
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCore-electron contributions to the magnetic response of molecules with heavy elements and their significance in aromaticity assessments†‡
Mesías
Orozco-Ic
 *a,
Luis
Soriano-Agueda
a,
Dage
Sundholm
b,
Eduard
Matito
*a and
Gabriel
Merino
*c
*a,
Luis
Soriano-Agueda
a,
Dage
Sundholm
b,
Eduard
Matito
*a and
Gabriel
Merino
*c
aDonostia International Physics Center (DIPC), 20018 Donostia, Euskadi, Spain
bDepartment of Chemistry, Faculty of Science, University of Helsinki, P.O. Box 55, A. I. Virtasen aukio 1, FIN-00014 Helsinki, Finland
cDepartamento de Física Aplicada, Centro de Investigación y de Estudios Avanzados, Unidad Mérida. Km 6 Antigua Carretera a Progreso. Apdo. Postal 73, Cordemex, 97310, Mérida, Yuc., Mexico. E-mail: mesias.orozco@dipc.org; ematito@dipc.org; gmerino@cinvestav.mx
First published on 8th July 2024
Abstract
This study delves into the magnetic response of core electrons and their influence on the global magnetic response of planar and three-dimensional systems containing heavy elements, employing the removing valence electron (RVE) approximation. We also explore electronic aromaticity indices to understand the potential role of core electrons on electron delocalization in the absence of an external perturbation. The study reveals that core electrons significantly contribute to the overall magnetic response, especially to the magnetic shielding, affecting the interpretation of aromaticity. In contrast, the calculation of the electronic aromaticity indices suggests a negligible participation of the core electrons on electron delocalization. Despite their widespread use, the study emphasizes caution in labeling systems as strongly aromatic based solely on shielding function computations. It is noteworthy to emphasize the limitations associated with each aromaticity criterion; particularly in the context of magnetic shielding function calculations, the core-electron effect contamination is undeniable. Hence, the integration of various criteria becomes imperative for attaining a comprehensive understanding of magnetic responses within complex systems.
Introduction
In recent years, aromaticity has been extensively used to understand the stability of organometallic or transition metal compounds containing heavy elements.1–10 Criteria based on the magnetic response of a molecule in the presence of an external magnetic field have become very popular among the experimental and theoretical communities due to their close relationship with nuclear magnetic resonance experiments.11,12 However, determining aromaticity of inorganic molecules from a magnetic perspective is challenging because of the multiple delocalization characters and the presence of heavy elements.13 As one moves down in the periodic table, several factors become critical in calculating the magnetic response, in particular the inclusion of relativistic corrections and spin–orbit (SO) effects for heavy element systems.14–19 Moreover, in non-planar molecules, the mixing of various orbitals can further complicate the task of separating the magnetic response into its orbital components.20,21It is crucial to recognize that the core electrons can significantly influence the magnetic response in molecules with heavy elements. This aspect has been previously highlighted, especially in widely used nucleus-independent chemical shift (NICS) calculations.22–24 The NICS25,26 index, computed at the center of the molecular rings, is strongly affected by the proximity of heavy nuclei, potentially leading to misinterpretations of aromaticity. This issue is particularly concerning for systems identified as having multiple types of aromaticity (σ, π, δ, etc.) based solely on NICS(0) values.27 A recent example is the K2[Th(C8H8)Cl2]3 cluster. While negative NICSzz values suggest σ-aromaticity;28 an in-depth current density analysis by Cuyacot and Foroutan-Nejad reveals non-aromatic character.29 Szczepanik further supported this by showing that the stability of K2[Th(C8H8)Cl2]3 arises from multi-center charge-shift bonding, not σ-aromaticity.30 His electron density of delocalized bonds31 (EDDB) calculations showed discontinuous isosurfaces on the Th–Th bond, indicating the lack of global delocalization along the Th3 ring, in line with the absence of a ring-current pattern from the current density calculations.29,30 This correlation between discontinuous EDDB isosurfaces and the absence of global ring currents has also been discussed in helicene-bridged systems.32 The core electrons may also shape the magnetically induced current density of systems with heavy elements.33 This can result in complex current–density pathways, making it difficult to determine whether the global currents are specific to ring currents or influenced by the heavy nuclei circulations. For example, a double three-dimensional magnetic aromaticity in icosahedral B12I12-based clusters with spherically concentric layers of boron and iodine, respectively, has been recently reported.34 Like icosahedral carbon fullerenes,35,36 it has been shown that they exhibit spherical-like currents, but in the case of B12I12-clusters, the spherical current is distorted by the local currents produced by the iodine atoms giving the impression of double concentric spherical currents, which could be misinterpreted as double 3D and concentrically aromatic,34 in agreement with the discontinuity shown in the EDDB isosurfaces of the iodine layer.34 Hence, the interpretation obtained from the magnetic criteria in all types of systems containing heavy elements is not trivial and the consideration of aromaticity criteria of a different nature can be insightful.32,37,38
Recently, we proposed an approach to obtain the magnetic response arising only from core electrons. This technique, removing the valence electrons (RVE) of a molecule and calculating its magnetic response,21 addresses inaccurate core-contribution values from orbital partitions, such as the natural chemical shielding39 (NCS) analysis. The RVE technique allows for straightforward corrections to both core- and σ-contributions to the magnetic shielding.21,40
For organic molecules, RVE calculations have shown that core-electron currents are primarily localized around the nuclei, exerting a negligible influence on magnetic shielding and ring-current strengths.21 The RVE approximation enables computation and visualization of the domain of these local core-electron (also called atomic) currents, but also estimates their strengths, which for carbon is approximately 4.4 nA T−1.21 Similar values have recently been reported for the methyl group of toluene at accurate levels of theory.41,42 Although the influence of core electrons due to strong local currents (and shielding) in heavy-nuclei systems is recognized,21,23,24,33 a quantitative description of these effects on the magnetic response is still lacking. Addressing this issue is of paramount importance for accurately determining and interpreting the aromaticity of inorganic systems.
In this study, we investigate the magnetic response of core electrons and its impact on the overall magnetic response of a series of systems containing heavy elements using the RVE approximation. Given the multidimensional nature of aromaticity,43 we also compute the electron delocalization between bonds via the delocalization index (DI)44,45 and expand our analysis to include electronic aromaticity indicators such as the Iring index46 and the multicenter index (MCI).47 Generally, discrepancies frequently arise between conclusions drawn from aromaticity criteria based on the magnetic response and electronic indices in systems with large rings and inorganic compounds.32,38 Therefore, we evaluate electron delocalization in the absence of an external field, thereby offering a way to determine whether the impact of core electrons affects response and intrinsic properties. For this study, we selected a variety of planar and three-dimensional molecular systems, each previously reported as highly aromatic and containing heavy elements (see Fig. 1). These systems include planar E42− (E = Al, Ga, In, Tl) clusters, icosahedral M122− (M = Ge, Sn, Pb) cages, the small planar Hf3 triangle,27,48 the organometallic platinabenzene,49,50 osmapyridinium51 and the monocationic osmapetalene,52 the planar pentacoordinate SrCs5+,53 and the cage-like Ge94− cluster.54 Given that the RVE approximation is valid in the presence of a sufficiently large energy gap between the lowest occupied σ-orbital and the highest occupied core-electron orbital (ΔLσ-Hcore),21 we also examine cases such as Ta3O3−, the gold-based Au3+ and W@Au12, and the case of K2[Th(C8H8)Cl2]3 (Fig. 1), which exhibit a ΔLσ-Hcore of less than 9 eV.
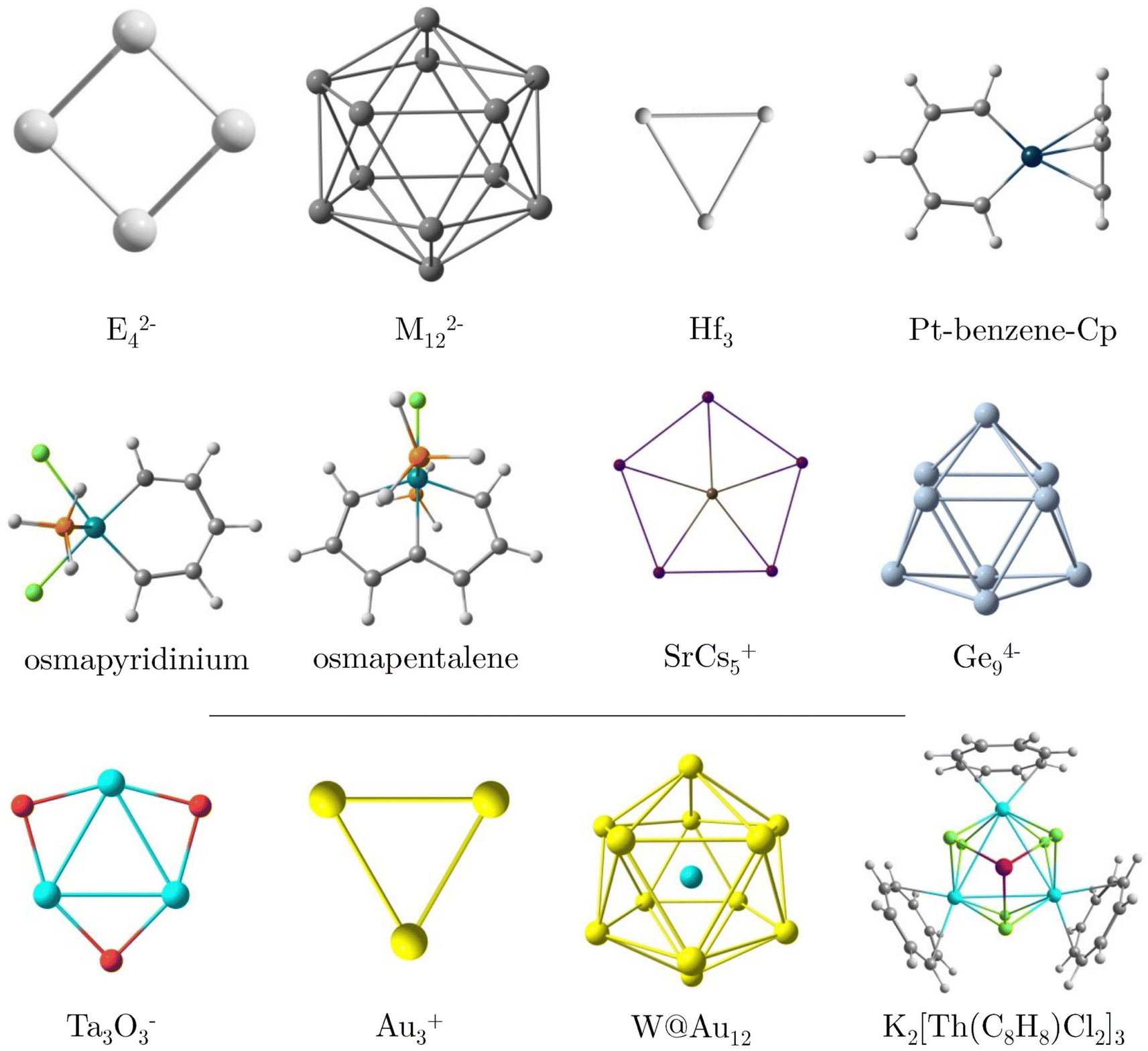 | ||
| Fig. 1 Molecules studied here. For E42−, E = Al, Ga, In, and Tl and for M122−, M= Ge, Sn, and Pb. | ||
Our computations show that in these inorganic systems, the magnetic response of core electrons can substantially influence the total system response, thereby impacting the chemical interpretation of aromaticity. This effect is particularly evident in magnetic shielding, which typically affects local indices such as NICS values. Furthermore, in systems with several heavy atoms, core electrons may contribute significantly to the total current–density, altering the values of ring-current strength in both planar and non-planar molecules. This aspect becomes critical when exploring systems characterized by multiple aromaticity.13 On the other hand, electronic indices for aromaticity suggest a consistent trend of negligible core-electron contribution to electron delocalization. Consequently, large shielding and ring-current strengths are often found in the realm of inorganic systems. However, determining and quantifying aromaticity through magnetic response calculations is a complex task that requires careful consideration of the substantial contributions from core electrons. The RVE approximation provides a simple and functional approximation for computing, excluding core-electron noise, but it is not universally applicable. Hence, caution is recommended when denoting a system as strongly aromatic based solely on shielding function computations.
Computational details
The inclusion of relativistic corrections plays a crucial role in determining the structural and magnetic response properties of molecules with elements beyond the sixth row in the periodic table.15–18,55,56 In this study, we incorporated relativistic and SO effects using the exact two-component theory57–61 (x2c) in the optimization. The TPSS62 functional, in conjunction with Grimme's dispersion correction with Beck-Johnson damping D3(BJ),63 was employed, along with the all-electron x2c-TZVPall-2c basis set for all atoms.64–66 The molecular geometries are provided in the ESI.‡ The magnetic response was assessed by computing the magnetically induced current density67–69 (Jind) and the induced magnetic field70–72 (Bind) using the GIMIC67–69 and Aromagnetic73 programs, respectively. Gauge-including atomic orbitals74,75 (GIAOs) within the Turbomole 7.7.1 software76 interface were employed for these calculations, including the x2c scalar-relativistic approach with the finite nucleus model based on a Gaussian charge distribution.77 The BHandHLYP78 functional was selected for both Jind and Bind calculations, as it has been shown to yield magnetic properties close to those obtained at the CCSD(T) level.79An external unit field (|Bext| = 1 T) parallel to the z-axis was applied, with the axis of the highest molecular symmetry. In planar molecules, the external field was oriented perpendicularly to the molecular plane. Under these circumstances, the z-component of Bind (Bzind) is typically the largest one, allowing the Bind analysis to focus on Bzind, which is equivalent to the zz-component of the nucleus-independent chemical shift (NICSzz).25,26,80 Aromagnetic was used to calculate z-profiles of NICS and Bzind, as well as two- and three-dimensional Bzind plots for visualizing the magnetic shielding in the molecular environment. GIMIC was employed to calculate Jind plots and ring-current strengths (Jind) by integrating the current density flowing through a plane intersecting one or more chemical bonds.67–69 The ring-current strength is commonly used to quantify aromaticity. Additionally, the current–density flux across different sections of the plane was determined by calculating the derivative of Jind (dJind/dr) relative to one of the plane coordinates (r).67–69 The integration plane extends eight bohr both above and below the molecular ring plane. The horizontal r-coordinate, starting at r = 0 on the horizontal axis at the molecular center, intersects the chemical bond and extends to the point where Jind vanishes. Ring-current strengths are reported in nA T−1, while NICS and Bzind are given in ppm.
To compute the core-electron contributions to Bind and Jind, the RVE approach was used.21 This involves calculating the magnetic response of highly charged molecules after removing their valence electrons, without relaxing the molecular structure.21 Consequently, the magnetic response of a system without its valence electrons corresponds to the magnetic response of the core electrons. The nature of the chemical bonding undergoes significant changes when the external magnetic field is of the order of 105 T,81,82 aligning with the criterion that ΔLσ-Hcore should be large enough to ensure that a small perturbation caused by a weak homogeneous external magnetic field does not modify the electronic structure of the core-electron orbitals. Simultaneously, this criterion guarantees an unambiguous differentiation between σ and core orbitals. Hence, ΔLσ-Hcore was reported for each molecule.
Electronic indices, such as Iring and the MCI, have been widely employed to estimate electron delocalization in planar molecules.83–85 The Iring index evaluates simultaneous electron sharing among various centers,46 with large positive values indicating aromaticity and negative MCI values suggesting antiaromatic behaviors in some cases.86 The MCI index,47 a generalization of Iring, considers delocalization both along and across the ring. These indices were calculated using the Bader87 partition performed by the AIMAll software,88 with atomic overlap matrices generated from Turbomole wavefunction files as input for the ESI-3D code.45,89 The accuracy of these calculations was ensured, with an electron count error of less than 0.01 a.u., and the maximum integrated Laplacian in the atomic domain below 10−4 a.u. These calculations were conducted at the same BHandHLYP/x2c-TZVPall-2c level, which is expected to reduce electron delocalization errors in both magnetic properties and electronic indices,90,91 and the contribution of core electrons to these indices was also obtained using the RVE approach. Furthermore, since ESI-3D enables direct dissection of electronic indices within its orbital contributions to planar molecules, we also tested the reliability of the RVE approximation by comparing their core-electron contributions with those obtained from ESI-3D's orbital decomposition in E42− systems obtained from the direct inspection of the orbital symmetry. In the case of K2[Th(C8H8)Cl2]3, all calculations were performed on a BHandHLYP level with an x2c-TZVPall-2c basis for all atoms except for Th, where the Stuttgart-Bonn relativistic effective-core potential and the associated segmented valence basis sets for thorium were used. These calcualtions were performed with Gaussian 16 using the geometry reported in ref. 30.
Results and discussion
E42− (E = Al, Ga, In, Tl)
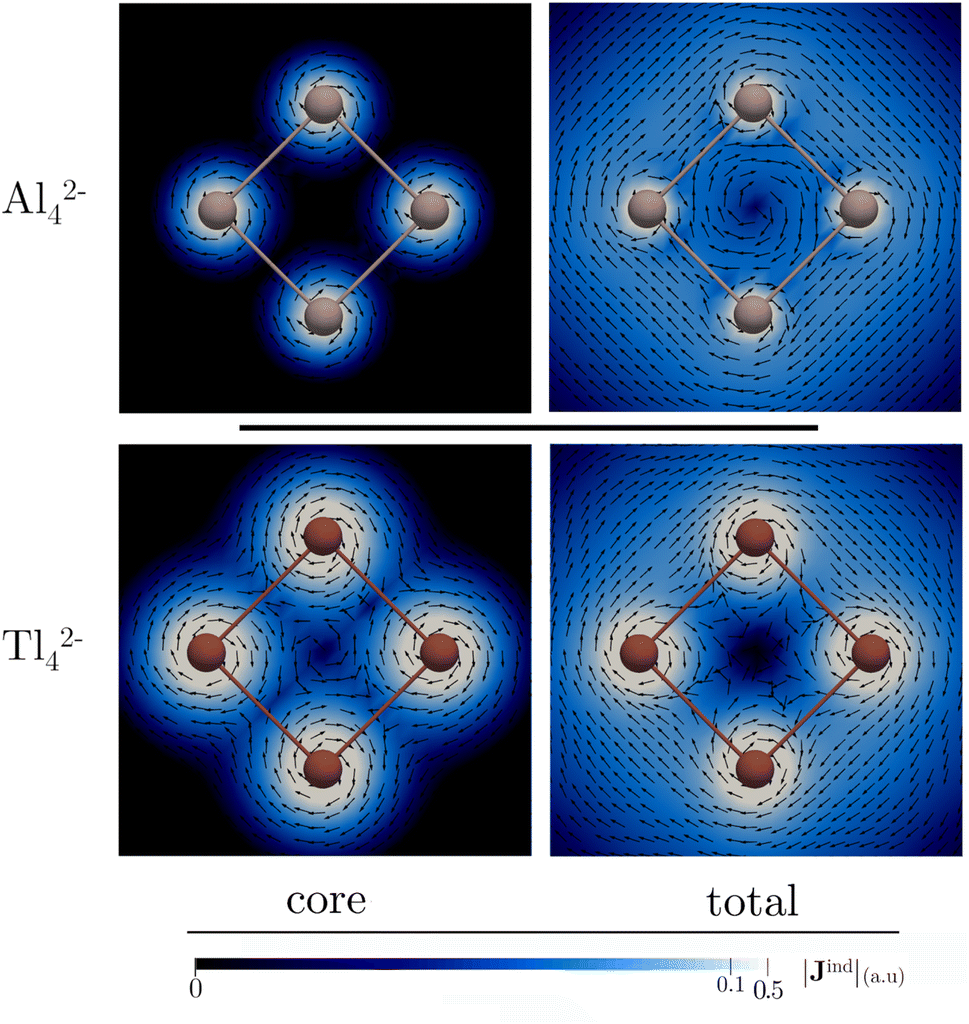
The detection in the gas phase of the Al42− cluster represents a pivotal advancement in the study of aromatic compounds.92 Its (σ + π)-aromatic character has been consistently confirmed through magnetic response calculations,93–97 particularly emphasizing the presence of a ring current flowing in a clockwise (diatropic) direction attributed to the delocalized σ-electrons, and to a lesser extent, π-electrons.93–95 This leads to pronounced σ- and π-shielding.96,97 Fowler and Havenith95 were the first to report the core-electron contribution to NICS(0) and Bzind(0) in the Al42− cluster, showing stronger values (0.1 and 2.7 ppm, respectively at the PW91 level) compared to benzene. However, the study did not extensively discuss the core-electron effects on the magnetic response.Fig. 2 depicts the contributions of core- and valence electrons to Bzind in the molecular plane and regions above and below it. The total Bzind shows a shielding cone over the ring, predominantly influenced by the σ-part. The RVE core-electron contribution reveals strong shielding around the Al nuclei, significantly affecting the total magnetic response. However, these effects are not directly associated with aromaticity since the core electrons generate local currents21,98 circulating around the nuclei. Interestingly, despite the visual prominence of core electrons on the Jind maps (Fig. 3), the integration of current density across an Al–Al bond yields a ring-current strength of 29.6 nA T−1 without core-electron contributions (Table 1). These core-electron currents can be visualized as small current loops circulating around the nuclei, leading to deshielded regions converging at the ring's center (Fig. 2). This phenomenon accounts for the presence of positive NICS and Bzind values at the ring center (Table 1) and along the z-axis (see Fig. S1 in ESI‡), consistent with the findings of Havenith and Fowler.95 Furthermore, we have calculated the ring-current strength profile across a plane intersecting an Al atom (Fig. S2‡). This essentially yields the same total value as integrating along the plane intersecting an Al–Al bond (28 nA T−1), as the core-electron current contributions effectively cancel out along the integration domain, resulting in a net zero-strength. The RVE approximation also allows an estimation of the core-electron current strength of 19 nA T−1 for Al, which is notably stronger than that of carbon in benzene (4.4 nA T−1).21
 | ||
| Fig. 2 Isolines of the core, valence, and total contributions to Bzind of Al42− and Tl42− plotted on (top) and in a slide orthogonal (bottom) to the molecular plane. These calculations were performed at the BHandHLYP/x2c-TZVPall-2c level. | ||
 | ||
| Fig. 3 Plots of the total and the core-electron contribution to Jind maps for Al42− and Tl42−. The arrows indicate the direction of the current density. The |Jind| scale is in atomic units (1 au = 100.63 nA T−1 Å−2). These calculations were performed at the BHandHLYP/x2c-TZVPall-2c level. | ||
| Molecule | ΔLσ-Hcore | Contribution | NICS(0) | B ind z (0) | J ind |
|---|---|---|---|---|---|
| Al42− | 70.16 | Total | −39.55 | −67.32 | 29.06 |
| Core | −0.12 | 3.0 | 0.0 | ||
| Valence | −39.43 | −70.32 | 29.06 | ||
| Ga42− | 15.62 | Total | −39.80 | −65.80 | 30.15 |
| Core | −0.55 | 12.72 | 0.10 | ||
| Valence | −39.25 | −78.52 | 30.05 | ||
| In42− | 15.02 | Total | −36.00 | −53.03 | 31.28 |
| Core | −0.29 | 17.71 | 0.13 | ||
| Valence | −35.71 | −70.75 | 31.15 | ||
| Tl42− | 11.15 | Total | −33.06 | −47.50 | 31.38 |
| Core | −0.87 | 23.76 | 0.37 | ||
| Valence | −32.19 | −71.26 | 31.01 |
Similar trends are found for Ga42− and In42−. In these instances, the core electrons shape the total shielding Bzind near the nuclei and in the molecular plane (Fig. S3‡). The current–density calculations reveal that the local nuclear loops cover large areas and display strong core-electron strengths of approximately 51 and 82 nA T−1, respectively (see Fig. S2 and S4‡). Nonetheless, their net impact on the ring-current strength is minimal, though not negligible (Table 1). This finding is further supported by the core-electron ring current strength profiles, which show slight fluctuations from Ga42− onwards (Fig. S2‡). This pattern indicates that core electrons make a small contribution to the global ring-current in these planar molecules containing heavy elements.
The Tl42− case (Fig. 3) is particularly noteworthy, as it exhibits a ring-current strength comparable to that in earlier examples (31.4 nA T−1). This value agrees well with that reported with a four-component Dirac–Coulomb treatment.16 Here, the core-electron contribution to Jind spans a large area due to the very strong core-electron currents flowing around Tl (134 nA T−1). The pronounced shielding cones near the nuclei and large positive Bzind values (>20 ppm) at the ring center are attributed to the formation of a deshielding cone over the ring (Fig. 2). The long-range deshielding effect exerted by core electrons extends beyond 1 Å above the ring along the z-axis (Fig. S1‡). Consequently, Bzind(1) (and even NICS(1)) calculations also reveal significant contamination from the local core-electron shielding, so, when quantifying aromaticity in these planar systems containing heavy atoms, it becomes more appropriate to calculate either the ring-current strength or the valence electron contribution to Bzind, aiming to mitigate the influence of core electrons.
To gain further insights into the impact of core electrons on electron delocalization in the absence of an external magnetic field, we computed the delocalization indices44,45 (DIs) for the E–E bonds of the clusters (Table 2). The DI quantifies the number of electron pairs shared between two atoms. In organic molecules like benzene, the total DI value for a C–C bond typically stands at 1.4, with 28% of this value attributed to π-electron delocalization99,100 and the remaining 72% to its σ-component. This aligns with the assumption that the core-electron contribution is negligible. For the E42− rings, the total DIs are approximately 1.08 (Al), 1.06 (Ga), 1.04 (In), and 1.01 (Tl). According to the RVE approach, the core-electron components are small in all cases (ranging from 0.0 to 0.12), which are practically the same values as those of the core-electron contributions provided by the ESI-3D orbital decomposition using the inspection of the orbital symmetry (Table 2). These findings are consistent with ring-current strength calculations, indicating that the total electron delocalization in the E–E bonds includes a small contribution from core electrons.
| Index | Contribution | Al42− | Ga42− | In42− | Tl42− |
|---|---|---|---|---|---|
| DI (E–E bond) | Total | 1.080 | 1.060 | 1.050 | 1.010 |
| Core | 0.000 (0.000) | 0.045 (0.000) | 0.060 (0.040) | 0.116 (0.078) | |
| Valence | 1.080 | 1.020 | 0.990 | 0.894 | |
| I ring | Total | 0.136 | 0.141 | 0.145 | 0.156 |
| Core | 0.000 (0.000) | 0.000 (0.002) | 0.000 (0.002) | 0.000 (0.005) | |
| Valence | 0.136 | 0.141 | 0.0145 | 0.156 | |
| MCI | Total | 0.366 | 0.381 | 0.394 | 0.434 |
| Core | 0.000 (0.000) | 0.000 (0.003) | 0.000 (0.005) | 0.000 (0.009) | |
| Valence | 0.366 | 0.381 | 0.394 | 0.434 |
Furthermore, the multicenter indices in the E42− clusters display aromatic characteristics, with total Iring and MCI values ranging between [0.14,0.16] and [0.37,0.43], respectively. As is common in inorganic compounds,38,101 these values are significantly larger than those reported for benzene (0.048, and 0.071, respectively).84 Additionally, it is also possible to compare normalized quantities in the electronic indices, taking their 1/n power (where n is the number of atoms).84 However, the trend remains the same with respect to benzene. Consistent with magnetic response computations, these findings also confirm the absence of a core-electron component (Table 2). Thus, while there is a minor contribution from core electrons to the delocalization in the E–E bonds, as indicated by DIs, it becomes evident that the core electrons do not play a significant role in contributing to the multicentric aromaticity.
M122− (M = Ge, Sn, Pb)
Icosahedral metal-spherenes, renowned for their highly symmetric structures and capacity to stabilize endohedral entities, have garnered attention for their potential three-dimensional aromaticity. The aromatic nature of these cages has been supported by NICS and Bzind calculations.102 Nonetheless, discussion of the impact of core electrons on the shielding effects within these systems has been relatively scarce, especially considering the small size of these metallic cages, which may lead to significant contamination of the NICS and Bzind values through contributions from core electrons inside the cage.Our calculations on Ge122−, Sn122−, and Pb122− reveal that these compounds show an inside paratropic and an outside spherical diatropic current, akin to all-carbon fullerenes.35,36 The core-electron contribution to the total current density is significant, particularly near the nuclei where the current density pathways are markedly distorted (Fig. 4). This is evident in the Bzind calculations, which show a considerable influence of core electrons on shielding. For example, in Ge122−, the shielding cones around the nuclei mask the deshielding effects from valence electrons within the interior of the cage, an effect not readily apparent from the total Bzind shielding alone (see Fig. 5). The influence of the core electron on Bzind values is less pronounced at the center of the cage but intensifies near the surface (see Fig. S5‡), indicating that the core-electron contribution strongly contaminates NICS and Bzind values calculated at the centre of the faces.
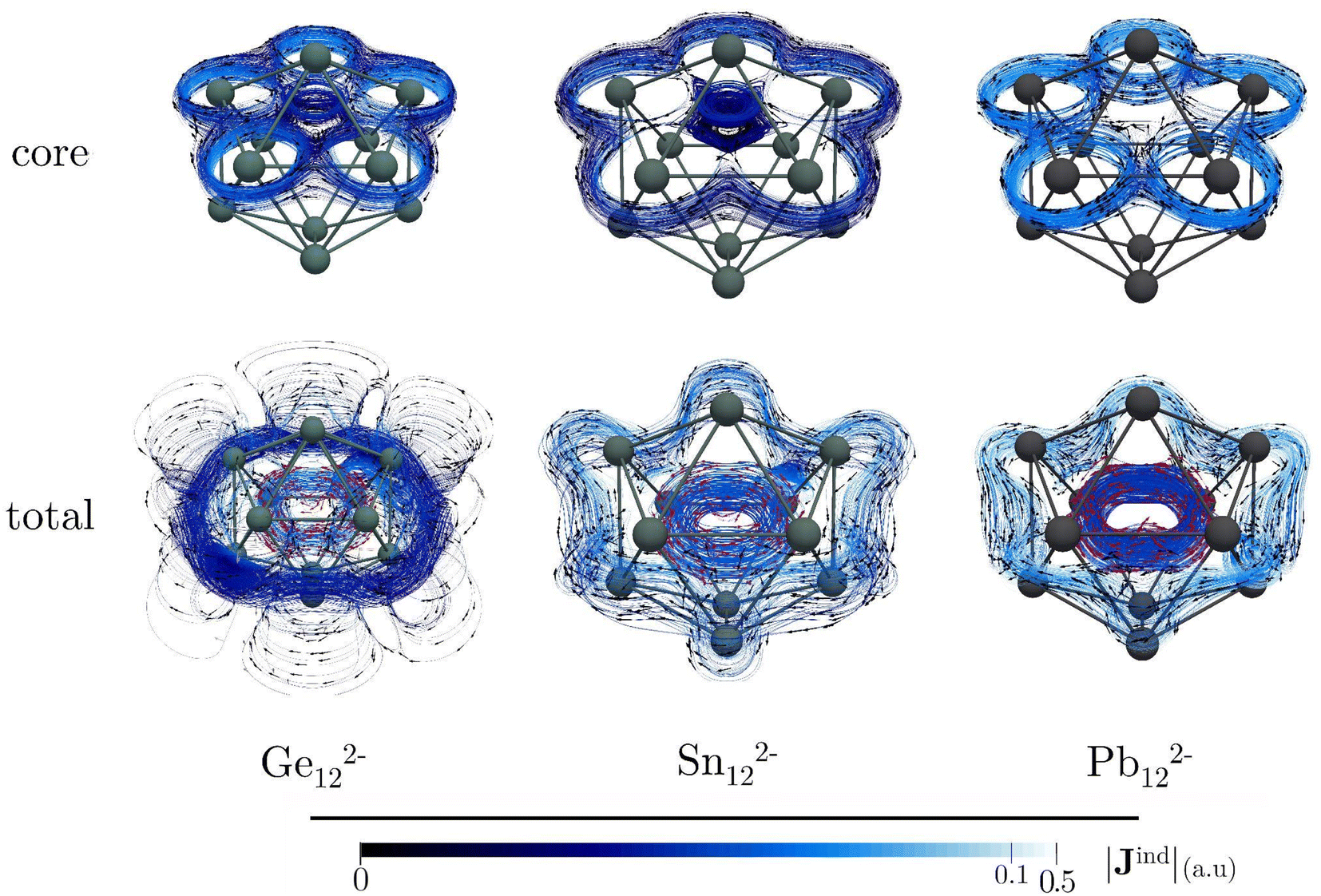 | ||
| Fig. 4 Plots of the total and the core-electron contribution to the Jind maps of the icosahedral M122− clusters. The outer diatropic and paratropic inner current flow on the current–density streamlines is represented by black and red arrows, respectively. The |Jind| scale is in atomic units (1 au = 100.63 nA T−1 Å−2). These calculations were performed at the BHandHLYP/x2c-TZVPall-2c level. | ||
 | ||
| Fig. 5 Isolines of the core, valence, and total contributions to Bzind for the icosahedral M122− clusters. These calculations were performed at the BHandHLYP/x2c-TZVPall-2c level. | ||
In icosahedral systems, the global ring-current strength is typically assessed by integrating Jind across a plane that begins at the symmetry C5-axis and extends until Jind vanishes, intersecting the icosahedral molecular surface in the process. This integration plane invariably intersects the nuclei at the icosahedral poles, whose para- and diatropic contributions do not cancel, leading to substantial core-electron contributions to the ring-current strength profiles (Fig. S6‡). The dJind/dr profiles reveal peaks between 2 and 3 Å, corresponding to intersections with an atom at one of the vertices. Unlike the initial decaying sharps attributed to the nuclei at the poles, the contribution from these peaks cancel when integrating the curren strength. However, note that the core-electron strengths are also diatropic but stronger than the total, indicating that the net ring-current strength derived from the valence electrons is paratropic (Table 3). Hence, it is mandatory to double-check the interpretation based on valence ring-current strengths, as they suggest antiaromatic features that diminish from Ge122− to Pb122−. Moreover, the Jind plots show an increase in the diatropic current outside the cages and a decrease in the paratropic current inside, a trend corroborated by the Bzind calculations. Thus, when analyzing non-planar entities, such as molecular cages containing heavy elements, it is imperative to discern the core-electron contributions from the total (shielding and ring-current strengths) to ensure a proper chemical interpretation.
| Molecule | ΔLσ-Hcore | Contribution | NICS(0) | B z ind(0) | J ind |
|---|---|---|---|---|---|
| Ge122− | 11.52 | Total | 7.33 [−10.01] | 7.32 [−11.64] | 66.55 |
| Core | 0.05 [−0.60] | 0.05 [3.80] | 102.95 | ||
| Valence | 7.28 [−9.41] | 7.28 [−15.44] | −36.40 | ||
| Sn122− | 11.45 | Total | −0.70 [−15.78] | −0.69 [−14.01] | 139.38 |
| Core | 0.20 [−0.54] | 0.20 [6.11] | 162.98 | ||
| Valence | −0.90 [−15.24] | −0.90 [−20.13] | −23.60 | ||
| Pb122− | 9.24 | Total | −19.91 [−26.14] | −19.92 [−23.80] | 256.41 |
| Core | −0.15 [−1.12] | −0.14 [7.30] | 268.07 | ||
| Valence | −19.76 [−25.02] | −19.77 [−31.10] | −11.66 |
In the case of non-planar molecules, our calculations of electronic criteria were focused on DI indices, given that the multicenter indices present significant computational challenges due to the myriad permutations involved. Table S1‡ shows that the DI values for the M–M bonds are approximately 0.57 for the three cages. The core-electron contributions account for 3.1%, 4.2%, and 8.17% of the total electron delocalization for Ge, Sn, and Pb, respectively. While these DI values hint at a modest core-electron contribution, such findings do not align directly with the insights obtained from magnetic response analyses, underscoring the complexity of accurately capturing the electronic behavior in these non-planar systems.
Hf3, Pt-benzene, SrCs5+, osmapyridinium, osmapentalene cations, and Ge94−
To explore the concept of higher-order multiple aromaticity, looking further into the periodic table is crucial, particularly focusing on transition metals with d-orbitals involved in chemical bonding. Averkiev and Boldyrev proposed the Hf3 cluster as the smallest planar homoatomic system exhibiting delocalization across σ-, π-, and δ-orbitals.27 This cluster, described as a triplet C3v with a multi-configurational character in its ground state, showcases a fascinating aspect of aromaticity.27,103,104 The closest singlet isomer, which adopts D3h symmetry but is stabilized by SO-effects,27,104 was highlighted for its triply aromatic nature. Bzind(0) and Bzind(1) calculations indicated strong aromaticity, but a contradictory shift in the sign from −50 ppm to 4 ppm was reported.27 An intriguing aspect of Hf3 is its compact ring size, with bond lengths of 2.65 Å, and the significant impact of heavy nuclei on magnetic shielding calculations, such as NICS(0). Multiple aromaticity and delocalized electrons in such systems suggest expectedly high shielding values and pronounced ring currents. However, the augmented number of electrons in the inner shells accentuates core-electron effects, challenging the interpretation of aromaticity based solely on local shielding calculations.At our level of theory, the singlet is just 6.6 kcal higher in energy than the triplet case. Our NICS(0) and Bzind(0) computations for Hf3 indicate large shielding values for both the triplet and the singlet state, especially for the singlet state (Table 4). The significant difference in magnitude between the NICS(0) and Bzind(0) values suggests that the x- and y-components, parallel to the plane at the center of the ring, are not negligible. Therefore, these results should not be interpreted as direct evidence of super-strong aromaticity in the metal clusters. Upon removing the core-electron response, the strongly diamagnetic features persist inside and above the ring in Bzind (Fig. 6). For the singlet, a combination of pronounced diamagnetic and paramagnetic responses appears over the ring (Fig. S7‡), explaining the transition from negative to positive NICSzz values along the z-axis, as reported by Averkiev and Boldyrev.27 The Bzind strengths for the singlet state of Hf3 are consistently strong in systems predisposed to open-shell features.105 The core electrons significantly shape the overall behavior of the current density around the nuclei (Fig. 7). Integrating the current–density across a Hf–Hf bond yields a diatropic strength of 9.82 and 6.55 nA T−1 for the triplet and singlet, respectively. These values do not surpass those for benzene, confirming that their aromaticity, as implied by NICS(0) and Bzind(0) values, is not remarkably large. Like the previous planar systems, the core-electron contribution to the ring-current strength is almost zero. This finding underscores that the shielding, strongly influenced by the local shielding cones of the heavy nuclei, is significantly affected. As expected, the responses from core electrons in the singlet and triplet structures are practically identical (Table 4). From a magnetic point of view, the triplet has an aromatic character when considering the valence set, whereas the singlet is non-aromatic. The alignment with electronic indices is consistent, showing aromatic features associated with both isomers and unveiling minor contributions from core electrons (Tables S2 and S3‡). The DI index in an Hf–Hf bond is 1.86 and 2.19 for triplet and singlet structures, respectively, highlighting a significant delocalization compared to benzene. The core-electron contributions for both cases are 0.059 (approximately 3.17 and 2.7%, respectively). Core electrons exhibit negligible contributions to the Iring and MCI indices, reinforcing that magnetic shielding of core electrons significantly distorts the magnetic interpretation of the aromaticity of metallic systems.
| Molecule | ΔLσ-Hcore | Contribution | NICS(0) | B z ind(0) | J ind |
|---|---|---|---|---|---|
| a Value computed in the center of a three-membered Cs–Sr–Cs ring. | |||||
| Hf3 | 19.86 (19.91) | Total | −841.21 (−21803.37) | −83.04 (−26.80) | 9.82 (6.55) |
| Core | −1.85 (−1.96) | 30.44 (31.03) | 0.04 (0.04) | ||
| Valence | −839.86 (−21801.41) | −113.48 (−57.83) | 9.78 (6.51) | ||
| Pt-benzene | 65.72 | Total | 14.74 | −6.30 | 8.44 |
| Core | 0.0 | 0.0 | 0.0 | ||
| Valence | 14.74 | −6.30 | 8.44 | ||
| Osmapyridinium | 43.27 | Total | −5.06 | −9.26 | 11.36 |
| Core | −0.10 | 4.21 | 0.0 | ||
| Valence | −5.16 | −5.05 | 11.36 | ||
| Osmapentalene | 47.57 | Total | −9.67 | −10.52 | 12.94 |
| Core | −0.14 | 4.71 | 0.0 | ||
| Valence | −9.81 | −5.81 | 12.94 | ||
| SrCs5+ | 9.05 | Total | −7.52a | −11.01a | 106.75 |
| Core | −2.42a | 13.58a | 83.78 | ||
| Valence | −5.10a | −24.59a | 22.97 | ||
| Ge94- | 10.87 | Total | −78.63 [−81.36] | −78.24 [−83.36] | 100.49 |
| Core | −0.18 [−0.79] | 0.54 [0.56] | 54.54 | ||
| Valence | −78.45 [−80.57] 100.49 | −78.78 [−83.92] | 45.95 | ||
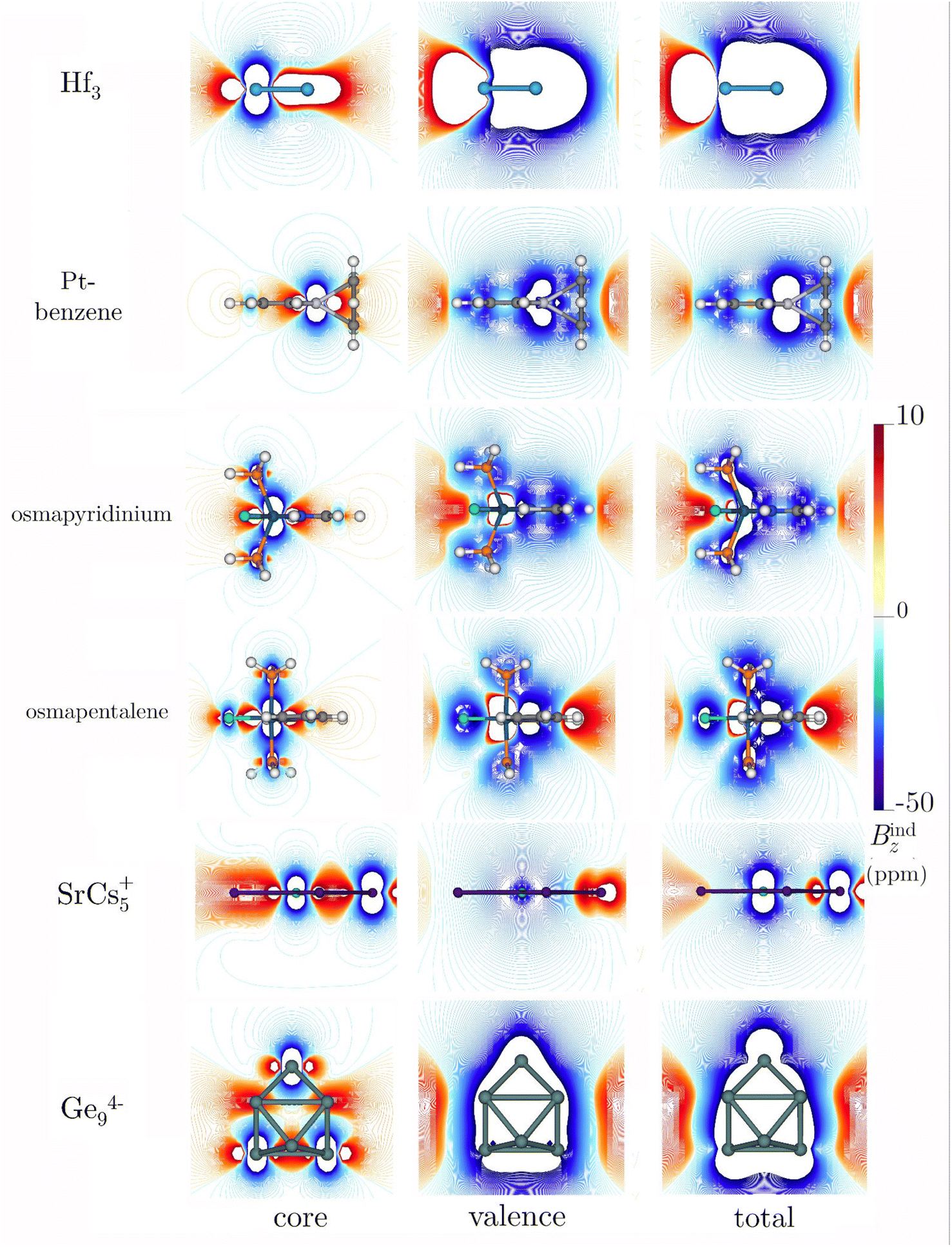 | ||
| Fig. 6 Isolines of the core, valence, and total contributions to Bzind of Hf3 (triplet), Pt-benzene, osmapyridinium, osmapentalene (monocation), SrCs5+, and Ge94− clusters. For Hf3 only, the color scale ranges from −100 to 100 ppm. These calculations were performed at the BHandHLYP/x2c-TZVPall-2c level. | ||
 | ||
| Fig. 7 Plots of the total and the core-electron contribution to Jind maps for Hf3 (triplet), Pt-benzene, osmapyridinium, osmapentalene (monocation), SrCs5+, and Ge94− clusters. The arrows indicate the direction of the current density. The paratropic inner current flow is represented by red arrows. The |Jind| scale is in atomic units (1 au = 100.63 nA T−1 Å−2). These calculations were performed at the BHandHLYP/x2c-TZVPall-2c level. | ||
The exploration of aromaticity in metallabenzene rings, particularly those containing heavy metals, initiated a critical examination of the impact of core-electron contamination on magnetic responses.22–24 The case of platinabenzene is especially intriguing due to the significant influence of SO effects, which account for nearly 70% of its ring-current strength.49 Our findings indicate that these effects are predominantly localized around the platinum atom (Fig. 6 and 7), leading to the conclusion that the net ring-current strength attributable to core electrons is zero. Similar to other planar rings containing a single heavy atom, osmapyridinium and osmapentalene monocations have been suggested to be aromatic in both their ground and triplet states.51,52 However, previous studies raised concerns about the reliability of magnetic criteria for these systems due to the presence of the Os atom.51,52 Our calculations indicate that these can indeed be considered aromatic. While the RVE approach shows that core-electron contributions to Bzind(0) are significantly stronger than those for platinabenzene, the integration across a C–C bond reveals zero contribution from the core electrons in both cases (Table 4). This is evident from Fig. 6 and 7, where the primary interference from core electrons is localized near the Os nucleus. Thus, for molecules with a single heavy atom on the ring, it appears sufficient to avoid an integration near its area of influence to obtain an accurate velue for the ring current. This approach effectively eliminates the misleading influence of core electrons and provides a more reliable assessment of aromaticity in such systems. Furthermore, various electronic indices corroborate this, indicating a negligible contribution from electron delocalization (Tables S2 and S3‡).
In contrast, the scenario presented by SrCs5+, featuring a heavy atom at its center, starkly differs.53 In such molecules, the presence of a central atom introduces a domain of core-electron current that can extend concentrically away from the nucleus, aligning with the ring current. This alignment is particularly pronounced in clusters with heavy nuclei. Consequently, our analysis reveals a substantial core-electron contribution to the NICS and Bzind values both in and above the molecular plane (Fig. 6). The Jind plots further illustrate the significant role of core electrons within the molecular ring, particularly around the central Sr atom (Fig. 7). This underscores the critical need to carefully consider or explicitly exclude core-electron currents when evaluating ring-current strength to avoid misinterpreting the aromatic or antiaromatic nature of the molecule.
Addressing this challenge requires using dJind/dr profiles, which allow the monitoring of Jind variations across the integration plane, thereby avoiding the influence of central current domains. This approach has been successfully applied in the analysis of planar hypercoordinate atoms106 and the [Th@Bi12]4− cluster.4 However, accurately defining the domain of each current–density pathway, as exemplified by the case of SrCs5+, remains a complex task.
We computed the core and total ring-current profiles originating from the ring center of SrCs5+, specifically from the Sr atom and extending across a Cs–Cs bond (Fig. 8). The initial decaying peak observed in this plane is attributed to the core-electron current circulating Sr. Note that the profiles of both the core and total ring currents exhibit a high degree of similarity, eventually vanishing at long distances (r > 10 Å) from the Cs–Cs bond. It becomes challenging to distinguish between the core-electron contribution and the ring-current strength originating in the peripheral five-membered ring, particularly due to the significant strength values associated with Sr. To address this challenge, we employed the RVE approach, which proved invaluable in quantifying the effect of this central core-electron current whose effect is barely present within the range of 0 and 2 Å (Fig. 8). This results in a core-electron strength of 83.78 nA T−1. By subtracting this value from the total ring-current strength (106.75 nA T−1), we derived a more refined estimation of the ring current attributable to valence electrons, approximately 23 nA T−1. This calculation suggests substantial aromaticity of the system.
 | ||
| Fig. 8 The dJind/dr profiles of the core-electron (RVE) contribution and total (all-electron) using a plane that starts at the Sr atom and intersects the Cs–Cs bond calculated at the BHandHLYP/x2c-TZVPall-2c level. The small inset box provides an enlarged view of the same plots between 0.5 and 3.5 Å. | ||
However, accurately determining the ring-current strength in molecules featuring central atoms presents notable challenges. It becomes imperative to disregard the effect of the central current to avoid overestimating electron delocalization. Our analysis, complemented by electronic indices, indicates small electron delocalization characteristics of SrCs5+ compared to benzene. Importantly, these indices confirm the absence of core-electron contributions to electron delocalization (as detailed in Tables S2 and S3‡), highlighting the relevance of dissecting the contributions of the core and valence electrons in such complex systems with an atom at ring centers.
Another nice example with remarkable shielding is Ge94−. This cluster is of interest because large negative NICSzz values at the cage center could indicate σ-aromaticity,54 but it also suggest a potential strong core-electron contamination in magnetic shielding. Upon computing NICS(0) and Bzind(0) both at the center and on one of the faces of the cage, it was noted that the core-electron contribution to the magnetic shielding is weak. Although the Bzind plots indicate a strong negative core-electron response around the nuclei (Fig. 6), they also reveal a deshielding region with positive Bzind values inside the cage, similar to the M122− clusters previously discussed. Nonetheless, the aromatic character of Ge94− is confirmed by the dominant diamagnetic response of the valence orbitals, which significantly influences the system's overall magnetic shielding. Jind maps further show a strong diatropic ring current near the cage surface, with an important core-electron contribution around the nuclei. Similar to the cases above, the presence of a central atom in the integration plane requires the elimination of core-electron contamination to quantify ring-current strength accurately. Thus, applying the RVE approach allows for a more precise quantification of electron delocalization in three-dimensional systems where atoms intersect the integration plane. This approach confirms a significant global current of approximately 46 nA T−1 attributed to valence electrons, reinforcing the concept of three-dimensional global aromaticity in this cluster. The total DI values for Ge94− suggest electronic delocalization comparable to, but not exceeding, that of benzene, with non-zero contributions from the nucleus across different bonds, albeit not surpassing 3.1% of the total value.
J ind maps reveal a strong diatropic ring current near the surface of the cage, influenced by the core-electron contribution around the nuclei (Fig. 7). Similar to the cases of SrCs5+ and M122− clusters, the presence of a central atom in the integration plane contains the central core-electron contribution when quantifying the ring-current strength. Consequently, employing the RVE approach to separate the core component offers an effective means to quantify electron delocalization in three-dimensional systems where intersecting atoms are present.107 This analysis reveals a significant global current of approximately 46 nA T−1 attributable to the valence electrons, confirming the presence of three-dimensional global aromaticity. In line with earlier cage-like structures, the total DI values for Ge94− fall between 0.24 and 0.74, indicating that electron delocalization is well below that of benzene. Like the M122− clusters, Ge94− exhibits non-zero nuclear contributions, which vary from bond to bond but do not exceed 3.1% of the total value. Therefore, it appears that the contribution of core electrons to the delocalization slightly increases in non-planar molecules (Table S2‡).
Ta3O3−, Au3+, W@Au12, and K2[Th(C8H8)Cl2]3
We have already shown the utility of the RVE approach in elucidating the role of core electrons in electron delocalization. The ΔLσ-Hcore gap dramatically decreases as one descends the periodic table, raising questions about the applicability of RVE as a reliable indicator of the core-electron component. During the course of our study we noticed that unreliable behaviors in the estimation of core-electron effects appeared in both the magnetic response and the electronic indices when ΔLσ-Hcore is less than 9 eV. Indeed, the systems we have discussed, displaying a ΔLσ-Hcore > 9 eV, allow for a distinction between core- and σ-orbitals, yielding significant insights. There are also trends when we extend to other clusters with small ΔLσ-Hcore. For example, the Ta3O3− cluster, known to exhibit δ-aromaticity with highly negative shielding values,108,109 has not been analyzed regarding ring-current strength or core-electron contribution. Our computations of the total magnetic response identified a pattern of strong local diatropic currents surrounding the Ta atoms, diverging from a typical ring-current type path (Fig. S8‡). Remarkable deshielding cones are located around the Ta atoms, with a zone of pronounced shielding occurring at the ring's center, explaining the extremely large values of NICS(0) and Bzind(0) (Fig. S9‡). Calculating the total ring-current strength yielded a strong value (ca. 49 nA T−1), supporting the notion of strong aromaticity. However, with a ΔLσ-Hcore of merely 4.7 eV, RVE current–density plots show an ill-defined irregular pattern (Fig. S8‡), with a ring-current strength of 224.93 nA T−1 (Table 5). Hence, the predictability and utility of the RVE approach in this context are questionable.| Molecule | ΔLσ-Hcore | Contribution | NICS(0) | B z ind(0) | J ind |
|---|---|---|---|---|---|
| Ta3O3− | 4.66 | Total | −174.12 | −50.02 | 4.53 |
| Core | −15025.58 | −44277.42 | 224.93 | ||
| Au3+ | 3.84 | Total | −33.40 | 8.90 | 9.96 |
| Core | 27.12 | 250.27 | −27.43 | ||
| W@Au12 | 2.39 | Total | −89.83 | −144.83 | 519.00 |
| Core | −43.27 | 3.86 | 927.78 |
Examining gold-based clusters also highlights limitations in the RVE approach, especially concerning core electrons. Au3+ and W@Au12 (ref. 110–113) are classified as aromatic, but both exhibit small ΔLσ-Hcore values (see Table 4), making the differentiation between core and valence-electron contributions challenging (Table 5). Au3+, characterized as σ-aromatic through NICS calculations,110 presents a diamagnetic response at NICS(0), yet shows a paramagnetic response at Bzind(0) (Table 5), underscoring the complexity of determining overall magnetic behavior solely from these metrics. The Bzind isolines reveal that the ring is shielded when subjected to an external magnetic field (Fig. S8‡). In line with this, Jind maps expose a total diatropic flux, albeit with highly pronounced local circulations due to the presence of Au atoms (Fig. S9‡). However, the RVE approach in Au3+ yields somewhat irregular maps for Jind and Bzind that lack clear patterns of core-electron currents, showing strong paratropic circulation along the bonds. Additionally, a strong paratropic circulation is observed flowing on the bonds. The integration of current density yields contrasting ring strengths for the total (9.96 nA T−1) and RVE (−27.43 nA T−1) calculations. Also, the Bzind(0) corresponding to the RVE is remarkably paramagnetic, indicating the RVE's inadequacy in isolating core-electron effects accurately. The applicability due to a small ΔLσ-Hcore gap has also been discussed in other gold-based planar systems, where an option to obtain an estimate of the ring-current strength is achieved by using an integration plane intersecting a gold nucleus, given that the para- and diatropic contributions due to core-electron currents are almost canceled.114
The scenario is further complicated in W@Au12, where the large nucleus within the cage amplifies core-electron contributions to the magnetic response. With a ΔLσ-Hcore of merely 2.39 eV, the RVE approach fails to distinguish core-electron currents; instead, Jind patterns suggest a strong, ring-current-like circulation and paratropic circulation near the central atom. The substantial total ring-current strength (519 nA T−1), exacerbated by intersections with various nuclei, is significantly overestimated by the RVE calculation (>900 nA T−1), showing the inapplicability of this approach for accurately assessing core-electron impacts in such systems.
This analysis also underscores the limitations of the RVE approach in dealing with systems where small ΔLσ-Hcore values obscure the distinction between core and valence-electron contributions. The resulting discrepancies, particularly in aromaticity assessment, emphasize the need for cautious interpretation of RVE-derived data in complex, heavy-element clusters. The reported delocalization and electronic indices further question the RVE's efficacy, with values exceeding expected contributions and sometimes surpassing the total DIs.
Based on our results, we now turn to the magnetic response of the K2[Th(C8H8)Cl2]3 cluster, a system with debated aromaticity.28 Unfortunately, RVE is inapplicable to this cluster due to its small ΔLσ-Hcore value (<1 eV at our level). However, considering that the Th3 ring consists solely of heavy elements, the reliability of local NICS(0) and Bzind(0) calculations for aromaticity assessment becomes questionable. In this study, these calculations yield values of −20.98 and −16.38 ppm, respectively. Even the values obtained at 1 and 2 Å above the Th3 ring might be influenced by the proximity of potassium core electrons (see Fig. S10‡). Consistent with Cuyacot and Foroutan-Nejad's results,29 the current–density pathways suggest a non-aromatic pattern, exhibiting local circular currents around the cores (Fig. S10‡). Estimating the Th3 ring ring-current strength is challenging as the integration plane may contain core-electron contributions from the potassium atoms. Cuyacot and Foroutan-Nejad acknowledged this by employing a plane intersecting a Th–Th bond and extending 1 Å above and 1 Å below the Th3 ring, leading to small negative values.29 Following this approach, we obtained a ring-current strength value of 2.04 nA T−1 at our level of theory, further supporting the non-aromatic nature of the system. The small DI values (0.27) in the Th–Th bonds (Table S4‡) also reinforce this conclusion. Increasing the integration plane height significantly increases the ring-current strength due to the inclusion of potassium core-electron currents. This highlights the importance of caution when choosing the integration plane to avoid incorporating contributions from neighboring atom core electrons.
Conclusions
We have examined the aromaticity through the lens of magnetic response in a range of planar and non-planar molecules containing heavy elements under an external magnetic field; we delved into the intricacies of core electrons' influence on these responses. Using the removing valence electrons (RVE) approximation, we aimed to elucidate the impact of core electrons on magnetic phenomena. Furthermore, we calculated multicenter electronic indices (Iring and MCI) to evaluate aromaticity, exploring the role of core electrons in electron delocalization in the presence and absence of external perturbations, focusing on their influence on magnetic responses. Our key findings are summarized as follows:1. Across all examined systems, the magnetic response attributable to core electrons predominantly manifests as local atomic current loops encircling the nuclei, resulting in extensive magnetic shielding cones. There is a clear increasing trend with respect to the size of the atom. This effect is particularly pronounced in systems featuring heavy atoms, especially from the fourth row onwards of the periodic table where atomic core-electron currents and their resultant shielding are accentuated. These local shielding effects of core electrons are significantly extended beyond the nuclei, especially in molecules with multiple heavy elements or those with a central atom embedded within a ring structure. Such phenomena profoundly affect local indices like NICS(0) and Bzind(0), and even NICS(1) and Bzind(1), indicating that reliance solely on magnetic shielding functions for conclusions is not recommended without supplementary criteria.
2. Assessing current densities highlights the significant role that core-electron effects play in shaping the overall current density profile. Moreover, determining ring-current strength is essential for quantifying the extent of core electron contributions. In instances such as E42− rings with heavier elements, minor core-electron contributions to the ring current were observed, potentially misleading interpretations of strong aromaticity. This emphasizes the importance of focusing on valence-electron values to assess aromaticity accurately.
3. For non-planar structures, particularly those cage-like molecules, quantifying aromaticity through ring-current strengths is challenging. The unavoidable intersection of integration planes with nuclei leads to substantial core-electron contributions, necessitating qualitative and quantitative analyses to interpret current–density pathways and shielding behaviors accurately.
4. The use of electronic indices to evaluate electronic delocalization has proven invaluable, suggesting that the effects of core electrons directly impact the magnetic response rather than other estimators of aromaticity. The DI, Iring, and MCI indices consistently showed minimal core-electron contributions across all systems, reinforcing the traditional association of aromaticity with valence electrons. Thus, discussions on aromaticity in metallic clusters are best conducted concerning valence magnetic responses. So, electronic aromaticity indices offer a valuable tool for studying delocalization in systems containing heavy elements. These indices are particularly advantageous because they demonstrably exclude contributions from core electrons, which can lead to misinterpretations in other approaches.
5. Lastly, our study utilized the RVE approach for analyzing core electrons' magnetic properties. This method, while applicable, requires careful application, mainly because it is useful only when there is a significant energy gap (>9 eV) between the lowest occupied σ orbital and the highest occupied core-electron orbital. Unfortunately, accurate estimation of core electron magnetic behaviors can be elusive, as demonstrated in the cases of Ta3O3− and gold-based clusters, where magnetic responses were poorly defined. This also implies acknowledging the limitations inherent in each aromaticity criterion; particularly regarding shielding function calculations, the contamination of the core-electron effects is undeniable. In such scenarios, incorporating various criteria is imperative for a comprehensive understanding of the magnetic responses in complex systems. Therefore, it is emphasized that being able to rule out the effects of core-electrons on the magnetic response to address aromaticity correctly is critical, reaffirming this recommendation for molecules containing heavy elements from the fourth row onwards of the periodic table. The RVE approach provides an unsophisticated option, but it should be noted that it is only a tool that works under certain conditions.
A recent report during our manuscript's peer-review described an all-metal σ-aromatic Bi4 ring system.10 The authors presented calculations of the current density on the Bi44+ cluster.10 Intrigued by this work, we applied the RVE approach to assess the magnetic response of this cluster. Integrating the current–density across a Bi–Bi bond, yielded a ring-current strength of 9.7 nA T−1 with a zero core-electron RVE contribution. We believe our findings hold significance for the qualitative and quantitative diagnosis of complex systems with heavy elements. These systems are of considerable interest within the growing field of aromaticity research and the broader chemical community.
Data availability
Extra data (Fig. S1–S10 and Tables S1–S4‡), and the Cartesian coordinates for all compounds are provided in the ESI‡ accompanying this paper.Author contributions
M. O.-I. and G. M. conceived the idea of the project. M. O.-I. and D. S. performed the current densities and ring-current strengths calculations. M. O.-I. performed the calculations of the induced magnetic field. L. S.-A. performed the calculations of the electronic indices. All authors discussed the results. M. O.-I., E. M., and G. M. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the computational resources and technical and human support provided by DIPC and the SGI/IZO-SGIker UPV/EHU. M. O.-I. acknowledges funding from the Juan de la Cierva Program. The authors also thank the DIPC for a research grant (DIPC_INV_003132). Grants PID2022-140666NB-C21 funded by MCIN/AEI/10.13039/501100011033 and “FEDER Una manera de hacer Europa”, and the grants funded by the Gobierno Vasco (IT1584-22, and PIBA_2023_1_0055) are also acknowledged. The work in Finland was supported by the Academy of Finland through project 340583 as well as by the Swedish Cultural Foundation in Finland. The authors also acknowledge the CSC–IT Center for Science, Finland, for computational resources.References
- A. I. Boldyrev and L.-S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 11589–11605 CAS.
- D. K. Seo and J. D. Corbett, Science, 2001, 291, 841–842 CrossRef CAS.
- C. Liu, I. A. Popov, Z. Chen, A. I. Boldyrev and Z.-M. Sun, Chem.–Eur. J., 2018, 24, 14583–14597 CrossRef CAS PubMed.
- A. R. Eulenstein, Y. J. Franzke, N. Lichtenberger, R. J. Wilson, H. L. Deubner, F. Kraus, R. Clérac, F. Weigend and S. Dehnen, Nat. Chem., 2021, 13, 149–155 CAS.
- D. Y. Zubarev, B. B. Averkiev, H.-J. Zhai, L.-S. Wang and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 257–267 RSC.
- F. Bigi, D. Cauzzi, N. Della Ca’, M. Malacria, R. Maggi, E. Motti, Y. Wang and G. Maestri, ACS Org. Inorg. Au, 2022, 2, 373–385 CrossRef CAS PubMed.
- X. Lin, W. Wu and Y. Mo, J. Am. Chem. Soc., 2023, 145, 8107–8113 CrossRef CAS.
- Y.-H. Xu, W.-J. Tian, A. Muñoz-Castro, G. Frenking and Z.-M. Sun, Science, 2023, 382, 840–843 CrossRef CAS.
- J. M. Mercero, A. I. Boldyrev, G. Merino and J. M. Ugalde, Chem. Soc. Rev., 2015, 44, 6519–6534 RSC.
- R. Yadav, A. Maiti, M. Schorpp, J. Graf, F. Weigend and L. Greb, Nat. Chem., 2024 DOI:10.1038/s41557-024-01530-z.
- R. H. Mitchell, Chem. Rev., 2001, 101, 1301–1315 CrossRef CAS PubMed.
- J. A. Pople, J. Chem. Phys., 1956, 24, 1111 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, in Nanoclusters – A Bridge across Disciplines, Elsevier, 2010, pp. 219–267 Search PubMed.
- L. Alvarado-Soto, L. Alvarez-Thon, R. Ramirez-Tagle and J. Math, Chem, 2014, 52, 1182–1190 CAS.
- L. Alvarez-Thon and N. Inostroza-Pino, J. Comput. Chem., 2018, 39, 862–868 CrossRef CAS.
- L. Alvarez-Thon and W. Caimanque-Aguilar, Chem. Phys. Lett., 2017, 671, 118–123 CrossRef CAS.
- R. Pino-Rios, A. Vásquez-Espinal, L. Alvarez-Thon and W. Tiznado, Phys. Chem. Chem. Phys., 2020, 22, 22973–22978 RSC.
- M. Orozco-Ic, J. Barroso, R. Islas and G. Merino, ChemistryOpen, 2020, 9, 657–661 CrossRef CAS PubMed.
- A. Muñoz-Castro and R. Arratia-Perez, Chem. Phys. Rev., 2023, 4, 021313 CrossRef.
- G. Monaco, F. F. Summa and R. Zanasi, J. Chem. Inf. Model., 2021, 61, 270–283 CrossRef CAS PubMed.
- M. Orozco-Ic, N. D. Charistos, A. Muñoz-Castro, R. Islas, D. Sundholm and G. Merino, Phys. Chem. Chem. Phys., 2022, 24, 12158–12166 RSC.
- C. Foroutan-Nejad, Theor. Chem. Acc., 2015, 134, 8 Search PubMed.
- Z. Badri, S. Pathak, H. Fliegl, P. Rashidi-Ranjbar, R. Bast, R. Marek, C. Foroutan-Nejad and K. Ruud, J. Chem. Theory Comput., 2013, 9, 4789–4796 CrossRef CAS.
- C. Foroutan-Nejad, J. Vícha and A. Ghosh, Phys. Chem. Chem. Phys., 2020, 22, 10863–10869 RSC.
- P. von R. Schleyer, P. von Ragué Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Ragué Schleyer, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef PubMed.
- B. B. Averkiev and A. I. Boldyrev, J. Phys. Chem. A, 2007, 111, 12864–12866 CrossRef CAS PubMed.
- J. T. Boronski, J. A. Seed, D. Hunger, A. W. Woodward, J. van Slageren, A. J. Wooles, L. S. Natrajan, N. Kaltsoyannis and S. T. Liddle, Nature, 2021, 598, 72–75 CrossRef CAS PubMed.
- B. J. R. Cuyacot and C. Foroutan-Nejad, Nature, 2022, 603, E18–E20 CrossRef CAS.
- D. W. Szczepanik, Angew Chem. Int. Ed. Engl., 2022, 61, e202204337 CrossRef CAS.
- D. W. Szczepanik, M. Andrzejak, J. Dominikowska, B. Pawełek, T. M. Krygowski, H. Szatylowicz and M. Solà, Phys. Chem. Chem. Phys., 2017, 19, 28970–28981 RSC.
- M. Orozco-Ic, L. Soriano-Agueda, S. Escayola, D. Sundholm, G. Merino and E. Matito, J. Org. Chem., 2024, 89, 2459–2466 CrossRef CAS.
- G. Periyasamy, N. A. Burton, I. H. Hillier and J. M. H. Thomas, J. Phys. Chem. A, 2008, 112, 5960–5972 CrossRef CAS.
- J. Poater, S. Escayola, A. Poater, F. Teixidor, H. Ottosson, C. Viñas and M. Solà, J. Am. Chem. Soc., 2023, 145, 22527–22538 CAS.
- M. Orozco-Ic and D. Sundholm, Phys. Chem. Chem. Phys., 2022, 24, 22487–22496 RSC.
- M. P. Johansson, J. Jusélius and D. Sundholm, Angew Chem. Int. Ed. Engl., 2005, 44, 1843–1846 CrossRef CAS PubMed.
- Q. Zhu, S. Chen, D. Chen, L. Lin, K. Xiao, L. Zhao, M. Solà and J. Zhu, Fundam. Res., 2023, 3, 926–993 CAS.
- M. Solà, F. Feixas, J. O. C. Jiménez-Halla, E. Matito and J. Poater, Symmetry, 2010, 2, 1156–1179 Search PubMed.
- J. A. Bohmann, F. Weinhold and T. C. Farrar, J. Chem. Phys., 1997, 107, 1173–1184 CrossRef CAS.
- E. Steiner and P. W. Fowler, Phys. Chem. Chem. Phys., 2004, 6, 261–272 RSC.
- M. Dimitrova, D. Jia, J. Manz and D. Sundholm, Phys. Rev. A, 2022, 106, 042801 Search PubMed.
- D. Jia, M. Dimitrova, Y. Man, D. Sundholm and Y. Yang, Phys. Rev. A, 2022, 106, 042802 CrossRef.
- M. Solà, Front. Chem., 2017, 5, 22 Search PubMed.
- X. Fradera, M. A. Austen and R. F. W. Bader, J. Phys. Chem. A, 1999, 103, 304–314 CrossRef CAS.
- E. Matito, M. Solà, P. Salvador and M. Duran, Faraday Discuss., 2007, 135, 325–345 RSC.
- M. Giambiagi, M. S. de Giambiagi and K. C. Mundim, Struct. Chem., 1990, 1, 423–427 CrossRef CAS.
- P. Bultinck, R. Ponec and S. Van Damme, J. Phys. Org. Chem., 2005, 18, 706–718 CrossRef CAS.
- H.-J. Zhai, B. B. Averkiev, D. Y. Zubarev, L.-S. Wang and A. I. Boldyrev, Angew Chem. Int. Ed. Engl., 2007, 119, 4355–4358 CrossRef.
- D. Arias-Olivares and D. Páez-Hernández, New J. Chem., 2022, 46, 16708–16716 RSC.
- V. Jacob, T. J. R. Weakley and M. M. Haley, Angew Chem. Int. Ed. Engl., 2002, 41, 3470–3473 CrossRef CAS.
- T. Shen, D. Chen, L. Lin and J. Zhu, J. Am. Chem. Soc., 2019, 141, 5720–5727 CrossRef CAS PubMed.
- D. Chen, D. W. Szczepanik, J. Zhu and M. Solà, Chem.–Eur. J., 2020, 26, 12964–12971 CrossRef CAS PubMed.
- M.-H. Wang, A. J. Kalita, M. Orozco-Ic, G.-R. Yan, C. Chen, B. Yan, G. Castillo-Toraya, W. Tiznado, A. K. Guha, S. Pan, G. Merino and Z.-H. Cui, Chem. Sci., 2023, 14, 8785–8791 RSC.
- N. V. Tkachenko and A. I. Boldyrev, Chem. Sci., 2019, 10, 5761–5765 RSC.
- R. Ramírez-Tagle, L. Alvarado-Soto, A. Villavicencio-Wastavino and L. Alvarez-Thon, Phys. Chem. Chem. Phys., 2016, 18, 25751–25755 RSC.
- M. Rauhalahti, S. Taubert, D. Sundholm and V. Liégeois, Phys. Chem. Chem. Phys., 2017, 19, 7124–7131 RSC.
- W. Liu and D. Peng, J. Chem. Phys., 2009, 131, 031104 CrossRef.
- D. Peng and M. Reiher, Theor. Chem. Acc., 2012, 131, 1081 Search PubMed.
- D. Peng and M. Reiher, J. Chem. Phys., 2012, 136, 244108 CrossRef.
- D. Peng, N. Middendorf, F. Weigend and M. Reiher, J. Chem. Phys., 2013, 138, 184105 CrossRef.
- M. Filatov, W. Zou and D. Cremer, J. Chem. Phys., 2013, 139, 014106 CrossRef.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- Y. J. Franzke, R. Treß, T. M. Pazdera and F. Weigend, Phys. Chem. Chem. Phys., 2019, 21, 16658–16664 RSC.
- Y. J. Franzke, L. Spiske, P. Pollak and F. Weigend, J. Chem. Theory Comput., 2020, 16, 5658–5674 CrossRef CAS PubMed.
- Y. J. Franzke and C. Holzer, J. Chem. Phys., 2023, 159, 184102 CrossRef CAS PubMed.
- J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC.
- D. Sundholm, H. Fliegl and R. J. F. Berger, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS.
- G. Merino, T. Heine and G. Seifert, Chem.–Eur. J., 2004, 10, 4367–4371 CrossRef CAS PubMed.
- T. Heine, R. Islas and G. Merino, J. Comput. Chem., 2007, 28, 302–309 CrossRef CAS PubMed.
- R. Islas, T. Heine and G. Merino, Acc. Chem. Res., 2012, 45, 215–228 CrossRef CAS PubMed.
- M. Orozco-Ic, J. L. Cabellos and G. Merino, Aromagnetic, Cinvestav-Mérida, Mexico., 2016 Search PubMed.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- Y. J. Franzke, C. Holzer, J. H. Andersen, T. Begušić, F. Bruder, S. Coriani, F. Della Sala, E. Fabiano, D. A. Fedotov, S. Fürst, S. Gillhuber, R. Grotjahn, M. Kaupp, M. Kehry, M. Krstić, F. Mack, S. Majumdar, B. D. Nguyen, S. M. Parker, F. Pauly, A. Pausch, E. Perlt, G. S. Phun, A. Rajabi, D. Rappoport, B. Samal, T. Schrader, M. Sharma, E. Tapavicza, R. S. Treß, V. Voora, A. Wodyński, J. M. Yu, B. Zerulla, F. Furche, C. Hättig, M. Sierka, D. P. Tew and F. Weigend, J. Chem. Theory Comput., 2023, 19, 6859–6890 CrossRef CAS.
- L. Visscher and K. G. Dyall, At. Data Nucl. Data Tables, 1997, 67, 207–224 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- S. Lehtola, M. Dimitrova, H. Fliegl and D. Sundholm, J. Chem. Theory Comput., 2021, 17, 1457–1468 CrossRef CAS.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS.
- K. K. Lange, E. I. Tellgren, M. R. Hoffmann and T. Helgaker, Science, 2012, 337, 327–331 CrossRef CAS.
- E. I. Tellgren, T. Helgaker and A. Soncini, Phys. Chem. Chem. Phys., 2009, 11, 5489–5498 RSC.
- I. Casademont-Reig, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, Molecules, 2020, 25, 711 CAS.
- E. Matito, Phys. Chem. Chem. Phys., 2016, 18, 11839–11846 RSC.
- F. Feixas, E. Matito, J. Poater and M. Solà, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- R. Ponec, P. Bultinck and A. G. Saliner, J. Phys. Chem. A, 2005, 109, 6606–6609 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press on Demand, 1994 Search PubMed.
- T. A. Keith, AIMAll (Version 14.11.23), 2014 Search PubMed.
- E. Matito, M. Duran and M. Solà, J. Chem. Phys., 2005, 122, 14109 CrossRef.
- I. Casademont-Reig, R. Guerrero-Avilés, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, Angew Chem. Int. Ed. Engl., 2021, 60, 24080–24088 CrossRef CAS PubMed.
- I. Casademont-Reig, T. Woller, J. Contreras-García, M. Alonso, M. Torrent-Sucarrat and E. Matito, Phys. Chem. Chem. Phys., 2018, 20, 2787–2796 RSC.
- X. Li, A. E. Kuznetsov, H.-F. Zhang, A. I. Boldyrev and L.-S. Wang, Science, 2001, 291, 859–861 CrossRef CAS PubMed.
- Y.-C. Lin, J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2005, 122, 214308 CrossRef PubMed.
- P. W. Fowler, R. W. A. Havenith and E. Steiner, Chem. Phys. Lett., 2002, 359, 530–536 CrossRef CAS.
- R. W. A. Havenith and P. W. Fowler, Phys. Chem. Chem. Phys., 2006, 8, 3383–3386 RSC.
- Z. Chen, C. Corminboeuf, T. Heine, J. Bohmann and P. R. Schleyer, J. Am. Chem. Soc., 2003, 125, 13930–13931 CrossRef CAS.
- R. Islas, T. Heine and G. Merino, J. Chem. Theory Comput., 2007, 3, 775–781 CrossRef CAS.
- M. Dimitrova and D. Sundholm, Current density, current–density pathways, and molecular aromaticity, in Aromaticity: Modern Computational Methods and Applications, Elsevier, 2021, pp. 155–194 Search PubMed.
- E. Matito, F. Feixas and M. Solà, Theochem, 2007, 811, 3–11 CrossRef CAS.
- F. Feixas, E. Matito, M. Solà and J. Poater, Phys. Chem. Chem. Phys., 2010, 12, 7126–7137 RSC.
- F. Feixas, E. Matito, J. Poater and M. Solà, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 105–122 CAS.
- A. Castro, E. Osorio, J. O. C. Jiménez-Halla, E. Matito, W. Tiznado and G. Merino, J. Chem. Theory Comput., 2010, 6, 2701–2705 CrossRef CAS PubMed.
- H. Wang, Z. Hu, H. Haouari, R. Craig, Y. Liu, J. R. Lombardi and D. M. Lindsay, J. Chem. Phys., 1997, 106, 8339–8343 CrossRef CAS.
- D. Dai, S. Roszak and K. Balasubramanian, Chem. Phys. Lett., 1999, 308, 495–502 CrossRef CAS.
- M. Orozco-Ic, A. Restrepo, A. Muñoz-Castro and G. Merino, J. Chem. Phys., 2019, 151, 014102 CrossRef PubMed.
- G. Castillo-Toraya, M. Orozco-Ic, E. Dzib, X. Zarate, F. Ortíz-Chi, Z.-H. Cui, J. Barroso and G. Merino, Chem. Sci., 2021, 12, 6699–6704 RSC.
- L.-X. Bai, J. Barroso, M. Orozco-Ic, F. Ortiz-Chi, J.-C. Guo and G. Merino, Chem. Commun., 2023, 59, 4966–4969 RSC.
- H.-J. Zhai, B. B. Averkiev, D. Y. Zubarev, L.-S. Wang and A. I. Boldyrev, Angew Chem. Int. Ed. Engl., 2007, 46, 4277–4280 CrossRef CAS.
- S.-D. Li, C.-Q. Miao and J.-C. Guo, Eur. J. Inorg. Chem., 2008, 2008, 1205–1209 CrossRef.
- Y. Li, V. Oliveira, C. Tang, D. Cremer, C. Liu and J. Ma, Inorg. Chem., 2017, 56, 5793–5803 CrossRef CAS PubMed.
- Y. Gao, S. Bulusu and X. C. Zeng, Chemphyschem, 2006, 7, 2275–2278 CrossRef CAS.
- M. P. Johansson, J. Vaara and D. Sundholm, J. Phys. Chem. C Nanomater. Interfaces, 2008, 112, 19311–19315 CrossRef CAS.
- J. Autschbach, B. A. Hess, M. P. Johansson, J. Neugebauer, M. Patzschke, P. Pyykkö, M. Reiher and D. Sundholm, Phys. Chem. Chem. Phys., 2004, 6, 11–22 RSC.
- S. Das, S. Sinha, G. Roymahapatra, M. Orozco-Ic, G. Chandra De and S. Giri, New J. Chem., 2024, 48, 4765–4771 RSC.
Footnotes |
| † Paper dedicated to Miquel Solà on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Fig. S1–S9, Tables S1–S4, and Cartesian coordinates of the optimized molecular structures. See DOI: https://doi.org/10.1039/d4sc02269f |
| This journal is © The Royal Society of Chemistry 2024 |