30369, DOI:10.1073/pnas.2008535117.
- A. Quijano-Rubio, H. W. Yeh, J. Park, H. Lee, R. A. Langan, S. E. Boyken, M. J. Lajoie, L. Cao, C. M. Chow, M. C. Miranda, J. Wi, H. J. Hong, L. Stewart, B. H. Oh and D. Baker, De novo design of modular and tunable protein biosensors, Nature, 2021, 591, 482–487, DOI:10.1038/s41586-021-03258-z.
- D. D. Sahtoe, F. Praetorius, A. Courbet, Y. Hsia, B. I. M. Wicky, N. I. Edman, L. M. Miller, B. J. R. Timmermans, J. Decarreau, H. M. Morris, A. Kang, A. K. Bera and D. Baker, Reconfigurable asymmetric protein assemblies through implicit negative design, Science, 2022, 375, eabj7662, DOI:10.1126/science.abj7662.
- A. C. Hunt, J. B. Case, Y. J. Park, L. Cao, K. Wu, A. C. Walls, Z. Liu, J. E. Bowen, H. W. Yeh, S. Saini, L. Helms, Y. T. Zhao, T. Y. Hsiang, T. N. Starr, I. Goreshnik, L. Kozodoy, L. Carter, R. Ravichandran, L. B. Green, W. L. Matochko, C. A. Thomson, B. Vögeli, A. Krüger, L. A. VanBlargan, R. E. Chen, B. Ying, A. L. Bailey, N. M. Kafai, S. E. Boyken, A. Ljubetič, N. Edman, G. Ueda, C. M. Chow, M. Johnson, A. Addetia, M. J. Navarro, N. Panpradist, M. Gale, Jr., B. S. Freedman, J. D. Bloom, H. Ruohola-Baker, S. P. J. Whelan, L. Stewart, M. S. Diamond, D. Veesler, M. C. Jewett and D. Baker, Multivalent designed proteins neutralize SARS-CoV-2 variants of concern and confer protection against infection in mice, Sci. Transl. Med., 2022, 14, eabn1252, DOI:10.1126/scitranslmed.abn1252.
- S. Yao, A. Moyer, Y. Zheng, Y. Shen, X. Meng, C. Yuan, Y. Zhao, H. Yao, D. Baker and C. Wu, De novo design and directed folding of disulfide-bridged peptide heterodimers, Nat. Commun., 2022, 13, 1539, DOI:10.1038/s41467-022-29210-x.
- Z. Chen, S. E. Boyken, M. Jia, F. Busch, D. Flores-Solis, M. J. Bick, P. Lu, Z. L. VanAernum, A. Sahasrabuddhe, R. A. Langan, S. Bermeo, T. J. Brunette, V. K. Mulligan, L. P. Carter, F. DiMaio, N. G. Sgourakis, V. H. Wysocki and D. Baker, Programmable design of orthogonal protein heterodimers, Nature, 2019, 565, 106–111, DOI:10.1038/s41586-018-0802-y.
- G. Ueda, A. Antanasijevic, J. A. Fallas, W. Sheffler, J. Copps, D. Ellis, G. B. Hutchinson, A. Moyer, A. Yasmeen, Y. Tsybovsky, Y. J. Park, M. J. Bick, B. Sankaran, R. A. Gillespie, P. J. Brouwer, P. H. Zwart, D. Veesler, M. Kanekiyo, B. S. Graham, R. W. Sanders, J. P. Moore, P. J. Klasse, A. B. Ward, N. P. King and D. Baker, Tailored design of protein nanoparticle scaffolds for multivalent presentation of viral glycoprotein antigens, eLife, 2020, 9, e57659, DOI:10.7554/eLife.57659.
- M. Steinegger and J. Söding, MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets, Nat. Biotechnol., 2017, 35, 1026–1028, DOI:10.1038/nbt.3988.
- A. G. Green, H. Elhabashy, K. P. Brock, R. Maddamsetti, O. Kohlbacher and D. S. Marks, Large-scale discovery of protein interactions at residue resolution using co-evolution calculated from genomic sequences, Nat. Commun., 2021, 12, 1396, DOI:10.1038/s41467-021-21636-z.
- D. M. Taverna and R. A. Goldstein, Why are proteins so robust to site mutations?, J. Mol. Biol., 2002, 315, 479–484, DOI:10.1006/jmbi.2001.5226.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02233e |
| This journal is © The Royal Society of Chemistry 2024 |